

Background.
The causes and circumstances surrounding death are poorly studied in patients with portopulmonary hypertension (PoPH). We sought to determine the specific reasons for dying and characteristics surrounding this process in patients with PoPH.
Methods.
All deaths of patients with PoPH followed in the Cleveland Clinic Pulmonary Vascular Program were prospectively reviewed by the pulmonary hypertension team between 1996 and 2020.
Results.
A total of 69 patients with PoPH (age 56.0 ± 8.9 y), with 49% females, were included. Causes of death were available in 52 (75%) patients, of these PoPH either directly or indirectly contributed to death in 13 of 52 (25%) of patients, meanwhile 39 of 52 (75%) of the patients died because of progressive liver disease and its related complications. Decompensated liver disease was the leading cause of death in this cohort 20 of 52 (38%), whereas 19 of 52 (37%) died because of conditions associated with liver disease. About half, 36 of 69 (52%) of patients died in a healthcare environment and 23 of 36 (64%) during a hospitalization at Cleveland Clinic. A total of 59 of 69 (74%) of patients received pulmonary arterial hypertension (PAH)-specific therapies. Six patients died after liver transplantation (in 3 death was related to PAH-related complications). Most of the patients in this cohort of PoPH patients were considered unsuitable for liver transplantation for a variety of reasons. Advanced healthcare directives were available in only 28% of patients.
Conclusions.
Most patients with PoPH died because of complications of their liver disease. PAH directly or indirectly contributed to death in a third of them. A quarter of them did not receive PAH-specific therapy before their death.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a chronic progressive disease characterized by elevated pulmonary vascular resistance (PVR) that eventually leads to right heart failure and premature death.1 Portopulmonary hypertension (PoPH) is a type of PAH associated with portal hypertension.2 PoPH is noted in 5%–6% of patients with liver cirrhosis undergoing orthotopic liver transplant (OLT) evaluation.3-5 The presence of PoPH is associated with worse outcomes such as higher liver transplant waitlist mortality.6 In addition, patients with PoPH have worse outcomes during transplant surgery, therefore a thorough evaluation, with careful consideration of risks and benefits, is performed before offering this life-saving intervention.7-9 Patients with PoPH are treated with PAH-specific therapies, with the goal of reducing the risk of PAH complications, improving functional parameters and hemodynamics, and ultimately facilitating OLT.7
Based on the United States Registry to Evaluate Early and Long-term PAH Disease Management (REVEAL Registry), patients with PoPH have worse 2- and 5-y survival compared with idiopathic or familial PAH (67% versus 85% and 40% versus 64%, respectively).9 This poor survival appears to improve with OLT. In fact, a series of patients with PoPH who underwent OLT (most continued to receive treatment for PAH) showed an 8-y survival rate of 85%.10 A cohort of patients with PoPH from the United States and France demonstrated a 3-y survival rate post-OLT of 63% and 77%, respectively.11,12 However, little is known about the natural history of patients with PoPH who are not able to get an OLT and subsequently die.
It remains unclear whether patients with PoPH die predominantly because of complications of their liver or pulmonary vascular disease. We have previously shown that most patients with PAH die from complications associated with their PAH,1 but this may be different in patients with PoPH. Despite significant advances in the diagnosis and treatment of PoPH, the cause and mode of death remain unexplored. This paucity of data stems from the rarity of the disease, relative short follow-up of clinical studies, and the predominant focus in OLT. We are in a good position to evaluate the reasons and circumstances of death in patients with PoPH because over the years, we have prospectively examined the cause of death in a relatively large number of patients with PoPH. We hypothesize that the majority of patients with PoPH die because of their advanced liver disease in a healthcare environment.
MATERIALS AND METHODS
This was a retrospective analysis of prospective data collected from February 1996 until April 2020 at the Cleveland Clinic, Ohio. Institutional review board approval was obtained and informed consent was waived. All patients had documented portal hypertension and precapillary PH defined by a mean pulmonary artery pressure (mPAP) ≥25 mm Hg with pulmonary artery wedge pressure ≤15 mm Hg and PVR ≥3 Wood units (WU).13
Patients underwent extensive investigations to exclude other causes of PAH, including blood work, pulmonary function, and 6-min walk tests, chest radiograph, ventilation-perfusion scan, echocardiography, and right heart catheterization. We performed further evaluations when tests were either positive or inconclusive. Using these data, 2 PH physicians (the primary PH physician plus a second reviewer) determined the cause of PAH. If disagreement occurred, consensus was achieved after a formal presentation in a weekly PH meeting.
Assessment of the Cause of Death in PoPH
The Cleveland Clinic Pulmonary Vascular team routinely reviews all deaths of patients with PH as part of quality improvement, using a structured Mortality Review Form. For this study, a minimum of 2 PH physicians and a PH nurse discussed and reached consensus on the likely cause of death.1 Relevant information was obtained from medical records, specialty pharmacies, and conversations with primary care physicians and family members.
Other Information Collected
We reviewed all patients’ medical records to collect pertinent clinical information during the last outpatient visit or the time of last hospitalization, for patients that died in the outpatient or inpatient settings, respectively. We recorded New York Heart Association functional class, use of intravenous (IV) prostacyclin analogs, liver transplant listing status, presence of advance directives, use of vasopressors or mechanical ventilation, advanced decision on receiving cardiopulmonary resuscitation (CPR), and choice to withhold active treatments (comfort care) before death. We also recorded the PH-specific treatments before death. If the patient opted for comfort or palliative care, we considered the PAH regimen provided immediately before this change in status.
Statistical Analysis
Continuous data are presented as mean ± SD or median (interquartile range [IQR]) as appropriate. Categorical data are summarized as discrete values and percentages (n, %). We used t test and Mann-Whitney U test as appropriate for comparison of continuous variables. The statistical analyses were performed using the statistical package IBM SPSS, version 22 (IBM; Armonk, NY).
RESULTS
Patient Characteristics
During the study period, we identified a total of 109 patients with PoPH, of whom 40 (37%) were alive and therefore excluded. Of the 40 patients that were excluded, 5 received liver transplantation and were alive at the time of the analysis. A total of 69 (63%) patients with PoPH died, including 6 patients who underwent OLT but continued to have PAH after this intervention. Mean age ± SD at the time of death was 56.0 ± 8.9 y, of whom 35 (50.7%) were males and 57 (83%) of White race. New York Heart Association functional class was III or IV in 54 (78%) patients (Table 1). All patients had precapillary PH; however, 17 (25%) had combined precapillary and postcapillary PH. One subject did not have PoPH on initial RHC but developed this condition over time.
TABLE 1.
Baseline patient characteristics
| Mean ± SD, median (IQR), n (%) | |
|---|---|
| N | 69 |
| Age at the time of death (y) | 56.0 ± 8.9 y |
| Gender (female) | 34 (49.3%) |
| Race White | 57 (83%) |
| Black | 9 (13%) |
| Hispanic/Latino | 2 (3%) |
| Asian/Pacific Islander | 1 (1%) |
| BMI (kg/m2) | 31 ± 7.5 |
| Interval between last clinic visit and death (mo) | 5.5 (1.0–12.0) |
| NYHA FC at last clinic visit or hospitalization | |
| I | 1 (1%) |
| II | 14 (21%) |
| III | 43 (62%) |
| IV | 11(16%) |
| Last BNP or NT-proBNP level | |
| BNP pg/mL (n = 43) | 135 (44–370) |
| NT-proBNP pg/mL (n = 14) | 1069.5 (340–4716) |
| Time from last BNP or NT-proBNP level (mo) | |
| BNP pg/mL (n = 43) | 3.0 (0.83–13) |
| NT-proBNP pg/mL (n = 14) | 4.5 (1.0–18.8) |
| Oxygen use (yes) | 9 (13%) |
| SpO2 (n = 68) | 95% (93–98) |
| Last 6MWD (m) (n = 51) | 306 (228–390) |
| Time last 6MWD to death (mo) (n = 51) | 9.0 (3.0–22.0) |
| REVEAL 2.0 score at the last clinic visit/hospitalization (n = 32) | 12.5 (9.2–14.0) |
| Last DLCO (% predicted) (n = 47) | 59 ± 18 |
| Number of all-cause hospitalizations in the 6 mo before death | 2.1 ± 2.0 |
6MWD, 6-min walk distance; BMI, body mass index; BNP, brain natriuretic peptide; DLCO, diffusing capacity for carbon monoxide; FC, functional capacity; IQR, interquartile range; NT-proBNP, N-terminal Probrain natriuretic peptide; NYHA, New York Heart Association; REVEAL, Registry to Evaluate Early and Long-term PAH Disease Management score version 2.0.
Median time between last office visit and death was 5.5 (IQR, 1.0–12) mo. REVEAL 2.0 score calculation (with the limitation that not all variables were available for all patients) at last clinic visit revealed a median score of 12.5 (IQR, 9.5–14), with all patients in the high-risk category. The median time from last echocardiography to death was 3 (IQR, 0.8–12.5) mo. The last echocardiography before death showed a right ventricular (RV) systolic pressure of 65 ± 22 mm Hg. RV function was normal in 28 of 69 (41%). Meanwhile, RV dysfunction was mild, moderate, and severe in 19 (28%), 12 (17%), and 10 (14%) patients, respectively. Of the 28 patients with normal RV function, 3 died of gastroenterology bleeding, 6 died of decompensated liver disease, 3 of sepsis, 2 with PoPH (including 1 with sudden circulatory death attributed to PH), and 12 had no available information. In patients with mild RV dysfunction, PH contributed to death in 1 patient, PH was directly related to death in another patient, and PH was unrelated to death in 3 patients (11 patients had no available information). In patients with moderate RV dysfunction, cause of death was directly related to PH in 2 patients and unrelated to PH in 4 (8 patients had not available information). In patients with severe RV dysfunction, PH was not related to death in 2 and contributed to death in 1 patient (8 patients had no available information on the cause of death).
The median time from the last RHC and death was 8 mo (IQR, 3.0–32.5). Table 2 shows pulmonary hemodynamic determinations at the time of PoPH diagnosis and last RHC. Mean PAP decreased by 2.4 ± 11.9 mm Hg (n = 54) and PVR by 0.76 ± 2.68 WU (n = 52) from diagnostic RHC to last RHC. Median survival from diagnostic RHC and death was 24 mo (IQR, 9.5–44.5) and from last RHC and death 8 mo (IQR, 3.0–32.5).
TABLE 2.
Hemodynamic determinations in patients with PoPH
| Baseline hemodynamics | Hemodynamics closest to the time of death | |
|---|---|---|
| Mean ± SD or median (IQR) | Mean ± SD or median (IQR) | |
| Time from RHC to death (mo) | 24 (IQR 9.5–44.5) | 8 (IQR 3.0–32.5) |
| RAP (mm Hg) (n = 67) | 10.5 ± 5.9 | 11.0 ± 6.7 |
| Mean PAP (mm Hg) (n = 69) | 46.7 ± 11.3 | 44.7 ± 11.6 |
| PAWP (mm Hg) (n = 68) | 13.7 ± 5.8 | 13.9 ± 5.5 |
| TD CO (L/min) (=69) | 6.3 ± 2.3 | 6.9 ± 2.8 |
| TD CI (n = 69) | 3.2 ± 1.0 | 3.5 ± 1.4 |
| PVR (WU) (n = 69) | 5.9 ± 2.8 | 5.5 ± 3.3 |
| MvO2 (n = 60) | 67.2 ± 9.4 | 68.3 ± 9.6 |
| TPG (n = 68) | 32.8 ± 11.7 | 30.2 ± 12.4 |
| DPG (n = 67) | 18.8 ± 9.3 | 17.5 ± 10.2 |
DPG, diastolic pulmonary gradient; IQR, interquartile range; MvO2, mixed venous oxygen saturation; PAP, pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PoPH, portopulmonary hypertension; PVR, pulmonary vascular resistance; RAP, right atrial pressure; RHC, right heart catheterization; TDCI, thermodilution cardiac index; TDCO, thermodilution cardiac output; TPG, transpulmonary gradient; WU, Wood units.
Before death, PAH-specific treatment included: phosphodiesterase-5 inhibitors (PDE5i) in 35 (51%) (sildenafil in 32 and tadalafil in 3), riociguat in 1, endothelin receptor antagonist in 6 (ambrisentan in all cases), and drugs targeting prostacyclin pathway in 22 (32%) patients (intravenous epoprostenol in 17 and treprostinil in 5 patients (delivered subcutaneously in 3, intravenously in 1, and orally in 1 patient). A total of 41 (60%) patients received PAH monotherapy, whereas 10 (14%) received dual combination treatment. Of patients on combination therapy, 3 were treated before the year 2010, while the rest after this year.
There were 32 of 69 (46%) patients with a mPAP > 45 mm Hg on the last RHC before their death (9 were not on PAH-specific treatment). Meanwhile, 20 patients had a mPAP between 35 and 45 mm Hg (4 patients were not on PAH-specific treatment) and 17 had a mPAP < 35 mm Hg (5 were not on PAH-specific treatment). Overall, a total of 18 (26%) patients did not receive PAH-specific therapy before death. In 4 of these patients, PoPH directly contributed to death (mPAP > 45 mm Hg in 2, and mPAP between 35 and 45 mm Hg in another 2). Interestingly, of the patients in whom PoPH was unrelated to the cause of death, 11 patients had a mPAP > 45 mm Hg and 9 had mPAP < 35 mm Hg). Among patients with combined pre- and postcapillary PH, 2 died of sepsis/hemorrhagic shock, 2 of progressive liver disease, and 1 of right heart failure.
Liver Disease in Patients With PoPH
All patients had cirrhosis, and the cirrhosis was associated with alcohol in 27 (39%), hepatitis C in 12 (17%), nonalcoholic steatohepatitis in 8 (12%), and other causes in 22 (32%) (Table 3). Comorbidities were common, including diabetes mellitus in 24 (35%), coronary artery disease in 4 (6%), atrial fibrillation in 9 (13%), and obesity in 36 (52%) patients. Only 21 (30%) patients were listed for OLT (Table 3). Six patients underwent OLT and continued to have PAH postliver transplant with a mPAP of 41.8 ± 6.8 mm Hg, pulmonary artery wedge pressure of 11.0 ± 3.0 mm Hg, cardiac output of 5.5 ± 2.4 L/min, and PVR of 6.5 ± 2.1 WU. The median duration from liver transplantation to death was 64 mo (IQR, 29.0–88.0).
TABLE 3.
Type of the liver disease and its complications
| Variables | Mean ± SD, median (IQR) or n (%) |
|---|---|
| Etiology of liver disease (n = 69) | |
| Alcohol cirrhosis | 27 (39%) |
| Hepatitis C cirrhosis | 12 (18%) |
| Cryptogenic cirrhosis | 10 (15%) |
| NASH cirrhosis | 8 (12%) |
| Primary biliary cirrhosis | 5 (7%) |
| Autoimmune hepatitis | 2 (3%) |
| Primary sclerosis cholangitis | 2 (3%)1 (1%) |
| Biliary cirrhosis | |
| Congenital hepatic fibrosis | 1 (1%) |
| Wilson’s disease | 1 (1%) |
| Presence of ascites (n = 68) | 50 (73%) |
| Bleeding varices (n = 68)Nonbleeding varices (n = 68) | 26 (38%)16 (23%) |
| Hepatic encephalopathy (n = 68) | 40 (58%) |
| Spontaneous bacterial peritonitis (n = 68) | 5 (7%) |
| Hepatopulmonary syndrome (n = 68) | 5 (7%) |
| Hepatorenal syndrome (n = 68) | 20 (29%) |
| History of TIPS procedure (n = 68) | 13 (19%) |
| Time from last MELD-Na score and death in mo (n = 43) (mo in median) | 0.6 (IQR, 0.3–2.0) |
| MELD-Na score closest to diagnostic RHC | 13 (IQR, 9–18) |
| MELD-Na score closest to death | 23 (IQR, 16–27) |
| OLT liver transplant listing (n = 69) | |
| Yes | 21 (30%) |
| No | 48 (70%) |
| Time from PoPH diagnosis to waitlist removal (mo) (n = 15) | 6.0 (IQR, 4.0–31.0) |
| Reasons for OLT listing removal (n = 21) | |
| Comorbidities | 5 (24%) |
| Died | 3 (14%) |
| Transplanted | 6 (29%) |
| Uncontrolled PAH | 7 (33%) |
IQR, interquartile range; MELD, model for end-stage liver disease; NASH, nonalcoholic steatohepatitis; OLT, orthotopic liver transplantation; PAH, pulmonary arterial hypertension; PoPH, portopulmonary hypertension; RHC, right heart catheterization; TIPS, transjugular intrahepatic portosystemic shunt.
The model for end-stage liver disease-sodium (MELD-Na) scores were 13 (IQR, 9–18) at the time of diagnostic RHC and 23 (IQR, 16–27) at the last clinic visit before death, indicating progression of liver disease. The MELD-Na score was 24 (IQR, 18–30) in patients that died from decompensated liver cirrhosis and 24 (IQR, 19–26) for the patients in whom PH directly or indirectly contributed to death. Table 4 shows a comparison of RHC hemodynamics among patients who died from decompensated liver failure and those where PoPH either directly caused death or contributed to death.
TABLE 4.
Hemodynamics and REVEAL 2.0 closest to time of death among those who died from decompensated liver failure or PAH
| Patients who died mainly from decompensated liver failure | Patients who died directly or indirectly from PAH (RVF/sudden death) | P (t test and Mann-Whitney U test) | |
|---|---|---|---|
| Mean ± SD, or n (%), | Mean ± SD, median (IQR), or n (%) | ||
| n | 20 (60%) | 13 (40%) | |
| RAP (mm Hg) | 10.8 ± 7.3 | 14.7 ± 7.3 | 0.15 |
| Mean PAP (mm Hg) | 43.5 ± 12.5 | 48.5 ± 11.6 | 0.26 |
| PAWP (mm Hg) | 15 ± 6.6 | 13.15 ± 3.9 | 0.36 |
| CO (L/min) | 5.9 (4–8.9) | 5.3 (4.5–7.5) | 0.86 |
| CI (L/min/m2) | 3 (2.2–4.8) | 2.8 (2.4–3.5) | 0.65 |
| PVR (WU) | 4.2 (3.2–6.7) | 6.5 (4.1–8.5) | 0.19 |
| Last REVEAL 2.0 score | 12.5 ± 1.9 | 13.6 ± 2.5 | 0.30 |
IQR, interquartile range; PAH, pulmonary arterial hypertension; PAP, pulmonary artery pressure; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance; RAP, right atrial pressure; REVEAL 2.0, Registry to Evaluate Early And Long-term PAH Disease Manage; TDCI, thermodilution cardiac index; TDCO, thermodilution cardiac output; WU, Wood units.
Concomitant Hepatopulmonary Syndrome
Concomitant hepatopulmonary syndrome (HPS) (defined as pulse oximetry measured saturation [SpO2] < 96%, partial pressure of O2 in arterial blood [PaO2] of <80 mm Hg, and A-a gradient of ≥15 mm Hg on room air, in the setting of late appearance of bubbles on echocardiogram, in the absence of any primary lung, or heart disease that could explain hypoxemia) was observed in 5 patients (7.2%). These 5 patients died of decompensated liver failure (n = 3), sepsis (n = 1), and gastrointestinal bleed (n = 1).
Cause and Place of Death in PoPH
Specific information on the cause of death was available in 52 (75%) patients (Figure 1). Of them, 39 of 52 (75%) died of causes unrelated to PH, with progressive liver disease being the leading cause 20 of 52 (38%). PoPH directly caused death in 9 of 52 (17%) but contributed to death in 4 (8%) patients. Six patients died after liver transplantation (4 of them died in intensive care unit [ICU] setting, 2 due to decompensated liver failure and 2 due to right heart failure; information in 2 patients was available). Only 21 patients were listed for transplant before dying (Table 3). Of these 21 patients, 5 were removed because of new comorbidities, 3 died before being transplantation, 7 were listed, but their PAH hemodynamic determinations remained above the cutoff used for our transplant center (PVR <3 WU), and 6 were transplanted.
FIGURE 1.
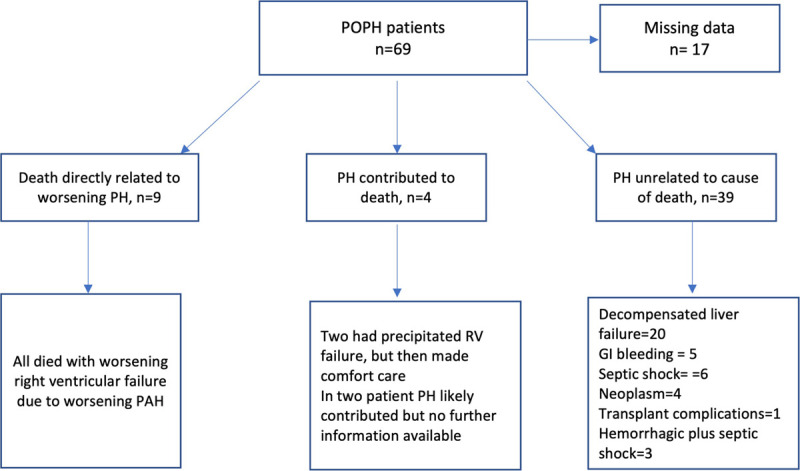
Flowchart of the causes of death in our cohort. GI, gastroenterology; PAH, pulmonary arterial hypertension; POPH, portopulmonary hypertension; PH, pulmonary hypertension; RV, right ventricular.
Specific location of death was available in 36 of 69 (52%) of patients, of which 23 of 36 (64%) patients died at Cleveland Clinic (21 [91%] in ICU, 1 in the emergency room and 1 in the operating room). Of the rest, 11 (30%) died at an outside hospital and 2 at a skilled nursing facility. Of the patients who died at Cleveland Clinic ICU, 11 (52%) received mechanical ventilatory support, 1 continuous veno-venous hemodialysis, and 5 vasopressors. Intravenous prostacyclin was initiated in only 1 patient in the ICU before death. Advanced directive information was available in 19 (28%) patients. Comfort care measures were taken in 22 (32%) patients. Do-not-resuscitate status was noted in 18 of 69 (26%) patients and of them 6 received cardiopulmonary resuscitation (CPR) before death.
DISCUSSION
Portopulmonary hypertension is one of the least studied subgroups of PAH and yet one with the highest mortality. PoPH is rare and likely underdiagnosed.14 Furthermore, PoPH tends to occur at late stages of liver disease in patients who have several comorbidities. In general, randomized studies using PAH-specific medications excluded patients with PoPH.15 Centers that reported their experience in managing PoPH particularly focused on describing treatments to facilitate liver transplantation. There is paucity of data on the causes and circumstances around death in patients with PoPH, particularly those who do receive OLT, an important information to better inform and treat patients who carry the burden of cirrhosis and PAH.
When compared with our prior study describing the causes and mode of death in patients with idiopathic and heritable PAH, we noted that in patients with PoPH the cause of death was elusive in a quarter of patients. This is because PoPH patients with advanced liver disease come to our institution for OLT evaluation, and if they are deemed unsuitable candidates for OLT, they return back to their referral institutions, complicating the adequate characterization of the cause of death. When the cause was identified, the majority (75%) of PoPH patients predominantly died of progressive liver disease. Unfortunately, only 30% of patients were listed for OLT and 9% (29% of the ones listed) were transplanted, reflecting the significant challenges in listing and transplanting these patients.
Nevertheless, the presence of PoPH impacts mortality either directly, because of right heart failure, or indirectly, by precluding patients from undergoing OLT, a life prolonging intervention for patients with advanced liver disease.6,11 In the present cohort, a significant number of PoPH patients received treatment to improve their pulmonary hemodynamics to facilitate transplantation but with variable success in achieving the hemodynamic goal (PVR <3 WU). The hemodynamic goals vary among transplant centers.11,16 Savale et al17 showed that OLT and PAH combination therapy prolonged survival in PoPH. Swanson et al proposed to divide PoPH patients based on their PAH severity. Those with stable liver disease and mild to moderate PAH could be treated with oral therapies, with drug escalation based on response. Meanwhile, patients with moderate to severe PAH, particularly those with unstable liver disease, need treatment with parenteral prostacyclin therapy to achieve the degree of pulmonary hemodynamic improvement required for OLT.18
At Cleveland Clinic, we consider every patient with advanced liver disease and PoPH for OLT. If the patient meets criteria for OLT and there are no major contraindications apart from PoPH, we treat the PAH with the goal of achieving a PVR <3 WU and normalizing the RV function.15 It remains unknown if PAH-specific therapies would provide a morbi-mortality benefit in patients who are not deemed candidates for OLT (eg, active alcohol intake, poor social support, morbid obesity). The relatively conservative PAH-treatment approach (a quarter of patient did not receive PAH treatment) in our cohort is explained by the broad duration of our study (1996–2020) that in part predated the results of the Ambition trial in 2015,19 and the lack of evidence showing a beneficial effect of PAH therapies in patients that are not candidates for OLT.20,21
More than half of the patients in our study died while under care in a healthcare facility, and one-third received circulatory or respiratory support before death, with high utilization of healthcare resources during the late stages of life, despite unproved beneficial effects. In a retrospective multicenter study, CPR was attempted in 26% of PAH patients and only 6% survived at 3 mo.22 In our cohort, less than a third of patients had advanced healthcare directives, a lower than ideal percentage in a population with a severe and frequently fatal disease, in part explained by the hope of receiving an OLT. These findings support a proactive approach that includes discussing end-of-life decisions during the evaluation of patients with PoPH, particularly in those who are not candidates for OLT.
Limitations of our study warrant comment. Because the study group consisted of patients seen at a large PH program, the results might not be generalizable to other centers. Our findings may be dissimilar to studies with different demographics,23 causes of portal hypertension, timing of PAH screening, and access to treatment including OLT. The broad duration of our study was a necessary limitation to include a relatively large number of patients; nevertheless, although treatment protocols and listing OLT criteria changed across this time span, patients continued to die as result of their liver disease. Despite these limitations, our study had a (1) consistent diagnosis of PoPH, (2) rigorous prospective evaluation of patients, and (3) meticulous review of information regarding cause and mode of death by PH specialists. Overall, as patients with PoPH die of their liver disease, it appears reasonable to screen and eventually treat PoPH more aggressively at an earlier stage, to potentially facilitate OLT, which remains the only life prolonging intervention. Similarly, palliative care should be involved earlier in the course of the disease, allowing patients to express their values and desires related to end-of-life care and providing relief from symptoms and stress of the disease.
CONCLUSIONS
In our cohort, most patients with PoPH died of their liver disease, independently to the presence of PAH. Roughly a third of our patients were not treated for their PoPH, their death was mainly caused by the liver disease highlighting the importance of liver transplantation in the management of this disease. Given known risks and comorbidities only a third of patients were listed and a few received OLT. Importantly, palliative care measures and advanced healthcare planning remain underutilized. Prospective studies are needed to elucidate whether earlier treatment of PAH facilitates OLT, reduces the utilization of healthcare resource and ultimately improves survival.
Footnotes
Published online 9 June, 2021.
A.R.T. is supported by NIH grand no. R01HL130307.
S.S. is the speaker and consultant for Actelion, Bayer, and United Therapeutics; advisor for Boehringer Ingelheim, Liquidia Technologies, Gossamer Bio, and Altavant Sciences. Clinical trial endpoint adjudication committee member for a GSK sponsored RCT, Research grant from ACCP CHEST Foundation. A.R.T. is the guarantor of the article, taking responsibility for the integrity of the work as a whole, from inception to published article. G.A.H. received personal fees for being a member in Bayer Healthcare – Advisory Board and Speaking. The other authors declare no conflicts of interest.
S.S. participated in writing and revising the article, concept design, analyzing the results, and performing statistical analyses. S.A.A. participated in the data collection, statistical analysis, interpretation of the results, writing and critical revision of the article for important intellectual content, and final approval of the article submitted. C.M. participated in the data collection, IRB application, and critical revision of the article for important intellectual content. J.N. participated in the data collection, and critical revision of the article for important intellectual content. R.A.D. participated in the interpretation of the results and critical revision of the article for important intellectual content and final approval of the article submitted. G.A.H. participated in the interpretation of the results and critical revision of the article for important intellectual content. A.R.T. participated in the design of the study, data collection, statistical analysis, interpretation of the results, writing and critical revision of the article for important intellectual content, and final approval of the article submitted.
REFERENCES
- 1.Tonelli AR, Arelli V, Minai OA, et al. Causes and circumstances of death in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2013; 188:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards BS, Weir EK, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol. 1987; 10:1233–1238. [DOI] [PubMed] [Google Scholar]
- 3.Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology. 2003; 37:401–409. [DOI] [PubMed] [Google Scholar]
- 4.Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl. 2005; 11:1107–1111. [DOI] [PubMed] [Google Scholar]
- 5.Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology. 2006; 44:1502–1510. [DOI] [PubMed] [Google Scholar]
- 6.Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transpl. 2000; 6:443–450. [DOI] [PubMed] [Google Scholar]
- 7.Martin P, DiMartini A, Feng S, et al. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the study of liver diseases and the American Society of transplantation. Hepatology. 2014; 59:1144–1165. [DOI] [PubMed] [Google Scholar]
- 8.DuBrock HM, Goldberg DS, Sussman NL, et al. Predictors of waitlist mortality in portopulmonary hypertension. Transplantation. 2017; 101:1609–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krowka MJ, Miller DP, Barst RJ, et al. Portopulmonary hypertension: a report from the US-based REVEAL registry. Chest. 2012; 141:906–915. [DOI] [PubMed] [Google Scholar]
- 10.Khaderi S, Khan R, Safdar Z, et al. Long-term follow-up of portopulmonary hypertension patients after liver transplantation. Liver Transpl. 2014; 20:724–727. [DOI] [PubMed] [Google Scholar]
- 11.Cartin-Ceba R, Burger CD, Swanson K, et al. Clinical outcomes after liver transplantation in patients with portopulmonary hypertension. Transplantation. 2020. doi:10.1097/TP.0000000000003490 [DOI] [PubMed] [Google Scholar]
- 12.Savale L, Sattler C, Coilly A, et al. Long-term outcome in liver transplantation candidates with portopulmonary hypertension. Hepatology. 2017; 65:1683–1692. [DOI] [PubMed] [Google Scholar]
- 13.Hoeper MM, Bogaard HJ, Condliffe R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013; 62(25 Suppl):D42–D50. [DOI] [PubMed] [Google Scholar]
- 14.Thomas C, Glinskii V, de Jesus Perez V, et al. Portopulmonary hypertension: from bench to bedside. Front Med (Lausanne). 2020; 7:569413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.AbuHalimeh B, Krowka MJ, Tonelli AR. Treatment barriers in portopulmonary hypertension. Hepatology. 2019; 69:431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DuBrock HM, Runo JR, Sadd CJ, et al. Outcomes of liver transplantation in treated portopulmonary hypertension patients with a mean pulmonary arterial pressure ≥35 mm Hg. Transplant Direct. 2020; 6:e630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savale L, Guimas M, Ebstein N, et al. Portopulmonary hypertension in the current era of pulmonary hypertension management. J Hepatol. 2020; 73:130–139. [DOI] [PubMed] [Google Scholar]
- 18.Swanson KL. Treatment of portopulmonary hypertension–isn’t it time to move forward? Liver Transpl. 2008; 14:270–271. [DOI] [PubMed] [Google Scholar]
- 19.Galiè N, Barberà JA, Frost AE, et al. ; AMBITION Investigators. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015; 373:834–844. [DOI] [PubMed] [Google Scholar]
- 20.Ghofrani HA, Galiè N, Grimminger F, et al. ; PATENT-1 Study Group. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med. 2013; 369:330–340. [DOI] [PubMed] [Google Scholar]
- 21.Sitbon O, Bosch J, Cottreel E, et al. Macitentan for the treatment of portopulmonary hypertension (PORTICO): a multicentre, randomised, double-blind, placebo-controlled, phase 4 trial. Lancet Respir Med. 2019; 7:594–604. [DOI] [PubMed] [Google Scholar]
- 22.Hoeper MM, Galié N, Murali S, et al. Outcome after cardiopulmonary resuscitation in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002; 165:341–344. [DOI] [PubMed] [Google Scholar]
- 23.Le Pavec J, Souza R, Herve P, et al. Portopulmonary hypertension: survival and prognostic factors. Am J Respir Crit Care Med. 2008; 178:637–643. [DOI] [PubMed] [Google Scholar]