


Abstract
Rat1 is a 5′→3′ exoribonuclease in budding yeast. It is a highly conserved protein with homologs being present in fission yeast, flies, worms, mice and humans. Rat1 and its human homolog Xrn2 have been implicated in multiple nuclear processes. Here we report a novel role of Rat1 in mRNA splicing. We observed an increase in the level of unspliced transcripts in mutants of Rat1. Accumulation of unspliced transcripts was not due to the surveillance role of Rat1 in degrading unspliced mRNA, or an indirect effect of Rat1 function in termination of transcription or on the level of splicing factors in the cell, or due to an increased elongation rate in Rat1 mutants. ChIP-Seq analysis revealed Rat1 crosslinking to the introns of a subset of yeast genes. Mass spectrometry and coimmunoprecipitation revealed an interaction of Rat1 with the Clf1, Isy1, Yju2, Prp43 and Sub2 splicing factors. Furthermore, recruitment of splicing factors on the intron was compromised in the Rat1 mutant. Based on these findings we propose that Rat1 has a novel role in splicing of mRNA in budding yeast. Rat1, however, is not a general splicing factor as it crosslinks to only long introns with an average length of 400 nucleotides.
INTRODUCTION
Rat1/Xrn2 belongs to the Xrn-family of nucleases (1). These are highly conserved proteins with homologs present in almost every eukaryote investigated so far (2–7). It has been found that Rat1/Xrn2 has pleiotropic roles in RNA metabolism in eukaryotes. Studies carried out in different eukaryotic systems have demonstrated involvement of Rat1/Xrn2 in RNA trafficking, RNA quality control, RNA processing, promoter-associated transcription, elongation and termination of transcription (8–15).
Rat1 was discovered in budding yeast as a factor required for nucleo-cytoplasmic transport of mRNA. In a temperature-sensitive mutant of Rat1, mRNA was cleaved and polyadenylated but failed to move out of the nucleus and hence was named RNA trafficking factor or Rat1 (8). Further investigation revealed that Rat1 and its higher eukaryotic homolog Xrn2 are involved in termination of RNAPII-mediated transcription in a manner dependent on their 5′→3′ exoribonuclease activity (13,15). The termination of transcription involves pausing of RNAPII beyond the poly(A)-site, which facilitates recruitment of CF1 and CPF 3′ end processing/termination factors in yeast (16). The transcribing mRNA is cleaved downstream of the poly(A)-site and is polyadenylated by poly(A)-polymerase (17,18). The monophosphorylated mRNA downstream of the cleavage site is still attached to the elongating RNAPII. Rat1 binds to the uncapped transcript and the 5′→3′ exoribonuclease activity digests the nascent RNA. Removal of the RNA helps disengage RNAPII from the DNA template (13). The Rat1 homolog Xrn2 facilitates termination of transcription in higher eukaryotes by a similar mechanism (15,19,20). Rat1/Xrn2-mediated termination is thus reminiscent of the ‘torpedo’ mechanism of termination of transcription in prokaryotes (21). The enzymatic activity of Rat1 is crucial but not sufficient for termination of transcription (22). Genomewide studies have revealed that Rat1 is not a universal termination factor like the CF1 subunit Pcf11 and CPF subunit Ysh1, but is required for termination of transcription of only 35% of RNAPII-transcribed genes in budding yeast (23).
Rat1 may also be involved in termination at the pre-cleavage/polyadenylation step in accordance with the ‘allosteric’ mechanism. The pausing of the polymerase near the poly(A)-site, which is critical for both cleavage/polyadenylation and termination, is dependent on Rat1. In the rat1-1 mutant, which exhibits a termination defect, there is hyperphosphorylation of CTD-serine-2 resulting in an increased elongation rate. The rat1-1 termination defect could be rescued by introduction of the rpb1-N488D allele, which causes a slower elongation rate (12). Furthermore, it was shown that Rat1-dependnence on termination is dependent on promoter-driven transcription (22). These findings strongly suggest that Rat1 has dual roles in termination; a pre-cleavage role in RNAPII pausing downstream of the poly(A)-site, and a post-cleavage role in degrading 5′ monophosphorylated RNA followed by dissociation of RNAPII from the DNA template. Another important implication is involvement of Rat1 in the elongation step of transcription through its influence on hyperphosphorylation of the CTD at serine-2 (24). Indeed, combining the rat1-1 allele with the rpb1-E1103G mutation, which causes a faster polymerase elongation rate, resulted in an enhanced growth defect (24). Depletion of Xrn2 in human cell lines resulted in promoter-proximal pausing, which also could be the consequence of the role of the protein in the early elongation step (25).
In multiple organisms, Rat1 function appears linked to the promoter-terminator crosstalk. In Caenorhabditis elegans, not all genes that exhibit 3′ end occupancy of Rat1 are dependent on this protein for termination. Whether Rat1 is required for termination is determined by the promoter element in worms (14). In budding yeast also, Rat1 could dismantle only the promoter-driven transcription complexes in an in vitro assay (22). In keeping with a role in gene looping, Rat1/Xrn2 have been found to bind both the 5′ and 3′ ends of genes (13,20,23,25). Localization of Rat1/Xrn2 in the promoter-proximal region gave rise to speculation that it plays a role in promoter-associated transcription. Experimental evidence has implicated Rat1/Xrn2 in the initiation-elongation transition (26). In the absence of Xrn2, a peak of promoter-proximally paused polymerase was observed, thereby suggesting that Xrn2 is involved in early stages of transcription. Xrn2 has also been found to function in promoter directionality as an increase in polymerase signal was observed in the region upstream of promoters in the absence of Xrn2 activity (10,20). A similar involvement of Rat1 in promoter directionality in budding yeast cannot be ruled out. Both Rat1 and Xrn2 have also been shown to play a role in mRNA quality control by degrading aberrant uncapped transcripts (10–12). In line with these results, human Xrn2 has been reported to coimmunoprecipitate with the Edc3, Dcp1a and Dcp2 capping proteins (10). No such interaction of Rat1 with capping enzymes has been observed in budding yeast, but Rat1 can similarly degrade uncapped yeast transcripts (11).
Rat1/Xrn2 have also been implicated in degradation of splicing-defective transcripts. Xrn2 was reported to degrade unspliced transcripts in human cells, while Rat1 in yeast was linked to degradation of unspliced transcripts in the mutant of splicing factor Prp2 (9). We discovered accumulation of unspliced transcripts in mutants of Rat1 with a wild type Prp2 allele. This increase was not due to stabilization of unspliced transcripts, or an indirect effect of defective termination or lowering of level of splicing factors, or due to an absence of polymerase pausing on intronic regions in the Rat1 mutant. We present evidence that Rat1 plays a direct role in splicing of precursor mRNA. We discovered a physical interaction of Rat1 with the intronic sequences as well as with the splicing factors of NineTeen complex (NTC). Furthermore, recruitment of splicing factors to the intron was compromised in the absence of functional Rat1. Our findings strongly suggest a novel role of Rat1 in splicing of primary transcripts of a subset of genes in budding yeast.
MATERIALS AND METHODS
Yeast strains
The yeast strains used in this study are listed in supplemental Table S1. ZA1, ZA2, ZD47 and ZD63 were constructed by replacing the entire ORF of targeted gene by TRP1 or HIS3 as described in Wach et al. (27). C-terminal TAP-tagged Rat1 (ZD7) and MYC-tagged Rat1 (ZD12) strains were derived from FY23 by transforming with a PCR product amplified from pBS1479 and pFA6a-13Myc-TRP1, respectively. C-terminal FRB-tagged Rat1 (ZD42) was constructed from HHY168 strain using PCR amplified product from pFA6a-FRB-GFP-KanMX6. C-terminal MYC-tagged Rat1 (ZD12), MYC-tagged Prp2 (ZD64), Clf1 (ZD76), MYC-tagged Isy1 (ZD75) strains were derived from FY23 and ZD42 strains by transforming with a PCR product amplified from pFA6a-13Myc-TRP1/HIS3. The C-terminal HA-tagged Yju2 (ZD70), Clf1 (ZD71), Sub2 (ZD72), Prp43 (ZD73) and Isy1 (ZD74) strains were derived from the ZD12 strain by transforming with a PCR product amplified from pFA6a-3HA-KanMX6. The primers used for the C-terminal tagging of proteins are listed in the supplemental Table S2. A 5′ gene-specific primer and a 3′ reverse primer within the tag was used to confirm that the tag was inserted at the correct genomic position. ZA1 and ZA2 were constructed by replacing the targeted gene by the TRP1 selection marker amplified from pRS414. For validation of a gene knockout, forward and reverse primer designed in the upstream and downstream regions of the open reading frame were used. Primers to confirm the strains by colony PCR are listed in Supplementary Table S2
Cell culture
A 5 ml culture was started in yeast-peptone-dextrose (YPD) medium using colonies from a freshly streaked plate. The culture was grown overnight at 30°C with constant shaking at 250 rpm. All Saccharomyces cerevisiae cell cultures were grown in YPD medium unless otherwise stated. All strains except the temperature-sensitive mutants were grown at 30°C and 250 rpm. Next morning, the overnight grown cultures were diluted 1:100 in YPD broth and allowed to grow at 30°C with constant shaking until the desired A600 was reached. Cells were centrifuged at 1860 × g for 3 min at 4°C and the cell pellets were used further for further experiments.
Cell culture for Anchor-away experiment
The starter culture Rat1-FRB-tagged strain was grown in YPD broth whereas strains with plasmids expressing wild type (pRS415-WT-Rat1) or catalytically inactive mutant of Rat1 (pRS415-D235A-Rat1) were grown in synthetic complete media without leucine. The cells were incubated overnight at 30°C and 250 rpm in an orbital shaker. The cells from the starter cultures were added in 1:100 dilution to the respective culture broths. Cultures were grown until A600 0.5–0.7 was reached. At this point, one-half of the culture was treated with rapamycin (final concentration 1 μg/ml) and the other was left untreated. Both the samples were grown for another 60 min. Cells were collected by centrifugation as described above and used for RT-PCR, TRO and ChIP analysis.
Cell culture of temperature-sensitive strains
Temperature-sensitive mutants of Rat1 (rat1-1), Rna14 (rna14-1) and Rna15 (rna15-2), were used in this study. The overnight cultures were grown as described above except that cells were grown at 25°C. Next morning, the overnight culture was diluted 1:50 in YPD broth. The cells were grown with gentle shaking at the permissive temperature (25°C) until the they reached mid-log phase. Cells were then split into two halves and grown at the permissive (25°C) or non-permissive temperature (37°C) for 3 h (rat1-1) or 1 hour (rna14-1 and rna15-2). An isogenic wild type strain was subjected to similar growth conditions and the final cell density was normalized considering the mutants strain cell density. Cells were harvested as described above and used for RT-PCR, TRO or RNA-IP analysis.
Isolation of RNA and RT-PCR analysis
Cells were grown and processed as discussed in previous section. RNA was extracted as described in McNeil and Smith, 1986 (28). 2 μg/μl of RNA was used for cDNA synthesis using M-MuLV reverse transcriptase (NEB) with oligo-dT, 5S, 3′ gene specific snRNA primers, respectively. cDNA was diluted ten times prior to PCR amplification by Taq DNA polymerase. The gene specific primers used for PCR reaction are listed in Supplementary Table S2. Each PCR signal was normalized with respect to 5S ribosomal RNA control. In parallel, a negative control without reverse transcriptase was run to ensure DNA contamination is not contributing to any RT-PCR signal. Each experiment was repeated with at least three independently grown cultures. Quantification of data was performed as described in detail in Support Protocol 1 of El Kaderi et al. (29).
Transcription run-on assay (TRO)
FRB-tagged strains were processed as discussed in previous section. The transcription run-on assay was performed as described in (30). For the transcription analysis, 500 ng/μl of RNA was used for cDNA synthesis with Superscript IV reverse transcriptase using oligo-dT and 5S 3′ primers. cDNA was diluted five times prior to PCR amplification by Platinum SuperFi DNA polymerase. The gene specific primers used for PCR reaction are listed in Supplementary Table S2. Each PCR was normalized with respect to 5S ribosomal RNA control. A sample without reverse transcriptase enzyme was run in parallel as a negative control. Each experiment was repeated with at least three independently grown cultures.
Chromatin immunoprecipitation (ChIP)
100 ml of cell culture was treated with 1% formaldehyde and 20 mM disuccinimidyl glutarate (DSG) (20593; Thermo Fisher Scientific) in 1X PBS with constant shaking for 20 min at 25°C. Crosslinking was quenched by the addition of 125 mM glycine and incubating for 5 min at 25°C. Cells were lysed and chromatin was isolated and sonicated as described previously (31). ChIP for Rat1-TAP was performed by incubating 500 μl of the solubilized chromatin sample with 20 μl of IgG Sepharose 6 Fast Flow (GE healthcare) for 3 h at 4°C. For RNAPII ChIP, 400 μl of the solubilized chromatin was incubated with 4 μl Anti-8WG16 (05-952-I, Millipore Sigma). For Prp2-Myc, Clf1-Myc and Isy1-Myc, 400 μl of the solubilized chromatin was pre-incubated with 5 μl Anti-Myc antibody (PA1-981; Invitrogen) for 2 h at 4°C. 20 μl of Magnetic Dynabeads Protein G was prewashed two times prior to use. Antibody–antigen complex was captured on magnetic Dynabeads (1003D) by incubating at room temperature for 15 min. The tubes were placed on a ThermoFisher Scientific Magjet rack, and supernatant containing unbound antigen and antibody was discarded. After removing the unbound material, the Sepharose or magnetic beads were subjected to a series of washing steps and processed to obtain DNA as described in El Kaderi et al. (31). Purified DNA from the input and ChIP fractions were used as a template for PCR using the primers specific for a gene. The primers used for ChIP-PCR analysis are listed in Supplementary Table S2. The association of a protein to a given genomic region was expressed as ChIP over input ratios. Quantification and statistical analyses were performed as described in (29,58). Each experiment was repeated with at least three independently grown cultures.
Data mining and re-analysis of published datasets
Published ChIP-Seq analysis dataset was retrieved through the Gene Expression Omnibus (GEO), GSE79222. Raw reads were further processed and analyzed using the Galaxy web platform at usegalaxy.org (32). Raw reads (50 bp paired end reads) were aligned to the S. cerevisiae genome (sacCer3, version 64.2.1) using the Bowtie (version 2.3.4.2) (33) following the parameters described in Baejen et al., 2017 (23). To normalize and compare two BAM files and obtain a log2 ratio of IP/Input in the form of a bigwig file, bamCompare (Galaxy Version 3.1.2.0.0) in deepTools (34) was used. The aligned reads with MAPQ alignment quality smaller than 7 (-q 7) were skipped. Signal extraction scaling (SES) factor was used for scaling and establishing ChIP enrichment profile over log2 scale with the options: –l 100 –n 100 000 and the bin size of 20. The subsequent data in the bigwig file was selectively enriched for the genomic regions in the BED file using computeMatrix function (Galaxy Version 3.3.0.0.0). Specifically, three BED files were used in this study; 280 intron-containing genes with TSS and TES coordinates, 280 intron-containing genes with intron start and intron end coordinates, and 2392 non-intronic genes with TSS and TES coordinates. Heatmap was plotted to visualize the score distributions of the transcription factors or RNAPII enrichment associated with genomic regions specified in the BED files using the plotHeatmap (Galaxy Version 3.3.0.0.1). The metagene plot of the transcription factors and RNAPII was drawn with the plotProfile function (Galaxy Version 3.3.0.0.0).
ChIP/Input data in bigwig files were imported to Integrated Genome Browser (IGB) (35) for visualization. In case of RNAP II heatmap and metagene profile, the occupancy pattern was plotted on the window of intron scaled to 400 bp and 0.4 kb upstream and 1.0 kb downstream of the intronic region. For transcription factors, Rat1 and Pcf11, the plot was constructed on the window of introns scaled to 400 bp and 0.1 kb upstream and 1 kbp downstream of the intronic region. A confidence interval of mean is computed at each bins (20 bp bin sizes) using bootstrap sampling or Rat1 and Pcf11 and is visually represented by a semi-transparent color with a width corresponding to one unit of the standard error. The correlation of Rat1 intron occupancy for the length of the intron, distance from 5′ splice site to branch point, and distance from 3′ splice site to branch point has been determined for 280 intronic genes using box-whisker plots. Mann–Whitney U test was used to determine statistical significance of Rat1 occupancy for the intron length, distance from 5′ or 3′ splice site to the branch point. The list of intron-containing genes with TATA box or TATA-less promoters were filtered from data file of yeast genes characterized into TATA-containing or TATA-less promoter (36). Distribution of TATA box or TATA-less promoter reflecting the percentage of intron containing genes with and without enrichment of Rat1 on the intronic region is shown using a pie chart. The statistical significance for the promoter preference and Rat1 occupancy was determined using Fisher's exact test. Statistical analysis was computed on Graphpad Prism 8.4.2. Gene Ontology (GO) analyses using SGID of the intronic genes with Rat1 occupancy was performed using PANTHER (http://geneontology.org/) using default options (37). Additionally, we performed GO ontology functional analysis and detection of protein class of the genes in the dataset. Benjamini-corrected P-value was used for ranking GO terms.
RNA immunoprecipitation
Mutant rat1-1 strain and the isogenic wild type strain were grown, and RNA was isolated as described previously. 2 μg/μl of RNA was set aside as input that was used to quantify the total amount of spliced and unspliced RNA in the samples. Briefly, cDNA synthesis of 2 μg/μl of RNA was performed using Superscript IV reverse transcriptase and oligo-dT 3′ primer. cDNA was diluted five times prior to use in PCR amplification using Platinum SuperFi DNA polymerase. The gene specific primers used for PCR reaction are listed in Supplementary Table S2.
The immunoprecipitation of the capped transcripts was performed by incubating 4 μl of Anti-7′-methylguanosine (m7G)-Cap mAb (RN016M; MBL) with 40 μg of total RNA adjusted to the total volume of 400 μl using RNA-IP binding buffer (20 mM HEPES–KOH, 10 mM MgCl2, 150 mM NaCl, 0.5 mM DTT, 80U RNase inhibitor). Immunoprecipitated complex was captured by incubating with 15 μl of prewashed magnetic Dynabeads for 15 min at room temperature. The beads were washed three times with 400 μl RNA-IP binding buffer. After the final wash, 100 μl of binding buffer was added, mixed to resuspend the beads, and transferred to a new tube. The buffer was removed as described above. Capped RNA bound to the Anti-7′-methylguanosine (m7G)-Cap mAb was released from beads by 100 μl of elution buffer (20 mM HEPES–KOH, 1% SDS, 2mM EDTA). RNA elution was performed by gently shaking tubes on a nutator for 5 min at room temperature. The eluant containing capped RNA was transferred into a new tube and subjected to phenol-chloroform extraction and ethanol precipitation. The RNA pellet was air-died and resuspended in 16 μl of RNase-free water. The concentration of RNA was determined using Nanodrop. Approximately 200 ng/μl of RNA was used for cDNA synthesis. cDNA synthesis from the input and immunoprecipiated RNA fraction was performed using Superscript IV reverse transcriptase and oligo-dT 3′ primer. cDNA was diluted five times prior to use in PCR amplification using Platinum SuperFi DNA polymerase. The gene specific primers used for PCR reaction are listed in Supplementary Table S2. A negative control without reverse transcriptase enzyme was carried out in parallel to rule out the possibility of DNA contamination.
Preparation of protein extract from yeast cells
The Rat1-TAP and an isogenic no-tag control strains were cultured in YPD media to an A600 of ∼4.0. Two liters of each cultures were harvested by centrifugation at 5000 rpm for 5 min at 4°C. Cell pellets were washed with 1× TBS and resuspended in 15 ml of lysis buffer (20 mM HEPES–KOH, 10 mM MgCl2, 150 mM NaCl, 1 mM PMSF, 0.5 mM DTT and 10% glycerol). The resultant cell suspension was flash frozen in liquid nitrogen and stored at −80°C.
Lysis of frozen cell was performed in Retsch MM301 mixer mill by repeating cycles of 3 mins at 15 Hz 15 times. Lysed cells were scrapped from the chambers and diluted with an additional 30 ml of lysis buffer. Cell lysate was centrifuged at 5000 rpm for 10 min at 4°C. The supernatant represents the whole cell extract. Soluble extract was obtained from the whole cell extract as described in Svejstrup et al. (38) and stored at −80°C until further use.
Purification of Rat1 complex and preparation of sample for mass spectrometry
The Rat1 complex was purified by TAP approach essentially as described in Puig et al. (39). The presence of Rat1-TAP at different steps of purification was analyzed using western blot analysis as described in El Kaderi et al. (31). Trichloroacetic acid (TCA) was added at a final concentration of 30% to the tandem affinity purified preparation. The samples were incubated at 4°C for 2 hours on ice. Samples were then centrifuged at 15 700 × g for 30 min at 4°C to pellet precipitated proteins. The supernatant was carefully removed, and any traces of TCA were removed by spinning again at 15 700 × g for 1 minute at 4°C. The TCA precipitated protein pellet was washed with 500 μl of 100% acetone. The acetone washed pellet was air-dried at room temperature for 10 min and then resuspended in 40 μl of buffer (100 mM Tris–HCl pH 8.8, 1% SDS).
Mass spectrometry analysis
The processing of samples from the wild type Rat1-TAP (test) and wild type no-tag (control) samples was performed at the proteomic facility at Wayne State University as described below. To gauge the background contamination due to sample processing, a background (bkg) tube without protein was processed in parallel with the sample tubes. Samples were precipitated with two volumes of ice-cold 100% methanol in 1 mM acetic acid at −20°C overnight. The next day, samples were spun at 17 000 × g for 20 min at 4°C and the supernatant was removed. The resultant pellets were rinsed with 100 μl methanol-acetic acid. Samples were dried in Speed-Vac for 3 min. The pellets were solubilized in 50 mM triethylammonium bicarbonate (TEAB) using Qsonica sonicator, then reduced with 5 mM DTT and alkylated with 15 mM iodoacetamide (IAA) under standard conditions. Excess IAA was quenched with an additional 5 mM DTT. The samples were then digested overnight in 50 mM TEAB with sequencing-grade trypsin (Promega). The next day, digests were acidified with 1% formic acid and a 10% aliquot of the supernatant was analyzed.
The peptides were separated by reversed-phase chromatography (Acclaim PepMap100 C18 column, Thermo Scientific), followed by ionization with the Nanospray Flex Ion Source (Thermo Scientific), and introduced into a Q Exactive™ mass spectrometer (Thermo Scientific). Abundant species were fragmented with high-energy collision-induced dissociation (HCID). Data analysis was performed using Proteome Discoverer 2.1 (Thermo Scientific) which incorporated the SEQUEST algorithm (Thermo Scientific). The Uniprot_Yeast_Compl_20160407 database was searched for yeast protein sequences and a reverse decoy protein database was run simultaneously for false discovery rate (FDR) determination. The data files were loaded into Scaffold (Proteome Software) for distribution. SEQUEST was searched with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 10 PPM. Carbamidomethylation of cysteine was specified in SEQUEST as a fixed modification. Deamidation of asparagine and glutamine, oxidation of methionine, and acetylation of the N-terminus were specified in SEQUEST as variable modifications. Minimum protein identification probability was set at ≤1.0% FDR with two unique peptides at ≤1.0% FDR minimum peptide identification probability. (0.5% protein decoy FDR, 0.16% peptide decoy FDR).
Co-immunoprecipitation
Cells expressing Rat1-C-MYC and Clf1/Isy1/Prp43/Sub2/Yju2-C-HA were grown in 100 ml of YPD to an OD600 of 1.2–1.4, harvested, washed with 1× Tris-buffer Saline, and then suspended in 1.0 ml of lysis buffer (20 mM HEPES–KOH, 10 mM MgCl2, 150 mM NaCl, 1 mM PMSF, 0.5 mM DTT and 10% glycerol). The cell suspension was flash frozen in liquid nitrogen and processed as described in El Kaderi et al. For immunoprecipitation, cell lysate was incubated with 20 μl of Anti-HA Agarose (26182; Pierce). Beads were washed thrice with lysis buffer and elution was carried out in SDS sample buffer lacking 2-mercaptoethanol. Western blot analysis was performed using anti-MYC (PA1-981; Invitrogen) to detect the presence of Rat1-C-MYC.
RESULTS
Unspliced mRNA accumulates in the absence of functional Rat1
Rat1 has been implicated in a variety of nuclear processes in yeast and higher eukaryotes. In order to have a comprehensive understanding of the role of Rat1 in RNA biogenesis in budding yeast, we monitored mRNA levels of selected genes in the temperature-sensitive rat1-1 mutant. RNA analysis was performed in cells grown at the permissive (25°C) and non-permissive (37°C) temperatures by RT-PCR. Briefly, the protocol involved isolation of total RNA from exponentially growing cells, reverse transcription of RNA using an oligo-dT, PCR amplification of resultant cDNA using gene-specific primers as described in Materials and Methods and quantification of RT-PCR products as described in El Kaderi et al. (29). Since Rat1 affects transcription of several mRNA species as well as 5.8S and 28S rRNAs, we used 5S rRNA as a normalization control in all experiments. In accord with the termination function of Rat1, a decrease in the mRNA level of most genes was observed upon shifting of cells to the non-permissive temperature. However, we observed an unusual result with the APE2 gene. A longer transcript appeared upon shifting of rat1-1 cells to the non-permissive temperature (Figure 1B, lane 4). APE2 is one of the few intron-containing genes in budding yeast, and sequencing of the PCR product revealed that the longer transcript was the unspliced mRNA. To determine if the accumulation of unspliced transcripts is unique to APE2, we examined several other intronic genes. Of all the genes that we analyzed, STO1, ASC1 and IMD4, exhibited a similar trend of increased unspliced mRNA in the rat1-1 mutant at the elevated temperature (Figure 1B, lane 4 and Figure 1C, black bars). The accumulation of unspliced transcripts was to a much lesser extent in the isogenic wild type cells at 37°C (Figure 1B, lane 2 and Figure 1C). RT-PCR performed using gene-specific RT primers gave identical results. To gauge the effect of Rat1 on accumulation of unspliced transcripts accurately, we calculated splicing efficiency by measuring percentage of total transcripts undergoing splicing. Splicing efficiency of APE2, STO1, ASC1 and IMD4 registered a 30–60% decline in the mutant at the non-permissive temperature (Figure 1D). Not all intron-containing genes, however, exhibit increased unspliced transcript levels in the mutant. Out of eight genes that we tested, four (APE2, STO1, ASC1 and IMD4) exhibited splicing defects, while four (KIN28, HPC2, REC114 and MCO76) did not show any splicing defect.
Figure 1.

Unspliced transcripts accumulate in the rat1-1 mutant at non-permissive temperature. (A) Schematic depiction of a gene showing the position of primers F and R used in RT-PCR analysis. (B) Gel pictures showing RT-PCR analysis of mRNA in wild type (WT) and rat1-1 mutant cells at the indicated temperatures. (C) Quantification of data shown in (B). 5S rRNA was used as normalization control. The results shown are an average of six independent PCRs from three separate RNA preparations from three independently grown cultures. The values and error bars for each condition indicate the mean ± standard deviation. Statistical significance (P-values) was determined using the two-tailed paired Student's t-test as described in (58). (D) Splicing efficiency representing the percentage of total transcripts undergoing splicing in WT and rat1-1 cells at the indicated temperatures.
To confirm that accumulation of unspliced transcripts in the rat1-1 strain was due to the inactivation of Rat1, and not due to a factor in the genetic background of the rat1-1 strain, we used the ‘anchor away’ approach. This technique involves selective depletion of a protein from the nucleus by anchoring it to ribosomes in the cytoplasm (40). The approach takes advantage of the rapamycin-dependent heterodimerization of FK506 binding protein (FKBP12) with FKBP12-rapamycin-binding (FRB) domain (41,42). We inserted the FRB domain at the carboxy-terminus of Rat1 in a strain that has FKBP12 fused to the ribosomal protein RPL13A, a fpr1 deletion and a tor1-1 mutation. The resultant strain was named Rat1-AA. In the presence of rapamycin, Rat1-FRB dimerized with ribosomal RPL13A-FKBP12 in the anchor away strain, leading to depletion of Rat1 from the nucleus within 60 minutes of addition of the antibiotic to the medium (Supplementary information, Figure S1). We monitored levels of spliced and unspliced transcripts in the Rat1-AA strain in the presence of rapamycin (Rat1 depleted from the nucleus) and in the absence of rapamycin (Rat1 present in the nucleus) by RT-PCR as described above. We observed a 2–4-fold increase in the level of unspliced APE2, STO1, MRK1, NMD2 and LSB4 transcripts upon depletion of Rat1 (Figure 2B, lane 4 and Figure 2C, red bars). There was no such rapamycin-dependent buildup of unspliced mRNA in the isogenic wild type strain (Figure 2B, lane 2 and Figure 2C, red bars). Splicing efficiency of four genes analyzed here decreased by about 25–50% in the presence of rapamycin (Figure 2D). We conclude that there is an accumulation of unspliced transcripts when Rat1 is inactivated or depleted from the nucleus. All intron-containing genes of budding yeast, however, do not exhibit accrual of unspliced RNA in the absence of Rat1. Out of eight genes that we tested, only four exhibited accumulation of unspliced transcripts in the absence of Rat1, while KIN28, HPC2, REC114 and MCO76 showed no difference.
Figure 2.

Accumulation of unspliced transcripts upon nuclear depletion of FRB-tagged Rat1 in the presence of rapamycin. (A) Schematic depiction of a gene showing the position of primers F and R used in RT-PCR analysis. (B) Gel pictures showing RT-PCR products for the indicated genes in FRB-tagged Rat1 strain and untagged strain (control) in the presence and absence of rapamycin. (C) Quantification of data in lanes 3 and 4 of (B). 5S rRNA was used as normalization control. The results shown are an average of six independent PCRs from three separate RNA preparations from three independently grown cultures. The values and error bars for each condition indicate the mean ± standard deviation. Statistical significance (P-values) was determined using the two-tailed paired Student's t-test as described in (58). (D) Splicing efficiency representing the percentage of total transcripts undergoing splicing in FRB-tagged Rat1 strain and untagged (control) strain in the presence and absence of rapamycin.
To explain the presence of unspliced mRNA in the absence of Rat1 activity, we formulated five hypotheses: (i) Rat1 plays a surveillance role and degrades unspliced transcripts by its exoribonuclease activity; (ii) accumulation of unspliced transcripts is the consequence of an indirect effect of Rat1-mediated termination on splicing; (iii) Rat1 affects splicing indirectly by adversely affecting the level of splicing factors; (iv) Rat1 effect on elongation facilitates pausing of the polymerase during transcription of intronic regions to enable completion of splicing reactions; (v) Rat1 has a novel role in splicing of primary transcripts.
We tested each of these hypotheses and the results are described below.
Rat1 is not the surveillance factor that degrades unspliced transcripts
The experiments described above monitored steady-state RNA levels, which is the net product of two opposing processes, transcription and degradation. To determine if Rat1 has a surveillance role in degrading unspliced mRNA by its exoribonuclease activity, we measured levels of unspliced and spliced transcripts by the transcription run-on (TRO) approach. The TRO assay detects nascent transcripts and therefore reflects transcription and rules out the contribution of RNA stability to the measured RNA content. The strand-specific TRO analysis was carried out using Br-UTP by the modification of the method described in Core et al. (43). Briefly, exponentially growing yeast cells were permeabilized with sarkosyl and allowed to resume transcription in the presence of Br-labelled UTP for 2–5 min. Br-UTP-labelled nascent RNA was immunopurified, reverse transcribed using oligo-dT and amplified by PCR as described in Dhoondia et al. (30). TRO analysis revealed a 2–7-fold increase in the levels of unspliced transcripts of APE2, STO1, MRK1 and LSB4 upon depletion of Rat1 in the Rat1-AA strain (Figure 3B and C).
Figure 3.

Transcription Run-On (TRO) analysis showing that accumulation of nascent unspliced transcripts upon nuclear depletion of FRB-tagged Rat1 in the presence of rapamycin is rescued by wild type Rat1 but not the catalytically inactive mutant. (A) Schematic depiction of a gene showing the position of primers F and R used in TRO analysis. (B) Gel pictures showing TRO data for the indicated genes in FRB-tagged Rat1 strain and untagged strain (control) in the presence and absence of rapamycin and upon complementation with WT Rat1 or catalytically inactive D235A Rat1 mutant. (C) Quantification of TRO signals shown in (B). 5S rRNA was used as normalization control. The results shown are an average of six independent PCRs from three separate RNA preparations from three independently grown cultures. The values and error bars for each condition indicate the mean ± standard deviation. Statistical significance (P-values) was determined using the two-tailed paired Student's t-test as described in (58).
Since splicing occurs cotranscriptionally in both yeast and higher eukaryotes, these findings suggest that Rat1 may be affecting cotranscriptional splicing of mRNA. The possibility of Rat1 playing a role in cotranscriptional degradation of unspliced transcripts, however, could not be ruled out. To resolve the issue, we transformed the Rat1-AA strain with a plasmid expressing either wild type Rat1 or a mutant form of Rat1 containing a point mutation at an evolutionarily conserved catalytic residue (D235A). Denoted as exo-Rat1, the mutant is deficient in exoribonuclease activity (13). TRO was repeated in the presence and absence of rapamycin. Expression of the wild type Rat1 rescued the accumulation of unspliced transcript, but the catalytically inactive exo-Rat1 mutant was unable to do so (Figure 3B and C). One interpretation is that Rat1 catalytic activity is required for degradation of unspliced transcripts, and therefore there is a buildup of unspliced mRNA in the exo-Rat1 mutant.
Rat1 is highly specific for 5′ monophosphorylated RNA substrates. 7′-methylguanosine capped mRNA is not a suitable substrate of the Rat1 nuclease. Thus, Rat1 will be able to degrade unspliced mRNA only if it is uncapped. We therefore examined the capping pattern of unspliced transcripts. RNA was isolated from mutant rat1-1 cells grown at 25°C and 37°C and subjected to immunoprecipitation (RNA-IP) using antibodies against the 7′-methylguanosine cap. Immunoprecipitated RNA was subjected to RT-PCR for APE2, STO1, RPS14A and RPS9B as described previously. 5S rRNA was used as a normalization control to ensure that equal amounts of RNA were used for immunoprecipitation. As expected, there was an approximate 7-fold increase in the amount of unspliced mRNA of APE2, STO1, RPS14A and RPS9B at 37°C compared to 25°C (Figure 4B) and there was no accumulation of unspliced transcripts with 7′-methyl guanosine caps in the wild type strain (Supplementary information, Figure S2). About 70–80% of both spliced and unspliced transcripts of the four genes analyzed could be immunoprecipitated by anti-7′-methylguanosine antibodies (Figure 4B). This demonstrates that the degree of capping of unspliced mRNA is similar to that of spliced mRNA, and therefore Rat1 enzymatic activity is unlikely to be directed to unspliced transcripts by decapping. We conclude that the accumulation of unspliced mRNA in the absence of Rat1 activity is not likely due to a surveillance role of Rat1 in degrading intron-containing transcripts.
Figure 4.
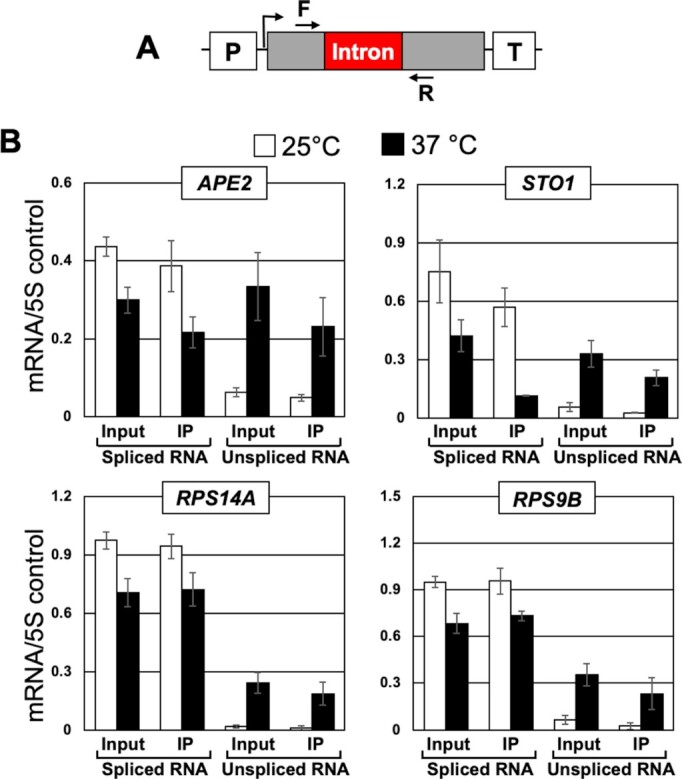
RNA immunoprecipitation analysis shows that unspliced transcripts in rat1-1 mutant are capped. (A) Schematic depiction of a gene showing the position of primers F and R used in RT-PCR analysis. (B) Affinity purified RNA using anti-m7G were reverse transcribed. Quantification of data shown in for the indicated genes in rat1-1 mutant at the indicated temperatures. 5S rRNA was used as normalization control. The error bars represent one unit of standard deviation. The results shown are an average of six independent immunoprecipitates from three separate RNA preparations from three independently grown cultures.
Accumulation of unspliced mRNA in the absence of Rat1 is not the consequence of defective termination
Rat1 and its homolog Xrn2 are transcription termination factors. We reasoned that if appearance of unspliced transcripts in the absence of Rat1 activity is due to an indirect effect of defective termination on splicing, other termination-defective mutants may also display a splicing defect.
Termination of transcription by RNAPII in yeast requires the CF1 and CPF complexes. Both complexes are required for cleavage and polyadenylation of the messenger RNA. The cleavage-polyadenylation of primary transcripts is coupled to the poly(A)-dependent termination of transcription. A recent genomewide analysis revealed that Rat1 is required for termination of nearly 35% of yeast genes (23). In contrast, CF1 and CPF subunits have a more robust role as they affect poly(A)-dependent termination of nearly 90% of genes (23). To determine if accumulation of unspliced transcripts is an indirect effect of defective termination in the Rat1 mutant, we monitored levels of spliced and unspliced mRNA of APE2, STO1, ASC1 and IMD4 in the termination-defective rna14-1 mutant. Rna14 is a subunit of CF1 complex and are essential for poly(A)-dependent termination (44). There was a decrease in the level of polyadenylated mRNA of APE2, STO1, ASC1 and IMD4 upon shifting of cells to the elevated temperature (Figure 5B, lane 4 and Figure 5C, black bars) in agreement with the need for termination to achieve optimal transcription. There was, however, no increase in the level of unspliced transcripts at the non-permissive temperature (Figure 5B, lane 4 and Figure 5C, black bars). The amount of unspliced mRNA in the mutant at 37°C was similar to that in the isogenic wild type strain (Figure 5C, black bars). To further probe the role of Rna14 in splicing, we measured splicing efficiency of APE2, STO1, ASC1 and IMD4 in the mutant at 37°C. Splicing of APE2 and ASC1 was not significantly affected in the mutant at 37°C, but STO1 and IMD4 exhibited about 20–30% decrease in splicing efficiency at the elevated temperature (Figure 5D). The possibility of Rna14 having a moderate effect on splicing of some genes therefore could not be ruled out. We therefore repeated the experiment with the mutant of Rna15, which is also a subunit of CF1A complex and is essential for poly(A)-dependent termination of transcription. In rna15-2 mutant, the level of spliced transcripts of IMD4, STO1, NMD2 and LSB4 decreased by 15–30% at 37°C (Supplementary information, Figure S3 B, black bars). There was, however, no appreciable increase in the level of unspliced transcripts at 37°C in the mutant (Supplementary information, Figure S3 B, black bars). Splicing efficiency of four genes analyzed here decreased by about 5–15% (Supplementary information, Figure S3C). This is in contrast to 25–60% decrease in splicing efficiency observed in the mutants of Rat1 (Figures 1 and 2). The defective termination in the Rna14 and Rna15 mutants therefore did not have an appreciable effect on splicing. The accumulation of unspliced mRNA in the Rat1 mutant thus may not be the consequence of defective termination.
Figure 5.

Defective termination in rna14-1 mutant has a moderate effect on splicing efficiency. (A) Schematic depiction of a gene showing the position of primers F and R used in RT-PCR analysis. (B) Gel pictures showing RT-PCR analysis of mRNA in wild type (WT) and rna14-1 mutant cells at the indicated temperatures. (C) Quantification of RT-PCR data for indicated genes in wild type (WT) and rna14-1 mutant at the indicated temperatures. 5S rRNA was used as normalization control. The results shown are an average of at least six independent PCRs from three separate RNA preparations from three independently grown cultures. The values and error bars for each condition indicate the mean ± standard deviation. Statistical significance (P-values) was determined using the two-tailed paired Student's t-test as described in (58). (D) Splicing efficiency representing the percentage of total transcripts undergoing splicing in WT and rna14-1 cells at the indicated temperatures.
Rat1 mediated effect on splicing is not by effecting the level of splicing factors
Rat1 may influence splicing indirectly by affecting the transcription of snRNAs or expression of protein factors involved in splicing. We therefore monitored the level of all five snRNA species and three splicing proteins, Prp2, Clf1 and Isy1 in Rat1-AA strain in the presence and absence of rapamycin. To measure levels of U1, U2, U4, U5 and U6 snRNAs, RNA was isolated from cell lysate and subjected to RT-PCR analyses. There was no appreciable change in the level of any of the five snRNA species in the presence of rapamycin in Rat1-AA strain (Supplementary information, Figure S4A, black bars). Isogenic wild type cells gave similar results. Next, we measured levels of Myc-tagged Prp2, Clf1 and Isy1 by Western blot in the cell lysate prepared from Rat1-AA strain. Again, there was no change in the level of three splicing proteins in the mutant in the presence of rapamycin (Supplementary information, Figure S4B). Rat1-dependent accumulation of unspliced transcripts, therefore, is not the consequence of an indirect effect of the protein on the level of splicing factors in the cell.
Does Rat1 facilitated pausing of polymerase during elongation of intronic regions enable the recruitment of splicing factors?
During cotranscriptional splicing, the polymerase slows down or pauses while transcribing intronic regions (45). This may enable recruitment of splicing factors, leading to efficient execution of the splicing reaction. Consequently, the increased elongation rate associated with some mutants of RNAPII produces splicing defects (45). The rat1-1 mutant also exhibits an enhanced RNAPII elongation rate by stimulating hyper-phosphorylation of CTD-serine-2 (24). The possibility of elongation speed contributing to the splicing defect observed in rat1-1 mutant thus could not be ruled out.
We therefore examined pausing of polymerase in the absence of Rat1 activity using Rpb1-ChIP-Seq data from Baejen et al. (23). This study analyzed genomewide transcription using Rpb1-ChIP-Seq approach following Rat1 depletion from the nucleus. We extracted Rpb1-ChIP-Seq data for 280 intron containing genes analyzed in this study. Rpb1-ChIP-Seq reads were aligned with respect to 5′ and 3′ splice sites of introns and included a 400 bp window upstream of the 5′ splice site and a 1 kbp window downstream of the 3′ splice site (Figure 6). There was no global change in polymerase density over the intron upon nuclear depletion of Rat1 (Figure 6A and B). Rpb1-ChIP profiles of individual genes confirms that the Rpb1 signal over the intron and splice sites is similar (Figure 6C). As expected, there was an increased RNAPII read-through signal in 1 kbp window downstream of termination sites upon nuclear depletion of Rat1, confirming a termination defect. These findings indicate that accumulation of unspliced transcripts in Rat1 mutants is not an indirect consequence of loss of pausing of the polymerase over intronic regions in the Rat1 mutant.
Figure 6.

RNAPII occupancy across the intronic genes after nuclear depletion of Rat1. (A) Heatmaps of Rpb1 occupancy levels calculated as the log2 IP/Input ratio in Rat1-FRB strain before (-rapamycin) and after (+rapamycin) nuclear depletion of Rat1 from ChIP-seq experiments performed in Baejen et.al., 2017. Genes are sorted in the descending order of Rpb1 occupancy. The y- axis indicates individual genes from the 280 intron containing genes and the x-axis shows relative position of 0.4 Kbp upstream and 1.0 kb downstream around 5′ ss and 3′ ss intronic sites. (B) Averaged normalized occupancy profiles from the ChIP-seq data of RNAPII. Metagene plot was designed with intron size averaged to 300 bp and plotting 0.4 Kbp upstream and 1Kbp downstream. The profiles are aligned at both the 5′ ss and 3′ ss of 280 intron containing genes. The shaded areas around the ChIP-seq profile represent standard error ±1. (C) IGB browser view of log2 IP/Input Rpb1 (RNAPII) read counts from ChIP-seq experiment performed in Rat1-FRB tagged strain before (blue) and after (red) nuclear depletion of Rat1. Four representative ORFs shown are YMR116C (ASC1), YHR203C (RPS4B), YML056C (IMD4), YCR031C (RPS14A).
Rat1 has a novel role in splicing of mRNA
None of the studies described above provide an explanation for accumulation of unspliced transcripts in the absence of functional Rat1. An alternative possibility is that Rat1 has a novel role in splicing of primary transcripts. Therefore, we examined the direct involvement of Rat1 in splicing by three parallel approaches. We reasoned that if Rat1 is involved in splicing, (1) it will physically interact with introns; (2) it will exhibit a transient or stable interaction with the splicing factors; and (3) it will facilitate the recruitment of splicing factors on the intron.
Rat1 is enriched over introns
We performed ChIP to determine if Rat1 is enriched over intronic DNA. A TAP-tag was inserted at the carboxy-terminus of Rat1. TAP-tag does not interfere with the termination function of Rat1. We used this strain for high resolution ChIP that employs extensive sonication as described in El Kaderi et al. (31). Splicing factors do not directly crosslink to the splice sites on DNA but interactions with the elongating transcript and CTD of RNAPII during cotranscriptional splicing provides sufficient indirect contacts to detect splicing factors by ChIP on DNA (46,47). To favor indirect contacts we used a more robust crosslinking approach, disuccinimidyl glutarate (DSG) together with formaldehyde, as described in GRID-Seq approach (48). We found that Rat1 crosslinks to the promoter and terminator regions of APE2 (Supplementary information, Figure S5, primers pairs A and E) in accordance with published findings (13,23). More importantly, Rat1 also localized to the intronic splice sites of APE2 (Supplementary information, Figure S5, primer pair C). The crosslinking of Rat1 to the splice sites suggest that Rat1 may have a direct role in splicing of APE2 mRNA.
We next examined if other intron-containing genes similarly exhibit crosslinking of Rat1 to the intron. We used ChIP-Seq data from Baejen et al. (23) for this analysis. We extracted data for intron-containing genes from this analysis. The Rat1 occupancy profile on 280 intron-containing genes, aligned to TSS and TES ± 400 bp, showed that Rat1 is localized at the terminator and promoter regions of genes (Supplementary information, Figure S6). We also analyzed occupancy of Pcf11, which is an essential termination factor and a component of CF1A 3′ end processing/termination complex, at TSS and TES as described above using the data from the same genomewide study. The Pcf11 and Rat1 occupancy profile were comparable at the termination region. They, however, differed considerably near the promoter. Rat1 occupancy was significantly higher than that of Pcf11 just downstream of the promoter (Supplementary information, Figure S6A). Most yeast introns are positioned near the TSS and the specific enrichment of Rat1 near the TSS might be due to its localization to the intron. We then examined the Rat1 and Pcf11 occupancy profiles of non-intronic genes and found them essentially identical (Supplementary information, Figure S6B). This suggests that the Rat1 enrichment at the TSS may be due in part to Rat1 crosslinking to the promoter-proximal intron.
Next, Rat1-ChIP-Seq reads were aligned with respect to the 5′ and 3′ splice sites of introns, encompassing a 100 bp window upstream of the 5′ splice sites and a 1 kbp window downstream of the 3′ splice sites as shown in Figure 7. Out of 280 intron-containing genes analyzed, 105 exhibited crosslinking of Rat1 to the intronic sequence. Thus, not all intron-containing genes show Rat1 occupancy. Unlike Rat1, Pcf11 does not exhibit significant localization to the intronic region (Figure 7A and B). These results indicate that Rat1 localizes to the introns of a subset of genes in budding yeast. We randomly selected five of the 105 intron-containing genes that exhibited Rat1-intron occupancy to test whether Rat1 occupancy correlated with Rat1-dependence on splicing. Out of these five genes, four; RPS11B, RPS9B, RPS4B and RPS14A were dependent on Rat1 for efficient splicing, while ECM33 tested negative (Supplementary information, Figure S7). Thus, not all introns that recruit Rat1 are dependent on Rat1 for splicing.
Figure 7.

Rat1 is selectively enriched in the intronic region of the genes. (A) Heatmaps of Rat1 and Pcf11 occupancy levels calculated as the log2 IP/Input ratio from ChIP-Seq experiments performed in Rat1-TAP and Pcf11-TAP strains, respectively. Genes are sorted in the descending order of the occupancy. The y- axis indicates individual genes from the 280 intron containing genes and the x-axis shows relative position of 0.1 kb upstream and 1.0 kb downstream around 5′ ss and 3′ ss intronic sites. (B) Averaged normalized occupancy profiles from the ChIP-Seq data of Rat1 (red) and Pcf11 (blue). The metagene plot was designed with an average intron size of 300 bp and plotting 0.1 kb upstream and 1Kbp downstream. The profiles are aligned at both the 5′ ss and 3′ ss of 280 intron containing genes. Wilcoxon test P-value (P = 1.22E–22). The shaded areas around the ChIP-Seq profile represent standard error ±1.
Next, we searched among the 105 Rat1-occupied introns for common features, which conferred potential dependence on Rat1 for their splicing. Rat1-occupied introns do not display common predicted structures or enriched sequence motifs. Of the features that we examined, only the size of the intron showed a significant correlation (P-value = 6.9 × 10–26). On average, introns that bound Rat1 were ∼400 bp long, while those not exhibiting Rat1 occupancy were ∼100 bp in length (Figure 8A). The distance between the branchpoint and 3′ splice site was the same for all introns (Figure 8B), but the distance between the 5′ splice site and branchpoint tended to be longer for Rat1-occupied introns (Figure 8C). Furthermore, Rat1 intron occupancy was not associated with the presence or absence of TATA-box in the promoter of the gene (P-value = 0.492) (Figure 8D). Ontological analysis revealed that the Rat1-occupied introns were enriched for ribosomal protein genes (Figure 8E).
Figure 8.

Rat1 crosslinked introns are longer. Genes with the presence and absence of Rat1 on intron are sorted using box-whisker plot according to intron length (A), distance between 3′ SS to branchpoint (B), distance between 5′ SS to branchpoint (C), and the promoter element TATA-box and TATA-less (D). Mann–Whitney U test was used to calculate P-value for A, B, and C. Fisher's exact test was used to calculate P-value for D. (E) Gene ontology analysis of intronic genes with Rat1 occupancy are enriched for translation/ribosomal proteins coding genes.
Rat1 interacts with splicing factors
If Rat1 i sinvolved in splicing, it may exhibit stable or transient interactions with the splicing machinery. To determine if splicing proteins interact with Rat1, we performed biochemical purification of Rat1 employing the Tandem Affinity Purification (TAP) approach. Purification was performed as described in Puig et al. (39) and the purified fraction was subjected to mass spectrometry (Supplementary information, Figure S8). A total of 1220 proteins were identified. Rai1 and Rtt103, as expected, were present in the purified fraction of Rat1. A significant finding was the identification of five splicing proteins, Clf1, Isy1, Yju2, Sub2 and Prp43 in the Rat1 preparation (Figure 9A). Clf1, Isy1 and Yju2, however, exhibited low spectral counts. To confirm the interaction, we HA-tagged the splicing factors Clf1, Isy1, Yju2, Sub2 and Prp43 and looked for Myc-tagged Rat1 in coimmunoprecipitated fraction by western blot. Myc-tagged Rat1 coimmunoprecipitated with all five splicing factors (Figure 9B). No signal for Rat1 was observed when the experiment was repeated in a strain without Myc-tagged Rat1. Reciprocal coimmunoprecipitation with Myc-tagged Rat1 produced similar findings but with high background. To further corroborate the interaction of Rat1 with the splicing factors described above, we repeated Rat1 coimmunoprecipitation for Prp2 and Prp16. These two yeast-specific splicing factors were not detected in our mass spectrometric analysis and no signal for Prp2 or Prp16 was detected following Rat1 pull down (data not presented). Of the five Rat1 interacting splicing factors, Clf1, Isy1, and Yju2 are the subunits of NineTeen complex (NTC). Clf1 and Isy1 are part of the core complex and Yju2 is an accessory protein. Clf1 is an essential splicing factor that serves as a scaffold in spliceosome assembly during assembly of the tri-snRNP (U4 U5.U6). It also interacts with the branch point binding proteins Mud2 and Prp40 (49). Isy1 is not an essential splicing factor but contributes to the fidelity of the splicing reaction (50). Yju2 is an essential splicing factor that functions after Prp2 to promote the first transesterification reaction (51). Sub2, a DEAD-box RNA helicase, is known to collaborate with Msl5 that interacts with branchpoint binding protein to promote the recruitment of U2 snRNP (52). Like Sub2, Prp43 is an RNA helicase; however, it functions post splicing in disassembling the U2, U5 and U6 snRNPs (53). The splicing factors, however, were not present in stoichiometric proportion to Rat1. This suggests that Rat1 is not in a stable complex with splicing factors, but transiently interacts with them during cotranscriptional splicing.
Figure 9.

Rat1 physically interacts with splicing factors. (A) Table from mass spectrometry analysis showing presence of Rat1-interacting proteins in tandem affinity-purified Rat1 preparation. Previously known interactors of Rat1 are shown in red (Rai1 and Rtt103) and purple (Las1 and Grc3). Splicing factors (Sub2, Prp43, Isy1, Yju2 and Clf1) are shown in blue. Rat1-interactors are listed relative to the levels of Rat1 in the preparation. (B) Coimmunoprecipitation followed by Western blot analysis confirmed Rat1 interaction with splicing factors Clf1, Isy1, Yju2, Sub2 and Prp43.
Recruitment of splicing factors is compromised in the absence of Rat1
Rat1 interaction with intronic sequences as well as its interaction with Isy1, Prp43, Clf1, Sub2 and Yju2 splicing factors strongly suggested a direct involvement of Rat1 in splicing of primary transcripts of a subset of genes. To further probe the role of Rat1 in splicing, we monitored the recruitment of splicing factors on introns. We reasoned that if Rat1 is indeed playing a role in splicing, recruitment of some splicing factors on the intron may be affected in the absence of Rat1. We chose Prp2 for three reasons; (i) it exhibits a genetic interaction with Rat1; (ii) it is recruited to the intron before Yju2 which is the Rat1 interacting factor identified above in our analysis and (iii) its recruitment on the intron can be detected by ChIP. We checked binding of Prp2 to the intron of APE2, ASC1, IMD4 by ChIP in the Rat1-AA strain in the presence and absence of rapamycin. Prp2-ChIP signal could be detected on the intron of APE2, ASC1 and IMD4 in the absence of rapamycin (Figure 10B, region B/C, white bars). But when Rat1 is depleted from the nucleus, the Prp2-ChIP signal registered a nearly 30–50% decline for all three genes (Figure 10B, region B/C, black bars). To further probe the role of Rat1 in recruitment of splicing factors, we monitored recruitment of two more splicing factors, Clf1 and Isy1 on intronic region of APE2, ASC1 and IMD4 in Rat1-AA strain in the presence and absence of rapamycin. There was nearly 25–70% decline in the recruitment of Clf1 and Isy1 on intron in the presence of rapamycin (Figure 10B, region B/C, black bars). There was, however, no change in the RNAPII signal over the intronic region of three genes (Supplementary information S9, regions B/C, black bars), thereby suggesting that reduced recruitment of splicing factors on the intron in the absence of Rat1 in the nucleus is not due to reduced transcription of these genes. Furthermore, reduced intronic recruitment of splicing factors was not due to a decrease in the level of the factors in the absence of a functional Rat1 in the cell as the levels of Prp2, Clf1 and Isy1 remained unchanged in the presence of rapamycin (Supplementary information, Figure S4B). These findings are in agreement with the observation that splicing of APE2, ASC1 and IMD4 introns is not completely dependent on Rat1 but decreases by about 50% in the absence of Rat1 activity. Taken together, these studies strongly suggest that Rat1 has a direct role in splicing. Furthermore, Rat1 is not an essential splicing factor but enhances the efficiency of splicing of a subset of yeast genes.
Figure 10.

Crosslinking of Prp2, Clf1 and Isy1 to intronic region is reduced in the absence of a functional Rat1 in the nucleus. (A) Schematic depiction of a gene indicating the position of ChIP primer pairs A, B, C and D. (B) ChIP analysis showing crosslinking of Prp2, Clf1 and Isy1 to different regions of APE2, ASC1 and IMD4 in FRB-tagged Rat1 strain in the presence and absence of rapamycin. Input controls were used to normalize the PCR amplification efficiency of each primer pair. The values and error bars for each condition indicate the mean ± standard deviation. Statistical significance (P-values) was determined using the two-tailed paired Student's t-test as described in (58).
DISCUSSION
Xrn2 has been implicated in degradation of unspliced mRNA in humans (54). A report suggested involvement of Rat1 in degrading unspliced transcripts in budding yeast as well (9). In a temperature-sensitive mutant of the essential splicing factor Prp2 called prp2-1, splicing was compromised, and unspliced transcripts could be detected at the elevated temperature. In the double mutant prp2-1/rat1-1, there was a further increase in the amount of unspliced transcripts of selected genes. The authors attributed this additional increase in the double mutant to the role of Rat1 in degrading unspliced transcripts by its exoribonuclease activity. Estimating the level of unspliced transcripts in the rat1-1 mutant alone, which is an important control, was not performed in this study. When we measured splicing in the rat1-1 mutant, we observed accumulation of unspliced transcripts at the non-permissive temperature even in cells with wild type Prp2. Nuclear depletion of Rat1 by anchor-away approach gave identical results indicating that the rat1-1 mutant does not have a secondary mutation in another splicing factor. We have three pieces of evidence to support that accumulation of unspliced transcripts in the absence of Rat1 in yeast cells is not due to the surveillance role of Rat1 in degrading unspliced transcripts. First, unspliced transcripts were capped. Capping of unspliced mRNA was almost to the same extent as that of spliced transcripts (Figure 4). Rat1 can degrade 5′ monophosphorylated transcripts, but not 7′-methyl guanosine capped transcripts. Second, Rat1 does not copurify with any known decapping protein. Xrn2 in humans coimmunoprecipitates with three decapping proteins; Edc3, Dcp1a and Dcp2 (10). Our mass spectrometric analysis of Rat1 did not yield any decapping proteins of yeast in affinity purified Rat1 preparation. Third, deletion of Rat1 interacting protein Rai1, which has been shown to remove the demethylated cap from mRNA in yeast under in vitro conditions (11), has absolutely no effect on the level of unspliced transcripts in yeast cells (Supplementary information, Figure S10). These results rule out the possibility of Rat1 degrading unspliced transcripts in budding yeast.
Rat1-dependent accumulation of unspliced mRNA can also be due to an indirect effect of Rat1 function in termination or elongation of transcription or on the level of splicing factors. We have four lines of evidence that unspliced transcripts are not produced because of defective termination in Rat1 mutants. First, unspliced transcripts were polyadenylated in rat1-1 as well as Rat1-AA cells (Figures 1, 2 and 3). Second, in the mutant of CF1 subunit Rna14 and Rna15, which affects the cleavage-polyadenylation step of termination, the amount of unspliced transcripts was similar to that in isogenic wild type cells and splicing efficiency was also not significantly affected. Third, in the absence of Rat1 interacting partner Rai1, which is not a termination factor itself but affects dissociation of the polymerase from the template by stimulating 5′→3′ exoribonuclease activity of Rat1, there was absolutely no increase in the level of unspliced mRNA over the wild type control (Supplementary information, Figure S10). Fourth, in the absence of Rtt103, which is not an essential termination factor but has been implicated in termination (13), there was no increase in the level of unspliced mRNA over the isogenic wild type strain (Supplementary information, Figure S10). Our results also demonstrate that generation of the unspliced transcript is not the result of an increased elongation rate associated with the rat1-1 mutant. In Rat1-AA strain, there was no reduction in polymerase signal over the intronic region, thereby suggesting that there was no decrease in pausing of the polymerase while transcribing the intronic region when Rat1 is nuclear depleted. Furthermore, there was no decrease in the steady state level of snRNA and splicing proteins in the absence of a functional Rat1 in the nucleus.
The results discussed above give credence to the hypothesis that Rat1 is playing a direct role in splicing of mRNA in budding yeast. We have three pieces of experimental evidence in support of involvement of Rat1 in splicing. First, genomewide ChIP analysis found Rat1 crosslinked to the intronic region of a number of intron-containing yeast genes (Figure 7). We expect a factor with a direct role in splicing to contact introns. Since splicing occurs cotranscriptionally, any protein that is involved in splicing gets indirectly crosslinked to the intronic regions on the gene through its direct contact with the transcribing RNA and the polymerase. Second, mass spectrometric analysis of tandem-affinity-purified Rat1 found at least five splicing factors, Sub2, Isy1, Prp43, Yju2 and Clf1, in the purified preparation (Figure 9). Three of these splicing proteins, Clf1, Isy1 and Yju2, are a part of the NineTeen complex that helps in activation of the assembled spliceosome. Third, recruitment of splicing factors Prp2, an essential DEAD-box containing splicing factor in yeast, as well as Clf1 and Isy1, which are part of the NineTeen complex, onto the intron is compromised in the absence of a functional Rat1 in the cell (Figure 10). Based on these results, we propose that Rat1 has a novel role in splicing of precursor mRNA in budding yeast. Rat1, however, is not an essential splicing factor as it crosslinks to less than 50% of intron containing yeast genes. Rat1 ChIP-Seq analysis revealed 105 out of 280 genes analyzed in the study showed crosslinking of Rat1 to introns. All of the genes that exhibited intronic Rat1 occupancy, however, are not dependent on the protein for removal of their introns. We believe that there are other factors that determine if Rat1 localization on the intron will require it to complete the splicing reaction. In C. elegans, several genes that exhibited 3′ end occupancy of Rat1 did not require Rat1 for termination. Rat1 facilitated termination was, in fact, dictated by the promoter of the gene (14). A similar mechanism might be determining if intron bound Rat1 plays a role in splicing in yeast. It is possible that either the promoter or terminator region of the gene determines the Rat1-dependence on splicing. Even considering the genes that require Rat1 for splicing, there is no complete loss of splicing in the absence of Rat1. On average, there is a 30–70% decrease in splicing efficiency in the absence of Rat1 for the genes that require it for removing an intron. These data suggest that Rat1 is not an essential splicing factor but has a rather stimulatory effect on the splicing process.
We searched for features of introns that made them dependent on Rat1 for efficient splicing. The 105 Rat1-occupying introns, which are relatively more dependent on Rat1 for splicing, were found to be approximately four-times longer in size compared to introns showing no Rat1 occupancy (Figure 8A). The longer size of Rat1-crosslinked introns was not due to a longer distance between the branchpoint and 3′ splice site, but due to a longer distance between the 5′ splice site and branchpoint (Figure 8B and C). These data suggest that Rat1 probably has a role in stabilizing the interaction of the branchpoint with 5′ splice sites of long introns, which is critical for the first transesterification reaction. We propose that Rat1 can do so due to its interaction with NineTeen complex. The distance between 5′ splice site and branchpoint, however, is not the only factor that determines Rat1-dependence on splicing as some introns with short 5′ splice site-branchpoint distance required Rat1 for splicing, while some introns with long 5′ splice site-branchpoint distance did not require Rat1 for splicing.
The observed effect of Rat1 on splicing is not due to Rat1-mediated degradation of unspliced transcripts. It is therefore surprising that Rat1 catalytic activity is required for its splicing function. The D235A mutation, which abolishes exoribonuclease activity of the protein, may be affecting some other property of Rat1 apart from its catalytic activity. The change from a negatively charged residue to a non-polar residue may bring about a change in the conformation of protein, which may affect its interaction with the spliceosomal complex. A thorough analysis is needed to uncover the role of D235A mutation in Rat1 splicing function.
Apart from Prp2, Rat1 is known to exhibit a genetic interaction with at least four splicing factors; Msl5, Yhc1, Sub2 and Prp46, in yeast thereby corroborating the role of Rat1 in splicing (9,55,56). A logical question is if Rat1 homolog Xrn2 plays a similar role in splicing in higher eukaryotes. Xrn2 is a component of in vivo assembled, purified splicing complexes in human HeLa cells and chicken DT-40 cells (57). These purified supraspliceosomes contained all five U-snRNPs and other known splicing proteins. In addition, they identified some novel proteins that are not known to play any role in splicing. Xrn2 was one such protein that was present in both human and chicken supraspliceosomes. The supraspliceosomes did not contain any other termination factor besides Xrn2. The possibility of Xrn2, like Rat1, playing a role in splicing of a subset of introns in higher eukaryotes therefore cannot be ruled out.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Claire Moore of Tufts University for kindly providing the plasmids expressing wild type and mutant Rat1. We acknowledge the assistance of the Wayne State University Proteomics Core that is supported through National Institutes of Health grants P30 ES020957, P30 CA 022453 and S10 OD010700. We are grateful to Dr Victoria Meller and Dr Jared Schrader of Wayne State University for critical reading of the manuscript. We thank members of our group, Katherine Dwyer and Michael O’ Brien for helpful suggestions. We thank Dan Spatt, Olga Viktorovskaya, and Catherine Weiner from Dr Fred Winston group for sharing reagents and materials for yeast. PGK1 and 8W16G antibodies was a kind gift from Winston group. We also thank Eric Wooten and Chris Nardone from Dr Stephen Elledge lab for sharing rapamycin. Lastly, we thank Hyuck Joon Kang and Dr Mitzi Kuroda for the necessary support at Brigham & Women's Hospital/Harvard Medical School in completing the experiments that are part of the revised manuscript. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Science Foundation.
Contributor Information
Zuzer Dhoondia, Department of Biological Sciences, Wayne State University, Detroit, MI 48202, USA.
Hesham Elewa, Department of Biological Sciences, Wayne State University, Detroit, MI 48202, USA.
Marva Malik, Department of Biological Sciences, Wayne State University, Detroit, MI 48202, USA.
Zahidur Arif, Department of Biological Sciences, Wayne State University, Detroit, MI 48202, USA.
Roger Pique-Regi, Center for Molecular Medicine and Genetics, Wayne State University School of Medicine, 540 East Canfield, Detroit, MI 48201, USA.
Athar Ansari, Department of Biological Sciences, Wayne State University, Detroit, MI 48202, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB1936030 to A.A.]. Funding for open access charge: National Science Foundation.
Conflict of interest statement. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
REFERENCES
- 1. Nagarajan V.K., Jones C.I., Newbury S.F., Green P.J.. XRN 5′→3′ exoribonucleases: structure, mechanisms and functions. Biochim. Biophys. Acta. 2013; 1829:590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson A.W. Rat1p and Xrn1p are functionally interchangeable exoribonucleases that are restricted to and required in the nucleus and cytoplasm, respectively. Mol. Cell. Biol. 1997; 17:6122–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kastenmayer J.P., Green P.J.. Novel features of the XRN-family in Arabidopsis: evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:13985–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Richter H., Katic I., Gut H., Großhans H.. Structural basis and function of XRN2-binding by XTB domains. Nat. Struct. Mol. Biol. 2016; 23:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shobuike T., Sugano S., Yamashita T., Ikeda H.. Characterization of cDNA encoding mouse homolog of fission yeast dhp1+ gene: structural and functional conservation. Nucleic Acids Res. 1995; 23:357–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sugano S., Shobuike T., Takeda T., Sugino A., Ikeda H.. Molecular analysis of the dhp1+ gene of Schizosaccharomyces pombe: an essential gene that has homology to the DST2 and RAT1 genes of Saccharomyces cerevisiae. Mol. Gen. Genet. 1994; 243:1–8. [DOI] [PubMed] [Google Scholar]
- 7. Zhang M., Yu L., Xin Y., Hu P., Fu Q., Yu C., Zhao S.. Cloning and mapping of the XRN2 gene to human chromosome 20p11.1–p11.2. Genomics. 1999; 59:252–254. [DOI] [PubMed] [Google Scholar]
- 8. Amberg D.C., Goldstein A.L., Cole C.N.. Isolation and characterization of RAT1: an essential gene of Saccharomyces cerevisiae required for the efficient nucleocytoplasmic trafficking of mRNA. Genes Dev. 1992; 6:1173–1189. [DOI] [PubMed] [Google Scholar]
- 9. Bousquet-Antonelli C., Presutti C., Tollervey D.. Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell. 2000; 102:765–775. [DOI] [PubMed] [Google Scholar]
- 10. Brannan K., Kim H., Erickson B., Glover-Cutter K., Kim S., Fong N., Kiemele L., Hansen K., Davis R., Lykke-Andersen J.et al.. mRNA decapping factors and the exonuclease Xrn2 function in widespread premature termination of RNA polymerase II transcription. Mol. Cell. 2012; 46:311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiao X., Xiang S., Oh C., Martin C.E., Tong L., Kiledjian M.. Identification of a quality-control mechanism for mRNA 5′-end capping. Nature. 2010; 467:608–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jimeno-González S., Haaning L.L., Malagon F., Jensen T.H.. The yeast 5′-3′ exonuclease Rat1p functions during transcription elongation by RNA polymerase II. Mol. Cell. 2010; 37:580–587. [DOI] [PubMed] [Google Scholar]
- 13. Kim M., Krogan N.J., Vasiljeva L., Rando O.J., Nedea E., Greenblatt J.F., Buratowski S.. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004; 432:517–522. [DOI] [PubMed] [Google Scholar]
- 14. Miki T.S., Carl S.H., Großhans H.. Two distinct transcription termination modes dictated by promoters. Genes Dev. 2017; 31:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. West S., Gromak N., Proudfoot N.J.. Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature. 2004; 432:522–525. [DOI] [PubMed] [Google Scholar]
- 16. Mischo H.E., Proudfoot N.J.. Disengaging polymerase: terminating RNA polymerase II transcription in budding yeast. Biochim. Biophys. Acta. 2013; 1829:174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Butler J.S., Platt T.. RNA processing generates the mature 3′ end of yeast CYC1 messenger RNA in vitro. Science. 1988; 242:1270–1274. [DOI] [PubMed] [Google Scholar]
- 18. Jenny A., Minvielle-Sebastia L., Preker P.J., Keller W.. Sequence similarity between the 73-kilodalton protein of mammalian CPSF and a subunit of yeast polyadenylation factor I. Science. 1996; 274:1514–1517. [DOI] [PubMed] [Google Scholar]
- 19. Eaton J.D., Davidson L., Bauer D.L.V., Natsume T., Kanemaki M.T., West S. Xrn2 accelerates termination by RNA polymerase II, which is underpinned by CPSF73 activity. Genes Dev. 2018; 32:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fong N., Brannan K., Erickson B., Kim H., Cortazar M.A., Sheridan R.M., Nguyen T., Karp S., Bentley D.L.. Effects of transcription elongation rate and Xrn2 exonuclease activity on RNA polymerase II termination suggest widespread kinetic competition. Mol. Cell. 2015; 60:256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin D.J., Burgess R.R., Richardson J.P., Gross C.A.. Termination efficiency at rho-dependent terminators depends on kinetic coupling between RNA polymerase and rho. Proc. Natl Acad. Sci. U.S.A. 1992; 89:1453–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pearson E.L., Moore C.L.. Dismantling promoter-driven RNA polymerase II transcription complexes in vitro by the termination factor Rat1. J. Biol. Chem. 2013; 288:19750–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baejen C., Andreani J., Torkler P., Battaglia S., Schwalb B., Lidschreiber M., Maier K.C., Boltendahl A., Rus P., Esslinger S.et al.. Genome-wide analysis of RNA polymerase II termination at protein-coding genes. Mol. Cell. 2017; 66:38–49. [DOI] [PubMed] [Google Scholar]
- 24. Jimeno-González S., Schmid M., Malagon F., Haaning L.L., Jensen T.H.. Rat1p maintains RNA polymerase II CTD phosphorylation balance. RNA. 2014; 20:551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nojima T., Gomes T., Grosso A.R.F., Kimura H., Dye M.J., Dhir S., Carmo-Fonseca M., Proudfoot N.J. Mammalian NET-Seq reveals genome-wide nascent transcription coupled to RNA processing. Cell. 2015; 161:526–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harlen K.M., Churchman L.S.. Subgenic Pol II interactomes identify region-specific transcription elongation regulators. Mol. Syst. Biol. 2017; 13:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wach A., Brachat A., Pöhlmann R., Philippsen P.. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast. 1994; 10:1793–1808. [DOI] [PubMed] [Google Scholar]
- 28. McNeil J.B., Smith M.. Transcription initiation of the Saccharomyces cerevisiae iso-1-cytochrome c gene: multiple, independent T-A-T-A sequences. J. Mol. Biol. 1986; 187:363–378. [DOI] [PubMed] [Google Scholar]
- 29. El Kaderi B., Medler S., Ansari A.. Analysis of interactions between genomic loci through Chromosome Conformation Capture (3C). Curr. Protoc. Cell Biol. 2012; doi:10.1002/0471143030.cb2215s56. [DOI] [PubMed] [Google Scholar]
- 30. Dhoondia Z., Tarockoff R., Alhusini N., Medler S., Agarwal N., Ansari A.. Analysis of termination of transcription using BrUTP-strand-specific transcription run-on (TRO) approach. J. Vis. Exp. 2017; doi:10.3791/55446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Kaderi B., Medler S., Raghunayakula S., Ansari A.. Gene looping is conferred by activator-dependent interaction of transcription initiation and termination machineries. J. Biol. Chem. 2009; 284:25015–25025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Afgan E., Baker D., van den Beek M., Blankenberg D., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Eberhard C.et al.. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016; 44:W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramírez F., Ryan D.P., Grüning B., Bhardwaj V., Kilpert F., Richter A.S., Heyne S., Dündar F., Manke T.. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016; 44:W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nicol J.W., Helt G.A., Blanchard S.G., Raja A., Loraine A.E.. The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009; 25:2730–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Basehoar A.D., Zanton S.J., Pugh B.F.. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004; 116:699–709. [DOI] [PubMed] [Google Scholar]
- 37. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T.et al.. Gene Ontology: tool for the unification of biology. Nat. Genet. 2000; 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Svejstrup J.Q., Petrakis T.G., Fellows J.. Purification of elongating RNA polymerase II and other factors from yeast chromatin. Methods Enzymol. 2003; 371:491–498. [DOI] [PubMed] [Google Scholar]
- 39. Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B.. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001; 24:218–229. [DOI] [PubMed] [Google Scholar]
- 40. Haruki H., Nishikawa J., Laemmli U.K.. The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell. 2008; 31:925–932. [DOI] [PubMed] [Google Scholar]
- 41. Belshaw P.J., Ho S.N., Crabtree G.R., Schreiber S.L.. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc. Natl Acad. Sci. U.S.A. 1996; 93:4604–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen J., Zheng X.F., Brown E.J., Schreiber S.L.. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc. Natl Acad. Sci. U.S.A. 1995; 92:4947–4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Core L.J., Waterfall J.J., Lis J.T.. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008; 322:1845–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minvielle-Sebastia L., Preker P.J., Wiederkehr T., Strahm Y., Keller W.. The major yeast poly(A)-binding protein is associated with cleavage factor IA and functions in premessenger RNA 3′-end formation. Proc. Natl Acad. Sci. U.S.A. 1997; 94:7897–7902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Braberg H., Jin H., Moehle E.A., Chan Y.A., Wang S., Shales M., Benschop J.J., Morris J.H., Qiu C., Hu F.et al.. From structure to systems: high-resolution, quantitative genetic analysis of RNA polymerase II. Cell. 2013; 154:775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herzel L., Ottoz D.S.M., Alpert T., Neugebauer K.M. Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol. 2017; 18:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nojima T., Rebelo K., Gomes T., Grosso A.R., Proudfoot N.J., Carmo-Fonseca M.. RNA polymerase II phosphorylated on CTD serine 5 interacts with the spliceosome during co-transcriptional splicing. Mol. Cell. 2018; 72:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li X., Zhou B., Chen L., Gou L.-T., Li H., Fu X.-D.. GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 2017; 35:940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chung S., McLean M.R., Rymond B.C.. Yeast ortholog of the Drosophila crooked neck protein promotes spliceosome assembly through stable U4/U6.U5 snRNP addition. RNA. 1999; 5:1042–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Villa T., Guthrie C.. The Isy1p component of the nineteen complex interacts with the ATPase Prp16p to regulate the fidelity of pre-mRNA splicing. Genes Dev. 2005; 19:1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu Y.-C., Chen H.-C., Wu N.-Y., Cheng S.-C.. A novel splicing factor, Yju2, is associated with NTC and acts after Prp2 in promoting the first catalytic reaction of pre-mRNA splicing. Mol. Cell. Biol. 2007; 27:5403–5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhang M., Green M.R.. Identification and characterization of yUAP/Sub2p, a yeast homolog of the essential human pre-mRNA splicing factor hUAP56. Genes Dev. 2001; 15:30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arenas J.E., Abelson J.N.. Prp43: An RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl Acad. Sci. U.S.A. 1997; 94:11798–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Davidson L., Kerr A., West S.. Co-transcriptional degradation of aberrant pre-mRNA by Xrn2. EMBO J. 2012; 31:2566–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Costanzo M., VanderSluis B., Koch E.N., Baryshnikova A., Pons C., Tan G., Wang W., Usaj M., Hanchard J., Lee S.D.et al.. A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016; 353:aaf1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. González-Aguilera C., Tous C., Gómez-González B., Huertas P., Luna R., Aguilera A.. The THP1-SAC3-SUS1-CDC31 complex works in transcription elongation-mRNA export preventing RNA-mediated genome instability. Mol. Biol. Cell. 2008; 19:4310–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen Y.-I.G., Moore R.E., Ge H.Y., Young M.K., Lee T.D., Stevens S.W.. Proteomic analysis of in vivo-assembled pre-mRNA splicing complexes expands the catalog of participating factors. Nucleic Acids Res. 2007; 35:3928–3944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Student The probable error of a mean. Biometrika. 1908; 6:1–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.