


Abstract
Sequence-based analysis and prediction are fundamental bioinformatic tasks that facilitate understanding of the sequence(-structure)-function paradigm for DNAs, RNAs and proteins. Rapid accumulation of sequences requires equally pervasive development of new predictive models, which depends on the availability of effective tools that support these efforts. We introduce iLearnPlus, the first machine-learning platform with graphical- and web-based interfaces for the construction of machine-learning pipelines for analysis and predictions using nucleic acid and protein sequences. iLearnPlus provides a comprehensive set of algorithms and automates sequence-based feature extraction and analysis, construction and deployment of models, assessment of predictive performance, statistical analysis, and data visualization; all without programming. iLearnPlus includes a wide range of feature sets which encode information from the input sequences and over twenty machine-learning algorithms that cover several deep-learning approaches, outnumbering the current solutions by a wide margin. Our solution caters to experienced bioinformaticians, given the broad range of options, and biologists with no programming background, given the point-and-click interface and easy-to-follow design process. We showcase iLearnPlus with two case studies concerning prediction of long noncoding RNAs (lncRNAs) from RNA transcripts and prediction of crotonylation sites in protein chains. iLearnPlus is an open-source platform available at https://github.com/Superzchen/iLearnPlus/ with the webserver at http://ilearnplus.erc.monash.edu/.
INTRODUCTION
High-throughput sequencing has significantly advanced and experienced widespread use over the past few decades, generating the unprecedented volume of the DNAs, RNAs and protein sequence data. With the fast accumulation of these data, effectively analyzing, mining and visualizing biological sequences have become a non-trivial task (1). Among a variety of computational solutions, machine-learning methods are a popular and efficient solution for the accurate function prediction/analysis for biological sequences (2–7). Many sequence-based machine-learning approaches have been proposed, contributing to a better understanding of the functions and structures of DNAs, RNAs and proteins (8–10), particularly in the context of human disease (11–13). Despite the diversity of machine-learning frameworks for the sequence analysis and prediction, in general, they follow the same set of five major steps after the sequence data was collected: feature extraction, feature analysis, classifier construction, performance evaluation, and data/result visualization, as demonstrated in Figure 1.
Figure 1.

A summary of the five major steps involved in the development of machine learning-based models for biological sequences analysis. These steps include feature extraction, feature analysis, classifier construction, performance evaluation, and data/result visualization.
Bioinformatics-driven data analysis is an essential part of biological studies. The sequence-based analysis and predictions often require complex processing steps, data science expertise, and access to sophisticated software. These requirements have become a significant hurdle, especially for biologists with limited bioinformatics expertise. Several web servers and standalone software packages for the sequence-based analysis and prediction have been recently developed to meet these needs. Representative tools include iFeature (14), iLearn (15), Selene (16), Kipoi (17), Janggu (18), BioSeq-Analysis (19) and BioSeq-Analysis2.0 (20). Selene is PyTorch-based deep-learning library for rapid development, training, and application of deep-learning models from biological sequences. Janggu is a python package that similarly focuses on the deep learning models. Kipoi is a collaborative initiative that defines standards and fosters reuse of trained models. However, these three tools cover only a portion of the complete pipeline outlined in Figure 1. We provide a detailed side-by-side comparison in Supplementary Table S1. BioSeq-Analysis is regarded as the first automated platform for machine learning-based bioinformatics analysis and predictions at the sequence level (19). Its subsequent version, BioSeq-Analysis2.0 covers residue-level analysis, further improving the scope of this platform (20). In 2018, we released the first computational pipeline, iFeature, that generates features for both protein and peptide sequences. Later, we extended iFeature to design and implement iLearn, which is an integrated platform and meta-learner for feature engineering, machine-learning analysis and modelling of DNA, RNA and protein sequence data. Both platforms, iFeature and iLearn, have been applied in many areas of bioinformatics and computational biology including but not limited to the prediction and identification of mutational effects (21), protein-protein interaction hotspots (22), drug-target interactions (23), protein crystallization propensity (24), DNA-binding sites (25) and DNA-binding proteins (26), protein families (27,28), and DNA, RNA and protein modifications (29–32). The breadth and number of these applications show a substantial need for such solutions. However, further work is needed. First, new platforms need to overcome limitations of the current solutions in terms of streamlining and easiness of use, so that they make a sophisticated machine-learning based analysis of biological sequence accessible to both experienced bioinformaticians and biologists with limited programming background. This means that the development of the complex predictive and analytical pipelines should be streamlined by providing one platform that handles and offers support for the entire computational process. Second, the current platforms offer limited facilities for feature extraction, feature analysis and classifier construction. This calls for new approaches that provide a more comprehensive and molecule-specific (DNA veruss RNA versus protein) set of feature descriptors, a broader range of tools for feature analysis, and which should ideally cover state-of-the-art machine-learning algorithms including deep learning.
To this end, we release a comprehensive and automated sequence analysis and prediction platform, iLearnPlus, implemented in Python/PyQt5. iLearnPlus works across all major operating systems (i.e. Windows, macOS and Linux). Our platform includes four modules: iLearnPlus-Basic, iLearnPlus-Estimator, iLearnPlus-AutoML and iLearnPlus-LoadModel. These modules support a wide range of functionality, such as feature extraction, feature analysis, construction of machine-learning framework, training of machine-learning models/classifiers, assessment of predictive performance for these models, statistical analysis, and data/result visualization. iLearnPlus is geared to be used by both experienced bioinformaticians and biologists with limited bioinformatics expertise. When compared to the currently available tools (Supplementary Table S2), iLearnPlus offers the following key advantages:
To the best of our knowledge, iLearnPlus is the first GUI-based platform that facilitates machine learning-based analysis and prediction of biological sequences;
iLearnPlus outperforms the existing platforms in the number of the available machine-learning algorithms and the coverage of features produced from the input sequences: 21 machine-learning algorithms (12 conventional machine-learning methods, two ensemble-learning frameworks and seven deep-learning approaches) and 19 classes of features that cover 147 feature sets;
iLearnPlus provides a variety of ways to visualize the user-defined data and prediction performance including scatter plots, ROC (Receiver Operating Characteristic) curves, PRC (Precision-Recall Curves), histograms, kernel density plots, heatmaps and boxplots;
iLearnPlus supports two popular statistical tests: the Student's t-test and bootstrap test (33), to assess the statistical significance of differences and improvements in the context of the model performance;
iLearnPlus provides the iLearnPlus-AutoML module for evaluating the prediction performance of different machine-learning models and selecting the best-performing model via automatic parameter optimization, to support less data science-savvy users in maximizing the predictive capability of machine-learning pipelines;
iLearnPlus facilitates the deployment of the developed models with the iLearnPlus-LoadModel module. This module applies the already trained machine-learning models on new data;
iLearnPlus provides more options for model integration by exploring possible combinations of the prediction outcomes of separate models as the input, and retrain another machine-learning model (excluding the deep-learning approaches), to assess if the prediction performance can be further improved; and
iLearnPlus provides auxiliary functionalities for data preprocessing, such as file format transformations and combination of multiple feature encodings into one file.
iLearnPlus offers user-friendly interface and integrates four functional modules that streamline the entire computational process related to analysis and sequence-based prediction of the DNA, RNA and protein sequences. This ‘one-stop’ solution facilitates generation of biological hypotheses by supporting the design, testing and deployment of accurate predictive models. Following, we describe the features and capabilities of our platform for each of the five major steps defined above. We also demonstrate its application with two case studies that concern the development and testing of novel machine-learning models for the predictions of long noncoding RNAs (lncRNAs) and crotonylation sites in protein chains.
MATERIALS AND METHODS
Feature extraction
The feature extraction functionality in the iLearnPlus-Basic module generates numeric vectors from biological sequences. These vectors encode biochemical, biophysical, and compositional properties of the input sequences in the format that is compatible with the subsequent machine-learning tasks. iLearnPlus incorporates 19 major classes of features for protein, RNA and DNA sequences (Tables 1 and 2). To compare, iLearnPlus outnumbers the current platforms, including iLearn (15), iFeature (14) and BioSeq-Analysis2.0 (20) by 50, 94 and 31 feature sets. Supplementary Table S2 provides a detailed side-by-side comparison of the feature set numbers that these platforms offer for the DNA, RNA and protein sequences. The input sequences of iLearnPlus are required to be in the FASTA format. We designed an extended version of the header line (standard FASTA format is accepted; refer to the online instructions for more detail) which is processed by the graphical input data explorer. The biological sequence type (i.e. DNA, RNA or protein) is detected automatically based on the input sequences. The lists of the feature sets are provided in Table 1 (for protein sequences) and Table 2 (for DNA and RNA sequences), and can be customized by users. The selected subset of feature sets is output with a convenient table widget, which includes the molecule name, molecule label, feature (column) names and the corresponding values. iLearnPlus supports four formats for saving the calculated features, including LIBSVM format, Comma-Separated Values (CSV), Tab Separated Values (TSV), and Waikato Environment for Knowledge Analysis (WEKA) format. This variety of popular formats facilitates direct use of the features in third-party computational tools, such as scikit-learn (34), WEKA (35) and its web interface.
Table 1.
Feature descriptors calculated by iLearnPlus for protein sequences
| Descriptor group | Descriptor (abbreviation) | Reference |
|---|---|---|
| Amino acid composition | Amino acid composition (AAC) | (37) |
| Enhanced amino acid composition (EAAC) | (14,15) | |
| Composition of k-spaced amino acid pairs (CKSAAP) | (38,39) | |
| Kmer (dipeptides and tripeptides) composition (DPC and TPC) | (37,40) | |
| Dipeptide deviation from expected mean (DDE) | (40) | |
| Composition (CTDC) | (41–45) | |
| Transition (CTDT) | (41–45) | |
| Distribution (CTDD) | (41–45) | |
| Conjoint triad (CTriad) | (46) | |
| Conjoint k-spaced Triad (KSCTriad) | (14,15) | |
| Adaptive skip dipeptide composition (ASDC) | (47) | |
| PseAAC of distance-pairs and reduced alphabet (DistancePair) | (20,48) | |
| Grouped amino acid composition | Grouped amino acid composition (GAAC) | (14,15) |
| Grouped enhanced amino acid composition (GEAAC) | (14,15) | |
| Composition of k-spaced amino acid group pairs (CKSAAGP) | (14,15) | |
| Grouped dipeptide composition (GDPC) | (14,15) | |
| Grouped tripeptide composition (GTPC) | (14,15) | |
| Autocorrelation | Moran (Moran) | (49,50) |
| Geary (Geary) | (51) | |
| Normalized Moreau-Broto (NMBroto) | (52) | |
| Auto covariance (AC) | (53–55) | |
| Cross covariance (CC) | (53–55) | |
| Auto-cross covariance (ACC) | (53–55) | |
| Quasi-sequence-order | Sequence-order-coupling number (SOCNumber) | (56–58) |
| Quasi-sequence-order descriptors (QSOrder) | (56–58) | |
| Pseudo-amino acid composition | Pseudo-amino acid composition (PAAC) | (59,60) |
| Amphiphilic PAAC (APAAC) | (59,60) | |
| Pseudo K-tuple reduced amino acids composition (PseKRAAC_type 1 to type 16) | (61) | |
| Residue composition | Binary - 20bit (binary) | (62,63) |
| Binary - 6bit (binary_6bit) | (20,64) | |
| Binary - 5bit (binary_5bit_type 1 and type 2) | (20,65) | |
| Binary - 3bit (binary_3bit_type 1 to type 7) | (47) | |
| Learn from alignments (AESNN3) | (20,66) | |
| Overlapping property features - 10 bit (OPF_10bit) | (47) | |
| Overlapping property features - 7 bit (OPF_7bit type 1 to type 3) | (47) | |
| Physicochemical property | AAIndex (AAIndex) | (67) |
| BLOSUM matrix | BLOSUM62 (BLOSUM62) | (68) |
| Z-Scale index | Z-Scale (Zscale) | (69) |
| Similarity-based descriptor | K-nearest neighbor (KNN) | (70) |
Table 2.
Feature descriptors calculated by iLearnPlus for DNA and RNA sequences
| Descriptor group | Descriptor (abbreviation) | Sequence type | Reference |
|---|---|---|---|
| Nucleic acid composition | Nucleic acid composition (NAC) | DNA/RNA | (15) |
| Enhanced nucleic acid composition (ENAC) | DNA/RNA | (15) | |
| k-spaced nucleic acid pairs (CKSNAP) | DNA/RNA | (15) | |
| Basic kmer (Kmer) | DNA/RNA | (71) | |
| Reverse compliment kmer (RCKmer) | DNA | (72,73) | |
| Accumulated nucleotide frequency (ANF) | DNA/RNA | (74) | |
| Nucleotide chemical property (NCP) | DNA/RNA | (74) | |
| The occurrence of kmers, allowing at most m mismatches (Mismatch) | DNA/RNA | (20) | |
| The occurrences of kmers, allowing non-contiguous matches (Subsequence) | DNA/RNA | (20) | |
| Adaptive skip dinucleotide composition (ASDC) | DNA/RNA | (47) | |
| Local position-specific dinucleotide frequency (LPDF) | DNA/RNA | (75) | |
| The Z curve parameters for frequencies of phase-specific mononucleotides (Z_curve_9bit) | DNA/RNA | (76) | |
| The Z curve parameters for frequencies of phase-independent dinucleotides (Z_curve_12bit) | DNA/RNA | (76) | |
| The Z curve parameters for frequencies of phase-specific dinucleotides (Z_curve_36bit) | DNA/RNA | (76) | |
| The Z curve parameters for frequencies of phase-independent trinucleotides (Z_curve_48bit) | DNA/RNA | (76) | |
| The Z curve parameters for frequencies of phase-specific trinucleotides (Z_curve_144bit) | DNA/RNA | (76) | |
| Residue composition | Binary (binary) | DNA/RNA | (62,63) |
| Dinucleotide binary encoding (DBE) | DNA/RNA | (75) | |
| Position-specific of two nucleotides (PS2) | DNA/RNA | (20,77) | |
| Position-specific of three nucleotides (PS3) | DNA/RNA | (20,77) | |
| Position-specific of four nucleotides (PS4) | DNA/RNA | (20,77) | |
| Position-specific tendencies of trinucleotides | Position-specific trinucleotide propensity based on single-strand (PSTNPss) | DNA/RNA | (78,79) |
| Position-specific trinucleotide propensity based on double-strand (PSTNPds) | DNA | (78,79) | |
| Electron-ion interaction pseudopotentials | Electron-ion interaction pseudopotentials value (EIIP) | DNA | (80,81) |
| Electron-ion interaction pseudopotentials of trinucleotide (PseEIIP) | DNA | (80,81) | |
| Autocorrelation and cross-covariance | Dinucleotide-based auto covariance (DAC) | DNA/RNA | (53–55) |
| Dinucleotide-based cross covariance (DCC) | DNA/RNA | (53–55) | |
| Dinucleotide-based auto-cross covariance (DACC) | DNA/RNA | (53–55) | |
| Trinucleotide-based auto covariance (TAC) | DNA | (53) | |
| Trinucleotide-based cross covariance (TCC) | DNA | (53) | |
| Trinucleotide-based auto-cross covariance (TACC) | DNA | (53) | |
| Moran (Moran) | DNA/RNA | (49,50) | |
| Geary (Geary) | DNA/RNA | (51) | |
| Normalized Moreau-Broto (NMBroto) | DNA/RNA | (52) | |
| Physicochemical property | Dinucleotide physicochemical properties (DPCP type 1 and type 2) | DNA/RNA | (82) |
| Trinucleotide physicochemical properties (TPCP type 1 and type 2) | DNA | (82) | |
| Mutual information | Multivariate mutual information (MMI) | DNA/RNA | (83) |
| Similarity-based descriptor | K-nearest neighbor (KNN) | DNA/RNA | (83) |
| Pseudo nucleic acid composition | Pseudo dinucleotide composition (PseDNC) | DNA/RNA | (53,84) |
| Pseudo k-tupler composition (PseKNC) | DNA/RNA | (53,84) | |
| Parallel correlation pseudo dinucleotide composition (PCPseDNC) | DNA/RNA | (53,84) | |
| Parallel correlation pseudo trinucleotide composition (PCPseTNC) | DNA | (53,84) | |
| Series correlation pseudo dinucleotide composition (SCPseDNC) | DNA/RNA | (53,84) | |
| Series correlation pseudo trinucleotide composition (SCPseTNC) | DNA | (53,84) |
Besides the data tables, iLearnPlus provides advanced facilities to visualize the data. For instance, it generates hybrid plots that overlay kernel density curves and histograms (Figure 2) that can be used to shed light on the statistical distributions of the extracted features. The histogram provides a visual representation of feature values grouped into discrete intervals, while the kernel density approach produces a smooth curve that represents the probability density function for continuous variables (36) (Figure 2A). The visualization can be conducted for a specific dataset as well as a selected subset of features in that dataset.
Figure 2.

An example of hybrid plots for the extracted features generated by iLearnPlus. The histogram and kernel density plot for visualization of data distribution (A), and the scatter plot for dimension reduction result (B). The N1-methyladenosine dataset from (30) was used to display the example plots.
Feature analysis
Feature analysis is an optional but highly-recommended step that helps to eliminate irrelevant, noisy, or redundant features from the original feature set, with the overarching goal to optimize the predictive performance of the subsequently used machine-learning algorithm(s) (32). iLearnPlus provides multiple options to facilitate feature analysis, including ten feature clustering, three dimensionality reduction, two feature normalization and five feature selection approaches (Table 3). Compared with iLearn, the currently most comprehensive platform in the context of feature analysis (Supplementary Table S1), iLearnPlus provides four additional clustering algorithms: the Mini Batch k-means Clustering (85,86), Markov Clustering (MCL) (87), Agglomerative Clustering (88), and Spectral Clustering (89). The feature analysis supports the same comprehensive list of the file formats as the feature extraction tools (i.e. LIBSVM format, CSV, TSV and WEKA format).
Table 3.
The feature analysis approaches provided in iLearnPlus
| Method | Algorithm (abbreviation) | Reference |
|---|---|---|
| Clustering | k-means (kmeans) | (85,86) |
| Mini-Batch K-means (MiniBatchKMeans) | (85,86) | |
| Gaussian mixture (GM) | (85,86) | |
| Agglomerative (Agglomerative) | (88) | |
| Spectral (Spectral) | (89) | |
| Markov clustering (MCL) | (87) | |
| Hierarchical clustering (hcluster) | (85,90) | |
| Affinity propagation clustering (APC) | (91) | |
| Mean shift (meanshift) | (92) | |
| DBSCAN (dbscan) | (93) | |
| Feature selection | Chi-square test (CHI2) | (38) |
| Information gain (IG) | (38,39) | |
| F-score value (FScore) | (94) | |
| Mutual information (MIC) | (95) | |
| Pearson's correlation coefficient (Pearson) | (96) | |
| Dimensionality reduction | Principal component analysis (PCA) | (97) |
| Latent dirichlet allocation (LDA) | (98) | |
| t-distributed stochastic neighbor embedding (t_SNE) | (99) | |
| Feature normalization | Z-Score (ZScore) | (15) |
| MinMax (MinMax) | (15) |
The clustering groups similar objects (molecules) in a given dataset described by a specific set of features. Upon completion of the clustering process, molecules are grouped, and each group is assigned with a cluster ID. The cluster IDs are displayed in the table widget. The feature selection and dimensionality reduction approaches serve to reduce the number of features, while potentially boosting the prediction performance by eliminating irrelevant (to a given predictive task) and redundant (mutually correlated) features. Finally, feature normalization rescales the feature values to a specific range, so different features can be used together in the same dataset. We provide two widely used normalization algorithms: Z-score normalization and MinMax normalization. In the Z-score normalization, features are rescaled to the normal distribution with the mean of 0 and the standard deviation of 1. In the MinMax normalization, features are scaled to the unit range between 0 and 1. In iLearnPlus, the results produced by the feature selection and normalization methods can be conveniently visualized using the hybrid plots, while a scatter plot can be used to display the outputs produced by the clustering and dimensionality reduction tools (Figure 2B).
Classifier construction and integration
Many objectives related to the analysis of the DNA/RNA/protein sequences can be formulated as a classification problem. Examples include the prediction of structures and functions of protein and nucleic acid sequences (19,100,101). iLearnPlus supports both binary classification (two outcomes) and multi-class classification (multiple outcomes). It offers 12 conventional machine-learning algorithms, two ensemble-learning frameworks, and seven deep-learning approaches (Table 4). This broad selection of algorithms is more comprehensive than what the current platforms offer, i.e. 21 versus 5 in iLearn and BioSeq-Analysis2.0 (Supplementary Table S2). We use the implementation from four popular third-party machine-learning platforms, including scikit-learn (34), XGBoost (102), LightGBM (103) and PyTorch (104). Deep-learning approaches are implemented using the PyTorch library, while LightGBM and XGBoost algorithms are implemented using the LightGBM and XGBoost package, respectively. The scikit-learn library is used to implement the remaining algorithms.
Table 4.
The integrated machine-learning and deep-learning algorithms in iLearnPlus
| Algorithm category | Algorithm | Reference |
|---|---|---|
| Conventional machine-learning algorithms | Random forest (RF) | (105) |
| Decision tree (DecisionTree) | (106) | |
| Support vector machine (SVM) | (107) | |
| K-nearest neighbors (KNN) | (108) | |
| Logistic regression (LR) | (109) | |
| Gradient boosting decision tree (GBDT) | (110) | |
| Light gradient boosting machine (LightBGM) | (111) | |
| Extreme gradient boosting (XGBoost) | (102) | |
| Stochastic gradient descent (SGD) | (34) | |
| Naïve Bayes (NaïveBayes) | (112) | |
| Linear discriminant analysis (LDA) | (113) | |
| Quadratic discriminant analysis (QDA) | (113) | |
| Ensemble-learning frameworks | Bagging (Bagging) | (114) |
| Adaptive boosting (AdaBoost) | (115) | |
| Deep-learning algorithms | Convolutional neural network (CNN) | (30) |
| Attention based convolutional neural network (ABCNN) | (116) | |
| Recurrent neural network (RNN) | (117) | |
| Bidirectional recurrent neural network (BRNN) | (118,119) | |
| Residual network (ResNet) | (120) | |
| Auto-encoder (AE) | (121) | |
| Multilayer perceptron (MLP) | (122) |
For the conventional classifiers, iLearnPlus supports automatic parameter optimization while still allowing users to specify their own parameters. We adopt the grid search strategy to automate the parametrization. For example, users can either directly specify the values of the penalty and gamma parameters for the RBF (Radial Basis Function) kernel of the SVM classifier, or select the ‘Auto optimization’ option to optimize these two parameters automatically. The default parameter search space, which for the gamma values ranges from 2−10 to 25, can be modified to a user-defined range. iLearnPlus also provides two classifier-dependent ensemble-learning frameworks: Bagging and AdaBoost. These frameworks are typically used to boost predictive performance. Importantly, iLearnPlus supports parallelization (via the use of multiple processors) to improve the computational efficiency for parallelable algorithms, such as RF, Bagging, XGBoost and LightGBM.
Another key advantage of iLearnPlus is the availability of multiple modern deep-learning classifiers. The deep-learning techniques rely on multi-layer (deep) neural networks (NNs) to train complex predictive models from high-dimensional data that can be produced from the biological sequences (123,124). To facilitate applications of deep-learning techniques in the analysis of DNA, RNA and protein sequences, iLearnPlus incorporates deep-learning architectures including convolutional NN, attention-based convolutional NN, recurrent NN, bidirectional recurrent NN, residual NN, auto-encoder NN and the traditional multilayer perceptron NN (Table 4). These frameworks rely on a wide range of recent advancements including the convolution operation, attention mechanism, stacked residual blocks, long short-term memory (LSTM) units, and gated recurrent units (GRUs). Their inclusions are motivated by recent successful applications to predict protein contact maps (125,126), protein function (127), DNA-protein binding (128) and compound-protein affinity (129), to name but a few examples. Details concerning the architectures and parameters of these deep-learning networks can be found in the iLearnPlus online manual. We highlight the fact that iLearnPlus automatically detects and uses GPU devices to optimize performance and reduce the computational burden. When training deep-learning models, our platform utilizes the following default parameters: cross-entropy as the loss function, learning rate set as 10−3, maximum number of epochs set as 1000, termination of training with no performance improvement within 100 epochs, and parameter optimization utilizing the widely used Adam algorithm (130). Alternatively, these parameters can also be configured manually by users.
iLearnPlus also provides an option to perform meta-learning (131), where results produced by multiple predictive models (so called base models) are used in tandem to train new machine-learning models; the deep-learning approaches are excluded from the meta-learning. The underlying objective is to improve predictive performance compared to the performance of the base models. iLearnPlus assesses the performance for the constructed meta-models and identifies the best one (i.e. model producing the best predictive performance).
Performance evaluation
iLearnPlus implements the K-fold cross-validation and independent test assessments to evaluate the performance of the constructed classifiers. The K-fold cross-validation test divides the dataset at random into K equally-sized subsets of sequences (i.e. folds). One of these folds is used as the validation dataset, and the remaining K – 1 folds are used as the training dataset to train the machine-learning model and optimize its parameters. After repeating this process K times, each fold is used once as the validation dataset (132). The independent test aims to evaluate and compare the predictive performance of multiple classifiers using a non-overlapping test dataset. This allows users to control the level of similarity between the training and test sequences. In iLearnPlus, the samples labeled as ‘training’ are used to implement the K-fold cross-validation test, while the samples labeled as ‘testing’ are used as the test dataset.
For a binary classification task, we provide eight commonly employed measures that quantify the predictive performance including sensitivity (Sn; Recall), specificity (Sp), accuracy (Acc), Matthews correlation coefficient (MCC), Precision, F1 score (F1), the area under ROC curve (AUROC) and the area under the PRC curve (AUPRC) (15,20,133,134), which are defined as:
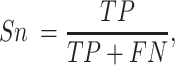 |
(1) |
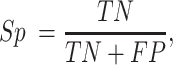 |
(2) |
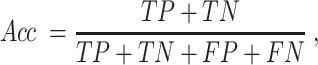 |
(3) |
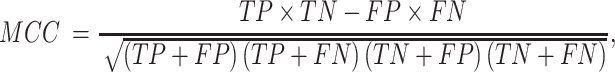 |
(4) |
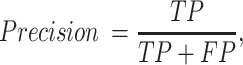 |
(5) |
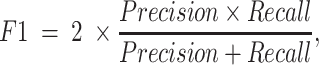 |
(6) |
where TP, FP, TN and FN represent the numbers of true positives, false positives, true negatives and false negatives, respectively. The AUROC and AUCPRC values, which range between 0 and 1, are calculated based on the receiver-operating-characteristic (ROC) curve and the precision–recall curve, respectively. The higher the AUROC and AUPRC values, the better predictive performance of the underlying model.
For a multi-class classification task, we implement the popular Acc measure, which is defined as (134,135):
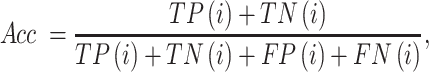 |
(7) |
where TP(i), FP(i), TN(i) and FN(i) represent the numbers of the samples (molecules) predicted correctly to be in the ith class, the total number of the samples in the ith class that are predicted as one of the other classes, the total number of the samples predicted correctly not to be in the ith class, and the total number of the samples not in the i-th class that are predicted as the ith class, respectively.
Data visualization and statistical analysis
iLearnPlus provides a wide range of tools to support analysis and visualization of the prediction results. It offers a variety of statistical plots including histograms, kernel density curves, heatmaps, boxplots, ROC and PRC curves, to assist users to interpret the prediction outcomes effectively (Table 5). As discussed above, histograms and kernel density plots are particularly suitable to visualize data distributions while the scatter plots should be used to analyze feature clustering and dimensionality reduction results. We supplement the predictive performance quantified with AUROC and AUPRC with the corresponding ROC and PRC curves. Users should employ boxplots to illustrate the distribution of the evaluation metrics values from the K-fold cross-validation experiments, allowing for comparison of predictive quality across different models (e.g. the performance based on different feature descriptors and/or machine-learning algorithms). We use the matplotlib library (136) to generate plots in iLearnPlus. The corresponding graphics can be saved using a variety of image formats (e.g. PNG, JPG, PDF, TIFF etc.).
Table 5.
Graphical display options and statistical analysis methods in iLearnPlus
| Category | Types | Purpose |
|---|---|---|
| Graphic display options | Histogram | Display data distribution |
| Kernel density plot | Display data distribution | |
| Heatmap | Display P-value/correlation matrix between different models | |
| Scatter plot | Display clustering and dimensionality reduction result | |
| Boxplot | Depict the group values in the K-fold cross-validation for each of the eight metrics | |
| ROC curve | Depict the overall performance of a model for balanced data | |
| PRC curve | Depict the overall performance of a model for un-balanced data | |
| Statistical analysis | Student's t-test | Compare the means of two evaluate metric |
| Bootstrap test | Evaluate the significance of performance difference between all pairs of ROC or PRC curves |
iLearnPlus supports two statistical tests that can be used to compare the predictive performance across different models or tests. The student's t-test compares the means of two sets of performance measures, typically obtained via the K-fold cross-validation test. The bootstrap test (33) is typically used to assess the significance of differences between data quantified with the ROC and PRC curves. For example, to compare the AUROC values, we apply the following formula:
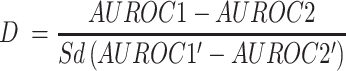 |
(8) |
where AUROC1 and AUROC2 denote the two original AUROC values, while AUROC1′ and AURCO2′ are the bootstrap resampled values of AUROC1 and AUROC2, respectively and Sd represents the standard deviation. By default, we perform 500 bootstrap replicates. In each replicate, we resample the original measurements with replacement to produce new ROC curves. After resampling, we compute AUROC1′, AUROC2′ and their difference (i.e. AUROC1′ – AUROC2′) and use these values to calculate P-values. We also visualize these results with a heatmap.
RESULTS AND DISCUSSION
The functions and modules in iLearnPlus
iLearnPlus covers the five major steps needed to build effective models for analysis and prediction of nucleic acid and proteins sequences: feature extraction, feature analysis, classifier construction, performance evaluation and data/result visualization (Figure 1). We implement these steps by developing four modules in iLearnPlus: iLearnPlus-Basic, iLearnPlus-Estimator, iLearnPlus-AutoML and iLearnPlus-LoadModel (Figure 3). The iLearnPlus-Basic module facilitates analysis and prediction using a selected feature-based representation of the input protein/RNA/DNA sequences (sequence descriptors) and a selected machine-learning classifier. This module is particularly instrumental when interrogating the impact of using different sequence feature descriptors and machine-learning algorithms on the predictive performance. The iLearnPlus-Estimator module provides a flexible way to perform feature extraction by allowing users to select multiple feature descriptors. The iLearnPlus-AutoML module focuses on automated benchmarking and maximization of the predictive performance across different machine-learning classifiers that are applied on the same set or combined sets of feature descriptors. In addition, by combining the iLearnPlus-Estimator and iLearnPlus-AutoML modules, users can conveniently and efficiently evaluate and compare the predictive quality across different selected sequence descriptors and different machine-learning algorithms. Moreover, models generated by iLearnPlus can be exported and saved as model files with the ‘.pkl’ extension in both the stand-alone software and using the web server. Using the iLearnPlus-LoadModel module, users can upload, deploy and test their models on new (test) data. Moreover, the saved models that rely on conventional machine-learning algorithms can be directly applied in the scikit-learn environment, whereas the exported deep-learning models can be applied using the PyTorch library. Section 8 of the user manual provides detailed instructions.
Figure 3.

The iLearnPlus architecture with four major built-in modules, including iLearnPlus-Basic, iLearnPlus-Estimator, iLearnPlus-AutoML, and iLearnPlus-LoadModel.
Building and customizing machine-learning pipelines using iLearnPlus
iLearnPlus makes it easy and straightforward to design and optimize machine-learning pipelines to achieve a competitive (if not the best) predictive performance. The design process typically boils down to two key objectives: extraction and selection of features, and selection and parametrization of machine-learning models, both of which are supported by iLearnPlus. Our platform tackles these objectives via a simple example procedure summarized in Figure 4. Users should first apply the iLearnPlus-Estimator module to generate multiple sequence descriptors (feature sets) from the input sequences and test them by constructing and evaluating a machine-learning model in a batch mode. This allows to establish a point of reference for subsequent optimization/parameterization of the model. The corresponding results and models can be saved for future reference. Subsequently, the iLearnPlus-Basic module should be used to analyze and rank the feature descriptors. Based on the ranking, users should select and evaluate a subset of well-performing features (e.g. a subset of top N features). Next, the evaluation should be performed with the help of the iLearnPlus-AutoML module that optimizes different machine-learning classifiers to the selected feature set. This module also performs statistical comparative analysis of the results and provides the option to save the best model.
Figure 4.

An example of building and customizing machine-learning pipelines using iLearnPlus. First, the iLearnPlus-Estimator module is used to evaluate the performance of multiple feature descriptors based on the input sequences. Next, the feature descriptors with satisfactory performance are selected and the iLearnPlus-Basic module is then used to select the top N important features and save the top N features into a file. Finally, users can upload the feature selection results to the iLearnPlus-AutoML module to evaluate the performance of the machine-learning algorithms of interests in an automated manner.
The iLearnPlus web server and source code
The full version of iLearnPlus that covers the four modules (iLearnPlus-Basic, iLearnPlus-Estimator, iLearnPlus-AutoML, and iLearnPlus-LoadModel) and a graphical user-interface is available on the GitHub repository at https://github.com/Superzchen/iLearnPlus/. The GUI for the four modules is shown in Figure 5.
Figure 5.

The screenshot showing the GUI version of iLearnPlus modules, including iLearnPlus-Basic module (A), iLearnPlus-Estimator module (B), iLearnPlus-AutoML module (C) and iLearnPlus-LoadModel module (D).
iLearnPlus is also freely available as a web server at http://ilearnplus.erc.monash.edu/. In this case, the calculations are performed on the server side, freeing the users from engaging their own computational resources. This server relies on the Nectar (The National eResearch Collaboration Tools and Resources, which is an online infrastructure that supports researchers to connect with colleagues in Australia and around the world) cloud computing infrastructure, which is managed by the eResearch Centre at Monash University. The iLearnPlus web server was implemented using the open-source web platform LAMP (Linux-Apache-MySQL-PHP) and is equipped with 16 cores, 64GB memory and 2TB hard disk. The server supports five popular web browsers including the Internet Explorer (≥v.7.0), Microsoft Edge, Mozilla Firefox, Google Chrome and Safari. Given the high computational cost, the web server runs only the iLearnPlus-Basic module that supports basic analysis and machine-learning modeling of DNA, RNA and protein sequence data. Figure 6 shows a screenshot of the main page of the iLearnPlus web server, where the inputs and parameters of the analysis are entered.
Figure 6.

A screenshot showing the web server version of iLearnPlus for analyzing DNA (A), RNA (B) and protein (C) sequences. For each sub-server, user can generate their desired analysis pipelines via the major panels marked with (i), (ii), (iii) and (iv). The example input sequences were extracted from the lncRNA dataset prepared by Han et al. (137). The training and validation datasets contain 4,200 lncRNA and 4200 mRNA sequences, while the test dataset includes 1800 lncRNA and 1800 mRNA chains from Mus musculus. The category ‘0’ refers to mRNA sequences, while the category ‘1’ denotes the lncRNA sequences.
Case studies
We showcase real-world applications of iLearnPlus with two bioinformatic scenarios: identification of the long noncoding RNAs (lncRNAs) and prediction of the protein crotonylation sites. We emphasize that the underlying objective is to illustrate how to use our platform for two such diverse applications, rather than securing the top predictive performance compared to the state-of-the-art.
The lncRNAs are the transcripts that are over 200 bp long which do not code for proteins (137). Approximately 70% of the noncoding sequences are transcribed into lncRNAs. They regulate a variety of biological processes and are linked to several human diseases (138,139). We applied the iLearnPlus-Basic module to extract the feature sets and train a classifier that accurately differentiated between lncRNAs and mRNA sequences. We used the datasets from a recent study by Han et al. (137). The training and validation datasets contain 4200 lncRNA and 4200 mRNA sequences, while the test dataset includes 1,800 lncRNA and 1,800 mRNA chains from Mus musculus. Several studies demonstrate that the distribution of adjoining bases is different for lncRNAs and mRNAs (140,141). Thus, we selected the ‘Kmer’ (size = 3) feature descriptor to extract the features. We applied the random forest algorithm (number of trees = 1000) to train the classifier using these features and optimized the classifier based on the 5-fold cross-validation test. This simple design secured AUC = 0.897, Acc = 81.89% and MCC = 0.642 on the test dataset. The entire process took about 10 mins to complete with the iLearnPlus web server using one CPU. Figures 6 and 7 show the parameter configurations and prediction results that we obtained using the online version of the iLearnPlus-Basic model. We note that the kernel density curves and histograms for distribution visualization of the extracted features that are included in Figure 7 are the new features of iLearnPlus that are not available in the other current tools. This case study demonstrates that the entire process that produces a simple and well-performing model can be conveniently completed in a matter of minutes. We used the entire datasets (a total of 12 000 sequences) with the iLearnPlus web server. However, considering the computational burden on the server side, we set the maximum number of sequences that can be submitted to the server to 2000. In cases where users need to process larger amount of sequence data, we encourage to use the GUI version of iLearnPlus which does not limit the number of input sequences.
Figure 7.
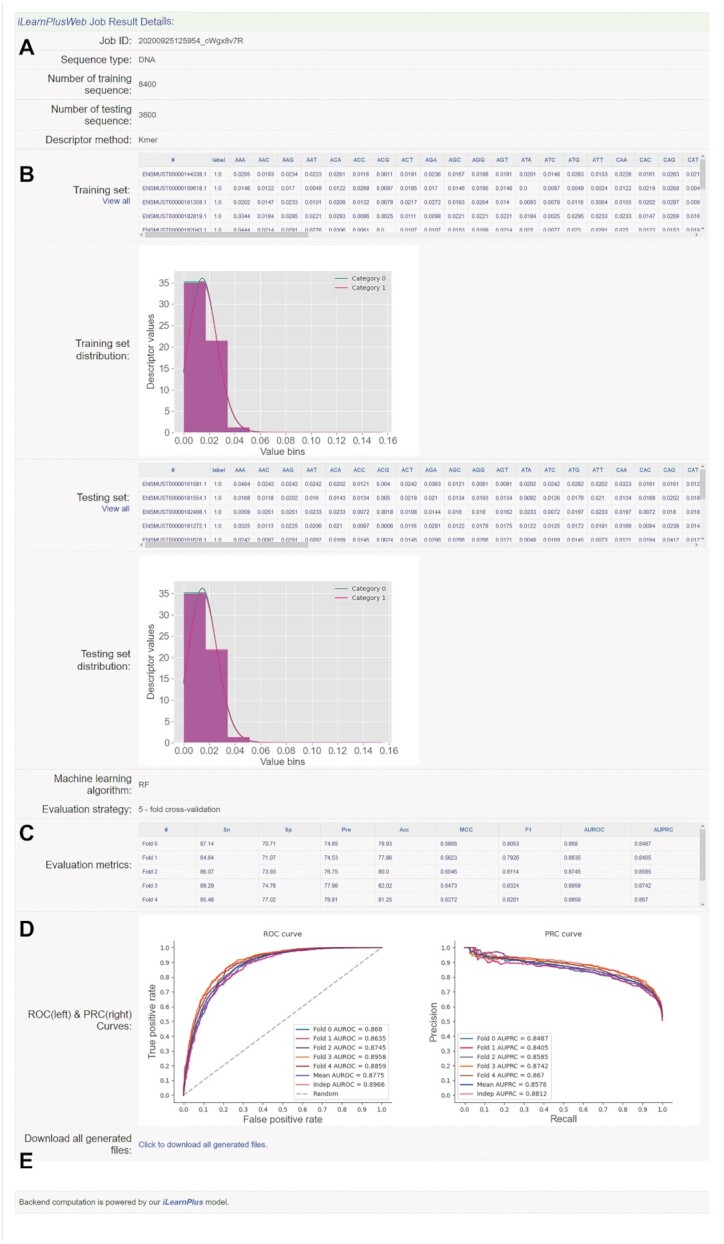
A screenshot demonstrating the result page of iLearnPlus for lncRNA prediction using the iLearnPlus web server. The result page includes the summary of basic information (A), including the input sequence type, number of sequences used for training and test, respectively, and the selected feature descriptor type, the generated features and feature analysis result (B), the selected machine-learning algorithm and the evaluation metrics listed for each fold and independent test (C), the ROC and PRC for demonstrating the prediction performance (D) and the hyperlink for downloading all the generated files including the generated feature encoding files, feature analysis result and plots, evaluation metrics matrix file, prediction scores, ROC and PRC curves, and the constructed models (E). The category ‘0’ refers to mRNA sequences, while the category ‘1’ denotes the lncRNA sequences.
Lysine crotonylation (Kcr) is a post-translational modification (PTM) that was originally found in the histone proteins (142). Recently, this PTM was discovered in non-histone proteins and was found to be involved in the regulation of cell cycle progression and DNA replication cell cycle (143,144). Here, we used the iLearnPlus-Estimator module to comparatively assess the performance of different feature sets using a dataset retrieved from (145,146). We used the data preprocessing strategy from Chen et al. (32) which was developed for a similar prediction problem. Inspired by a recent study by Chen et al. (147), we collected 6687 Kcr sites (positives) and 67 240 non-Kcr sites (negatives) using the sequence segments of 29 residues. We used the iLearnPlus-Estimator module in the standalone GUI version (Figure 8A) to load the data, produced seven feature sets (AAC, EAAC, EGAAC, DDE, binary, ZScale and BLOSUM) and selected a machine-learning algorithm. Similar to the other case study, we used the random forest algorithm (with the default setting of 1000 trees) to construct the classifier via 10-fold cross-validation. We show the corresponding setup in Figure 8A. Figure 8B summarizes the predictive performance quantified with AUROC and AUPRC for models that used each of the seven selected feature sets as inputs. This analysis reveals that the model built utilizing the EGAAC feature descriptors achieved the best performance. This also shows how easy it is to use iLearnPlus to rationally select a well-performing feature encoding for the input sequences. Next, we used the iLearnPlus-AutoML module to comparatively evaluate the predictive performance across seven machine-learning algorithms: SGD, LR, XGBoost, LightGBM, RF, MLP and CNN. We used the bootstrap tests to assess statistical significance of the differences between the ROC curves produced by these algorithms. Figure 9 summarizes the corresponding performance evaluation. More specifically, it shows the evaluation metrics in terms of Sn, Sp, Pre, Acc, MCC and F1 (panel A) whose calculations were based on the default threshold values, correlation matrix that quantifies mutual correlations between classifiers (panel B), ROC (panel C) and PRC (panel D) curves, and boxplots that are used to compare results between classifiers (panel E) directly. We found that the deep-learning model, CNN, achieved the best predictive performance among all the seven machine-learning algorithms, with Acc = 85.4% and AUC = 0.823. Overall, this case study demonstrates how to effectively and efficiently address the two key objectives that lead to designing accurate models: extraction and selection of useful features and selection of the best-performing machine-learning models. We considered seven different feature sets, selected the best set and comparatively evaluated seven machine-learning models using a broad and informative set of metrics. This ultimately led to an informed selection of an accurate solution. We emphasize that four of the seven selected algorithms (i.e. CNN, SGD, XGBoost and LightBGM), ability to run statistical tests, and key methods for visualization of results (i.e. boxplots and heatmaps of the correlations between the models) are among the new features offered by iLearnPlus that are not available in the current platforms.
Figure 8.

The prediction results for protein crotonylation sites using the selected feature descriptors and the local GUI version of iLearnPlus, including a screenshot of the parameter setup using iLearnPlus-Estimator module (A), and the ROC and PRC curves of the seven RF models using different feature descriptors (B).
Figure 9.

An illustration of the prediction results generated by different machine-learning algorithms using the iLearnPlus-AutoML module for identification of protein crotonylation sites, including the evaluation metrics showing the predictive performance in terms of eight evaluation metrics (A), the correlation matrix of seven selected classifiers (B), ROC curves (C), PRC curves (D) and the boxplots (E) of eight metrics for comparative performance assessment of all the seven selected machine-learning algorithms.
These case studies demonstrate that iLearnPlus is a comprehensive platform for the design, evaluation and analysis of the predictive models for both nucleic acid and protein sequences. It can be used to produce an accurate model effectively and, at the same time, to run a fully-fledged design protocol that encompasses feature extraction, feature selection, model selection, comprehensive comparative assessment, and result visualization. Moreover, the trained models can be exported and deployed on new data using the iLearnPlus-LoadModel module.
CONCLUSION
Massive accumulation of sequence data calls for the equally aggressive efforts to develop computational models that can analyze and make an inference from these data. In this article, we addressed this need by delivering a comprehensive automated pipeline, iLearnPlus, which provides ‘one-stop’ services for machine learning-based predictions from the DNA, RNA and protein sequence data. iLearnPlus includes four built-in modules, calculates a variety of feature set and provides 21 machine-learning algorithms including seven popular and modern deep-learning methods. Our platform offers a diverse collection of strategies to conceptualize, design, test, comparatively assess and deploy predictive models. iLearnPlus caters to a broad range of users, including biologists with limited bioinformatics expertise who can benefit from the easy-to-use web server. We provide two case studies using iLearnPlus: predictions of lncRNAs and protein crotonylation sites. The first highlights the fact that our platform supports rapid development of accurate models, while the second demonstrates a sophisticated process that performs feature and classifier selection to maximize the predictive performance of the constructed model. We conclude that iLearnPlus is an effective tool for the design, testing and deployment of machine-learning pipelines for analysis and prediction of the rapidly increasing volume of sequence data for biologists and bioinformaticians.
Supplementary Material
Contributor Information
Zhen Chen, Collaborative Innovation Center of Henan Grain Crops, Henan Agricultural University, Zhengzhou 450046, China.
Pei Zhao, State Key Laboratory of Cotton Biology, Institute of Cotton Research of Chinese Academy of Agricultural Sciences (CAAS), Anyang 455000, China.
Chen Li, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia.
Fuyi Li, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Centre for Data Science, Faculty of Information Technology, Monash University, Melbourne, VIC 3800, Australia; Department of Microbiology and Immunology, The Peter Doherty Institute for Infection and Immunity, The University of Melbourne, Melbourne, Victoria 3000, Australia.
Dongxu Xiang, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Centre for Data Science, Faculty of Information Technology, Monash University, Melbourne, VIC 3800, Australia.
Yong-Zi Chen, Laboratory of Tumor Cell Biology, Key Laboratory of Cancer Prevention and Therapy, National Clinical Research Center for Cancer, Tianjin Medical University Cancer Institute and Hospital, Tianjin Medical University, Tianjin 300060, China.
Tatsuya Akutsu, Bioinformatics Center, Institute for Chemical Research, Kyoto University, Kyoto 611-0011, Japan.
Roger J Daly, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia.
Geoffrey I Webb, Monash Centre for Data Science, Faculty of Information Technology, Monash University, Melbourne, VIC 3800, Australia.
Quanzhi Zhao, Collaborative Innovation Center of Henan Grain Crops, Henan Agricultural University, Zhengzhou 450046, China; Key Laboratory of Rice Biology in Henan Province, Henan Agricultural University, Zhengzhou 450046, China.
Lukasz Kurgan, Department of Computer Science, Virginia Commonwealth University, Richmond, VA, USA.
Jiangning Song, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC 3800, Australia; Monash Centre for Data Science, Faculty of Information Technology, Monash University, Melbourne, VIC 3800, Australia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Health and Medical Research Council of Australia (NHMRC) [APP1127948, APP1144652]; Young Scientists Fund of the National Natural Science Foundation of China [31701142]; National Natural Science Foundation of China [31971846]; Australian Research Council [LP110200333, DP120104460]; National Institute of Allergy and Infectious Diseases of the National Institutes of Health [R01 AI111965]; Major Inter-Disciplinary Research project awarded by Monash University, and the Collaborative Research Program of Institute for Chemical Research, Kyoto University; Fundamental Research Funds for the Central Universities [3132020170, 3132019323]; National Natural Science Foundation of Liaoning Province [20180550307]; C.L. is supported by an NHMRC CJ Martin Early Career Research Fellowship [1143366]; L.K. is supported in part by the Robert J. Mattauch Endowment funds. Funding for open access charge: Grants in support of this submission.
Conflict of interest statement. None declared.
REFERENCES
- 1. Toronen P., Medlar A., Holm L.. PANNZER2: a rapid functional annotation web server. Nucleic Acids Res. 2018; 46:W84–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen H., Li F., Wang L., Jin Y., Chi C.H., Kurgan L., Song J., Shen J.. Systematic evaluation of machine learning methods for identifying human-pathogen protein-protein interactions. Brief. Bioinform. 2020; doi:10.1093/bib/bbaa068. [DOI] [PubMed] [Google Scholar]
- 3. Bonetta R., Valentino G.. Machine learning techniques for protein function prediction. Proteins. 2020; 88:397–413. [DOI] [PubMed] [Google Scholar]
- 4. Wei L., Zou Q.. Recent progress in machine learning-based methods for protein fold recognition. Int. J. Mol. Sci. 2016; 17:2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xie J., Ding W., Chen L., Guo Q., Zhang W.. Advances in protein contact map prediction based on machine learning. Med. Chem. 2015; 11:265–270. [DOI] [PubMed] [Google Scholar]
- 6. Sacar M.D., Allmer J.. Machine learning methods for microRNA gene prediction. Methods Mol. Biol. 2014; 1107:177–187. [DOI] [PubMed] [Google Scholar]
- 7. Walia R.R., Caragea C., Lewis B.A., Towfic F., Terribilini M., El-Manzalawy Y., Dobbs D., Honavar V.. Protein-RNA interface residue prediction using machine learning: an assessment of the state of the art. BMC Bioinformatics. 2012; 13:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhou Y., Zeng P., Li Y.H., Zhang Z., Cui Q.. SRAMP: prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016; 44:e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim N., Kim H.K., Lee S., Seo J.H., Choi J.W., Park J., Min S., Yoon S., Cho S.R., Kim H.H.. Prediction of the sequence-specific cleavage activity of Cas9 variants. Nat. Biotechnol. 2020; 38:1328–1336. [DOI] [PubMed] [Google Scholar]
- 10. Jia C., Bi Y., Chen J., Leier A., Li F., Song J.. PASSION: an ensemble neural network approach for identifying the binding sites of RBPs on circRNAs. Bioinformatics. 2020; 36:4276–4282. [DOI] [PubMed] [Google Scholar]
- 11. Zhou J., Theesfeld C.L., Yao K., Chen K.M., Wong A.K., Troyanskaya O.G.. Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nat. Genet. 2018; 50:1171–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou J., Troyanskaya O.G.. Predicting effects of noncoding variants with deep learning-based sequence model. Nat. Methods. 2015; 12:931–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhou J., Park C.Y., Theesfeld C.L., Wong A.K., Yuan Y., Scheckel C., Fak J.J., Funk J., Yao K., Tajima Y.et al.. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019; 51:973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhou C., Wang C., Liu H., Zhou Q., Liu Q., Guo Y., Peng T., Song J., Zhang J., Chen L.et al.. Identification and analysis of adenine N(6)-methylation sites in the rice genome. Nat. Plants. 2018; 4:554–563. [DOI] [PubMed] [Google Scholar]
- 15. Chen Z., Zhao P., Li F., Marquez-Lago T.T., Leier A., Revote J., Zhu Y., Powell D.R., Akutsu T., Webb G.I.et al.. iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data. Brief. Bioinform. 2020; 21:1047–1057. [DOI] [PubMed] [Google Scholar]
- 16. Chen K.M., Cofer E.M., Zhou J., Troyanskaya O.G.. Selene: a PyTorch-based deep learning library for sequence data. Nat. Methods. 2019; 16:315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Avsec Ž., Kreuzhuber R., Israeli J., Xu N., Cheng J., Shrikumar A., Banerjee A., Kim D.S., Beier T., Urban L., Kundaje A., Stegle O., Gagneur J.. TheKipoirepository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 2019; 37:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kopp W., Monti R., Tamburrini A., Ohler U., Akalin A.. Deep learning for genomics using Janggu. Nat. Commun. 2020; 11:3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu B. BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Brief. Bioinform. 2017; 4:1280–1294. [DOI] [PubMed] [Google Scholar]
- 20. Liu B., Gao X., Zhang H.. BioSeq-Analysis2.0: an updated platform for analyzing DNA, RNA and protein sequences at sequence level and residue level based on machine learning approaches. Nucleic Acids Res. 2019; 47:e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodrigues C.H.M., Myung Y., Pires D.E.V., Ascher D.B.. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 2019; 47:W338–W344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Q., Chen P., Wang B., Zhang J., Li J.. Hot spot prediction in protein-protein interactions by an ensemble system. BMC Syst. Biol. 2018; 12:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahmud S.M.H., Chen W., Meng H., Jahan H., Liu Y., Hasan S.M.M.. Prediction of drug-target interaction based on protein features using undersampling and feature selection techniques with boosting. Anal. Biochem. 2020; 589:113507. [DOI] [PubMed] [Google Scholar]
- 24. Zhu Y.H., Hu J., Ge F., Li F., Song J., Zhang Y., Yu D.J.. Accurate multistage prediction of protein crystallization propensity using deep-cascade forest with sequence-based features. Brief. Bioinform. 2020; doi:10.1093/bib/bbaa076. [DOI] [PubMed] [Google Scholar]
- 25. Zhu Y.H., Hu J., Song X.N., Yu D.J.. DNAPred: accurate identification of DNA-binding sites from protein sequence by ensembled hyperplane-distance-based support vector machines. J. Chem. Inf. Model. 2019; 59:3057–3071. [DOI] [PubMed] [Google Scholar]
- 26. Zhou L., Song X., Yu D.J., Sun J.. Sequence-based detection of DNA-binding proteins using multiple-view features allied with feature selection. Mol Inform. 2020; 39:e2000006. [DOI] [PubMed] [Google Scholar]
- 27. Zhang D., Kabuka M.R.. 2018 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). 2018; 2390–2393. [Google Scholar]
- 28. Zhang D., Kabuka M.. Multimodal deep representation learning for protein interaction identification and protein family classification. BMC Bioinformatics. 2019; 20:531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu H., Jia P., Zhao Z.. Deep4mC: systematic assessment and computational prediction for DNA N4-methylcytosine sites by deep learning. Brief. Bioinform. 2020; doi:10.1093/bib/bbaa099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen Z., Zhao P., Li F., Wang Y., Smith A.I., Webb G.I., Akutsu T., Baggag A., Bensmail H., Song J.. Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences. Brief. Bioinform. 2019; 21:1676–1696. [DOI] [PubMed] [Google Scholar]
- 31. Chen Z., Liu X., Li F., Li C., Marquez-Lago T., Leier A., Akutsu T., Webb G.I., Xu D., Smith A.I.et al.. Large-scale comparative assessment of computational predictors for lysine post-translational modification sites. Brief. Bioinform. 2019; 20:2267–2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Z., He N., Huang Y., Qin W.T., Liu X., Li L.. Integration of a deep learning classifier with a random forest approach for predicting malonylation sites. Genomics Proteomics Bioinformatics. 2018; 16:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hanley J.A., McNeil B.J.. A method of comparing the areas under receiver operating characteristic curves derived from the same cases. Radiology. 1983; 148:839–843. [DOI] [PubMed] [Google Scholar]
- 34. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V.et al.. Scikit-learn: Machine learning in python. 2011; 12:2825–2830. [Google Scholar]
- 35. Hall M., Frank E., Holmes G., Pfahringer B., Reutemann P., Witten I.H.. The WEKA data mining software: an update. 2009; 11:10–18. [Google Scholar]
- 36. Howell D.C. Statistical Methods for Psychology. 2002; Pacific Grove, CA: Duxbury/Thomson Learning. [Google Scholar]
- 37. Bhasin M., Raghava G.P.. Classification of nuclear receptors based on amino acid composition and dipeptide composition. J. Biol. Chem. 2004; 279:23262–23266. [DOI] [PubMed] [Google Scholar]
- 38. Chen K., Jiang Y., Du L., Kurgan L.. Prediction of integral membrane protein type by collocated hydrophobic amino acid pairs. J. Comput. Chem. 2009; 30:163–172. [DOI] [PubMed] [Google Scholar]
- 39. Chen K., Kurgan L.A., Ruan J.. Prediction of flexible/rigid regions from protein sequences using k-spaced amino acid pairs. BMC Struct. Biol. 2007; 7:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Saravanan V., Gautham N.. Harnessing computational biology for exact linear B-cell epitope prediction: a novel amino acid composition-based feature descriptor. OMICS. 2015; 19:648–658. [DOI] [PubMed] [Google Scholar]
- 41. Cai C.Z., Han L.Y., Ji Z.L., Chen X., Chen Y.Z.. SVM-Prot: web-based support vector machine software for functional classification of a protein from its primary sequence. Nucleic Acids Res. 2003; 31:3692–3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cai C.Z., Han L.Y., Ji Z.L., Chen Y.Z.. Enzyme family classification by support vector machines. Proteins. 2004; 55:66–76. [DOI] [PubMed] [Google Scholar]
- 43. Dubchak I., Muchnik I., Holbrook S.R., Kim S.H.. Prediction of protein folding class using global description of amino acid sequence. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:8700–8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dubchak I., Muchnik I., Mayor C., Dralyuk I., Kim S.H.. Recognition of a protein fold in the context of the structural classification of proteins (SCOP) classification. Proteins. 1999; 35:401–407. [PubMed] [Google Scholar]
- 45. Han L.Y., Cai C.Z., Lo S.L., Chung M.C., Chen Y.Z.. Prediction of RNA-binding proteins from primary sequence by a support vector machine approach. RNA. 2004; 10:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shen J., Zhang J., Luo X., Zhu W., Yu K., Chen K., Li Y., Jiang H.. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:4337–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wei L., Zhou C., Chen H., Song J., Su R.. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics. 2018; 34:4007–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu B., Xu J., Lan X., Xu R., Zhou J., Wang X., Chou K.C.. iDNA-Prot|dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS One. 2014; 9:e106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feng Z.P., Zhang C.T.. Prediction of membrane protein types based on the hydrophobic index of amino acids. J. Protein Chem. 2000; 19:269–275. [DOI] [PubMed] [Google Scholar]
- 50. Lin Z., Pan X.M.. Accurate prediction of protein secondary structural content. J. Protein Chem. 2001; 20:217–220. [DOI] [PubMed] [Google Scholar]
- 51. Sokal R.R., Thomson B.A.. Population structure inferred by local spatial autocorrelation: an example from an Amerindian tribal population. Am. J. Phys. Anthropol. 2006; 129:121–131. [DOI] [PubMed] [Google Scholar]
- 52. Horne D.S. Prediction of protein helix content from an autocorrelation analysis of sequence hydrophobicities. Biopolymers. 1988; 27:451–477. [DOI] [PubMed] [Google Scholar]
- 53. Liu B., Liu F., Fang L., Wang X., Chou K.C.. repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects. Bioinformatics. 2015; 31:1307–1309. [DOI] [PubMed] [Google Scholar]
- 54. Dong Q., Zhou S., Guan J.. A new taxonomy-based protein fold recognition approach based on autocross-covariance transformation. Bioinformatics. 2009; 25:2655–2662. [DOI] [PubMed] [Google Scholar]
- 55. Guo Y., Yu L., Wen Z., Li M.. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008; 36:3025–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chou K.C. Prediction of protein subcellular locations by incorporating quasi-sequence-order effect. Biochem. Biophys. Res. Commun. 2000; 278:477–483. [DOI] [PubMed] [Google Scholar]
- 57. Chou K.C., Cai Y.D.. Prediction of protein subcellular locations by GO-FunD-PseAA predictor. Biochem. Biophys. Res. Commun. 2004; 320:1236–1239. [DOI] [PubMed] [Google Scholar]
- 58. Schneider G., Wrede P.. The rational design of amino acid sequences by artificial neural networks and simulated molecular evolution: de novo design of an idealized leader peptidase cleavage site. Biophys. J. 1994; 66:335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chou K.C. Prediction of protein cellular attributes using pseudo-amino acid composition. Proteins. 2001; 43:246–255. [DOI] [PubMed] [Google Scholar]
- 60. Chou K.C. Using amphiphilic pseudo amino acid composition to predict enzyme subfamily classes. Bioinformatics. 2005; 21:10–19. [DOI] [PubMed] [Google Scholar]
- 61. Zuo Y., Li Y., Chen Y., Li G., Yan Z., Yang L.. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics. 2017; 33:122–124. [DOI] [PubMed] [Google Scholar]
- 62. Chen Z., Chen Y.Z., Wang X.F., Wang C., Yan R.X., Zhang Z.. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS One. 2011; 6:e22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Chen Z., Zhou Y., Song J., Zhang Z.. hCKSAAP_UbSite: improved prediction of human ubiquitination sites by exploiting amino acid pattern and properties. Biochim. Biophys. Acta. 2013; 1834:1461–1467. [DOI] [PubMed] [Google Scholar]
- 64. Wang J.T.L., Ma Q., Shasha D., Wu C.H.. New techniques for extracting features from protein sequences. 2001; 40:426–441. [Google Scholar]
- 65. White G., Seffens W.. Using a neural network to backtranslate amino acid sequences. Electron. J. Biotechnol. 1998; 12:196–201. [Google Scholar]
- 66. Lin K., May A.C., Taylor W.R.. Amino acid encoding schemes from protein structure alignments: multi-dimensional vectors to describe residue types. J. Theor. Biol. 2002; 216:361–365. [DOI] [PubMed] [Google Scholar]
- 67. Tung C.W., Ho S.Y.. Computational identification of ubiquitylation sites from protein sequences. BMC Bioinformatics. 2008; 9:310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lee T.Y., Chen S.A., Hung H.Y., Ou Y.Y.. Incorporating distant sequence features and radial basis function networks to identify ubiquitin conjugation sites. PLoS One. 2011; 6:e17331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen Y.Z., Chen Z., Gong Y.A., Ying G.. SUMOhydro: a novel method for the prediction of sumoylation sites based on hydrophobic properties. PLoS One. 2012; 7:e39195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen X., Qiu J.D., Shi S.P., Suo S.B., Huang S.Y., Liang R.P.. Incorporating key position and amino acid residue features to identify general and species-specific Ubiquitin conjugation sites. Bioinformatics. 2013; 29:1614–1622. [DOI] [PubMed] [Google Scholar]
- 71. Lee D., Karchin R., Beer M.A.. Discriminative prediction of mammalian enhancers from DNA sequence. Genome Res. 2011; 21:2167–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Noble W.S., Kuehn S., Thurman R., Yu M., Stamatoyannopoulos J.. Predicting the in vivo signature of human gene regulatory sequences. Bioinformatics. 2005; 21:i338–343. [DOI] [PubMed] [Google Scholar]
- 73. Gupta S., Dennis J., Thurman R.E., Kingston R., Stamatoyannopoulos J.A., Noble W.S.. Predicting human nucleosome occupancy from primary sequence. PLoS Comput. Biol. 2008; 4:e1000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Chen W., Tran H., Liang Z., Lin H., Zhang L.. Identification and analysis of the N(6)-methyladenosine in the Saccharomyces cerevisiae transcriptome. Sci. Rep. 2015; 5:13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Qiang X., Chen H., Ye X., Su R., Wei L.. M6AMRFS: robust prediction of N6-methyladenosine sites with sequence-based features in multiple species. Front Genet. 2018; 9:495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gao F., Zhang C.T.. Comparison of various algorithms for recognizing short coding sequences of human genes. Bioinformatics. 2004; 20:673–681. [DOI] [PubMed] [Google Scholar]
- 77. Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R.et al.. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016; 34:184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Cursons J., Pillman K.A., Scheer K.G., Gregory P.A., Foroutan M., Hediyeh-Zadeh S., Toubia J., Crampin E.J., Goodall G.J., Bracken C.P.et al.. Combinatorial targeting by microRNAs co-ordinates post-transcriptional control of EMT. Cell Syst. 2018; 7:77–91. [DOI] [PubMed] [Google Scholar]
- 79. He W., Jia C., Zou Q.. 4mCPred: machine learning methods for DNA N4-methylcytosine sites prediction. Bioinformatics. 2018; 35:593–601. [DOI] [PubMed] [Google Scholar]
- 80. Lalovic D., Veljkovic V.. The global average DNA base composition of coding regions may be determined by the electron-ion interaction potential. Biosystems. 1990; 23:311–316. [DOI] [PubMed] [Google Scholar]
- 81. Nair A.S., Sreenadhan S.P.. A coding measure scheme employing electron-ion interaction pseudopotential (EIIP). Bioinformation. 2006; 1:197–202. [PMC free article] [PubMed] [Google Scholar]
- 82. Manavalan B., Basith S., Shin T.H., Lee D.Y., Wei L., Lee G.. 4mCpred-EL: an ensemble learning framework for identification of DNA N(4)-methylcytosine sites in the mouse genome. Cells. 2019; 8:1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wei L., Su R., Luan S., Liao Z., Manavalan B., Zou Q., Shi X.. Iterative feature representations improve N4-methylcytosine site prediction. Bioinformatics. 2019; 35:4930–4937. [DOI] [PubMed] [Google Scholar]
- 84. Liu B., Liu F., Wang X., Chen J., Fang L., Chou K.C.. Pse-in-one: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015; 43:W65–W71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jain A.K., Murty M.N., Flynn P.J.. Data clustering: a review. ACM Comput. Surv. 1999; 31:264–323. [Google Scholar]
- 86. Rokach L., Maimon O.. Maimon O., Rokach L.. Data Mining and Knowledge Discovery Handbook. 2005; Boston, MA: Springer US; 321–352. [Google Scholar]
- 87. Enright A.J., Van Dongen S., Ouzounis C.A.. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002; 30:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Theodoridis S., Koutroumbas K.. Theodoridis S., Koutroumbas K.. Pattern Recognition. 2009; 4th ednBoston: Academic Press; 653–700. [Google Scholar]
- 89. Filippone M., Camastra F., Masulli F., Rovetta S.. A survey of kernel and spectral methods for clustering. Pattern Recognit. 2008; 41:176–190. [Google Scholar]
- 90. Jain A.K. Data clustering: 50 years beyond K-means. Pattern Recognit. Lett. 2010; 31:651–666. [Google Scholar]
- 91. Frey B.J., Dueck D.. Clustering by passing messages between data points. Science. 2007; 315:972–976. [DOI] [PubMed] [Google Scholar]
- 92. Cheng Y.Z. Mean shift, mode seeking, and clustering. IEEE Trans. Pattern Anal. Mach. Intell. 1995; 17:790–799. [Google Scholar]
- 93. Ester M., Kriegel H.-P., Sander r., Xu X.. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining. 1996; Portland, Oregon: AAAI Press; 226–231. [Google Scholar]
- 94. Chen J., Guo M., Wang X., Liu B.. A comprehensive review and comparison of different computational methods for protein remote homology detection. Brief. Bioinform. 2018; 19:231–244. [DOI] [PubMed] [Google Scholar]
- 95. Peng H.C., Long F.H., Ding C.. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005; 27:1226–1238. [DOI] [PubMed] [Google Scholar]
- 96. Stigler S.M. Francis Galton's account of the invention of correlation. Stat. Sci. 1988; 4:73–86. [Google Scholar]
- 97. Pearson K. LIII. On lines and planes of closest fit to systems of points in space. London Edinburgh Dublin Philos. Mag. J. Sci. 1901; 2:559–572. [Google Scholar]
- 98. Blei D.M., Ng A.Y., Jordan M.I.. Latent dirichlet allocation. 2003; 3:993–1022. [Google Scholar]
- 99. Maaten L.V.D. Accelerating t-SNE using tree-based algorithms. J. Mach. Learn. Res. 2014; 15:3221–3245. [Google Scholar]
- 100. Larranaga P., Calvo B., Santana R., Bielza C., Galdiano J., Inza I., Lozano J.A., Armananzas R., Santafe G., Perez A.et al.. Machine learning in bioinformatics. Brief. Bioinform. 2006; 7:86–112. [DOI] [PubMed] [Google Scholar]
- 101. Libbrecht M.W., Noble W.S.. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015; 16:321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Chen T., Guestrin C.. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016; San Francisco, California: Association for Computing Machinery; 785–794. [Google Scholar]
- 103. Ke G., Meng Q., Finley T., Wang T., Chen W., Ma W., Ye Q., Liu T.-Y.. Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017; Long Beach, California: Curran Associates Inc; 3149–3157. [Google Scholar]
- 104. Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., Killeen T., Lin Z., Gimelshein N., Antiga L.et al.. Wallach H., Larochelle H., Beygelzimer A., d’Alche-Buc F., Fox E., Garnett R.. PyTorch: An Imperative Style, High-Performance Deep Learning Library. Advances in Neural Information Processing Systems 32. 2019; 32:Curran Associates, Inc; 8024–8035. [Google Scholar]
- 105. Breiman L. Random forests. Mach. Learn. 2001; 45:5–32. [Google Scholar]
- 106. Breiman L., Friedman J., Stone C.J., Olshen R.A.. Classification and Regression Trees. 1984; Wadsworth, Belmont, CA: Taylor & Francis. [Google Scholar]
- 107. Cortes C., Vapnik V.. Support-vector networks. Mach. Learn. 1995; 20:273–297. [Google Scholar]
- 108. Altman N.S. An introduction to kernel and nearest-neighbor nonparametric regression. Am. Stat. 1992; 46:175–185. [Google Scholar]
- 109. Freedman A.D. Statistical models: theory and practice. Technometrics. 2006; 48:315–315. [Google Scholar]
- 110. Friedman J.H. Greedy function approximation: a gradient boosting machine. Ann. Stat. 2001; 29:1189–1232. [Google Scholar]
- 111. Ke G., Meng Q., Finley T., Wang T., Chen W., Ma W., Ye Q., Liu T.-Y.. LightGBM: a highly efficient gradient boosting decision tree. Proceedings of the 31st International Conference on Neural Information Processing Systems. 2017; Long Beach, California: Curran Associates Inc; 3149–3157. [Google Scholar]
- 112. Rennie J.D.M., Shih L., Teevan J., Karger D.R.. Tackling the poor assumptions of naive Bayes text classifiers. Proceedings of the 20th International Conference on International Conference on Machine Learning. 2003; 616–623. [Google Scholar]
- 113. McLachlan G.J. Discriminant Analysis and Statistical Pattern Recognition. 1992; NY: John Wiley & Sons. [Google Scholar]
- 114. Breiman L. Bagging predictors. Mach. Learn. 1996; 24:123–140. [Google Scholar]
- 115. Rojas R. AdaBoost and the Super Bowl of Classifiers A Tutorial Introduction to Adaptive Boosting. 2009; Berlin, Tech Rep: Freie University. [Google Scholar]
- 116. Wang D., Zeng S., Xu C., Qiu W., Liang Y., Joshi T., Xu D.. MusiteDeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics. 2017; 33:3909–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hochreiter S., Schmidhuber J.. Long short-term memory. Neural Comput. 1997; 9:1735–1780. [DOI] [PubMed] [Google Scholar]
- 118. Hanson J., Yang Y., Paliwal K., Zhou Y.. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinformatics. 2017; 33:685–692. [DOI] [PubMed] [Google Scholar]
- 119. Heffernan R., Yang Y., Paliwal K., Zhou Y.. Capturing non-local interactions by long short-term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure, backbone angles, contact numbers and solvent accessibility. Bioinformatics. 2017; 33:2842–2849. [DOI] [PubMed] [Google Scholar]
- 120. He K., Zhang X., Ren S., Sun J.. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). 2016; 770–778. [Google Scholar]
- 121. Yu N., Yu Z., Pan Y.. A deep learning method for lincRNA detection using auto-encoder algorithm. BMC Bioinformatics. 2017; 18:511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wang S.-C. Interdisciplinary Computing in Java Programming. 2003; Boston, MA: Springer US; 3–15. [Google Scholar]
- 123. LeCun Y., Bengio Y., Hinton G.. Deep learning. Nature. 2015; 521:436–444. [DOI] [PubMed] [Google Scholar]
- 124. Min S., Lee B., Yoon S.. Deep learning in bioinformatics. Brief. Bioinform. 2017; 18:851–869. [DOI] [PubMed] [Google Scholar]
- 125. Jones D.T., Kandathil S.M.. High precision in protein contact prediction using fully convolutional neural networks and minimal sequence features. Bioinformatics. 2018; 34:3308–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Li Y., Hu J., Zhang C., Yu D.J., Zhang Y.. ResPRE: high-accuracy protein contact prediction by coupling precision matrix with deep residual neural networks. Bioinformatics. 2019; 35:4647–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Zhang F., Song H., Zeng M., Li Y., Kurgan L., Li M.. DeepFunc: a deep learning framework for accurate prediction of protein functions from protein sequences and interactions. Proteomics. 2019; 19:e1900019. [DOI] [PubMed] [Google Scholar]
- 128. Zeng H., Edwards M.D., Liu G., Gifford D.K.. Convolutional neural network architectures for predicting DNA-protein binding. Bioinformatics. 2016; 32:i121–i127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Karimi M., Wu D., Wang Z., Shen Y.. DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics. 2019; 35:3329–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kingma D.P., Ba J.. Adam: a method for stochastic optimization. 2014; arXiv doi:30 January 2017, preprint: not peer reviewedhttps://arxiv.org/abs/1412.6980v1.
- 131. Lemke C., Budka M., Gabrys B.. Metalearning: a survey of trends and technologies. Artif Intell Rev. 2015; 44:117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Lopez Y., Dehzangi A., Lal S.P., Taherzadeh G., Michaelson J., Sattar A., Tsunoda T., Sharma A.. SucStruct: prediction of succinylated lysine residues by using structural properties of amino acids. Anal. Biochem. 2017; 527:24–32. [DOI] [PubMed] [Google Scholar]
- 133. Liu B., Fang L., Long R., Lan X., Chou K.C.. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016; 32:362–369. [DOI] [PubMed] [Google Scholar]
- 134. Liu B., Yang F., Huang D.S., Chou K.C.. iPromoter-2L: a two-layer predictor for identifying promoters and their types by multi-window-based PseKNC. Bioinformatics. 2018; 34:33–40. [DOI] [PubMed] [Google Scholar]
- 135. Feng P.M., Chen W., Lin H., Chou K.C.. iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013; 442:118–125. [DOI] [PubMed] [Google Scholar]
- 136. Hunter J.D. Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 2007; 9:90–95. [Google Scholar]
- 137. Han S., Liang Y., Ma Q., Xu Y., Zhang Y., Du W., Wang C., Li Y.. LncFinder: an integrated platform for long non-coding RNA identification utilizing sequence intrinsic composition, structural information and physicochemical property. Brief. Bioinform. 2019; 20:2009–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Yang Q., Zhang S., Liu H., Wu J., Xu E., Peng B., Jiang Y.. Oncogenic role of long noncoding RNA AF118081 in anti-benzo[a]pyrene-trans-7,8-dihydrodiol-9,10-epoxide-transformed 16HBE cells. Toxicol. Lett. 2014; 229:430–439. [DOI] [PubMed] [Google Scholar]
- 139. Yao R.W., Wang Y., Chen L.L.. Cellular functions of long noncoding RNAs. Nat. Cell Biol. 2019; 21:542–551. [DOI] [PubMed] [Google Scholar]
- 140. Sun L., Luo H., Bu D., Zhao G., Yu K., Zhang C., Liu Y., Chen R., Zhao Y.. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013; 41:e166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wang L., Park H.J., Dasari S., Wang S., Kocher J.P., Li W.. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013; 41:e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N.et al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011; 146:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Wei W., Mao A., Tang B., Zeng Q., Gao S., Liu X., Lu L., Li W., Du J.X., Li J.et al.. Large-Scale identification of protein crotonylation reveals its role in multiple cellular functions. J. Proteome Res. 2017; 16:1743–1752. [DOI] [PubMed] [Google Scholar]
- 144. Huang H., Wang D.L., Zhao Y.. Quantitative crotonylome analysis expands the roles of p300 in the regulation of lysine crotonylation pathway. Proteomics. 2018; 18:e1700230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Xu W., Wan J., Zhan J., Li X., He H., Shi Z., Zhang H.. Global profiling of crotonylation on non-histone proteins. Cell Res. 2017; 27:946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Wu Q., Li W., Wang C., Fan P., Cao L., Wu Z., Wang F.. Ultradeep lysine crotonylome reveals the crotonylation enhancement on both histones and nonhistone proteins by SAHA treatment. J. Proteome Res. 2017; 16:3664–3671. [DOI] [PubMed] [Google Scholar]
- 147. Zhao Y., He N., Chen Z., Li L.. Identification of protein lysine crotonylation sites by a deep learning framework with convolutional neural networks. IEEE Access. 2020; 8:14244–14252. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.