


Abstract
Single-nucleotide variant (SNV) detection in the genome of single cells is affected by DNA amplification artefacts, including imbalanced alleles and early PCR errors. Existing single-cell genotyper accuracy often depends on the quality and coordination of both the target single-cell and external data, such as heterozygous profiles determined by bulk data. In most single-cell studies, information from different sources is not perfectly matched. High-accuracy SNV detection with a limited single data source remains a challenge. We developed a new variant detection method, SCOUT (Single Cell Genotyper Utilizing Information from Local Genome Territory), the greatest advantage of which is not requiring external data while base calling. By leveraging base count information from the adjacent genomic region, SCOUT classifies all candidate SNVs into homozygous, heterozygous, intermediate and low major allele SNVs according to the highest likelihood score. Compared with other genotypers, SCOUT improves the variant detection performance by 2.0–77.5% in real and simulated single-cell datasets. Furthermore, the running time of SCOUT increases linearly with sequence length; as a result, it shows 400% average acceleration in operating efficiency compared with other methods.
INTRODUCTION
Genome variation is the source of distinct cell phenotypes. The high intra-tumour heterogeneity (ITH) of the cancer genome increases the difficulty of studying tumour occurrence and development (1,2). Many rare mutations with a low variant allele frequency (VAF) are unable to be confidently detected using bulk whole-genome sequencing methods (3). Single-cell DNA sequencing (scDNA-seq) has emerged as a powerful tool to resolve ITH and trace clonal lineages during tumourigenesis (4). Recent applications of scDNA-seq have revealed the evolutionary process of tumour subpopulations and showed an association between genome variations and cancer phenotypes, such as chemoresistance (5) and metastasis (6). However, the accurate detection of single-cell variations is still a challenge in practice.
To generate a sufficient DNA library for each single cell, a whole-genome amplification (WGA) procedure is required, which introduces amplification artefacts. For example, because only one template is available initially, the PCR errors introduced at early amplification cycles accumulate and dominate (7). In addition, the scDNA-seq data are likely to suffer from an imbalanced amplification of the alleles, where reads from paternal and maternal alleles are amplified disproportionately. These amplification artefacts lead to a poor performance of genotyping methods designed for bulk sequencing data (8).
Several genotyping methods that are specifically designed for single-cell sequencing data have improved genotyping accuracy by leveraging information from other cells or bulk samples. Monovar (9) assumes that different loci are independent and corrects for the uneven coverage of two alleles by integrating the base count across multiple cells. SCcaller (10) and SCAN-SNV (11) apply information from adjacent heterozygous SNVs identified in the matched bulk sample. When the tissue is highly homogeneous, the true genotypes of most cells in the tested sample are convergent. Therefore, utilizing information of other cells or bulk can well calibrate the genotyping error in the single cell. However, these strategies lose their advantage when information from other single cells or bulk samples is no longer reliable, such as a single cell from a minor clone or rare mutations in specific genomic regions that are difficult to observe in the external data.
To solve this problem, we directly modelled the sequencing data from one cell. We hypothesized that the data distribution of adjacent nucleotides is likely to be more similar and, therefore, could correct each other when there is amplification bias within local genomic regions. Our statistical discriminant model calculates the likelihood of each genotype and artefact status by taking advantage of base counts from both homozygous and heterozygous loci around candidate SNVs. We implemented the model into a single-cell genotyping software named SCOUT (Single Cell Genotyper Utilizing Information from Local Genome Territory). In real and simulated datasets, SCOUT improved the F1 score by 2–77.5% compared with GATK, SCcaller, and Monovar, even with complete external data. Furthermore, because of the application of linear calculation process, the calculation efficiency of the open-source software SCOUT was increased by 400% on average compared with other single-cell genotypers.
MATERIALS AND METHODS
Modelling single-cell DNA-seq data
We introduce a conditional local-smoothing mixture generative model to describe the allele fraction data,
 |
(1) |
where 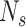 is the total read count of alleles in locus
is the total read count of alleles in locus 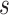 and S is the length of the whole genome.
and S is the length of the whole genome. 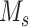 is the candidate SNV reported by sequencing in locus
is the candidate SNV reported by sequencing in locus  , which follows a multinomial distribution with success probability
, which follows a multinomial distribution with success probability 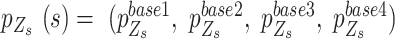 where the components,
where the components, 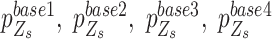 , are sorted by their values and represent the success probability of the first, second, third and fourth alleles with largest counts observed at locus
, are sorted by their values and represent the success probability of the first, second, third and fourth alleles with largest counts observed at locus  . The components of the same rank in
. The components of the same rank in 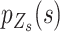 are similar in the neighbourhood and could be estimated based on alleles of the same rank from the adjacent SNVs with the same status
are similar in the neighbourhood and could be estimated based on alleles of the same rank from the adjacent SNVs with the same status 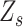 .
. 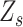 is a latent variable indicating the status of locus
is a latent variable indicating the status of locus 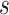 and follows multinomial distribution with parameters
and follows multinomial distribution with parameters 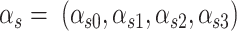 .
. 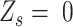 indicates that
indicates that  is a homozygous locus;
is a homozygous locus; 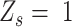 indicates that
indicates that  is a heterozygous locus, and the other values of
is a heterozygous locus, and the other values of 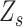 represent error statuses due to errors such as early PCR and amplification bias, which cause the major allele frequency of
represent error statuses due to errors such as early PCR and amplification bias, which cause the major allele frequency of 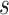 to fall in between homozygous and heterozygous, ‘intermediate SNV’ (
to fall in between homozygous and heterozygous, ‘intermediate SNV’ ( ), or much lower than heterozygous, ‘low major allele SNV’ (
), or much lower than heterozygous, ‘low major allele SNV’ (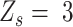 ). The exact genotypes of these loci need further identification.
). The exact genotypes of these loci need further identification.
We suggest that the distributions of  that are generated due to PCR errors can be divided into two categories. In the setting of equation (1),
that are generated due to PCR errors can be divided into two categories. In the setting of equation (1), 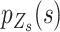 ,
, 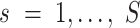 cannot be identified because there is no information for
cannot be identified because there is no information for 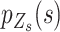 that illustrates which
that illustrates which 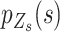 belongs to which status. Therefore, we assume that an ordered relationship existing between the success probability of the major alleles
belongs to which status. Therefore, we assume that an ordered relationship existing between the success probability of the major alleles 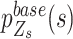 such that
such that 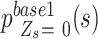 is the largest, followed by
is the largest, followed by  ,
, 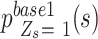 , and
, and  . According to Murphy et al (12), the estimation of
. According to Murphy et al (12), the estimation of 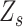 is then calculated as follows:
is then calculated as follows:
 |
(2) |
The success probabilities  ,
, 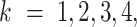
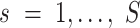 , and
, and 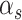 , in Equation (2) are unknown and need to be estimated. We assumed that each
, in Equation (2) are unknown and need to be estimated. We assumed that each 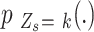 is a smoothing enough function for
is a smoothing enough function for  , which has been extensively studied and could be well estimated (13) by local linear fitting. This assumption allows for the local allele information of locus
, which has been extensively studied and could be well estimated (13) by local linear fitting. This assumption allows for the local allele information of locus 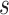 to be borrowed to estimate
to be borrowed to estimate  . The parameter
. The parameter 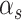 represents the probability of the four states. Technically,
represents the probability of the four states. Technically,  should be calculated by the local distribution of SNVs with
should be calculated by the local distribution of SNVs with 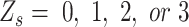 . However, the frequency of error sites in the local genome could not be estimated directly in practice. Therefore, we set
. However, the frequency of error sites in the local genome could not be estimated directly in practice. Therefore, we set 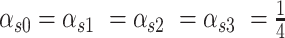 in each
in each 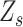 . Our estimation placed more emphasis on the first half of equation (2), i.e. the likelihood score. Although this process brings some bias, we found that the influence is acceptable. As expected, according to the real and simulated data, this procedure maintains the high accuracy of the calculation. The detailed method used for the estimation is displayed in Equations (3) and (4).
. Our estimation placed more emphasis on the first half of equation (2), i.e. the likelihood score. Although this process brings some bias, we found that the influence is acceptable. As expected, according to the real and simulated data, this procedure maintains the high accuracy of the calculation. The detailed method used for the estimation is displayed in Equations (3) and (4).
Identification of candidate SNVs
Raw reads were aligned to human reference hg19 using bwa mem version (0.7.12-r1039). The reads were sorted, duplicates were marked, and the base recalibrated according to GATK best practices pipeline (version 4.1.1.0). When no candidate SNVs were provided, the base count and coverage of each candidate SNV were calculated using the pysam package. SCOUT selects genomic loci with a sequencing depth > 10 and > 1 variant read as candidate SNVs. The other loci with high coverage (depth ≥ 10) and low variant base count (alternative base count ≤ 1) were considered non-SNV control sites. Then, SCOUT individually sorts all alleles in descending order according to their counts for each locus. The top 4 alleles of each candidate SNVs and non-SNV control sites are recorded in two data frames for further calculation. In a diploid genome, the proportion of the top 2 alleles, major and minor alleles, are the most important for genotype determination. To set initial genotype values for all candidate SNVs locally, SCOUT splits the whole region into small segments whose length is equal to the window size parameter (-W; by default, 30 kb). For each segment, we classify all candidate SNVs into two categories with hierarchical clustering using the Ward agglomeration method based on the Euclidian distance of major allele frequencies. Candidate SNVs with lower minor allele frequencies are labelled as heterozygous SNVs, while the others are labeled as homozygous SNVs.
Estimating the success probability for four genome status
For each candidate SNV, we estimate the success probability  and standard variation
and standard variation 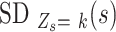 of homozygous (
of homozygous ( ), heterozygous (
), heterozygous (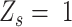 ), and intermediate SNV stages (
), and intermediate SNV stages ( ) by integrating adjacent SNVs
) by integrating adjacent SNVs  (the range of adjacent SNVs was set to 30 kb, both upstream and downstream from the
(the range of adjacent SNVs was set to 30 kb, both upstream and downstream from the  by default):
by default):
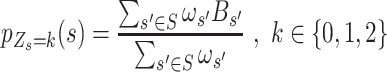 |
(3) |
 |
(4) |
where the S represents the set of adjacent candidate SNVs with the same genotype stages according to the initialization described in the previous section. 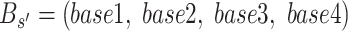 is a vector representing the fraction of the four alleles with largest counts in
is a vector representing the fraction of the four alleles with largest counts in  . Similar to the basic idea of k-means algorithm, we estimate the success probability of each status using loci with the same genotype. For intermediate SNV stages (
. Similar to the basic idea of k-means algorithm, we estimate the success probability of each status using loci with the same genotype. For intermediate SNV stages ( ), an equal number of adjacent homozygous and heterozygous SNVs was leveraged in the estimation, as it represents an uncertain status falling in between homozygous and heterozygous SNVs. We weighed every adjacent SNVs with parameter
), an equal number of adjacent homozygous and heterozygous SNVs was leveraged in the estimation, as it represents an uncertain status falling in between homozygous and heterozygous SNVs. We weighed every adjacent SNVs with parameter  to assign higher weights to closer genome loci:
to assign higher weights to closer genome loci:
 |
(5) |
where 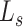 , and
, and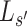 represent the genomic locations of
represent the genomic locations of 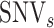 and
and  , respectively. Throughout this study, the parameter
, respectively. Throughout this study, the parameter 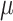 , which determines the decay rate as a function of distance from the centre
, which determines the decay rate as a function of distance from the centre  , is set to
, is set to 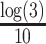 so that SNVs 10 kb from the estimation centre have a weight of
so that SNVs 10 kb from the estimation centre have a weight of 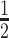 .
.
Different from these three statuses, the success probability of ‘low allele frequency SNVs’ (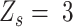 ), representing the lower outlier of heterozygous, is unable to be estimated due to the lack of adjacent data. As an approximation, following Seo et al (14) , we calculate the success probability for
), representing the lower outlier of heterozygous, is unable to be estimated due to the lack of adjacent data. As an approximation, following Seo et al (14) , we calculate the success probability for 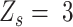 with standard deviation methods:
with standard deviation methods:
 |
(6) |
The probability of the top allele is considered in the calculation of likelihood in ‘low allele frequency SNVs’ status.
Calculating the likelihood of genotype and error stages
Based on the success probability estimated above, SCOUT calculated 3 likelihood values of homozygous ( ), heterozygous (
), heterozygous (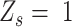 ) and ‘intermediate SNVs’ (
) and ‘intermediate SNVs’ (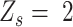 ) for each candidate
) for each candidate 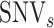 :
:
 |
(7) |
In contrast to the other three genotype statuses, the likelihood of ‘low allele frequency SNVs’ status (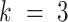 ), representing the lower outlier of heterozygous SNVs, is approximately calculated as follows:
), representing the lower outlier of heterozygous SNVs, is approximately calculated as follows:
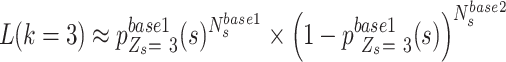 |
(8) |
All candidate SNVs are labeled with a status with the largest likelihood value. In the final output, SNVs with  or
or 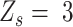 are classified as homozygous or heterozygous with an error flag according to the second maximum likelihood score. Finally, the candidate SNV would be labelled with the ‘ADO’ flag, representing the potential allele dropout SNV, if there is no heterozygous genome loci or over 40% genome loci labelled with error flags in their adjacent genome regions (1 window size up and downstream of the candidate SNV)
are classified as homozygous or heterozygous with an error flag according to the second maximum likelihood score. Finally, the candidate SNV would be labelled with the ‘ADO’ flag, representing the potential allele dropout SNV, if there is no heterozygous genome loci or over 40% genome loci labelled with error flags in their adjacent genome regions (1 window size up and downstream of the candidate SNV)
Data processing
Single-cell WGS and matched bulk data were downloaded from the SRA database (10,15) and GigaDB (16), as described in Supplementary Table S1. We must emphasize the single-cell WGS dataset for IL-11, IL-12 and their unamplified kindred bulk clone, IL-1c constructed by Xiao Dong et al (10). Because these samples are clones from the same single cell, we believe that the mutations in these samples are the same. To evaluate the accuracy of each software, we used matched unamplified bulk data (IL-1c) as the benchmark. We mapped clean reads to human reference build hg19 with the Burrows-Wheeler Aligner (bwa, version 0.7.12-r1039) (17). Aligned reads were sorted and indexed using SAMtools version 1.2.1 (18), and the Genome Analysis Toolkit (GATK, version 4.1.1.0) (19) was used to realign reads to the genome and eliminate PCR duplicates.
Running SCOUT
SCOUT is implemented using Python 3.7, and all dependencies can be installed by Conda with no external databases required. SCOUT implements multi-processing by cutting the whole genome into 2-MB fragments and analysing the fragments separately using a multiprocessing package to improve the calculation speed.
Running other genotypers
SCcaller version 2.0.0 was run as recommended in the GitHub README (https://github.com/biosinodx/SCcaller) with heterozygous SNVs precalled from matched bulk samples and dbSNP 138 common variants, respectively.
Monovar was downloaded from bitbucket (https://bitbucket.org/hamimzafar/monovar/src/master/). Single-cell BAMs were input into SAMtools version 1.35 with options -BQ0 -d 10000 -q 40, which were piped into the monovar.py script with parameters -p 0.002 -a 0.2 -t 0.05 -m 15. SNVs without the ‘PASS’ flag in the vcf output or any other SNVs within 10 bp were filtered.
The best practices workflows of GATK version 4.1.1.0 were run on all single-cell and bulk WGS data.
Determining true-positive, false-negative and false-positive mutations
To assess the performance of genotypers, the results of all genotypers were compared with the matched bulk data. Single-cell detectable SNVs (base quality > 17 and depth ≥ 10) that were verified in matched bulk data with the GATK best practice pipeline were defined as gold standard SNVs. The true-positive rate (TPR) of each dataset is defined as follows:
 |
(9) |
In the results of Monovar, SCcaller, and GATK, ‘true-positive SNVs’ (TP) represented SNVs with the same genotype in single-cell and bulk data. For SCOUT, we also included SNVs with error flags as ‘true-positive SNVs’.
The false-positive SNVs (FP SNVs) represented variants detected in single-cell data with different genotypes in bulk data, and the false-positive rate (FPR) was calculated as follows:
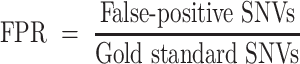 |
(10) |
We also calculated the precision, recall rate and F1 score to evaluate the performance of each genotyper on different single-cell datasets:
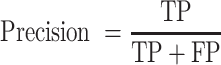 |
(11) |
 |
(12) |
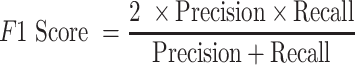 |
(13) |
Detection of single-cell errors
All SNVs with sequence depth larger than 10× in the single-cell and matched bulk data were isolated. Then, for each SNV, we compared the GATK best practice result of the single-cell and matched bulk data. All SNVs with different genotype in single-cell and bulk were labelled as SNV errors. Error SNVs with homozygous genotype in bulk were marked as ‘homozygous errors’, and the rest ‘heterozygous errors’. The ‘heterozygous errors’ were labelled as a ‘Hetero-to-Homo’ block if there were more than three ‘heterozygous errors’ in a row, and the distance between the two was less than 10 kb.
RESULTS
Genotype errors affected the precision of single-cell WGS
The analysis of single-cell genetic variants suffers from whole genome amplification bias. Traditional genotypers designed for bulk variant detection often do not account for this issue, which results in genotyping errors. In published WGS data obtained from cell lines (10,15–16) (Supplementary Table S1), the single-cell WGS data showed 5.93–47.64% different genotypes using the GATK4 best-practice pipeline compared with the matched bulk sample (20) (Supplementary Table S2). These single-cell errors are widely distributed in the genome and affect the determination of both heterozygous and homozygous SNVs (Figure 1A). The wrong genotypes of heterozygous SNVs (Hetero-to-Homo) account for 36.22–85.71% of all single-cell errors (Supplementary Table S2), and a nonnegligible portion of heterozygous errors occurred continuously to form homozygous mutation-enriched genomic regions (29.92–79.17%, Figure 1B and Supplementary Table S2), designated as the ‘Hetero-to-Homo’ error blocks (Figure 1B). Based on previous research, the ‘Hetero-to-Homo’ error blocks are mainly caused by an imbalanced replication of the two alleles in the single-cell whole genome amplification process (7). Sequencing reads of the affected genomic regions would be mainly derived from the over-amplified allele, leading to the high major allele frequency of heterozygous SNVs (Supplementary Figure S1A). This incorrect information could produce an inaccurate genotyping result in the single-position based genotyper (Figure 1C). However, these errors are not impossible to correct. We were able to obtain information for both alleles in some ‘Hetero-to-Homo’ error SNVs (Figure 1D). When the sequencing depth was greater than 10X, the base information of the minor allele was detectable in 1.8–17.1% of heterozygous SNVs (hSNVs) located in the ‘Hetero-to-Homo’ error blocks, and these loci were incompletely lost heterozygous SNVs (Figure 1E and Supplementary Table S2). The remaining 18.93–75.73% of hSNVs in the ‘Hetero-to-Homo’ error blocks showed base information from only one allele and completely formed the allele drop out error (ADO) (7,21). Unfortunately, the occurrence of amplification bias is unpredictable. Even in the two single-cell WGS of the same cell line from the same batch, there is a huge difference in the affection of amplification bias. In some cells, a severe allele imbalance even caused a bimodal distribution of variant allele frequency in heterozygous SNVs (Supplementary Figure S1B).
Figure 1.

Imbalanced amplification in scDNA-seq data. (A) Bar plot showing the percentage of single-cell genotyping errors affecting the homozygous SNVs (blue) and heterozygous SNVs (red) in published datasets. (B) Bar plot showing the percentage of heterozygous SNV errors located within ‘Hetero-to-Homo’ error blocks (green) and appearing independently (purple). (C) An example of a single-cell DNA allelic amplification imbalance where heterozygous SNVs are outlined by red boxes, and the homozygous SNV is outlined by the blue box. The detailed coverage information is labelled around each SNV locus. GT: genotype. (D) Bar plot showing the fraction of mutation-supporting reads (red, Alt) and reference-supporting reads (blue, Ref) of continuous SNVs around the genomic region shown in Figure 1C, and SNVs associated with Figure 1C are framed out. (E) Bar plot showing the percentage of all ‘Hetero-to-Homo’ error block SNVs with a >1 minor allele count in each sample.
Except for the ‘Hetero-to-Homo’ error blocks, the remaining single-cell errors tended to appear independently, including both heterozygous (17.34–45.53%) and homozygous (14.29–63.78%) SNV errors (Supplementary Table S2). This type of SNV error may originate from the early PCR process, where the mismatched base pair in the early DNA replication cycle accumulated and ultimately was unignorable in the sequencing data (Supplementary Figure S1A).
Adjacent information is useful to correct single-cell genotyping error
Although amplification errors seriously affected the accuracy of single-cell genotyping, it is possible to correct these errors using information from adjacent SNVs. According to the published cell line WGS data, 85.30–86.57% of SNVs have at least three homozygous and three heterozygous SNVs located within 30 kb (Supplementary Figure S2). The major allele frequencies of heterozygous SNVs fluctuated in different genome intervals where the heterozygous SNVs far apart may harbour a substantial difference in the major allele frequencies (Figure 2A). In some cases, the major allele frequencies of heterozygous SNVs were even close to homozygous SNVs due to seriously imbalanced allele amplification (Figure 2B). Genotypers, such as GATK, relying on the information from each candidate SNV locus may not be suitable for this situation, producing very different genotyping results between single-cell data and the matched bulk sample (Figure 2C). With local hierarchical clustering based on the MAF value (Materials and Methods), we were able to reduce the inconsistency of the genotyping between single-cell and matched bulk data, and thereby distinguished homozygous and heterozygous SNVs more effectively (Figure 2C). Furthermore, we also observed that some SNVs suffered from a ‘Homo-to-Hetero’ genotyping result compared with the matched bulk sample harbouring a very extreme major allele frequency. These mis-genotyped SNVs were classified into two types: intermediate SNVs with a MAF falling between homozygous and heterozygous SNVs (Figure 2D); and low major allele SNVs with a MAF lower than adjacent heterozygous SNVs (Figure 2E). As we mentioned above, these errors may arise from the PCR process, and we marked them based on their unique MAF distribution. In summary, all candidate SNVs have four possible genotype stages.
Figure 2.

Typical error types identified in single-cell WGS data. (A) Scatter plot showing the major allele frequency distribution of homozygous (blue) and heterozygous (red) SNVs located within chr1: 55185298–55234632 from the single-cell WGS MDA-2_46 dataset. The black curve represents the fitted curve of the major allele frequency of the heterozygous SNVs. (B) Density plot showing the major allele frequency distribution of homozygous (blue) and heterozygous (red) SNVs located within the allele imbalanced region, chr1: 55208299–55215523. (C) The hierarchical clustering analysis using the Ward agglomeration method based on the Euclidian distance of the major allele frequency shows the re-genotyped homozygous (blue) and heterozygous (red) SNVs compared with the bulk and GATK output results. (D, E) Box plots showing the major allele frequency of the intermediate SNV chr1:55192785 (D), the low allele frequency SNVs chr1:55204934, chr1:55205974 (E), and their surrounding correct SNVs. The major allele frequency of intermediate SNVs are marked with a black frame.
According to our analysis, we proposed a local-smoothing mixture generative model to genotype the single-cell genome independently. Our model is built on the hypothesis that similar sequencing information of each state is shared in each local genomic region. Allele count data from the closer loci with the same genotype have similar distribution patterns. We hypothesized that the observed base count distribution of each candidate SNV is generated from a Bernoulli distribution, with the four results representing the major allele, the minor allele, and two additional allele counts caused by sequencing noise (Figure 3A). Four different Bernoulli distributions and the related likelihood functions are defined to represent the genotype stages of homozygous, heterozygous, intermediate SNV and low major allele SNVs (Matrials and Methods).
Figure 3.

The description of the model. (A) The data extraction process for SNV candidates from single-cell WGS sequencing datasets. (B) Schematic and scatter plot showing the initial genotyping of candidate SNVs with local hierarchical classification based on the Euclidian distance of major allele frequency. (C) Schematic diagram showing the parameter estimations of four different genotype stages. Information from the adjacent heterozygous and homozygous SNVs are color-coded as described in (B). The thickness of the linking curves represents the weight of adjacent information. The parameters of the ‘low major allele SNV’ are estimated using outliner methods (see the Materials and Methods). (D) Schematic and bar plot showing the likelihood score of the target SNV marked in (B). (E) Schematic and scatter plot showing the genotyping result obtained using SCOUT. Dots and lines are color-coded in the figure. Blue: homozygous SNVs, red: heterozygous SNVs, green: low major allele frequency SNVs, purple: intermediate SNV, grey zone: potential allele dropout region. The associated likelihood values are displayed around each typical SNVs as bar plots.
To estimate the parameters of each Bernoulli distribution, we introduced a locus-by-locus strategy called SCOUT. First, all candidate SNVs in each local genomic region are classified into homozygous and heterozygous based on their major allele frequency with local classification (Figure 3B, Materials and Methods). Then, the associated parameter, success probabilities, of each Bernoulli distribution was estimated by the allele frequency data for adjacent candidate SNVs with the same genotype stage through weighted local constant fitting (22,23) with adaptive bandwidth (Figure 3C, Materials and Methods). Neither error stage-associated SNVs, namely, intermediate SNVs and low major allele SNVs, were labelled in the initialization hieratical classification step, and their parameters were only obtained with an approximate calculation. For the intermediate SNVs, we estimated their parameters with the mixture of both homozygous and heterozygous SNVs in the local genome region (Figure 3C, Materials and Methods). For the low major allele SNVs, we approximated them as a lower outliner of adjacent heterozygous SNVs and estimated their parameters based on the heterozygous SNVs (Figure 3C, Methods). Finally, the stage of each candidate SNV was identified by the largest likelihood value (Figure 3D, Materials and Methods). The parameters of each genotype stage were different in candidate SNVs, but they were uniformly represented by a set of smoothing functions with small disturbance in the local genome region (Figure 3E). Using this approach, the similarity of parameters at local genomic locations is depicted, and our model maintains the difference to provide a flexible fitting space for exploring nonlinear relationships between the allele count data distributions (13).
SCOUT performs better than other genotypers in single-cell WGS data
We compared the performance of SCOUT and other genotypers using a single-cell WGS dataset published by Xiao Dong et al., which contains two SCMDA-amplified single-cell regions, IL-11 and IL-12, and their unamplified kindred bulk clone, IL-1c (10). SNVs were considered validated if they were called in IL-1c with the GATK best practices pipeline (Materials and Methods). In total, 2724817 and 2282756 SNVs in IL-11 and IL-12, respectively, were compared. All of them had a definite genotype in the bulk sample, proper depth and count of variant reads with high base quality in the single-cell data (Methods). Notably, SCOUT performed better than SCcaller, Monovar and GATK, with higher true-positive rates (SCOUT: 98.79% for IL-11 and 92.98% for IL-12; SCcaller 76.58% for IL-11 and 69.67% for IL-12; Monovar: 88.96% for IL-11 and 95.66% for IL-12; GATK: 95.06% for IL-11 and 87.67% for IL-12) and lower false-positive rates (SCOUT: 11.91% for IL-11 and 17.60% for IL-12; SCcaller 15.86% for IL-11 and 20.43% for IL-12; Monovar: 27.04% for IL-11 and 27.95% for IL-12; GATK: 16.10% for IL-11 and 21.32% for IL-12, Figure 4A and Supplementary Figure S3A). We next compared the performance of these algorithms in analysing other published single-cell datasets generated from the YH (16), and BJ (15) cell lines. SCOUT achieved a similar excellent performance in all single-cell datasets based on both MDA and LIANTI (Supplementary Figure S3B). In the SW480 dataset, despite showing high false-positive rates, SCOUT still performed better in terms of the true-positive and false-negative rates (Supplementary Figure S3B).
Figure 4.

Software performance and error analysis. (A) Box plots show the true positive, false positive and F1 score for software performance on single-cell data for IL-11 and IL-12. Different software packages are colour coded at the bottom. Each dot represents for result from one chromosome. (B) Scatter plot showing the major allele frequency of all heterozygous SNVs located in chr1: 88169929–88194929 for IL-12. The fit curve is coloured in black with the 95% confidence interval shadowed in grey. The largest success probability, 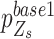 of four genotype statuses estimated by SCOUT are colour-coded in the figure. (C) Bar plot showing the true positive rate of each 2.5-kb window located within the genome region shown in Figure 4B. Different software packages are colour-coded in the figure. (D) IGV screens show the base count of the heterozygous genome loci, chr1: 6764690, in two single-cell samples, IL-11 and IL-12, and the matched bulk sample, IL-1c. The labelled genotypes of IL-11 and IL-12 are provided by Monovar. Bar plot showing the allele frequency of the SNV loci and joint allele frequency as an input data for Monovar. Ref: Fraction of reference-supporting reads, Alt: fraction of alternative-supporting reads. (E) The IGV screen shows the base count data for chr1:3772055–chr1:3775226 from a single-cell sample, IL-12, and matched bulk sample, IL-1c. The error point, chr1:3773034, is indicated with a red arrow. Adjacent heterozygous SNVs are indicated with blue arrows. The surrounding homozygous SNVs are labelled with yellow dots.
of four genotype statuses estimated by SCOUT are colour-coded in the figure. (C) Bar plot showing the true positive rate of each 2.5-kb window located within the genome region shown in Figure 4B. Different software packages are colour-coded in the figure. (D) IGV screens show the base count of the heterozygous genome loci, chr1: 6764690, in two single-cell samples, IL-11 and IL-12, and the matched bulk sample, IL-1c. The labelled genotypes of IL-11 and IL-12 are provided by Monovar. Bar plot showing the allele frequency of the SNV loci and joint allele frequency as an input data for Monovar. Ref: Fraction of reference-supporting reads, Alt: fraction of alternative-supporting reads. (E) The IGV screen shows the base count data for chr1:3772055–chr1:3775226 from a single-cell sample, IL-12, and matched bulk sample, IL-1c. The error point, chr1:3773034, is indicated with a red arrow. Adjacent heterozygous SNVs are indicated with blue arrows. The surrounding homozygous SNVs are labelled with yellow dots.
Interestingly, although GATK achieved a high true-positive rate in IL-11, only 3.73% lower than the median true-positive rate of SCOUT, the accuracy declined sharply in the IL-12 dataset. The IL-12 dataset suffers from a substantial amplification imbalance, where most of the heterozygous SNVs preferentially showed read information for only one allele (Supplementary Figure S1B). These artefacts are difficult to adjust using a base-independent genotyper, such as GATK. In contrast, SCOUT maintained a higher accuracy than GATK in these bias regions, such as the ‘Region 6’, chr1:88169929–88194929 in IL-12 (Figure 4B and C), suggesting that the use of adjacent information may result in better genotyping performance. Unlike GATK, Monovar obtains a higher true-positive rate in the imbalanced single-cell data (IL-12) when running jointly with IL-11, suggesting the positive role of information from other single-cell data in correcting errors. However, adjustment based on another cell may not always be successful. Monovar showed substantially poorer performance than GATK and SCOUT for IL-11, which is fewer affected by the imbalanced amplification artefacts (Figure 4A). This phenomenon may be due to misleading additional information from IL-12. For example, for the heterozygous SNV chr1: 6764690, appearing with the major allele ‘C’ (14 out of 23, 60.9%) and minor allele ‘T’ (9 out of 23, 39.1%) in bulk data (IL-1C), both IL-12 and IL-11 showed a low minor allele frequency at this locus, suggesting an imbalanced amplification error in both cells. Monovar identified them as homozygous SNVs due to their low total percentage of minor allele frequency (9 out of 94, 9.6%, Figure 4D). This treatment is reasonable in some cases, as some other allele information may also appear at the homozygous SNVs because of the influence of sequencing errors and PCR errors. By using the allele number distribution information of the adjacent SNVs, SCOUT avoids such wrong SNV identification. Similarly, additional data from matched bulk samples may also affect the outcome of SCcaller. For example, on chr1:3773034 of IL-12, the nearest heterozygous SNV obtained 0 variant reads because of heavy amplification bias, and the candidate position obtained only 1 variant read, which caused SCcaller to identify the wrong genotype at this site (Figure 4E). These errors indicate that information derived from bulk or another cell may not necessarily play a reliable role in single-cell genotype estimation.
SCOUT makes better use of the adjacent information
Unlike the other neighbourhood genome-based single-cell genotypers, which estimate allelic bias according to known heterozygous SNVs from matched bulk samples, SCOUT does not rely on external information when genotyping. Leveraging information from adjacent genome loci of the target single-cell data only would make the method more applicable for different types of datasets that are faster and more stable in the output (Supplementary Note, Supplementary Figure S4). However, the initialization step of SCOUT introduces errors as it identifies heterozygous SNVs with local hierarchical clustering (Methods). In the published data, 5.4–30.6% of heterozygous SNVs and 6.7–20.22% of homozygous SNVs were not correctly utilized in our model (Supplementary Figure S5A). The correct SNVs still dominated the parameter estimation for most of the candidate SNVs (Figure 5A and Supplementary Figure S5B), which ensured the accuracy. Compared with SCcaller, SCOUT also included homozygous SNVs into the genotyping process for parameter estimation (Figure 5B). Different from the hSNVs reflecting the amplification bias, the two alleles of homozygous SNVs are identical, but their major allele frequency in the single-cell data would be disturbed due to errors caused by PCR or sequencing noise. Using our model, we were able to mark the candidate SNVs that may suffer from amplification errors as intermediate SNVs or low major allele frequency SNVs in different single-cell WGS datasets (Supplementary Figure S6A). Furthermore, we also provided a method to mark the allele drop out error (ADO) in SCOUT, and we labelled some obvious ADO regions in single-cell WGS data (Supplementary Figure S6B). These error flags would be marked in the final output together with the genotyping result, and could be used by other models for further improvement.
Figure 5.

Genomic information utilized by SCOUT. (A) Density plots showing the distribution of the weight percentage of correct SNVs in the parameter estimation of IL-11 and IL-12. (B) Bar plot showing the percentage of homozygous and heterozygous SNVs used by SCOUT in each single-cell sample.
We next explore the effect of parameters on the performance of SCOUT. The alterable parameters include the window size (-W), determining the length of each local genomic region, and the weight parameter (-M), reflecting the decay speed of weight value according to the distance between adjacent SNVs and candidate SNV of interest (Methods). The window size parameter mainly affects the number of adjacent SNVs to be considered in the calculation. When the window size is smaller than 20 kb, the number of considered adjacent SNVs varies greatly but gradually increases as the length of window increases (Supplementary Figure S2), and the performance of our software improves, especially the amplification imbalanced samples (Supplementary Figure S7). When the window size is larger than 20 kb, the growth rate of the candidate SNVs with over three homozygous and three heterozygous in the local genomic region slowed down and stabilized (Supplementary Figure S2). In the meantime, the improvement of SCOUT also slows (Supplementary Figure S7), suggesting the nearby adjacent genome loci have stabilized the parameter estimation in most of candidate SNVs due to their high proportion of weight. Different from the window size parameter directly determined the number adjacent loci, the weight parameter affected the deviation between the estimated success probability and the true allele frequency (Supplementary Figure S8A). However, the performance of SCOUT appears to not be sensitive to the weight parameter (Supplementary Figure S8B).
DISCUSSION
Detecting SNVs in single-cell DNA-seq data presents many challenges. The main artefacts of single-cell DNA-seq are caused by imbalanced amplification of alleles and early PCR error. Standard genotypers designed for estimating the genotype of each individual candidate SNV from bulk data, such as GATK (20), do not perform correctly in allele-imbalanced regions. Previous research developed two types of single-cell genotypers to overcome this problem by means of additional information. The first type of software, such as Monovar (9), borrowed VAF information from the other single-cell data to improve accuracy; the second type of software, such as SCcaller (10) and SCAN-SNV (11), estimated allelic bias using nearby known heterozygous SNVs that are usually detected in the matched bulk sample. To some extent, these methods mitigate single-cell artefacts, but excessive dependence on other information may decrease the performance and usability of the software when facing unreliable or even unavailable external information. Over reliance on external data is not helpful to the robustness of the algorithm.
In this study, we developed a new genotyper that estimated the genotype of each SNV candidate with the aid of adjacent genome positions. In contrast to the other single-cell genotypers SCcaller, SCAN-SNV, and Monovar, which rely on external information from matched bulk or single-cell samples, SCOUT performs genotype estimation with target single-cell data only. In addition, SCOUT integrates adjacent base count data from heterozygous, homozygous and even ref-homozygous genome loci from the single-cell data. Except for the estimation of homozygous and heterozygous genotypes, SCOUT also calculates the likelihood of ‘intermediate SNVs’, ‘low allele frequency SNVs’, and ‘allele drop out’ status for all candidate SNVs to label the potential single-cell artefacts caused by early PCR error and allelic imbalance. By comprehensively integrating the adjacent single-cell data, SCOUT performs better and is more robust than other genotypers in different single-cell datasets and simulated heterogeneous data.
One of the limitations of our study is that we built up our model on the diploid genome, and the current version of SCOUT is not yet ready for an aneuploid genome. A simple improvement plan is to extend the output genotype stages of our model to twice the chromosome ploidy. With the development of the third-generation sequencing, it is possible to further apply phasing information into our model in the future. We mainly tested our tool on MDA-amplified single-cell datasets because this method is widely utilized to identify single-cell SNVs and is likely to be used for high-throughput scWGS (24). We only tested a small number of single-cell datasets (the SW480 dataset generated by MALBAC and the LIANTI_BJ generated by LIANTI) using other scWGS methods, such as MALBAC (25) and LIANTI (15). Our method displayed relatively good performance in analysing the LIANTI dataset, but it may not be very applicable to the MALBAC dataset. This limited applicability may be caused by the limited length of amplicons and poor data quality generated in the early MALBAC dataset, where the adjacent information were unable to be well utilized. Since the majority of scWGS is based on MDA, our method could still be widely applied. Furthermore, we have to point out that whether SCOUT is useful for a single-cell cancer genome is unknown in the current study because the genome of cancer cells is more complicated than that of the cell lines we used in this paper. We did not test our tool on single-cell data from real tumours, as the gold standard data necessary to evaluate the precision and accuracy are difficult to obtain.
In summary, we developed a new single-cell genotyper, SCOUT, which leverages base count information from the nearby local genomic region. By focusing on the target single-cell data and one step local estimation (26,27), SCOUT uses substantially less computing resources than conventional methods (9,10). In addition, it makes full use of adjacent information and performs with linear complexity in terms of the calculation efficiency, while maintaining a high accuracy in SNV locus detection.
DATA AVAILABILITY
SCOUT is an open source collaborative initiative available in the GitHub repository (https://github.com/Goatofmountain/SCOUT).
Single-cell sequencing data and matched bulk data from human dermal fibroblast cells and BJ cells were downloaded from the Sequence Read Archive under accession numbers SRP067062 and SRP102259, respectively. The raw sequence data of the YH and SW480 cell lines were downloaded from GigaScience Repository, GigaDB. All single-cell datasets are listed in Supplementary Table S1.
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: W.H. and D.X. designed and supervised the research; K.T. developed the software. K.T., K.L. and Q.Z. analysed the data. K.T., K.L., W.H. and D.X. wrote the manuscript.
Contributor Information
Kailing Tu, National Frontier Center of Disease Molecular Network, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
Keying Lu, National Frontier Center of Disease Molecular Network, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
Qilin Zhang, National Frontier Center of Disease Molecular Network, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
Wei Huang, School of Mathematics and Statistics, Key Laboratory for Applied Statistics of the Ministry of Education, Northeast Normal University, 130024, Changchun, China.
Dan Xie, National Frontier Center of Disease Molecular Network, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Natural Science Foundation of China [91631111, 31571327, 31771426 to D.X., 11671073, 11690012 to W.H.]. Funding for open access charge: Chinese National Natural Science Foundation [91631111].
Conflict of interest statement. None declared.
REFERENCES
- 1. McGranahan N., Swanton C.. Clonal heterogeneity and tumor evolution: past, present, and the future. Cell. 2017; 168:613–628. [DOI] [PubMed] [Google Scholar]
- 2. Dagogo-Jack I., Shaw A.T.. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018; 15:81. [DOI] [PubMed] [Google Scholar]
- 3. Xu C. A review of somatic single nucleotide variant calling algorithms for next-generation sequencing data. Comput. Struct. Biotechnol. J. 2018; 16:15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang X., Marjani S.L., Hu Z., Weissman S.M., Pan X., Wu S.. Single-cell sequencing for precise cancer research: progress and prospects. Cancer Res. 2016; 76:1305–1312. [DOI] [PubMed] [Google Scholar]
- 5. Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E.. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018; 173:879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leung M.L., Davis A., Gao R., Casasent A., Wang Y., Sei E., Vilar E., Maru D., Kopetz S., Navin N.E.. Single-cell DNA sequencing reveals a late-dissemination model in metastatic colorectal cancer. Genome Res. 2017; 27:1287–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spits C., Le Caignec C., De Rycke M., Van Haute L., Van Steirteghem A., Liebaers I., Sermon K.. Whole-genome multiple displacement amplification from single cells. Nat. Protoc. 2006; 1:1965. [DOI] [PubMed] [Google Scholar]
- 8. Gawad C., Koh W., Quake S.R.. Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 2016; 17:175. [DOI] [PubMed] [Google Scholar]
- 9. Zafar H., Wang Y., Nakhleh L., Navin N., Chen K.. Monovar: single-nucleotide variant detection in single cells. Nat. Methods. 2016; 13:505–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dong X., Zhang L., Milholland B., Lee M., Maslov A.Y., Wang T., Vijg J.. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods. 2017; 14:491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Luquette L.J., Bohrson C.L., Sherman M.A., Park P.J.. Identification of somatic mutations in single cell DNA-seq using a spatial model of allelic imbalance. Nat. Commun. 2019; 10:3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Murphy K.P. Machine Learning: A Probabilistic Perspective. 2012; MIT Press. [Google Scholar]
- 13. Fan J., Gijbels I.. Local Polynomial Modelling and Its Applications: Monographs on Statistics and Applied Probability 66. 1996; CRC Press. [Google Scholar]
- 14. Seo S. A Review and Comparison of Methods for Detecting Outliers in Univariate Data Sets. 2006; University of Pittsburgh, (Unpublished)Master's Thesishttp://d-scholarship.pitt.edu/id/eprint/7948. [Google Scholar]
- 15. Chen C., Xing D., Tan L., Li H., Zhou G., Huang L., Xie X.S.. Single-cell whole-genome analyses by linear amplification via transposon insertion (LIANTI). Science. 2017; 356:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hou Y., Wu K., Shi X., Li F., Song L., Wu H., Dean M., Li G., Tsang S., Jiang R.. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. Gigascience. 2015; 4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H., Durbin R.. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M.. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010; 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., Del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauri A., Lazzari G., Galli C., Lagutina I., Genzini E., Braga F., Mariani P., Williams J.L.. Assessment of MDA efficiency for genotyping using cloned embryo biopsies. Genomics. 2013; 101:24–29. [DOI] [PubMed] [Google Scholar]
- 22. Nadaraya E.A. On estimating regression. Theor. Probabil. Applic. 1964; 9:141–142. [Google Scholar]
- 23. Watson G.S. Smooth regression analysis. Sankhyā: The Indian Journal of Statistics, Series A. 1964; 26:359–372. [Google Scholar]
- 24. Fu Y., Zhang F., Zhang X., Yin J., Du M., Jiang M., Liu L., Li J., Huang Y., Wang J.. High-throughput single-cell whole-genome amplification through centrifugal emulsification and eMDA. Commun Biol. 2019; 2:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zong C., Lu S., Chapman A.R., Xie X.S.. Genome-Wide Detection of Single-Nucleotide and Copy-Number Variations of a Single Human Cell. Science. 2012; 338:1622–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bickel P.J. One-step Huber estimates in the linear model. J. Am. Statist. Assoc. 1975; 70:428–434. [Google Scholar]
- 27. Robinson P.M. The stochastic difference between econometricstatistics. Econometrica: Journalof the Econometric Society. 1988; 56:531–548. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
SCOUT is an open source collaborative initiative available in the GitHub repository (https://github.com/Goatofmountain/SCOUT).
Single-cell sequencing data and matched bulk data from human dermal fibroblast cells and BJ cells were downloaded from the Sequence Read Archive under accession numbers SRP067062 and SRP102259, respectively. The raw sequence data of the YH and SW480 cell lines were downloaded from GigaScience Repository, GigaDB. All single-cell datasets are listed in Supplementary Table S1.