


Abstract
RNA–RNA inter- and intramolecular interactions are fundamental for numerous biological processes. While there are reasonable approaches to map RNA secondary structures genome-wide, understanding how different RNAs interact to carry out their regulatory functions requires mapping of intermolecular base pairs. Recently, different strategies to detect RNA–RNA duplexes in living cells, so called direct duplex detection (DDD) methods, have been developed. Common to all is the Psoralen-mediated in vivo RNA crosslinking followed by RNA Proximity Ligation to join the two interacting RNA strands. Sequencing of the RNA via classical RNA-seq and subsequent specialised bioinformatic analyses the result in the prediction of inter- and intramolecular RNA–RNA interactions. Existing approaches adapt standard RNA-seq analysis pipelines, but often neglect inherent features of RNA–RNA interactions that are useful for filtering and statistical assessment. Here we present RNAnue, a general pipeline for the inference of RNA–RNA interactions from DDD experiments that takes into account hybridisation potential and statistical significance to improve prediction accuracy. We applied RNAnue to data from different DDD studies and compared our results to those of the original methods. This showed that RNAnue performs better in terms of quantity and quality of predictions.
INTRODUCTION
The ability of RNA to base-pair with itself and other RNAs is crucial for its function in vivo. For example, many non-coding RNAs (ncRNAs) are post-transcriptional regulators of gene expression that act through base-pairing with their target mRNA (1). Others are involved in central cellular processes such as splicing, RNA editing and others. Often, ncRNAs confer their function with the help of proteins or as parts of large ribonucleoprotein complexes. These RNA-protein interactions can be studied with methods targeting the protein part. For example, CLIP (crosslinking immunoprecipitation) is based on UV-Crosslinking of proteins to fixate RNA-protein complexes in vivo that are then immunoprecipitated. The RNA part of the latter is finally analysed to identify the binding partners of the respective proteins. In combination with high-throughput sequencing, HITS-CLIP or CLIP-Seq is able to detect genome-wide RNA-protein interaction maps. RNA-binding proteins can have several domains that bind single-stranded RNA (ssRNA) and double-stranded RNA (dsRNA), allowing to capture tripartite protein-RNA–RNA complexes. Therefore, profiling of protein-RNA interactions can also detect the corresponding RNA–RNA interaction (2). For this, CLASH has been proposed as a method for the transcriptome-wide profiling of RNA–RNA interactions (3–5). It’s experimental steps are similar to HITS-CLIP but optimized for the recovery of RNA–RNA duplexes. Several studies, e.g. using the RNA chaperone Hfq (RIL-Seq; (6,7)), RNase E (RNase E-CLASH; (8)) or ProQ (RIL-Seq; (9)) where performed in Escherichia coli. However, in a typical CLASH experiment only 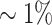 of the sequencing reads provide information about RNA–RNA interactions (8).
of the sequencing reads provide information about RNA–RNA interactions (8).
A more holistic approach was proposed with the concept of RNA Proximity Ligation (RPL) (10). In order to capture in vivo RNA–RNA interactions the biochemical reactions are carried out in the crude cell extract. First, ssRNAs are depleted by Nuclease digestion, RNA duplexes ligated, the so called Proximity Ligation step, and subsequently sequenced. Chimeric reads, which contain the inter- and intramolecular interaction partners, are detected bioinformatically to decipher the RNA–RNA interactome. Recently, the RPL approach has been extended by Psoralen-mediated crosslinking and adapted independently to human, mouse and yeast in different studies, termed Direct Duplex Detection (DDD) methods (11): LIGR-Seq (12), SPLASH (13) and PARIS (14). Additionally, a DDD experiment in E.coli (15) will be referred to as mCLASH in the following.
The methods differ in the experimental protocols (reviewed in (16)) and also in their bioinformatics analyses, although the input data is basically the same, namely sequencing reads with a fraction of chimeras. According to (16) the latter is in the range of ∼0.5–3.9%. In the following we will show that this is partly the result of inadequate algorithms for primary data analysis, e.g. read mapping and that the quality of the predictions in general can be enhanced by apropriate filtering, statistical assessment and annotation-independent clustering. We compiled all this into our tool RNAnue, compared it to the existing pipelines and can show that it is superior in terms of quality and competitive in terms of speed. Although RNAnue has been primarily designed for the analysis of DDD data, it can also be used on CLASH, HITS-CLIP or CRAC data, which also consist of chimeric reads.
MATERIALS AND METHODS
Preprocessing
RNAnue utilizes the Boyer-Moore string-search algorithm (17) to remove adapter contamination from the sequence reads. The algorithm is based on the idea that by matching the pattern from the right rather than from the left, regions containing no matches can be quickly identified and skipped, which results in a significant speed-up. However, this turns out to be less efficient on small alphabets (e.g. DNA), because substrings re-occur frequently. As a result skips get shorter. (18) introduced a variant of the algorithm that also works efficiently on small alphabets by memorizing the last two matched blocks and, thus, facilitating longer shifts. 5′, as well as 3′ adapters, can be trimmed, and also partial adapter sequences are removed. Additionally, 3′ adapter sequences that occur within a read are recognized, and the corresponding reads trimmed at the first position of the adapter. The algorithm is implemented as a finite automaton using a smart transition table that takes over the bookkeeping of the pattern. We further modified the algorithm to allow mismatches in the search pattern (option --mmrate). Finally, reads with an average Phred score quality below a user-defined cutoff (option --avgqual) or below a minimum length (option --minlen) are dropped. In the case of paired-end reads, RNAnue determines the longest common substring using a generalized suffix array to merge the read pairs into a single longer read.
Split read mapping
We use Segemehl (19) to align reads to the reference genome, because it supports (multiple) split read alignment. A read that can not be aligned due to insufficient accuracy (option --accuracy) will be subjected to the split read alignment. As a consequence, the higher the accuracy parameter is set, the more reads will be probed for split read alignment. In order to be reported as a split match, each fragment needs to have a minimum score (option --minfragsco) and a minimum length (option --minfraglen). Furthermore, the fragments need to cover a user defined fraction of the original read (option --minsplicecov). For eukaryotic and archaeal data sets RNAnue needs to take into account splicing (option --splicing).
Clustering & annotation
We expect to find several split reads for an individual interaction, and we call a group of such split reads an interaction. Such an interaction is described by a pair of non-overlapping genomic segments. To derive interactions we cluster detected split reads, if both their pairs of locations on the genome overlap. One or both segments of the interactions may overlap with annotated genomic features, e.g. exons and ncRNAs. In this case, we further group interactions into so called transcript interactions. In more detail, we cluster the split reads into interactions as follows: Let split reads and clusters be given by pairs of mapping coordinates (a, b): (c, d). Two split reads, a split read and a cluster, or two clusters (a1, b1): (c1, d1) and (a2, b2): (c2, d2) are merged if, both, dab = max (a1 − b2, a2 − b1) and dcd = max (c1 − d2, c2 − d1) do not exceed a threshold δ, i.e. max (dab, dcd) ≤ δ. By default δ equals 0, such that a minimum overlap of 1 nt in both segments is required for merging. The resulting cluster is assigned the coordinates (min (a1, a2), max (b1, b2)): (min (c1, c2), max (d1, d2)). The procedure is shown schematically in Figure 1. Setting δ to values greater than 0, which can be done via the --clustdist parameter of RNAnue, merges also clusters/split reads in close proximity (≤δ). The resulting interactions are compared with the existing genome annotation based on the locations of their segments. If an interaction segment overlaps with an annotated feature, it is assigned to the respective feature. An interaction segment that does not overlap with any annotated feature is treated as a putative new feature and assigned a unique ID. As a result, transcript interactions may consist of two annotated transcripts, one annotated and one new transcript, or two new transcripts. Efficient matching to the annotation is done with the help of a modified interval B+ tree that is pre-filled with all annotations.
Figure 1.

Clustering of the split reads according to the start position of both segments. Black arcs connect the start positions of the segments within a split read. Overlaps between the segments of individual split reads determine the affiliation to a cluster. Colored bands indicate the clusters, that span from the start to the end of each segment. Clusters (A), (B) and (B), (C) differ in the second and first segment, respectively. (D) consists of a single split read (singleton) and (E) occurs isolated from other split reads.
Statistical analysis
In order to assess the significance of detected interaction features RNAnue adopts the strategy of (12) to estimate the likelihood of ligation by chance. We use the multinomial distribution (k = 2) to model the discrete probability distribution for the ligation by chance of an transcript interaction between two transcripts tx and ty. The probability for success (ligation by chance) is proportional to the relative abundances of each of the transcripts. We define the joint probability density function for a random ligation event between the transcripts tx and ty with rx and ry reads, respectively, as
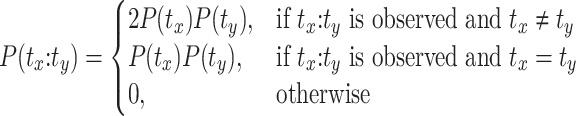 |
(1) |
where
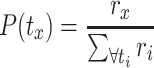 |
(2) |
For pairs tx: ty that have not been observed we explicitly set the probability to zero, because we cannot faithfully decide if they are missing because they are impossible or have simply not been observed, e.g., due to insufficient sequencing depth. As a result, we have to re-normalize the P(tx:ty) to sum up to 1. The number of split reads X for an interaction tx:ty is modeled as
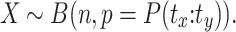 |
(3) |
For each interaction, we apply a binomial test to generate a p-value and apply the Benjamini-Hochberg adjustment to correct for multiple testing and apply a standard α value of 0.1.
Complementarity
The complementarity of two putative interaction sites is computed as the fraction of matches in a modified local alignment procedure, where A aligns with U and G aligns with C and U. Matches are scored with 1, mismatches with –1 and gap open and extension with –3 and –2, respectively. This scoring scheme is inspired by (20), where these scores proved to be optimal for sequences with 75% sequence conservation, which is in the range that we expect for the complementarity of interactions. Furthermore, this favors contiguous over fragmented alignments, a typical feature of the seed region of interactions (21,22). In principle, we use the Waterman-Eggert algorithm to compute the alignments between the segments of all k split reads, while considering the opposing segment in reverse order. As this reports also suboptimal alignment, we select the one with the highest ratio between the number of matches and the length of the alignment, that satisfies the alignment to read length ratio. Assuming that the alignment of all k split reads results in j optimal/suboptimal alignments, then the sets 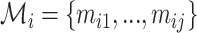 and
and 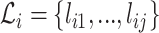 for split read i correspond to the number of matches in the respective alignment and the alignment length, respectively. We define the complementarity ci for split read i as follows:
for split read i correspond to the number of matches in the respective alignment and the alignment length, respectively. We define the complementarity ci for split read i as follows:
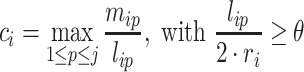 |
(4) |
ri corresponds to the length of read i. θ is a user-defined cutoff (parameter --sitelenratio) for the aligned portion of a read. On the level of transcript interactions we have to summarize the complementarity information of several split reads and for this we introduce the global complementarity score gcs. Let 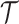 be a transcript interaction that contains k split reads withcomplementarity scores
be a transcript interaction that contains k split reads withcomplementarity scores 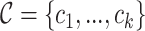 , we define the gcs as follows:
, we define the gcs as follows:
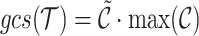 |
(5) |
where 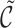 denotes the median of
denotes the median of 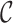 . In addition to the gcs we report the fraction of reads that pass θ and the ratio of unaligned to total read length cutoffs.
. In addition to the gcs we report the fraction of reads that pass θ and the ratio of unaligned to total read length cutoffs.
Hybridisation energy and probability
The interaction of two RNAs is driven by the thermodynamics of the hybridisation reaction, resulting in the loss of free energy. We use RNAlib v2.4.14 (23) to estimate the minimum free energy hybrid structure and its probability in the ensemble of all possible interactions. To be precise we compute ΔΔG = ΔGp + ΔGu, where ΔGp is the free energy loss of the hybridisation and ΔGu the free energy gain needed to unpair the interacting sites. Similar to the complementarity, we also provide a summarised score for transcript interactions that we termed global hybridisation score 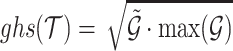 , where
, where 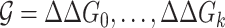 and k is the number of split reads that support the interaction. It is noted that ΔΔGi ≤ 0,
and k is the number of split reads that support the interaction. It is noted that ΔΔGi ≤ 0, 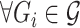 , otherwise RNAnue discards the split read. Similarly, the probability of the hybridisation is computed as the product of the probabilities of the two interactions to be unpaired times the probability of the hybridisation. Accordingly, for probabilities
, otherwise RNAnue discards the split read. Similarly, the probability of the hybridisation is computed as the product of the probabilities of the two interactions to be unpaired times the probability of the hybridisation. Accordingly, for probabilities 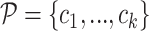 , we define the global probability score
, we define the global probability score 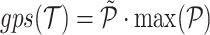 . In addition the fraction of discarded reads is reported.
. In addition the fraction of discarded reads is reported.
Data
We obtained the following method specific data sets: LIGR-seq (GEO: GSE80167), splash (SRA: PRJNA318958), paris (GEO: GSE74353) and mCLASH (SRA: SRP103891). These include experiments in human embryonic kidney (HEK) 293T cells (LIGR-seq, paris), HeLa cells (paris), Lymphoblastoid cells and human embryonic stem (hES) cells as well as retinoic acid (RA) differentiated ES cells (splash). Please note that the SPLASH datasets are already pre-processed with SeqPrep (https://github.com/jstjohn/SeqPrep) using undisclosed parameter settings. Nevertheless, the intrinsic pre-processing of RNAnue was also used for these. Furthermore, we analyzed data from wild-type and a Prp43 helicase mutant of S. cerevisiae (splash) and mouse embryonic stem (mES) cells (paris). The following reference genome sequences from NCBI RefSeq (24) where used: human genome release GRCh38.p13 (RefSeq assembly: GCF_000001405.39), mouse genome release GRCm38.p6 (RefSeq assembly: GCF_000001635.26) and genome release S. cerevisiae S288C (RefSeq assembly: GCF_000146045.2). The respective genome annotations were further complemented with information from LNCipedia 5 (25), snoDB (26) and miRTarBase 7.0 (27).
Implementation
RNAnue complies to the C++17 standard. Segemehl v0.3.4 is used for split read mapping. Furthermore, it relies on Boost C++ libary v1.72.0, SeqAn v3.0.1 and RNAlib v2.4.14. RNAnue can be configured, both, with a configuration file or through the command line, where the latter has precedence. As it provides the complete workflow starting from raw sequencing reads, they have to be arranged into a predefined folder structure. The main output of RNAnue consists of the split read alignments together with the scores for complementarity and hybridisation. These are reported in SAM format with custom tags for complementarity (XC) and hybridization (XE) scores. The results of the clustering, their overlap with existing annotations and the interactions are then reported in a comprehensive tab-delimited interaction file and individual read lists for each interaction. Furthermore, RNAnue can produce additional output formats for further downstream analyses. In particular, count tables (--outcnt) for differential expression analyses and JSON graph format files (http://jsongraphformat.info/) for visualization (--outjgf). For that, we recently introduced VisualGraphX (28) that allows interactive visualization of large-scale graphs. RNAnue can be installed using platform specific installer or build using CMake starting with v3.4 (29). Additionally, we provide a Docker container of RNAnue with all its dependencies at Dockerhub (https://hub.docker.com/r/cobirna/rnanue).
Benchmarking procedures
We benchmarked RNAnue and the original data analysis pipeline based on experimentally validated targets from miRTarBase 7.0 (27) and snoDB 1.2.1 (26). For that, we extracted all intermolecular interactions that involve microRNAs and snoRNAs and compared them to miRTarBase 7.0 and snoDB 1.2.1, respectively. Based on this we compute the positive predictive values (PPV) as the ratio of the number of detected interactions that match the respective database (true positives) to all detected interactions involving microRNAs and snoRNAs, respectively. Mayer et al. (30) argue that ∼18 nt are required for an unambigious alignment against the human reference genome. As a consequence, RNAnue was configured to select all reads that pass a minimum length of 36 nt and an average Phred score of 20. In the alignment procedure, reads were identified as split reads whose fragment length is at least 18 nt with the whole split read being covered by at least 50%. The considered transcript interactions surpass a gcs of 0.75 with the complementarity covering at least 50% of the split read. Analyses were carried out on a system with 2x Intel Xeon CPU E5-2697 v2 @ 2.70 and 378 GB DDR3 SDRAM. We used GNU Time to measure the runtime (user + sys) and space requirements of the respective methods. The runtime was normalized by the read count and scaled to minutes per million reads. In additon, the alignment tools were assessed using chimeric and regular RNA-seq datasets. Artifical chimeric reads were created using a regular RNA-seq dataset (31) by concatenating individual reads.
RESULTS AND DISCUSSION
We developed RNAnue, a comprehensive software package that performs the full analysis of DDD data from quality and adapter trimming, over read mapping to interaction prediction including statistical assessment. An overview of the RNAnue workflow is shown in Figure 2 and details about the individual steps can be found in the Materials and Methods section. The most important steps in this workflow with respect to the special nature of the data are to our opinion the read mapping, clustering & annotation and evaluation. Almost every study based on DDD experiments used its own combination of algorithms for these steps, especially none of them use the same pre-processing or mapping tools. Furthermore, study-specific adaptations hamper their application in a general sense. Table 1 lists these computational methods and gives an overview of their differences compared to RNAnue. We investigated each of the steps and strived to find an optimal solution, which we describe in the following. Please note that we will not consider the data and scripts of Mclash in the following, because the amount of data is very small. Even more important is that it was performed for the prokaryote E. coli, while all other methods were applied to eukaryotes.
Figure 2.

Schematic overview of RNAnue. Sequence reads are either preprocessed (clipped, trimmed and merged) or directly subjected to split read detection. This includes mapping, calculation of filter scores (e.g. complementarity, hybridization energy) and is followed by the clustering of the identified split reads. Clusters are merged with overlapping annotated genome features to so called transcript interactions. These are evaluated statistically and the p-value together with the global filtering scores is reported in the transcript interaction table.
Table 1.
Overview of computational methods for DDD data analysis
| Method | Preprocessing | Mapping | Aggregation by | Statistical assessment | Filtering |
|---|---|---|---|---|---|
| Aligater | - | Bowtie2 | Annotation | Binomial test | None |
| Mclash scripts | Flexbar | Blast | Annotation | Fisher’s exact test | None |
| paris scripts | Trimmomatic | Star | Annotation | None | Coverage |
| RNAnue | 2Block-based | Segemehl | Clustering & annotation | Binomial test | Complementarity & hybridization energy |
| splash scripts | SeqPrep | Bwa-mem, star | Annotation | None | None |
Pre-processing and mapping of chimeric reads
The first step in any sequencing data analysis is a proper quality control. This comprises quality trimming and adapter clipping, and in the case of paired reads may include the merging of read pairs. Although many tools for this pre-processing are available we decided to implement a custom workflow, because the existing ones did not seamlessly integrate into our pipeline. The pre-processing step of RNAnue performs quality trimming, adapter clipping and read merging for paired reads (see section Preprocessing for details). In the respective DDD studies, the transcripts have been sequenced as single-end (LIGR-seq, paris) and paired-end reads (splash, mCLASH) with some of them already being pre-processed (LIGR-seq, splash). Nevertheless, we applied the pre-processing procedure of RNAnue to all datasets to ensure identical cutoffs (e.g., minimum read length, average quality score). This results in 83.1% (LIGR-seq), 96.7% (splash) and 94.3% (PARIS) of the original reads in the human datasets (see Supplementary Tables S3–S5 for details).
In DDD experiments, the interesting fraction of reads are those that contain parts from two interacting RNA strands, so called chimeric reads. Popular read mappers like Bowtie2 (32) are not capable of assigning a read to several locations on the reference sequence and, thus, suboptimal mappings have to be inspected to find a compatible pair that represents the individual mappings of the parts. This is computationally very expensive. The problem of chimeric read mapping is similar to the problem of mapping RNA-seq reads that cross exon-exon boundaries. For this purpose, several mapping tools have been developed, e.g., Tophat2 (33), Hisat2 (34) and BBMap (https://github.com/BioInfoTools/BBMap) but they rely on splicing-specific features, such as donor- and acceptor-sites, which renders them unsuited for general purpose chimeric read mapping. Bwa-mem2 (35), star and Segemehl offer direct chimeric read mapping. Based on the performance of the aligners in the detection of split reads (see Supplementary Results S2.1, Supplementary Figure S1 and Tables S1 and S2) we selected Segmehl for the integration into RNAnue. We applied RNAnue to the human data sets from the studies listed in table 1. RNAnue settings were adapted as far as possible to the settings used in the original analysis pipelines, e.g. length cut-offs for read mapping and others. Comparing the alignment results, RNAnue retrieves as many or more aligned reads than the respective native methods. Solely in the case of splash there are slightly less aligned reads. Anyway, the number of split mappings is more important and here RNAnue identifies substantially (1.5–5 times) more than any other tool (see Supplementary Figure S2, Tables S6– S8), which can mainly be attributed to the superior split mapping performance of segemehl.
Filtering
One of the hallmarks of RNA–RNA interactions that all DDD approaches rely on is the formation of base pairs. As a result, the interacting parts represented by the chimeric reads should show a reasonable degree of complementarity. Furthermore, we expect that interactions are thermodynamically favourable, e.g. associated with a loss of free energy (ΔG). For these reasons, RNAnue includes filtering steps for complementarity and hybridisation energy. These filters can be configured or totally switched off by the user in order to adapt the analysis to special properties of the data to analyse.
Clustering
In order to assess abundances we have to cluster interactions that originate from the same transcripts. This can be done based on gene annotation or in a location based fashion. In RNAnue we use both, because the latter is more reliable, especially for non-model organisms whose genome annotation is often patchy, and the first provides more information. The clustering procedure we implemented is based on the mapping positions of both parts of the chimeric reads and requires overlaps in both for merging. The resulting clusters represent interactions, which can be further merged to transcript interactions based on the annotation (see Section Clustering for details). The final outcome of the clustering can hold split reads (singletons), interactions (clusters not overlapping any annotated feature) and transcript interactions.
Statistical assessment
The mapped chimeric reads ideally represent the interacting strands of one (structure) ore two (interaction) RNA molecules. But due to the complex experimental procedures, we have to account for artifacts. The major source of artifacts is the Proximity Ligation step, where the two paired strands are ligated. The used ligases usually catalyze the ligation of two single-stranded RNAs. For this reason we expect that in this step non-interacting RNAs are ligated by chance and that the likelihood of such events depends on the abundance of the RNAs in the ligation reaction. For statistical assessment, we argue that for two non-interacting RNAs the interaction count only depends on the individual abundances of these two. We get these abundances from the total number of mapped reads and use them to parameterize a binomial test to assess the significance of the transcript interaction.
Detected interactions
On the level of predicted interactions the comparison of RNAnue to the original analysis pipelines on the respective data sets is summarized in Figure 3. Except for paris, RNAnue recalls 88–97% of the originally predicted interactions. paris is special, because the analysis pipeline does neither perform a statistical assessment nor a rigid filtering. An interaction has to be supported by two or more chimeric reads, only. Figure 3 also shows that RNAnue captures novel interactions (LIGR-seq: ∼29%, splash: ∼43%, paris: ∼12%). Among these, 2.5–7.6% involve transcripts that do not overlap any annotation (numbers in brackets) and could therefore only be detected due to RNAnue annotation independent clustering procedure. Other reasons for the increased number of predictions by RNAnue are the generally higher number of mapped chimeras (see Supplementary Figure S2) and a more flexible filtering, especially in the case of paris.
Figure 3.

Detected interactions of the corresponding datasets in human samples using RNAnue in comparison to the original analysis. Numbers in brackets indicate interactions without annotated features.
Benchmarks
We benchmarked RNAnue in comparison to the original data analysis pipelines based on experimentally validated targets from miRTarBase 7.0 and snoDB 1.2.1. MiRTarBase classifies interactions into strong or weak, depending on their experimental support. To identify potential differences based on this classification, we carried out benchmarks for both classes and the results are shown in Figure 4. Interestingly, for the class with weak support RNAnue achieves a lower PPV compared to the original analysis pipeline of LIGR-seq, but higher values for the other two. For those with strong support, RNAnue outperforms the other methods. It does not only achieve higher PPVs, but also larger absolute numbers of true positives. Taken both classes together, RNAnue achieves a PPV of 0.74, compared to 0.70, 0.44 and 0.67 for LIGR-seq, paris and splash, respectively. For snoRNA-rRNA interactions, RNAnue always achieves higher PPVs (between 0.55 and 0.72) than the original tools, up to twice as high as the competitors. Except for the SPLASH data, it also performs better in terms of total number of true positives. The very low numbers of snoRNA-rRNA interactions within the LIGR-seq data are mainly the result of the ribosomal RNA depletion that is part of the library preparation protocol.
Figure 4.

Performance of RNAnue in comparison to the original analyses. The positive predictive value (PPV) corresponds to the fraction of detected interactions involving microRNAs, that are listed in miRTarBase 7.0 and snoRNA-rRNA interactions listed in snoDB 1.2.1. Numbers within bars represent total number of true positives.
In order to compare runtime and memory consumption of RNAnue to its competitors, we analyzed the human datasets (HEK293T, Lymphoblast) with the original analysis pipelines and RNAnue. Figure 5 shows the runtime of the individual phases (e.g. preprocessing, alignment, detection). RNAnue is faster than Aligater but slower than the pipelines from SPLASH and PARIS. Here, the alignment step is one of the main causes in all cases. The extensive filtering, statisticel assessment and the additional clustering step additionally increase the computation time of RNAnue. Nevertheless, it is only 2.4 times slower in the worst case. The upper chart in Figure 5 displays the time needed to build the genome indexes for the respective mapping tools. Although these are one-time costs and heavily depend on the size of the genome to be indexed, they may significantly impact the total time of analysis. The maximum resident set size (max. RSS) was 183GB, compared to 3.9GB (aligater), 4.7GB (SPLASH) and 11.3GB (PARIS). In all cases the alignment tools, due to the in-memory indices, are responsible for the peak memory consumption. In the case of segemehl, and likely also the other tools, the peak is reached during index building. This step needs to be done only once per genome and can also be carried out independently on a large memory server. Without index building the maximum memory consumption of segemehl drops to 60GB. Due to the fact, that modern HPC servers commonly carry ≥128GB RAM, we do not think that the extensive memory requirements of segemehl, and thus of RNAnue, are a major problem. Furthermore, the numbers above are for the human genome. Smaller genomes have a smaller memory footprint.
Figure 5.

Comparison of the runtime of RNAnue and the original methods for the different analysis steps. The upper graph shows the CPU time needed for building the genome index (GRCh38) of the respective method. It is to be noted that the individual steps correspond to the workflow in the original analyses. Therefore, not all analyses include the same steps (e.g. clustering)
CONCLUSION
We present a general bioinformatics pipeline to infer RNA–RNA interactions from raw DDD data. Compared to existing tools we could improve the efficiency in terms of detected chimeras, and the specificity due to complementarity and thermodynamic filtering as well as a thorough statistical assessment. Finally, our method includes the detection if interactions of so far un-annotated transcripts, which is especially important for studies of non-model organisms whose genome annotation is often sparse. In summary, we a think that RNAnue is currently the most comprehensive method for the analysis of DDD data.
In addition, RNAnue can also process data from other methods, such as CLASH, RIL-seq or CRAC. For these kind of data, the statistical filtering should not be used, because the protein based enrichment used in these methods introduces a protein-specific bias, for which a universal statistical model is not appropriate. Nevertheless, filtering by complementarity and hybridisation energy are still valid, such that we expect reasonable performance also on these kind of data.
The results of an RNAnue analysis are, among others, interaction counts, which are similar to read counts used in differential gene expression (DGE) analysis. However, whether statistical methods used for DGE prediction are applicable to analyse differential interaction needs to be thoroughly investigated.
DATA AVAILABILITY
RNAnue is distributed under the GNU GPLv3 (General Public License). We provide the source code, binaries and documentation with sample data are available at https://github.com/Ibvt/RNAnue.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Christoph Schaal and Brigitte Schönberger for valuable discussions on the manuscript. Furthermore, we would like to thank the authors of splash for providing us with the data.
Contributor Information
Richard A Schäfer, University of Stuttgart, Computational Biology, Institute of Biochemical Engineering, Allmandring 31, 70569 Stuttgart, Germany.
Björn Voß, University of Stuttgart, Computational Biology, Institute of Biochemical Engineering, Allmandring 31, 70569 Stuttgart, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Ministry of Education and Research grants RNAProNet [031L0164A to B.V] and inteRNAct [031A310 to B.V.]. Funding for open access charge: German Ministry of Education and Research.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cech T.R., Steitz J.A.. The noncoding RNA revolution—trashing old rules to forge new ones. Cell. 2014; 157:77–94. [DOI] [PubMed] [Google Scholar]
- 2. Sanford J.R., Wang X., Mort M., VanDuyn N., Cooper D.N., Mooney S.D., Edenberg H.J., Liu Y.. Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts. Genome Res. 2009; 19:381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kudla G., Granneman S., Hahn D., Beggs J.D., Tollervey D.. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:10010–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Helwak A., Kudla G., Dudnakova T., Tollervey D.. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013; 153:654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Helwak A., Tollervey D.. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat. Protoc. 2014; 9:711–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Melamed S., Peer A., Faigenbaum-Romm R., Gatt Y.E., Reiss N., Bar A., Altuvia Y., Argaman L., Margalit H.. Global mapping of small RNA-target interactions in bacteria. Mol. Cell. 2016; 63:884–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Melamed S., Faigenbaum-Romm R., Peer A., Reiss N., Shechter O., Bar A., Altuvia Y., Argaman L., Margalit H.. Mapping the small RNA interactome in bacteria using RIL-Seq. Nat. Protoc. 2018; 13:1–33. [DOI] [PubMed] [Google Scholar]
- 8. Waters S.A., McAteer S.P., Kudla G., Pang I., Deshpande N.P., Amos T.G., Leong K.W., Wilkins M.R., Strugnell R., Gally D.L.et al.. Small RNA interactome of pathogenic E. coli revealed through crosslinking of RNase E. EMBO J. 2017; 36:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Melamed S., Adams P.P., Zhang A., Zhang H., Storz G.. RNA–RNA interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol. Cell. 2020; 77:411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramani V., Qiu R., Shendure J.. High-throughput determination of RNA structure by proximity ligation. Nat. Biotechnol. 2015; 33:980–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weidmann C.A., Mustoe A.M., Weeks K.M.. Direct duplex detection: an emerging tool in the RNA structure analysis toolbox. Trends. Biochem. Sci. 2016; 41:734–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharma E., Sterne-Weiler T., O’Hanlon D., Blencowe B.J.. Global mapping of human RNA–RNA interactions. Mol. Cell. 2016; 62:618–626. [DOI] [PubMed] [Google Scholar]
- 13. Aw J.G.A., Shen Y., Wilm A., Sun M., Lim X.N., Boon K.-L., Tapsin S., Chan Y.-S., Tan C.-P., Sim A.Y.L.et al.. In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell. 2016; 62:603–617. [DOI] [PubMed] [Google Scholar]
- 14. Lu Z., Zhang Q.C., Lee B., Flynn R.A., Smith M.A., Robinson J.T., Davidovich C., Gooding A.R., Goodrich K.J., Mattick J.S.et al.. RNA duplex map in living cells reveals higher-order transcriptome structure. Cell. 2016; 165:1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu T., Zhang K., Xu S., Wang Z., Fu H., Tian B., Zheng X., Li W.. Detecting RNA–RNA interactions in E. coli using a modified CLASH method. BMC Genomic. 2017; 18:343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schönberger B., Schaal C., Schäfer R., Voß B.. RNA interactomics: recent advances and remaining challenges. F1000Research. 2018; 7:1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boyer R.S., Moore J.S.. A fast string searching algorithm. Commun. ACM. 1977; 20:762–772. [Google Scholar]
- 18. Sustik M.A., Moore J.S.. String Searching over Small Alphabets. 2007; Department of Computer Sciences, University of Texas at Austin; Technical Report TR-07-62. [Google Scholar]
- 19. Hoffmann S., Otto C., Kurtz S., Sharma C.M., Khaitovich P., Vogel J., Stadler P.F., Hackermüller J.. Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLOS Comput. Biol. 2009; 5:e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. States D.J., Gish W., Altschul S.F.. Improved sensitivity of nucleic acid database searches using application-specific scoring matrices. Methods. 1991; 3:66–70. [Google Scholar]
- 21. Papenfort K., Bouvier M., Mika F., Sharma C.M., Vogel J.. Evidence for an autonomous 5′ target recognition domain in an Hfq-associated small RNA. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:20435–20440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fabian M.R., Sonenberg N., Filipowicz W.. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010; 79:351–379. [DOI] [PubMed] [Google Scholar]
- 23. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA Package 2.0. Algorithm. Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pruitt K.D., Tatusova T., Maglott D.R.. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005; 33:D501–D504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Volders P.-J., Anckaert J., Verheggen K., Nuytens J., Martens L., Mestdagh P., Vandesompele J.. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019; 47:D135–D139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bouchard-Bourelle P., Desjardins-Henri C., Mathurin-St-Pierre D., Deschamps-Francoeur G., Fafard-Couture É., Garant J.-M., Elela S.A., Scott M.S.. snoDB: an interactive database of human snoRNA sequences, abundance and interactions. Nucleic Acids Res. 2020; 48:D220–D225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chou C.-H., Shrestha S., Yang C.-D., Chang N.-W., Lin Y.-L., Liao K.-W., Huang W.-C., Sun T.-H., Tu S.-J., Lee W.-H.et al.. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res. 2018; 46:D296–D302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schäfer R.A., Voß B.. VisualGraphX: interactive graph visualization within Galaxy. Bioinformatics. 2016; 32:3525–3527. [DOI] [PubMed] [Google Scholar]
- 29. Martin K., Hoffman B.. An open source approach to developing software in a small organization. IEEE Softw. 2007; 24:46–53. [Google Scholar]
- 30. Mayer A., Churchman L.S.. Genome-wide profiling of RNA polymerase transcription at nucleotide resolution in human cells with native elongating transcript sequencing. Nat. Protoc. 2016; 11:813–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Seo S.W., Kim D., Latif H., O’Brien E.J., Szubin R., Palsson B.O.. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 2014; 5:4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L., Kim D., Pertea G., Trapnell C., Pimentel H.et al.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L.. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019; 37:907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vasimuddin M., Misra S., Li H., Aluru S.. Efficient architecture-aware acceleration of BWA-MEM for multicore systems. 2019 IEEE International Parallel and Distributed Processing Symposium (IPDPS). 2019; 314–324. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAnue is distributed under the GNU GPLv3 (General Public License). We provide the source code, binaries and documentation with sample data are available at https://github.com/Ibvt/RNAnue.