

Abstract
Endosomal escape is a critical step in the intracellular delivery of biomacromolecular drugs, but a quantitative, high-throughput study of endosomal-vesicle disruption remains elusive. We designed two genetically encoded split-luciferase turn-on reporter assays that can be measured rapidly in well plates on live cells using a luminometer. Both systems use nonluminescent N-terminal and C-terminal luciferase fragments that can reconstitute a functional luminescent enzyme when they are colocalized by their fusion partners. The first system uses luciferase-fragment fusion to Galectin 8 (Gal8) and CALCOCO2. Gal8 and CALCOCO2 interact following endosomal-vesicle disruption to facilitate luciferase complementation into the active enzyme, enabling a luminescence readout (G8C2 system). The second system expresses the N-terminal carbohydrate recognition domain (N-CRD) of Gal8 fused to each luciferase fragment (G8G8 system). Following endosome disruption, G8-NCRD binds to exposed glycans inside endosomes, concentrating both fragments in close proximity and reconstituting active luciferase. The G8G8 system emerged as the lead reporter candidate and was further characterized by comparing it to previously reported Gal8-YFP tracking using microscopy. We also characterized the G8G8 system response to several commercial and research drug-delivery reagents: DOTAP lipid, JetPEI, Lipofectamine 2000, and a library of polymers with known endosomal-escape activity, revealing dose-dependent increases in luminescence due to endosomal disruption. These new reporters provide a first-in-class luminescent assay to rapidly detect endosome disruption in a high-throughput format while excluding toxic formulations. Endosome-disruption screening with these turn-on assays has the potential to accelerate and to improve the rigor of programs focused on the discovery and development of intracellular biologic drug-delivery formulations.
Keywords: endosomal escape, high-throughput screening, intracellular drug delivery, split luciferase, endosome disruption
Graphical Abstract
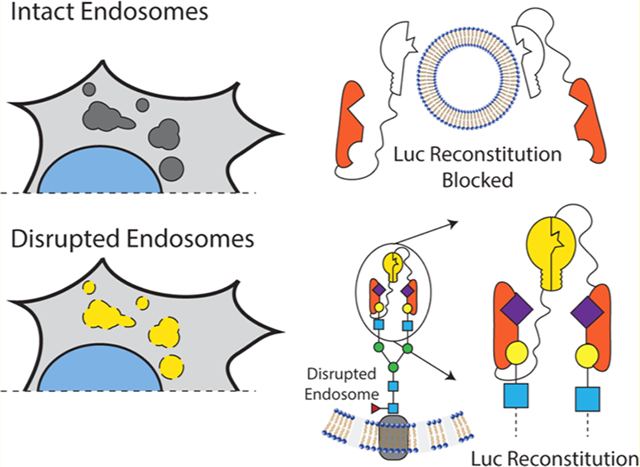
The intracellular endosomal membrane remains a critical barrier to large-molecule drugs with intracellular mechanisms of action. While there are now several exciting Food and Drug Administration (FDA)-approved large-molecule drugs that act inside the cell, including an siRNA-lipid-complex nanomedicine that uses active endosome-disruption technology,1–3 the broad-scale clinical utilization of intracellular-acting biologic medicines has been hindered by challenging pharmacokinetics in the circulatory system, specific tissue-level barriers to delivery,4,5 and barriers at the cellular and subcellular level. At the cellular level, large, hydrophilic molecules such as nucleic acids and peptides are internalized by endocytosis, which compartmentalizes them away from cytosolic drug targets and normally leads to lysosomal trafficking and degradation or recycling exocytosis. A common strategy to promote cell entry and endosomal escape is formulation into nanoscale drug-delivery systems, which are often engineered to have pH-responsiveness that is activated by endosome acidification to promote endosomal-membrane disruption and translocation.
While endosome disruption is an important feature for nanoscale drug-delivery systems, this process is difficult to measure rapidly and quantitatively. Several methods, including subcellular fractionation6 and transmission electron microscopy,7 can be used to assess the cytosolic distribution and endosomal disruption in a low-throughput format. We and others have recently investigated tracking of intracellular galectins as a method to assess endosomal integrity8 in live cells;9–12 this method has been applied by us and others to assess endosome-disrupting nanomedicine formulations.9–20 Galectins are a family of carbohydrate-binding proteins that serve to mediate cell–cell and cell–matrix interactions, modulate immune cell functions, and restrict intracellular pathogens.21 Glycosylation on the inner leaflet of endosomes becomes accessible to cytosolic Galectin-3, -8, and -9, which bind and concentrate there following endosomal membrane disruption by pathogens8 or drug carriers.9–11 By quantitative tracking of fluorescence redistribution from diffuse (in the cytosol) to punctate (inside disrupted endosomes), the extent of endosomal disruption by pathogens and drug carriers can be assessed.
We have been particularly interested in the development of Galectin 8-based (Gal8) assays to monitor endosomal disruption in the context of subcellular trafficking of drug carriers.9–11 Gal8 is specifically involved in the detection of endosomal disruption and is critical to the restriction of intracellular infection by pathogens.8 Gal8 is a tandem-repeat lectin, containing two carbohydrate recognition domains (CRD) at the N and C termini with dissimilar substrate specificities. The N-terminal CRD (G8-NCRD) binds to host rather than pathogen glycans8 (Figure 1A), specifically to sialylated glycans containing α-2-3-sialylated or 3′ sulfated β-galactosides,22 thus providing Gal8 a natural affinity for the inside of endosomes. Meanwhile, the C-terminal CRD mediates trafficking of damaged endosomes to an autophagy pathway by interacting with the C-terminus of CALCOCO2, which recruits the adapter protein LC3 to induce macroautophagy of the damaged vesicle.8,9,23
Figure 1.
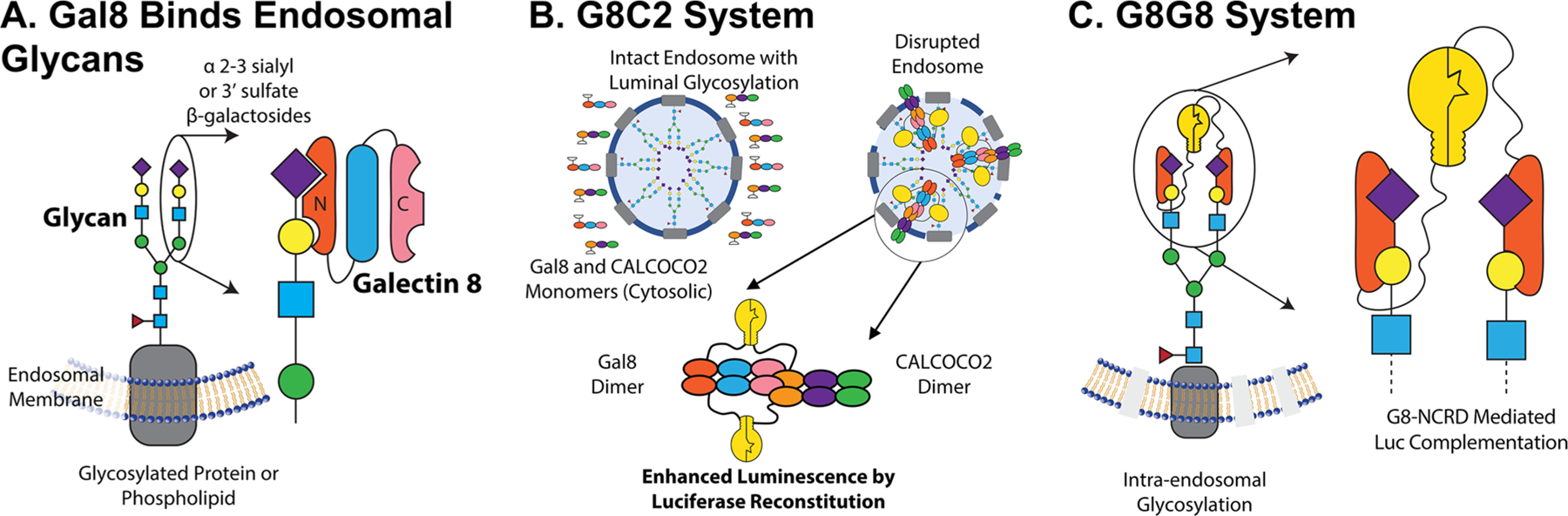
Overview of Gal8 endogenous function and engineered endosome disruption reporters. (A) Gal8 binds to α-2-3 sialyl or 3′ sulfated β-glycans inside endosomes through its N-terminal carbohydrate recognition domain (CRD) upon disruption of the endosomal membrane. (B) Following the endosomal disruption, Gal8 binds to glycans inside endosomes and recruits CALCOCO2 through a dimer–dimer complex to initiate macroautophagy of the damaged vesicle. The G8C2 system was designed to detect this protein–protein interaction, with full-length Gal8 constructed as an N-terminal fusion to the N-terminal luciferase fragment and full-length CALCOCO2 as a C-terminal fusion to the C-terminal luciferase fragment. (C) G8G8 system uses the specific binding of G8-NCRD to concentrate and facilitate the reconstitution of NLuc and CLuc fragments on the interior leaflet of the disrupted endosomal membrane. The N-terminal and C-terminal luciferase fragments were fused to the N-terminus and C-terminus, respectively, of the G8-NCRD fragment.
We previously demonstrated a method of measuring endosomal disruption by quantifying the total recruited Gal8 on a per cell basis using automated fluorescent microscopy and computational image analysis. We showed that this method correlated to cytosolic siRNA bioavailability and that it was more predictive of cytosolic drug bioactivity than methods traditionally used to measure endosome disruption.11 However, this method required high-content microscopy and computational image analysis capable of screening and quantifying thousands of cells for robust analysis. This image analysis technique is also prone to artifacts from cell death and extracellular debris, as bright spots can be interpreted as endosomal disruption in automated analysis algorithms.
The fluorescent-Gal8-tracking method was also applied for in vivo screening using MDA-MB-231 orthotopic tumors formed with fluorescent-Gal8-reporter-transduced cells. Because subcellular imaging through the skin is difficult, tumors were excised from the mice for imaging, precluding the ability to perform kinetic analyses. Though subcellullar Gal8 redistribution was difficult to image in tissue ex vivo, tumors extracted from mice that had been treated intravenously with an endosomolytic nanocarrier showed statistically significant Gal8 redistribution indicative of endosomal disruption compared to vehicle control injections. However, based on the promise of this method combined with the technical challenges that it presented, we sought a turn-on assay with faster data acquisition, the potential for simple and longitudinal intravital measurements, and the capability to be implemented without requiring advanced microscopy or computational image analysis.
In the current work, we developed two turn-on-style endosome-disruption reporters based on a split-luciferase system, wherein the 550-amino-acid firefly luciferase is split into two overlapping fragments (A.A. 1–398 and 394–550). This split-luciferase system was chosen because it is a low-affinity pair suitable for detecting transient protein–protein interactions by producing a significant increase in luminescent signal when the two fragments of the enzyme are brought into close proximity, facilitating their complementation.24 The use of a luciferase-based system, as opposed to a fluorescent reporter, was attractive because firefly luciferase is widely used in vitro and in vivo and utilizes the inexpensive and nontoxic substrate D-luciferin. Firefly-luciferase signal (unlike renilla or related coelenterazine-dependent luciferases) also requires ATP, which is released from cells upon loss of viability, reducing the chances of identifying cytotoxic formulations as false-positive assay hits.
We designed two split-luciferase-reconstitution systems to test as reporters for high-throughput measurement of endosomal disruption. The first design, called G8C2, was constructed to measure the protein–protein interactions of full-length copies of Gal8 and CALCOCO2 (also known as NDP52), which interact downstream of Gal8 clustering to initiate LC3-mediated macroautophagy of damaged endosomes.8 Full-length copies of Gal8 and CALCOCO2 were connected N-terminal to human-codon-optimized Luc2 amino acids 1–398 (NLuc398) and C-terminal to luciferase amino acids 394–550 (CLuc394), respectively, by a 3×(GGGGS) linker. An internal ribosomal entry site was used to drive the expression of both partners from the same mRNA transcript (Figure S1A). Our design was informed by the known arrangement of the Gal8/CALCOCO2 interaction, such that NLuc398 and CLuc394 would be in close proximity and facilitate active-enzyme reconstitution upon the dimer–dimer interaction of Gal8/Gal8 and CALCOCO2/CALCOCO2 complexes (Figure 1B). This design is based on the understanding that the C-terminal carbohydrate-recognition domain of Gal8 interacts with an N-terminal region of CALCOCO2,23 an event that mediates trafficking of damaged endosomes to autophagosomes.8
The second reporter design, called G8G8, was based on the hypothesis that the single carbohydrate-binding domain on the N-terminus of Gal8, G8-NCRD, which mediates Gal8 binding to damaged endosomes,22 is sufficient to drive accumulation and complementation of both NLuc and CLuc fragments on the inner leaflet of disrupted endosomes. Thurston et al.8 showed G8-NCRD is responsible for Gal8 accumulation at damaged endosomes and that an R69H mutation within G8-NCRD ablates this accumulation. Mimicking our earlier design, NLuc398 was connected as an N-terminal fusion to G8-NCRD, while CLuc394 was connected as a C-terminal fusion to G8-NCRD, both using a 3×(GGGGS) linker (Figure S1B). Upon concentration inside endosomes by binding to glycans, NLuc and CLuc fragments could reconstitute the active luciferase enzyme on the glycocalyx of the inner membrane of endosomes. A schematic of the anticipated interaction is shown in Figure 1C.
Herein, we implement both of these reporter systems in vitro through a single lentiviral transfer vector. After creating a series of stable cell populations, we assessed assay turn-on upon treatment with well-validated endosomolytic reagents such as anionic poly(propylacrylic acid) (PPAA),10,17,25–29 commercial cationic lipids 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) and Lipofectamine 2000, and the in vivo cationic polymer transfection reagent JetPEI. We also cross-validated results with the previously established Gal8-YFP method and by screening a series of polymers known as “50B” (diblock structures of PEG and a random copolymer of 50 mol % of the basic monomer dimethylaminoethyl methacrylate (DMAEMA) and 50 mol % of the hydrophobic monomer butyl methacrylate (BMA)).11,30 These polymers provide a platform for a robust demonstration that the lead split-luciferase assay can detect differences among polymers previously validated to produce varied levels of endosome disruption, including a smaller-molecular-weight, negative-control “50B S” polymer that is inert, does not disrupt endosomes, and does not produce intracellular bioactivity of delivered cargo. These collective studies were designed to provide proof of concept that endosomal disruption can be sensitively measured in high throughput with split-luciferase turn-on systems.
RESULTS AND DISCUSSION
Response of G8C2 and G8G8 Systems to PPAA.
To validate that endosomal disruption induces luciferase reconstitution in these systems, we treated cells with a range of concentrations of an anionic pH-responsive polymer PPAA, a well-studied intracellular-delivery reagent,10,17,25–29 that we have previously shown induces rapid endosomal disruption that is detectable using the microscopy-based Gal8-YFP recruitment assay within 30 min.10,17,18 We tested five different populations each of HEK 293T cells which were transduced using a dilution series of either G8C2 or G8G8 system-encoding lentivirus to assess the relative effect of transduction ratio on luminescent response due to PPAA treatment (Figures S2 and S3, left). We varied the viral-transduction multiplicity of infection (MOI) to experimentally determine whether varied levels of overexpression would negatively impact the system, reasoning that high cytosolic concentrations of NLuc and CLuc may cause spontaneous reconstitution while low cytosolic concentration may limit the magnitude of both the baseline and the endosome-disruption-associated turn-on signal. In this initial screen, two cell populations expressing the G8C2 system and all five cell populations expressing the G8G8 system produced statistically significant dose responses to PPAA treatment. After normalizing the luminescence data to the average of buffer controls, we plotted fold-response curves (Figures S2 and S3, right). The G8C2 system transfected at the highest MOI had the highest overall fold-change response (Figures 2A and S2). However, the absolute luminescence was lower and the intrasample variance was higher for the G8C2 system. By contrast, all G8G8 cell populations except one show significant and broadly similar responses to a wide range of PPAA doses, ranging from the best-responsive population (Pop4) which had significant and dose-dependent responses from 19.5 to 312.5 μg/mL PPAA to the least-responsive system (Pop5) which had significant responses only at 156.25 and 312.5 μg/mL (Figure S3). Cells transduced at the highest MOI (G8G8 Pop1, Figure S3A) also showed low fold-change, suggesting that high overexpression may not consistently produce a higher signal. It is also observable from these data that PPAA consistently induces cytotoxicity at 1250 and 625 μg/mL dosing (Figure 2A,B), which is detected as luminescence signal below baseline (vehicle-treated cells). Cytotoxicity in this dose range was also cross-validated with the resazurin cytotoxicity assay (Figure S4).
Figure 2.
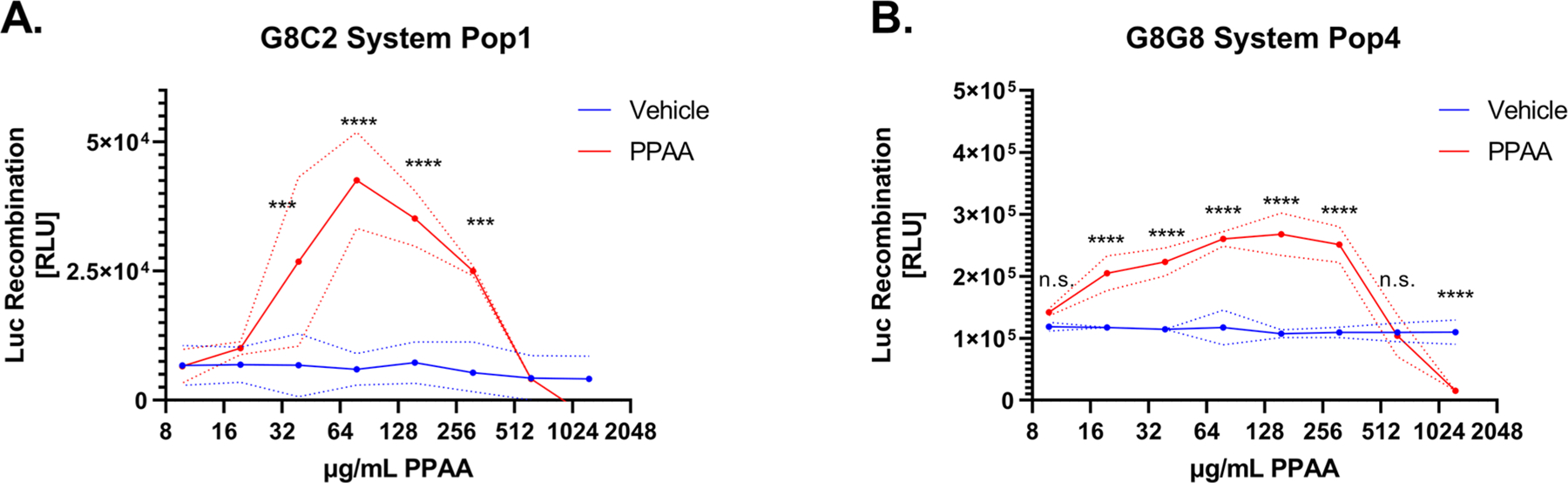
Lead candidate G8C2 and G8G8 cell populations demonstrate robust luciferase reconstitution following the endosomal disruption. G8C2 and G8G8 HEK 293T cells were incubated with a twofold dilution series from 1250 to 9.8 μg/mL PPAA for 2 h. Both systems show cell toxicity at the two highest doses tested (1250 and 625 μg/mL), visible as luminescence at or below vehicle-treated cell baseline. Both systems also generate a significant luminescent response at doses from 313 to 39 μg/mL for G8C2 population 1 (Pop1) and from 313 to 20 μg/mL for G8G8 population 4 (Pop4). Dashed lines represented the standard deviation. Additional information on statistical tests is provided in Supporting Methods, and full statistical test results are reported in Supporting Tables 1 and 2.
Because all but one G8G8 system population showed broadly similar responses, we reasoned that it was less sensitive to MOI or selection pressure, and thus was likely more facile to implement by other groups, compared to the G8C2 system, which requires high MOI. The G8G8 Pop4 cells in particular were promising, as they were generated using a low MOI and showed a measurable and differential response over a broad range of endosome-disruptive-polymer concentrations. Making a series of cell populations with varied MOIs was initially motivated by the idea that cytosolic luciferase complementation may occur and create baseline signal (without coconcentration of the reporter proteins into disrupted endosomes) if the reporter proteins were overexpressed at too high of a level. However, higher MOI correlated with better readouts with the G8C2 system. In this system, the interaction between NLuc-Gal8 and CALCOCO2-CLuc is in competition with their respective binding partners being endogenously produced within the cell, as it is expected that each of the two split-luciferase-tagged proteins also forms nonluminescent complexes with the endogenous protein binding partners. However, the G8G8 split-luciferase-system responsivity was not sensitive to the transduction ratio. In the G8G8 system, both Gal8-CLuc and Gal8-NLuc, as well as endogenous Gal8, are competing for binding to glycans that cover the internal leaflet of the endosomal membrane. We hypothesize that the competition with endogenous Gal8 in this system may be less impactful because the luminal surface of the endosome provides a larger reaction surface area available to coordinate luciferase reconstitution, versus the fixed conformation of the dimer–dimer interaction of CALCOCO2 and Gal8. The specific expression systems that we used to create these reporters may limit direct comparisons between the two, however, as they have different versions of the EF1A promoter. Furthermore, the fusion proteins produced with each reporter system may have different inherent protein-stability profiles (half-lives) in the cytosol, which would also affect MOI optimization.
Another potential advantage of the G8G8 reporter is that it uses a minimal N-terminal protein domain derived from Gal8, which is in opposition to the G8C2 system that expresses full-length Gal8 and CALCOCO2. As a result, the G8G8 design uses a shorter DNA sequence (3318 base pairs for G8G8 versus 4344 base pairs for the G8C2 system), which allows additional elements to be introduced into the same lentiviral vector (which are limited to approximately 6400 base pairs). For example, here we additionally incorporated an EGFP element to assess transduction efficiency by microscopy into the G8G8 system. The use of the minimal N-terminal Gal8 system may also be less disruptive to normal cellular processes, as the effector functions of Gal8 signaling as an endosomal-damage sensor occur through its C-terminal carbohydrate recognition domain,23 which is not expressed by our G8G8 construct. For these reasons, we focused our subsequent, deeper characterization and benchmarking on the G8G8 system, specifically Pop4.
Comparisons to Validated Gal8-YFP Endosomal Escape Assay.
A HEK 293T Gal8-YFP cell line was generated based on our previous work done in other cell lines11 to serve as a benchmark. Gal8-YFP tracking shows the same overall trend for PPAA in HEK 293T cells, with a strong dose response to PPAA treatment (Figure S5). In comparing the Gal8-YFP system to our lead cell line, G8G8 Pop4, we observed that the split-luciferase G8G8 system better excludes high, toxic doses of PPAA, as occurs at 625 μg/mL (Figure 3). When the loss of cellular metabolic activity occurs, the G8G8 system showed a loss of luminescent signal while the Gal-YFP assay shows a very high Gal8-YFP response. While the Gal-YFP assay must be used with cytotoxicity measurements in parallel, the G8G8 system gives a positive response only on live and metabolically active cells due to the ATP-dependence of firefly luciferase and shows similar sensitivity for detection of the effects of cell treatment with low concentrations of endosomolytic polymer. In our earlier, optimized Gal8-YFP assay, we see higher overall fold-change, relatively lower intra-and intersample variability, and very low baseline in control wells as shown by relatively small standard deviations in Figures 3 and S5B. However, the G8G8 split-luciferase assay requires less technical expertise than Gal8-YFP recruitment assays, which require microscopists to image and process data from dozens or hundreds of wells, which may preclude assay adoption by users who lack equipment or expertise for this more technically challenging and time-consuming assay. We believe these orthogonal measurements complement each other and prove useful as initial screening tools prior to additional confirmation using low-throughput methodologies, such as bioactivity assays, cellular fractionation, or transmission electron microscopy.
Figure 3.
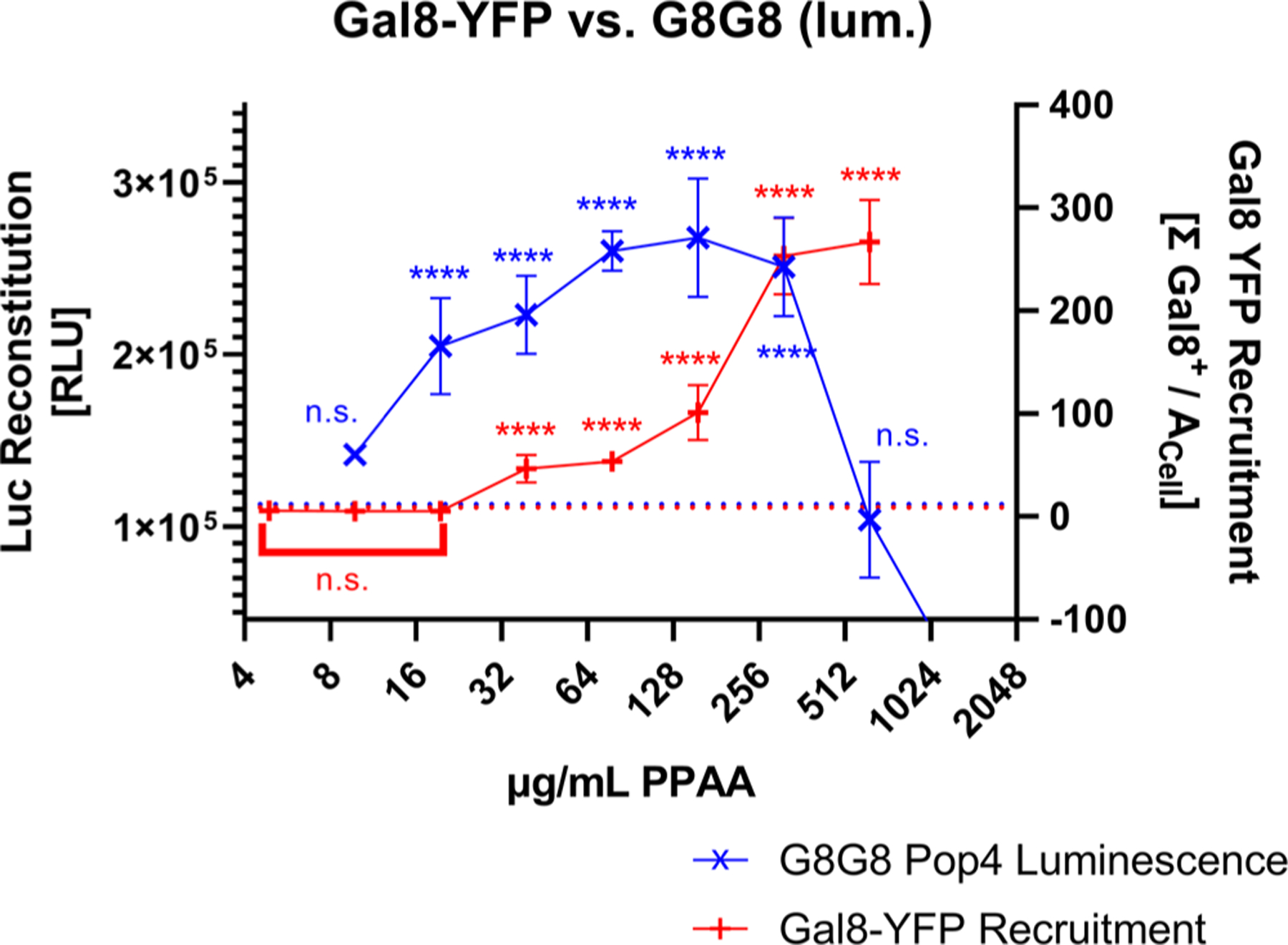
G8G8 cross-validation with the Gal8-YFP assay. Gal8-YFP and G8G8 HEK-293-T cells were treated with a twofold dilution series from 1250 to 9.8 μg/mL PPAA for 2 h. G8G8 luminescence data are plotted in blue as × on the left y-axis. Gal8-YFP response data are plotted in red as + on the right y-axis. Buffer treatment control responses for both assays were aligned and plotted as a dotted line; maximal responses were also aligned to compare responses. Error bars represent standard deviation.
G8G8 Assay Response to a Broad Set of Transfection Reagents.
Next, we assessed the ability of our lead G8G8 Pop4 cells to detect endosome disruption caused by several other transfection reagents. We treated cells with the commercial cationic transfection lipids DOTAP and Lipofectamine 2000, as well as the in vivo transfection reagent JetPEI. We also screened four additional cationic siRNA-delivery 50B polymers that we previously developed,30 characterized, and screened11 for endosomal disruption (using Gal8-YFP recruitment and several other gold standard assays) and siRNA-delivery efficiency. The 50B polymers screened have the same composition but varied molecular weights. Molecular weight has a significant correlation to endosome disruption and intracellular bioavailability with this delivery system11 (data reproduced as Figure S6 with 50B-S, -L, -2XL, and -4XL having increasing molecular weights), ranging from no endosomal disruption and no siRNA delivery for 50B-S to significant and increasing endosomal disruption and gene-silencing bioactivity across 50B-L, 50B-2XL, and 50B-4XL.
We show that G8G8 cells produce a significant increase in cellular luminescence for all of the agents tested except the control polymer 50B-S, which was tested as a negative control that does not induce endosomal disruption as validated in earlier studies.11 DOTAP, JetPEI, and Lipofectamine 2000 all showed significant increases in luminescence within 2 h of treatment, and, similar to the results with PPAA, allowed for the identification of toxicity at the highest doses (Figure 4A–C). Because DOTAP was formulated in dimethyl sulfoxide (DMSO) (100 mg/mL stock in DMSO) and Lipofectamine 2000 is formulated in ethanol (proprietary concentration), we also ran separate control experiments to show that neither DMSO nor ethanol treatment is sufficient to induce increases in luminescence in this assay format, showing only cytotoxicity at high doses (Figure S7).
Figure 4.
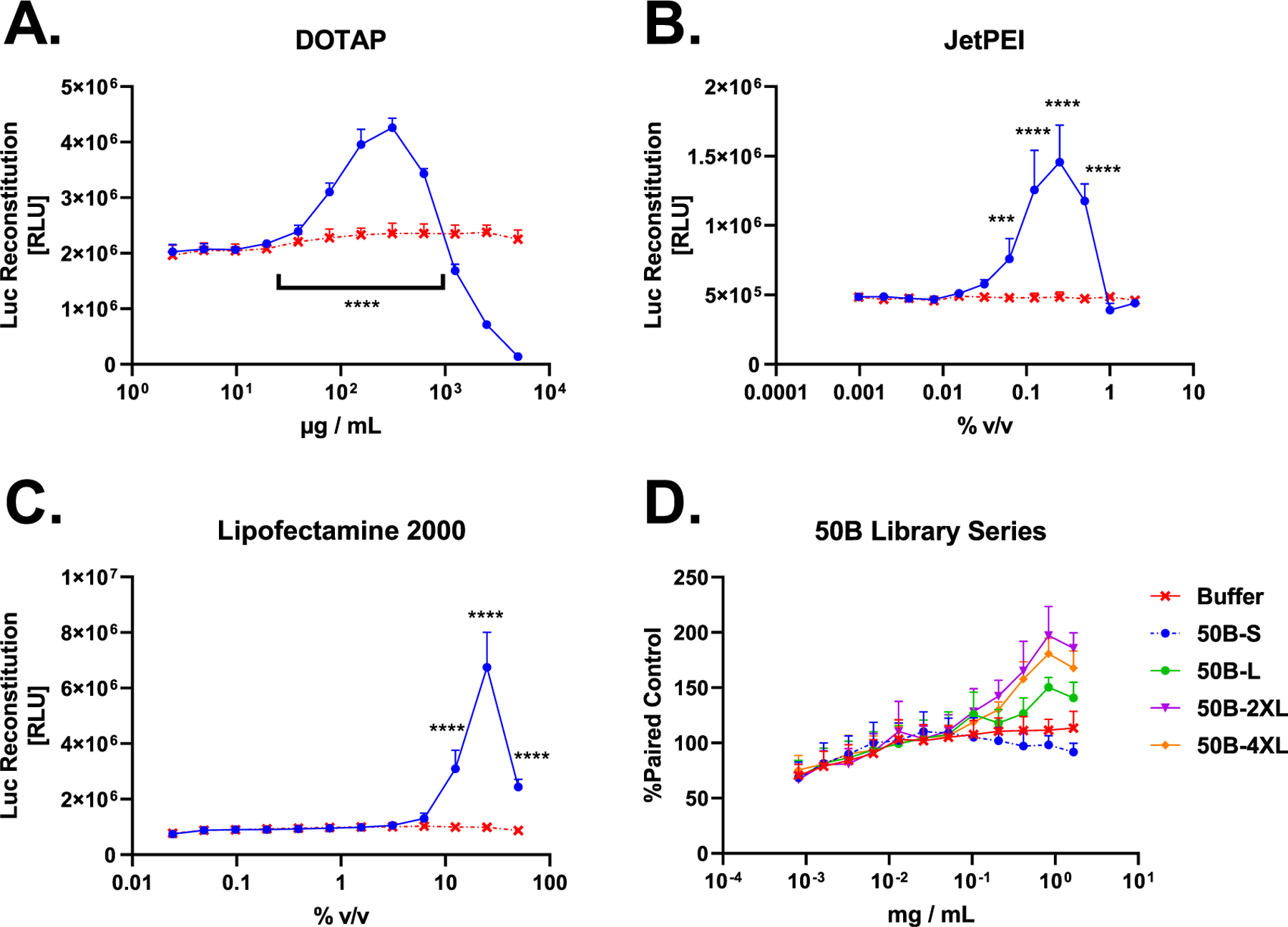
G8G8 Pop4 detects endosomal disruption from several commercial and investigational transfection compounds. (A) HEK 293T G8G8 Pop4 cells were treated for 2 h with a twofold dilution series of DOTAP lipid in DMSO, starting at 5 mg/mL–2 μg/mL. Toxicity is observed at doses higher than 625 μg/mL, which shows a significant luminescent response. Signal returns to baseline at 39 μg/mL DOTAP and below. DMSO alone does not cause luciferase reconstitution (Figure S7). (B) HEK 293T G8G8 Pop4 cells were treated for 2 h with a twofold dilution series of JetPEI as formulated by the manufacturer, starting at 2.0–0.001% v/v. Baseline response is observed at 2.0 and 1.0%; significant luciferase reconstitution is observed from 0.5 to 0.06%, which shows a significant luminescent response. Signal returns to baseline at 0.03% and below. (C) HEK 293T G8G8 Pop4 cells were treated for 2 h with a twofold dilution series of Lipofectamine 2000 as formulated by the manufacturer, starting at 50–0.02% v/v. At 50, 25, and 12.5% v/v, significant luciferase reconstitution is observed, which peaks at 25%. Signal returns to baseline at 6.25% and below. (D) HEK 293T G8G8 Pop4 cells were treated for 6 h with a twofold dilution series of 50B polymers, starting at 1.665 mg/mL–800 ng/mL. Responses were normalized to the same plate buffer readings and plotted. Significant responses were observed for 50B-4XL, 50B-2XL, and 50B-L. 50B-S did not produce a significant response. Significant toxicity was not observed in this dose and time regime. For full statistical analysis for this figure, see Supporting Tables 3 and 4. Error bars represent standard deviation.
When we screened four members of our 50B library for G8G8 luminescence, we found that 50B-L, -2XL, and -4XL all showed significant luminescence, while 50B-S, a nonendosomolytic control polymer, did not produce significant luminescence (Figure 4D). This mirrors our earlier bioactivity, transmission electron microscopy, and Gal8-YFP recruitment data for these polymers11 (partially reproduced in Figure S6), which showed that the 50B-S polymer did not cause endosomal disruption, did not recruit Gal8, and did not cause siRNA-mediated gene knockdown, in contrast to the larger-molecular-weight polymers. The lack of response to dose-matched 50B-S versus other 50B-library members is an important validation of the mechanism, as 50B-S does not disrupt endosomes, but otherwise shares the same composition as the other polymers. Furthermore, the assay fully recapitulated the rank-order of endosome disruption among this polymer series as previously measured by Gal8-YFP, where the greatest disruption is seen for 50B-2XL and 4XL, followed by -L, with 50B-S showing no activity.
DOTAP and JetPEI showed luminescent responses in vitro at manufacturer recommended ranges, which are 50–200 μg/mL and 0.2–0.4% v/v, respectively. We did not, however, initially detect endosomal disruption at the supplier-recommended dose of Lipofectamine 2000 (0.2–0.5%). Further exploration of a time course following cell treatment with 0.5% v/v Lipofectamine at 0, 2, 8, and 24 h revealed that this reagent/dose peaks later, at 8 h for the time points that we measured (Figure S8), and showed statistically significant luminescence increases relative to control at all three time points tested. Notably, Lipofectamine 2000 produced a measurable endosomal disruption response at 0.5% at the 2 h time point in the follow-up experiment shown in Figure S8; this study used a newer stock of Lipofectamine 2000 than the Figure 4C study, in which the particular stock of older Lipofectamine 2000 may have partially lost its activity. We likewise conducted a similar time-course study with PPAA (Figure S9) at 15, 30, 60, and 120 min, which revealed that high doses initially show high luminescence but cause toxicity at longer time points, while lower doses show endosomal disruption that peaks after about 30 min but remains strong for at least 120 min. This run showed a higher baseline signal compared to earlier experiments (Figure 3) but showed similar fold-increase luminescence relative to internal buffer controls as other assay runs. These experiments demonstrate the ability of the G8G8 assay to dissect concentration-dependent endosome-disruption kinetics.
CONCLUSIONS
The development of intracellular-acting large-molecule biologic drugs remains limited by cellular barriers to intracellular delivery, especially the plasma and endosomal membranes. While several assays have been developed to assess intracellular drug delivery, assays to specifically measure endosomal disruption are cumbersome, indirect, and susceptible to artifacts. Here, two proof-of-concept, first-in-class, genetically encoded, split-luciferase-based, turn-on assays are introduced that enable the use of luminescence as a rapid and sensitive measurement of endosomal disruption in live cells. Like the previous fluorescent-Gal8 microscopy assay, these new assays are carrier- and drug-agnostic and do not require labeling of either drug or carrier with tracers, dyes, or synthetic fluorophores that could alter cellular uptake or trafficking. We show that our lead system, G8G8, has significant, dose-dependent luminescent responses to PPAA, Lipofectamine 2000, DOTAP, JetPEI, and 50B polymers. This assay also has the advantage of having an inherent capacity to rule out cytotoxic treatments because the signal generation is dependent on the presence of intracellular ATP.
Despite their utility shown herein, these new G8C2 and G8G8 endosome-disruption reporters do have opportunities for further exploration and improvement in future studies. For example, the level of luminescence increase in these systems may provide insufficient sensitivity for in vivo applications. Therefore, it will be of interest to determine which reporter design components dictate levels of basal reconstitution and magnitude of endosome-disruption-associated turn-on luminescent signal. Specifically, new reporter variants should be developed and tested using different split-luciferase fragments24 with varied binding affinities and/or using different gene promoters that allow variation of reporter-expression levels over a broader range. Future studies should also characterize the sources of variance in these assays due to factors such as the timing of substrate addition, temperature, lot of D-luciferin, user, and cell-passage number, which were accounted for herein by comparison to buffer-treated controls within each experiment. In all, the systems presented here are first-in-class turn-on luminescence-based assays that directly, rapidly, and quantitatively measure endosomal disruption in live cells without requiring the addition of labels to carriers or cargo.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the United States National Institutes of Health (Grants R01HL122347, R01EB019409, and R01CA224241) to C.L.D.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssensors.0c00103.
Graphical overview of the G8C2 and G8G8 transcripts; G8C2 and G8G8 population screen with PPAA; HEK 293T PPAA viability assay; HEK 293T cell line establishment; G8G8 DMSO and ethanol control studies; time point testing of Lipofectamine and PPAA with G8G8 assay system; G8C2 and G8G8 plasmid map; materials and methods (PDF)
Supplementary statistical analysis for Figures S2, S3, and 4A–D; summary of Supporting Table 2 (XLSX)
The authors declare no competing financial interest.
Plasmids encoding the G8C2 (plasmid ID 128387, https://www.addgene.org/128387/) and G8G8 (plasmid ID 128386, https://www.addgene.org/128386/) systems are available from AddGene.org to nonprofit requesters. For requests of cell lines, please contact the corresponding author.
An earlier version of this manuscript is available as a formatted pre-print: https://doi.org/10.1101/2020.01.16.906180.
Complete contact information is available at: https://pubs.acs.org/10.1021/acssensors.0c00103
REFERENCES
- (1).Alnylam Pharmaceuticals, Inc. Alnylam Announces First-Ever FDA Approval of an RNAi Therapeutic, ONPATTRO (patisiran) for the Treatment of the Polyneuropathy of Hereditary Transthyretin-Mediated Amyloidosis in Adults. https://investors.alnylam.com/press-release?id=22946 (accessed January 15, 2020).
- (2).FDA. Package Insert—ONPATTRO. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210922Orig1s000lbl.pdf (accessed January 15, 2020).
- (3).Jayaraman M; Ansell SM; Mui BL; Tam YK; Chen J; Du X; Butler D; Eltepu L; Matsuda S; Narayanannair JK; Rajeev KG; Hafez IM; Akinc A; Maier MA; Tracy MA; Cullis PR; Madden TD; Manoharan M; Hope MJ Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem., Int. Ed 2012, 51, 8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Marwick C First “Antisense” Drug Will Treat CMV Retinitis. J. Am. Med. Assoc 1998, 280, 871. [PubMed] [Google Scholar]
- (5).Adams D; Gonzalez-Duarte A; O’Riordan WD; Yang C-C; Ueda M; Kristen AV; Tournev I; Schmidt HH; Coelho T; Berk JL; Lin K-P; Vita G; Attarian S; Planté-Bordeneuve V; Mezei MM; Campistol JM; Buades J; Brannagan TH; Kim BJ; Oh J; et al. Patisiran, an RNAi Therapeutic, for Hereditary Transthyretin Amyloidosis. N. Engl. J. Med 2018, 379, 11–21. [DOI] [PubMed] [Google Scholar]
- (6).Shi J; Chou B; Choi JL; Ta AL; Pun SH Investigation of Polyethylenimine/DNA Polyplex Transfection to Cultured Cells Using Radiolabeling and Subcellular Fractionation Methods. Mol. Pharm 2013, 10, 2145–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gilleron J; Querbes W; Zeigerer A; Borodovsky A; Marsico G; Schubert U; Manygoats K; Seifert S; Andree C; Stöter M; Epstein-Barash H; Zhang L; Koteliansky V; Fitzgerald K; Fava E; Bickle M; Kalaidzidis Y; Akinc A; Maier M; Zerial M Image-Based Analysis of Lipid Nanoparticle-Mediated siRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat. Biotechnol 2013, 31, 638–646. [DOI] [PubMed] [Google Scholar]
- (8).Thurston TLM; Wandel MP; Von Muhlinen N; Foeglein Á; Randow F Galectin 8 Targets Damaged Vesicles for Autophagy to Defend Cells against Bacterial Invasion. Nature 2012, 482, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wittrup A; Ai A; Liu X; Hamar P; Trifonova R; Charisse K; Manoharan M; Kirchhausen T; Lieberman J Visualizing Lipid-Formulated siRNA Release from Endosomes and Target Gene Knockdown. Nat. Biotechnol 2015, 33, 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kilchrist KV; Evans BC; Brophy CM; Duvall CL Mechanism of Enhanced Cellular Uptake and Cytosolic Retention of MK2 Inhibitory Peptide Nano-Polyplexes. Cell. Mol. Bioeng 2016, 9, 368–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kilchrist KV; Dimobi SC; Jackson MA; Evans BC; Werfel TA; Dailing EA; Bedingfield SK; Kelly IB; Duvall CL Gal8 Visualization of Endosome Disruption Predicts Carrier-Mediated Biologic Drug Intracellular Bioavailability. ACS Nano 2019, 13, 1136–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Steinauer A; LaRochelle JR; Knox SL; Wissner RF; Berry S; Schepartz A HOPS-Dependent Endosomal Fusion Required for Efficient Cytosolic Delivery of Therapeutic Peptides and Small Proteins. Proc. Natl. Acad. Sci. U.S.A 2019, 116, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Werfel TA; Wang S; Jackson MA; Kavanaugh TE; Joly MM; Lee LH; Hicks DJ; Sanchez V; Ericsson PG; Kilchrist KV; Dimobi SC; Sarett SM; Brantley-Sieders DM; Cook RS; Duvall CL Selective MTORC2 Inhibitor Therapeutically Blocks Breast Cancer Cell Growth and Survival. Cancer Res 2018, 78, 1845–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wang Y; Xie Y; Kilchrist KV; Li J; Duvall CL; Oupický D Endosomolytic and Tumor-Penetrating Mesoporous Silica Nanoparticles for siRNA/miRNA Combination Cancer Therapy. ACS Appl. Mater. Interfaces 2020, 12, 4308–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Rui Y; Wilson DR; Choi J; Varanasi M; Sanders K; Karlsson J; Lim M; Green JJ Carboxylated Branched Poly(β-Amino Ester) Nanoparticles Enable Robust Cytosolic Protein Delivery and CRISPR-Cas9 Gene Editing. Sci. Adv 2019, 5, No. eaay3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hendershot J; Smith AE; Werfel TA Preparation of Neutrally-Charged, pH-Responsive Polymeric Nanoparticles for Cytosolic siRNA Delivery. J. Vis. Exp 2019, 147, No. e59549. [DOI] [PubMed] [Google Scholar]
- (17).Evans BC; Fletcher RB; Kilchrist KV; Dailing EA; Mukalel AJ; Colazo JM; Oliver M; Cheung-Flynn J; Brophy CM; Tierney JW; Isenberg JS; Hankenson KD; Ghimire K; Lander C; Gersbach CA; Duvall CL An Anionic, Endosome-Escaping Polymer to Potentiate Intracellular Delivery of Cationic Peptides, Biomacromolecules, and Nanoparticles. Nat. Commun 2019, 10, No. 5012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mukalel AJ; Evans BC; Kilchrist KV; Dailing EA; Burdette B; Cheung-Flynn J; Brophy CM; Duvall CL Excipients for the Lyoprotection of MAPKAP Kinase 2 Inhibitory Peptide Nano-Polyplexes. J. Controlled Release 2018, 282, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Blersch J; Francisco V; Rebelo C; Jiménez-Balsa A; Antunes H; Gonzato C; Pinto S; Simões S; Liedl K; Haupt K; Ferreira L A Light-Triggerable Nanoparticle Library for the Controlled Release of Non-Coding RNAs. Angew. Chem 2020, 132, 2001–2007. [DOI] [PubMed] [Google Scholar]
- (20).Dingjan I; Verboogen DRJ; Paardekooper LM; Revelo NH; Sittig SP; Visser LJ; Von Mollard GF; Henriet SSV; Figdor CG; Ter Beest M; Van Den Bogaart G Lipid Peroxidation Causes Endosomal Antigen Release for Cross-Presentation. Sci. Rep 2016, 6, No. 22064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Vasta GR Galectins as Pattern Recognition Receptors: Structure, Function, and Evolution. Adv. Exp. Med. Biol 2012, 946, 21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ideo H; Seko A; Ishizuka I; Yamashita K The N-Terminal Carbohydrate Recognition Domain of Galectin-8 Recognizes Specific Glycosphingolipids with High Affinity. Glycobiology 2003, 13, 713–723. [DOI] [PubMed] [Google Scholar]
- (23).Kim BW; Hong SB; Kim JH; Kwon DH; Song HK Structural Basis for Recognition of Autophagic Receptor NDP52 by the Sugar Receptor Galectin-8. Nat. Commun 2013, 4, No. 1613. [DOI] [PubMed] [Google Scholar]
- (24).Paulmurugan R; Gambhir SS Combinatorial Library Screening for Developing an Improved Split-Firefly Luciferase Fragment-Assisted Complementation System for Studying Protein-Protein Interactions. Anal. Chem 2007, 79, 2346–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cheung CY; Murthy N; Stayton PS; Hoffman AS A pH-Sensitive Polymer That Enhances Cationic Lipid-Mediated Gene Transfer. Bioconjugate Chem 2001, 12, 906–910. [DOI] [PubMed] [Google Scholar]
- (26).Foster S; Duvall CL; Crownover EF; Hoffman AS; Stayton PS Intracellular Delivery of a Protein Antigen with an Endosomal-Releasing Polymer Enhances CD8 T-Cell Production and Prophylactic Vaccine Efficacy. Bioconjugate Chem 2010, 21, 2205–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Crownover E; Duvall CL; Convertine A; Hoffman AS; Stayton PS RAFT-Synthesized Graft Copolymers That Enhance pH-Dependent Membrane Destabilization and Protein Circulation Times. J. Controlled Release 2011, 155, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Evans BC; Hocking KM; Osgood MJ; Voskresensky I; Dmowska J; Kilchrist KV; Brophy CM; Duvall CL MK2 Inhibitory Peptide Delivered in Nanopolyplexes Prevents Vascular Graft Intimal Hyperplasia. Sci. Transl. Med 2015, 7, No. 291ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Evans BC; Hocking KM; Kilchrist KV; Wise ES; Brophy CM; Duvall CL Endosomolytic Nano-Polyplex Platform Technology for Cytosolic Peptide Delivery to Inhibit Pathological Vasoconstriction. ACS Nano 2015, 9, 5893–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Nelson CE; Kintzing JR; Hanna A; Shannon JM; Gupta MK; Duvall CL Balancing Cationic and Hydrophobic Content of PEGylated siRNA Polyplexes Enhances Endosome Escape, Stability, Blood Circulation Time, and Bioactivity in Vivo. ACS Nano 2013, 7, 8870–8880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.