

Abstract
The last decade has seen development of oral, small molecule therapies that address the basic cystic fibrosis transmembrane conductance regulator (CFTR) protein defect. Highly effective modulator treatment (HEMT) that is efficacious for a large majority of people living with cystic fibrosis (CF) promises to change the landscape of this chronic life-limiting disease. Some people living with CF have a CFTR genotype that renders them eligible for HEMT, but also have comorbidities that excluded them from the original Phase III clinical trials that led to US Food and Drug Administration approval. The purpose of this review is to address the use of HEMT in challenging situations, including initiation for those with advanced CF lung disease, and use after solid organ transplant, during pregnancy, and for individuals with CFTR-related disorders without a definitive diagnosis of CF.
Introduction
Oral cystic fibrosis transmembrane conductance regulator (CFTR) protein modulator treatment has revolutionized the management of cystic fibrosis (CF). The first approved CFTR modulator was ivacaftor (IVA) for treatment of the approximately 4% of individuals with CF with the Gly551Asp (G551D) mutation.(1) The substantial improvements conferred by IVA in CFTR function (as measured by change in sweat chloride concentration) and pulmonary function (as measured by change in percent predicted forced expiratory volume in one second, ppFEV1) established the benchmark for highly effective modulator treatment (HEMT). Improvement in ppFEV1 and sweat chloride concentration with the use of elexacaftor/tezacaftor/ivacaftor (ETI) for individuals with CF with at least one copy of Phe508del (F508del) is comparable to, or better than, gains observed for those treated with IVA (Figure 1).(2, 3) Additional benefits of HEMT include decreased frequency of pulmonary exacerbations, and improved nutritional status (as measured by body mass index (BMI)) and respiratory health-related quality of life.(1–3) The development of HEMT that is efficacious for a large majority of people living with CF promises to change the landscape of this chronic life-limiting disease.
Figure 1: CFTR modulator treatment for patients with cystic fibrosis and effect on lung function and sweat chloride concentration.
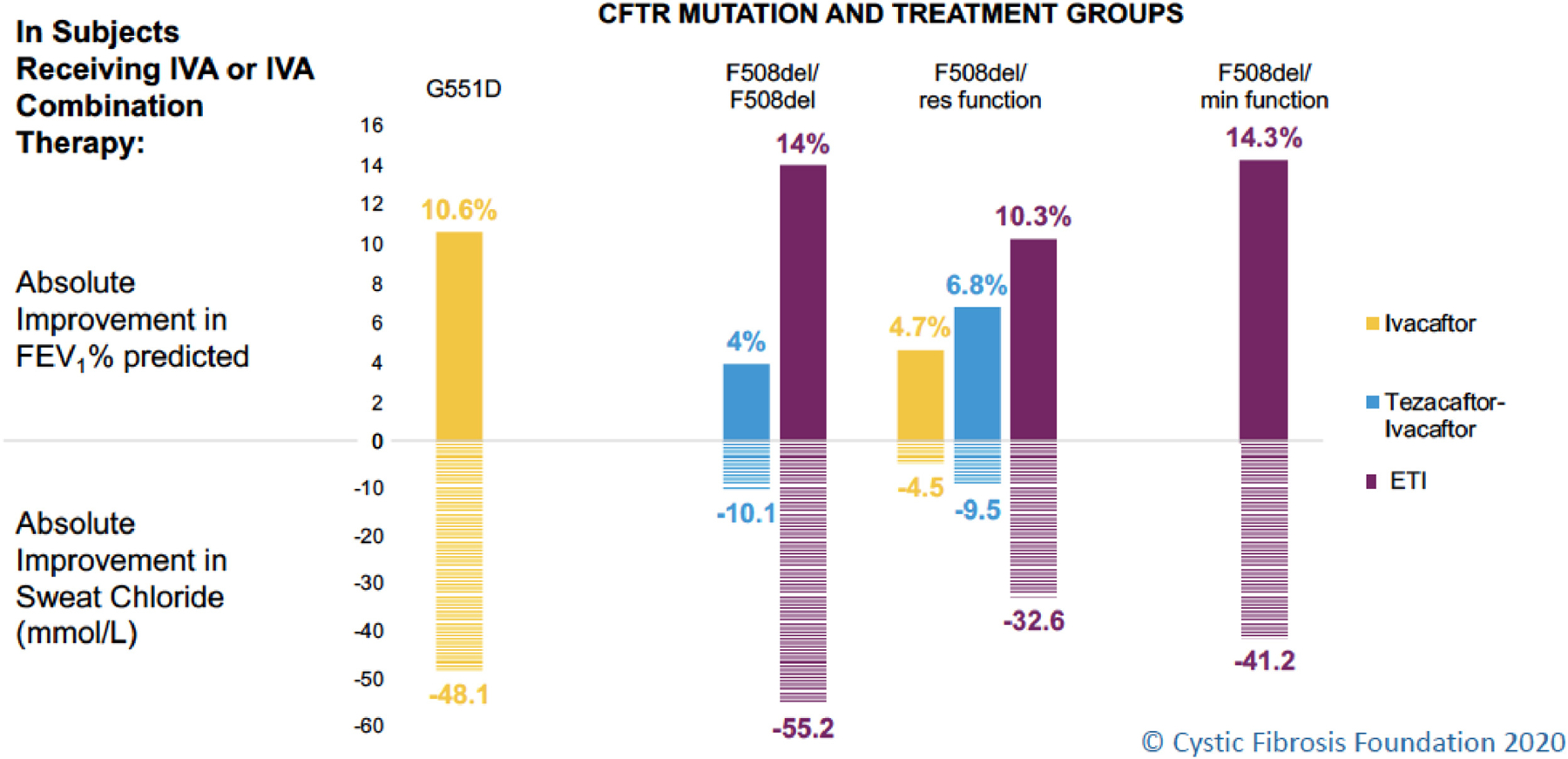
Abbreviations: CFTR = cystic fibrosis transmembrane conductance regulator; IVA = ivacaftor; FEV1 = forced expiratory volume in one second; ETI = elexacaftor/tezacaftor/ivacaftor; Res function=residual function (mutation allows some CFTR function and/or is responsive to CFTR modulators in vitro); Min function=minimal function (mutation produces no CFTR protein and/or does not respond to CFTR modulators in vitro)
Changes in the graph for ETI use in patients with CF with mutations for which there were approved therapies in the U.S. (F508del/F508del and F508del/res function) reflect total predicted change compared to placebo. For example, the ETI bar for those homozygous for F508del reflects the addition of the effect of tezacaftor/ivacaftor versus placebo of 4% in the phase III(45) plus the effect of the addition of elexacaftor in those already on tezacafator/ivacaftor of 10% in the phase III trial(3).
In December 2020, the US Food and Drug Administration (FDA) approved ETI for people living with CF, age 12 years and older, with at least one copy of an additional 177 rare CFTR mutations.(4) The effectiveness of ETI for these additional mutations is unclear, so comments on HEMT below refer primarily to individuals with a F508del mutation, not to all individuals who are eligible for ETI.
Some people living with CF have a CFTR genotype that is eligible for HEMT, but also have comorbidities that would have excluded them from the original Phase III clinical trials that led to FDA approval (Table 1).(1–3) The risk to benefit ratio in these unstudied or understudied populations is unclear and few published studies exist to support the role of ETI in these patients. Given the remarkable effects of HEMT seen in clinical trials and in clinical practice, expanding access to other individuals who have the potential to benefit is a priority for the CF community. The purpose of this review is to address the use of HEMT in challenging situations, including initiation for those with advanced CF lung disease, and use after solid organ transplantation, during pregnancy, and for individuals with CFTR-related disorders without a clear diagnosis of CF. While the focus of the review is on the clinical risks and benefits of HEMT, issues of cost and access to HEMT are also briefly addressed.
Table 1.
Major exclusion criteria of common conditions in people with cystic fibrosis for the elexacaftor/tezacaftor/ivacaftor trials.(2, 3)
| ■ppFEV1 <40 |
| ■Clinically significant cirrhosis with or without portal hypertension |
| ■Solid organ or hematological transplantation |
| ■Pregnant or nursing females |
| ■Lung infection with Burkholderia cenocepacia, B. dolosa, and Mycobacterium abscessus |
| ■Single copy of F508del without a second identified CFTR mutation |
ppFEV1 = percent predicted forced expiratory volume in one second; CFTR = cystic fibrosis transmembrane conductance regulator
Advanced CF lung disease: initiation of HEMT
As in the case of most Phase III trials of CFTR modulators in people with CF, individuals with advanced CF lung disease were excluded from the Phase III clinical trials that led to FDA approval of ETI (Table 1).(1–3, 5) Previous experience with the use of IVA in people with advanced CF lung disease suggested more modest clinical benefits in lung function, BMI and exacerbation reduction than were seen in the pivotal trials with a similar safety profile.(5) This experience provides some reassurance for use of ETI in those with advanced CF lung disease. Furthermore, a small number of individuals whose ppFEV1 fell to <40% of predicted between enrollment and randomization were included in the Phase III studies of HEMT, and these people had similar benefit in lung function compared to the overall cohort during the clinical trials.(2, 5)
Although no randomized trials have been conducted that have intentionally included those with advanced disease, in a single-center open-label cohort study conducted by investigators in Ireland, ppFEV1, BMI, and pulmonary exacerbation frequency improved dramatically among patients with ppFEV1 <40% who were treated with ETI, from a mean ppFEV1 of 27% to 36%.(6) Additionally, in a multicenter retrospective cohort study, Bermingham et al reported that in 64 patients with baseline ppFEV1 <40%, there was an average increase in ppFEV1 of 8.2% (compared to approximately 14% in the Phase III trials) with an improvement in BMI of 0.65 kg/m2.(7) Early clinical experience suggests varied outcomes for people with advanced CF lung disease treated with ETI, with most experiencing an improvement in BMI, exacerbation frequency and cough, but variable effects on lung function. From the available clinical trial data for ETI, adverse event rates were similar for individuals with ppFEV1 <40% compared to those with higher lung function.(2, 5) Collective anecdotal experience suggests that some individuals with advanced CF lung disease have severe cough and copious sputum production in the first few days after initiation, suggesting that counseling on early symptom changes is prudent. However, experience to date does not suggest that hospitalization of those with advanced disease is warranted solely for the purpose of initiating ETI. Further studies of ETI for those with advanced CF lung disease are warranted, but initiation of ETI appears to be safe and effective for most patients who are otherwise good candidates for treatment.
An evaluation of data from the US and United Kingdom (UK) registries demonstrated that IVA use was associated with decreased risk of death and need for lung transplant in those treated CF populations.(8) However, it is important to recognize that HEMT does not “cure” CF, and consideration of lung transplant for people with advanced CF lung disease on HEMT is an essential component of comprehensive CF care. When individuals with advanced CF lung disease meet lung transplant referral criteria despite HEMT use, it is appropriate to refer them to a lung transplant program for evaluation. The decision to list for lung transplant will be more complex in the HEMT era, as we lack data about long-term outcomes for a large population treated with HEMT. Early experience has demonstrated that a significant number of persons with CF who were listed for lung transplant improved sufficiently to be removed from the transplant waiting list. Transplant teams are encouraged to consider each patient’s clinical and physiological trajectory, in addition to relevant markers of disease severity (such as oxygen requirement, exacerbation frequency, and presence of hypercapnia and pulmonary hypertension), when determining the timing of listing for lung transplant or when deciding about removal from the transplant waitlist. Also, frank discussion regarding the unknown risks and benefits of ETI post-transplant should occur prior to transplant.
Solid organ transplant: To continue or initiate HEMT
Non-lung solid organ transplant
Individuals with CF who have undergone non-lung solid organ transplant, most often isolated liver transplant, continue to have CF lung disease and treatment with HEMT is indicated to improve pulmonary outcomes, including ppFEV1 and pulmonary exacerbation frequency. Initiation of ETI in transplant recipients requires consultation with the transplant team. There may be complications in the graft that could prohibit use of HEMT for some period of time following transplant. The risk for liver toxicity due to ETI, especially in the setting of liver transplant, is an important consideration and warrants increased monitoring for elevation of transaminases. Because organ transplant recipients require multiple medications for immunosuppression, attention to drug-drug interactions is critical. Interactions of ETI with cytochrome P450 enzymes leads to induction or inhibition of multiple medications, including azole antifungal agents (e.g. fluconazole, posaconazole), calcineurin inhibitors (e.g. tacrolimus, cyclosporine), mTOR inhibitors (e.g. sirolimus, everolimus), and others that may be used in the transplant setting.(4) Dose reductions of ETI and/or the transplant medication may be necessary, and consultation with the transplant team and a pharmacist is recommended.(4) There are currently no clinical standards for assessing adequate ETI levels at baseline nor in the setting of dose reduction to correct for drug-drug interactions.
Lung transplant
Lung transplant recipients no longer have CF lung disease. FDA approval of HEMT was based on significant improvement in the primary endpoint of ppFEV1 in the pivotal Phase III trials.(1–3) Therefore, lung transplant recipients with CF will not derive the primary benefit of this treatment. HEMT, including ETI, has effects on important non-pulmonary manifestations of CF, including nutritional outcomes (e.g. BMI)(1–3), sinus disease (9–11), and the severity of CF-related abnormalities in glucose metabolism (12–14). IVA may influence antibiotic responsiveness of Pseudomonas aeruginosa, an important CF pathogen. (15)
For lung transplant recipients with CF, extra-pulmonary manifestations of CF may contribute to development of chronic lung allograft dysfunction (CLAD)(16), otherwise known as “chronic rejection.” CLAD is the largest barrier to long-term survival after lung transplant (16, 17) and reducing the risk of CLAD could drastically impact lung transplant recipient longevity. There are multiple known risk factors for CLAD. Acute cellular rejection is the biggest risk factor for CLAD (18, 19), and antibody mediated rejection (20) is increasingly recognized as a risk factor.(16) Non-immune causes of epithelial injury and subsequent CLAD include: acute infections, specifically with viruses (21, 22); gastroesophageal reflux disease, where a growing body of literature shows an association with CLAD, and anti-reflux surgery may improve ppFEV1 or prevent progression of CLAD (23, 24); chronic sinus disease and chronic airway infection (18). Airway inflammation likely plays an important role in the development of CLAD, and multiple inflammatory markers have been associated with airway injury in the lung allograft [IL-6, IL-8, TGF-β, IL-17, PMNs](25–27).
Chronic sinus disease is thought to be a risk factor for colonization of the lung allograft with P. aeruginosa and methicillin-resistant Staphylococcus aureus.(28, 29) P. aeruginosa colonization in the lung allograft has been linked to increased risk for development of donor specific HLA antibodies, antibody mediated rejection, and CLAD.(18, 30) Surgical treatment of sinus disease has been associated with decreased risk of pulmonary infections in lung transplant recipients with CF.(31) There have been conflicting results in small studies of sinus surgery for CF chronic rhinosinusitis and effect on lung transplant outcomes.(32–34) One study showed that for patients in whom sinus pathogens were eradicated, there was an improvement in 5-year survival and reduced rates of CLAD.(34) A separate study showed that IVA enhances fluoroquinolone activity against P. aeruginosa.(15) For individuals with refractory symptomatic sinus disease, colonization with P. aeruginosa, and/or difficult to manage chronic rhinosinusitis, use of HEMT after lung transplant has the potential for benefit, but requires further study.
Malnutrition is commonly present for lung transplant recipients with CF.(35, 36) While survival after lung transplant may be worse for malnourished CF recipients compared to CF recipients with normal weight,(36) there are no data about targeted treatment, such as HEMT, to improve BMI after transplant. From clinical experience, CF recipients tend to gain weight after lung transplant due to a combination of corticosteroids, reduced caloric needs, and decreased pulmonary symptoms. Whether HEMT will prove to be beneficial for individuals who struggle with refractory malnutrition after lung transplant is unknown, however, a trial of HEMT may be reasonable for some individuals with refractory malnutrition.
CF lung transplant recipients are at an increased risk of gastrointestinal malignancies (37) and post-transplant lymphoproliferative disorder (PTLD)(38). There is evidence that CFTR protein acts as a tumor suppressor (39) and CFTR dysfunction could play a role in CF recipients’ risk for malignancy after lung transplant. Determining whether HEMT might prevent malignancy will require long-term observation of individuals on treatment for years after lung transplant.
Just as in non-lung solid organ transplant, there are risks associated with the use of HEMT after lung transplant. The potential for drug-drug interactions (4), side effects, and toxicity from HEMT adds additional complexity for a patient population with potentially tenuous clinical status. Further, it is not clear whether the lung transplant population will derive meaningful benefit beyond what can be achieved with traditional therapies for sinus disease or malnutrition. In truth, we are still learning about the many systemic implications of HEMT in the PROMISE (NCT04038047), RECOVER (NCT04602468), and other ongoing clinical studies (NCT04056702). HEMT should be studied in a randomized controlled trial to determine benefits and harms in the lung transplant population.
Pregnancy: To stop or continue HEMT
As women with CF live longer and healthier lives, an increased number are contemplating pregnancy (40, 41); consequently, the number of pregnancies in women with CF is increasing (42). In order to protect the unborn fetus from known or unknown impacts of new therapeutics, pregnancy is an exclusion criterion for most Phase III studies. Pregnancy was an exclusion for enrollment in the studies that led to approval of ETI, as well as for all of the previously approved CFTR modulators (Table 1).(1–3, 43–45) Animal studies, case reports and an international survey have suggested no major harm from use of CFTR modulators during all or part of pregnancy; however, human data are relatively limited.(46–53) Thus, when a woman with CF using CFTR modulators presents to discuss the potential of pregnancy or presents with pregnancy, the risks and benefits must be weighed. There are several considerations, including 1) the benefits to the mother (particularly improved lung function and weight and decreased pulmonary exacerbations) 2) the risk to the mother of stopping (decreased lung function and weight, inability to gain adequate weight, increased pulmonary exacerbations, and in rare cases, death) (54, 55) and 3) the unknown risk to the developing fetus of continuation of the drug (4, 56) (Figure 2).
Figure 2. Risks of continuation or discontinuation of highly effective modulator treatment (HEMT) during pregnancy.
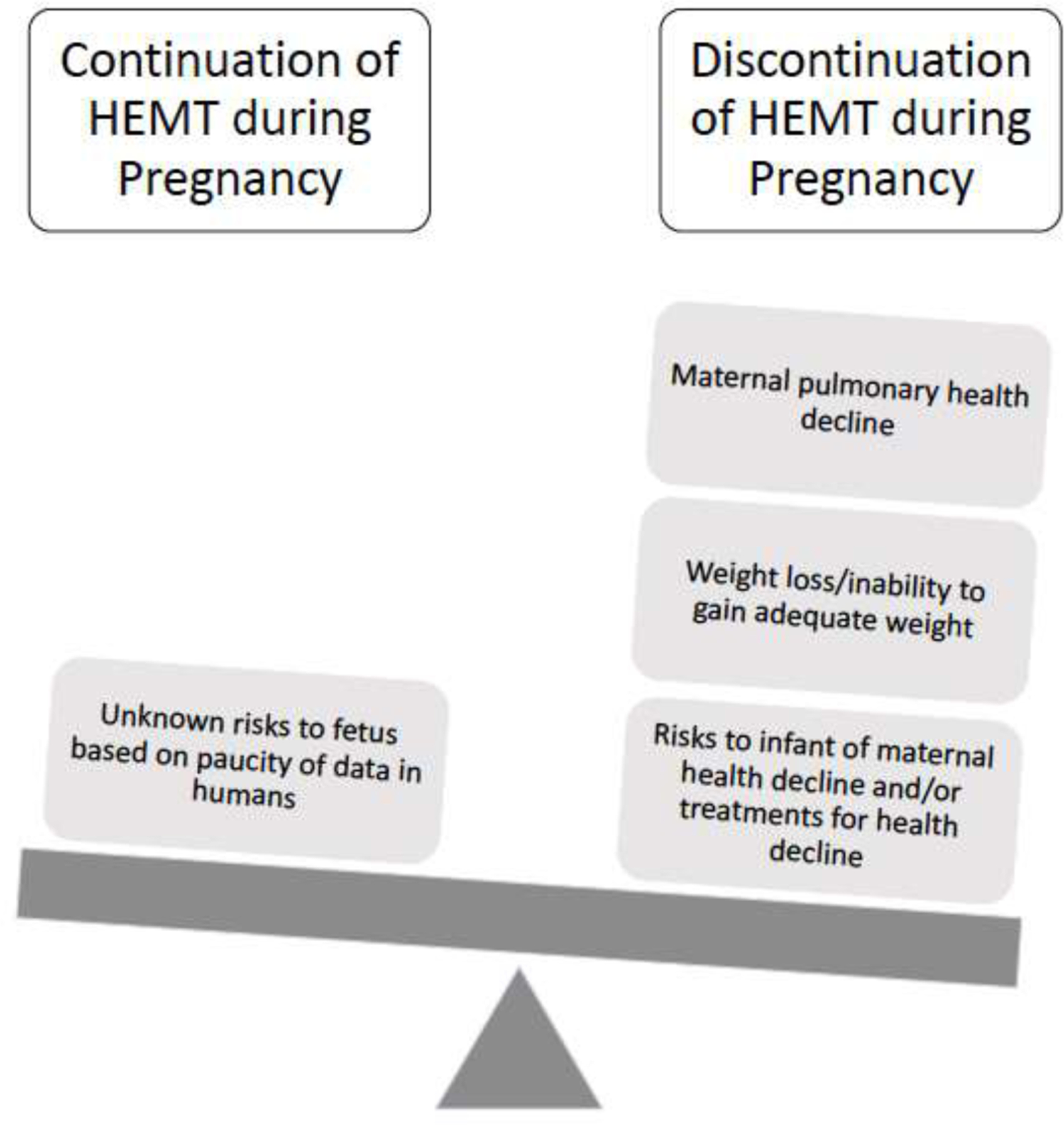
The unknown risk to the fetus includes the potential risk of the development of cataracts based on demonstration of cataract formation in juvenile rats who were administered ivacaftor.(56)
Consider the following scenarios:
In spite of counseling about the possibility of increased fertility in women with CF using CFTR modulators (46–48), a G0P0 35 year-old married woman with CF who is heterozygous for F508del and Gly542X (G542X) whose baseline ppFEV1 is 65% postinitiation of ETI presents to CF clinic with a positive home pregnancy test 3 months after beginning ETI. Prior to beginning ETI, her baseline ppFEV1 was 50% and she was experiencing approximately 3–4 pulmonary exacerbations per year, including at least 1 that required intravenous antibiotics. She and her husband are planning to continue the pregnancy, and she would like an opinion about whether to continue or stop ETI therapy during pregnancy.
A 25-year-old woman is referred to CF clinic with a new diagnosis of CF. She had originally presented to the obstetrics clinic for her first evaluation during which she underwent genetic screening which revealed that she was heterozygous for F508del and 3120 +1G>A. The husband is also determined to be a F508del carrier. The woman and her husband elect to forego amniocentesis because they plan to continue with the pregnancy regardless of the testing outcome. She and her husband would like to know if there are any therapies she can start to ensure her optimal health during her pregnancy that are also safe for her baby.
When considering the risk to the unborn child, sponsors for each new therapy that is being considered by the FDA for approval must evaluate the impact of the therapy in an animal reproduction model.(57) Although none of the approved CFTR modulators were tested in combination in animal reproduction models, IVA, lumacaftor, tezacaftor and elexacaftor were tested in such models individually.(4, 56, 58, 59) Data from animal testing indicated that each drug was transferred across the placenta, but led to no genotoxicity at any tested dose. Further, there was no impact on fetal organogenesis or survival when normal human doses were administered. These data in animals are reassuring. The limited data in women with CF who continued CFTR modulators during pregnancy were reviewed by a task force of the European Respiratory Society/Thoracic Society of Australia and New Zealand. The reproduction and pregnancy in women with airways diseases management task force deemed the approved CFTR modulators as “probably safe” during pregnancy.(60)
When counseling women with CF regarding the risks and benefits of continuing CFTR modulators during pregnancy, care teams must also consider the risk to the mother of discontinuation of CFTR modulator therapy. It is clear from Phase II studies of CFTR modulators that the rapid gains from CFTR modulators are quickly lost following CFTR modulator cessation.(55) Furthermore, Trimble et al. reported the occurrence of “ivacaftor withdrawal syndrome” following abrupt discontinuation of IVA after long-term use; two people experienced rapid and severe decline in lung function and one person died.(54) Finally, in a report of 61 women with CF who continued CFTR modulators during all or part of pregnancy, 9 women experienced clinical decline after cessation of modulator therapy that led to re-initiation of therapy.(53) Severe and/or rapid loss of lung function or other health decline in the mother may also pose a risk to the fetus (Figure 2), thus these risks of modulator discontinuation must be taken into account.
In the reports of women with CF who continued modulators during pregnancy to date (46–53), none have born children with CF. Currently, IVA is approved down to 4 months of age in the US.(56) Based on the data from trials of CFTR modulators in young children, it is clear that some aspects of the complications of CFTR dysfunction, such as pancreatic insufficiency, are completely or partially reversible with early administration.(61–63) These data have sparked interest in whether even earlier administration, at the time of diagnosis, or in utero in a pregnant woman with CF known to be carrying a child with CF would prevent complications in CF.
To evaluate the impact of IVA in utero on CF outcomes in the fetus, Sun and colleagues administered IVA (VX770) to pregnant ferrets generated to be homozygous for G551D, or heterozygous for G551D and a CFTR knock out mutation.(64) In IVA-treated animals homozygous for G551D, investigators demonstrated rescue of multiple downstream consequences of CFTR dysfunction including pancreatic dysfunction and absence of the vas deferens and epididymis in the kits. Furthermore, even in treated pregnant ferrets heterozygous for G551D and a knockout mutation, in utero administration of IVA led to reduction in the incidence of meconium ileus and pancreatic dysfunction in the kits. Thus, it is possible that treatment of a pregnant woman with CF known to be carrying a child with CF could markedly improve outcomes for her child. While treatment of a pregnant woman with CF who is carrying a child with CF might seem an obvious choice to some because of the clear potential benefits to both mother and child, the discussion of treating a mother who is an asymptomatic CF carrier who is discovered to be carrying a child with CF will be more challenging. Treating a mother who is an asymptomatic carrier would offer her no known benefit in the setting of only one abnormal CFTR allele. Furthermore, the costs for a medication not covered by insurance because of lack of indication for the pregnant woman would likely be prohibitive.
The data demonstrating rescue of pancreatic function in pregnant animal models of CF not only have potential implications for mothers with CF whose unborn infant is known to have CF, but also for newborn screening programs. Newborn screening (NBS) programs are now common both in and out of the US. The testing used for newborn screening varies widely. For example, in some states immunoreactive trypsinogen (IRT), a measure of pancreatic function, is used as the initial test. In other states, genetic testing for F508del alone, or for a limited number of mutations, is utilized. Finally, some states use a combination of IRT and genetic testing. Based on data from the CF ferret model, an infant born to a mother with CF treated with HEMT throughout pregnancy might be partially or completely pancreatic sufficient. In such a case, it is highly likely that a NBS test solely relying on IRT for diagnosis would produce a false negative result. Therefore, it will be critical to perform CFTR genotyping for infants born to these mothers in states that rely on IRT for NBS.
People with CFTR-related disorders: a role for HEMT?
Mutations in the CFTR gene that produce lower levels of CFTR function are associated with worsened CF phenotypes. In the US, there are more than 10 million CF carriers, people with only one mutation in CFTR, and they are routinely counseled that they are not at an increased risk of disease. CF carriers may, however, have a form of CFTR haploinsufficiency (the normal CFTR allele produces functioning CFTR protein but the total amount of CFTR protein does not reach a sufficient level of function to allow for a normal phenotype). In a study by Miller et al., investigators aimed to determine whether CF carriers had increased risk for CF-related conditions.(65) The investigators identified a CF carrier cohort and a CF cohort via ICD-9/ICD-10 codes in a large commercial claims research database. They generated age- and sex-matched cohorts at a 5:1 ratio for comparison. The investigators estimated the odds of individuals being diagnosed with a CF-related condition relative to people in their matched cohort using conditional logistic regression. CF carriers had an increased risk for nearly all 59 tested CF-related conditions, across multiple organ systems, when compared to matched controls. The relative log odds ratios across the 59 conditions were correlated between CF carriers and subjects with CF. As the relative odds of a given condition increased among subjects with CF compared to their matched controls, so did the corresponding relative odds for carriers. Multiple sensitivity analyses showed robust results demonstrating increased relative risk for CF carriers compared to matched controls. The authors stressed that the absolute risk for a majority of the CF-related conditions in CF carriers remains low. CF carriers are at increased risk for most conditions that commonly occur in people with CF, such as sinusitis, bronchiectasis and pancreatitis, amongst others which may have implications for screening, prevention, and/or treatment of those conditions.
Variants of uncertain clinical significance or variants of varying clinical consequence pose a similar challenge for clinicians considering treatment for CF-related conditions or a CF phenotype. When an individual with potential CFTR-related haploinsufficiency and a CF phenotype has normal sweat chloride testing and/or only one mutation identified by whole genome sequencing, the use of HEMT has biological plausibility for potential clinical benefit to the individual. Given the high cost of CFTR modulators, it will be cost prohibitive to treat CF carriers, or individuals who do not meet diagnostic criteria for CF, who have CF-related conditions unless there are robust clinical trial data to support the practice, which in turn could lead to insurance coverage of the medications.
Conclusions
While there are anecdotal clinical experiences with the use of ETI in unstudied or understudied populations, there are little to no data published to determine the role of HEMT in most of the challenging situations described herein. Given the potential for tremendous benefit with the use of HEMT in people with native CF lung disease (e.g. advanced CF lung disease, pregnant women with CF, non-lung solid organ transplant recipients), the potential benefits of initiation/continuation of HEMT warrant shared decision making between patients and providers. For populations in which the pulmonary benefit of HEMT may not be experienced (e.g. lung transplant recipients, CF carriers, those without a clear diagnosis of CF), caution is advised until there are data to support the clinical use of these treatments. HEMT is rapidly transforming the severity of many CF manifestations, and the careful consideration of this therapy for populations with the most favorable risk to benefit ratio is critical.
Highlights:
Highly effective modulator treatment (HEMT) may be indicated in complex settings
People with native CF lung disease are likely to benefit from initiation of HEMT
Benefits and harms of HEMT after lung transplant are uncertain and should be studied
In pregnancy, maternal risks and benefits weigh against uncertain risks to the fetus
Cost of HEMT may limit access despite biologic plausibility of clinical benefit
Acknowledgements:
The authors thank the Cystic Fibrosis Foundation for the use of a modified version of the copyrighted graphic in Figure 1. This graphic was originally presented by Dr. Jennifer Taylor-Cousar in the 2020 North American Cystic Fibrosis Conference Plenary “Defining the New CF in the Era of Highly Effective Modulators.”
Funding:
KJR receives funding from the Cystic Fibrosis Foundation (RAMOS17A0, RAMOS20A0-KB, LEASE16A3, RAMOS20Y5), the National Institutes of Health (K23-HL138154), and the CHEST Foundation (in partnership with Vertex Pharmaceuticals) for work that is outside the scope of this manuscript. JMP receives funding from the Cystic Fibrosis Foundation (PILEWS17AB0, BOMBER19R0, ZEMKE19A0, Morrell17A1, ELWYN18QI0), NIH, Vertex Pharmaceuticals, Laurent Pharmaceuticals, Celtaxsys, Breath Therapeutics, Incyte Corporation, and Translate Bio for work that is outside the scope of this manuscript. JLTC receives funding from the Cystic Fibrosis Foundation (TAYLOR19Y3, TAYLOR19A0, JAIN19AO, GODFRE19AO, HARRIS19A0, NICK14Y0), and conducts clinical trials, and provides clinical trial development consultation for Vertex pharmaceuticals, but received no funding to complete this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: Dr. Kathleen Ramos reports grants from Cystic Fibrosis Foundation, and a grant from the CHEST Foundation in partnership with Vertex Pharmaceuticals, outside the submitted work. Dr. Joseph Pilewski reports grants from the CF Foundation, Vertex, Breath Therapeutics, Celtaxsys, Incyte, Laurent and Translate Bio for research or clinical trial work, outside the submitted work and without personal fees. Dr. Jennifer Taylor-Cousar reports grants and personal fees from Gilead, grants from N30, grants and personal fees from Vertex, grants and personal fees from Proteostasis, grants from Bayer, personal fees from Novartis, personal fees from Genentech, personal fees from Protalix, personal fees from Santhera, personal fees from 4DMT, personal fees from Polarean Imaging, personal fees from Insmed, personal fees from Abbvie, grants and personal fees from Celtaxys, outside the submitted work; Service on the CF TDN Clinical Research Executive Committee; and Service as the Chair of the ATS Clinical Problems Assembly Program Committee.
References
- 1.Ramsey BW, Davies J, McElvaney NG, Tullis E, Bell SC, Dřevínek P, Griese M, McKone EF, Wainwright CE, Konstan MW. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. New England Journal of Medicine 2011; 365: 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Middleton PG, Mall MA, Dřevínek P, Lands LC, McKone EF, Polineni D, Ramsey BW, Taylor-Cousar JL, Tullis E, Vermeulen F. Elexacaftor–tezacaftor–ivacaftor for cystic fibrosis with a single Phe508del allele. New England Journal of Medicine 2019; 381: 1809–1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heijerman HG, McKone EF, Downey DG, Van Braeckel E, Rowe SM, Tullis E, Mall MA, Welter JJ, Ramsey BW, McKee CM. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: a double-blind, randomised, phase 3 trial. The Lancet 2019; 394: 1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Food and Drug Administration (FDA). TRIKAFTA drug insert. 2020. Jan 12, 2021]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/212273s002lbl.pdf.
- 5.Shteinberg M, Taylor-Cousar JL. Impact of CFTR modulator use on outcomes in people with severe cystic fibrosis lung disease. European Respiratory Review 2020; 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Shea KM, O’Carroll OM, Carroll C, Grogan B, Connolly A, O’Shaughnessy L, Nicholson T, Gallagher CG, McKone EF. The efficacy of Elexacaftor/Tezacaftor/Ivacaftor in patients with cystic fibrosis and advanced lung disease. European Respiratory Journal 2020. [DOI] [PubMed]
- 7.Bermingham B, Rueschhoff A, Ratti G, NeSmith A, Flume P, Solomon G, Cohen L, Garcia B. A multicenter retrospective analysis of the clinical efficacy of elexacaftor-tezacaftor-ivacaftor in patients with advanced lung disease. Abstract #645. 2020 North American Cystic Fibrosis Conference Pediatric Pulmonology: WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2020. p. S290–S290. [Google Scholar]
- 8.Bessonova L, Volkova N, Higgins M, Bengtsson L, Tian S, Simard C, Konstan MW, Sawicki GS, Sewall A,Nyangoma S. Data from the US and UK cystic fibrosis registries support disease modification by CFTR modulation with ivacaftor. Thorax 2018; 73: 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMango E, Overdevest J, Keating C, Francis SF, Dansky D, Gudis D. Effect of highly effective modulator treatment on sinonasal symptoms in cystic fibrosis. Journal of Cystic Fibrosis 2020. [DOI] [PubMed]
- 10.Beswick D, Humphries S, Vladar E, Strand M, Balkissoon C, Lynch D, Taylor-Cousar J. Impact of TC CFTR therapy on sinus disease: quantitative sinus CT and PROs. Abstract #436. 2020 North American Cystic Fibrosis Conference Pediatric Pulmonology: WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2020. p. S208–S209. [Google Scholar]
- 11.Stapleton A, Kimple A, Goralski J, Nouraie M, Shaffer A, Pilewski J, Senior B, Lee S, Zemke A. Elexacaftor-ivacaftor-tezacaftor improves chronic rhinosinusitis symptoms in cystic fibrosis. 2020 North American Cystic Fibrosis Conference Pediatric Pulmonology: WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA; 2020. p. S292–S292. [Google Scholar]
- 12.Bellin MD, Laguna T, Leschyshyn J, Regelmann W, Dunitz J, Billings J, Moran A. Insulin secretion improves in cystic fibrosis following ivacaftor correction of CFTR: a small pilot study. Pediatric diabetes 2013; 14: 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly A, De Leon DD, Sheikh S, Camburn D, Kubrak C, Peleckis AJ, Stefanovski D, Hadjiliadis D, Rickels MR, Rubenstein RC. Islet hormone and incretin secretion in cystic fibrosis after four months of ivacaftor therapy. American journal of respiratory and critical care medicine 2019; 199: 342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dagan A, Cohen-Cymberknoh M, Shteinberg M, Levine H, Vilozni D, Bezalel Y, Sarouk I, Ashkenazi M, Lavie M, Tsabari R. Ivacaftor for the p. Ser549Arg (S549R) gating mutation–The Israeli experience. Respiratory Medicine 2017; 131: 225–228. [DOI] [PubMed] [Google Scholar]
- 15.Cho D-Y, Lim DJ, Mackey C, Skinner D, Zhang S, McCormick J, Woodworth BA. Ivacaftor, a cystic fibrosis transmembrane conductance regulator potentiator, enhances ciprofloxacin activity against Pseudomonas aeruginosa. American Journal of Rhinology & Allergy 2019; 33: 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verleden GM, Glanville AR, Lease ED, Fisher AJ, Calabrese F, Corris PA, Ensor CR, Gottlieb J, Hachem RR, Lama V. Chronic lung allograft dysfunction: Definition, diagnostic criteria, and approaches to treatment―A consensus report from the Pulmonary Council of the ISHLT. The Journal of Heart and Lung Transplantation 2019; 38: 493–503. [DOI] [PubMed] [Google Scholar]
- 17.Chambers DC, Cherikh WS, Harhay MO, Hayes D, Hsich E, Khush KK, Meiser B, Potena L, Rossano JW, Toll AE. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-sixth adult lung and heart–lung transplantation Report—2019; Focus theme: Donor and recipient size match. The Journal of Heart and Lung Transplantation 2019; 38: 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kulkarni HS, Tsui K, Sunder S, Ganninger A, Tague LK, Witt CA, Byers DE, Trulock EP, Nava R, Puri V. Pseudomonas aeruginosa and acute rejection independently increase the risk of donor-specific antibodies after lung transplantation. American Journal of Transplantation 2020; 20: 1028–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burton CM, Iversen M, Carlsen J, Mortensen J, Andersen CB, Steinbrüchel D, Scheike T. Acute cellular rejection is a risk factor for bronchiolitis obliterans syndrome independent of post-transplant baseline FEV1. The Journal of heart and lung transplantation 2009; 28: 888–893. [DOI] [PubMed] [Google Scholar]
- 20.Itoda Y, Sato M, Thuita L, NIikawa H, Ayyat K, Okamoto T, Farver C, Zhang A, Budev M, Balckstone E. Impact for Survival and Chronic Lung Allograft Dysfunction of ISHLT Consensus of Antibody Mediated Rejection after Lung Transplantation. The Journal of Heart and Lung Transplantation 2019; 38: S405. [Google Scholar]
- 21.Peghin M, Los-Arcos I, Hirsch HH, Codina G, Monforte V, Bravo C, Berastegui C, Jauregui A, Romero L, Cabral E. Community-acquired respiratory viruses are a risk factor for chronic lung allograft dysfunction. Clinical Infectious Diseases 2019; 69: 1192–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fisher CE, Preiksaitis CM, Lease ED, Edelman J, Kirby KA, Leisenring WM, Raghu G, Boeckh M, Limaye AP. Symptomatic respiratory virus infection and chronic lung allograft dysfunction. Clinical Infectious Diseases 2016; 62: 313–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davidson JR, Franklin D, Kumar S, Mohammadi B, Dawas K, Eaton S, Curry J, De Coppi P, Abbassi-Ghadi N. Fundoplication to preserve allograft function after lung transplant: Systematic review and meta-analysis. The Journal of thoracic and cardiovascular surgery 2020; 160: 858–866. [DOI] [PubMed] [Google Scholar]
- 24.Lo W-K, Goldberg HJ, Boukedes S, Burakoff R, Chan WW. Proton pump inhibitors independently protect against early allograft injury or chronic rejection after lung transplantation. Digestive diseases and sciences 2018; 63: 403–410. [DOI] [PubMed] [Google Scholar]
- 25.Berastegui C, Gómez-Oll-s S, Sánchez-Vidaurre S, Culebras M, Monforte V, López-Meseguer M, Bravo C, Ramon MA, Romero L, Sole J. BALF cytokines in different phenotypes of chronic lung allograft dysfunction in lung transplant patients. Clinical transplantation 2017; 31: e12898. [DOI] [PubMed] [Google Scholar]
- 26.Slebos D-J, Postma DS, Koëter GH, van der Bij W, Boezen M, Kauffman HF. Bronchoalveolar lavage fluid characteristics in acute and chronic lung transplant rejection. The Journal of heart and lung transplantation 2004; 23: 532–540. [DOI] [PubMed] [Google Scholar]
- 27.Hodge G, Hodge S, LI-LIEW C, Reynolds PN, Holmes M. Increased natural killer T-like cells are a major source of pro-inflammatory cytokines and granzymes in lung transplant recipients. Respirology 2012; 17: 155–163. [DOI] [PubMed] [Google Scholar]
- 28.Choi KJ, Cheng TZ, Honeybrook AL, Gray AL, Snyder LD, Palmer SM, Abi Hachem R, Jang DW. Correlation between sinus and lung cultures in lung transplant patients with cystic fibrosis. International forum of allergy & rhinology: Wiley Online Library; 2018. p. 389–393. [DOI] [PMC free article] [PubMed]
- 29.Holzmann D, Speich R, Kaufmann T, Laube I, Russi EW, Simmen D, Weder W, Boehler A. Effects of sinus surgery in patients with cystic fibrosis after lung transplantation: a 10-year experience. Transplantation 2004; 77: 134–136. [DOI] [PubMed] [Google Scholar]
- 30.Van Herck A, De Muynck B, Sacreas A, Heigl T, Kaes J, Vanstapel A, Ambrocio G, Verleden S, Neyrinck A, Ceulemans L. Successful Pseudomonas aeruginosa Eradication Improves Outcomes after Lung Transplantation. The Journal of Heart and Lung Transplantation 2020; 39: S303. [DOI] [PubMed] [Google Scholar]
- 31.Cheng TZ, Choi KJ, Honeybrook AL, Zakare-Fagbamila RT, Gray AL, Snyder LD, Palmer SM, Abi-Hachem R, Jang DW. Decreased antibiotic utilization after sinus surgery in cystic fibrosis patients with lung transplantation. American journal of rhinology & allergy 2019; 33: 354–358. [DOI] [PubMed] [Google Scholar]
- 32.Leung M-K, Rachakonda L, Weill D, Hwang PH. Effects of sinus surgery on lung transplantation outcomes in cystic fibrosis. American journal of rhinology 2008; 22: 192–196. [DOI] [PubMed] [Google Scholar]
- 33.De Muynck B, Van Herck A, Sacreas A, Heigl T, Kaes J, Vanstapel A, Verleden SE, Neyrinck AP, Ceulemans LJ, Van Raemdonck DE. Successful Pseudomonas aeruginosa eradication improves outcomes after lung transplantation: a retrospective cohort analysis. European Respiratory Journal 2020. [DOI] [PubMed]
- 34.Vital D, Hofer M, Benden C, Holzmann D, Boehler A. Impact of sinus surgery on pseudomonal airway colonization, bronchiolitis obliterans syndrome and survival in cystic fibrosis lung transplant recipients. Respiration 2013; 86: 25–31. [DOI] [PubMed] [Google Scholar]
- 35.Ramos KJ, Kapnadak SG, Bradford MC, Somayaji R, Morrell ED, Pilewski JM, Lease ED, Mulligan MS, Aitken ML, Gries CJ. Underweight patients with cystic fibrosis have acceptable survival following lung transplantation: a united network for organ sharing registry study. Chest 2020. [DOI] [PMC free article] [PubMed]
- 36.Lederer DJ, Wilt JS, D’Ovidio F, Bacchetta MD, Shah L, Ravichandran S, Lenoir J, Klein B, Sonett JR, Arcasoy SM. Obesity and underweight are associated with an increased risk of death after lung transplantation. American journal of respiratory and critical care medicine 2009; 180: 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fink AK, Yanik EL, Marshall BC, Wilschanski M, Lynch CF, Austin AA, Copeland G, Safaeian M, Engels EA. Cancer risk among lung transplant recipients with cystic fibrosis. Journal of Cystic Fibrosis 2017; 16: 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lowery EM, Adams W, Grim SA, Clark NM, Edwards L, Layden JE. Increased risk of PTLD in lung transplant recipients with cystic fibrosis. Journal of Cystic Fibrosis 2017; 16: 727–734. [DOI] [PubMed] [Google Scholar]
- 39.Scott P, Anderson K, Singhania M, Cormier R. Cystic Fibrosis, CFTR, and Colorectal Cancer. International journal of molecular sciences 2020; 21: 2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kazmerski TM, Gmelin T, Slocum B, Borrero S, Miller E. Attitudes and decision making related to pregnancy among young women with cystic fibrosis. Maternal and child health journal 2017; 21: 818–824. [DOI] [PubMed] [Google Scholar]
- 41.Kazmerski TM, Sawicki GS, Miller E, Jones KA, Abebe KZ, Tuchman LK, Ladores S, Rubenstein RC, Sagel SD, Weiner DJ. Sexual and reproductive health behaviors and experiences reported by young women with cystic fibrosis. Journal of Cystic Fibrosis 2018; 17: 57–63. [DOI] [PubMed] [Google Scholar]
- 42.Registry CFFP. Cystic Fibrosis Foundation Patient Registry Annual Data Report 2019. 2020.
- 43.Wainwright CE, Elborn JS, Ramsey BW, Marigowda G, Huang X, Cipolli M, Colombo C, Davies JC, De Boeck K, Flume PA, Konstan MW, McColley SA, McCoy K, McKone EF, Munck A, Ratjen F, Rowe SM, Waltz D, Boyle MP. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N Engl J Med 2015; 373: 220–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowe SM, Daines C, Ringshausen FC, Kerem E, Wilson J, Tullis E, Nair N, Simard C, Han L, Ingenito EP, McKee C, Lekstrom-Himes J, Davies JC. Tezacaftor-Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N Engl J Med 2017; 377: 2024–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taylor-Cousar JL, Munck A, McKone EF, van der Ent CK, Moeller A, Simard C, Wang LT, Ingenito EP, McKee C, Lu Y, Lekstrom-Himes J, Elborn JS. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N Engl J Med 2017; 377: 2013–2023. [DOI] [PubMed] [Google Scholar]
- 46.Jones GH, Walshaw MJ. Potential impact on fertility of new systemic therapies for cystic fibrosis. Paediatr Respir Rev 2015; 16 Suppl 1: 25–27. [DOI] [PubMed] [Google Scholar]
- 47.Ladores S, Kazmerski TM, Rowe SM. A Case Report of Pregnancy During Use of Targeted Therapeutics for Cystic Fibrosis. J Obstet Gynecol Neonatal Nurs 2017; 46: 72–77. [DOI] [PubMed] [Google Scholar]
- 48.Ladores SBL, Brown J. Two Unanticipated Pregnancies While on Cystic Fibrosis Gene-Specific Drug Therapy. Journal of Patient Experience 2020; 7: 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vekaria S, Popowicz N, White SW, Mulrennan S. To be or not to be on CFTR modulators during pregnancy: Risks to be considered. J Cyst Fibros 2019. [DOI] [PubMed]
- 50.Trimble A, McKinzie C, Terrell M, Stringer E, Esther CR Jr. Measured fetal and neonatal exposure to Lumacaftor and Ivacaftor during pregnancy and while breastfeeding. J Cyst Fibros 2018; 17: 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mainz JG, Michl RK, Beiersdorf N, Lorenz M, Schneider U, Groten T, Jaudszus A. Successful Pregnancy of a Patient with Cystic Fibrosis Genotype F508del/ F508del and Progressed Pulmonary Destruction on lumacaftor/ivacaftor. Klin Padiatr 2019; 231: 271–273. [DOI] [PubMed] [Google Scholar]
- 52.Kaminski R, Nazareth D. A successful uncomplicated CF pregnancy while remaining on Ivacaftor. J Cyst Fibros 2016; 15: 133–134. [DOI] [PubMed] [Google Scholar]
- 53.Nash EF, Middleton PG, Taylor-Cousar JL. Outcomes of pregnancy in women with cystic fibrosis (CF) taking CFTR modulators - an international survey. J Cyst Fibros 2020. [DOI] [PubMed]
- 54.Trimble AT, Donaldson SH. Ivacaftor withdrawal syndrome in cystic fibrosis patients with the G551D mutation. J Cyst Fibros 2018; 17: e13–e16. [DOI] [PubMed] [Google Scholar]
- 55.Carpino EAFR, Uluer AZ, Sawicki GS. Acute Clinical Outcomes Following Participation in Short-Term CFTR Modulator Trials in Adults with Cystic Fibrosis: A Retrospective Chart Review. Pediatr Pulmonol 2018; 53: 260–261. [Google Scholar]
- 56.Food and Drug Administration (FDA). KALYDEKO drug insert. 2012.
- 57.Food and Drug Administration (FDA). Pregnancy and Lactation Labeling Rule.
- 58.Food and Drug Administration (FDA). SYMDEKO drug insert. 2018. December 9, 2020]. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210491lbl.pdf.
- 59.Food and Drug Administration (FDA). ORKAMBI drug insert. 2015.
- 60.Middleton PG, Gade EJ, Aguilera C, MacKillop L, Button BM, Coleman C, Johnson B, Albrechtsen C, Edenborough F, Rigau D, Gibson PG, Backer V. ERS/TSANZ Task Force Statement on the management of reproduction and pregnancy in women with airways diseases. Eur Respir J 2020; 55. [DOI] [PubMed] [Google Scholar]
- 61.Davies JC, Cunningham S, Harris WT, Lapey A, Regelmann WE, Sawicki GS, Southern KW, Robertson S, Green Y, Cooke J, Rosenfeld M, Group KS. Safety, pharmacokinetics, and pharmacodynamics of ivacaftor in patients aged 2–5 years with cystic fibrosis and a CFTR gating mutation (KIWI): an open-label, single-arm study. Lancet Respir Med 2016; 4: 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hamilton JL, Zobell JT, Robson J. Pancreatic insufficiency converted to pancreatic sufficiency with ivacaftor. Pediatr Pulmonol 2019; 54: 1654. [DOI] [PubMed] [Google Scholar]
- 63.Hutchinson I, McNally P. Appearance of Pancreatic Sufficiency and Discontinuation of Pancreatic Enzyme Replacement Therapy (PERT) in Children with Cystic Fibrosis on Ivacaftor. Ann Am Thorac Soc 2020. [DOI] [PubMed]
- 64.Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, Evans TIA, Rotti PG, Yang Y, Gray JS, Park SY, Zhou W, Zhang Y, Moll SR, Woody L, Tran DM, Jiang L, Vonk AM, Beekman JM, Negulescu P, Van Goor F, Fiorino DF, Gibson-Corley KN, Engelhardt JF. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med 2019; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller AC, Comellas AP, Hornick DB, Stoltz DA, Cavanaugh JE, Gerke AK, Welsh MJ, Zabner J, Polgreen PM. Cystic fibrosis carriers are at increased risk for a wide range of cystic fibrosis-related conditions. Proc Natl Acad Sci U S A 2020; 117: 1621–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]