

Abstract
Genome editing has therapeutic potential for treating genetic diseases and cancer. However, the currently most practicable approaches rely on the generation of DNA double-strand breaks (DSBs), which can give rise to a poorly characterized spectrum of chromosome structural abnormalities. Here, using model cells and single-cell whole genome sequencing, as well as by editing at a clinically relevant locus in clinically relevant cells, we show that CRISPR-Cas9 editing generates structural defects of the nucleus—micronuclei and chromosome bridges—that initiate a mutational process called chromothripsis. Chromothripsis is extensive chromosome rearrangement restricted to one or a few chromosomes that can cause human congenital disease and cancer. These results demonstrate that chromothripsis is a previously unappreciated on-target consequence of CRISPR-Cas9-generated DSBs. As genome editing is implemented in the clinic, the potential for extensive chromosomal rearrangements should be considered and monitored.
CRISPR-Cas9 is directed to its target-site by a guide RNA (gRNA), creating specific DNA double-strand breaks (DSBs) almost anywhere in the genome1,2. Error-prone DNA repair by non-homologous end joining (NHEJ) of Cas9-generated DSBs can create small insertions and deletions, which can be exploited therapeutically by disrupting protein coding or DNA regulatory sequences. A particularly promising application of this approach is for autologous hematopoietic stem cell (HSC) therapy of common β-hemoglobinopathies including sickle cell disease and β-thalassemia. Specifically, NHEJ-mediated disruption of DNA regions that are required for the repression of fetal hemoglobin (HbF) expression in red blood cell progenitors can alleviate the symptoms of severe β-hemoglobinopathies3–6. Cas9 can also be used to install precise nucleotide substitutions by homology-directed repair (HDR) for correction of monogenic diseases, including reversion of the mutant sickle cell disease codon1,2,7–10. Several promising CRISPR-based strategies that do not require DSB intermediates have been described, but are at earlier stages of development and have not yet been advanced to clinical trials11–13. Moreover, these strategies generate single strand DNA nicks that, at relatively low frequency, can be converted into DSBs.
It is important to understand the genotoxicities associated with therapeutic CRISPR-Cas9 genome editing. While much attention has been paid to unintended, “off-target” DSBs14, this outcome can be reduced by utilizing more specific gRNAs, high-specificity Cas-nucleases, or other gene editing strategies such as the use of double-nickases14. Less is known about potential detrimental consequences that arise from on-target genome editing-mediated DSBs. On-target DNA breakage can induce the TP53 tumor suppressor, which in principle might create selective pressure for TP53 loss, and thus potentially support tumorigenesis15–18. Additionally, on-target genome editing can cause local DNA rearrangements and deletions up to several kilobases in length19–24, megabase-scale deletions telomeric to the CRISPR-Cas9 cut site23,25,26 and loss of the entire cleaved chromosome23. The mechanisms leading to these DNA alterations remain poorly defined. Moreover, genome editing protocols that induce more than one on-target DSB can lead to incorrect DNA end-joining and chains of chromosome translocations that can persist at low levels for months in treated patients27. Reassuringly, to date, these translocations have not been linked to deleterious consequences27.
Here, using a variety of approaches including the combination of imaging and single-cell whole genome sequencing (Look-Seq)28,29, we report that chromothripsis is a previously unrecognized consequence of on-target Cas9-mediated DNA breakage. This occurs because in actively dividing cells, genome editing with Cas9 causes up to a 20-fold increase in the formation of micronuclei and/or chromosome bridges, aberrant nuclear structures that can initiate chromothripsis. In addition to causing rare human congenital disease30,31, chromothripsis is common in cancer, where it is well established to generate tumor suppressor loss, fusion oncogenes, or oncogene amplification through the formation of circular double minute chromosomes32–36. Unlike chromothripsis during tumorigenesis, CRISPR-Cas9-induced chromothripsis occurs on targeted chromosomes, meaning its carcinogenic potential will likely depend upon the set of genes on the targeted chromosome arm, and whether rearrangements occurring after the initial cut cause those genes to be deleted, fused, or amplified. Our findings reveal that initial errors from on-target genome editing can be amplified into far more extensive genetic alterations in subsequent cell cycles via the generation of micronuclei and chromosome bridges.
Results
CRISPR-Cas9 genome editing generates micronuclei
Cas9 generates a DSB that cleaves the targeted chromosome into two segments: one with the centromere region (the “centric” fragment) and one without (the “acentric” fragment). If the DSB is not repaired prior to cell division, the acentric fragment lacking a functional centromere can missegregate, forming a micronucleus (Fig. 1a)37–39.
Figure 1. Micronucleation is an on-target consequence of CRISPR-Cas9 genome editing.
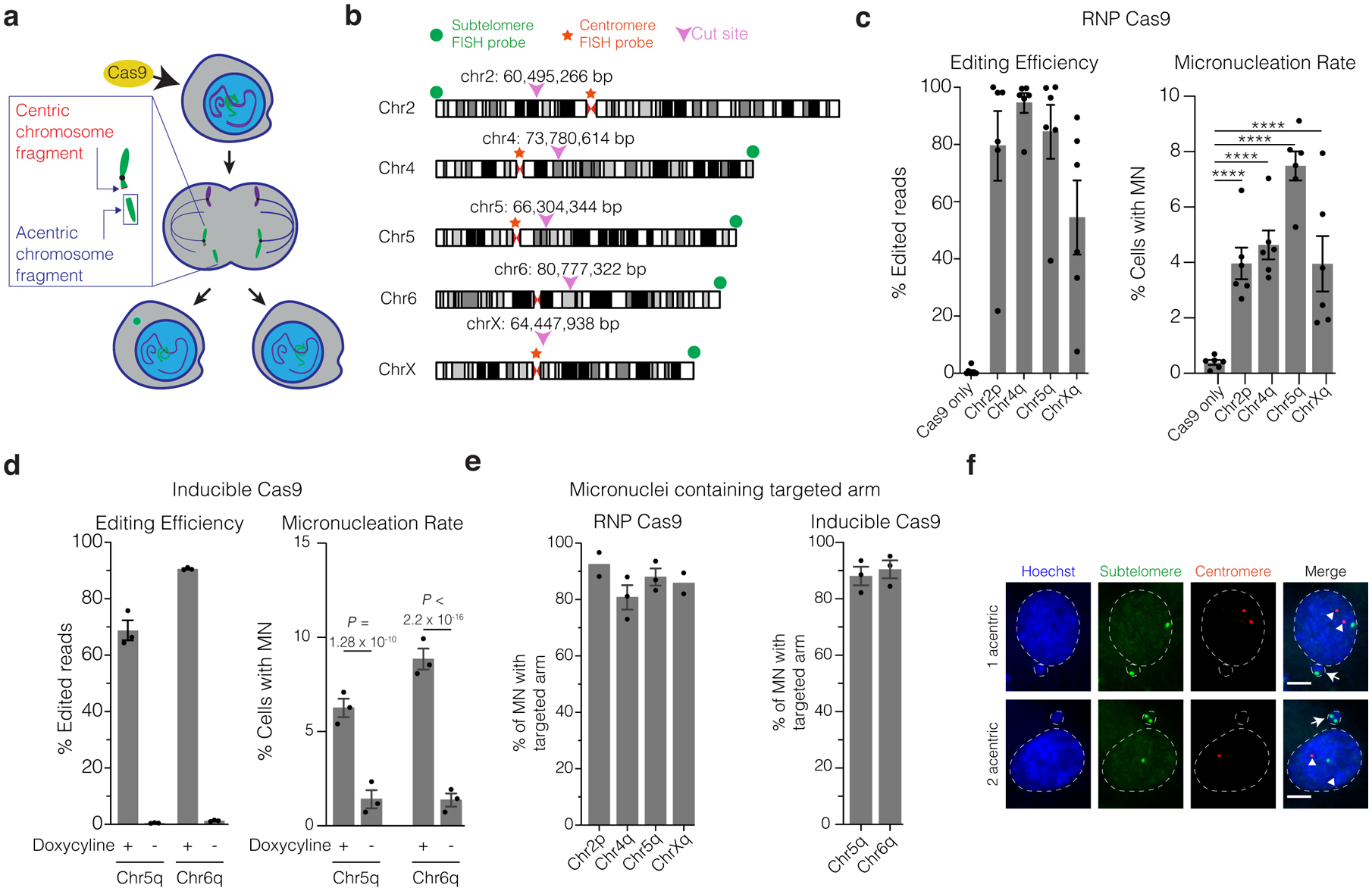
(a) Schematic of how Cas9 DNA cleavage of a chromosome arm can generate micronuclei. In the shown example cleavage of one sister chromatid occurs in during G2. The centric fragment segregates properly into a daughter nucleus whereas the acentric fragment that cannot be segregated by the spindle is partitioned into a micronucleus. Variations on this outcome include cleavage in G1, cleavage of both sisters in a G2 cell, and cleavage of both homologous chromosomes.
(b) Chromosome locations of gRNAs and FISH probes. Magenta arrowheads and numerical coordinates indicate the cut site for specific gRNAs. Green dot: acentric fragment FISH probe locations; red star: centric fragment FISH probe locations.
(c) The frequency of micronucleation after CRISPR-Cas9 RNP transfection in p53-proficient RPE-1 cells. Left, editing efficiency after Cas9/gRNA RNP transfection. Right, frequency of micronucleation for these transfections, 46 h after release of RPE-1 cells from a G1 block. (n = 3 experiments with 5311, 5451, 5144, 4555, 5272 cells scored, left to right). Error bars: mean +/− SEM, **** p < 2.2 × 10−16, two-tailed Fisher’s exact test.
(d) As in panel C, but for doxycycline-inducible CRISPR-Cas9 with constitutively expressed gRNA. p53 siRNA treatment was performed prior to doxycycline treatment. (n = 3 experiments with 1265, 1261, 1244, 1239 cells scored for micronucleation, left to right). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(e) Percentage of MN containing the targeted chromosome arm. Left, RNP transfection (n = 2 experiments with 64 and 96 micronuclei scored for chr2p, chrXq, respectively, n = 3 experiments with 83 and 116 micronuclei scored for chr4q, chr5q, respectively). Right, RPE-1 cells with inducible-Cas9 and constitutively expressed gRNA (n = 3 experiments with 168 micronuclei scored for each). Error bars: mean +/− SEM.
(f) Example images of FISH analysis after Cas9/gRNA RNP transfection from data in panel (e) (single plane from a confocal imaging stack). Red: centric fragment probe; green: acentric fragment probe; blue: Hoechst stain (DNA); white arrows: micronuclei; white arrowheads: centromeres; dashed white line: outline of Hoechst (DNA) label. Scale bar 5 μm.
We evaluated this possibility in genetically stable human retinal pigment epithelial cells (hTERT RPE-1). To estimate the rate of micronucleation in a single cell cycle, we synchronized cells with a serum starvation-block and release protocol followed by transfection with a Cas9/gRNA ribonucleoprotein (RNP) complex shortly before the next cell division (22 hours after release, approximately during S/G2 [Extended Data Fig. 1a]). We used single guide RNAs (gRNAs), each targeting unique genomic sites on four different chromosomes (Fig. 1b and Supplementary Table 1). All gRNAs targeted intergenic sequences, except one that disrupts the erythroid-specific enhancer of the BCL11A gene on chromosome 2 (“chr2p”), according to therapeutic strategies to induce fetal hemoglobin to treat β-thalassemia or sickle cell disease5,6,40 (NCT03655678, NCT03745287). The BCL11A gene encodes a transcriptional repressor protein that silences γ-globin expression postnatally in red blood cells.
CRISPR-Cas9 cutting at individual target sites induced micronucleation at frequencies of 4.0 – 7.5 %, 10.2 to 19.3-fold higher than controls (hereafter “CRISPR-MN”, [Fig. 1c]). Similar results were obtained in asynchronous cells (Extended Data Fig. 1b), and in cells that constitutively express gRNAs (targeting chr5q and chr6q) where Cas9 was expressed from a third-generation doxycycline-inducible promoter (Fig. 1b,d and Extended Data Fig. 1a,c)41. The frequencies of genome editing and micronucleation correlated in general, although not with a strict 1:1 correspondence. Other factors, including locus-specific differences in DNA repair efficiency42, may impact micronucleation rates independent of editing efficiency.
Fluorescence in situ hybridization (FISH) established that 81 – 92 % of CRISPR-MN contained the chromosome arm targeted by the specific gRNAs (Fig. 1e,f and Extended Data Fig. 1d). Most micronuclei contained two copies of the targeted chromosome segment, which could result from either cleavage of both homologous chromosomes in a cell, or from cleavage of both sister chromatids of one homolog in G2 phase (Fig. 1f and Extended Data Fig. 1d). Co-staining with centromere-specific FISH probes confirmed that CRISPR-MN are mostly acentric chromosome fragments, as expected (Fig. 1f). Similar results for micronucleus formation and chromosome arm copy number alterations were also obtained in BJ foreskin fibroblasts (Extended Data Fig. 1e,f).
Allele-specific genome editing has numerous medical applications43. In principle, DSBs on one homolog might lead to less frequent micronucleation due to the potential for homology directed repair from the intact homolog. Using gRNAs that target only one allele due to a PAM site-polymorphism, we detected CRISPR-Cas9 editing events exclusively on the targeted homolog (Extended Data Fig. 1g,h). These editing events were associated with a 2.7 and 12.0–fold increase in micronucleation frequency for chr1p and chr5q-targeting gRNAs respectively (Extended Data Fig. 1i). Allele-specific gRNAs primarily generated CRISPR-MN with two copies of the targeted chromosome (Extended Data Fig. 1j). Therefore, allele-specific guides do not eliminate genome editing-induced micronucleus formation in actively dividing cells, consistent with findings that homologous chromosomes are poor DSB repair substrates in mitotic cells44.
Importantly, CRISPR-MN exhibited characteristic functional defects, including spontaneous nuclear envelope rupture (Extended Data Fig. 2a), defective DNA replication (Extended Data Fig. 2b), and the accumulation of DNA damage (Extended Data Fig. 2c–e)45–52. Therefore, CRISPR-Cas9 genome editing can generate micronuclei containing the acentric fragment of the targeted chromosome, which is then subject to extensive DNA damage.
Chromothripsis as a consequence of Cas9 genome editing
Generation of micronuclei after a CRISPR-Cas9 induced DSB suggested that chromothripsis might be an unrecognized, on-target consequence of CRISPR-Cas9 genome editing. To directly test this hypothesis, we used “Look-Seq”, a procedure combining long-term live-cell imaging with single-cell whole-genome sequencing of the imaged cells28,29. CRISPR-MN were generated in daughter cells as above, using three different gRNAs, including the chr2p guide targeting the erythroid-specific BCL11A enhancer5,40. Because Cas9 genome editing or division of the resulting micronucleated cells could be limited by p53 induction15–18, we transiently depleted p53 by siRNA-mediated knockdown prior to inducing CRISPR-MN. Micronucleated daughter cells were allowed to divide, and their progeny (granddaughter cells) were then isolated for single-cell sequencing.
In total, we sequenced 18 granddaughter pairs derived from micronucleated daughter cells. The targeted chromosome arm exhibited several patterns of copy number alterations which may explained as follows: Cas9 can cleave one or both homologous chromosomes and one or both sister chromatids; acentric fragments can be distributed in any combination to granddaughter cells; and/or the micronuclear DNA can be severely under-replicated (in most cases, DNA replication in micronuclei is highly inefficient)37,45,51. We observed examples consistent with each of the above scenarios (Fig. 2) and additional events from co-occurring chromosome bridges, as discussed later.
Figure 2. Summary of genomic outcomes after the division of 18 micronucleated cells.
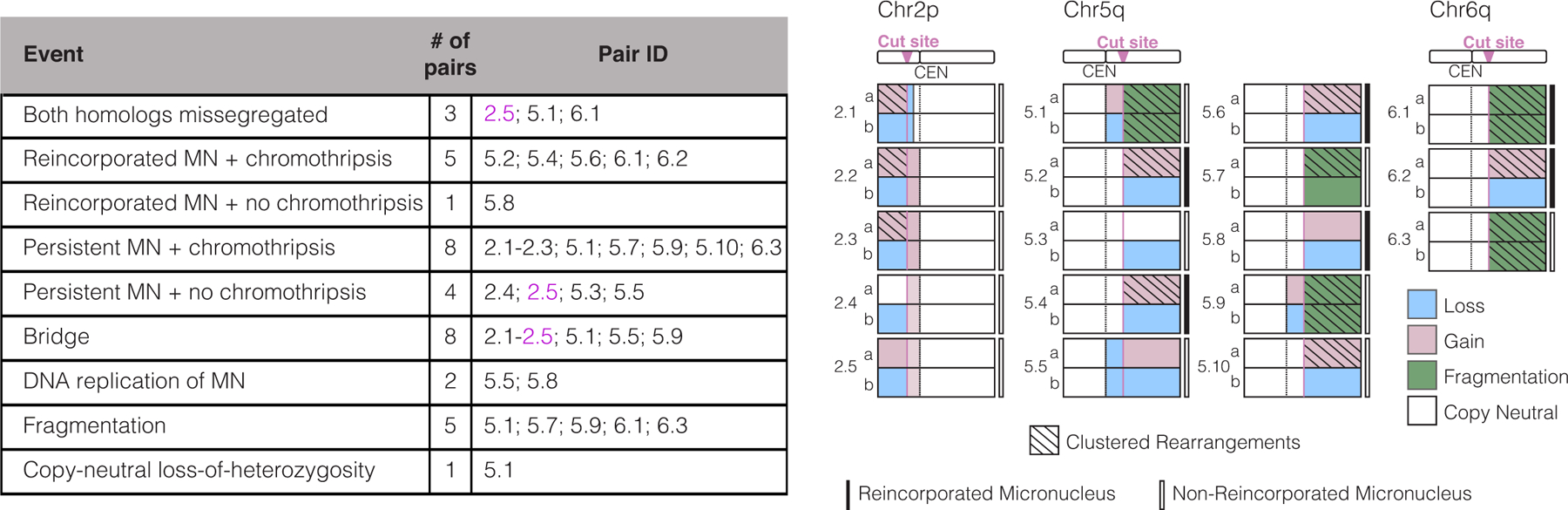
(a) Left, summary table. Reincorporation of the MN DNA was inferred from the absence of detectable extranuclear GFP-H2B signal in either granddaughter. Bridges were inferred from the DNA sequence analysis. Replication of the MN or copy-number neutral LOH was inferred from haplotype-resolved DNA copy number. Fragmentation is evident from reciprocal changes in the DNA copy number along the chromosome arm when comparing the two granddaughters. Multiple classes of genomic events can occur in a single sample, highlighted by sample 2.5 in magenta text. Right, schematic summary of each of the 18 granddaughter cell pairs. Number to the left of the schematics is an ID: first number is the targeted chromosome; second number is a sample identifier for that chromosome. These experiments were performed using inducible Cas9 (chr5q and chr6q gRNAs) and RNP (chr2p gRNA).
Haplotype-specific copy number analysis showed that in 15 of 18 granddaughter pairs, the acentric arm from one homologous chromosome was missegregated, whereas both homologs were missegregated in the remaining 3 pairs (Fig. 2 and Extended Data Fig. 3, pairs 2.5, 5.1, 6.1). In one notable example, missegregation of the acentric chromosome fragments occurred in a “swapped” manner where both paternal copies of the acentric fragment segregated to one daughter and both maternal copies segregated to the other daughter. This generated copy-neutral loss-of-heterozygosity (LOH) for the chromosome segment telomeric to the CRISPR-Cas9 cut site (i.e., uniparental disomy for the acentric fragment in both granddaughter cells, pair 5.1 in Extended Data Fig. 3). Copy-neutral LOH is common in cancer and can result in tumor suppressor inactivation53. Our findings potentially provide a simple mechanistic explanation for similar LOH patterns that have been noted, but not explained, after CRISPR-Cas9 genome editing24,54,55. In summary, on-target Cas9 genome editing can generate micronuclei, which in turn can induce arm-level DNA copy number alterations as well as copy-number neutral LOH.
Chromothripsis is extensive chromosomal rearrangements that are clustered on one or a few chromosomes or chromosome arms and are commonly accompanied by oscillations between two or three DNA copy number levels32,37,56. We identified the characteristic clustering of rearrangements on the acentric segment of the Cas9-targeted chromosome arm in 13 of 18 granddaughter pairs sequenced (Figs. 2,3 and Extended Data Figs. 3,4, p ≤ 7.3 × 10−6, one-sided Poisson test, Supplementary Table 2). The most striking example was a targeted chr6q arm in which we detected 646 intrachromosomal breakpoints distributed between the two granddaughter cells (Extended Data Figs. 3,4, pair 6.1). By a one-sided Poisson test with Bonferroni correction28, no enrichment of intrachromosomal rearrangements was identified on any chromosome arm other than the arm targeted by CRISPR-Cas9 (Supplementary Table 2). If we include interchromosomal rearrangements, we find a single non-targeted arm with a p-value of 0.02 (whereas targeted arms have p ≤ 7.3 × 10−6 after Bonferroni correction).
Figure 3. CRISPR-Cas9 genome editing can cause chromothripsis.
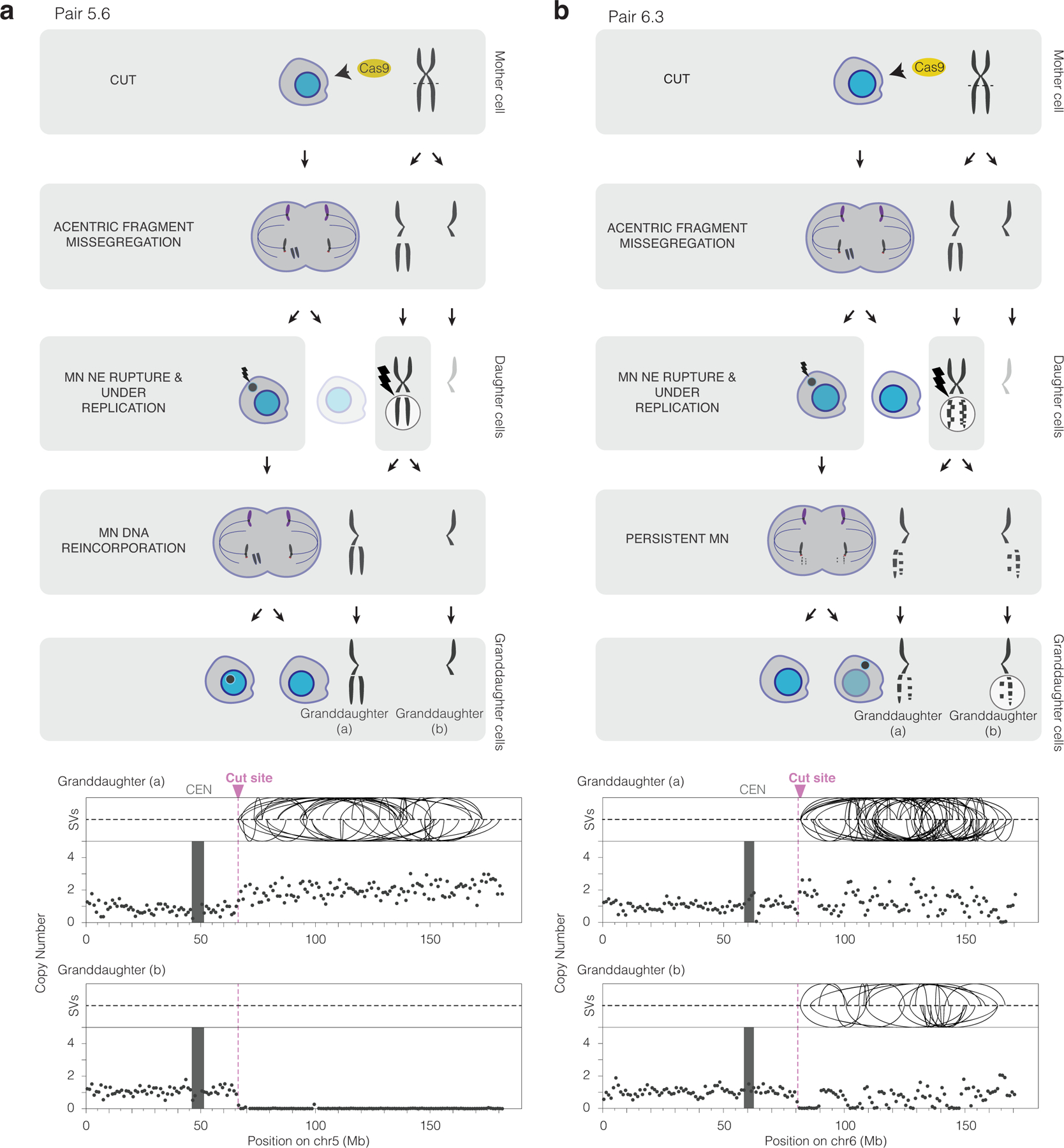
(a) Chromothripsis after a micronucleus is reincorporated into a granddaughter cell. Left, cartoon depicting the cellular events leading to the genomic outcomes for CRISPR-Cas9 pair 5.6 (Extended Data Fig. 3). Cells are on the left and chromosomes are depicted on the right. In the first generation, both sisters from one homolog were cleaved in a G2 cell (horizontal dashed line) that divides to generate a micronucleated daughter (left) and a non-micronucleated daughter (right, faded cell not subsequently followed). DNA in the micronucleus is poorly replicated. In the second cell division, the micronuclear chromosome is reincorporated into a granddaughter cell’s primary nucleus. Lightning bolt: DNA damage. Right, plots showing structural variants (SVs) and DNA copy number for haplotype of the cleaved chromosome. Top, intrachromosomal SVs (> 1 Mb) are show by the curved lines. Bottom: copy number plot (1 Mb bins). CEN: centromere.
(b) Chromothripsis after the bulk of a micronuclear chromosome fails to be reincorporated into a granddaughter cell primary nucleus for pair 6.3 (Extended Data Fig. 3). Cartoon (left) and SV and copy number plots (right) as in (b). In this example, the two arms from cleaved sister chromatids are fragmented, generating chromothripsis in both daughters.
Haplotype copy number analysis also demonstrated fragmentation of the targeted acentric chromosome fragment. If a chromosome from a micronucleus is cleaved into fragments that are distributed randomly between the granddaughter cells, the granddaughter cells will display a mirror image DNA copy number pattern that oscillates between two levels28. In the simplest case, one homolog is replicated and segregated normally. However, fragmentation of the other homolog, which is severely underreplicated, can generate oscillations between zero copies and one copy in each daughter. The regions with one copy of the fragmented homolog will retain heterozygosity, leading to islands of heterozygosity interspersed within regions of LOH, one criterion for chromothripsis56. In five of 18 pairs, haplotype copy number analysis identified fragmentation (Fig. 2,3b and pairs 5.1, 5.7, 5.9, 6.1, 6.3 in Extended Data Fig. 3). In eight of 18 pairs, there were clustered rearrangements on the targeted arm without detectable copy number oscillations, producing copy-neutral chromothripsis whereby the acentric segment was fragmented but most fragments were inherited by only one granddaughter. Copy-neutral chromothripsis is frequently observed in human congenital disease, an observation that is likely explained by the strong selection against gene copy number imbalance during human development31.
Micronuclei that spontaneously lose their nuclear envelope integrity demonstrate defects in nuclear functions, including impaired DNA replication49,51. Accordingly, we and others previously hypothesized that the DNA ligation required to generate chromothripsis would only occur after mitosis and upon reincorporation of the micronuclear chromosome into a nucleus with functional DNA end-joining28,50,57. However, in many cases, micronuclear chromosomes fail to reincorporate into a primary nucleus and are again partitioned into micronuclei39,48. Furthermore, micronuclei that lack kinetochores, like CRISPR-MN, are rarely reincorporated39,57.
We tested whether bulk chromosome reincorporation is required to generate chromothripsis by sequencing granddaughter cells with micronuclear chromosomes present in the cytoplasm. Surprisingly, of the 12 CRISPR-generated granddaughter pairs with a persistent micronucleus, eight showed chromothripsis involving the targeted chromosome arm (Fig. 2,3b, Extended Data Fig. 3 and Supplementary Videos 1,2). Chromothripsis in these samples could either be due to end-joining of chromosome fragments in the cytoplasm, aberrant mitotic DNA synthesis29,58, or ligation of a subset of chromosome fragments that might have been incorporated into the granddaughter nucleus after the division of a micronucleated cell. It was recently reported that spontaneously arising micronuclei in mouse embryonic cells often fail to be reincorporated, which was hypothesized to reflect a mechanism to prevent chromothripsis during embryo development57. However, our data establish that chromothripsis can occur even without visible reincorporation of the bulk of the micronuclear chromosome.
The CRISPR-MN results provide an important validation of our previous single-cell analysis showing that micronuclei can cause chromothripsis. In this prior work, we used random mitotic errors to generate micronuclei and then inferred the identity of the micronuclear chromosome based on it being the only underreplicated chromosome28. The current results, in which the identity of the micronuclear chromosome is known a priori, confirm that the micronuclear chromosome is the one that undergoes chromothripsis. Moreover, in 16 of the 18 samples (all but pairs 5.5 and 5.8), haplotype-resolved DNA copy number analysis demonstrated that the micronuclear chromosome showed little detectable replication, again orthogonally validating our prior analysis28. Together with other recent work examining clonal cell populations after the induction of micronuclei, it is now clear that these structures generate chromothripsis at remarkably high rates34,50,52.
The above findings bring up questions about whether cells with fragmented micronuclear chromosomes could undergo p53-dependent cell cycle arrest or cell death. We investigated this by using live-cell imaging to compare the division rates of control and micronucleated RPE-1 cells, with or without p53 knockdown. Cells were synchronized by serum starvation, and chr5q CRISPR-MN were induced after release from the G1 block (as in Extended Data Fig. 1a, bottom scheme). As expected, p53 loss did not affect the rate of micronucleation (Fig. 4a). As shown in the lifetime plots (Fig. 4b), cells lacking micronuclei divided efficiently, with or without p53 knockdown. However, cells with micronuclei remained in interphase for ~1.5 h longer than controls regardless of p53 status. Furthermore, of the micronucleated cells, 8 % of cells with p53 knockdown failed to undergo cell division. By contrast, 54 % of micronucleated p53-proficient cells failed to divide, however, the remaining 46 % of these cells divided successfully. Therefore, intact p53 suppresses (~2-fold), but does not prohibit the division of micronucleated cells generated by CRISPR-Cas9. These findings help explain data from human patients with congenital disease or cancer, where chromothripsis can frequently be observed without p53 loss30,31,36.
Figure 4. The impact of p53-status on the ability of micronucleated cells to undergo division.
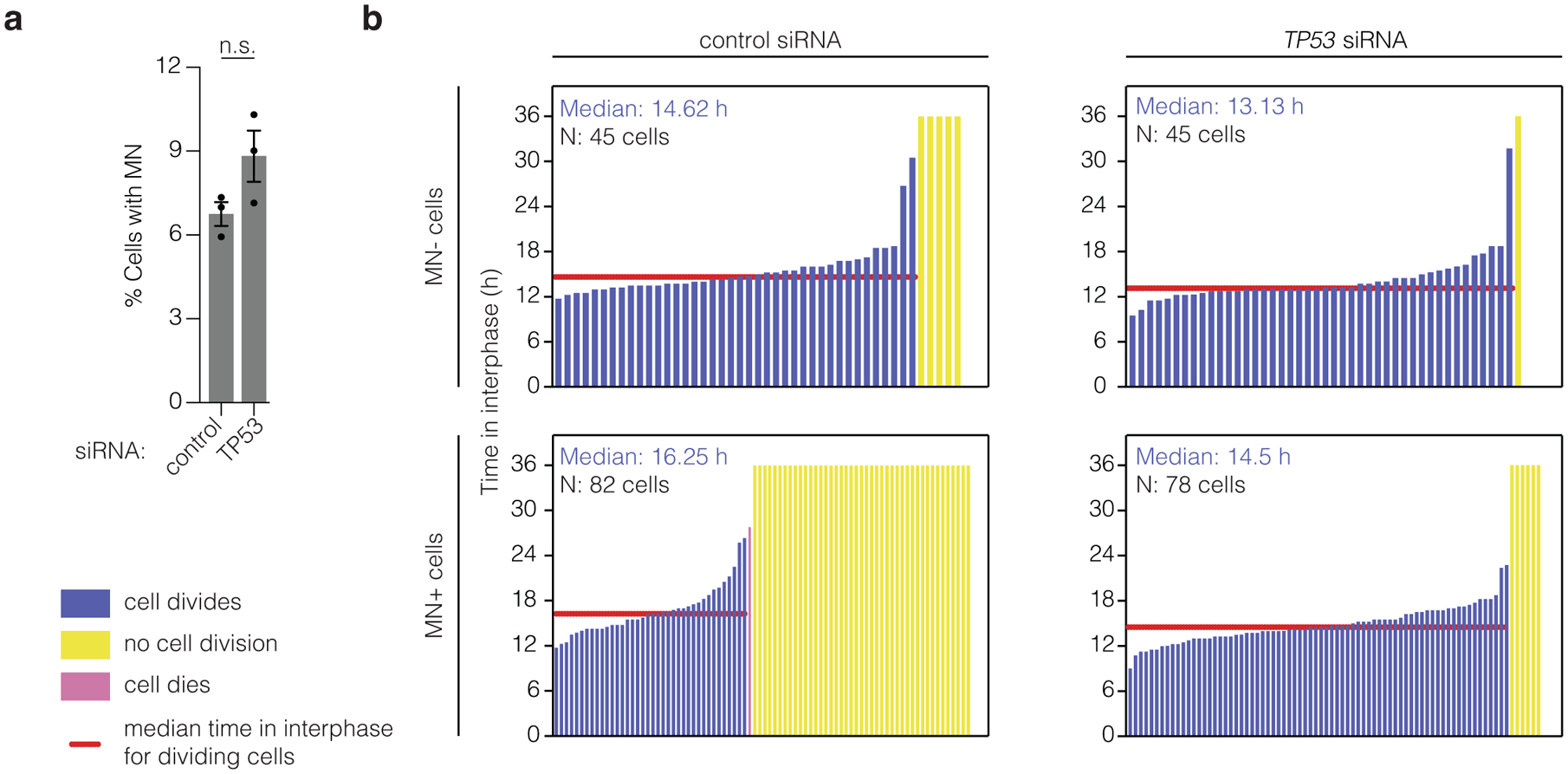
(a) p53 loss does not affect the frequency of micronucleation after CRISPR-Cas9 genome editing. Cells, with or without p53 RNAi, were synchronized, released from a G1 block, and chr5q CRISPR-MN were generated by doxycycline-induced Cas9 expression as in Extended Data Fig. 1a bottom scheme. RFP-H2B labeled micronucleated cells were identified ~40 h after release [n = 3 experiments with 1186 and 1234 cells scored, left to right, knockdown of 22.2 – 87.2 % (mean 44.7 %; standard deviation 36.8) of total p53 at 48 h after release from G1 block]. Error bars: mean +/− SEM, p = 0.0801, two-tailed Fisher’s exact test.
(b) The results of long-term live-cell imaging is shown as lifetime plots of control and micronucleated cells, with or without p53 knockdown. (n = 3 experiments). Profiles only include cells whose complete cell cycle starting from mitosis was viewed.
Because fragmented chromosomes or chromosome arms are inherently unstable, our findings also leave questions about how functional chromosomes are established after chromothripsis. Recent experiments have shown that after micronucleation or chromosome bridge formation, stable chromosomes with chromothripsis can indeed be established in clonal cell populations during long-term culture29,34,50,52,59–61. Stable chromosomes must have only one centromere and the chromosome ends need to be capped with telomeres. After CRISPR-Cas9 cleavage, a chromothriptic acentric fragment can be stabilized by re-ligation to the centric portion of the broken chromosome, or by translocation to another chromosome (acrocentric chromosomes are a common translocation recipient29). In principle, de novo telomere addition could also stabilize chromothriptic chromosomes, but this occurs at low frequencies62. Therefore, our data, together with that of others31,36,52, indicates that at least some micronucleated cells can divide and expand into a clonal population with stably propagating chromothriptic chromosomes, independent of p53 status.
CRISPR-Cas9 editing generates chromosome bridges
In addition to the formation of micronuclei, Cas9-generated DNA breaks can lead to dicentric chromosome bridge formation due to ligation of the centric fragments of Cas9-cleaved sister chromatids29,63. We recently identified a series of mechanistic steps through which chromosome bridges, like micronuclei, induce chromothripsis29.
In eight out of 18 pairs (pairs 2.1–5, 5.1, 5.5, 5.9) derived from CRISPR-MN cells, we identified DNA copy number signatures of bridge formation, which added complexity to the copy number patterns resulting from the missegregation of acentric fragments. All of these samples involve two cell divisions during which bridges could form (Extended Data Fig. 5a,b): bridges can form during the first division, when the micronucleus forms; or during the second division, when the micronucleated daughter cell divides. If the bridge forms and breaks in the first cell division, the two granddaughters descended from the micronucleated daughter will exhibit shared segmental gains or losses on the centromeric side of the Cas9 cut, as seen in six of 18 granddaughter pairs (Fig. 5a and Extended Data Fig. 5a, pairs 2.1–2.5, 5.5). If the bridge forms and breaks in the second cell division, the cells will display reciprocal gain and loss of DNA sequence on the centromeric side of the cut site, as observed in two of 18 granddaughter pairs (Fig. 5b and Extended Data Fig. 5b, pairs 5.1 and 5.9). Note that the megabase-scale copy number loss on the centromeric side of the cut, which we attribute to bridge breakage, cannot be explained by DNA resection from the cut site because resection is generally limited to several kilobases64. Moreover, resection cannot explain copy number gains on the centromeric side of the breaks. Instead, segmental gains are a sequence signature of the chromosome breakage-fusion-bridge cycle, a common mutational process in cancer that generates gene amplification29,60,65–67. Finally, the chromosome that was inferred to form a bridge shared the same haplotype as the micronuclear chromosome, in agreement with the expectation that dicentric bridges and acentric micronuclei can arise simultaneously from the same Cas9 cut.
Figure 5. CRISPR Cas9-genome editing induces chromosome bridge formation, adding to the genome complexity from micronuclei.

(a) Evidence for genome editing-induced chromosome bridge in pair 5.5 (Extended Data Fig. 3). Scheme as in Fig. 3. CRISPR-Cas9 cut site is indicated by the dashed line and relevant segments of chr5 are indicated by letters A-C. In the first division, the DNA break on sister chromatids results in the formation of a micronucleus with the acentric portions of chr5 (segment C). At the same time, the sister centric fragments (AB) fuse, generating a dicentric bridge concomitantly with the formation of the micronucleus. Asymmetric breakage of the bridge leads to the loss of the “B” segment from the bridge chromosome in the micronucleated daughter. Faded cell inferred to contain two copies of the B segment was not followed further. DNA copy number analysis indicated that in this example the chromosome fragments in the micronucleus underwent DNA replication. This region showed no detectable rearrangements. Note that the acentric fragments of chr5 were not reincorporated into a daughter primary nucleus in the second division. A (purple): p-arm; B (black): centromere to cut site, inferred to reside in the bridge; C (teal): cut site to the telomere. Bottom: Copy number and rearrangement plots of cells from above, as in Fig. 3.
(b) Bridge formation, micronucleation, chromosome fragmentation and chromothripsis from CRISPR-Cas9 genome editing in pair 5.1 (Extended Data Fig. 3). In this sample both homologs were cleaved. The acentric arm of homolog 1 (blue allele) missegregates into a micronucleus in the first generation. The centric fragments of homolog 1 fuse, resulting in a dicentric bridge in the second cell division. After the second cell division, the cell that inherited the acentric fragment of homolog 2 (red) was found to have few SVs or copy number alterations, suggesting it was partitioned into the primary nucleus as indicated in the scheme. By contrast, the acentric segments of homolog 1 were fragmented. Bottom: copy number and rearrangement plots of cells shown above, as in Fig. 3.
Support for chromosome bridge formation also came from fluorescence imaging, which showed that 13.8 % of cell divisions that formed micronuclei after CRISPR-Cas9 cutting also formed visibly detectable chromosome bridges (Extended Data Fig. 5c) after p53 knockdown. Micronucleation physically separates the centric and acentric sides of the CRISPR-generated DNA break, preventing the acentric fragment from being used as a ligation partner for the centric fragment of the broken chromosome. We therefore hypothesized that the presence of a micronucleus would bias for ligation of the centric fragments of the broken chromosome to each other, leading to elevated rates of dicentric bridge formation in the granddaughters. Accordingly, the frequency of bridge formation was higher still after the division of micronucleated cells (22.4 % of divisions) [Extended Data Fig. 5c]). Similar results were obtained without p53 knockdown (Extended Data Fig. 5c). Thus, CRISPR-Cas9 genome editing is accompanied by chromosome bridge formation in addition to micronucleation, both of which can trigger ongoing cycles of genome instability.
Micronucleation from therapeutically relevant genome editing
We next investigated our findings in the context of a therapeutic genome editing approach to induce HbF for the treatment of β-hemoglobinopathies3–5. We electroporated normal donor human CD34+ hematopoietic stem and progenitor cells (HSPCs) with a Cas9/gRNA RNP complex targeting the erythroid-specific BCL11A enhancer on chromosome 2p5,40 (see the Supplementary Note for further discussion). The on-target editing efficiency was 89.4 % with a 4.9-fold increase of HbF in erythroid progeny compared to unedited cells (Fig. 6a,b). Similar to our observations in cell lines, the frequency of micronucleation increased 16-fold by 24 hours after RNP transfection (Fig. 6c). Using FISH probes surrounding the RNP-induced DSB, we found that over 80 % of cells containing micronuclei exhibited copy number alterations affecting the acentric fragment of the targeted chromosome (Fig. 6d,e). Moreover, 7.3 % of cells without micronuclei exhibited abnormal numbers of this chromosome arm, indicating that cutting was followed by missegregation of the acentric fragment to the primary nucleus (Fig. 6d,e). Some of these TP53-intact cells were capable of entering mitosis with an unrepaired DSB, as 3.25 % of cells analyzed had breaks in chr2p detected by spectral karyotyping (SKY) 24 h after Cas9 treatment (Fig. 6f,g). We also detected high-level phosphorylation of histone H2AX in 12.9 % of micronuclei (Fig. 6h,i), indicating extensive DNA damage.
Figure 6. Hallmark cytological features of chromothripsis after a genome editing approach for the treatment of sickle cell disease.
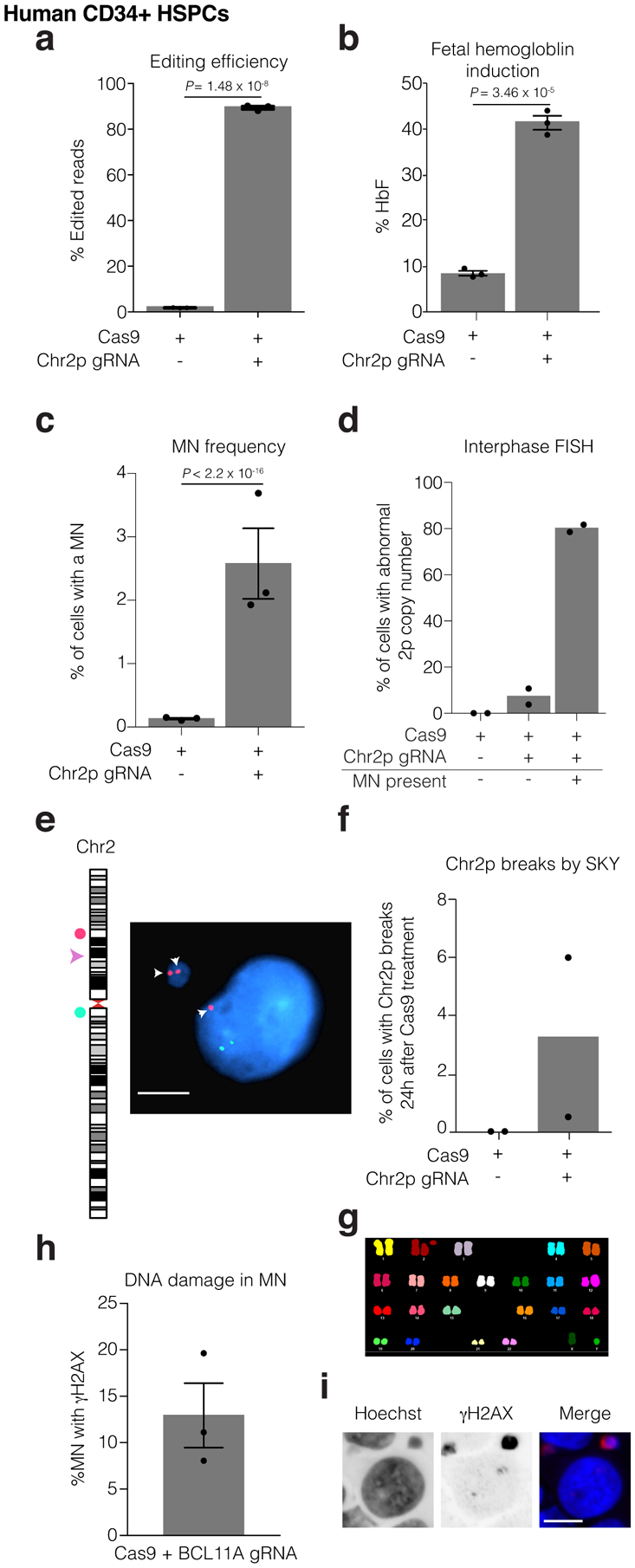
Human CD34+ HSPCs were electroporated with Cas9/gRNA RNP targeting the erythroid-specific enhancer of BCL11A. Microscopic analysis of micronucleation was performed 24 h post electroporation.
(a) Editing efficiency of BCL11A determined from amplicon sequencing. n = 3 experiments, Error bars: mean +/− SEM, two-tailed unpaired t-test).
(b) Fetal hemoglobin (HbF) levels were measured by HPLC in erythroid-differentiated CD34+ HSPCs as a functional readout of successful editing of BCL11A 10 days after RNP electroporation. n = 3 experiments, Error bars: mean +/− SEM, two-tailed unpaired t-test).
(c) Percent of cells with a micronucleus (n = 3 experiments with 7827 and 6480 cells counted, left to right). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(d) Percent of cells with aberrant 2p copy number assayed by FISH (n = 2 experiments with 1957, 1926, 74, cells counted, left to right).
(e) Representative FISH image of data in (d). Cut site is represented by a pink arrowhead; DNA is blue; telomere proximal probe is red, and marked by arrows; centromere proximal probe is green. Shown is a micronucleated cell with 3 copies of the cut arm, two of which are in the micronucleus. Scale bar 5 μm.
(f) Chr2p breaks present 24 hours after electroporation in metaphase visualized by SKY (n = 2 experiments, 400 spreads per condition).
(g) Sample SKY image from (f).
(h) Percent of CD34+ CRISPR-MN with extensive DNA damage covering the DNA present in the micronucleus by γH2AX-labeling (n = 3 experiments, 135 micronuclei scored). Error bars: mean +/− SEM.
(i) Representative image of data in (h). Scale bar 5 μm.
Non-adherent cells such as HSPCs are not amenable to our combined imaging and single-cell genomic analysis, precluding direct detection of chromothripsis. Using a PCR-based method, we did not detect LOH suggestive of chromothripsis in hematopoietic colonies derived from genome edited single-cell clones (Extended Data Fig. 6). However, micronucleus formation is expected to have occurred in only approximately 2.5 % of clones analyzed, only a small fraction of which would be expected to undergo chromothripsis and remain viable. Thus, we have limited detection sensitivity for this event by analyzing hundreds or even thousands of clones. By contrast, patient-scale treatments are predicted to contain millions of micronucleated cells, meaning that low frequency micronucleation in this setting could still be consequential. Together, our results establish that HSPCs acquire hallmark cytological features associated with chromothripsis following CRISPR-Cas9 genome editing.
Discussion
Here we demonstrate that on-target CRISPR-Cas9 genome editing can induce the formation of micronuclei and chromosome bridges in dividing cells, leading to copy number alterations of large chromosomal segments and chromothripsis. These findings provide potential mechanisms for the recently observed large chromosomal deletions or loss of heterozygosity surrounding on-target DSBs following genome editing in embryos23,25,55,68,69. Moreover, they raise a new potential concern for therapeutic genome editing strategies that require DSB formation, because chromothripsis can drive the rapid acquisition of multiple cancer-causing mutations simultaneously. Chromothripsis can promote tumorigenesis in many tissue types, including ones relevant for therapeutic editing, even in cells with intact p5332,36,37,70.
To date, malignant transformation or abnormal clonal cell expansion following genome editing has not been observed in animal studies, including non-human primate models71–73, nor in a relatively small number of human subjects who have participated in clinical trials and were monitored for relatively short periods of time6,27,74. In quantitative terms, the clinical risks associated with nuclease-based genome editing therapies in human subjects remains unclear. In particular, the rates of forming micronuclei or chromosome bridges followed by chromothripsis, as well as the frequency at which affected cells will expand, are unknown for any therapeutic application. These outcomes will likely differ according to the target locus and its efficiency of DNA repair, the density of oncogenes and tumor suppressors on the targeted arm and the target cell type. For example, the erythroid-specific enhancer of BCL11A might fortuitously be a favorable site for editing in HSPCs, because breaks in the enhancer that trigger large-scale alterations will disrupt the BCL11A gene. Based on prior work showing that single copy loss of BCL11A impairs the ability of HSCs to repopulate the bone marrow75, it is expected that arm-level copy number losses and/or chromothripsis at this locus should be subject to strong negative selection.
Additionally, the rates of chromothripsis and its antecedent aberrations may be dependent on the specific editing protocols used which differ greatly between individual research laboratories and also for large scale therapeutic applications. For example, longer culture times or the use of HSC expanding reagents prior to editing may drive cells into cycle and increase the probability of cell division with a broken chromosome. One important question for the field is whether quantifying micronucleus formation, chromosome bridges and chromothripsis during therapeutic protocol development with attention to minimizing these events will enhance safety outcomes.
Numerous studies have established that micronucleation or chromosome bridge formation can trigger chromothripsis followed by clonal expansion29,34,50,52,60,61,67,76. However, many chromothripsis events are expected to compromise cell fitness, leading to senescence or cell death. Nevertheless, even rare events that facilitate clonal expansion could be impactful at clinical scale genome editing. Current protocols infuse approximately 3–16 × 106 genome edited CD34+ cells per kilogram6, which, based on micronucleation frequencies we observed in Fig. 6, include approximately 106 micronucleated cells. Nonetheless, the clinical outcome of such events is unknown. Ultimately, the potential for clinically deleterious chromothripsis caused by therapeutic genome editing must be assessed by long-term monitoring of individuals enrolled in clinical trials.
Although TP53 loss likely promotes the survival of cells with chromothripsis33, chromothripsis can occur and persist in TP53 proficient cells, as indicated by several lines of evidence31,33,36. First, this study and prior literature provide evidence that an appreciable fraction of p53 proficient cells with CRISPR-generated micronuclei are capable of division, albeit at a somewhat reduced frequency. Second, patients with clonal chromothripsis causing congenital disease do not have loss of TP5331. Moreover, chromothripsis has been observed to occur and persist in p53-proficient hematopoietic stem cells. Here, chromothripsis followed by clonal expansion of a stem cell resulted in the spontaneous cure of WHIM-syndrome via the loss of a dominant mutation77. Third, clonal expansion of p53-proficient malignant cells with chromothripsis is common; across all human tumors, the incidence of chromothripsis is only enriched 1.5-fold in those that contain inactivating mutations in TP5336. Finally, it has recently been shown that CRISPR-Cas9 editing in p53-proficient human embryos can generate large, cut site-associated deletions that can be propagated to at least the cleavage stage23. Therefore, performing CRISPR-Cas9 genome editing in p53 proficient cells does not guarantee that clones with chromothripsis, or other large-scale chromosome alterations, will be unable to develop.
Our results have several practical implications. Efficient Cas9-mediated HDR requires cells to be actively dividing whereas NHEJ does not. Therefore, therapeutic genome editing via NHEJ in non-dividing cells, such as retinal photoreceptors78, should not produce micronuclei. Conversely, efforts to specifically edit dividing cells in order to enhance HDR rates, for example, by using a modified Cas9 with reduced activity in non-dividing cells79, may enhance micronucleation and its downstream consequences, such as chromothripsis. Accordingly, for therapeutic NHEJ editing of HSCs, it may be beneficial to maintain HSC quiescence. Some CD34+ HSPC editing protocols appear to favor quiescent or G1 HSCs, whereas other protocols cause a higher frequency of editing in cycling or G2 HSCs, sometimes unintentionally5,8,72,79–81. Additionally, we suggest that for NHEJ applications, fusion of Cas9 to a G1-specific Cdt1 segment could be employed to restrict editing to G1 cells79,82, thereby minimizing the probability of micronucleus formation and the deleterious downstream toxicities. Screening for micronucleation and/or chromothripsis in clinical protocols is expected to become more feasible as high-throughput and low-cost methods for single-cell genome sequencing are developed76. Finally, our study further motivates the development of genome editing strategies that do not generate double-stranded DNA breaks2,11–13,83, which in principle should minimize the potential for inducing chromothripsis.
Methods
Cell culture and generation of cell lines
Cells were cultured at 37 °C in 5 % CO2. Telomerase-immortalized RPE-1 retinal pigment epithelium (CRL-4000) and BJ-5ta foreskin fibroblasts (CRL-4001) from ATCC were grown in Delbucco’s Modified Eagle Medium/F12 (1:1) (Gibco) with 10 % FBS, 100 IU/ml penicillin and 100 μg/ml streptomycin. RPE-1 cells expressing Cas9 under a doxycycline-inducible promoter (gift from I. Cheeseman41) were grown using tetracycline-free FBS (X&Y Cell Culture).
Mobilized peripheral blood CD34+ cells were obtained from three de-identified healthy donors (Key Biologics, Lifeblood) and enriched by immunomagnetic bead selection using an AutoMACS instrument (Miltenyi Biotec). Cryopreserved CD34+ cells were thawed and pre-stimulated for 48 h in StemSpan SFEM (StemCell Technologies) supplemented with 100 ng/mL SCF, FLT3-L, and TPO (R&D Systems). CD34+ cells were maintained in complete SFEM post-electroporation for 1–5 days or subject to erythroid differentiation. Erythroid differentiation was induced using a two-phase protocol. Phase 1 (days 0–5): IMDM (Thermo) supplemented with 20 % FBS, 1 % penicillin/streptomycin, 20 ng/mL SCF, 1 ng/mL IL-3 (R&D Systems), and 2 U/mL EPO (Amgen). Phase 2 (days 5–10): IMDM supplemented with 20 % FBS, 1 % penicillin/streptomycin, 2 U/mL EPO, and 0.2 mg/mL holo-transferrin (Millipore Sigma).
RPE-1 cells expressing H2B-eGFP, RFP-H2B, TDRFP-NLS, and eGFP-BAF were created by transduction of lentivirus or retrovirus vectors containing the genes of interest as previously described29.
Cas9 RNP transfection in immortalized cell lines
sgRNAs were synthesized with the Trueguide Synthetic gRNA platform (Thermo Fisher Scientific) as chemically modified custom oligos, where the final 3 bases on both the 5’ and 3’ end of the sgRNA are 2’-O-Methyl bases and the linkages between them are phosphorothioates, in order to increase editing efficiency and protect from nuclease degradation. Their sequences are listed in Supplementary Table 1.
RNP complexes were prepared following a modified version of the suggested manufacturer’s protocol. Briefly, gRNA/Cas9 complexes were formed by incubating 250 ng of the gRNA with 1 μg of purified Cas9 protein (TrueCut Cas9 Protein v2, Invitrogen) in OptiMEM (Invitrogen). Cells were seeded on 12-well dishes, #1.5 glass coverslips (fixed imaging experiments), or 35-mm gridded ibiTreat dishes (ibidi) (Look-Seq), were synchronized by serum starvation in 0.1 % FBS-containing media for 24 h where applicable, and subjected to Cas9 RNP transfection 22 h upon release from the block. Transfection of ribonucleoprotein complexes was performed using Lipofectamine CRISPRMAX Reagent (Invitrogen). Cells were fixed 46 h after release from block to measure the percentage of cells with micronuclei and 35 – 40 h after release for the FISH experiments.
Editing of CD34+ HSPCs
Purified recombinant Cas9 protein was obtained from Berkeley Macrolabs. Chemically modified single guide RNAs (sgRNA) were synthesized by Synthego with 2′-O-methyl 3′-phosphorothioate modifications between the 3 terminal nucleotides at both the 5′ and 3′ ends. Ribonucleoprotein complexes (RNPs) were formed by incubating Cas9 (32 pmol/100,000 cells) with sgRNAs at a 1:2 molar ratio. CD34+ cells were washed in PBS, resuspended in the manufacturer provided buffer for primary cells, mixed with RNPs, and electroporated using program 24 of a Neon Transfection System (Thermo Fisher Scientific). Editing efficiency was determined as described previously80,84 using primers in Supplementary Table 3.
Doxycycline-inducible Cas9 treatments
Cas9 expression in the doxycycline-inducible system was validated by Western Blotting with an antibody against Cas9 (Cell Signaling Technology #14697S, clone 7A9–3A3, 1:1000) and a-Tubulin loading control (Sigma #T9026, clone DM1A 1:10000). Cells were trypsinized, pelleted, washed with PBS, and lysed at 4 °C in RIPA Buffer (Boston Bioproducts) supplemented with cOmplete mini protease inhibitor (Millipore Sigma), PhosSTOP protease inhibitor (Roche), 1 mM DTT, and 1 mM PMSF. Samples were centrifuged at 17,000 × g for 30 min at 4 °C and the supernatant was run on a 10 % Mini-PROTEAN TGX precast polyacrylamide gel (BioRad). Protein was transferred to a PVDF membrane using the iBlot 2 (Life Technologies). The membrane was blocked with 5 % milk in TBST for 1 h at room temperature, followed by incubation with primary antibodies overnight at 4 °C. Three washes were performed with TBST followed by 1 h incubation with secondary antibody (ECL, HRP linked, GE Healthcare) and another series of washes. Membranes were imaged using an ImageQuant LAS 4000 (GE Healthcare).
sgRNAs (Supplementary Table 1) were cloned into pLenti-Guide-Puro (Addgene) and delivered to hTERT-immortalized RPE-1 cells carrying a tetracycline-inducible promoter by lentiviral transduction, as above. Starting 24 h after transduction the population of cells was selected for one week in 12 μg/ml puromycin. Cells were treated with 40 nM ON-TARGETplus siRNA SMARTpool L-003329-00-0050 (Dharmacon) or non-targeting control siRNA D-001810-10-05 (Dharmacon) to deplete p53 in experiments using the doxycycline-inducible system. siRNA was transfected using Lipofectamine 3000 (Life Technologies) according to manufacturer’s instructions. p53 knockdown was validated in select experiments by Western Blot as above, with antibodies against p53 (Cell Signaling Technology #48818S, clone DO-7, 1:1000) and GAPDH loading control (Abcam ab9485, 1:5000). 6 h after siRNA treatment cells were synchronized by serum starvation in 0.1 % FBS-containing media. 24 h later, cells were released from block into complete medium containing 0.5 μg/ml doxycycline, which was washed out 15 h later 5 times. When MPS1 inhibitor (1 μM NMS-P715, EMD Millipore) was used to produce micronuclei from mitotic errors, cells were released without doxycycline, and MPS1 inhibitor was added ~18 h after release, before the next cell division. MPS1 inhibitor was washed out by 5 washes with complete medium 20 h later. Cells were then transferred to coverslips or dishes for immunofluorescence or FISH experiments, live-imaging experiments, or plated for Look-Seq.
Measurement of editing efficiency in immortalized cells
DNA was isolated 48 h after RNP transfection or doxycycline washout using the PureLink Genomic DNA kit (Invitrogen) according to manufacturer’s instructions. PCR was performed using Q5 High-Fidelity DNA Polymerase for 35 cycles after an initial 30 s denaturation step at 98 °C [5 s 98 °C, 10 s 60 °C, 15 s 72 °C for doxycycline treated samples and 10 s 98 °C, 15 s 55 °C, 20 s 72 °C for RNP treated RPE-1 cells] and a 2 min final extension at 72 °C with 2.5 mM dNTP, 10 μM forward and reverse primers, 10 μL Q5 Reaction Buffer, and at least 20 ng of genomic DNA. 2 % agarose gels were run in TAE buffer on an aliquot of PCR product to ensure production of a unique PCR product of the appropriate size. Primer pairs utilized are contained in Supplementary Table 3. PCR products were purified using the QIAquick PCR purification kit (QIAGEN) and diluted to 20 ng/μL, as measured by Qubit dsDNA HS Assay Kit (Invitrogen). Samples were then submitted to Genewiz for Amplicon-EZ sequencing or to the Center for Computational and Integrative Biology DNA core facility of Massachusetts General Hospital for amplicon next generation sequencing. Analysis of the raw data for detecting CRISPR variants from NGS reads was performed with the algorithms from Genewiz Amplicon-EZ service or the MGH core and meta-analysis to estimate the percentage of editing efficiency was performed manually by the users.
Fluorescence in situ hybridization of RPE-1 and BJ cells
FISH probes utilized in this study were as follows: Chr1p Subtelomere (Cytocell, LPT 01PG-A); Chr1 Centromere (Abbott Laboratories); Chr2p Subtelomere (Cytocell, LPT 02PG-A); Chr2 Centromere (Cytocell, LPE 002R-A); Chr4q Subtelomere (Cytocell, LPT 04QG-A); Chr4 Centromere (Cytocell, LPE 004R-A); Chr5q Subtelomere (Cytocell, LPT 05QG/R-A); Chr5 Centromere (Cytocell, LPE 005R-A); Chr6q Subtelomere (Cytocell LPT 06QR-A); ChrXq Subtelomere (Cytocell, LPT XYQG-A); ChrXq Centromere (Cytocell LPE 0XR-A)
Cells were seeded on #1.5 glass coverslips and were transfected with Cas9 RNP, as described. Cells were fixed ~35 hours after release from starvation media, in the first interphase where the cells have formed micronuclei. Prior to fixation the coverslips were swelled in pre-warmed 75 mM KCl and incubated at 37 °C for 20 min. Fixation was performed by dropwise addition of 0.5 volume of −20 °C Carnoy’s solution (3:1 methanol:acetic acid). After 5 minutes, the solution was exchanged for fresh −20 °C Carnoy’s solution twice more. Coverslips were then air dried for 48 h. Coverslips were warmed in 2X SSC + 0.5 % NP-40 at 37 °C for 30 min, and then dehydrated in ice cold solutions of 70, 85, and 100 % ethanol for 2 minutes each. Subtelomere-specifc or centromeric probes were diluted 1:5 – 1:10 in hybridization buffer B (Cytocell, purchased by Rainbow Scientific # HB1000L) and applied to the samples after air drying. Coverslips were then sealed onto glass slides with rubber cement, denatured at 73 °C for 2 min, and hybridized in a humidified chamber at 37 °C for two days. After hybridization, coverslips were floated from the slides in a PBD solution composed of 0.1 M Na2HPO4, 0.1 M NaH2PO4, and 0.1 % NP-40 for 3 min at RT. Samples were washed in 72 °C 0.5× SSC + 0.1 % SDS for 5 min, and then transferred to 2.5 μg/ml Hoechst 33342 (Life Technologies) dissolved in PBD solution for 10 min. Coverslips were then air dried and mounted on clean glass coverslips using ProLong Gold antifade (Life Technologies) or Vectashield Antifade Mounting Medium with DAPI (H-1200, Vector laboratories). Denaturation and wash steps were performed using a HybEZ II Hybridization system (ACD). Samples were imaged by confocal microscopy, as described below.
Fluorescence in situ hybridization of CD34+ HSPCs
For detection of chr2 abnormalities, two BAC clones were used as probes, one located distal to the BCL11A locus (2p21) as the telomeric marker and a clone from 2q11.2 as the centromeric marker. The telomeric BAC DNA (hg19 chr2:47612794 – 47782780) was labeled with a red-dUTP (AF594, Molecular Probes) by nick translation and the centromeric BAC DNA (hg19 chr2:99969552 – 100200667) was labeled with a green-dUTP (AF488, Molecular Probes). Both labeled probes were combined with sheared human DNA and hybridized in a solution containing 50 % formamide, 10 % dextran sulfate, and 2X SSC. The cells were then stained with 4,6-diamidino-2-phenylindole (DAPI) and imaged using a Nikon Eclipse 80i with a 100x/1.40 NA Plan Apo objective and Cytovision version 7.7 (Leica Biosystems).
SKY of CD34+ HSPCs
Day 1 post-electroporation, CD34+ cells were harvested by routine cytogenetic methods after a 4 h colcemid incubation. Commercially prepared SKY probes and protocols from Applied Spectral Imaging (Carlsbad, CA) were used for the hybridization and detection steps. Mitotic spreads were analyzed and chr2p breaks were quantified.
Indirect immunofluorescence microscopy
Immunofluorescence was performed as described28,51. Primary antibodies: γH2AX (1:400–500, MilliporeSigma, 05–636-I, clone JBW301), LBR (1:100, Abcam, ab32535, clone E398L). Secondary antibodies: Alexa Fluor, 488 (A11029), 568 (A11031) and 647 (A21236) (1:1000, Life Technologies). EdU was added 5 h before fixation.
Confocal images were collected using a Nikon Ti-E inverted microscope with a Yokogawa CSU-22 spinning disk head with the Borealis modification. Z-stacks were collected for 9 images at 0.4–0.6 μm spacing using a CoolSnap HQ2 CCD camera (Photometrics) and a 60x/1.40 NA Plan Apo oil immersion objective (Nikon) using Metamorph Software 7.10.2.240 (Molecular Devices). Alternatively, a Ti2 inverted microscope fitted with a CSU-W1 spinning disk system (Nikon) was used. Z-stacks were collected to cover the whole volume of cells at 0.4–0.6 μm spacing using a Zyla 4.2 sCMOS camera (Andor) and a 60x/1.40 NA Plan Apo λ oil objective and NIS-Elements 5.11.03 AR software (Nikon).
Indirect immunofluorescence microscopy of CD34+ HSPCs
1.5 × 105 CD34+ cells were deposited on glass slides using a Cytospin™ 4 cytocentrifuge (Thermo Scientific) for 5 min at 800 rpm. Fixation and indirect immunofluorescence were performed as above. Images were acquired with single-plane widefield illumination on a Nikon Eclipse Ni microscope using Nikon NIS-Elements software and a 40x/0.75 Plan Fluor objective. Antibodies are listed above.
HbF Quantification
Fetal hemoglobin quantification by ion-exchange high-performance liquid chromatography was performed and analyzed using LabSolutions v 5.81 SP1 software as previously described80.
Live-cell imaging
Live-cell imaging was performed as described previously with minor modifications28,51, which are elaborated upon in the Supplementary Note.
Image analysis
NIS-Elements AR 5.20.00 (Nikon) was used to analyze live-cell imaging videos, and ImageJ (v 1.51) was used to create annotated videos. Quantitative image analysis for fixed-cell experiments was performed using ImageJ. Briefly, nuclear segmentation was performed on maximum intensity projections based on Hoechst staining. This segmentation was used as a mask, and, if necessary, the mask was manually refined by the “Watershed”, “Erode”, or “Draw” functions. These masks were then applied to maximum intensity projections of other channels to measure the mean fluorescence intensity of channel. Background subtraction was performed by measuring the mean fluorescence intensity of a square region near the primary nucleus and micronucleus. Analysis of micronucleus formation and DNA damage in CD34+ HSPCs was performed qualitatively by sample-blinded individuals for the presence or absence of a single large focus of γH2AX signal covering most of the micronucleus. Graphical data and statistical analyses were calculated using Graphpad Prism 7.0d (GraphPad Software Inc) and R 3.4.2.
Look-Seq
Look-Seq was performed as previously described28,29, and is discussed in more detail in the Supplementary Note.
Quality assessment of sequencing libraries
Library quality assessment was performed as described previously28,29. Briefly, before deep-sequencing libraries were subjected to low-pass sequencing (~0.1× genome coverage) by the MiSeq platform (Illumina). From this we visually assessed library quality by the uniformity of whole-genome amplification in 10 Mb bins. Low-pass sequencing was used to assess haplotype-specific DNA copy number in order to identify cells with missegregation of the targeted chromosome. Libraries that passed quality checks were subjected to deep sequencing (8–47× genome coverage; 19× mean coverage, 11× median coverage) on the NovaSeq 6000 (Illumina).
Sequencing analysis and allelic copy-number calculation
Sequencing data processing and haplotype-specific DNA copy-number analysis were carried out using the same bioinformatic pipeline and computational workflow as described previously, including alignment by BWA 0.7.12-r1039 and duplicate marking by Picard software suite v 2.2.428,29.
Structural variant (SV) detection in single-cell genomes
Structural variants were detected using our previously described pipeline28,29, and are further described in the Supplementary Note.
Poisson tests and definition of fragmentation
We performed two-sample one-sided Poisson tests to determine whether SVs are enriched on the CRISPR-targeted segment compared to the background rate of SVs across the genome. We calculated this statistic relative to the depth of sequencing coverage in the targeted region (as a control, we also performed a similar test for enrichment on all non-targeted chromosome arms, Supplementary Table 2) as follows. For each pair of granddaughter cells (a, b), we calculated the fraction of reads (ra, rb) mapping to the genomic interval telomeric to the CRISPR cut in each sample. The null hypothesis is that breakpoints are drawn according to a Poisson process in which the expected density of breakpoints in a given genomic interval is proportional to the fraction of sequencing reads mapping to that interval. We computed the conditional probability of observing at least as many breakpoints as were detected on the targeted segment, given the total number of breakpoints in the genome. The test was implement as a one-sided, one-sample binomial test P(X≥k), where n = total breakpoints observed across the pair, p = (ra + rb) /2, X ~ Binomial(n,p), and k = breakpoints observed on the targeted segment. We note that this test yields the same outcome, whether we looked for enrichment of rearrangements on the entire targeted chromosome arm (Supplementary Table 2) or whether we confined the analysis to the sequence telomeric to the cut site (Extended Data Fig. 4). The test is implemented as a one-sided, one-sample binomial test P(X ≥ k) where n = total breakpoints observed across the pair, p = (ra + rb) / 2, and k = breakpoints observed on the targeted segment.
Segments were considered fragmented by visual inspection of copy number plots for the presence stretches of allele-specific reciprocal copy number change between daughter cells or many rearrangements on the targeted arm in both daughters. For Fig. 2, individual cells were marked as having ‘clustered rearrangements’ if there was significant enrichment by the Poisson test, the daughter did not lose the missegregated allele, and there was at least one rearrangement found in the cell in cases of fragmentation.
Extended Data
Extended Data Fig. 1. Micronucleus formation after CRISPR-Cas9 genome editing in several cell lines.

(a) Experimental schemes. Top, RNP transfection. Bottom, inducible Cas9 expression with constitutive expression of gRNAs (RPE-1 cells). G0 cell cycle block was by serum starvation. Dividing cell cartoon represents approximate time of cell division.
(b) Micronucleation frequency after CRISPR-Cas9 RNP transfection in asynchronous cells. Left, editing efficiency. Right, frequency of micronucleation for these RNP transfections. (n = 3 experiments with 1339, 1231, 1220, 1236, and 1237 cells scored, left to right). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(c) Representative Western blot of Cas9 levels at the indicated times after induction with doxycycline. 1st division is 24 hours after serum starve release, and 2nd division is 48 hours after release. Dox is doxycycline. n = 3 experiments.
(d) Number of cleaved chromosome arms contained within micronuclei for the indicated gRNAs and Cas9 expression strategies (RPE-1 cells) determined by FISH to detect the centromere (RNP Cas9) and/or subtelomere of the targeted chromosome (RNP Cas9 and Dox-inducible Cas9). RNP Cas9: for 2p: n = 2 experiments with 64 micronuclei counted, 4q: n = 2 experiments with 58 micronuclei counted, 5q: n= 3 experiments with 116 micronuclei counted, Xq: n = 2 experiments with 96 micronuclei counted; (Dox) Doxycycline-inducible Cas9; n = 3 experiments; 168 micronuclei counted per condition.
(e) Frequency of micronucleation in synchronized BJ fibroblasts after RNP transfection; (n = 3 experiments with 2378, 2487, 2423, 2714 cells, left to right). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(f) Left, percentage of MN containing the targeted chromosome arm for the chr5q-targeting gRNA in BJ cells, as counted using subtelomeric FISH probes. Right, the number of chr5q chromosome arms per micronucleus in BJ cells, determined from centromere-specific and subtelomere-specific FISH probes. (n = 2 experiments counting 109 micronuclei).
(g) Cut site and FISH probe locations for allele-specific gRNA experiments. PAM sequence is in bold, with the polymorphic site in red. Orange star is the centromere FISH probe and green circle the subtelomere FISH probe. gRNAs target the reference allele.
(h) Editing efficiency after Cas9/gRNA RNP transfection with allele-specific gRNAs. (n = 3 experiments). Error bars: mean +/− SEM.
(i) Micronucleation frequency from samples in (h). (n = 3 experiments with 7066, 7041, 7253, cells scored for micronucleation, left to right). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(j) Left, percentage of MN containing the targeted chromosome arm for the allele-specific gRNAs, as scored using subtelomeric FISH probes. Right, pie chart of the number of targeted arms per micronucleus in RPE-1 cells, as determined from subtelomere-specific FISH probes. (n = 3 experiments counting 123 and 184 micronuclei, left to right) Error bars: mean +/− SEM.
Extended Data Fig. 2. DNA damage, nuclear envelope rupture and reduced DNA replication in CRISPR-MN.
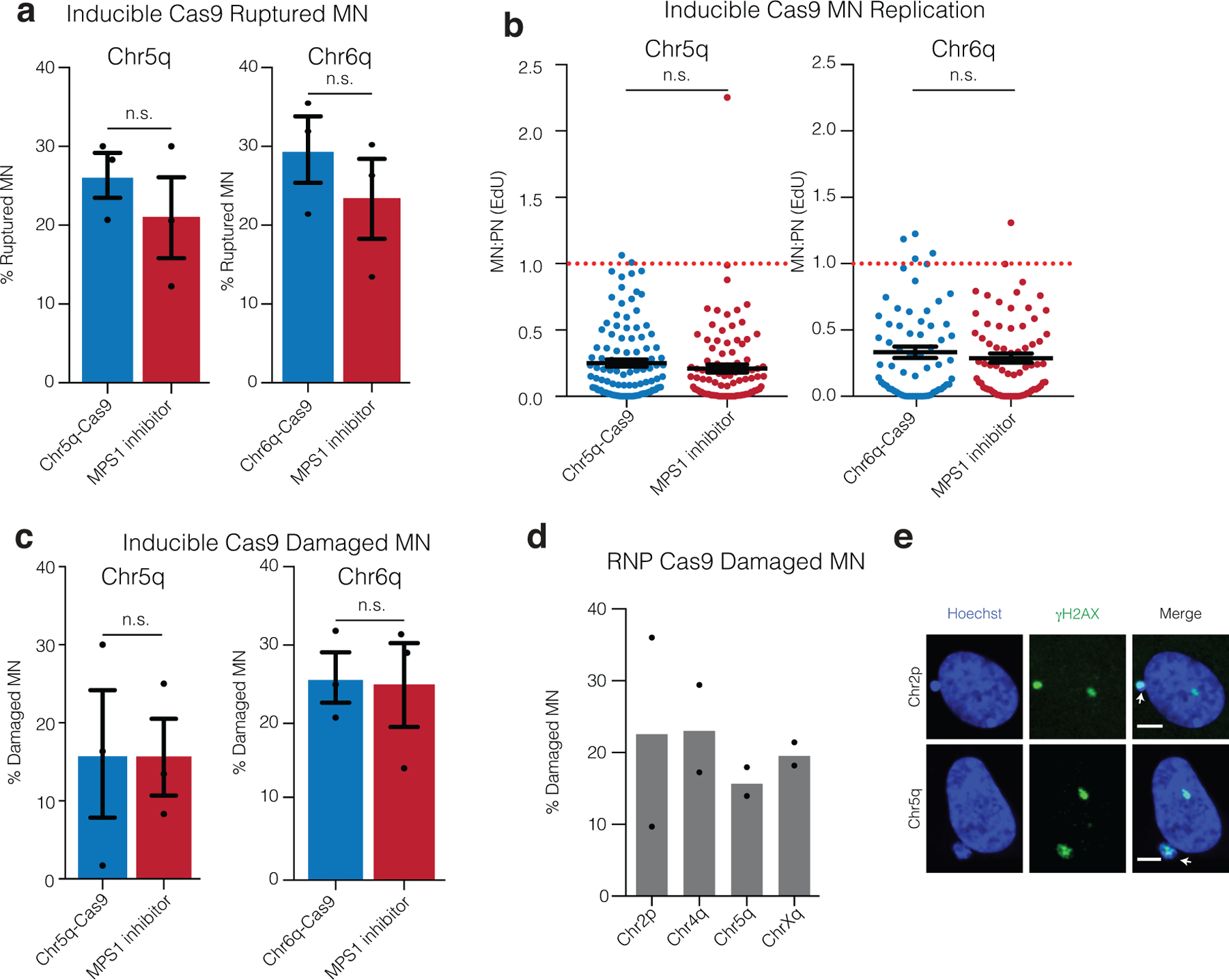
(a) Nuclear envelope rupture frequency for CRISPR-MN as compared to spindle checkpoint inhibitor-induced micronuclei. Rupture was defined as an MN:PN ratio of lamin B receptor (LBR)49 intensity > 3 (n = 3 experiments with 201 and 167 micronuclei analyzed for chr5q, p = 0.2216 and 165 and 152 micronuclei counted for chr6q, p = 0.2034). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(b) DNA replication defect of CRISPR-MN. EdU fluorescence intensity was measured after a 5-hour pulse. Only cells that had entered S-phase were scored (>150 a.u. EdU signal in primary nucleus). Dotted red line is normal levels of DNA replication in the micronucleus relative to the primary nucleus (n = 3 experiments with 109 and 97 micronucleated cells analyzed for chr5q, p = 0.1698 and 65 and 73 micronucleated cells analyzed for chr6q, p = 0.6948). Error bars: mean +/− SEM; two-tailed Mann-Whitney U-test.
(c) CRISPR-MN acquire DNA damage. Shown is the frequency of γH2AX positive micronuclei (> 3 standard deviations above mean signal in primary nuclei) for the indicated gRNAs using the inducible Cas9 system (n = 3 experiments with 203 and 184 micronucleated cells analyzed for chr5q, p = 0.6870 and 175 and 169 cells analyzed for chr6q, p = 0.8053). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
(d) CRISPR-MN acquire DNA damage (RNP Cas9 system). Shown is the frequency of γH2AX positive micronuclei for the indicated gRNAs (n = 2 experiments with 56, 46, 82, and 50 micronucleated cells analyzed, left to right).
(e) Example images of data from panel (d) showing γH2AX labeling. White arrows: micronuclei. Scale bars, 5 μm. The γH2AX focus in the primary nucleus likely decorates the centric portion of the broken chromosome. Alternatively, or additionally, it may label a DNA break on the homolog.
Extended Data Fig. 3. Haplotype copy number and SVs for the targeted chromosome for each sample in the paper.
Haplotype-resolved copy number and structural variant analysis for the targeted chromosome for each granddaughter pair. Red and blue dots represent 1 Mb copy number bins for each homolog, and curved lines represent structural variants of ≥ 1 Mb that could be on either homolog. Top, ‘granddaughter a’; middle, ‘granddaughter b’; bottom, sum copy number for each homolog for the pair of cells. Note that in most cases there should be a total of two red and two blue copies per granddaughter pair, and deviation from this represents certain missegregation or events, such as first-generation bridge formation. Copy number alterations occurring only in one daughter without a corresponding or reciprocal change in the other daughter were attributed to random noise due to variability in genome amplification quality. Text: inferred most likely explanation for each copy number and rearrangement profile. Note that alternative explanations exist for many samples, such as a G1 cut followed by replication of the cut chromosome.
Extended Data Fig. 4. Clustering of DNA breakpoints, indicative of chromothripsis, on the telomeric side of the CRISPR-Cas9-targeted cut site.
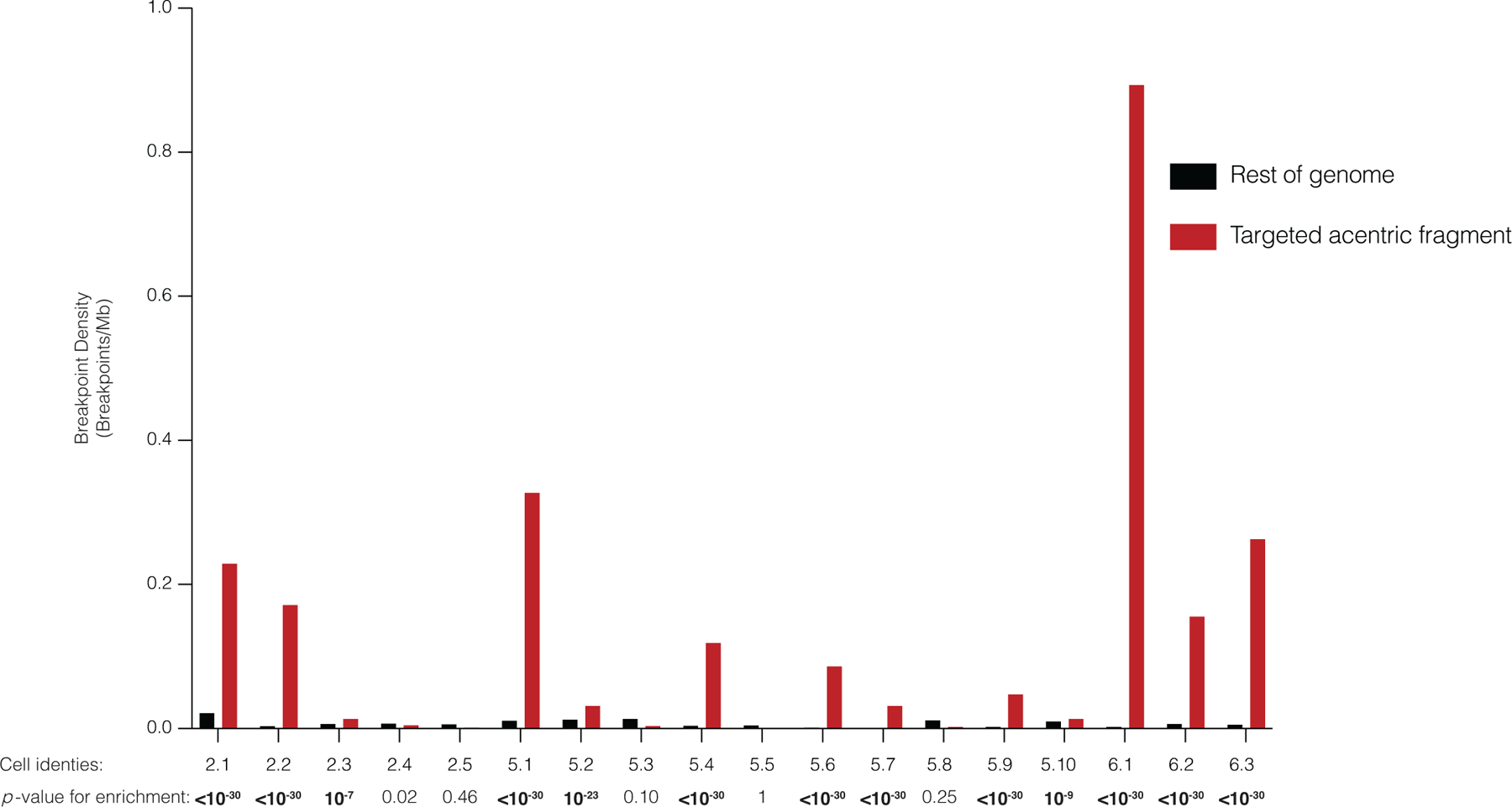
Breakpoint density for each daughter pair telomeric of the cut-site (red), relative to the rest of the genome (gray), normalized by read depth. Data include both inter- and intra-chromosomal rearrangements. Significance is derived from a one-sided Poisson test (Zhang et al., 2015). p – values are rounded to the nearest exponent, except for those <10−30. Bolded p - values denote significance after Bonferroni correction. Bonferroni-corrected a = 0.0028.
Extended Data Fig. 5. Chromosome bridge formation after CRISPR-Cas9 genome editing.

a) A bridge formed during the first cell division after Cas9 addition yields shared losses (left granddaughter pair) or gains (right granddaughter pair) depending upon how the bridge breaks. This copy number alteration will be on the centromeric side of the CRISPR-Cas9 break. Cells and chromosomes are depicted as in Fig. 3. The non-micronucleated daughter cell is faded and not followed. In this example, the micronuclear chromosome from the first division is not reincorporated and becomes a micronucleus in one granddaughter.
b) A bridge formed in the second cell division yields reciprocal copy number gains and losses centromeric of the break (comparing the granddaughters). The non-micronucleated daughter cell is faded and not followed.
c) The frequency of detectable chromosome bridges by live-cell imaging after CRISPR-Cas9 genome editing in RPE-1 cells expressing a fluorescence reporter that marks chromosome bridges efficiently (GFP-BAF). DNA breaks were induced with the Chr5q-targeting inducible Cas9 system after treatment with siRNA against TP53 or non-targeting siRNA. Chromosome bridges frequently arise when a micronucleus forms in at least one daughter cell in the first division (MN+), whereas when a micronucleus is not formed, bridge formation is uncommon (MN-). In the second division, micronucleated cells are more prone to bridge formation (MN+) as compared to non-micronucleated cells (MN-). Bridge formation is more frequent in the second division, which may be explained by isolation of the acentric arm from the centric fragment of the chromosome (p53 siRNA: n = 6 experiments with 175 and 172 cell divisions imaged [division 1] and 136 and 132 divisions imaged [division 2]; non-targeting siRNA: n = 3 experiments with 89 and 90 cell divisions imaged [division 1] and 43 and 58 divisions imaged [division 2]). Error bars: mean +/− SEM, two-tailed Fisher’s exact test.
Extended Data Fig. 6. Allele ratios of heterozygous SNPs from CD34+ HSPC colonies after editing.
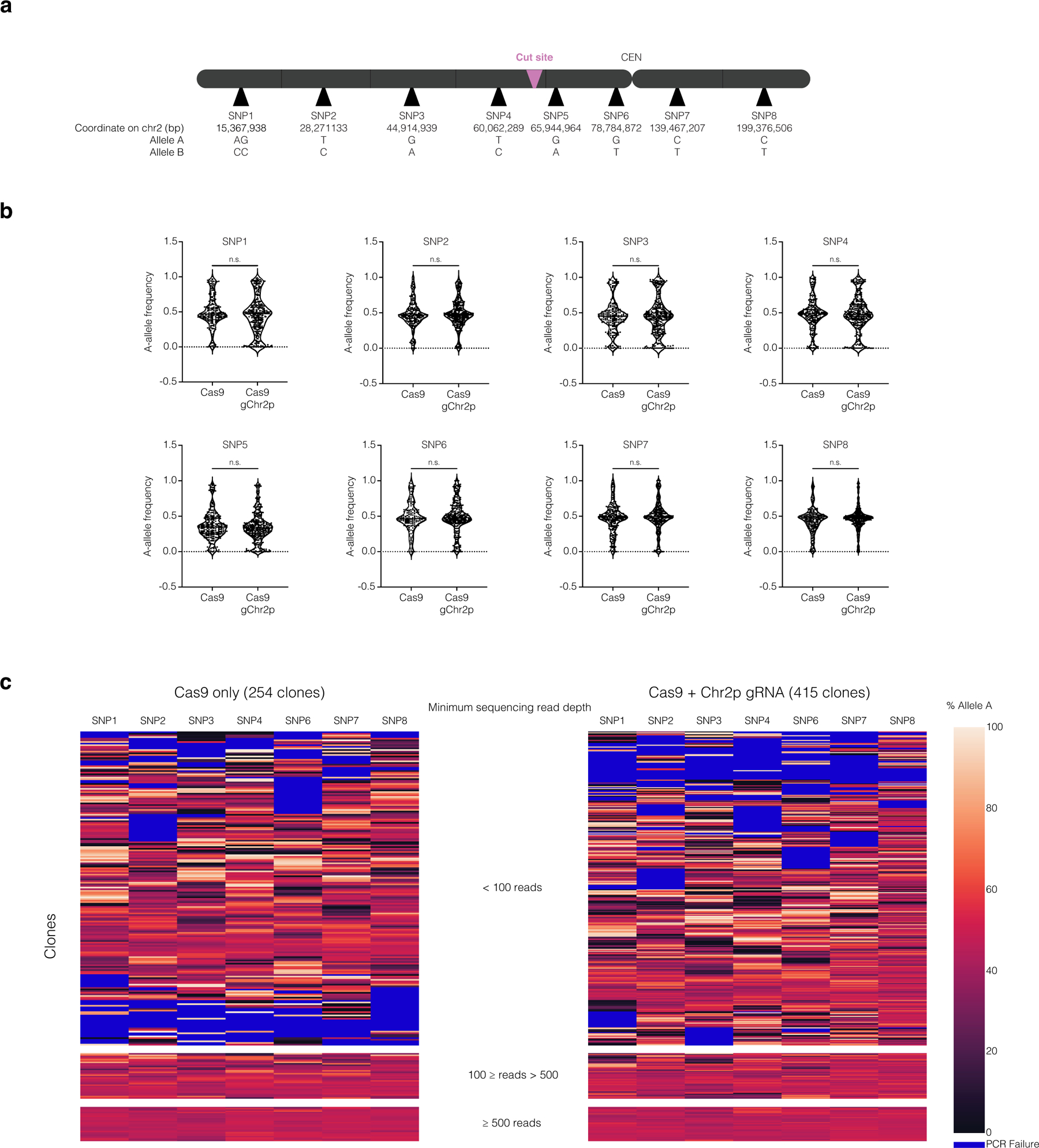
(a) Map of SNP locations, cut site, and the centromere (CEN) on chromosome 2 (not to scale).
(b) The distribution of A-allele frequencies for samples where A-allele and B-allele frequencies comprise greater than 90 % of the sequence reads. The p-values for SNPS 1–8 are p = 0.1089, 0.3140, 0.9967, 0.7792. 0.2751, 0.4659, 0.3178, and 0.2239 respectively (two-tailed Mann-Whitney U test). SNP5 exhibited a strong deviation from a 50:50 allelic ratio even in unedited controls, which may reflect a PCR amplification artifact. Because of this, SNP5 was excluded from subsequent analysis.
(c) Heatmap of allele frequency data for all samples (Cas9, left; Cas9 + Chr2p gRNA, right). The heatmap is divided into sections based on the minimum sequencing read depth. Minimum sequencing read depth was defined by the SNP with the lowest number of reads in the sample. Samples with low read depth exhibited high variability in allelic ratios, likely reflecting low input DNA from small colonies. Because we lack phasing information, any deviation from a 50:50 allele ratio for multiple adjacent SNPs suggests segmental copy number alterations.
See Supplementary Note for methods and additional discussion. For this experiment, only several hundred clones could feasibly be grown and analyzed, whereas patients will receive tens to hundreds of millions of edited cells. From the several hundred clones in our experiment, we only expect ~20 cells containing micronuclei based on micronucleation rates measured in Fig. 6. Extrapolating from these data, patients will receive millions of micronucleated cells, each one with the potential to undergo chromothripsis and grow into a clone. We note that this assay will not detect copy-number neutral chromothripsis nor chromothripsis that maintains copy number and heterozygosity at the assayed SNPs, with rearrangements located on other segments of the edited chromosome. Moreover, this approach has a limited ability to detect copy number gains or subclonal events that result from ongoing genomic instability triggered by micronucleation or bridging derived from the initial editing.
Supplementary Material
Acknowledgements
We are grateful to R. Jaenisch, S. Markoulaki, A. Spektor and members of the Pellman and Weiss laboratories for discussions, I. Cheeseman for the doxycycline-inducible Cas9 RPE-1 cell line, D. Cullins for assistance in single-cell sorting of HSPCs, and N. Mynhier for help with data visualization. This work was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE1144152 (M.L.L.), National Cancer Institute (NCI) career transition award K22CA216319 (C.-Z.Z.), Howard Hughes Medical Institute (D.P.), NIH grant R01 CA213404 (D.P.), F32 DK118822 (P.A.D), NIH grant P01 HL053749 (M.J.W.), the Assisi Foundation (M.J.W.), the Doris Duke Charitable Foundation (M.J.W.), and St. Jude/ALSAC. The St. Jude Cytogenetic and Center for Advanced Genome Engineering Shared Resource Laboratories are supported by NIH grant P30 CA21765 and by St. Jude/ALSAC. We would like to thank the members of the St. Jude Children’s Research Hospital Center for Advanced Genome Engineering and Cytogenetics core facilities.
Footnotes
Competing interests
M.J.W. is a consultant for Rubius Inc., Cellarity Inc., Beam Therapeutics, and Esperion; none of the consulting work is relevant to the current project. C.-Z. Z. is a scientific adviser for Pillar BioSciences. D.P. is a member of the Volastra Therapeutics scientific advisory board. All other authors declare no competing interests.
Data availability
Whole images of cells presented in Figs. 1f, 6e,i, and Extended Data Fig. 2e, and filtered SV calls are available under doi:10.5281/zenodo.4533300. Source data for Figs. 1c–e, 4, 6a–d,f,h and Extended Data Figs. 1b–f,h–j, 2a–d, 4, 5c, and 6 are provided with the paper, including the unprocessed Western blot from Extended Fig. 1c. Original images and videos that contribute to analyses in Figs. 1e, 4, 6c,h, Extended Data Figs. 1d, 2, 5c, and Look-Seq experiments (Figures 2, 3, 5, Extended Data Fig. 3) are not published due to constraints of file size, but are available upon reasonable request. CD34+ HSPC-derived FISH and SKY images and analyses (Fig. 6d–g) were generated by The St. Jude Cytogenetic Shared Resource Laboratory and derived data supporting the findings in Fig. 6d–g are available from the corresponding author upon request. Sequence read data are available in the Sequencing Read Archive (SRA) under Bioproject PRJNA676146.
Code availability
Scripts used for sequencing data analysis (allelic copy-number calculation and rearrangement detection), and for image analyses performed in Extended Data Fig. 2 are available at https://github.com/chengzhongzhangDFCI/CN_and_SV.
References
- 1.Hsu PD, Lander ES & Zhang F Development and applications of CRISPR-Cas9 for genome engineering. Cell 157, 1262–78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doudna JA The promise and challenge of therapeutic genome editing. Nature 578, 229–236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu J et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science 334, 993–6 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orkin SH & Bauer DE Emerging Genetic Therapy for Sickle Cell Disease. Annu Rev Med 70, 257–271 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Wu Y et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat Med 25, 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frangoul H et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N Engl J Med 384, 252–260 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Dever DP et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeWitt MA et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med 8, 360ra134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Richardson CD et al. CRISPR-Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat Genet 50, 1132–1139 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Romero Z et al. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol Ther 27, 1389–1406 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim D, Luk K, Wolfe SA & Kim JS Evaluating and Enhancing Target Specificity of Gene-Editing Nucleases and Deaminases. Annu Rev Biochem 88, 191–220 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Haapaniemi E, Botla S, Persson J, Schmierer B & Taipale J CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med 24, 927–930 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24, 939–946 (2018). [DOI] [PubMed] [Google Scholar]
- 17.van den Berg J et al. A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res 46, 10132–10144 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enache OM et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat Genet (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitworth KM et al. Use of the CRISPR/Cas9 system to produce genetically engineered pigs from in vitro-derived oocytes and embryos. Biol Reprod 91, 78 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin HY et al. CRISPR/Cas9 targeting events cause complex deletions and insertions at 17 sites in the mouse genome. Nat Commun 8, 15464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adikusuma F et al. Large deletions induced by Cas9 cleavage. Nature 560, E8–E9 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Kosicki M, Tomberg K & Bradley A Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol 36, 765–771 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zuccaro MV et al. Allele-Specific Chromosome Removal after Cas9 Cleavage in Human Embryos. Cell (2020). [DOI] [PubMed] [Google Scholar]
- 24.Weisheit I et al. Detection of Deleterious On-Target Effects after HDR-Mediated CRISPR Editing. Cell Rep 31, 107689 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Alanis-Lobato G et al. Frequent loss-of-heterozygosity in CRISPR-Cas9-edited early human embryos. bioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cullot G et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun 10, 1136 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stadtmauer EA et al. CRISPR-engineered T cells in patients with refractory cancer. Science 367(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang CZ et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umbreit NT et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 368(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu P et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146, 889–903 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kloosterman WP & Cuppen E Chromothripsis in congenital disorders and cancer: similarities and differences. Curr Opin Cell Biol 25, 341–8 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Stephens PJ et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rausch T et al. Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ly P et al. Chromosome segregation errors generate a diverse spectrum of simple and complex genomic rearrangements. Nat Genet 51, 705–715 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium ITP-CA o.W.G. Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortes-Ciriano I et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat Genet 52, 331–341 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leibowitz ML, Zhang CZ & Pellman D Chromothripsis: A New Mechanism for Rapid Karyotype Evolution. Annu Rev Genet 49, 183–211 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Ly P & Cleveland DW Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol 27, 917–930 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soto M, Garcia-Santisteban I, Krenning L, Medema RH & Raaijmakers JA Chromosomes trapped in micronuclei are liable to segregation errors. J Cell Sci 131(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKinley KL & Cheeseman IM Large-Scale Analysis of CRISPR/Cas9 Cell-Cycle Knockouts Reveals the Diversity of p53-Dependent Responses to Cell-Cycle Defects. Dev Cell 40, 405–420 e2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brinkman EK et al. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol Cell 70, 801–813 e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu J, Tang B & Tang Y Allele-specific genome targeting in the development of precision medicine. Theranostics 10, 3118–3137 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stark JM & Jasin M Extensive loss of heterozygosity is suppressed during homologous repair of chromosomal breaks. Mol Cell Biol 23, 733–43 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rao PN, Johnson RT & Sperling K Premature chromosome condensation : application in basic, clinical, and mutation research, xvi, 381 p. (Academic Press, New York, 1982). [Google Scholar]
- 46.Hoffelder DR et al. Resolution of anaphase bridges in cancer cells. Chromosoma 112, 389–97 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Terradas M, Martin M, Tusell L & Genesca A DNA lesions sequestered in micronuclei induce a local defective-damage response. DNA Repair (Amst) 8, 1225–34 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Crasta K et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatch EM, Fischer AH, Deerinck TJ & Hetzer MW Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ly P et al. Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19, 68–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S et al. Nuclear envelope assembly defects link mitotic errors to chromothripsis. Nature 561, 551–555 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kneissig M et al. Micronuclei-based model system reveals functional consequences of chromothripsis in human cells. Elife 8(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Priestley P et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikeda K et al. Efficient scarless genome editing in human pluripotent stem cells. Nat Methods 15, 1045–1047 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Liang D et al. Frequent gene conversion in human embryos induced by double strand breaks. bioRxiv (2020). [Google Scholar]
- 56.Korbel JO & Campbell PJ Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–36 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Vazquez-Diez C, Yamagata K, Trivedi S, Haverfield J & FitzHarris G Micronucleus formation causes perpetual unilateral chromosome inheritance in mouse embryos. Proc Natl Acad Sci U S A 113, 626–31 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minocherhomji S et al. Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–90 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Cleal K, Jones RE, Grimstead JW, Hendrickson EA & Baird DM Chromothripsis during telomere crisis is independent of NHEJ, and consistent with a replicative origin. Genome Res 29, 737–749 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maciejowski J, Li Y, Bosco N, Campbell PJ & de Lange T Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maciejowski J et al. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat Genet 52, 884–890 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ribeyre C & Shore D Regulation of telomere addition at DNA double-strand breaks. Chromosoma 122, 159–73 (2013). [DOI] [PubMed] [Google Scholar]
- 63.Maciejowski J & de Lange T Telomeres in cancer: tumour suppression and genome instability. Nat Rev Mol Cell Biol 18, 175–186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Canela A et al. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell 63, 898–911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McClintock B The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 26, 234–82 (1941). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Campbell PJ et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–13 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 508, 98–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ma H et al. Correction of a pathogenic gene mutation in human embryos. Nature 548, 413–419 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Egli D et al. Inter-homologue repair in fertilized human eggs? Nature 560, E5–E7 (2018). [DOI] [PubMed] [Google Scholar]
- 70.Finn JD et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep 22, 2227–2235 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Humbert O, Peterson CW, Norgaard ZK, Radtke S & Kiem HP A Nonhuman Primate Transplantation Model to Evaluate Hematopoietic Stem Cell Gene Editing Strategies for beta-Hemoglobinopathies. Mol Ther Methods Clin Dev 8, 75–86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Humbert O et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci Transl Med 11(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Demirci S et al. BCL11A enhancer-edited hematopoietic stem cells persist in rhesus monkeys without toxicity. J Clin Invest (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lu Y et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med 26, 732–740 (2020). [DOI] [PubMed] [Google Scholar]
- 75.Luc S et al. Bcl11a Deficiency Leads to Hematopoietic Stem Cell Defects with an Aging-like Phenotype. Cell Rep 16, 3181–3194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanders AD et al. Single-cell analysis of structural variations and complex rearrangements with tri-channel processing. Nat Biotechnol 38, 343–354 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McDermott DH et al. Chromothriptic cure of WHIM syndrome. Cell 160, 686–699 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Maeder ML et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat Med 25, 229–233 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Lomova A et al. Improving Gene Editing Outcomes in Human Hematopoietic Stem and Progenitor Cells by Temporal Control of DNA Repair. Stem Cells 37, 284–294 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Metais JY et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood Adv 3, 3379–3392 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weber L et al. Editing a gamma-globin repressor binding site restores fetal hemoglobin synthesis and corrects the sickle cell disease phenotype. Sci Adv 6, eaay9392 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Howden SE et al. A Cas9 Variant for Efficient Generation of Indel-Free Knockin or Gene-Corrected Human Pluripotent Stem Cells. Stem Cell Reports 7, 508–517 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Connelly JP & Pruett-Miller SM CRIS.py: A Versatile and High-throughput Analysis Program for CRISPR-based Genome Editing. Sci Rep 9, 4194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.