

Abstract
Metabolic disorders result from dysregulation of central nervous system and peripheral metabolic energy homeostatic pathways. To maintain normal energy balance, neural circuits must integrate feedforward and feedback signals from the internal metabolic environment to orchestrate proper food intake and energy expenditure. These signals include conserved meal and adipocyte cues such as glucose and leptin, respectively, in addition to more novel players including brain-derived neurotrophic factor (BDNF). In particular, BDNF’s two receptors, tropomoysin related kinase B (TrkB) and p75 neurotrophin receptor (p75NTR), are increasingly appreciated to be involved in whole body energy homeostasis. At times, these two receptors even seem to functionally oppose one another’s actions, providing the framework for a potential neurotrophin mediated energy regulatory axis, which we explore further here.
Keywords: metabolism, neurotrophins, hypothalamus, BDNF, p75NTR, TrkB
Connections Between Body Weight Homeostasis and Neurotrophins
Determinants of body weight such as food intake and energy expenditure are under tight homeostatic control. Insufficient or excess energy states can lead to detrimental effects, including starvation and obesity, respectively. To avoid these consequences, organisms must strongly regulate their food intake to correspond to their energy output [1]. Moreover, they must be responsive to changes in their external energy environment, appropriately recognizing and compensating for changes in food type or availability, or in locomotor activity and thermoregulatory control [2]. Many of the mechanisms through which organisms adapt to their environment exist within the central nervous system, largely in the hypothalamus, and are in constant communication with feedforward and feedback mechanisms from peripheral metabolic organs [3]. How each of these anatomic sites function independently and in combination, especially in response to changing environments, remain active areas of investigation.
One emerging mediator of energy homeostasis are neurotrophin family members and their receptors. Neurotrophins are canonically recognized as molecules critical in the control of neurodevelopment, and in the maintenance of neuronal plasticity [4]. The neurotrophin family consists of four structurally related growth factors: nerve growth factor (NGF), brain derived neurotrophic factor (BDNF), neurotrophin 3 (NT-3), and neurotrophin 4/5 (NT-4/5) [5]. Each of these neurotrophins binds to a specific cognate tyrosine receptor kinase (Trk A, B, or C) and all can bind to the more promiscuous p75 neurotrophin receptor (p75NTR) [6–8]. The scope of study of these neurotrophins and their receptors is both broad and deep, and we refer the interested reader to several excellent reviews that cover myriad functions of these molecules [8–10]. Here, we will focus on the relation of neurotrophins and their receptors in the control of whole organism energy metabolism, in particular the relationship between BDNF and its two receptors, TrkB and p75NTR. For more in-depth coverage of the biochemical pathways involved in these processes, we suggest other reviews on the topic [11,12]. BDNF, TrkB, and p75NTR have been implicated in several aspects of metabolism, with roles in energy intake and expenditure in both the central nervous system and peripheral metabolic organs [11,12]. Of note, data suggests these roles are important during development and in the adult state, though the implications of malfunctions at specific times have not been elucidated. Notably, TrkB largely appears to act anorexigenically, opposing weight gain, whereas p75NTR promotes weight gain on a high fat diet and food consumption after a fast [13–15]. We suggest this functionally antagonistic dynamic of TrkB and p75NTR position them as molecular/trophic rheostats that may be well suited to regulate a range of energy homeostatic processes.
BDNF Mediated Control Of Energy Balance Is Clinically Significant
Following initial studies in mice (Box 1), clinical investigations have identified critical roles for BDNF and TrkB in human energy homeostasis. The first of these was identified in 2004, in a young male who presented with severe obesity due to hyperphagia, along with impairments in short term memory and nociception[16]. Sequencing the functional exons of NTRK2, the gene encoding TrkB, revealed a specific partial loss-of-function mutation in the activation loop of the catalytic domain of the receptor (Y722C), part of the intrinsic activation mechanism of TrkB [16]. Subsequent mutations in other individuals with obesity have also been identified, including ones within the leucine rich repeat domain in the extracellular portion of TrkB (I98V, P204H), within the tyrosine kinase domain (P660L, R691H, R696K, S714F, R715Q, R715W), or near the SH2 interaction site of PLCγ (T821A, P831L) [17,18]. Of note, only the mutations in the catalytic domain have been conclusively shown to alter downstream TrkB signaling properties, leaving open the question of whether and how other mutations of TrkB may promote obesity [17]. One intriguing possibility yet to be tested lies in whether these mutations alter interactions with co-receptors of TrkB, including p75NTR [19]. These mutations might reveal novel regulatory dynamics of TrkB and/ or p75NTR function, especially in response to changing environmental states, and provide improved understanding of the clinical significance of these mutations.
Box 1. First Reports of Neurotrophins In Body Weight Homeostasis.
BDNF was first purified from mammalian brains in 1982 by Yves-Alain Barde and Hans Thoenen, and was shown to have a growth promoting function on sensory neurons [79]. Subsequent work has identified roles for BDNF in learning and memory, synaptic transmission and plasticity, and several neurodegenerative and neuropsychiatric disorders [80]. Since the 1990s, BDNF has been shown to have additional roles in the central regulation of feeding. While investigating BDNF as a potential therapy for Alzheimer’s, rats treated with BDNF by intracerebroventricular (icv) infusion appeared hypophagic, with weight loss of 10–15% over two weeks [81,82]. Notably, when tested in mouse models of obesity, icv BDNF retained these anorexigenic effects, significantly reducing food intake in both obese high fat diet fed mice and leptin receptor deficient db/db mice [48,49].
These data have since been confirmed and extended through at least two independently generated models of BDNF deficient mice. Among mice heterozygous for BDNF (as homozygotes are embryonically lethal), half develop severe, late onset, hyperphagic obesity [59]. Notably, body weight can be reduced (by ~30%) to wild type levels following chronic central administration of BDNF in adult BDNF heterozygous mice [59]. Importantly, these data demonstrate that BDNF deficiency is modifiable in the adult, and suggest that the underlying role of BDNF in body weight regulation is not solely due to a defect during development, but rather a necessary role in adult metabolic regulation. However, there may be additional developmental and environmental effects, given the heterogeneity in development of obesity in the BDNF heterozygous mice.
Many of these metabolic roles are mediated via BDNF binding to at least one of its receptors, which have been identified as TrkB and p75NTR [8]. While germline TrkB knockout mice are non-viable past day 1 of life due to a lack of feeding, TrkB hypomorphs with a near 75% knockdown of TrkB were found to be obese due to increased food intake [15,83]. Further evidence for the importance of TrkB in feeding comes from the fact that either TrkB ligand, BDNF or NT-4/5, can reduce weight by nearly 30% in obese BDNF heterozygous mice, and indeed NT-4/5 can also reduce body weight in diet-induced obese mice and in db/db mice [84]. This provides strong evidence that TrkB signaling, even independent of BDNF, is likely a key driver of energy homeostasis.
These findings in patients with TrkB mutations prompted a subsequent study to identify whether mutations in BDNF alter body weight in humans. One case report had laid the groundwork for this possibility, identifying a chromosomal inversion of a region involving the BDNF gene that led to hyperphagia induced obesity [20]. More conclusive evidence of BDNF’s involvement in human obesity emerged from follow up studies of patients with WAGR syndrome, caused by contiguous deletions on chromosome 11p of varying size. In one notable study, all patients with a partial chromosome 11p deletion that also functionally altered BDNF were clinically obese, compared to only 20% of patients with a partial chromosome 11p deletion where BDNF was intact [21]. This work demonstrated a strong association with BDNF gene deletion and obesity in this cohort [21]. Further studies have reinforced this finding, identifying individual SNPs in BDNF that are associated with increased risk of obesity [22–24].Several other diseases of obesity, including Prader-Willi syndrome (PWS; due to loss of paternally expressed genes on chromosome 15q) and Smith-Magenis syndrome (SMS; due to deletion of chromosome 17p11.2), also have reduced levels of BDNF expression [25,26]. While it is unknown how these syndromes lead to obesity, it has been suggested that they may broadly alter gene expression through loss of the small nucleolar RNA SNORD116 (in PWS), or the transcription factor RAI1 (in SMS) [27,28]. Some have proposed that downregulation of BDNF induced by loss of these factors may perturb normal hypothalamic development, which in turn could underlie the observed metabolic phenotypes [27,29]. Further work is needed to clarify the level of involvement of BDNF in these syndromes.
Taken together, there is now clear evidence of a critical function of human BDNF and TrkB in body weight and energy regulation. Work has also begun to elucidate roles for BDNF signaling in weight loss success on the Mediterranean diet, and in levels of food craving [30,31]. Less clear is the involvement of p75NTR in human energy metabolism. While one genome wide association study identified a polymorphism of p75NTR in human height regulation, there are no case reports of mutations of p75NTR, nor its involvement in human weight regulation [32]. This is in line with mouse phenotypes, where even homozygous deletion of p75NTR leads to a very limited set of obvious deficits. Instead, it is seemingly only a combination of genetic andenvironmental perturbations, such as energy excess or deficit, that has revealed a homeostatic role for p75NTR in rodent models. Given this, it is possible that p75NTR may play an as yet unidentified role in complex polygenic or gene:environment disorders. Indeed, there has been previous suspicion that p75NTR, in addition to BDNF, may contribute towards the pathophysiology of Prader-Willi syndrome [33]. This syndrome also involves loss-of-expression of genes on chromosome 15, including necdin and necdin-like proteins, and among other effects leads to obesity and hypothalamic changes in human patients [34]. Notably, p75NTR interacts with necdin and related proteins, and alteration of their binding has been shown to alter cell viability.p75NTR has also been shown to be robustly expressed in the human hypothalamus, including the paraventricular and mediobasal regions [35,36]. This leaves open the question of whether p75NTR may play a role in the hypothalamic and phenotypic changes observed in Prader-Willi syndrome, or in other complex disorders of energy balance.
Neurotrophins Control Feeding Throughout the Hypothalamus
It is increasingly recognized that the anatomic origins of many metabolic disorders lie in the brain, specifically within the hypothalamus. This is no exception for the obesity caused by mutations in BDNF and TrkB, nor for the altered feeding of p75NTR mutants. Figure 1 shows schematically, and Table 1 lists, each of the regions of the hypothalamus and the relative expression of BDNF and its receptors according to in situ hybridization data from the Allen Brain Atlas of cells in a given region. Of the regions listed, neurons of the dorsomedial (DMH), ventromedial (VMH), paraventricular (PVH) and arcuate (ARH) hypothalamic regions have been most extensively characterized. Interestingly, other neurotrophins, including TrkC and NT4, can be found in some hypothalamic regions, though their functions in metabolism have gone largely unexplored (Box 2).
Figure 1: Schematic representation of key hypothalamic regions and the expression of BDNF, p75NTR, and TrkB.
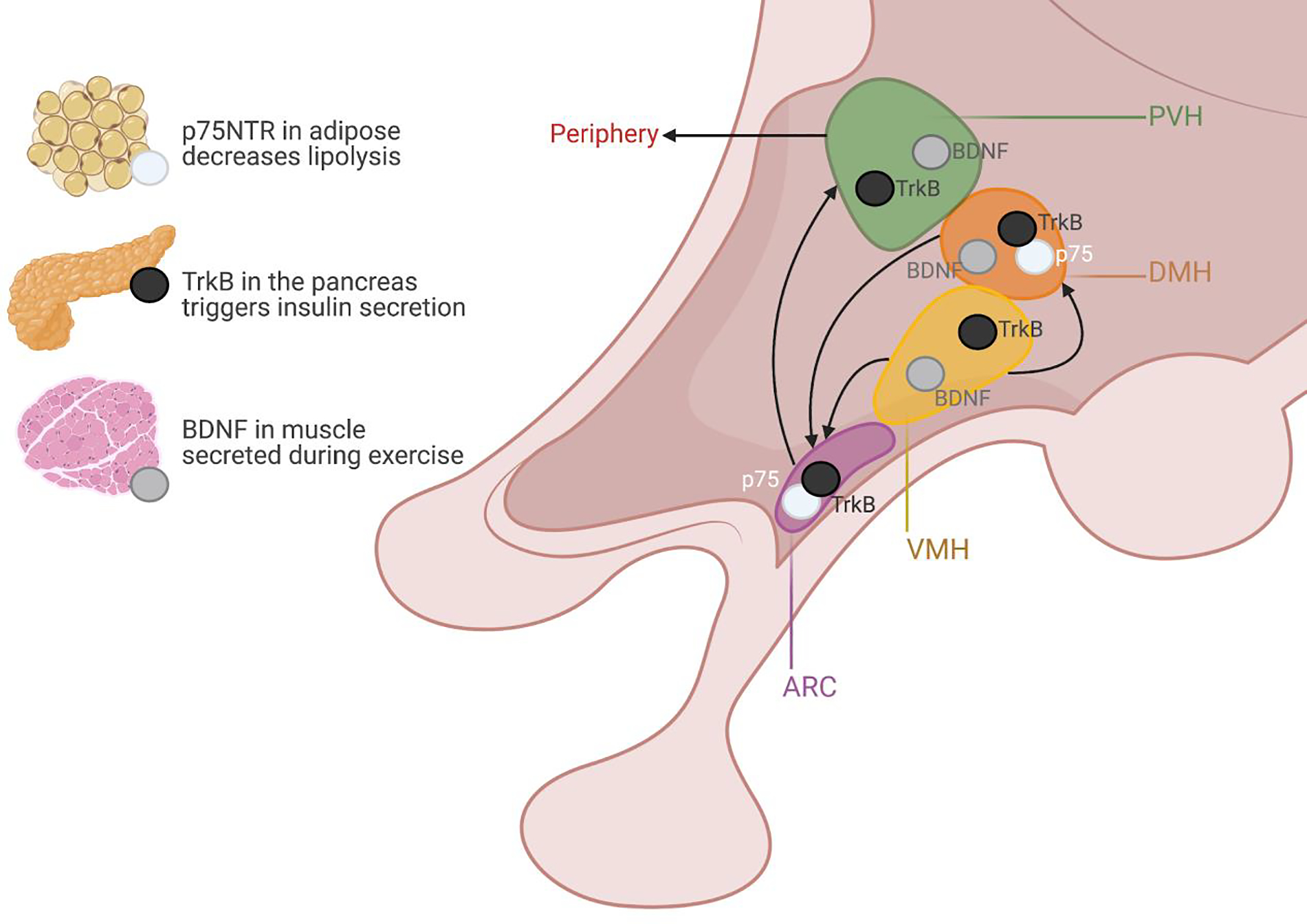
Arrows represent general connectivity between regions, and do not specify neurotrophin specific connections. Whether p75NTR and TrkB exist as independent or overlapping cell populations is unknown.
Table 1:
Hypothalamic expression of BDNF and it’s receptors*
| NGFR (p75NTR) | NTRK2 (TrkB) | BDNF | Refs | |
|---|---|---|---|---|
| Paraventricular hypothalamic nucleus (PVH) | - | Moderate | Moderate | TrkB: [42] BDNF: [43,46] |
| Periventricular hypothalamic nucleus (PV) | - | Low | - | |
| Arcuate hypothalamic nucleus (ARC) | Low | Moderate | Not expressed in ARC, but has been shown to alter ARC neuron structure | p75NTR: [14] BDNF: [46] |
| Anteroventral periventricular nucleus (AVPV) | - | Moderate | High | |
| Dorsomedial nucleus of the hypothalamus (DMH) | Moderate to Low | Moderate | Moderate | TrkB: [40] BDNF: [37] |
| Medial preoptic area (MPO) | - | Low | High | BDNF: [44] |
| Periventricular hypothalamic nucleus, posterior part (PVp) | Moderate | Moderate | - | |
| Suprachiasmatic nucleus (SCH) | - | Moderate | - | |
| Mammillary body (MBO) | - | - | Moderate to Low | |
| Ventral premammillary nucleus (PMv) | Moderate | Moderate | - | |
| Ventromedial hypothalamic nucleus (VMH) | - | Low | Moderate | BDNF: [37] |
| Posterior hypothalamic nucleus (PH) | - | Low | Moderate | |
| Lateral hypothalamic area (LHA) | - | Low | Low | |
| Parasubthalamic nucleus (PSTN) | Low | - | Low | |
| Retrochiasmatic area (RCH) | - | Moderate | - | |
| Tuberal nucleus (TU) | Moderate to Low | Moderate | - | |
| Zona incerta (ZI) | - | Low | - |
NGFR- nerve growth factor receptor, gene name for p75NTR. NTRK2- neurotrophin receptor tyrosine kinase 2, gene name for TrkB. (-) indicates expression not observed.
Box 2. More Than Just p75NTR and TrkB.
Beyond p75NTR and TrkB, other neurotrophin receptors have been implicated in aspects of metabolic homeostasis (Table I). A family-based association study of neurotrophin genes has suggested that TrkC, in addition to BDNF and TrkB, may be involved in the heritability of eating disorders including bulimia and anorexia nervosa [85]. These data, coupled with our emerging understanding of the varied metabolic roles of neurotrophins, may suggest that further study of neurotrophins in eating disorders may reveal valuable insights into their pathogenesis. Further, expression of TrkA, one of the receptors for NGF, has been demonstrated to be modifiable by glucose levels, and to interact with the insulin receptor in vitro [86,87]. Later work has since identified that NGF-TrkA activation is necessary for glucose-induced insulin secretion, with loss of TrkA leading to decreased insulin secretion and impaired glucose tolerance [88]. Further, TrkA may be of interest in regulating centrally mediated food intake, as early investigations of NGF as a potential therapy for Alzheimer’s led to gaunt and hypophagic rats [89]. It is unclear, however, whether this NGF effect is mediated via TrkA. These studies hint at further undiscovered roles for neurotrophin receptors in metabolic processes, and more work is needed to understand the mechanisms by which p75NTR and the Trk receptors may function in energy balance.
Table I:
Summary of Neurotrophins' Metabolic Roles
| NGF (TrkA, p75NTR) | BDNF (TrkB, p75NTR) | NT3 (TrkC, p75NTR) | NT4 (TrkB, p75NTR) | Refs | |
|---|---|---|---|---|---|
| Central administration | Reduces feeding | Reduces feeding | Unknown | Reduces Feeding | NGF: [82,89] BDNF: [82] NT4: [59] |
| Mouse mutant | Unknown | Obese and hyperphagic (same in humans) | Normal weight | Normal weight | BDNF: [59], [21] NT3: [59] NT4: [59] |
| TrkA (NGF) | TrkB (BDNF, NT4) | TrkC (NT3) | p75NTR (NGF, BDNF, NT3, NT4) | ||
| Human mutant | Unknown | Obese and hyperphagic | Association with eating disorders | Unknown | TrkB: [16] TrkC: [85] |
| Mouse mutant | Impaired insulin secretion and glucose tolerance | Obese and hyperphagic | Normal weight | Resistance to weight gain on HFD; decreased refeeding after fasting | TrkA: [88] TrkB: [15] TrkC: [59] p75NTR: [13,14] |
Dorsomedial/ ventromedial hypothalamus
Among the highest expressing hypothalamic regions for BDNF is the VMH. Early reports showed that BDNF expression in the VMH could be upregulated by meal signals such as glucose and leptin, and downregulated in response to fasting [15,37,38]. It was also demonstrated that VMH BDNF expression was regulated downstream of activation of the melanocortin-4-receptor (MC4R) [15]. Of note, whether this regulation of BDNF signaling is happening (1) as a cell autonomous event, whereby intracellular signaling of glucose, leptin, or MC4R is directly altering BDNF expression, or (2) whether this is a circuit level effect, whereby BDNF expressing neurons are activated or inhibited by an upstream projection, is an open question [38]. However, these 2 possibilities are not mutually exclusive, and regulation is likely multifaceted.
Based on these preliminary findings, cre lox mediated deletion of BDNF in the VMH and DMH in adult mice was then achieved, resulting in hyperphagia and obesity with normal locomotion, and revealing for the first time a critical site for BDNF expression [37]. Interestingly, there was no effect on body weight when BDNF was deleted from the VMH during embryogenesis, suggesting developmental compensation in hypothalamic wiring [39]. Follow-up work has subsequently investigated the role of DMH TrkB, finding its deletion similarly promotes hyperphagia and obesity, but with decreased locomotor activity and energy expenditure [40]. These data suggest that DMH TrkB may receive inputs from BDNF beyond the VMH/DMH that control locomotion. While DMH expression of p75NTR is among the highest of the hypothalamus, a functional role for p75NTR there has yet to be defined.
Paraventricular hypothalamus
In response to BDNF administration to the hypothalamic third ventricle, neurons of the PVH showed among the strongest induction of neural activity, and BDNF microinjection directly to the PVH led to significantly decreased food intake and body weight [37,41]. It was later shown that TrkB is highly expressed in the PVH, and its deletion either through Sim1-cre or adult stereotactic excision led to hyperphagic obesity with decreased locomotor activity [42]. While these PVH-TrkB neurons were shown to project to several brain regions, deletion of TrkB in only those neurons that project to the VMH similarly leads to hyperphagia and obesity [42]. This is noteworthy as it may suggest a means for BDNF produced locally in the VMH to act on TrkB-expressing axons that originated from the PVH, though it remains unknown precisely how BDNF acts at long distances on TrkB bearing neurons.
BDNF is also expressed in the PVH, and its deletion via Sim1-cre or stereotactic viral injection leads to hyperphagia and obesity, along with reduced energy expenditure and activity [43]. Of note, BDNF neurons of the posterior PVH were shown to project polysynaptically to brown adipose tissue, likely contributing towards its influence over thermogenic behaviors [43]. BDNF expressing neurons of the preoptic area have similarly been demonstrated to be involved in temperature regulation [44]. More recent work has also identified these PVH-BDNF neurons as essential players in leptin stimulated innervation of adipose depots, and show that they receive direct inputs from agouti related peptide (AgRP) neurons of the ARC [45].
Arcuate hypothalamus
While not as broadly characterized with respect to BDNF and TrkB, there is evidence that the arcuate hypothalamus may be a critical site of their function. Loss of the 3’ UTR BDNF mRNA leads to alterations of the projection patterns of AgRP and proopiomelanocortin (POMC) expressing neurons to the PVH and DMH, respectively, as well as an altered balance of excitatory and inhibitory inputs to these cell types [46]. This is similar to what is known about hypothalamic circuits in the leptin deficient ob/ob mouse model, and may partially underlie the feeding defects observed in mouse models of BDNF deficiency [47]. Further, TrkB has been shown to be expressed in the ARH in both known and novel cell types, but its function there is unknown [46]. We have recently demonstrated that p75NTR is also expressed in the ARC, and that it functions within AgRP neurons to promote feeding behavior following a fast [14]. It is intriguing to speculate that p75NTR and TrkB may even be expressed in some of the same cells within the ARC, and may function as part of a BDNF signaling axis to dynamically regulate neuron activity in response to changing energy states.
Metabolic Functions Beyond Feeding are Influenced by Neurotrophin Signaling
Glucose and insulin homeostasis
While much has been reported about neurotrophins and their receptors in control of feeding and body weight, they also appear to have independent functions in other metabolic systems. For example, BDNF administration to pair-fed diet induced obese mice reveals an improvement in glucose tolerance compared to vehicle treated pair-fed controls [48]. Similarly, pair-feeding and BDNF administration also reduce serum glucose and insulin in obese db/db mice [49]. These results suggest that BDNF administration itself can improve glucose metabolism, independent of its anorexigenic effect. Of note, peripheral glucose administration also causes a rapid increase of BDNF transcript levels in regions of the hypothalamus, suggesting that BDNF may serve as an important feedback molecule during hyperglycemia [37]. In addition to central regulation of glucose metabolism, more recent work has defined a role for BDNF to act on pancreatic-islet-expressedTrkB to promote peripheral insulin secretion [50]. It was further identified that the source of BDNF that acts on the pancreas may be skeletal muscle, suggesting that promotion of BDNF following exercise may be responsible for normalizing hyperglycemia [50].
In addition to BDNF and TrkB, the first report of p75NTR in a metabolic process documented its involvement in glucose homeostasis and insulin sensitivity. Whereas BDNF and TrkB deficient mice showed impaired glucoregulation, loss of p75NTR led to improvements of glucose and insulin tolerance [51]. In contrast to pancreatic control, these improvements were suggested to be mediated via adipocytes and skeletal muscle, with loss of p75NTR increasing insulin stimulated glucose uptake specifically in those tissues, and hence reducing serum glucose concentrations [51]. When p75NTR was conditionally deleted in adipocytes using the fabp4-cre, a similar phenotype of increased glucose homeostasis on high fat diet was observed, though this may have been secondary to reduced weight gain on a high fat diet compared to controls [13]. These data should be interpreted with caution, however, as the Fabp4-cre is known to produce ectopic expression in non-adipose tissues, including, significantly, the brain. This is made most clear in a report attempting to knockdown TrkB in adipocytes, where Fabp4-cre mediated TrkB knockout mice were reported to be obese, hyperphagic, and hyperaggressive- roles presumed to be centrally mediated [52]. Adipose TrkB deletion using the Adipoq-cre line, however, did not lead to hyperphagia or obesity, nor did it significantly affect glucose homeostasis, suggesting a limited role of TrkB in adipocyte regulated glucose homeostasis [52]. These data imply a more significant role for p75NTR in adipocytes than TrkB, but leave open the question of whether p75NTR may regulate metabolic processes in other peripheral organs where TrkB functions, including the pancreas.
Energy expenditure and Thermogenesis
While hyperphagic, the obesity observed in BDNF and TrkB deficient mouse models could also be a result of changes in energy expenditure. However, long term pair-feeding studies of BDNF heterozygotes reveal that they maintain body weight similar to controls with intact BDNF, suggesting they have normal baseline energy expenditure [53]. Despite this, acute administration of BDNF in obese db/db mice increases oxygen consumption and body temperature [54,55]. These data suggest that BDNF can increase energy expenditure and thermogenesis, and would align with the general anorexigenic functions previously reported. Recent work has also suggested a role for BDNF expressed from neurons of the lateral hypothalamus in thermogenic regulation [56]. While TrkB hypomorphs have not been studied in the same way, loss of TrkB in the majority of the hypothalamus using an Nkx2.1-cre reveals that female mice have increased food intake on normal chow and decreased core body temperature, suggesting a decrease in energy expenditure in line with previous reports of BDNF administration [57]. This effect may largely be mediated via TrkB neurons in the DMH, which have been shown to be temperature responsive and required for cold induced thermogenesis [58]. Interestingly, BDNF deletion using the same Nkx2.1-cre strategy reveals a similar increase in body weight, but only due to decreased energy expenditure, without a change in food intake [39]. For p75NTR, the resistance of knockouts to gain weight on a high fat diet has been attributed to an increase in energy expenditure due to increased fat oxidation and lipolysis, as well as increased thermogenesis, again revealing an opposing metabolic function of p75NTR [13].
Locomotor activity
Among the first reports of the heterozygous BDNF deletion mouse were data that suggested loss of BDNF led to hyperactivity in addition to hyperphagia. Interestingly, however, this hyperactivity was only seen in normal weight BDNF heterozygote mice, whereas obese BDNF heterozygote mice had wildtype levels of activity [59]. As the authors and others have suggested, normal activity for an obese mouse may in fact reveal an underlying elevation, as most obese mouse models are hypoactive [59]. Interestingly, to our knowledge there does not appear to be published data of any TrkB knockout model where locomotor activity is similarly increased, and in fact most reports show it as lower [40,42,57]. This suggests that there is an as yet unknown site where BDNF may work to mediate its control of locomotor activity, and may reveal further circuit based regulation of locomotor activity, for which data is lacking. Loss of p75NTR, on the other hand, does not appear to alter locomotor activity at baseline, but rather decreases activity during energy deficit states [14]. Whether TrkB and/or BDNF alter their regulation of locomotor activity based on energy state has not yet been investigated.
Signaling through p75NTR and TrkB
The determinants of whether BDNF will signal through TrkB or p75NTR in metabolic contexts are currently unknown. While it is plausible that specificity could be determined as a result of cell-type specific expression patterns or the differing affinities with which TrkB (high-affinity) and p75NTR (low-affinity) bind to BDNF, a third possibility exists in the post-translational processing of BDNF. BDNF is first secreted as an immature precursor, proBDNF, which exhibits exclusive binding to p75NTR, and acts in some cases to oppose BDNF-TrkB activity [60]. It could be conceptualized that this “default” original state (proBDNF-p75NTR signaling) is to promote orexigenic behavior. Then, when energy supplies grow to be sufficient or in excess, proBDNF could be cleaved to BDNF, which could convert orexigenic activity to anorexigenic. This proposes that a system could quickly switch from p75NTR activity to predominantly TrkB activity through regulation of a single cleavage event, converting proBDNF to BDNF.
Interestingly, a single nucleotide polymorphism in the pro domain of BDNF (Val66Met) associated with susceptibility to neuropsychiatric disorders in humans has been suggested to work partially via p75NTR [61,62]. This SNP leads to altered trafficking of BDNF within neurons, and decreased activity dependent secretion of mature BDNF [61]. Notably, this variant has been suggested to increase feeding behavior in mice, in line with the orexigenic activity of p75NTR [63]. Perhaps most strikingly, however, is that the Val66Met variant has been shown to alter anxiety levels in mice and humans, and combining the Val66Metvariant with a peri-pubertal stress leads to an Anorexia-Nervosa-like mouse model [64,65]. Based on these reports it is tempting to speculate that proBDNF may allow for greater dynamic control of energy regulation, and that it may afford the opportunity to investigate the links between anxiety and feeding.
Downstream of ligand activation, TrkB and p75NTR are known to engage disparate signaling pathways. TrkB promotes MAPK/ERK, PI3K, and PLC-γ1, pathways, while p75NTR promotes NF-κB, JNK and Rho pathways [12,66–69]. These differences in signaling activation have subsequently been demonstrated to lead to differential outcomes in neuronal survival and growth [70,71]. While these canonical neurotrophin pathways have been well-characterized over the years, the signaling mechanisms engaged to exert metabolic effects have not been as well-characterized, and it is unknown which signaling cascades are required for regulation of metabolism. Intriguingly, the few reports that have examined these pathways suggest that metabolic effects of p75NTR may instead act via PKA and CREB in peripheral and central metabolic control, respectively [13,14,51]. Separately, the effect of BDNF on metabolic signaling may also extend beyond the conventional full-length form of TrkB. In pancreatic β-cells, BDNF has been shown to act via a truncated form of TrkB, TrkB.T1, which lacks the catalytic tyrosine kinase domain, to promote calcium transients and insulin release [50]. These examples suggest the existence of largely unexplored avenues to modulate intracellular signaling pathways to achieve improved metabolic regulation.
Importantly, p75NTR and TrkB don’t exist in isolation. p75NTR can act as a co-receptor for TrkB, altering its binding affinity for BDNF [19,72]. Strikingly, this interaction can be synergistic or antagonistic depending on context. For example, culture systems have suggested that p75NTR can reduce TrkB activation in response to BDNF [73], but when extracellular and intracellular domains of p75NTR are liberated by alpha and gamma secretases the remaining p75NTR transmembrane portion can promote TrkB activation [74]. Given that levels of p75NTR, TrkB, and BDNF can be modulated in response to stimuli, it’s likely that strong dynamic regulation could be achieved through p75NTR tamping down or ameliorating BDNF-TrkB signaling, and that this could be cell-type dependent. Beyond p75NTR, TrkB can also be activated through interactions with other molecular players. For instance, it can be transactivated by GPCRs, including the receptors for adenosine and PACAP, leading to an alteration of the canonical neurotrophin signaling dynamics [75,76]. Interestingly, PACAP expressing hypothalamic neurons have been implicated in whole body energy homeostasis, and it would be intriguing to know whether PACAP interacts with hypothalamic TrkB [77]. Additionally, glucocorticoid binding to the glucocorticoid receptor can phosphorylate and activate TrkB independent of BDNF, suggesting another possibility for crosstalk between conventional metabolic signals and neurotrophin signaling pathways [78].
Concluding Remarks
We synthesize here a growing body of work delineating that BDNF, and potentially other neurotrophins, can exhibit potent and discriminating effects to regulate energy intake and expenditure through biased action on one of its two receptors. Whereas TrkB exhibits strong anorexigenic effects at baseline, p75NTR appears to function antagonistically as a countermeasure against severe energy deficit. While this is generally supported, further parsing cell type specific roles for TrkB and p75NTR may reveal more nuanced levels of regulation. While we have characterized many of the critical sites of action, more are likely to be uncovered by further site specific manipulations of neurotrophins and their receptors. Further, mapping the circuit interactions between these regions will be critical for increasing our understanding of how crosstalk between peripheral and central mechanisms contribute towards whole body energy homeostasis.
Our understanding of energy balance, including the interactions of central and peripheral systems, has advanced dramatically over the last several years. The study and inclusion of neurotrophins and their receptors in this work has opened the door to novel pathways and mechanisms by which these systems can develop and be dynamically regulated. Why should neurotrophins be involved in energy homeostasis? It is possible that the metabolic roles of neurotrophins are incidental, and their roles in adult energy balance may be holdouts of some earlier role in neurodevelopment. However, there is increasing evidence that homeostatic feeding and energy circuits are highly plastic in response to changing energy environments, which necessitates signaling pathways that can mediate dynamic structural rearrangements. The findings presented here pose intriguing questions (see Outstanding Questions) regarding how neurotrophins may aid in the adaptation to changing metabolic environments, and interplay with other critical responses to learning and stress, and whether these longer-term alterations may be amenable to targeting in chronic metabolic diseases (Box 3).
OUTSTANDING QUESTIONS.
How does BDNF differentially act on p75NTR and TrkB, and is this state dependent (i.e. sated vs fasted)? p75NTR and TrkB have different affinities for binding to BDNF and differ in their response to proBDNF. Additionally, BDNF levels have been shown to be induced by various metabolic states, including fasting and exercise. How these alterations affect p75NTR and TrkB signaling has yet to be fully elucidated.
Do p75NTR and TrkB functionally interact in cell type specific manners, and with other energy regulatory signaling pathways? Determining the extent of co-expression of p75NTR and TrkB within the same cells is a necessary first step to address possible interactions. Follow up biochemical and behavioral assays will be needed to parse out functional roles.
Does BDNF signaling through p75NTR/TrkB lead to long term structural remodeling of hypothalamic and other energy regulatory circuits, akin to other known neurotrophic roles? BDNF deficiency has been shown to alter hypothalamic structure, but whether this requires p75NTR and TrkB, or is merely a secondary consequence of the metabolic changes seen with loss BDNF, is unknown. This promises to reveal novel aspects of metabolic regulation, and may be an underrecognized component of defending changes in body weight.
Do other neurotrophins, including NGF, NT-4, TrkA, and TrkC, have further unexplored roles in metabolism? Early studies make it likely that some other neurotrophins may play roles in metabolism, but might have a less overt phenotype. Nevertheless, these roles may help elucidate poorly understood metabolic disorders, including potentially the metabolic contributions of some psychometabolic diseases.
Can neurotrophin signaling be leveraged to modify hunger and energy homeostasis in metabolic disease? Altering the activation ratio of neurotrophin receptors may yield the ability to bidirectionally modify hunger, and warrants future study.
Box 3. Small molecule modulation of TrkB and p75NTR signaling.
Whether TrkB and p75NTR may be targetable to ameliorate metabolic disorders is an area currently under investigation. Several selective modulators that are specific for either TrkB or p75NTR have been developed, and have begun to be tested as therapeutic approaches to several neurological disorders [90]. Weight gain of wildtype mice fed a high-fat-diet was attenuated compared to controls in pre-clinical trials testing the metabolic effects of one such TrkB activating compound, orally administered 7,8-DHF [91]. Interestingly, this effect appears solely to be due to alterations of peripheral energy expenditure without an effect on food intake, suggesting there may be biased activity for this agonist on specific TrkB functions. One possibility is that the locus of action of the agonist matters. For example, a different TrkB antibody agonist had a similar weight reducing effect in obese mice, but this was accompanied by a reduction of food intake when administered centrally [84]. However, this same compound tested in non-human primates showed a divergent effect on weight change when administered peripherally [92]. Lastly, a small molecule agonist of p75NTR, LM11A-31, which is currently in Phase 2a clinical trials for treatment of Alzheimer’s disease, has been extensively tested in wildtype mice [93]. While no change in body weight was observed following oral delivery over 2 months, further metabolic characterization of this ligand, nor testing in mouse models of metabolic disease, has been conducted to date [94].
HIGHLIGHTS.
BDNF signaling plays a significant role from mice to humans in controlling energy homeostasis. Loss of function of BDNF leads to hyperphagia and obesity.
BDNF signals through two receptors, TrkB and p75NTR, both of which have been shown to play roles in defined hypothalamic nuclei and cell types in controlling feeding.
Many of the feeding roles for p75NTR and TrkB are in opposition, with TrkB suppressing, and p75NTR promoting, feeding.
A variant of BDNF, Val66Met, that alters the immature precursor of BDNF, proBDNF, may preferentially activate p75NTR and promote feeding behavior.
TrkB and p75NTR, in addition to other neurotrophins, have emerging roles in the periphery, especially in glucose regulation. These roles for TrkB and p75NTR also appear to be functionally opposed.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Morton GJ et al. (2006) Central nervous system control of food intake and body weight. Nature 443, 289–295 [DOI] [PubMed] [Google Scholar]
- 2.Andermann ML and Lowell BB (2017) Toward a wiring diagram understanding of appetite control. Neuron 95, 757–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Woods SC et al. (1998) Signals that regulate food intake and energy homeostasis. Science 280, 1378–1383 [DOI] [PubMed] [Google Scholar]
- 4.Lewin GR and Barde YA (1996) Physiology of the neurotrophins. Annu. Rev. Neurosci. 19, 289–317 [DOI] [PubMed] [Google Scholar]
- 5.Bibel M and Barde YA (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 14, 2919–2937 [DOI] [PubMed] [Google Scholar]
- 6.Chao MV (1994) The p75 neurotrophin receptor. J. Neurobiol. 25, 1373–1385 [DOI] [PubMed] [Google Scholar]
- 7.Chao MV and Hempstead BL (1995) p75 and Trk: a two-receptor system. Trends Neurosci. 18, 321–326 [PubMed] [Google Scholar]
- 8.Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4, 299–309 [DOI] [PubMed] [Google Scholar]
- 9.Park H and Poo M (2013) Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7–23 [DOI] [PubMed] [Google Scholar]
- 10.Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos. Trans. R. Soc. Lond. B. Biol. Sci 361, 1545–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rios M (2013) BDNF and the central control of feeding: accidental bystander or essential player? Trends Neurosci. 36, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu B and Xie X (2016) Neurotrophic factor control of satiety and body weight. Nat. Rev. Neurosci. 17, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baeza-Raja B et al. (2016) p75 neurotrophin receptor regulates energy balance in obesity. Cell Rep. 14, 255–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Podyma B et al. (2020) The p75 neurotrophin receptor in AgRP neurons is necessary for homeostatic feeding and food anticipation. elife 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu B et al. (2003) Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat. Neurosci. 6, 736–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeo GSH et al. (2004) A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat. Neurosci. 7, 1187–1189 [DOI] [PubMed] [Google Scholar]
- 17.Gray J et al. (2007) Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int J Obes (Lond) 31, 359–364 [DOI] [PubMed] [Google Scholar]
- 18.Sonoyama T et al. (2020) Human BDNF/TrkB variants impair hippocampal synaptogenesis and associate with neurobehavioural abnormalities. Sci. Rep. 10, 9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bibel M et al. (1999) Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 18, 616–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gray J et al. (2006) Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes 55, 3366–3371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han JC et al. (2008) Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 359, 918–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mou Z et al. (2015) Human Obesity Associated with an Intronic SNP in the Brain-Derived Neurotrophic Factor Locus. Cell Rep. 13, 1073–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan AS et al. (2008) A DRD4/BDNF gene-gene interaction associated with maximum BMI in women with bulimia nervosa. Int. J. Eat. Disord. 41, 22–28 [DOI] [PubMed] [Google Scholar]
- 24.Han JC (2016) Rare Syndromes and Common Variants of the Brain-Derived Neurotrophic Factor Gene in Human Obesity. Prog. Mol. Biol. Transl. Sci. 140, 75–95 [DOI] [PubMed] [Google Scholar]
- 25.Burns B et al. (2010) Rai1 haploinsufficiency causes reduced Bdnf expression resulting in hyperphagia, obesity and altered fat distribution in mice and humans with no evidence of metabolic syndrome. Hum. Mol. Genet. 19, 4026–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han JC et al. (2010) Lower brain-derived neurotrophic factor in patients with prader-willi syndrome compared to obese and lean control subjects. J. Clin. Endocrinol. Metab. 95, 3532–3536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cassidy SB et al. (2012) Prader-Willi syndrome. Genet. Med. 14, 10–26 [DOI] [PubMed] [Google Scholar]
- 28.Carmona-Mora P et al. (2012) RAI1 transcription factor activity is impaired in mutants associated with Smith-Magenis Syndrome. PLoS ONE 7, e45155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bochukova EG et al. (2018) A Transcriptomic Signature of the Hypothalamic Response to Fasting and BDNF Deficiency in Prader-Willi Syndrome. Cell Rep. 22, 3401–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Primo D et al. (2021) Brain derived neurotrophic factor (BDNF) polymorphism rs 10767664 affects metabolic parameters after weight loss secondary to high fat hypocaloric diet with Mediterranean pattern. Eur. Rev. Med. Pharmacol. Sci. 25, 1944–1953 [DOI] [PubMed] [Google Scholar]
- 31.Bumb JM et al. (2020) BDNF influences neural cue-reactivity to food stimuli and food craving in obesity. Eur. Arch. Psychiatry Clin. Neurosci. DOI: 10.1007/s00406-020-01224-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kichaev G et al. (2019) Leveraging polygenic functional enrichment to improve GWAS power. Am. J. Hum. Genet. 104, 65–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schor NF (2005) The p75 neurotrophin receptor in human development and disease. Prog. Neurobiol. 77, 201–214 [DOI] [PubMed] [Google Scholar]
- 34.Muscatelli F et al. (2000) Disruption of the mouse Necdin gene results in hypothalamic and behavioral alterations reminiscent of the human Prader-Willi syndrome. Hum. Mol. Genet. 9, 3101–3110 [DOI] [PubMed] [Google Scholar]
- 35.Kuwako K et al. (2004) Necdin-related MAGE proteins differentially interact with the E2F1 transcription factor and the p75 neurotrophin receptor. J. Biol. Chem. 279, 1703–1712 [DOI] [PubMed] [Google Scholar]
- 36.Moga MM and Duong T (1997) p75 neurotrophin receptor immunoreactivity in the aged human hypothalamus. Neurosci. Lett. 231, 9–12 [DOI] [PubMed] [Google Scholar]
- 37.Unger TJ et al. (2007) Selective deletion of Bdnf in the ventromedial and dorsomedial hypothalamus of adult mice results in hyperphagic behavior and obesity. J. Neurosci. 27, 14265–14274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Komori T et al. (2006) Induction of brain-derived neurotrophic factor by leptin in the ventromedial hypothalamus. Neuroscience 139, 1107–1115 [DOI] [PubMed] [Google Scholar]
- 39.Yang H et al. (2016) Regulation of energy balance via BDNF expressed in nonparaventricular hypothalamic neurons. Mol. Endocrinol. 30, 494–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liao G-Y et al. (2019) TrkB-expressing neurons in the dorsomedial hypothalamus are necessary and sufficient to suppress homeostatic feeding. Proc Natl Acad Sci USA 116, 3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C et al. (2007) Brain-derived neurotrophic factor in the hypothalamic paraventricular nucleus increases energy expenditure by elevating metabolic rate. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R992–1002 [DOI] [PubMed] [Google Scholar]
- 42.An JJ et al. (2020) TrkB-expressing paraventricular hypothalamic neurons suppress appetite through multiple neurocircuits. Nat. Commun. 11, 1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.An JJ et al. (2015) Discrete BDNF neurons in the paraventricular hypothalamus control feeding and energy expenditure. Cell Metab. 22, 175–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tan CL et al. (2016) Warm-Sensitive Neurons that Control Body Temperature. Cell 167, 47–59.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang P et al. (2020) A leptin-BDNF pathway regulating sympathetic innervation of adipose tissue. Nature 583, 839–844 [DOI] [PubMed] [Google Scholar]
- 46.Liao G-Y et al. (2015) Brain-derived neurotrophic factor is required for axonal growth of selective groups of neurons in the arcuate nucleus. Mol. Metab. 4, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bouret SG et al. (2004) Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304, 108–110 [DOI] [PubMed] [Google Scholar]
- 48.Nakagawa T et al. (2003) Anti-obesity and anti-diabetic effects of brain-derived neurotrophic factor in rodent models of leptin resistance. Int. J. Obes. Relat. Metab. Disord. 27, 557–565 [DOI] [PubMed] [Google Scholar]
- 49.Nonomura T et al. (2001) Brain-derived neurotrophic factor regulates energy expenditure through the central nervous system in obese diabetic mice. Int. J. Exp. Diabetes Res. 2, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fulgenzi G et al. (2020) Novel metabolic role for BDNF in pancreatic β-cell insulin secretion. Nat. Commun. 11, 1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baeza-Raja B et al. (2012) p75 neurotrophin receptor regulates glucose homeostasis and insulin sensitivity. Proc Natl Acad Sci USA 109, 5838–5843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakagomi A et al. (2015) Role of the central nervous system and adipose tissue BDNF/TrkB axes in metabolic regulation. npj Aging Mech. Dis. 1, 15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coppola V and Tessarollo L (2004) Control of hyperphagia prevents obesity in BDNF heterozygous mice. Neuroreport 15, 2665–2668 [DOI] [PubMed] [Google Scholar]
- 54.Tsuchida A et al. (2001) Acute effects of brain-derived neurotrophic factor on energy expenditure in obese diabetic mice. Int. J. Obes. Relat. Metab. Disord. 25, 1286–1293 [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa T et al. (2000) Brain-derived neurotrophic factor regulates glucose metabolism by modulating energy balance in diabetic mice. Diabetes 49, 436–444 [DOI] [PubMed] [Google Scholar]
- 56.You H et al. (2020) A subpopulation of Bdnf-e1-expressing glutamatergic neurons in the lateral hypothalamus critical for thermogenesis control. Mol. Metab. 31, 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ozek C et al. (2015) Ablation of intact hypothalamic and/or hindbrain TrkB signaling leads to perturbations in energy balance. Mol. Metab. 4, 867–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Houtz J et al. (2021) Discrete TrkB-expressing neurons of the dorsomedial hypothalamus regulate feeding and thermogenesis. Proceedings of the National Academy of Sciences [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kernie SG et al. (2000) BDNF regulates eating behavior and locomotor activity in mice. EMBO J. 19, 1290–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Teng HK et al. (2005) ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J. Neurosci. 25, 5455–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Egan MF et al. (2003) The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269 [DOI] [PubMed] [Google Scholar]
- 62.Anastasia A et al. (2013) Val66Met polymorphism of BDNF alters prodomain structure to induce neuronal growth cone retraction. Nat. Commun. 4, 2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ieraci A et al. (2020) BDNF Val66Met polymorphism alters food intake and hypothalamic BDNF expression in mice. J. Cell. Physiol. 235, 9667–9675 [DOI] [PubMed] [Google Scholar]
- 64.Chen Z-Y et al. (2006) Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314, 140–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Madra M and Zeltser LM (2016) BDNF-Val66Met variant and adolescent stress interact to promote susceptibility to anorexic behavior in mice. Transl. Psychiatry 6, e776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zampieri N and Chao MV (2006) Mechanisms of neurotrophin receptor signalling. Biochem. Soc. Trans. 34, 607–611 [DOI] [PubMed] [Google Scholar]
- 67.Lin Z et al. (2015) Structural basis of death domain signaling in the p75 neurotrophin receptor. elife 4, e11692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang EJ and Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 [DOI] [PubMed] [Google Scholar]
- 69.Yamashita T et al. (1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24, 585–593 [DOI] [PubMed] [Google Scholar]
- 70.Harrington AW et al. (2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J. Neurosci. 22, 156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yoshii A and Constantine-Paton M (2010) Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 70, 304–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zaccaro MC et al. (2001) p75 Co-receptors regulate ligand-dependent and ligand-independent Trk receptor activation, in part by altering Trk docking subdomains. J. Biol. Chem. 276, 31023–31029 [DOI] [PubMed] [Google Scholar]
- 73.Vesa J et al. (2000) p75 reduces TrkB tyrosine autophosphorylation in response to brain-derived neurotrophic factor and neurotrophin 4/5. J. Biol. Chem. 275, 24414–24420 [DOI] [PubMed] [Google Scholar]
- 74.Saadipour K et al. (2017) The transmembrane domain of the p75 neurotrophin receptor stimulates phosphorylation of the TrkB tyrosine kinase receptor. J. Biol. Chem. 292, 16594–16604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee FS et al. (2002) Distinctive features of Trk neurotrophin receptor transactivation by G protein-coupled receptors. Cytokine Growth Factor Rev. 13, 11–17 [DOI] [PubMed] [Google Scholar]
- 76.Lee FS et al. (2002) Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J. Biol. Chem. 277, 9096–9102 [DOI] [PubMed] [Google Scholar]
- 77.Krashes MJ et al. (2014) An excitatory paraventricular nucleus to AgRP neuron circuit that drives hunger. Nature 507, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Arango-Lievano M et al. (2015) Neurotrophic-priming of glucocorticoid receptor signaling is essential for neuronal plasticity to stress and antidepressant treatment. Proc Natl Acad Sci USA 112, 15737–15742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barde YA et al. (1982) Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1, 549–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Binder DK and Scharfman HE (2004) Brain-derived neurotrophic factor. Growth Factors 22, 123–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pelleymounter MA et al. (1995) Characteristics of BDNF-induced weight loss. Exp. Neurol. 131, 229–238 [DOI] [PubMed] [Google Scholar]
- 82.Lapchak PA and Hefti F (1992) BDNF and NGF treatment in lesioned rats: effects on cholinergic function and weight gain. Neuroreport 3, 405–408 [DOI] [PubMed] [Google Scholar]
- 83.Klein R et al. (1993) Targeted disruption of the trkB neurotrophin receptor gene results in nervous system lesions and neonatal death. Cell 75, 113–122 [PubMed] [Google Scholar]
- 84.Tsao D et al. (2008) TrkB agonists ameliorate obesity and associated metabolic conditions in mice. Endocrinology 149, 1038–1048 [DOI] [PubMed] [Google Scholar]
- 85.Mercader JM et al. (2008) Association of NTRK3 and its interaction with NGF suggest an altered cross-regulation of the neurotrophin signaling pathway in eating disorders. Hum. Mol. Genet. 17, 1234–1244 [DOI] [PubMed] [Google Scholar]
- 86.Raile K et al. (2006) Glucose regulates expression of the nerve growth factor (NGF) receptors TrkA and p75NTR in rat islets and INS-1E beta-cells. Regul. Pept. 135, 30–38 [DOI] [PubMed] [Google Scholar]
- 87.Geetha T et al. (2013) Nerve growth factor receptor TrkA, a new receptor in insulin signaling pathway in PC12 cells. J. Biol. Chem. 288, 23807–23813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Houtz J et al. (2016) Neurotrophin Signaling Is Required for Glucose-Induced Insulin Secretion. Dev. Cell 39, 329–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Williams LR (1991) Hypophagia is induced by intracerebroventricular administration of nerve growth factor. Exp. Neurol. 113, 31–37 [DOI] [PubMed] [Google Scholar]
- 90.Longo FM and Massa SM (2013) Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat. Rev. Drug Discov. 12, 507–525 [DOI] [PubMed] [Google Scholar]
- 91.Wood J et al. (2018) BDNF mimetic alleviates body weight gain in obese mice by enhancing mitochondrial biogenesis in skeletal muscle. Metab. Clin. Exp. 87, 113–122 [DOI] [PubMed] [Google Scholar]
- 92.Lin JC et al. (2008) Appetite enhancement and weight gain by peripheral administration of TrkB agonists in non-human primates. PLoS ONE 3, e1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Massa SM et al. (2006) Small, nonpeptide p75NTR ligands induce survival signaling and inhibit proNGF-induced death. J. Neurosci. 26, 5288–5300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simmons DA et al. (2014) A small molecule p75NTR ligand, LM11A-31, reverses cholinergic neurite dystrophy in Alzheimer’s disease mouse models with mid- to late-stage disease progression. PLoS ONE 9, e102136. [DOI] [PMC free article] [PubMed] [Google Scholar]