

Abstract
Temporal lobe epilepsy (TLE) is characterized by recurrent spontaneous seizures and behavioral comorbidities. Reduced hippocampal theta oscillations and hyperexcitability that contribute to cognitive deficits and spontaneous seizures are present beyond the sclerotic hippocampus in TLE. However, the mechanisms underlying compromised network oscillations and hyperexcitability observed in circuits remote from the sclerotic hippocampus are largely unknown. Cholecystokinin (CCK)-expressing basket cells (CCKBCs) critically participate in hippocampal theta rhythmogenesis, and regulate neuronal excitability. Thus, we examined whether CCKBCs were vulnerable in nonsclerotic regions of the ventral hippocampus remote from dorsal sclerotic hippocampus using the intrahippocampal kainate (IHK) mouse model of TLE, targeting unilateral dorsal hippocampus. We found a decrease in the number of CCK+ interneurons in ipsilateral ventral CA1 regions from epileptic mice compared to those from sham controls. We also found that the number of boutons from CCK+ interneurons was reduced in the stratum pyramidale, but not in other CA1 layers, of ipsilateral hippocampus in epileptic mice, suggesting that CCKBCs are vulnerable. Electrical recordings showed that synaptic connectivity and strength from surviving CCKBCs to CA1 pyramidal cells (PCs) were similar between epileptic mice and sham controls. In agreement with reduced CCKBC number in TLE, electrical recordings revealed a significant reduction in amplitude and frequency of IPSCs in CA1 PCs evoked by carbachol (commonly used to excite CCK+ interneurons) in ventral CA1 regions from epileptic mice versus sham controls. These findings suggest that loss of CCKBCs beyond the hippocampal lesion may contribute to hyperexcitability and compromised network oscillations in TLE.
Keywords: disinhibition, perisomatic inhibition, basket cells, cognitive impairment
1. Introduction
Temporal lobe epilepsy (TLE) is the most common type of focal epilepsy and affects more than 60% of all epilepsy patients (Téllez-Zenteno and Hernández-Ronquillo, 2012). TLE is characterized by recurrent spontaneous seizures and associated comorbidities (Amlerova et al., 2013; Bui et al., 2018; Cánovas et al., 2011; Elger et al., 2004; Holmes, 2015; Kim et al., 2020). Although antiepileptic drugs (AEDs) are commonly used to control seizures, one third of epilepsy patients are refractory to AEDs (Kalilani et al., 2018). Temporal lobectomy is the most effective treatment for TLE when seizures are uncontrolled by AEDs, though there are risks and complications related to surgical therapy (Thom et al., 2010). The mechanisms underlying hyperexcitability, seizures, and impaired memory in TLE are not fully understood. Better understanding of such mechanisms will be critical for development of target molecules, cells, and circuits for therapeutic intervention in patients with refractory TLE.
Cholecystokinin-positive (CCK+) interneurons are a major group of GABAergic interneurons in the hippocampus that comprise the perisomatic CCK+ interneurons (i.e., CCK+ basket cells, CCKBCs) and dendritic CCK+ interneurons (e.g., Schaffer collateral-associated cells, SCA cells) (Bezaire and Soltesz, 2013; Lee et al., 2010). CCKBCs regulate excitability of hippocampal circuits and are critically involved in coordinated network oscillations in the hippocampus (e.g., theta rhythms), which support key functions of the hippocampus (e.g., episodic memory; Bezaire et al., 2016; Colgin, 2016; Freund and Katona, 2007). CCKBCs are vulnerable and CCK+ interneuron-mediated inhibition of principal cells is downregulated in animal models of TLE (Sun et al., 2014; Wyeth et al., 2010). Such changes in CCK+ interneuron circuits in the hippocampus may contribute to spontaneous seizures and memory deficits in TLE.
The unilateral intrahippocampal kainate (IHK) model of TLE is commonly used because it reproduces key features of human TLE: Spontaneous seizures controlled by AEDs, hippocampus-dependent memory deficits, and histopathological changes (e.g., hippocampal sclerosis; Rattka et al., 2013). Unlike animal models of TLE induced by systemic chemoconvulsant administration (e.g., pilocarpine or kainate) showing bilateral hippocampal sclerosis, the unilateral IHK model of TLE produces hippocampal sclerosis more restricted to the targeted ipsilateral hippocampal region, whereas the ipsilateral hippocampal region and contralateral hippocampus remote from the sclerotic region of the hippocampus appear to be relatively intact. Importantly, unilateral hippocampal sclerosis is observed in approximately 80% of patients with mesial temporal sclerosis (Alarcón and Valentín, 2010; Thom, 2014). Although most seizures arise from the sclerotic hippocampus in TLE patients with unilateral hippocampal sclerosis, the remaining seizures arise from other brain regions, including contralateral hippocampus (Bragin et al., 1999). Furthermore, reduced hippocampal theta network oscillations arise from ventral hippocampal circuits remote from the sclerotic region of the hippocampus in the dorsal unilateral IHK mouse model (Dugladze et al., 2007). These results collectively suggest that reorganization of neuronal circuits remote from hippocampal sclerosis may contribute to spontaneous seizures and compromised network oscillations in TLE. Indeed, a subset of hippocampal interneurons remote from the hippocampal lesion is vulnerable to injury in the IHK model (Marx et al., 2013), and neuronal degeneration in the contralateral dentate gyrus has been described (Groticke et al., 2008). However, it is unclear whether hippocampal CCK+ interneurons remote from the hippocampal lesion are vulnerable or whether surviving CCK+ interneurons manifest abnormal activity in the unilateral IHK mouse model.
In this study, we used immunostaining, confocal imaging, whole-cell patch-clamp recordings, video-EEG monitoring, and behavioral tests in a well-established unilateral IHK mouse model of TLE targeting dorsal hippocampus to test our hypothesis that ventral hippocampal CCK+ interneuron circuits remote from hippocampal sclerosis are compromised in the dorsal IHK mouse model. We focused on three main questions: 1) Is CCK+ interneuron number reduced in the ventral hippocampus in the dorsal IHK model; 2) do surviving CCKBCs in the IHK model manifest abnormal intrinsic and synaptic properties; and 3) does the IHK model result in downregulated CCK+ interneuron-mediated inhibition?
2. Materials and methods
2.1. Animals
Young adult C57BL/6J mice (Jackson laboratory, Bar Harbor, ME, USA) were maintained at ambient temperature and humidity with a 14-hour light/10-hour dark cycle and fed standard chow ad libitum. Similar numbers of males and females were used for the study, thus reducing sex bias (Will et al., 2017). Thus, the combined data from both sexes are presented in this study. All animal procedures were approved by the Institutional Animal Care and Use Committees of the University of Kentucky and the University of Arkansas for Medical Sciences (UAMS).
2.2. IHK mouse model of TLE and video monitoring for behavioral seizures
Six to eight-week-old males and females (49 ± 1 day-old mice, n = 111 total) were anesthetized under isoflurane and unilaterally injected with 50–100 nl (85.6 ± 1.2 nl) of sterile 0.9% saline (42 mice; 26 females and 16 males) or kainate (69 mice; 35 females and 34 males; KA, 20 mM in saline; Tocris Biosciences, Minneapolis, MN, USA) using the following stereotaxic coordinates relative to the bregma (anterior-posterior, −2.0 mm; medial-lateral, −1.25 mm; dorsal-ventral, −1.60 mm) as described previously (Krook-Magnuson et al., 2013). In order to minimize losses of mice caused by status epilepticus (>30 minutes of Racine stage 3–5 seizure; Trinka et al., 2015), diazepam (7.5 mg/kg) was injected intraperitoneally 1 hour after the first Racine stage 5 seizure, characterized by tonic-clonic convulsions, rearing, running, jumping and falling (Racine, 1972). Saline-injected mice (i.e., sham controls) also received intraperitoneal injections of diazepam. All postsurgical mice were individually housed and recovered in the vivarium. Three to four weeks after KA (n = 60, 29 females and 31 males) or saline (n = 42, 26 females and 16 males) injection, spontaneous behavioral activities were video-recorded continuously for 24 hours a day for 3 days using video cameras and Sirenia software (version 1.7.10; Pinnacle Technology, Lawrence, KS, USA) without implanting EEG electrodes. IHK mice that displayed Racine stage 3–5 seizures at least two times during the 3-day video recordings were defined as “epileptic” mice and used in this study.
2.3. Video-EEG recordings
Four to five weeks after KA (n = 8) or saline (n = 4) injection, mice were anesthetized with isoflurane and ipsilaterally implanted with a 2-channel depth electrode (Plastics One, Roanoke, VA, USA) in stereotaxic coordinates (anterior-posterior −2.5 mm, medial-lateral −1.75 mm, dorsal-ventral −1.25 mm with respect to bregma) as described in previous studies (Krook-Magnuson et al., 2013). Implanted electrodes were firmly secured on the skull using mounting screws and dental cement. One week after the surgery, the mice were monitored by synchronized video-EEG recording for 24 hours a day for 3 days. Electrodes were connected to the 3-pin head mount (Plastics One), which was connected to the customized Mouse Preamplifier (Pinnacle Technology). EEG data were acquired at a 500 Hz sampling frequency with 500 Hz low pass filter and 1.0 Hz high pass filter using 8401-HS Data Conditioning and Acquisition System (Pinnacle Technology). LED-equipped video cameras were projected from the top of cages for video recordings. Synchronized video-EEG files were acquired with Sirenia Acquisition software (version 1.7.10) and 8400 Tethered Mouse System (Pinnacle Technology), and analyzed using Sirenia Seizure Pro software (version 1.8.0; Pinnacle Technology).
2.4. Behavioral tests: Open field test and object location memory test
Four weeks after injection, IHK (n = 9) and sham control (n = 10) mice were subjected to the open field test followed by the object location memory (OLM) test. All behavioral tests were performed in a dedicated quiet room, where light illumination and temperature were maintained at 45–48 Lux and 20–22°C. Open field test was performed to examine anxiety-like behaviors based on locomotive activities. Mice were brought in their home cages from the vivarium to the testing room and acclimated at least 1 hour prior to the test. A clear acrylic arena (40 width × 40 length × 35 height cm) was placed on a table where the testing area was separated from experimenters by curtains. Mice were placed in the open arena one at a time, and their locomotive activities were digitally recorded for 5 minutes using the GigE Basler acA1300–60gc camera (Noldus, Wageningen, Netherlands) and analyzed by the video tracking software, Ethovision XT 13.0 (Noldus). The area of the arena was digitally divided into 25 equal squares (5 × 5) and the 9 center squares were defined as the center zone. Each video file was encrypted and analyzed to acquire total traveled distance, mean speed of movement, and cumulative time spent in the center zone during the 5 minutes. The arena was cleaned with 10% ethanol and water using a paper towel after each use.
We performed the OLM test, known as a hippocampal-dependent learning paradigm, to determine whether IHK mice displayed hippocampus-dependent memory impairment as reported earlier (Bui et al., 2018; Kim et al., 2020). Based on the published protocol (Vogel-Ciernia and Wood, 2014), after mice were handled for 5 days (2 minutes per day), they were habituated in an empty open field arena (white acrylic, 23 width × 30 length × 23 height cm) filled with regular bedding for 6 days (5 minutes per day). On the following day, mice were trained for 10 minutes by placing them in the arena where two identical objects were symmetrically placed in two locations (i.e., familiar locations). 24 hours after the training, mice were tested for 5 minutes by placing them back in the arena, where one of the two identical objects was randomly moved to a novel location. Locomotive activities during the 10-minute training and 5-minute testing were digitally recorded using the GigE Basler acA1300–60gc camera (Noldus). All objects were sanitized with 10% ethanol and water between animals. Video files were encrypted for unbiased analysis. Exploration time on each object during the training and the testing periods were measured using the manual scoring mode of Ethovision XT 13.0 (Noldus), while excluding behaviors such as rearing or mounting on the objects, or digging on the ground near the objects. The discrimination index (DI) was calculated as: (time exploring the novel object − time exploring the familiar object) / (time exploring the novel object + time exploring the familiar object) × 100. We excluded data from mice that failed to explore both objects for >3 seconds during the training or testing, or if animals displayed DI values less than −20 or greater than +20 due to a significant location bias during the training session, as previously described (Vogel-Ciernia and Wood, 2014).
2.5. Whole-cell patch-clamp recordings
Horizontal hippocampal slices (300 μm) from ventral hippocampi were prepared from both sexes of C57BL/6J mice 8–9 weeks after IHK or saline injection. All data in this study originating from the ventral hippocampus were from a restricted area (dorsal-ventral: approximately −2.4 to −3.6 mm from Bregma; 4 ipsilateral and 4 contralateral slices from each mouse). The slices were assigned for electrophysiology or immunohistochemistry (1:1). For electrophysiology, brain slices were incubated in sucrose-containing artificial cerebrospinal fluid (ACSF) for one hour at 33°C, and then stored at room temperature until electrical recording. ACSF contained, in mM, 85 NaCl, 75 sucrose, 25 glucose, 24 NaHCO3, 4 MgCl2, 2.5 KCl, 1.25 NaH2PO4, and 0.5 CaCl2. For immunohistochemistry, brain slices were fixed immediately in 4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer (pH 7.4). Hippocampal slices destined for electrophysiology were transferred to a recording chamber in ACSF containing, in mM, 126 NaCl, 26 NaHCO3, 10 glucose, 2.5 KCl, 2CaCl2, 2 MgCl2, and 1.25 NaH2PO4. Slices were visualized with an upright microscope (Eclipse FN1; Nikon Instruments Inc., Tokyo, Japan) with infrared-differential interference contrast optics.
To examine intrinsic properties of CCKBCs, whole-cell patch-clamp recordings from CCKBCs were performed in current-clamp configuration using the pipette solution containing (in mM) 126 K-gluconate, 4 KCl, 10 HEPES, 4 ATP-Mg, 0.3 GTP-Na, 10 phosphocreatine, and 5.4 biocytin with a pH of 7.2 and osmolarity of 290–295 mOsm. Patch pipettes had resistances of 3–5 MΩ. All electrical recordings were made at 33°C using a MultiClamp 700B amplifier (Molecular Devices, San Jose, CA, USA). Electrical signals were filtered at 10 kHz using a Bessel filter and digitized at 20 kHz with a Digidata 1440A analog-digital interface (Molecular Devices). Series resistance was carefully monitored; electrical recordings were discarded if the series resistance changed >20% or reached 25 MΩ. The recorded traces were analyzed using Clampfit software (Molecular Devices, version 10.7.0) and Mini Analysis (Synaptosoft Inc., Fort Lee, NJ, USA; version 6.0.7). Subthreshold and suprathreshold membrane responses of CCKBCs were evoked from resting membrane potential by 1 second current steps (−200 to +300 pA, +50 pA increments). To examine connections of CCKBC with CA1 PCs, we performed paired whole-cell patch-clamp recordings from presynaptic CCKBCs and postsynaptic CA1 PCs as previously described (Lee et al., 2017, 2015, 2014, 2010). Membrane potential of presynaptic CCKBCs was adjusted to −60 mV. Postsynaptic CA1 PCs were recorded in voltage clamp at a holding potential of −70 mV. The CA1 PC pipette solution contained (mM) 90 K-gluconate, 40 CsCl, 10 phosphocreatine, 10 HEPES, 3.5 KCl, 2 Mg-ATP, 1.8 NaCl, 1.7 MgCl2, 0.4 Na2GTP, and 0.05 EGTA, with a pH 7.2, and osmolarity of 290 mOsm. Trains of action potentials (APs) in presynaptic CCKBCs were evoked in current clamp by 2 millisecond depolarizing current steps (+2 nA, 50 current steps at 10 Hz) every minute to evoke unitary IPSCs (uIPSCs) in CA1 PCs.
To examine overall CCK+ interneuron-mediated inhibition of CA1 PCs, we used carbachol, which is known to excite CCKBCs, but not parvalbumin-expressing basket cells (Karson et al., 2008), as previously described (Wyeth et al., 2010). To examine the carbachol-induced increase in amplitude and frequency of IPSCs in CA1 PCs, we recorded from CA1 PCs in voltage-clamp configuration at a holding potential of −70 mV. The pipette solution contained (in mM) 90 K-gluconate, 40 CsCl, 10 phosphocreatine, 10 HEPES, 5 QX314, 3.5 KCl, 2 Mg-ATP, 1.8 NaCl, 1.7 MgCl2, 0.4 Na2GTP, and 0.05 EGTA, with a pH 7.2, and osmolarity of 290 mOsm. QX314 included in the pipette solution blocks muscarinic acetylcholine receptor (mAChRs)-mediated current in CA1 PCs (e.g., persistent sodium current; Yamada-Hanff and Bean, 2013). NBQX (10 μM) and APV (40 μM) were included in the ACSF to block excitatory synaptic transmission mediated by ionotropic glutamate receptors. High-speed perfusion of the ACSF (7–8 ml/minute) was used, since high oxygen supply is critical for carbachol-induced CCK+ interneuron network activity (Hájos and Mody, 2009).
Intrinsic and synaptic properties of CCKBCs were measured as we previously described (Armstrong et al., 2016; Kang et al., 2018; Lee et al., 2017, 2015). Briefly, intrinsic properties were measured from their voltage responses to a series of hyperpolarizing and depolarizing current steps from the resting membrane potential. The following properties were examined using Clampfit software (Molecular Devices): (1) Resting membrane potential was measured from the average voltage after an equilibration period. (2) Input resistance was measured from voltage responses to small current steps (1 second duration; −50 to +50 pA in 50 pA increments). The slope of current-voltage curve was calculated at steady state (0.8–1.0 second from the start of current steps). (3) Membrane time constant was measured from voltage responses to 1 second hyperpolarizing steps (−50 pA). Single exponential functions were fitted to the voltage responses from the start of current step to the hyperpolarization peak to measure decay time constant. (4) Sag amplitude was measured from the voltage responses to 1 second hyperpolarizing steps (−300 pA). Sag was defined as the difference between the hyperpolarization peak and the average voltage at steady state (0.8–1.0 second from the start of current step). (5) AP threshold, amplitude, and half-width were measured from the first three APs evoked by a 1 second depolarizing current step (+200 pA). AP threshold was defined as the point at which the derivative of the membrane potential (dV/dt) first exceeded its mean by two standard deviations during the interspike interval. AP amplitude was measured from threshold to peak; half-width was defined as the AP duration at one half AP amplitude.
CCKBC-mediated inhibition in CA1 PCs was calculated using current responses in postsynaptic CA1 PCs to APs generated in presynaptic CCKBCs. uIPSC amplitude and success rate were measured from 2–3 trains of 50 APs (10 Hz) in CCKBCs (1 minute interval between each train of 50 APs) using Clampfit software (Molecular Devices). All current responses in CA1 PCs were visually inspected and inward current responses were defined as uIPSCs only if they started within 3 ms from the peak of presynaptic APs and their amplitude was greater than three times the root mean square (RMS) noise level. In case of CCKBC-CA1 PC pairs showing no uIPSCs (i.e., “not connected pairs”) we examined all current responses in CA1 PCs immediately after evoked APs in CCKBCs and found no uIPSCs that were relatively well timed locked to the peak of presynaptic APs in connected pairs as we previously described (Lee et al., 2017, 2015, 2014, 2010). We averaged current responses to trains of 50 APs (2–3 trains for each “not connected pair”) to further examine whether there were missed uIPSCs when each sweep was examined individually. All current averages from not connected pairs revealed no deviation from baseline, as expected. In contrast, the current averages showed prominent inward currents (>3x RMS) when both successful events and not successful events were used to generate current averages (Fig. 6D). Connection probability was calculated as the number of connected pairs/the number of tested pairs.
Figure 6.
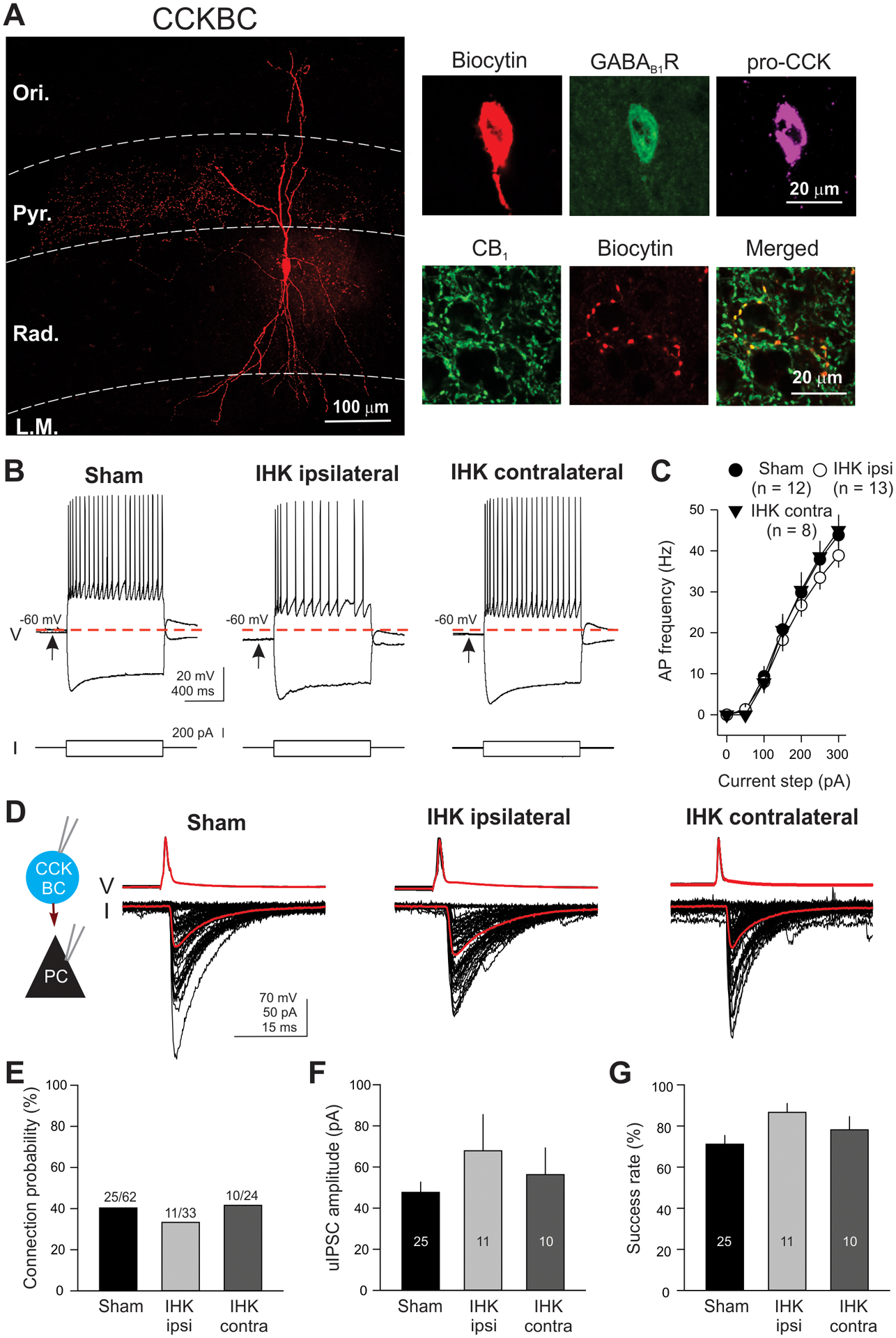
Intrinsic and synaptic properties of surviving CCKBCs in IHK mice. (A) A representative image of a CCKBC. The CCKBC was filled with biocytin during electrical recordings and was imaged using Zeiss confocal microscope. Axon terminals were localized in the stratum pyramidale, whereas dendrites covered all CA1 layers. The CCKBC expressed GABAB1R, pro-CCK, and CB1. (B) AP firing properties of CCKBCs. Evoked APs and voltage responses in CCKBCs from sham controls and IHK mice by 1 second depolarizing current step (+150 pA) and hyperpolarizing current step (−200 pA). Note that resting membrane potential of the CCKBC from the ipsilateral hippocampus of IHK mouse was more negative than that of the CCKBC from the sham control. (C) AP firing current-frequency relationship. The summary of AP firing frequency of CCKBCs from sham controls and IHK mice. “n” represents the number of CCKBCs. (D) Examples of uIPSCs in CA1 PCs evoked by APs in CCKBCs (50 APs at 10 Hz). APs in CCKBCs were evoked by 2 millisecond depolarizing current steps (2 nA) from a membrane potential of −60 mV. uIPSCs in CA1 PCs were measured at a holding potential of −70 mV. Black traces show individual currents in CA1 PCs and APs in CCKBCs and red traces show the mean of all currents and APs. (E, F, G) Summary of connection probability, uIPSC amplitude, and success rate of CCKBC-to-CA1 PC pairs. The numbers in E represent connected pairs/tested pairs. The numbers in the bars (F and G) represent the numbers of pairs.
Amplitude and frequency of spontaneous IPSCs (sIPSCs) were measured using Mini Analysis. A 2-minute control recording before bath application of carbachol and an additional 5-minute recording in carbachol for each CA1 PC were used for measuring amplitude and frequency of sIPSCs and CCK+ interneuron-mediated IPSCs, respectively. The IPSCs were manually detected using Mini Analysis and included for this study only if amplitudes of the synaptic currents were greater than three times RMS noise level as previously described (Butler et al., 2016).
2.6. Interneuron identification
All CCKBCs were identified based on firing properties (i.e., accommodating AP firing pattern), post hoc anatomical properties (i.e., preferential location of axon terminals in the CA1 PC layer), and expression of an immunohistochemical marker (i.e., cannabinoid type 1 receptor, CB1) in axon terminals, as we previously described (Kang et al., 2018; Lee et al., 2017, 2015, 2014, 2010; Lee and Soltesz, 2011). We also used other immunohistochemical markers (i.e., CCK and GABAB receptor 1, GABAB1R) and found that all tested CCKBCs (n = 8) manifested immunopositivity for CCK and GABAB1R, which is in agreement with previous findings (Lee et al., 2010; Lee and Soltesz, 2011).
Briefly, interneurons were first selected under infrared-differential interference contrast based on soma location in the stratum radiatum, large cell bodies and multipolar shape, and accommodating firing pattern. After electrical recordings with pipettes containing biocytin, the slices were fixed in a solution containing 4% paraformaldehyde and 0.2% picric acid in 0.1 M phosphate buffer (pH 7.4) and stored at 4°C for 24–48 hours. The slices were washed in phosphate buffer and cryopreserved with 30% sucrose solution. They were embedded and frozen in OCT compound (Fisher Scientific, Hampton, NH, USA), and cryosectioned using Leica Cryostat CM-1950 (Leica Biosystems, Nussloch, Germany) at 60 μm thickness. Biocytin-filled cells were stained with streptavidin conjugated with Rhodamine Red-X (Table 1; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) and subjected to acquire tile scans of Z-stacks using Airyscan mode of a Zeiss LSM880 confocal microscope (Zeiss, Jena, Germany) at the Digital Microscopy Core Facility at UAMS. Using Zen 2.3 (Blue Edition; Zeiss) Z stack images were projected two-dimensionally and stitched together in order to reconstruct biocytin-filled neurons. The sections were then subjected to immunohistochemistry using primary and secondary antibodies shown in Table 1. For primary antibodies against CB1, pro-CCK, and GABAB1R, tissues were pre-treated with 0.2 mg/ml pepsin in 0.2 N HCl at 37°C for 10–15 minutes and washed in phosphate buffer. All tissues were blocked in 5–10% normal goat serum (NGS) in Tris-buffered saline (TBS, pH 7.4) containing 0.3% Triton-X 100 for 1 hour at room temperature and followed by incubations in primary and secondary antibodies diluted in TBS with 0.3% Triton-X 100 and 1% NGS. After washing thoroughly in TBS with 0.05% Tween-20, the sections were mounted in Vectashield medium and subjected to Z-stack image acquisitions using a Nikon C1 confocal microscope (Nikon Instruments Inc.) equipped with S Fluor 40x NA 1.3 objective lens.
Table 1.
List of reagents used for fluorescence microscopy
| Reagent type | Product name | Manufacturer | Catalog number | Dilution |
|---|---|---|---|---|
| Primary antibody | Mouse anti-NeuN | Millipore | MAB377 | 1:100 |
| Rabbit anti-GFAP | Abcam | Ab7260 | 1:500 | |
| Guinea pig anti-CB1 | Frontier Institute Co. | CB1-GP-Af530 | 1:2,000 | |
| Rabbit anti-Calbindin | Swant | CB-38a | 1:500 | |
| Rabbit anti-vGAT | Synaptic Systems | 131003 | 1:1,000 | |
| Rabbit anti-proCCK | Frontier Institute Co. | CCK-pro-Rb-Af350 | 1:1,000 | |
| Mouse anti-GABAB1R | Abcam | Ab55051 | 1:5,000 | |
| Secondary antibody | Goat anti-mouse IgG (H+L), AF 488 | Invitrogen | A11001 | 1:500 |
| Goat anti-mouse IgG2a, AF 555 | Invitrogen | A21137 | 1:500 | |
| Goat anti-mouse IgG (H+L), AF 594 | Invitrogen | A11005 | 1:500 | |
| Goat anti-rabbit IgG (H+L), AF 488 | Invitrogen | A11008 | 1:500 | |
| Goat anti-rabbit IgG (H+L), AF 594 | Invitrogen | A11012 | 1:500 | |
| Goat anti-rabbit IgG (H+L), CF 633 | Biotium | 20122 | 1:500 | |
| Goat anti-Guinea Pig IgG (H+L), AF 488 | Invitrogen | A11073 | 1:500 | |
| Other | DAPI | Invitrogen | D1306 | 1:1,000 |
| Rhodamine Red-X (RRX) Streptavidin | Jackson ImmunoResearch | 016-290-084 | 1:500 |
2.7. Quantification of CCK+ interneuron somata and presynaptic terminals in the CA1 region of the hippocampus
A. Immunohistochemistry and image acquisition.
Since we performed electrical recordings using a restricted area of the ventral hippocampus (dorsal-ventral: approximately −2.4 to −3.6 mm from Bregma), we performed quantification of CCK+ interneuron number using a similar area of the ventral hippocampus from 10 sham control and 8 IHK mice. Horizontal ventral hippocampal slices (300 μm thickness) destined for immunohistochemistry were placed in fixative solution (4% paraformaldehyde, 0.2% picric acid in 0.1 M phosphate buffer, pH 7.4) and stored at 4°C for 24–48 hours. After washing in phosphate buffer, fixed brain slices were embedded in OCT Compound (Fisher Scientific) and resectioned at 30 μm thickness using a cryostat (Leica CM-1950). The sections were then subjected to immunohistochemistry using primary and secondary antibodies shown in Table 1. For primary antibodies against NeuN, glial fibrillary acidic protein (GFAP), pro-CCK, and GABAB1R, the sections were pre-treated with 0.2 mg/ml pepsin in 0.2 N HCl at 37°C for 10–15 minutes and washed in phosphate buffer. All tissues were blocked in 5–10% NGS in TBS (pH 7.4) containing 0.3% Triton-X 100 for 1 hour at room temperature, followed by incubations in primary and secondary antibodies diluted in TBS with 0.3% Triton-X 100 and 1% NGS. After washing thoroughly in TBS with 0.05% Tween-20, the sections were mounted in Vectashield medium and subjected to Z-stack image acquisitions using a Nikon C1 confocal microscope (Nikon Instruments Inc.) or a Zeiss LSM 880 inverted laser scanning confocal microscope (Zeiss). Images were analyzed with Fiji (version 1.0), ImageJ (version 1.50i), or Zen 2.3 (Blue Edition; Zeiss) software.
Immunohistochemical data were obtained from ventral hippocampal slices that were fixed by immersion and resectioned. Similar resectioning of fixed brain slices was used for quantification of GABAergic interneurons in animal models of TLE (Armstrong et al., 2016; Cossart et al., 2001). Importantly, a similar degree of CA1 GABAergic interneuron loss in animal models of TLE was reported, regardless of fixation method. For example, immunohistochemical results showed 42% and 46% loss of CA1 somatostatin-positive interneurons in a pilocarpine model of TLE when brains were fixed by transcardiac perfusion (Dinocourt et al., 2003) and when brain slices were fixed by immersion and resectioned (Cossart et al., 2001), respectively. Thus, immunostaining of interneuron markers on resectioned hippocampal slices for interneuron quantification in TLE is a well-validated quantification method.
B. Quantification of CCK+/GABAB1R+ interneuron somata in the CA1 region.
Three to four 30 μm-thick hippocampal sections were obtained from each sham control and IHK mouse. Those sections were stained with anti-pro-CCK and GABAB1R antibodies as described in section A. Using a 20x objective (Plan-Apochromat 20x/0.8, M27) controlled by a Zeiss LSM880 confocal microscope, 3×3 tile-scanned Z stack images (1275 μm × 1275 μm) of the CA1 region were acquired at 3 μm intervals (total 4 sections) and projected into 2-dimensions. Pro-CCK+/GABAB1R+ neurons from sham controls and TLE mice were manually counted in a double-blinded manner. The volume of the CA1 region used for soma quantification was measured and the number of counted neurons was normalized to tissue volume. These image analyses were conducted using the “Polygon Contour” and “Marker” modules in Zen 2.3 software (Blue Edition; Zeiss). The mean of CCK+ interneuron numbers within each animal was used to determine whether there was a difference in CCK interneuron numbers between sham control and IHK mice.
C. Quantification of CB1+/vGAT+ boutons in CA1 layers.
Three to four 30 μm-thick resectioned ventral hippocampal sections from each sham control (n = 10) and IHK mouse (n = 8) were stained with anti-CB1 and vesicular GABA transporter (vGAT) antibodies as described in section A. Using a Zeiss LSM 880 inverted confocal microscope equipped with a 100x oil objective (Plan-Apo 100x/1.46 oil DIC III), Z-stack image series were acquired for the CA1 layers (i.e., stratum oriens, stratum pyramidale, stratum radiatum, and stratum lacunosum moleculare) at 0.4 μm Z-interval per resected slice. Due to high densities of CB1+/vGAT+ boutons in the stratum pyramidale, cropped Z stack images (40 μm × 40 μm × 1.2 μm in the x, y, and z axes) were used for bouton quantification in the stratum pyramidale. Larger Z stack images (85 μm × 85 μm × 1.2 μm in the x, y, and z axes) were used for CB1+/vGAT+ bouton quantification in other CA1 layers (i.e., stratum oriens, stratum radiatum, and stratum lacunosum moleculare). The number of CB1+/vGAT+ boutons within Z-stack images of each layer was manually counted in a double-blinded manner using “Cell Counter” Plugins available in Fiji or ImageJ. The mean of CCK+ axon terminal numbers within each animal was used to determine whether there was a difference in the number of axon terminals of CCK interneurons between sham control and IHK mice.
D. Quantifying somatically projecting CB1+ axon terminals closely juxtaposed to CA1 PCs.
30 μm-thick ventral hippocampal sections from sham controls (n = 6) and IHK mice (n = 5) were stained with anti-CB1 (for CCK+ interneurons) and anti-calbindin antibodies (for CA1 PCs) as described in section A and used to generate Z-stack image series. Using a Zeiss LSM 880 inverted confocal microscope with a 100x oil objective (Plan-Apo 100x/1.46 oil DIC III), Z-stack images (0.4 μm interval) were obtained from the stratum pyramidale. As previously described (Armstrong et al., 2016), 2 image sections (85 μm × 85 μm) that were separated at least by 2.4 μm were chosen for the analysis from each Z-stack in order to avoid double counting of CB1+ boutons juxtaposed to calbindin+ somata of CA1 PCs. Encrypted images were analyzed by researchers blind to animal treatment or identity using the “Marker” and “Polygon Contour” analysis modules available in Zen 2.3 software. The sum of perimeters of calbindin+ CA1 PCs was obtained and used to normalize the number of juxtaposed CB1+ boutons in each analyzed image.
2.8. Statistical analysis
Unpaired two-tailed Student’s t-tests were used to compare two groups when the data showed a normal distribution on the basis of the Shapiro–Wilk test (Fig. 3). If the data did not show a normal distribution, a Mann–Whitney Rank Sum test (for unpaired data) was used (Fig. 3). One-way or two-way ANOVAs were followed by Tukey or Dunn’s tests (Figs. 4, 5, 6 and 7). Fisher’s exact test was used for connection probability between presynaptic CCKBCs and postsynaptic CA1 PCs (Fig. 6E). Two-sample Kolmogorov-Smirnov (K–S) test was used with Bonferroni–Holm multiple corrections (Fig. 7B, D). Two-way repeated measures ANOVA was followed by Tukey tests for mean comparisons (Fig. 8). Data were presented as mean ± SEM. A p value less than 0.05 was considered significant (*p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant). Statistical analyses were performed using SigmaPlot 14 (Systat Software Inc., San Jose, CA, USA) or Prism 9 (GraphPad Software, San Diego, CA, USA).
Figure 3.
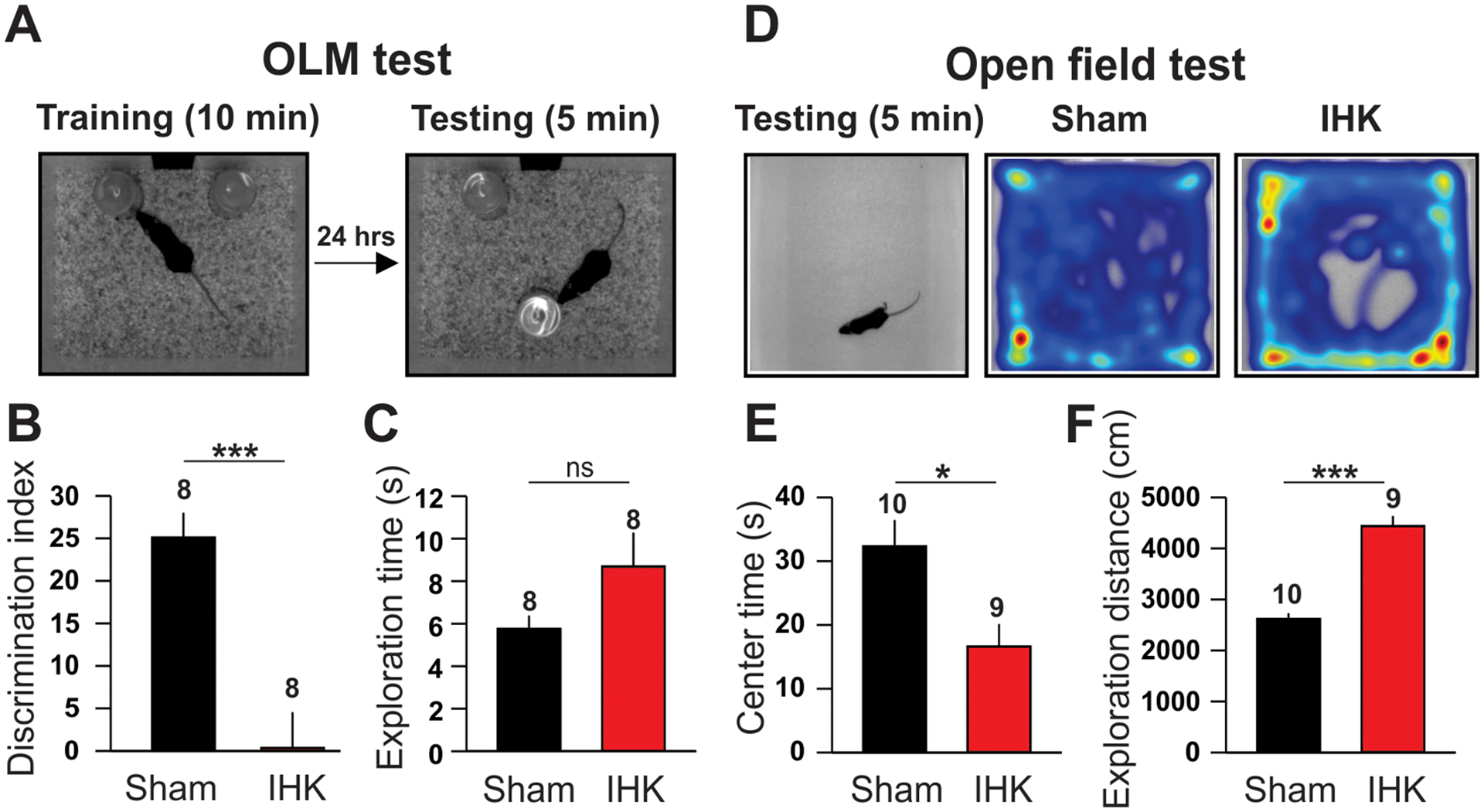
IHK mice manifest elevated anxiety-like behaviors and memory impairment. (A, B, C) Object location memory (OLM) test. A sham control mouse that explored objects during training and testing (A). Summary of DI (B) and exploration time (C). (D, E, F) Open field test. Heatmap of location of a sham control and an IHK mouse during testing period (D). Summary of center time (E) and exploration distance (F). The numbers above the bars indicate the number of mice used for OLM test or open field test; * indicates p < 0.05; *** indicates p < 0.001; ns, not significant.
Figure 4.
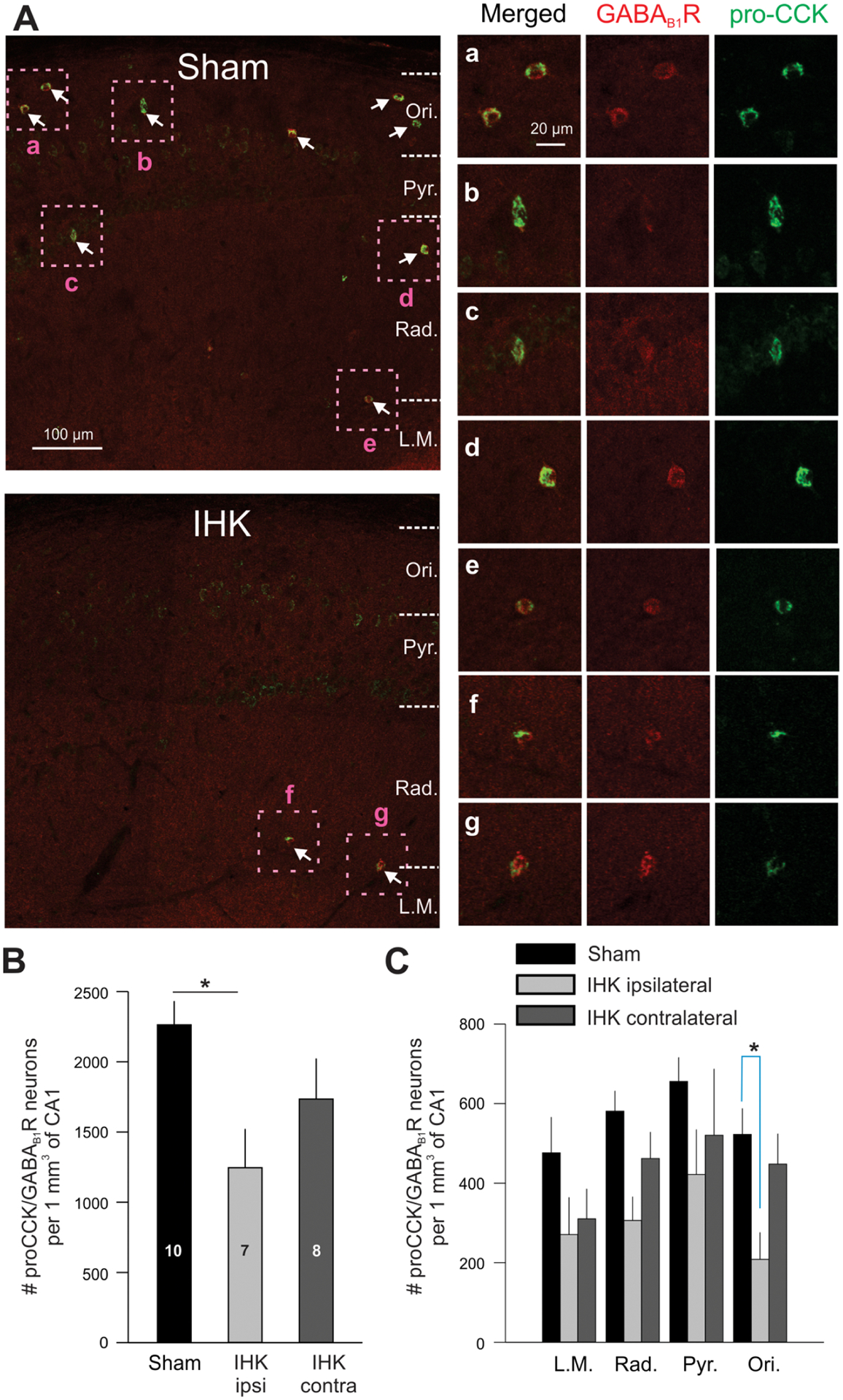
Reduced number of CCK+ interneurons in the CA1 regions of ventral hippocampi from IHK mice. (A) CCK+ interneurons were identified based on double immunopositivity for pro-CCK (green)/GABAB1R (red). Representative confocal images of the CA1 region of ipsilateral hippocampi from a sham control (Sham) or IHK mouse (IHK). In sham control, somata of pro-CCK+/GABAB1R+ neurons (marked with arrows) widely distributed throughout most CA1 layers. The number of pro-CCK+/GABAB1R+ neurons was greatly reduced in TLE mouse. Magnified boxed areas (a-g) display immunoreactivities for pro-CCK and GABAB1R. Ori, stratum oriens; Pyr., stratum pyramidale; Rad, stratum radiatum; L.M., stratum lacunosum moleculare. (B) Summary of the total numbers of interneurons double immunopositive for pro-CCK/GABAB1R in the CA1 region. Note that we quantified those from similarly sized CA1 volumes (no statistical differences in CA1 volumes between sham control and IHK mice for both ipsi- and contralateral hippocampi). The numbers within the bars indicate the number of animals used for these experiments. (C) When analyzed for each CA1 layer, the number of pro-CCK/GABAB1R double positive interneurons was significantly lower in the stratum oriens of ipsilateral hippocampi of IHK mice relative to sham controls. The ipsi- and contralateral data from sham control mice were combined. * indicates p < 0.05.
Figure 5.
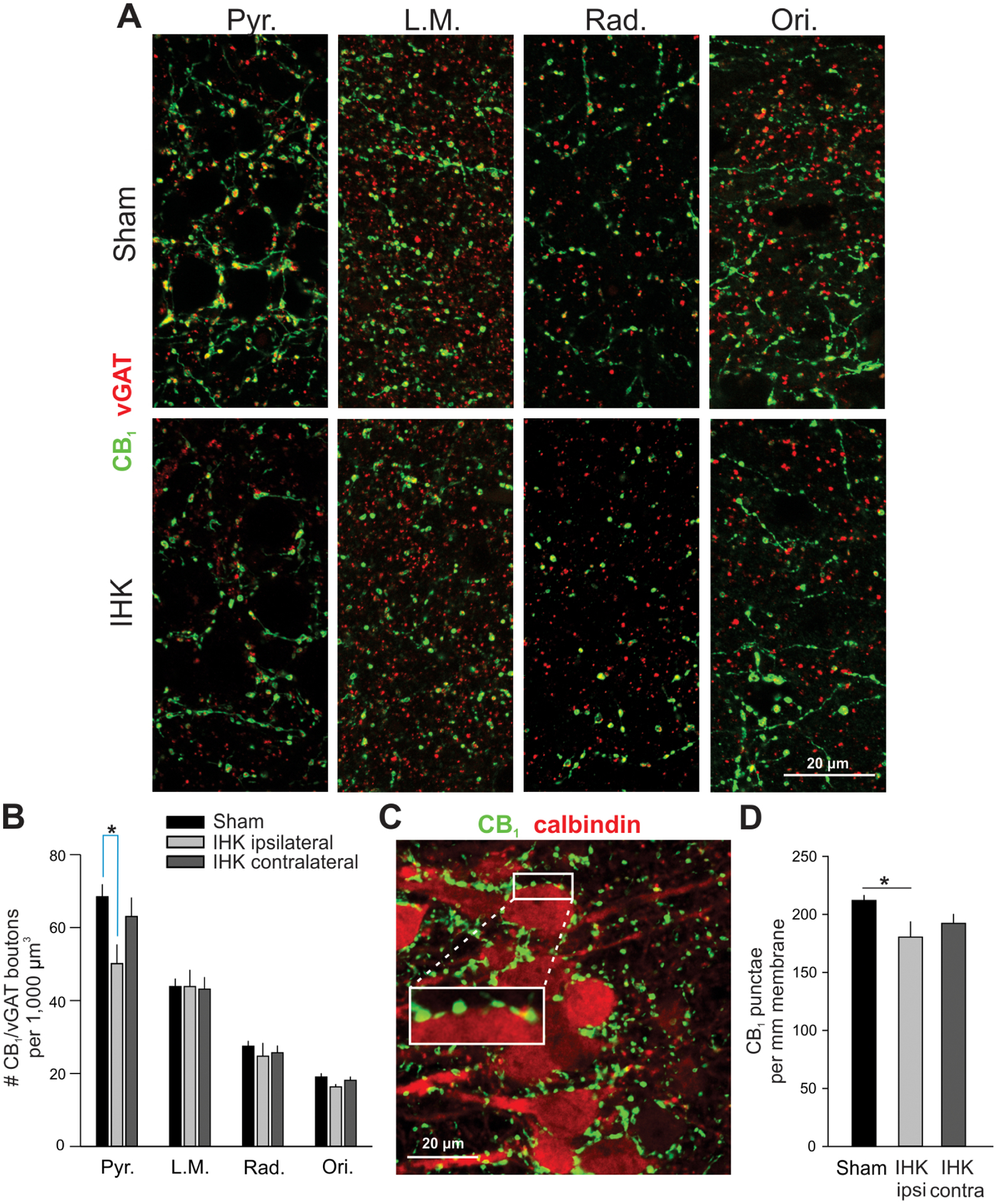
Reduced perisomatic innervation of CA1 PCs by CCK+ interneurons in IHK mice. (A) Representative confocal images showing immunostaining of CB1 and vGAT in ipsilateral ventral hippocampi of sham control and IHK mice. (B) Summary of the number of axon terminals double immunopositive for CB1/vGAT in all CA1 layers; 36–38, 14–15, and 14–16 resected hippocampal slices were used for sham control, IHK (ipsilateral), and IHK (contralateral) hippocampi, respectively, from 10 sham control and 8 IHK mice. Pyr., stratum pyramidale; L.M., stratum lacunosum moleculare; Rad, stratum radiatum; Ori, stratum oriens. (C) A representative Z-stack projection image of CA1 stratum pyramidale in the ventral ipsilateral hippocampus from a sham control mouse showing immunostaining of CB1 (green) and calbindin (red). CB1+ boutons were closely juxtaposed on the surface of somata of calbindin+ CA1 PCs. The area of white rectangle was magnified to show juxtapositions of CB1+ boutons along the surface of CA1 PCs. (D) Summary of the number of CB1+ axon terminals per mm membrane of CA1 PCs. The numbers of CB1+ punctae were normalized by perimeters of CA1 PCs for comparisons. The mean of the numbers of CB1+ terminals within each resected hippocampal slice was used to determine whether there was a difference in the number of CB1+ terminals among sham controls (n = 44), IHK ipsilateral (n = 16), and IHK contralateral (n = 20). * indicates p < 0.05.
Figure 7.
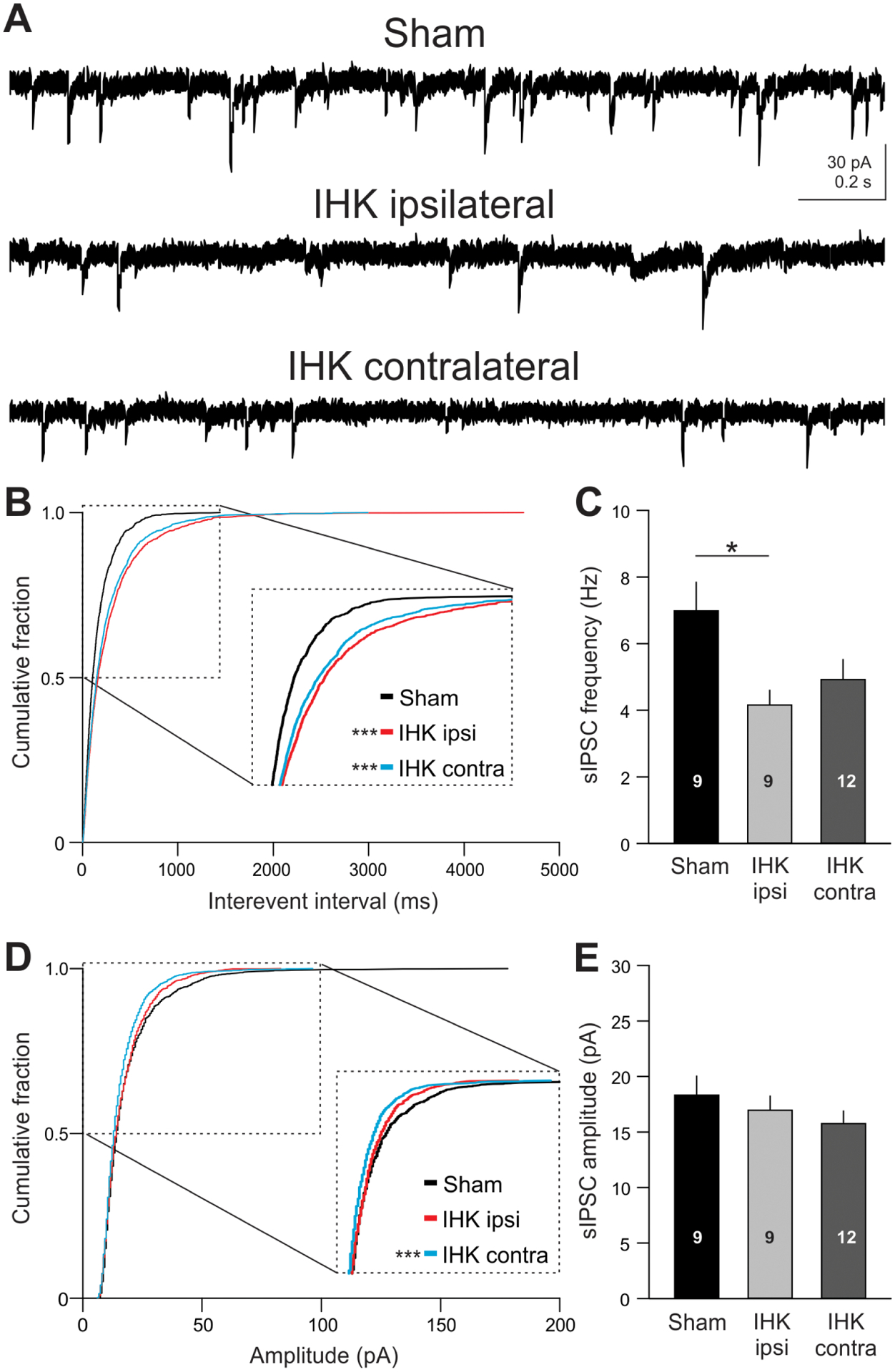
Reduced frequency and amplitude of sIPSCs in CA1 PCs in IHK mice. (A) Examples of sIPSCs in CA1 PCs in ventral hippocampi from sham control and IHK mice. (B, D) Cumulative probability plots of interevent interval and amplitude of individual sIPSCs in CA1 PCs from sham controls (1539 sIPSCs from 9 CA1 PCs), IHK ipsi- (1539 sIPSCs from 9 CA1 PCs), and IHK contralateral (2052 sIPSCs from 12 CA1 PCs). (C, E) Summary of sIPSC frequency and amplitude of each cohort using the means of sIPSC frequency and amplitude measured within each CA1 PC, respectively. * and *** indicate p < 0.05 and p < 0.001, respectively, for IHK ipsi- or contralateral vs sham controls.
Figure 8.
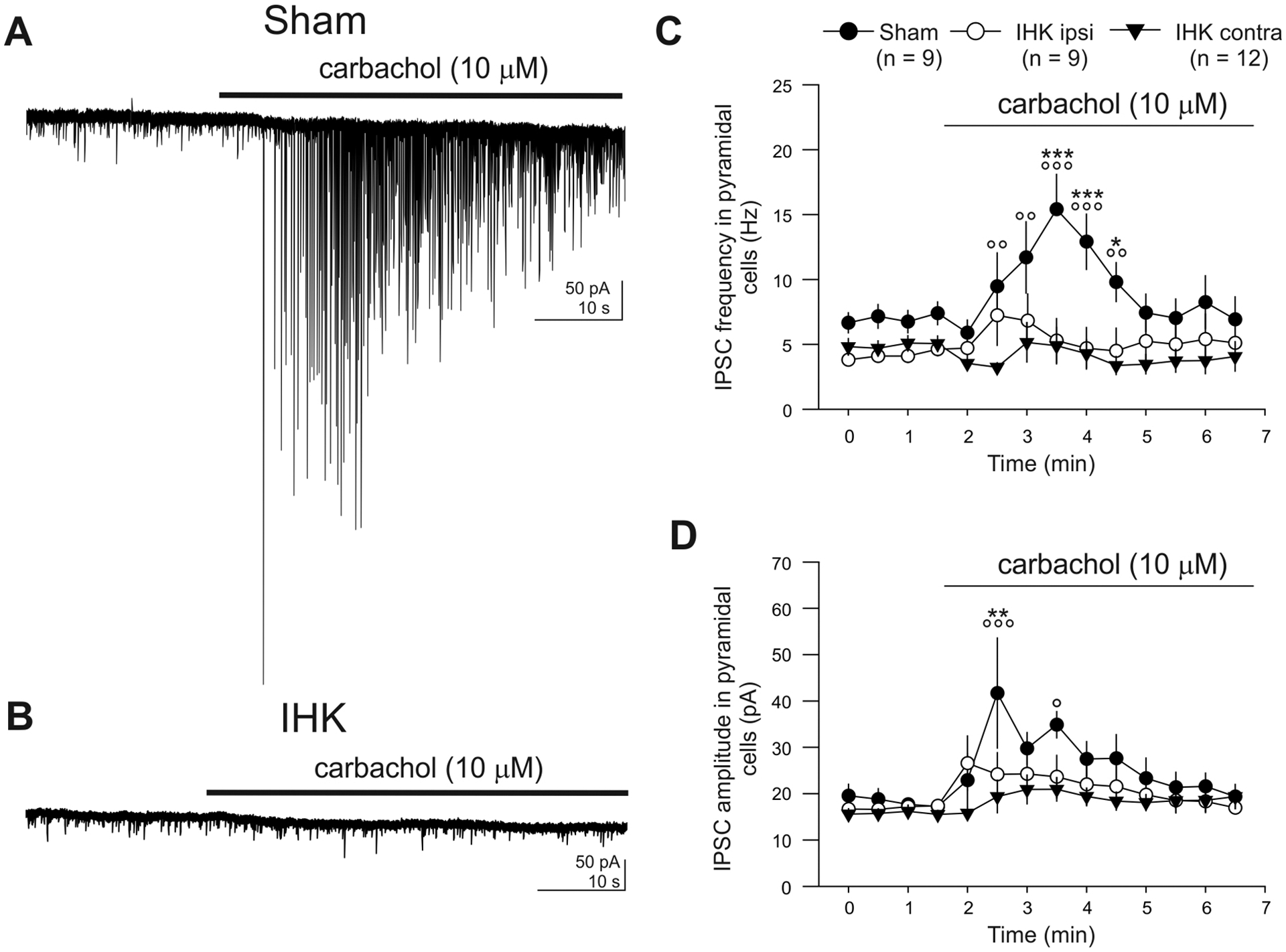
A lack of carbachol-mediated increases in IPSC frequency and amplitude in CA1 PCs in IHK mice. (A, B) Current records in CA1 PCs before and during carbachol (10 μM) application made in ipsilateral hippocampal slices from a sham control or an IHK mouse. Note that carbachol caused an increase in frequency and amplitude of IPSCs in the CA1 PC from the sham control, but not from the IHK mouse. (C, D) Summary of frequency and amplitude of IPSCs before and during carbachol application. The numbers for sham (solid circles), IHK (ipsi; open circles), and IHK (contra; solid triangles) indicate the numbers of CA1 PCs tested for these experiments. Two-way ANOVA with repeated measures were followed by post hoc Tukey tests for mean comparisons. *, **, and *** indicate p < 0.05, p < 0.01, and p < 0.001 vs IHK ipsilateral, respectively; °, °°, and °°° indicate p < 0.05, p < 0.01, and p < 0.001 vs IHK contralateral, respectively.
3. Results
3.1. IHK mouse model of TLE reproduced key properties of human TLE
The IHK mouse model of TLE reproduces key features of human TLE: Hippocampal sclerosis, behavioral and electrographic seizures, and cognitive dysfunction (Bui et al., 2018; Krook-Magnuson et al., 2013; Levesque and Avoli, 2013). In this study, we first determined whether our model reproduced previous findings. Behavioral seizures were observed after IHK-injected mice recovered from anesthesia. The mice initially displayed low levels of motor seizures such as repetitive mouth movement, head twitching, unilateral arm and leg clonus, tail stiffening, and freezing behaviors. Within 1 to 2 hours after the surgery, 84% of KA-injected mice (58/69) developed their first Racine stage 5 seizure, and periodically experienced recurring Racine stage 5 seizures within an hour after the first Racine stage 5 seizure. 10.1% of IHK mice (7/69) died from status epilepticus on the day of IHK injection (Fig. 1A). To examine whether spontaneous behavioral seizures developed over time in IHK mice, we video-monitored mice continuously for 3 days at around 3 weeks after IHK injection, and found that 90.0% of IHK mice (54 out of 60 mice) displayed two or more spontaneous Racine stage 3–5 seizures. These mice were referred as to epileptic mice. All 54 mice manifested Racine stage 4–5 seizures at least once (4.3 ± 0.6 Racine stage 4–5 seizures over 3 days; n = 54). In order to determine whether spontaneous recurrent behavioral seizures in epileptic mice were accompanied by electrographic seizures, we employed continuous 3-day video-EEG monitoring using a separate group of IHK mice (n = 8) and saline-injected sham controls (n = 4) that were implanted with intrahippocampal EEG electrodes. In five IHK mice, Racine stage 4–5 behavioral seizures were accompanied by electrographic seizures displaying large-amplitude, high-frequency polyspikes followed by postictal depression (Fig. 1B; Twele et al., 2017). The remaining three IHK mice did not display Racine stage 4–5 behavioral seizures. However, they manifested high-voltage sharp waves and hippocampal paroxysmal discharges, which are the most common types of focal seizures in the IHK mouse model of TLE (Twele et al., 2017, 2016; Zeidler et al., 2018). In contrast, none of the sham controls showed electrographic or behavioral seizures, hippocampal paroxysmal discharges, or high-voltage sharp waves. Thus, our IHK mouse model of TLE developed spontaneous, severe, and recurrent behavioral and electrographic seizures by three weeks after IHK injection.
Figure 1.
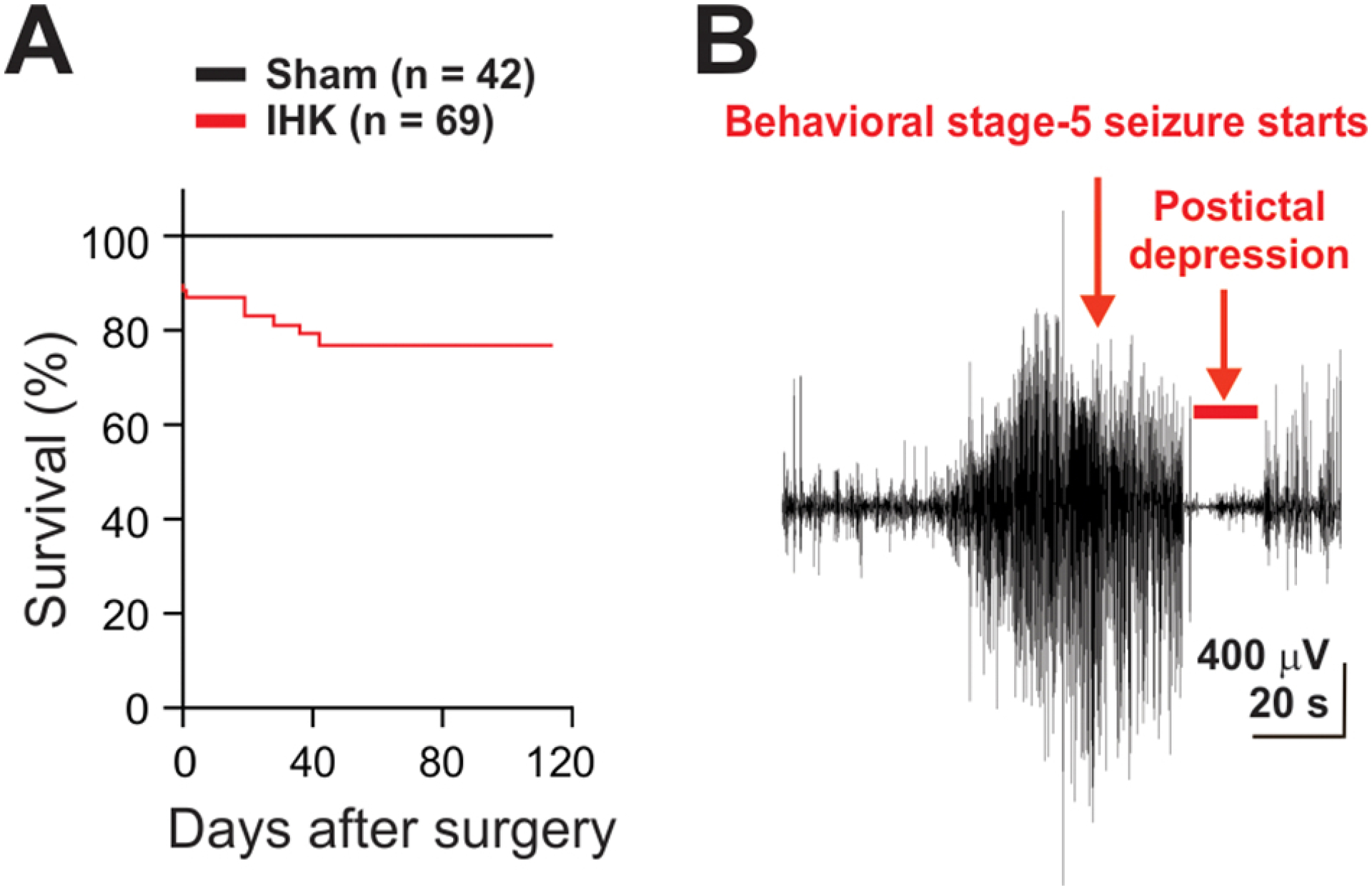
IHK mouse model of temporal lobe epilepsy. (A) Percent rate of survival was monitored for 4 months after an intrahippocampal saline or KA injection. (B) A representative example of a generalized electrographic seizure observed in an IHK mouse, accompanied by severe convulsive (Racine stage 5) seizures.
We next turned to histopathology associated with TLE. Hippocampal sclerosis, characterized by severe loss of CA1/CA3c PCs, granule cell dispersion, and gliosis, was observed in the ipsilateral, but not contralateral, dorsal hippocampi of IHK mice (n = 5 IHK mice; Fig. 2B, C). Such severe neuropathological deficits were not present in the ipsilateral ventral hippocampus of IHK mice (Fig. 2E). A mild increase in GFAP immunostaining was observed in the contralateral dorsal hippocampi of IHK mice (Fig. 2C). As expected, bilateral hippocampi from sham controls showed no histopathological abnormalities (n = 3 saline injected mice; Fig. 2A, D). These results indicated that our IHK mouse model reproduced the typical pathology of TLE, as previously described (Andersson et al., 1991; Groticke et al., 2008; Krook-Magnuson et al., 2013; Zeidler et al., 2018).
Figure 2.
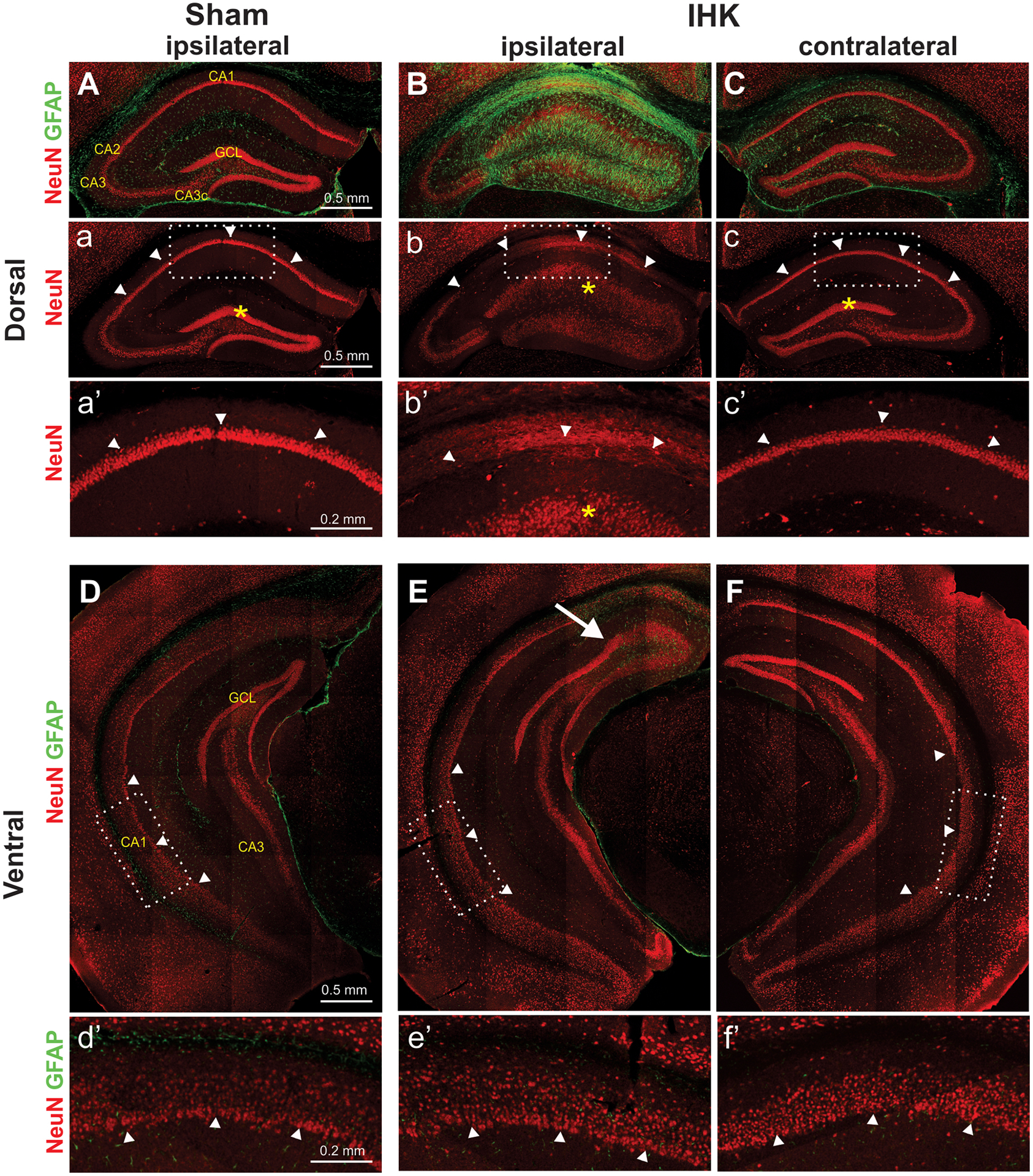
Neuropathological findings in the unilateral dorsal IHK mouse model. Double immunostaining of NeuN and GFAP of hippocampi from sham control and TLE mice revealed that severe hippocampal sclerosis occurred mostly in the ipsilateral dorsal hippocampus. (A, B, C) Severe damage such as loss of CA1 and CA3c pyramidal cells, granule cell dispersion in the dentate gyrus, and GFAP-labeled gliosis was found only in the ipsilateral dorsal hippocampus of IHK mice (B, b, b’), but not in the ipsilateral hippocampus of sham controls (A, a, a’) or in the contralateral hippocampus of IHK mice (C, c, c’). These images of dorsal hippocampi were from the KA injection site (anterior-posterior: −2.0 mm from the bregma). (D, E, F) The bilateral ventral hippocampi of IHK mice appeared to be well preserved without significant sclerotic damages. There was no obvious loss of CA1 PCs in the ipsilateral ventral hippocampus from IHK mice. The images of the ventral hippocampus were taken approximately 3.2 mm posterior to bregma. The rectangular areas in “a” to “c” and “D” to “F” were enlarged and displayed in a’ to c’ and d’ to f’, respectively. Arrowheads indicate pyramidal cell layers of the CA1 region, and asterisks indicate the granule cell layer (GCL) of the dentate gyrus. The arrow in E indicates the border of a sclerotic lesion expanded from the KA injection site.
Epilepsy is characterized not only by spontaneous seizures, but also by its cognitive/behavioral comorbidities (e.g., impaired memory, anxiety, and depression; Amlerova et al., 2013; Boro and Haut, 2003; Cánovas et al., 2011; Elger et al., 2004). The dorsal hippocampus IHK mouse model displays impaired hippocampus-dependent memory (Bui et al., 2018; Groticke et al., 2008; Zeidler et al., 2018). Thus, we examined whether our IHK mouse model of TLE manifested impaired hippocampus-dependent memory using the OLM test (Bui et al., 2018; Zeidler et al., 2018). Sham controls and IHK mice spent similar amounts of time exploring objects during the 5-minute testing period. The ability of mice to discriminate between novel and unchanged object position (DI) in the OLM test was significantly lower in IHK mice compared to sham controls (p < 0.001; Fig. 3B; DI of IHK mice, from −28.1 to +9.7, n = 8; DI of sham control mice, from +14.3 to +36.9, n = 8), suggesting that IHK mice were less able to discriminate novelty of object locations. Interestingly, an IHK mouse that displayed an extremely negative DI value (−28.1), a sign of behavioral abnormality with extreme avoidance of novel locations called ‘neophobia’, died 42 days after IHK injection. Next, we examined whether epileptic mice exhibited anxiety-like behaviors using the open field test (Prut and Belzung, 2003). When exposed to the open field, IHK mice displayed increased locomotive activity, indicative of elevated anxiety-like behaviors, compared to sham controls (Fig. 3D, 3F). Specifically, IHK mice traveled longer distances at a faster speed (p < 0.001), while exploring the center of the field significantly less than sham controls did (p < 0.05; Fig. 3E). These results suggest that our epileptic mice have both cognitive dysfunction and anxiety-like behaviors.
In summary, our IHK mouse model of TLE reproduced key properties of TLE: Spontaneous behavioral and electrographic seizures, hippocampal sclerosis, elevated anxiety-like behaviors, and hippocampus-dependent memory dysfunction.
3.2. Loss of CA1 CCK+ interneurons in the ventral hippocampus of dorsal IHK mice
Spontaneous seizures and compromised hippocampal network oscillations in TLE can arise from neuronal circuits remote from the epileptic focus (Bragin et al., 1999; Dugladze et al., 2007), suggesting long-term neuronal circuit plasticity of remote brain regions in TLE that may play a causal role in hyperexcitability and cognitive dysfunction. However, reorganization of neuronal circuits remote from the seizure focus in TLE is largely unknown. Unlike other commonly used rodent models of TLE induced by systemic injection of chemoconvulsants (e.g., pilocarpine and kainate), the unilateral dorsal IHK mouse model produced localized hippocampal sclerosis that was limited to ipsilateral dorsal hippocampus, without involving the ventral or contralateral hippocampus (Fig. 2). However, prior studies showed that ventral hippocampi manifested reduced theta rhythms in the dorsal IHK mouse model (Dugladze et al., 2007), suggesting that the ventral hippocampus, remote from the sclerotic dorsal hippocampus, also undergoes neuronal circuit reorganization. Since CCKBCs critically participate in hippocampal theta rhythmogenesis and regulate neuronal excitability (Bezaire et al., 2016; Kang et al., 2018; Klausberger et al., 2005), we used the dorsal IHK mouse model to determine whether CCKBCs were vulnerable in the ventral hippocampus.
We employed GABAB1R and pro-CCK immunostaining in ventral hippocampi to identify CCK+ interneurons, since both are expressed in the somata of CCK+ cells, including CCKBCs (Booker et al., 2017; Sloviter et al., 1999). We found that GABAB1R+/pro-CCK+ positive neurons were localized in all layers of the ventral CA1 (Fig. 4). Significantly fewer GABAB1R+/pro-CCK+ neurons were seen in the ventral CA1 region as a whole in ipsilateral, but not contralateral, hippocampi of IHK mice compared to sham controls (Fig. 4A, B; sham controls vs IHK ipsilateral, p < 0.05; sham controls vs IHK contralateral, p = 0.224). Next, we examined whether the reduced cell number was limited to specific ventral CA1 layers. Our immunostaining results showed that the overall number of GABAB1R+/pro-CCK+ neurons was reduced in all layers of the ventral CA1 region of both ipsilateral and contralateral hippocampi from IHK mice compared to sham controls (Fig. 4C). Cell numbers in the ipsilateral stratum oriens from IHK mice were significantly different from those from sham controls (p < 0.05; Fig. 4C). These results suggest loss of CCK+ interneurons in the ventral hippocampi from unilateral dorsal IHK mice.
3.3. Reduced innervation of CA1 stratum pyramidale by CCK+ interneurons in IHK mice
In order to examine changes in CCK+ interneuron circuits of IHK mice, ipsilateral and contralateral ventral hippocampi were stained with antibodies against CB1 and vGAT, since CB1 receptors are selectively expressed in CCK+ interneurons, but not other types of hippocampal GABAergic interneurons (e.g., parvalbumin-expressing basket cells; Katona et al., 1999; Lee et al., 2014). We blindly quantified the number of CB1+/vGAT+ boutons in ventral CA1 layers of IHK mice and sham controls. CB1+/vGAT+ boutons were found in all CA1 layers, most densely concentrated in the stratum pyramidale (Fig. 5A, B). Our imaging revealed a significant reduction in the number of CB1+/vGAT+ boutons in the stratum pyramidale of ipsilateral hippocampi from IHK mice, compared to sham controls (p < 0.05; Fig. 5A, B). There was a slight but statistically non-significant reduction in the number of CB1+/vGAT+ boutons in the stratum pyramidale of contralateral hippocampi from epileptic mice compared to sham controls (p = 0.815; Fig. 5B). In contrast to the stratum pyramidale, other CA1 layers of both ipsilateral and contralateral hippocampi (i.e., stratum oriens, stratum radiatum, and stratum lacunosum moleculare) displayed similar numbers of CB1+ inhibitory boutons between sham control and IHK mice (Fig. 5B).
Given the reduction in the number of CB1+ inhibitory boutons in the stratum pyramidale in the ipsilateral ventral CA1 region of IHK mice, we expected that ipsilateral CA1 PCs would be innervated by fewer CB1+ inhibitory terminals in IHK mice than in sham controls. To assess this, we stained the somata of CA1 PCs with antibodies against calbindin, and counted the number of CB1+ punctae along the surfaces of calbindin+ somata of CA1 PCs (Fig. 5C). As expected, the number of CB1+ punctae per given length of circumference of ipsilateral, but not contralateral, CA1 PCs was significantly lower in IHK mice compared to sham controls (p < 0.05; Fig. 5D). These anatomical results were generated using 556 CA1 PCs from 6 sham control mice, 187 ipsilateral CA1 PCs from 4 IHK mice, and 235 contralateral CA1 PCs from 5 IHK mice. These results are consistent with the hypothesis that perisomatic innervation of CA1 PCs by CCKBCs is reduced in the ipsilateral, but not contralateral, ventral hippocampi of IHK mice compared to sham controls.
3.4. Intrinsic and synaptic properties of surviving CCKBCs in IHK mice
To examine whether surviving CCKBCs in the ventral CA1 region of IHK mice show major changes in intrinsic biophysical properties, APs in CCKBCs were evoked by 1 second depolarizing current steps (from +50 pA to +300 pA, +50 pA increments), when they were at their resting membrane potential. The mean AP firing frequency of CCKBCs was measured (Fig. 6A, B, and C). There was no significant difference in AP firing frequency among sham controls, IHK ipsilateral, and IHK contralateral groups (sham controls: 9.4 ± 2.5 Hz at +100 pA, 43.8 ± 3.1 Hz at +300 pA, n = 12; IHK ipsilateral: 8.0 ± 2.4 Hz at +100 pA, 38.4 ± 2.9 Hz at +300 pA, n = 13; IHK contralateral: 7.9 ± 2.6 Hz at +100 pA, 45.0 ± 3.8 Hz at +300 pA, n = 8; p > 0.05). Next, we examined whether surviving CCKBCs exhibit significant changes in subthreshold membrane responses evoked by 1 second hyperpolarizing and depolarizing current steps. We found that there were no differences in input resistance, membrane time constant, or sag amplitude between sham controls and IHK mice (Table 2). However, the resting membrane potential of CCKBCs from the ipsilateral, but not contralateral, ventral hippocampi of IHK mice was significantly hyperpolarized compared to sham controls (Fig. 6B and Table. 2; sham controls: −60.0 ± 1.0 mV, n = 12; IHK ipsilateral: −64.2 ± 1.2 mV; n = 13; IHK contralateral: −62.6 ± 1.1 mV, n = 8; sham controls vs IHK ipsilateral, p < 0.05). Thus, we further examined whether surviving CCKBCs manifest alterations in other intrinsic properties. We found that the threshold, amplitude, and half-width of APs in CCKBCs were not significantly different among sham control, IHK ipsilateral, and IHK contralateral groups (Table 2). These results collectively showed that surviving CCKBCs in the ventral CA1 did not undergo major changes in their intrinsic properties.
Table 2.
Intrinsic properties of CCKBCs in sham control and IHK mice
| Animal group | Sham (n=12) | IHK (Ipsilateral; n=13) | IHK (Contralateral; n=8) | Significant differences One-way ANOVA |
|---|---|---|---|---|
| RMP (mV) | −60.0 ± 1.0 | −64.2 ± 1.2* | −62.6 ± 1.1 | p < 0.05 |
| Rinput (MΩ) | 189.4 ± 15.0 | 189.6 ± 15.6 | 197.6 ± 28.9 | ns |
| τmembrane (ms) | 17.6 ± 1.6 | 21.1 ± 1.9 | 16.9 ± 1.4 | ns |
| Sag amplitude (mV) | 8.8 ± 1.0 | 8.2 ± 1.1 | 7.9 ± 1.1 | ns |
| AP threshold (mV) | −37.7 ± 1.4 | −41.1 ± 1.1 | −39.0 ± 1.1 | ns |
| AP amplitude (mV) | 61.7 ± 2.7 | 68.1 ± 2.2 | 59.3 ± 3.5 | ns |
| AP half-width (ms) | 0.71 ± 0.03 | 0.66 ± 0.03 | 0.69 ± 0.03 | ns |
| AP frequency (Hz) | 43.8 ± 3.1 | 38.8 ± 2.9 | 45.0 ± 3.8 | ns |
See the methods section for descriptions of each of the measured intrinsic properties. The n values represent number of CCKBCs. AP frequency was measured from APs evoked by 1 s depolarizing current steps (300 pA). Abbreviations: RMP, resting membrane potential; Rinput, input resistance; τmembrane, membrane time constant; ns, not significant.
significantly different vs sham controls (post hoc Tukey test).
Next, we performed paired recordings from CCKBCs and CA1 PCs to examine whether surviving CCKBCs in the ventral CA1 region have altered synaptic connectivity properties in IHK mice. Paired recordings revealed that there were no differences in proportion of presynaptic CCKBC and postsynaptic CA1 PC pairs showing uIPSCs between sham controls and IHK mice (i.e., connection probability, Fig. 6D, E; sham controls: connected/tested = 25/62; IHK ipsilateral: 11/33; IHK contralateral: 10/24; p > 0.05 for both sham controls vs IHK ipsilateral and sham controls vs IHK contralateral). In connected pairs of CCKBCs and CA1 PCs, APs in CCKBCs produced similar amplitude of uIPSCs between sham controls and IHK mice (Fig. 6F; sham controls: 47.7 ± 5.2 pA, n = 25; IHK ipsilateral: 67.9 ± 17.8 pA, n = 11; IHK contralateral: 56.3 ± 13.2 pA, n = 10; p > 0.05). The proportion of APs in each CCKBC producing uIPSCs in each CA1 PC (i.e., success rate) was similar between sham controls and IHK mice (Fig. 6G; sham controls: 71.1 ± 4.4 %, n = 25; IHK ipsilateral: 86.6 ± 4.5 %, n = 11; IHK contralateral: 78.1 ± 6.6 %, n = 10; p > 0.05), suggesting that there were no changes in probability of AP-induced GABA release from CCKBCs in IHK mice. These results showed that surviving CCKBCs did not develop major changes in intrinsic and synaptic properties in concert with development of TLE.
3.5. Reduced inhibition of CA1 PCs in IHK mice
How does the reduction in CCK+ interneuron number (Fig. 4) and their innervation of CA1 PCs (Fig. 5) translate into functional changes in CCK+ interneuron-mediated inhibition of CA1 PCs in IHK mice? We performed whole-cell patch-clamp recordings to measure frequency and amplitude of sIPSCs in CA1 PCs of the ventral hippocampus. Interevent interval of sIPSCs in both ipsilateral and contralateral CA1 PCs from IHK mice was significantly increased compared to sham controls (Fig. 7A, B; p < 0.001 for IHK ipsi- and contralateral, K–S test). In order to ensure that there was no bias towards CA1 PCs with higher sIPSC frequency, we identified the cell with the smallest number of sIPSCs (n = 171) and used the same number of randomly selected sIPSCs from each CA1 PC from sham controls (n = 9, 1539 sIPSCs), IHK ipsilateral (n = 9, 1539 sIPSCs), and IHK contralateral (n = 12, 2052 sIPSCs), respectively, to generate cumulative distribution comparisons (Fig. 7B). In addition, we measured the mean sIPSC frequency from each CA1 PC. As expected, sIPSC frequency in ipsilateral CA1 PCs from IHK mice was significantly lower than that from sham controls (Fig. 7C; sham controls: 6.9 ± 0.8 Hz, n = 9; IHK ipsilateral: 4.1 ± 0.4 Hz, n = 9; p < 0.05). sIPSC frequency in contralateral CA1 PCs from IHK mice was numerically lower compared to sham controls, but the difference did not reach statistical significance (Fig. 7C; IHK contralateral: 4.9 ± 0.6 Hz, n = 12; p = 0.087). Cumulative distributions of sIPSC amplitude were also compared among sham controls, IHK ipsilateral, and IHK contralateral. sIPSC amplitude in CA1 PCs from IHK contralateral, but not from IHK ipsilateral, was significantly smaller than that in sham controls (Fig. 7D; p < 0.001 for IHK contralateral vs sham controls, K–S test). The mean sIPSC amplitude in CA1 PCs from sham controls was numerically greater than that in ipsilateral and contralateral CA1 PCs from IHK mice, but the difference did not reach statistical significance (Fig. 7E; sham controls: 18.3 ± 1.7 pA, n = 9; IHK ipsilateral: 16.9 ± 1.3 pA, n = 9; IHK contralateral: 15.7 ± 1.1 pA, n = 12; p > 0.05). These results are consistent with our anatomical evidence showing loss of CCK+ interneurons in IHK mice.
We next used carbachol to determine whether there was a global reduction in CCK+ interneuron-mediated inhibition of CA1 PCs in the ventral hippocampus from IHK mice compared to sham controls, since carbachol excites CCK+ interneurons, but not other CA1 interneuron types (Cea-Del Rio et al., 2011; Karson et al., 2008; Wyeth et al., 2010). Carbachol induced increases in IPSC frequency and amplitude in CA1 PCs from sham controls (Fig. 8A). In contrast, carbachol produced no increase in IPSC frequency and amplitude in CA1 PCs from IHK mice (Fig. 8B). As for the effect of carbachol on IPSC frequency, two-way ANOVA analysis with repeated measures showed a significant IHK treatment main factor (p < 0.01), time main factor (p < 0.001), and IHK treatment × time interaction (p < 0.001). Post hoc Tukey test revealed that sham controls transiently showed higher IPSC frequency compared to both IHK ipsi- and contralateral (Fig. 8C). As for the effect of carbachol on IPSC amplitude, two-way ANOVA analysis with repeated measures showed a significant time main factor (p < 0.001) and IHK treatment × time interaction (p < 0.05), but not for IHK treatment main factor (p = 0.15). Post hoc Tukey test revealed that sham controls transiently showed larger IPSC amplitude in the beginning of carbachol application compared to both IHK ipsi- and contralateral (Fig. 8D). These findings suggest that CCK+ interneurons are partially lost and their loss contributes to an overall decrease in CCK+ interneuron-mediated functional inhibition of CA1 PCs in the ventral hippocampus from dorsal IHK mice, but synaptic connectivity and strength from surviving CCKBCs to CA1 PCs were not altered.
4. Discussion
In this study, we found both structural and functional loss of CCK+ interneurons in ventral hippocampi in the dorsal IHK mouse model of TLE. The main findings in epileptic mice are: 1) CCK+ interneuron numbers were reduced in ventral hippocampi; 2) Axon terminals from CCKBCs targeting perisomatic regions of CA1 PCs were reduced in ventral hippocampi; 3) Surviving CCKBCs showed no major changes in their intrinsic and synaptic properties or connectivity with CA1 PCs; and 4) Carbachol-induced CCK+ interneuron-mediated inhibition of CA1 PCs in ventral hippocampi was reduced. Together, these novel results demonstrate vulnerability of CCK+ interneurons remote from the sclerotic region of the hippocampus in TLE.
4.1. Cholinergic modulation of CCK+ interneurons and TLE
In agreement with previous studies (Wyeth et al., 2010) we showed that the amplitude and frequency of carbachol-induced IPSCs in CA1 PCs were reduced in TLE (Fig. 8). It is likely that loss of CCK+ interneurons and terminals in dorsal IHK mice (Fig. 4, 5) contributes to the decrease in carbachol-induced IPSCs. We could not exclude the possibility that changes in cholinergic excitation of CCK+ interneurons (e.g., reduced cholinergic excitation of CCK+ interneurons; see below for known cholinergic modulation of CCK+ interneurons) contribute to the decrease in frequency and amplitude of carbachol-induced IPSCs, since a significant portion of CCK+ interneurons in the CA1 (55.0%) survive in dorsal IHK mice, compared to those in sham controls (Fig. 4), and surviving CCKBCs do not exhibit major changes in their intrinsic and synaptic properties (Fig. 6 and Table 2).
Activation of acetylcholine receptors in CCK+ interneurons produces an increase in membrane potential, AP duration, and the frequency of IPSCs (Alger et al., 2014; Lawrence, 2008; McQuiston, 2014). Given that acetylcholine- or carbachol-induced IPSCs in principal cells (e.g., CA1 PCs) are sensitive to CB1 agonists (Karson et al., 2008; Pitler and Alger, 1992; Trettel et al., 2004; Wyeth et al., 2010) excitatory effects of acetylcholine or carbachol on CCK+ interneurons are likely a primary cause of the induced IPSCs, since CCK+ interneurons, but not other types of interneurons, express CB1 receptors on axon terminals (Katona et al., 1999; Lee et al., 2010; Soltesz et al., 2015). Specifically, CCKBCs and SCA cells are a major target of acetylcholine modulation, since they strongly express M1 and M3 receptors (Cea-del Rio et al., 2010; Cea-Del Rio et al., 2011). A subset of CCK+ interneurons express nicotinic acetylcholine receptor (nAChR) α7 subunit-encoding mRNA (Morales et al., 2008). Thus, nAChRs in CCK+ interneurons may play a role in cholinergic modulation. Activation of mAChRs in CCKBCs and SCA cells increases firing frequency, AP duration, and membrane potential along with mAChR-mediated conversion of the spike afterhyperpolarization to a spike afterdepolarization (Cea-del Rio et al., 2010; Cea-Del Rio et al., 2011). SCA cells manifest biphasic changes (i.e., initial outward current followed by inward current) when a general muscarinic agonist is applied, whereas the muscarinic response of CCKBCs is depolarization (Cea-del Rio et al., 2010; Cea-Del Rio et al., 2011), suggesting differential cholinergic modulation among CCK+ interneuron subtypes.
Reorganization of cholinergic circuits in the hippocampus has been observed in several models of TLE. For example, septohippocampal cholinergic terminals are downregulated in the hilus, whereas the cholinergic terminals are upregulated in the inner and outer molecular layers of the dentate gyrus (Holtzman and Lowenstein, 1995; Soares et al., 2017). In addition, downregulation of hippocampal M1 and M3 receptors has been observed in animal models of TLE (Farivar et al., 2016; but see Mingo et al., 1998). However, how these changes in cholinergic signaling contribute to functional roles of CCK+ interneurons in TLE remains unclear. Since CCKBCs and SCA cells likely play a critical role in muscarinic modulation of coordinated hippocampal network oscillations and cognitive behaviors in TLE (Alger et al., 2014), it will be important in future studies to determine whether there is impaired cholinergic signaling at CCKBCs and SCA cells in animal models of TLE.
4.2. Reorganization of CCK+ interneuron circuits in TLE and functional relevance
CCK+ interneurons are a major group of GABAergic interneurons that regulate excitability of PCs, the excitatory neurons in the CA1 projecting to multiple brain regions (e.g., prefrontal cortex, amygdala, and medial entorhinal cortex; Lee et al., 2014). Our anatomical and electrophysiological results showed that CCK+ interneurons in the ventral hippocampus in our dorsal IHK TLE model were vulnerable, most likely due to status epilepticus and spontaneous seizures. This study is the first report of loss of CCK+ interneurons in the non-sclerotic, ventral CA1 regions in the dorsal IHK mouse model, which has clinical, pathological, and physiological features that closely resemble those of TLE patients diagnosed with classical unilateral hippocampal sclerosis (Thom, 2014). Importantly, our IHK model of TLE manifests hippocampal sclerosis, spontaneous seizures, and hippocampus-dependent memory deficits.
Previous studies using the dorsal IHK model showed that parvalbumin+ interneurons were reduced near the injection site (i.e., dorsal hippocampus), and the interneuron loss was also found in the intermediate hippocampus, whereas loss of interneurons expressing neuropeptide Y was limited to the region of the injection site in the dorsal hippocampus (Marx et al., 2013). These results suggest differential vulnerability of heterogeneous GABAergic interneurons in TLE. Our current studies show that carbachol-induced CCK+ interneuron-mediated inhibition is reduced in ventral CA1 regions, both ipsi- and contralaterally, which is consistent with our results from immunostaining of CCK+ interneuron markers. Our results thus agree with those of Marx et al. (2013) showing that reorganization of GABAergic inhibitory circuits extends beyond dorsal sclerotic hippocampus in IHK mice.
It is important to note that this investigation, like previous studies, employed immunostaining (e.g., parvalbumin and CCK) to count the number of specific types of interneurons. We cannot rule out the possibility that a decrease in the number of interneurons is due to downregulation of marker protein expression in addition to loss of interneurons (Alexander et al., 2016; Ruden et al., 2021). For example, the number of somata expressing parvalbumin is reduced in the non-sclerotic CA1 region of the hippocampus from patients with TLE (Wittner et al., 2005), whereas the number of parvalbumin+ axon terminals is not reduced, raising the possibility that expression of cellular markers used for interneuron identification is reduced in somata, but not in axon terminals, or that axons of surviving parvalbumin+ interneurons sprout. Thus, we counted not only somata, but also axon terminals expressing cellular markers that are selectively localized in CCK+ interneurons. Given that both somata and axon terminals of CCK+ interneurons were reduced in this study, the reductions are most likely due to loss of CCK+ interneurons, and not due to downregulation of CCK protein expression in CCK+ interneuron somata. Furthermore, our electrophysiological results demonstrating reduced carbachol-induced synaptic inhibition of CA1 PCs are consistent with a loss of CCK+ interneurons and reduced innervation of CA1 PCs. In agreement with our results, prior studies using other mouse models of TLE (Khan et al., 2018; Sayin et al., 2003; Sun et al., 2014; Wyeth et al., 2010) also showed that CCK+ interneurons were lost in the CA1 region of the hippocampus and the dentate gyrus. Thus, these results suggest that CCK+ interneurons are vulnerable in TLE, including in areas remote from the IHK lesion, and their loss likely contributes to hyperexcitability and spontaneous seizures along with loss of other types of GABAergic interneurons (Cossart et al., 2001; Dinocourt et al., 2003).
Previously, it was unclear whether surviving CCKBCs manifest normal activity, impaired activity, or otherwise altered function to support compensatory mechanisms in TLE. Our results demonstrate that surviving CCKBCs manifest no major changes in their intrinsic and synaptic properties in IHK mice, including the connectivity patterns of individual CCKBCs with CA1 PCs. Based on our current results showing a decrease in the number of CCK+ interneurons in the stratum radiatum and the stratum oriens in the dorsal IHK model, which was also shown in a rat model of TLE induced by systemic KA injection (Khan et al., 2018), we expected that the number of axon terminals from CCK+ interneurons would be reduced not only in the stratum pyramidale, but also in other CA1 layers, since loss of dendritically targeting CCK+ interneurons in the stratum radiatum (e.g., SCA cells; Bezaire and Soltesz, 2013) would contribute to the decrease in the number of CCK+ interneurons. However, we instead found similar numbers of axon terminals of CCK+ interneurons in other CA1 layers (except the stratum pyramidale) between sham controls and IHK mice. These findings suggest that surviving CCK+ dendritically-projecting interneurons may develop axon sprouting to compensate for interneuron loss in TLE. Indeed, dendritically projecting interneurons (e.g., somatostatin+ interneurons) develop axon sprouting in the dentate gyrus in TLE (Zhang et al., 2009). On the other hand, surviving CCK+ interneurons show abnormal anatomical properties in TLE (e.g., a decrease in soma size and dendritic complexity; Khan et al., 2018). How the loss of CCK+ interneurons and the morphological changes in surviving CCK+ interneurons are linked to pathological circuit activity in TLE will need to be addressed in future studies. Normal wiring of CCKBCs in the hippocampal circuit is critical to generate theta oscillations, which are downregulated in TLE (Bezaire et al., 2016; Chauviere et al., 2009; Dugladze et al., 2007; Shuman et al., 2017), and required to successfully process spatial memory (Del Pino et al., 2017). In conclusion, our findings suggest that the overall reduction in functional inhibition of CA1 PCs by surviving CCKBCs in areas remote from the primary seizure focus may contribute to spontaneous seizures and cognitive comorbidities in TLE.
Highlights.
Unilateral dorsal IHK mouse model of TLE reproduced key properties of human TLE.
The IHK model reduced CCK+ interneurons in the ventral hippocampus.
The IHK model reduced the number of CCKBC boutons in the ventral hippocampus.
The IHK model didn’t alter intrinsic and synaptic properties of surviving CCKBCs.
The IHK model reduced overall CCK+ interneuron-mediated inhibition.
Acknowledgments
We thank H. E. S. Lewis and M.W. Young for technical assistance with the analysis of behavioral seizures, and Dr. M.B. Halmos for statistical advice. This work was supported by the College of Medicine, UAMS (startup funding to S.-H.L), Core Facilities of the Center for Translational Neuroscience at UAMS, Award P30 GM110702 from the IDeA program at NIGMS, and R01 NS092552 (to B.N.S). The sponsors had no role in study design, data collection, analysis and interpretation, or writing of this manuscript.
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AED
antiepileptic drug
- AP
action potential
- APV
2-amino-5-phosphonopentanoic acid
- CB1
cannabinoid type 1 receptor
- CCK
cholecystokinin
- CCKBC
cholecystokinin-expressing basket cell
- DI
discrimination index
- GABAB1R
GABAB receptor 1
- IHK
intrahippocampal kainate
- KA
kainate
- K–S test
Kolmogorov–Smirnov test
- mAChR
muscarinic acetylcholine receptor
- nAChR
nicotinic acetylcholine receptor
- NBQX
2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo (F) quinoxaline
- NGS
normal goat serum
- OLM
object location memory
- PC
pyramidal cell
- RMS
root mean square
- sIPSC
spontaneous IPSCs
- TBS
Tris-buffered saline
- TLE
temporal lobe epilepsy
- UAMS
University of Arkansas for Medical Sciences
- uIPSC
unitary IPSC
- vGAT
vesicular GABA transporter
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
None.
References
- Alarcón G, Valentín A, 2010. Mesial Temporal Lobe Epilepsy with Hippocampal Sclerosis, in: Panayiotopoulos CP (Ed.), Atlas of Epilepsies. Springer, London, pp. 1171–1175. 10.1007/978-1-84882-128-6_173 [DOI] [Google Scholar]
- Alexander A, Maroso M, Soltesz I, 2016. Organization and control of epileptic circuits in temporal lobe epilepsy. Prog. Brain Res 226, 127–154. 10.1016/bs.pbr.2016.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alger BE, Nagode DA, Tang AH, 2014. Muscarinic cholinergic receptors modulate inhibitory synaptic rhythms in hippocampus and neocortex. Front. Synaptic Neurosci 6, 18. 10.3389/fnsyn.2014.00018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amlerova J, Laczo J, Vlcek K, Javurkova A, Andel R, Marusic P, 2013. Risk factors for spatial memory impairment in patients with temporal lobe epilepsy. Epilepsy Behav. 26, 57–60. 10.1016/j.yebeh.2012.10.025 [DOI] [PubMed] [Google Scholar]
- Andersson PB, Perry VH, Gordon S, 1991. The kinetics and morphological characteristics of the macrophage-microglial response to kainic acid-induced neuronal degeneration. Neuroscience 42, 201–214. 10.1016/0306-4522(91)90159-L [DOI] [PubMed] [Google Scholar]
- Armstrong C, Wang J, Lee SY, Broderick J, Bezaire MJ, Lee S-H, Soltesz I, 2016. Target-Selectivity of Parvalbumin-Positive Interneurons in Layer II of Medial Entorhinal Cortex in Normal and Epileptic Animals. Hippocampus 26, 779–793. 10.1002/hipo.22559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire MJ, Raikov I, Burk K, Vyas D, Soltesz I, 2016. Interneuronal mechanisms of hippocampal theta oscillations in a full-scale model of the rodent CA1 circuit. Elife 5, e18566. 10.7554/eLife.18566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezaire MJ, Soltesz I, 2013. Quantitative assessment of CA1 local circuits: knowledge base for interneuron-pyramidal cell connectivity. Hippocampus 23, 751–85. 10.1002/hipo.22141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booker SA, Althof D, Gross A, Loreth D, Müller J, Unger A, Fakler B, Varro A, Watanabe M, Gassmann M, Bettler B, Shigemoto R, Vida I, Kulik Á, 2017. KCTD12 Auxiliary Proteins Modulate Kinetics of GABAB Receptor-Mediated Inhibition in Cholecystokinin-Containing Interneurons. Cereb. Cortex 27, 2318–2334. 10.1093/cercor/bhw090 [DOI] [PubMed] [Google Scholar]
- Boro A, Haut S, 2003. Medical comorbidities in the treatment of epilepsy. Epilepsy Behav. 4, S2–S12. 10.1016/j.yebeh.2003.07.002 [DOI] [PubMed] [Google Scholar]
- Bragin A, Engel J, Wilson CL, Vizentin E, Mathern GW, 1999. Electrophysiologic analysis of a chronic seizure model after unilateral hippocampal KA injection. Epilepsia 40, 1210–1221. 10.1111/j.1528-1157.1999.tb00849.x [DOI] [PubMed] [Google Scholar]
- Bui AD, Nguyen TM, Limouse C, Kim HK, Szabo GG, Felong S, Maroso M, Soltesz I, 2018. Dentate gyrus mossy cells control spontaneous convulsive seizures and spatial memory. Science 359, 787–790. 10.1126/science.aan4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler CR, Boychuk JA, Smith BN, 2016. Differential effects of rapamycin treatment on tonic and phasic GABAergic inhibition in dentate granule cells after focal brain injury in mice. Exp. Neurol 280, 30–40. 10.1016/j.expneurol.2016.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cánovas R, León I, Serrano P, Roldán MD, Cimadevilla JM, 2011. Spatial navigation impairment in patients with refractory temporal lobe epilepsy: Evidence from a new virtual reality-based task. Epilepsy Behav. 22, 364–369. 10.1016/j.yebeh.2011.07.021 [DOI] [PubMed] [Google Scholar]
- Cea-Del Rio CA, Lawrence JJ, Erdelyi F, Szabo G, Mcbain CJ, 2011. Cholinergic modulation amplifies the intrinsic oscillatory properties of CA1 hippocampal cholecystokinin-positive interneurons. J. Physiol 589, 609–627. 10.1113/jphysiol.2010.199422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cea-del Rio CA, Lawrence JJ, Tricoire L, Erdelyi F, Szabo G, McBain CJ, 2010. M3 muscarinic acetylcholine receptor expression confers differential cholinergic modulation to neurochemically distinct hippocampal basket cell subtypes. J. Neurosci 30, 6011–6024. 10.1523/JNEUROSCI.5040-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauviere L, Rafrafi N, Thinus-Blanc C, Bartolomei F, Esclapez M, Bernard C, 2009. Early deficits in spatial memory and theta rhythm in experimental temporal lobe epilepsy. J Neurosci 29, 5402–5410. 10.1523/JNEUROSCI.4699-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgin LL, 2016. Rhythms of the hippocampal network. Nat. Rev. Neurosci 17, 239–249. 10.1038/nrn.2016.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben-Ari Y, Esclapez M, Bernard C, 2001. Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat. Neurosci 4, 52–62. 10.1038/82900 [DOI] [PubMed] [Google Scholar]
- Del Pino I, Brotons-Mas JR, Marques-Smith A, Marighetto A, Frick A, Marín O, Rico B, 2017. Abnormal wiring of CCK+ basket cells disrupts spatial information coding. Nat. Neurosci 20, 784–792. 10.1038/nn.4544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinocourt C, Petanjek Z, Freund TF, Ben-Ari Y, Esclapez M, 2003. Loss of interneurons innervating pyramidal cell dendrites and axon initial segments in the CA1 region of the hippocampus following pilocarpine-induced seizures. J. Comp. Neurol 459, 407–425. 10.1002/cne.10622 [DOI] [PubMed] [Google Scholar]
- Dugladze T, Vida I, Tort AB, Gross A, Otahal J, Heinemann U, Kopell NJ, Gloveli T, 2007. Impaired hippocampal rhythmogenesis in a mouse model of mesial temporal lobe epilepsy. Proc. Natl. Acad. Sci. U. S. A 104, 17530–17535. 10.1073/pnas.0708301104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M, 2004. Chronic epilepsy and cognition. Lancet Neurol. 3, 663–672. 10.1016/S1474-4422(04)00906-8 [DOI] [PubMed] [Google Scholar]
- Farivar TN, Nassiri-Asl M, Johari P, Najafipour R, Hajiali F, 2016. The effects of kainic acid-induced seizure on gene expression of brain neurotransmitter receptors in mice using RT2 PCR array. Basic Clin. Neurosci 7, 292–298. 10.15412/J.BCN.03070402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, 2007. Perisomatic Inhibition. Neuron 56, 33–42. 10.1016/j.neuron.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Groticke I, Hoffmann K, Loscher W, 2008. Behavioral alterations in a mouse model of temporal lobe epilepsy induced by intrahippocampal injection of kainate. Exp. Neurol 213, 71–83. 10.1016/j.expneurol.2008.04.036 [DOI] [PubMed] [Google Scholar]
- Hájos N, Mody I, 2009. Establishing a physiological environment for visualized in vitro brain slice recordings by increasing oxygen supply and modifying aCSF content. J. Neurosci. Methods 183, 107–113. 10.1016/j.jneumeth.2009.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes GL, 2015. Cognitive impairment in epilepsy: the role of network abnormalities. Epileptic Disord. 17, 101–116. 10.1684/epd.2015.0739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Lowenstein DH, 1995. Selective inhibition of axon outgrowth by antibodies to NGF in a model of temporal lobe epilepsy. J. Neurosci 15, 7072–7070. 10.1523/jneurosci.15-11-07062.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalilani L, Sun X, Pelgrims B, Noack-Rink M, Villanueva V, 2018. The epidemiology of drug-resistant epilepsy: A systematic review and meta-analysis. Epilepsia 59, 2179–2193. 10.1111/epi.14596 [DOI] [PubMed] [Google Scholar]
- Kang Y-J, Lewis HES, Young MW, Govindaiah G, Greenfield LJ, Garcia-Rill E, Lee S-H, 2018. Cell type-specific intrinsic perithreshold oscillations in hippocampal GABAergic interneurons. Neuroscience 376, 80–93. 10.1016/j.neuroscience.2018.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karson MA, Whittington KC, Alger BE, 2008. Cholecystokinin inhibits endocannabinoid-sensitive hippocampal IPSPs and stimulates others. Neuropharmacology 54, 117–128. 10.1016/j.neuropharm.2007.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF, 1999. Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci 19, 4544–4558. 10.1523/jneurosci.19-11-04544.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Shekh-Ahmad T, Khalil A, Walker MC, Ali AB, 2018. Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model. Br. J. Pharmacol 175, 2097–2115. 10.1111/bph.14202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HK, Gschwind T, Nguyen TM, Bui AD, Felong S, Ampig K, Suh D, Ciernia AV, Wood MA, Soltesz I, 2020. Optogenetic intervention of seizures improves spatial memory in a mouse model of chronic temporal lobe epilepsy. Epilepsia 61, 561–571. DOI: 10.1111/epi.16445. 10.1111/epi.16445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausberger T, Marton LF, O’Neill J, Huck JHJ, Dalezios Y, Fuentealba P, Suen WY, Papp E, Kaneko T, Watanabe M, Csicsvari J, Somogyi P, 2005. Complementary roles of cholecystokinin- and parvalbumin-expressing GABAergic neurons in hippocampal network oscillations. J. Neurosci 25, 9782–93. 10.1523/JNEUROSCI.3269-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krook-Magnuson E, Armstrong C, Oijala M, Soltesz I, 2013. On-demand optogenetic control of spontaneous seizures in temporal lobe epilepsy. Nat. Commun 4, 1376. 10.1038/ncomms2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JJ, 2008. Cholinergic control of GABA release: emerging parallels between neocortex and hippocampus. Trends Neurosci. 31, 317–27. 10.1016/j.tins.2008.03.008 [DOI] [PubMed] [Google Scholar]
- Lee S-H, Dudok B, Parihar VK, Jung KM, Zöldi M, Kang YJ, Maroso M, Alexander AL, Nelson GA, Piomelli D, Katona I, Limoli CL, Soltesz I, 2017. Neurophysiology of space travel: energetic solar particles cause cell type-specific plasticity of neurotransmission. Brain Struct. Funct 222, 2345–2357. 10.1007/s00429-016-1345-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Földy C, Soltesz I, 2010. Distinct endocannabinoid control of GABA release at perisomatic and dendritic synapses in the hippocampus. J. Neurosci 30, 7993–8000. http://www.jneurosci.org/cgi/doi/10.1523/JNEUROSCI.6238-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Ledri M, Toth B, Marchionni I, Henstridge CM, Dudok B, Kenesei K, Barna L, Szabo SI, Renkecz T, Oberoi M, Watanabe M, Limoli CL, Horvai G, Soltesz I, Katona I, 2015. Multiple Forms of Endocannabinoid and Endovanilloid Signaling Regulate the Tonic Control of GABA Release. J. Neurosci 35, 10039–10057. 10.1523/JNEUROSCI.4112-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I, 2014. Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron 82, 1129–1144. 10.1016/j.neuron.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-H, Soltesz I, 2011. Requirement for CB1 but not GABAB receptors in the cholecystokinin mediated inhibition of GABA release from cholecystokinin expressing basket cells. J. Physiol 589, 891–902. http://doi.wiley.com/10.1113/jphysiol.2010.198499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque M, Avoli M, 2013. The kainic acid model of temporal lobe epilepsy. Neurosci. Biobehav. Rev 37, 2887–2899. 10.1016/j.neubiorev.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx M, Haas CA, Häussler U, 2013. Differential vulnerability of interneurons in the epileptic hippocampus. Front. Cell. Neurosci 7, 167. 10.3389/fncel.2013.00167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuiston AR, 2014. Acetylcholine release and inhibitory interneuron activity in hippocampal CA1. Front. Synaptic Neurosci 6, 20. 10.3389/fnsyn.2014.00020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingo NS, Cottrell GA, Mendonça A, Gombos Z, Eubanks JH, Burnham WMI, 1998. Amygdala-kindled and electroconvulsive seizures alter hippocampal expression of the m1 and m3 muscarinic cholinergic receptor genes. Brain Res. 810, 9–15. 10.1016/S0006-8993(98)00748-3 [DOI] [PubMed] [Google Scholar]
- Morales M, Hein K, Vogel Z, 2008. Hippocampal interneurons co-express transcripts encoding the α7 nicotinic receptor subunit and the cannabinoid receptor 1. Neuroscience 152, 70–81. 10.1016/j.neuroscience.2007.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler T, Alger B, 1992. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slice. J. Physiol 450, 127–142. 10.1113/jphysiol.1992.sp019119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prut L, Belzung C, 2003. The open field as a paradigm to measure the effects of drugs on anxiety-like behaviors: A review. Eur. J. Pharmacol 463, 3–33. 10.1016/S0014-2999(03)01272-X [DOI] [PubMed] [Google Scholar]
- Racine RJ, 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr. Clin. Neurophysiol 32, 281–294. 10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- Rattka M, Brandt C, Löscher W, 2013. The intrahippocampal kainate model of temporal lobe epilepsy revisited: Epileptogenesis, behavioral and cognitive alterations, pharmacological response, and hippoccampal damage in epileptic rats. Epilepsy Res. 103, 135–152. 10.1016/j.eplepsyres.2012.09.015 [DOI] [PubMed] [Google Scholar]
- Ruden JB, Dugan LL, Konradi C, 2021. Parvalbumin interneuron vulnerability and brain disorders. Neuropsychopharmacology 46, 279–287. 10.1038/s41386-020-0778-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayin U, Osting S, Hagen J, Rutecki P, Sutula T, 2003. Spontaneous Seizures and Loss of Axo-Axonic and Axo-Somatic Inhibition Induced by Repeated Brief Seizures in Kindled Rats. J. Neurosci 23, 2759–2768. 10.1523/JNEUROSCI.23-07-02759.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman T, Amendolara B, Golshani P, 2017. Theta Rhythmopathy as a Cause of Cognitive Disability in TLE. Epilepsy Curr. 17, 107–111. 10.5698/1535-7511.17.2.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS, Ali-Akbarian L, Elliott RC, Bowery BJ, Bowery NG, 1999. Localization of GABA(B) (R1) receptors in the rat hippocampus by immunocytochemistry and high resolution autoradiography, with specific reference to its localization in identified hippocampal interneuron subpopulations. Neuropharmacology 38, 1707–1721. 10.1016/S0028-3908(99)00132-X [DOI] [PubMed] [Google Scholar]
- Soares JI, Valente MC, Andrade PA, Maia GH, Lukoyanov NV, 2017. Reorganization of the septohippocampal cholinergic fiber system in experimental epilepsy. J. Comp. Neurol 525, 2690–2705. 10.1002/cne.24235 [DOI] [PubMed] [Google Scholar]
- Soltesz I, Alger BE, Kano M, Lee S, Lovinger DM, Ohno-shosaku T, Watanabe M, 2015. Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci 16, 264–277. 10.1038/nrn3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Sun J, Erisir A, Kapur J, 2014. Loss of cholecystokinin-containing terminals in temporal lobe epilepsy. Neurobiol. Dis 62, 44–55. 10.1016/j.nbd.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Téllez-Zenteno JF, Hernández-Ronquillo L, 2012. A Review of the Epidemiology of Temporal Lobe Epilepsy. Epilepsy Res. Treat 2012, 630853. 10.1155/2012/630853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, 2014. Review: Hippocampal sclerosis in epilepsy: A neuropathology review. Neuropathol. Appl. Neurobiol 40, 520–543. 10.1111/nan.12150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Mathern GW, Cross JH, Bertram EH, 2010. Mesial temporal lobe epilepsy: How do we improve surgical outcome? Ann. Neurol 684, 424–434. 10.1002/ana.22142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel J, Fortin DA, Levine ES, 2004. Endocannabinoid signalling selectively targets perisomatic inhibitory inputs to pyramidal neurones in juvenile mouse neocortex. J. Physiol 556, 95–107. 10.1113/jphysiol.2003.058198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinka E, Cock H, Hesdorffer D, Rossetti AO, Scheffer IE, Shinnar S, Shorvon S, Lowenstein DH, 2015. A definition and classification of status epilepticus -Report of the ILAE Task Force on Classification of Status Epilepticus. Epilepsia 56, 1515–1523. 10.1111/epi.13121 [DOI] [PubMed] [Google Scholar]
- Twele F, Schidlitzki A, Töllner K, Löscher W, 2017. The intrahippocampal kainate mouse model of mesial temporal lobe epilepsy: Lack of electrographic seizure-like events in sham controls. Epilepsia Open 2, 180–187. 10.1002/epi4.12044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twele F, Töllner K, Brandt C, Löscher W, 2016. Significant effects of sex, strain, and anesthesia in the intrahippocampal kainate mouse model of mesial temporal lobe epilepsy. Epilepsy Behav. 55, 47–56. 10.1016/j.yebeh.2015.11.027 [DOI] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA, 2014. Examining object location and object recognition memory in mice. Curr. Protoc. Neurosci 69, 1–17. 10.1002/0471142301.ns0831s69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will TR, Proaño SB, Thomas AM, Kunz LM, Thompson KC, Ginnari LA, Jones CH, Lucas SC, Reavis EM, Dorris DM, Meitzen J, 2017. Problems and progress regarding sex bias and omission in neuroscience research. eNeuro 4, ENEURO.0195–19.2020. 10.1523/ENEURO.0278-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Eross L, Czirják S, Halász P, Freund TF, Maglóczky Z, 2005. Surviving CA1 pyramidal cells receive intact perisomatic inhibitory input in the human epileptic hippocampus. Brain 128, 138–152. 10.1093/brain/awh339 [DOI] [PubMed] [Google Scholar]
- Wyeth MS, Zhang N, Mody I, Houser CR, 2010. Selective Reduction of Cholecystokinin-Positive Basket Cell Innervation in a Model of Temporal Lobe Epilepsy. J. Neurosci 30, 8993–9006. 10.1523/jneurosci.1183-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada-Hanff J, Bean BP, 2013. Persistent sodium current drives conditional pacemaking in CA1 pyramidal neurons under muscarinic stimulation. J. Neurosci 33, 15011–21. 10.1523/JNEUROSCI.0577-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler Z, Brandt-Fontaine M, Leintz C, Krook-Magnuson C, Netoff T, Krook-Magnuson E, 2018. Targeting the mouse ventral hippocampus in the intrahippocampal kainic acid model of temporal lobe epilepsy. eNeuro 5, e0158–18.2018. 10.1523/ENEURO.0158-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yamawaki R, Wen X, Uhl J, Diaz J, Prince DA, Buckmaster PS, 2009. Surviving hilar somatostatin interneurons enlarge, sprout axons, and form new synapses with granule cells in a mouse model of temporal lobe epilepsy. J. Neurosci 29, 14247–14256. 10.1523/JNEUROSCI.3842-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]