

Summary
Blockade of the inhibitory receptor TIM-3 shows efficacy in cancer immunotherapy clinical trials. TIM-3 inhibits production of the chemokine CXCL9 by XCR1+ classical dendritic cells (cDC1), thereby limiting antitumor immunity in mammary carcinomas. We found that increased CXCL9 expression by splenic cDC1s upon TIM-3 blockade required type I interferons and extracellular DNA. Chemokine expression as well as combinatorial efficacy of TIM-3 blockade and paclitaxel chemotherapy were impaired by deletion of Cgas and Sting. TIM-3 blockade increased uptake of extracellular DNA by cDC1 through an endocytic process that resulted in cytoplasmic localization. DNA uptake and efficacy of TIM-3 blockade required DNA binding by HMGB1, while galectin-9-induced cell surface clustering of TIM-3 was necessary for its suppressive function. Human peripheral blood cDC1s also took up extracellular DNA upon TIM-3 blockade. Thus, TIM-3 regulates endocytosis of extracellular DNA and activation of the cytoplasmic DNA sensing cGAS-STING pathway in cDC1s, with implications for understanding the mechanisms underlying TIM-3 immunotherapy.
Keywords: TIM-3, dendritic cells, CD103+ cDC1, cGAS, STING, DNA, type I interferon, CXCL9
Graphical Abstract

eTOC Blurb
Blockade of the inhibitory receptor TIM-3 shows efficacy in cancer immunotherapy clinical trials. de Mingo Pulido et al. provide insight into the underlying mechanisms by revealing that TIM-3 suppresses HMGB1-dependent endocytosis of extracellular DNA and the subsequent activation of the cGAS-STING pathway in intra-tumoral dendritic cells.
Introduction
T-cell immunoglobulin and mucin domain containing (TIM)-3 was originally identified as a molecule selectively expressed on CD4+ Th1 and CD8+ cytotoxic T cells (Monney et al., 2002), and has since been described as a marker of T cell exhaustion in models of chronic viral infection and cancer, particularly in combination with high expression of programmed death (PD)-1 (Anderson et al., 2016). TIM-3 blocking antibodies (αTIM-3) partially reverse this exhausted phenotype, resulting in improved expression of interferon (IFN)-γ and suppressed tumor growth in multiple preclinical models (Ngiow et al., 2011). Efficacy is even more pronounced when αTIM-3 is combined with αPD-1 (Kurtulus et al., 2019; Ngiow et al., 2011; Sakuishi et al., 2010), or when it is used sequentially in αPD-1 resistant tumors (Koyama et al., 2016). As a result of these studies there are now several antibodies against TIM-3 (e.g. TSR-022/Cobolimab, MBG453, LY3321367, BMS986258) being evaluated in early phase clinical studies, mostly in combination with agents targeting the PD-1 and PD-ligand (PD)-L1 pathway.
TIM-3 is also constitutively expressed by a number of other leukocyte populations, including natural killer cells, mast cells, monocytes, macrophages, and classical dendritic cells (cDCs), usually at higher levels than observed on T cells (Anderson et al., 2007; Gleason et al., 2012; Nakayama et al., 2009; Ndhlovu et al., 2012; Phong et al., 2015). The relevance of this expression is unclear, but as TIM-3 is generally thought to act as a negative regulator of activation, it is possible these cells may be involved in mediating therapeutic response to αTIM-3. Indeed, αTIM-3 improves response to paclitaxel (PTX) chemotherapy in murine models of mammary carcinoma, despite limited expression of TIM-3 on T cells (de Mingo Pulido et al., 2018). In this setting, therapeutic efficacy is contingent upon the Batf3- and Irf8-dependent subset of cDCs (cDC1), a population of cells important for systemic as well as local anti-tumor immunity (Chow et al., 2019; Garris et al., 2018; Roberts et al., 2016). TIM-3 blockade enhances the expression of the chemokines Cxcl9 and Cxcl10 by tumor CD103+ cDC1s, thereby driving T cell effector function and response to chemotherapy in a manner dependent upon the receptor CXCR3 (de Mingo Pulido et al., 2018).
Here we examined the mechanism by which TIM-3 regulates cDC activation in tumors. We found that TIM-3 restricted extracellular DNA uptake through an HMGB1-dependent mechanism. Blocking TIM-3 promoted activation of the cyclic GMP-AMP synthase (cGAS) and stimulator of interferon genes (STING) pathway, expression of type I interferons (IFNs), and secondary expression of chemokines, all of which were necessary for the therapeutic efficacy of αTIM-3 and PTX chemotherapy in a mammary carcinoma model. These results delineate a mechanism by which TIM-3 suppresses DNA sensing through the cGAS-STING pathway, with potential implications for the design of clinical trials involving TIM-3 antibodies.
Results
αTIM-3 promotes type I IFN and CXCL9 expression by cDC1s
TIM-3 blockade enhances CXCL9 expression by splenic cDCs in the presence of tumor cell debris generated by irradiation or heat shock (HS), mirroring the increase in Cxcl9 expression observed in tumor cDCs following blockade of TIM-3 in vivo (de Mingo Pulido et al., 2018). We therefore sought to use this in vitro system to identify the signaling pathways regulated by αTIM-3. CXCL9 levels were first measured in a time course experiment wherein increased protein expression was observed by 4 hr after treatment with HS and αTIM-3, as compared to the control group treated with HS and an IgG2a isotype control (Figure 1A). We then purified CD8α+ cDC1 and CD11b+ cDC2 subsets after a 2 hr incubation and confirmed increased mRNA expression of Cxcl9 and Cxcl10 by RT-PCR (Figure S1A) prior to an analysis using a 770-gene Nanostring panel. Using a false discovery rate (FDR) of <0.05 we observed increased expression of 9 genes within the cDC1 subset (Figure 1B), with genes whose expression increased in cDC1s by >4-fold reflecting type I IFNs (Ifnb1), IFN-responsive chemokines (Cxcl9, Cxcl10, Cxcl11), or other IFN-responsive elements (Ifnl2) (Figure S1B). Expression of only a single gene in the cDC2 subset (Ifnl2) was increased, consistent with the reduced impact of αTIM-3 on CXCL9 expression by cDC2s. Using a less stringent cutoff of p<0.05 (Figure S1C–D) we also performed a pathway analysis using MetaCore. Both the Process Networks and Pathway Maps pointed to regulation of the type I IFN response as being the primary impact of blocking TIM-3 (Figure 1C). These in vitro results are consistent with in vivo findings that TIM-3 blockade increased expression of Cxcl9 and Cxcl10 by tumor CD103+ cDC1s without altering surface expression of activation markers (de Mingo Pulido et al., 2018).
Figure 1. TIM-3 blockade induces a type I IFN response in splenic cDC1s.
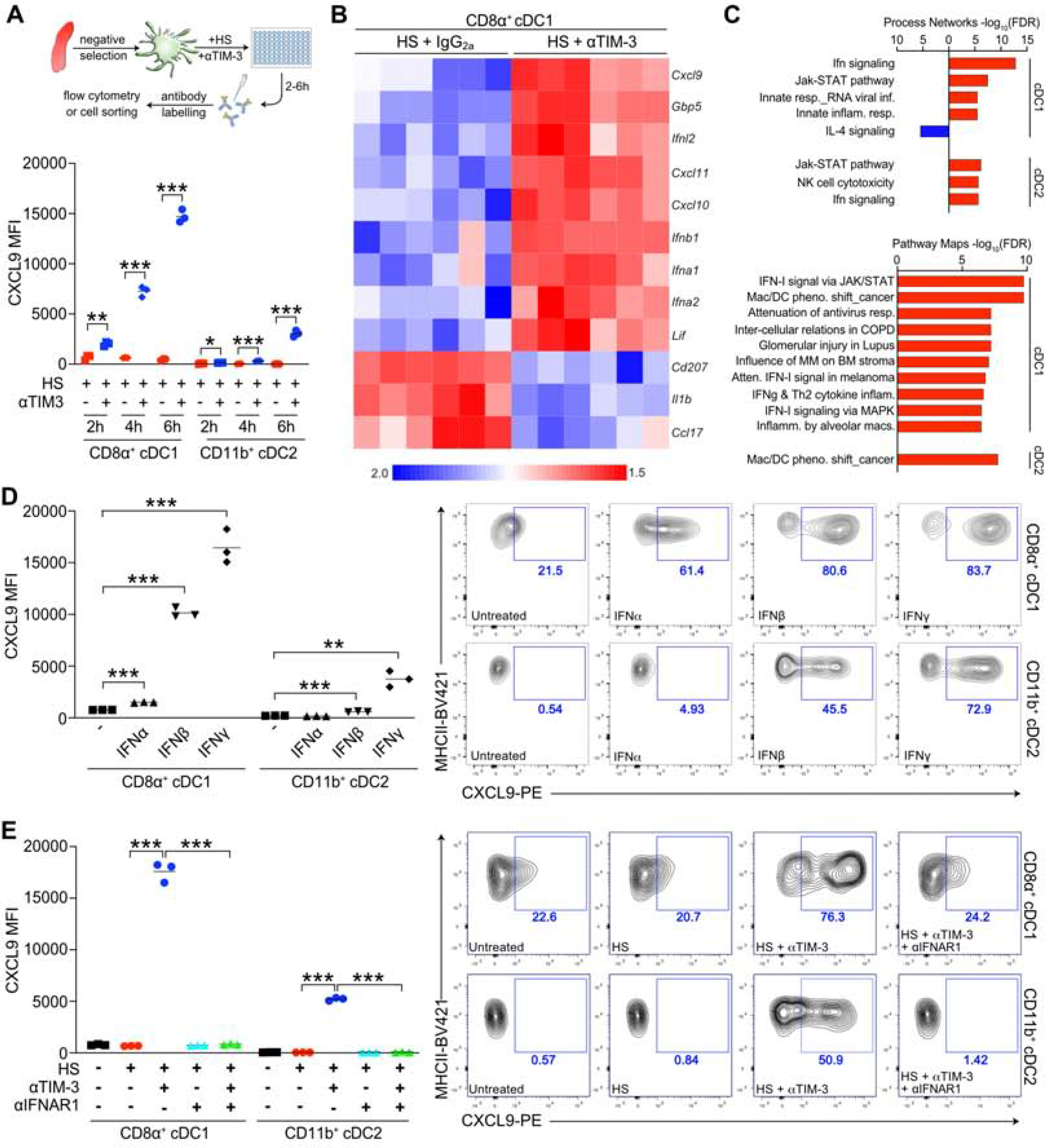
(A) Intracellular flow cytometric detection of CXCL9 in splenic cDCs following a 2–6 hr incubation with tumor cell debris generated by heat shock (HS), either in the presence of a rat IgG2a isotype control or the RMT3–23 TIM-3 blocking antibody (αTIM-3). n=3 technical replicates, with one of three independent experiments shown. (B) Significant (Z<0.05) gene expression changes in splenic CD8α+ cDC1 following stimulation with HS and αTIM-3, compared to HS and IgG2a. n=6 biological replicates, data compiled from two independent experiments. (C) Pathway analysis of significantly (p<0.05) altered genes from B. (D) Intracellular flow cytometric detection of CXCL9 in splenic cDCs following a 6 hr stimulation with 10 ng/ml IFN-γ, IFN-α, or IFN-β. n=3 technical replicates, with one of four independent experiments shown. (E) Intracellular flow cytometric detection of CXCL9 in splenic cDCs following a 6 hr stimulation with HS, αTIM-3, or αIFNAR1. n=3 technical replicates, with one of three independent experiments shown. For A, D and E data reflect the mean; significance determined by an unpaired t test (A) or one-way ANOVA (D, E) and shown as *p<0.05, **p <0.01, ***p<0.001. See also Figure S1.
Based upon the above results, and the ability of type I and type II IFNs to preferentially induce CXCL9 expression in cDC1s (Figure 1D), we evaluated whether blocking the type I IFN receptor (IFNAR1) was sufficient to prevent increased chemokine expression, focusing on CXCL9 due to antibody availability and the specific increase in Cxcl9 observed in tumor cDCs following TIM-3 blockade (de Mingo Pulido et al., 2018). αIFNAR1 abrogated the increase in CXCL9 expression normally observed during incubation of splenic CD8α+ cDC1 with αTIM-3 and HS (Figure 1E). αIFNAR1 also blocked the smaller increase in expression by splenic CD11b+ cDC2. Together these results indicate that TIM-3 suppresses the ability of cDCs to become activated by cellular debris, thereby limiting their expression of type I IFNs and subsequent induction of IFN-responsive genes and chemokines.
CXCL9 expression is dependent upon extracellular DNA and STING
Type I IFNs are strongly associated with a response against viruses or intracellular pathogens via recognition of nucleic acids, including non-eukaryotic structures such as dsRNA or the abnormal localization of dsDNA in the cytoplasm (Schlee and Hartmann, 2016). We therefore added DNase or RNase during the incubation of splenic cDCs with HS and αTIM-3 to determine if nucleic acids were required for activation of cDCs, using expression of CXCL9 as a secondary readout of type I IFN expression. As shown in Figure 2A, the addition of DNase completely abrogated the ability of αTIM-3 and HS to increase CXCL9 expression in CD8α+ cDC1, whereas RNase had little to no impact on the expression level.
Figure 2. CXCL9 expression is dependent upon extracellular DNA and STING.

(A-F) Intracellular flow cytometric detection of CXCL9 in splenic cDCs following a 6 hr stimulation with supernatant generated by heat shock (HS). (A) Splenic cDCs incubated with HS ± αTIM-3, with DNase (50 U/ml) or RNase (10 μg/ml) added to the supernatant 15 min prior to stimulation as indicated. (B) Splenic cDCs stimulated with 10 μg/ml of 2’3’-cGAMP or 3’3’-cGAMP in the presence or absence of αIFNAR1. (C) C57BL6/J or STING-deficient CD8α+ cDCs incubated with HS ± αTIM-3. (D) C57BL6/J, MyD88-deficient, TRIF-deficient, or MAVS-deficient CD8α+ cDCs incubated with HS ± αTIM-3. (E) C57BL6/J or Cgas-deficient CD8α+ cDCs incubated with HS ± αTIM-3. (F) CD8α+ cDCs stimulated with HS generated using PyMT cells deficient in Sting or Cgas. (G) Intracellular flow cytometric detection of CXCL9 in iCD103+ BMDCs incubated for 24 hr with HS ± αTIM-3. (H) Western blot of nuclear pIRF3 or pTBK1 in iCD103+ BMDC lysate following a 3 hr incubation with HS ± αTIM-3. The STING agonist DMXAA was used as a positive control. Nuclear p84, as well as total IRF3, TBK1, vinculin and β-actin were used as loading controls. For A-G, data reflect technical replicates and the mean, with one of three (A, C, G, H) or one of two (B, D, E, F) representative experiments shown. Significance was determined by a one-way ANOVA (A-E, G) and is shown as ***p<0.001. See also Figure S2.
The major pathway responsible for recognizing dsDNA in the cytoplasm involves activation of cGAS, which leads to production of 2’3’-cGAMP and activation of STING (Barber, 2015). The addition of eukaryotic 2’3’-cGAMP or bacterial 3’3’-cGAMP induced CXCL9 expression in a type I IFN-dependent manner (Figure 2B, S2A). CXCL9 expression in cDC2s was not observed following stimulation with cGAMP (Figure 2B), possible due to lower responsiveness to IFN stimulation (Figure 1D). We therefore evaluated splenic CD8α+ cDC1s from Sting-deficient mice and found that these failed to express CXCL9 during incubation with HS and αTIM-3 (Figure 2C, S2B). In contrast, splenic cDC1s deficient in the TLR signaling adapters (TRIF, MyD88) or a key molecule in RNA sensing (mitochondrial antiviral signaling protein, MAVS) expressed equivalent levels of CXCL9 after incubation with αTIM-3 and HS (Figure 2D).
Tumor-derived 2’3’-cGAMP can directly activate STING in CD11b+ myeloid cells independently of cGAS (Marcus et al., 2018), with transportation through the cytoplasmic membrane via the channel SLC19A1 in the human THP-1 myeloid cell line (Luteijn et al., 2019; Ritchie et al., 2019) or via the channel P2X7R in murine macrophages (Zhou et al., 2020). We thus evaluated CXCL9 expression in cGAS-deficient splenic cDCs and found these cells also failed to respond to αTIM-3 and HS (Figure 2E, S2C). This was not due to a defect in the ability of cGAS- or STING-deficient splenic cDC1s to express CXCL9 following stimulation with IFNs (Figure S2D). To further evaluate a role for tumor-derived 2’3’-cGAMP on CXCL9 expression we generated cGAS- or STING-deficient PyMT cells and used these to create tumor cell debris by HS (Figure S2E). HS from either cGAS- or STING-deficient tumor cells were able to induce CXCL9 expression in the presence of the TIM-3 blocking antibody (Figure 2F, S2F), supporting intrinsic activation of cGAS in cDC1s as being necessary to activate STING and subsequent expression of CXCL9. This difference between the role of cGAS in cDC1s versus macrophages may reflect a lack of P2X7R expression by tumor and splenic cDC1s, as compared to high expression by tumor macrophages (Figure S2G–H).
Splenic cDC preparations contain a mixed population of cells, and we therefore sought to replicate our findings in a relatively pure population of bone marrow (BM)-derived DCs (BMDCs). As FLT-3 Ligand (FLT-3L)-induced BMDCs, which are predominantly CD11b+, express minimal amounts of TIM-3 (de Mingo Pulido et al., 2018), we turned to iCD103+ DCs generated using a combination of human FLT-3L-Ig and GM-CSF (Mayer et al., 2014). These cells displayed consistent expression of TIM-3 by the majority of CD103+CD11c+MHCII+ cells (Figure S2I). Importantly, while incubating iCD103+ DCs with HS for 24 hr had no impact on CXCL9 expression, the addition of αTIM-3 led to a significant increase in expression compared to the single agent controls (Figure 2G). We next used these iCD103+ DCs to confirm that the STING pathway was activated following TIM-3 blockade by evaluating phosphorylation of interferon regulatory transcription factor 3 (IRF3) via Western blot (Figure 2H) and intracellular flow cytometry (Figure S2J). As expected, phosphorylation of IRF3, along with the upstream kinase serine/threonine-protein kinase (TBK1), was only observed following the addition of αTIM-3 and HS (Figure 2H). This was equivalent to the level of phosphorylation observed with the cell-permeable, murine STING agonist DMXAA. Cumulatively these data demonstrate that TIM-3 expression by cDCs negatively regulates activation of the cGAS-STING pathway in the presence of extracellular dsDNA.
STING expression by cDC1s is required for efficacy of TIM-3 blockade during chemotherapy
As with transgenic MMTV-PyMT mice on the FVB/NJ background, mice on the C57BL6/J background did not respond to αTIM-3 alone (Figure S3A), but did respond to the combination of αTIM-3 and PTX (Figure S3B). We also noted that changing the isotype of the TIM-3 antibody had no impact on therapeutic efficacy, indicating that binding to the Fc receptors was inconsequential in this model system (Figure S3C). In order to determine the relevance of STING activation for the therapeutic efficacy of TIM-3 blockade we turned to BM chimeric C57BL6/J mice orthotopically implanted with PyMT mammary carcinoma cells (Figure 3A), thereby allowing us to generate a sufficient number of age-matched animals lacking the gene of interest within leukocytes. As shown in Figure 3B, tumor-bearing BM chimeric mice reconstituted with wild-type (WT) BM displayed significantly reduced tumor growth in response to αTIM-3 and PTX, as compared to those treated with PTX and rat IgG2a. However, αTIM-3 had no impact on tumor response to PTX in mice reconstituted with Sting-deficient BM (Figure 3B). Sting-deficiency did not impact TIM-3 expression (Figure S3D), tumor growth in the absence of therapy (Figure S3E), response to PTX alone (Figure S3E), or the prevalence of cDCs, monocytes or macrophages in the tumors (Figure S3F). Cgas-deficiency also prevented αTIM-3 from improving the efficacy to PTX (Figure 3C) without impacting the density of myeloid cells in tumors (Figure S3G). In contrast, the absence of MyD88, TRIF, or MAVS did not reduce the therapeutic efficacy of αTIM-3 and PTX (Figure 3D–F). Thus, we observed consistent results between the inability of αTIM-3 to induce CXCL9 expression by Sting-deficient and Cgas-deficient splenic cDC1s in the presence of HS (Figure 2C–E) and the inability of Sting-deficient and Cgas-deficient hosts to respond to combination therapy with αTIM-3 and PTX in vivo (Figure 3B–C).
Figure 3. STING expression by cDC1s is required for efficacy of αTIM-3/PTX.

(A) Treatment schematic for creation of BM chimeras, PyMT tumor implantation, and treatment with paclitaxel (PTX) and αTIM-3. (B-F) Relative tumor volume in chimeric C57BL/6J animals reconstituted with BM from wild type (WT) C57BL/6J mice or Sting-deficient (B), Cgas-deficient (C), Trif-deficient (D), Myd88-deficient (E), or Mavs-deficient (F) animals. For A-F, data reflect the mean ± SEM, with n=6–10 mice per group, and one of two representative experiments shown. (G) Relative tumor volume in mixed BM chimeric animals after the administration of DT to deplete Xcr1-DTR+ cDC1s just prior to treatment with PTX ± αTIM-3. Data reflect the mean ± SEM, with n=8–10 mice per group, merged from two independent experiments. (H) Percentage of cDC1s within tumors from F, using flow cytometry to distinguish Xcr1-DTR+ cDC1s by expression of Venus. Data reflect the mean ± SEM, with 4–5 mice per group. Significance was determined by a two-way (A-G) or one-way (H) ANOVA and is shown as *p<0.05, ***p<0.001. See also Figure S3.
Although these results demonstrated that host expression of cGAS and STING were required for the combinatorial efficacy of TIM-3 blockade and PTX, they did not distinguish between a role of STING expression by leukocyte subsets or other BM-derived populations. To specifically evaluate the importance of STING expression by cDC1s we acquired Xcr1-DTRvenus animals (Xcr1-DTR), which permit the selective depletion of the cDC1 subset following administration of diphtheria toxin (DT). As expected, depletion of cDC1s abrogated response to αTIM-3 and PTX, while control animals not exposed to DT responded normally (Figure S3H). To determine the relevance of STING expression by cDC1s we next created mixed BM chimeras using a 50/50 mixture of Xcr1-DTR with either WT or Sting−/− BM. This experimental setup allowed tumors to develop in the presence of STING-proficient cDC1s, with the removal of these cells initiated by DT just prior to therapy. Importantly, mice reconstituted with 50% WT BM responded to αTIM-3 and PTX, whereas those reconstituted with 50% Sting−/− BM did not (Figure 3G). As a control for STING expression by other leukocytes we also evaluated efficacy in the absence of DT, and observed the expected reduction in tumor growth in response to αTIM-3. This occurred despite the reduced presence of Xcr1-DTR CD103+ cDC1s within tumors, likely as a result of XCR1 haploinsufficiency and the importance of XCR1 for recruitment of cDC1s into tumors (Bottcher et al., 2018). Thus, STING expression specifically by cDC1s is required for the therapeutic efficacy of TIM-3 blockade combined with PTX chemotherapy.
TIM-3 suppresses endocytosis of extracellular DNA by cDCs
Our results suggested a role for TIM-3 in regulating cGAS-STING activation, either through direct suppression of STING signaling, or indirect regulation of DNA uptake or intracellular localization. To address the first possibility, we measured the impact of TIM-3 blockade on the activation of splenic cDC1s by IFN-β or 3’3’-cGAMP, but found no discernible effect (Figure S4A). We next sought to track extracellular DNA uptake by generating tumor cell debris following a 24 hr incubation with 5-ethynyl-2-deoxyuridine (EdU). Using FLT-3L-induced BMDCs (which express minimal surface TIM-3) we were able to detect intracellular EdU in the majority of cells after a 2 hr incubation (Figure S4B). Critically, the EdU signal was lost if DNase was added to tumor cell debris for the duration of the 2 hr incubation, but was not impacted when DNase was added for the final 30 minutes, indicative of cellular uptake. Using the same approach, we evaluated the ability of splenic cDC1s to uptake tumor DNA and found that this was highly increased by the addition of αTIM-3 (Figure 4A). As before, DNase prevented detection of EdU in splenic cDCs only when added at the start of the 2 hr incubation period, consistent with intracellular localization. DNA was also detected within the cDC2 subset following TIM-3 blockade, suggesting that poor CXCL9 expression by this population is due to downstream factors. DNA uptake in splenic cDCs was not impacted by Sting deficiency (Figure S4C).
Figure 4. TIM-3 suppresses endocytosis of extracellular DNA by cDCs.

(A) Intracellular flow cytometric detection of tumor cell DNA (EdU-labeled) within splenic cDCs after a 2 hr incubation with HS ± αTIM-3. DNase (50 U/ml) was added at the start of incubation to prevent uptake, or after 90 min to demonstrate intracellular localization. Data reflect the mean of 3 technical replicates, with one of three representative experiments shown. Significance was determined by one-way ANOVA and is shown as *p<0.05, **p <0.01, ***p<0.001. (B) Same as A, but using image cytometry to detect intracellular localization of tumor cell-derived DNA. Images are representative of three independent experiments. (C) Stacked confocal microscopy images displaying EdU-labeled exogenous DNA (red), MHCII (blue) and GAPDH (green) in iCD103+ BMDCs treated with HS ± αTIM-3 for 2 hrs. DNase was added for the final 15 min to digest remaining extracellular DNA. Two representative images from one of 4 independent experiments are shown. Analysis of 9 images per group is shown on the right, quantifying the detection of EdU within 20 individual cells (top) and the percent of EdU colocalized with GAPDH (bottom). Data reflects the mean, with significance determined by an unpaired t test and shown as **p <0.01, ***p<0.001. (D) Impact of Ciliobrevin D (Cilio. D) on EdU-labelled DNA uptake or phagocytosis of pHrodo Deep Red E. coli BioParticles by FLT-3L BMDCs. (E) Impact of Dynasore on EdU-labelled DNA uptake or endocytosis of pHrodo Red Transferrin by FLT-3L BMDCs. For D-E, data reflect the mean ± SD, significance determined by an unpaired t test and shown as ***p<0.001, with one of three independent experiments shown. See also Figure S4.
To confirm that TIM-3 was preventing the uptake of extracellular DNA we next measured EdU within splenic cDCs by Imagestream. As shown in Figure 4B, EdU was almost undetectable when cDCs were incubated with HS and IgG2a, but was apparent within the majority of cDC1s and cDC2s upon the addition of αTIM-3. Similar results were obtained with iCD103+ BMDCs (Figure S4D–E). As activation of cGAS requires cytoplasmic localization of dsDNA, we performed confocal microscopy on iCD103+ BMDCs incubated with HS and either IgG2a or αTIM-3, marking the surface membrane with MHCII and the cytoplasm with GAPDH. Consistent with DNA uptake and activation of the cGAS-STING pathway (Figure 2H), only following TIM-3 blockade did we observe substantial colocalization of EdU with GAPDH (Figure 4C). DNA uptake was not a result of changes in efferocytosis, as TIM-3 blockade neither altered the uptake of a fluorescent model tumor antigen in vivo (Figure S4F), nor the ability of iCD103+ BMDCs to efferocytose apoptotic thymocytes in vitro (Figure S4G). Inhibiting phagocytosis with the dynein inhibitor Ciliobrevin D also had no impact on DNA uptake (Figure 4D). In contrast, DNA uptake was blocked by Dynasore, an inhibitor of dynamin that selectively prevents endocytosis (Figure 4E). Together these data demonstrate that TIM-3 suppresses the ability of cDCs to endocytose extracellular dsDNA.
Uptake of extracellular DNA by cDCs is HMGB1-dependent
Of the four identified ligands for TIM-3, only HMGB1 is associated with DNA. HMGB1 also facilitates the escape of LPS from phagolysosomes into the cytosol (Deng et al., 2018) and can alter DNA structure to enhance cGAS recognition in tumor cells (Andreeva et al., 2017). Together these point to an important role for HMGB1 in regulating cytoplasmic localization and cGAS activation, and identify multiple stages at which TIM-3 binding to HMGB1 could alter cDC activation. Consistent with this, we employed a neutralizing HMGB1 antibody (Zhou et al., 2009) that prevented HMGB1 from binding to TIM-3 in vitro (Figure S5A), and found that it completely blocked the increased expression of CXCL9 induced by αTIM-3 when splenic cDCs were incubated with HS (Figure 5A) or tumor cell debris generated by irradiation (Figure S5B). We next evaluated whether HMGB1 was required for DNA uptake, or was instead important for downstream processes, and found that αHMGB1 prevented DNA uptake during TIM-3 blockade (Figure 5B). Similar results were obtained for CD103+ cDC1s following the intratumoral injection of HS into mice dosed with αTIM-3 (Figure S5C).
Figure 5. Uptake of extracellular DNA by cDCs is HMGB1-dependent.

(A) Intracellular flow cytometric detection of CXCL9 in CD8α+ splenic cDC1s following a 6 hr stimulation with tumor debris generated by heat shock (HS) or irradiation (IR). αTIM-3 and a neutralizing antibody against HMGB1 were used as indicated. (B) Intracellular flow cytometric detection of tumor cell DNA (EdU-labeled) within splenic cDCs after a 2 hr incubation with HS, αTIM-3, or αHMGB1. (C) Flow cytometry detection of synthetic, rhodamine-labelled B-DNA in iCD103+ BMDCs after a 2 hr incubation in the presence or absence of αTIM-3. HMGB1 was admixed with B-DNA at a 1:1 ratio (w/w) for 15 min prior to the incubation as indicated. For A-C, data reflect the mean of 3 technical replicates, with one of two (A) or three (B, C) representative experiments shown. Significance was determined by one-way ANOVA and is shown as ***p<0.001. (D) Relative volume of PyMT tumors in C57BL/6J animals treated with PTX, αTIM-3, or αHMGB1. Data reflect the mean ± SEM, with n=7–10 per group, and one of two representative experiments shown. Significance was determined by two-way ANOVA and is shown as ***p<0.001. See also Figure S5.
Purified DNA is unable to enter BMDCs; thus, activation of the STING pathway by DNA requires transfection (Woo et al., 2014). As we observed uptake of unpurified DNA, and HMGB1 was required for this process, we sought to evaluate if adding HMGB1 to pure DNA was sufficient to allow uptake by DCs. As shown in Figure S5D, the addition of rhodamine-labeled synthetic B-DNA did not result in DNA uptake by FLT-3L-induced CD11b+ BMDCs; however, admixing recombinant murine HMGB1 with B-DNA prior to incubation led to detectable levels of uptake. Similar studies were therefore conducted with iCD103+ BMDCs, which were able to take up B-DNA mixed with HMGB1 following TIM-3 blockade (Figure 5C). The presence of recombinant HMGB1 also greatly enhanced the ability of B-DNA to bind to TIM-3 coated beads in vitro (Figure 5SE), suggesting that TIM-3 might negatively regulate DNA uptake by competing for binding with HMGB1. Consistent with the critical role of HMGB1 in promoting DNA uptake and CXCL9 expression following TIM-3 blockade, neutralizing HMGB1 in vivo blocked the ability of αTIM-3 to improve the efficacy of PTX (Figure 5D).
Galectin-9 regulates TIM-3 clustering and function
A galectin-9 neutralizing antibody that prevented binding to TIM-3 (Figure S6A) was equivalent to TIM-3 blockade in terms of enhancing the therapeutic efficacy of PTX (Figure 6A) and inducing CXCL9 expression by splenic cDC1s in the presence of HS (Figure 6B). Tumor and splenic cDCs also had high levels of galectin-9 on the cell surface (Figure 6C). Given that HMGB1 was necessary for DNA uptake, this suggested to us that galectin-9 might enhance the ability of TIM-3 to function as a negative regulator of this process. In support of this, DNA uptake by splenic cDCs was increased by the addition of the galectin-9 neutralizing antibody (Figure 6D).
Figure 6. Galectin-9 regulates TIM-3 clustering and function.

(A) Relative tumor volume in mice bearing orthotopic PyMT tumors treated with PTX and IgG2a, αTIM-3, or αGalectin-9 (αGal-9). Treatment was initiated when tumors reached ~100 mm3. Data reflect the mean ± SEM, with n=11–12 mice per group compiled from 2 separate experiments. Significance was determined by two-way ANOVA and is shown as *p<0.05. (B) Intracellular flow cytometric detection of CXCL9 in splenic CD8α+ cDC1s following a 6 hr incubated with HS ± αTIM-3 or αGal-9. Data reflects the mean of 3 technical replicates, with one of two experiments shown. Significance determined by one-way ANOVA and is shown as ***p<0.001. (C) Representative histograms displaying surface expression of galectin-9 on splenic or tumor cDCs, as determined by flow cytometry. One of two experiments is shown. (D) Intracellular flow cytometric detection of tumor cell DNA (EdU-labeled) within splenic cDCs after a 2 hr incubation with HS, αTIM-3, or αGal-9 (clone RG9–1). Data reflect the mean of 3 technical replicates, with one of three experiments is shown. Significance was determined by one-way ANOVA and is shown as *p<0.05; ***p<0.001. (E) Surface expression of TIM-3 or galectin-9 on iCD103+ BMDCs, either untreated (black) or treated with 2 μg/ml recombinant murine galectin-9 (rmGal-9) for 30 min (red). Data reflect the mean of 3 technical replicates, with one of three experiments is shown. Significance was determined by t test and is shown as **p<0.01. (F) Stacked confocal microscopy images displaying TIM-3 (green) and DAPI (blue) in iCD103+ BMDCs, either untreated or treated with rmGal-9 ± αGal-9 (RG9–1) for 30 min. Analysis of images is shown to the right, quantifying the number of TIM-3 clusters per cell and shown as the average per field of view (FOV) from one of three experiments. Significance was determined by one-way ANOVA and is shown as ***p<0.001. See also Figure S6.
Galectin-9 might regulate TIM-3 function either by inducing a conformational change or by enhancing clustering on the cell surface (Wolf et al., 2020). To address the first possibility, we evaluated the ability of TIM-3 to bind HMGB1 in vitro, but found that adding recombinant galectin-9 had no impact (Figure S6B). To address the second possibility, we utilized iCD103+ BMDCs, which expressed low levels of galectin-9 on the surface, thereby allowing us to artificially increase this level through the addition of recombinant murine galectin-9 (Figure 6E). The addition of galectin-9 did not alter the level of TIM-3 on the surface of the cells as measured by flow cytometry (Figure 6E), but led to an increase in the detection of TIM-3 clusters by immunofluorescent microscopy (Figure 6F). Thus, galectin-9 regulates TIM-3 clustering on the cell surface, supporting a role for galectin-9 in promoting the inhibitory functions of TIM-3, and providing a potential explanation for how both αTIM-3 and αGalectin-9 produce similar increases in DNA uptake, CXCL9 expression, and the efficacy of PTX.
αTIM-3 increases DNA uptake and chemokine expression by human cDCs
To determine if TIM-3 had a comparable role in regulating the activation of human DCs we utilized peripheral blood cDCs enriched by negative selection from healthy donors (Figure S7A), which express high levels of TIM-3 (de Mingo Pulido et al., 2018), along with cellular debris generated from the MDA-MB-231 breast cancer cell line. Incubation with HS alone led to detectable DNA in only a small population of CD141+ cDC1s or CD1c+ cDC2s (Figure 7A). However, the addition of either the commercially available αTIM-3 clone F38–2E2, or the clinical agent TSR-022, led to DNA uptake by over 90% of cells from each individual donor (Figure 7B–C). Similar results were obtained using a third αTIM-3 antibody (clone TSR-A7). We next evaluated the impact of this DNA uptake on gene expression and observed increased expression of several genes associated with a type I IFN response in both the CD141+ cDC1 and CD1c+ cDC2 populations (Figure 7D–E, S7B–C), including a significant increase in CXCL9, CXCL10 and CXCL11 (Figure S7D–E). In addition, TIM-3 blockade increased the percentage of CD141+ cDC1s expressing CXCL10 at the protein level, although expression was highly variable between donors (Figure 7F–G). A small increase in the percentage of CXCL10+ CD1c+ cDC2s was also noted (Figure 7H). CXCL9 was difficult to detect in CD141+ cDC1s, despite high gene expression, though an increase was still observed following TIM-3 blockade (Figure S7F–H).
Figure 7. TIM-3 blockade increases DNA uptake and chemokine expression by human cDCs.

(A-C) Intracellular flow cytometric detection of EdU-labelled DNA in human peripheral blood cDCs isolated by negative selection from healthy donors. cDCs were incubated with MDA-MB-231 cellular debris generated by heat shock (HS) and TIM-3 blocking antibodies for 2 hr as indicated. (A) Representative density plots for CD141+ cDC1s. (B) Percentage of EdU positive CD141+ cDC1s. (C) Percentage of EdU positive CD1c+ cDC2s. (D) Significant (p<0.05) gene expression changes in peripheral blood CD141+ cDC1 following stimulation with HS and αTIM-3 for 24 hrs, compared to HS and IgG1. n=5 biological replicates, data compiled from two independent experiments. (E) Pathway analysis of significantly altered genes from D, showing Process Networks with a false discovery rate (FDR) > 4, and Pathway Maps with a FDR > 5. (F-H) Intracellular flow cytometric detection of CXCL10 in human peripheral blood cDCs following a 24 hr incubation with HS ± αTIM-3. (F) Representative density plots for CD141+ cDC1s. (G) Percentage of CXCL10 positive CD141+ cDC1s. (H) Percentage of CXCL10 positive CD1c+ cDC2s. Data reflect the mean of 2 technical replicates from 6 individual donors. Data compiled from three independent experiments. Significance was determined by a ratio paired t test and is shown as *p<0.05, **p <0.01, ***p<0.001. See also Figure S7.
Discussion
Our results demonstrate that TIM-3 suppresses endocytosis of extracellular dsDNA by cDCs, thereby preventing cytoplasmic localization, activation of cGAS-STING, expression of type I IFNs, and production of CXCL9 or CXCL10. During chemotherapy TIM-3 blockade therefore indirectly supports the effector function of intratumoral CD8+ T cells (de Mingo Pulido et al., 2018), possibly by improving spatial localization of T cells with cDC1s and increasing exposure to interleukin (IL)-12 (Ruffell et al., 2014). IL-12 and CXCL9 expression by cDC1s are also important for the function of T cells during αPD-1 therapy (Chow et al., 2019; Garris et al., 2018). Thus the activity of cDC1s within tumors is emerging as a critical determinant of an effective T cell response (Gardner et al., 2020), in addition to their role in transporting and cross-presenting tumor antigens in the lymph nodes (Roberts et al., 2016; Theisen et al., 2018).
Host expression of STING is critical for spontaneous and radiation-induced anti-tumor immunity (Deng et al., 2014; Woo et al., 2014) and intratumoral injection of STING agonists are efficacious in preclinical models (Corrales et al., 2015; Demaria et al., 2015; Li et al., 2016; Wang et al., 2017). However, systemic administration of STING agonists lacks tumor selectivity, and intrinsic activation of STING within T cells and cancer cells can induce cell death (Larkin et al., 2017) and promote metastasis (Bakhoum et al., 2018), respectively, potentially limiting the clinical utility of this approach. Our finding that αTIM-3 selectively enhances DNA uptake by cDCs bypasses these restrictions. Presumably cDC activation will be most prominent in tissues with a high degree of cell death, although an extensive evaluation of cDC activation in normal tissues following administration of αTIM-3 remains to be conducted. Most importantly, systemic administration of αTIM-3 produces no evidence of toxicity in mice, even when used in combination with chemotherapy (de Mingo Pulido et al., 2018). TIM-3 blockade also preferentially induces chemokine expression in cDCs, despite TIM-3 expression by other phagocytic populations, possibly due to higher expression of DNase II in macrophages (Ahn et al., 2018). That said, the benefit of an improved safety profile for αTIM-3 would likely be sacrificing efficacy, given the ability of STING agonists to activate type I IFN expression in multiple cell types within tumors.
Our data suggest that αTIM-3 will prove most useful during periods of cell death and a corresponding release of DNA and HMGB1 into the surrounding environment. Release of HMGB1 is not agnostic to the type of cytotoxic agent employed as immunogenic cell death results from irradiation and a limited set of chemotherapies, including doxorubicin, oxaliplatin, and mitoxantrone (Galluzzi et al., 2017). Conversely, cisplatin can sequester HMGB1 in the nucleus (Cardinal et al., 2009), is poorly immunogenic (Tesniere et al., 2010), and the similar compound carboplatin shows minimal combinatorial efficacy with αTIM-3 (de Mingo Pulido et al., 2018). Most relevant to our studies, PTX induces an inconsistent amount of HMGB1 release in lung cancer, while chemotherapy combinations can elicit synergistic HMGB1 release and an anti-tumor immune response (Pfirschke et al., 2016). A similar approach in breast cancer is warranted to determine the optimum chemotherapeutic regimen to employ with αTIM-3.
How the TIM-3 ligands galectin-9, HMGB1, CEACAM-1, or PS regulate the activity of TIM-3, especially when multiple ligands are present simultaneously, remains an area of active investigation. Despite TIM-3 binding to phosphatidylserine (PS) (Nakayama et al., 2009), TIM-3 blockade did not interfere with efferocytosis or alter acquisition of a model tumor antigen. In contrast, galectin-9 neutralization and TIM-3 blockade acted similarly in promoting DNA uptake, CXCL9 expression, and response to chemotherapy. Rather than a being a ligand for TIM-3 per se, our data indicate that the dual binding domains of galectin-9 induce TIM-3 clustering, thereby enhancing its negative regulatory functions, as has previously been hypothesized (Sabatos-Peyton et al., 2018). Consistent with this, most TIM-3 blocking antibodies do not inhibit binding of galectin-9, but rather block the ligand-binding domain shared by PS, CEACAM-1 and HMGB1 (Chiba et al., 2012; Nakayama et al., 2009; Sabatos-Peyton et al., 2018). The most likely explanation for our results is therefore that galectin-9-induced clustering of TIM-3 enhances binding to HMGB1-DNA complexes, thereby interfering with the ability of HMGB1 to interact with an endocytic receptor on the surface of cDCs.
Despite the importance of host expression of cGAS and STING for the induction of an anti-tumor immune response, it is unclear how DNA is able to enter the cytosol, or even what cell type(s) are required to express cGAS or STING. Tumor-derived 2’3’-cGAMP can transfer to macrophages and lead to activation of STING and production of type I IFN (Marcus et al., 2018), possibly through exosomes (Diamond et al., 2018), gap junctions (Ablasser et al., 2013), or the release of extracellular cGAMP (Luteijn et al., 2019; Ritchie et al., 2019; Zhou et al., 2020). Thus, whether DNA uptake by cDCs cells drives spontaneous activation of the cGAS-STING pathway remains unclear. It is true that purified DNA does not enter BMDCs, and activation of STING in vitro requires transfection protocols; however, DNA uptake by myeloid cells can occur in vivo following injection into tumors (Woo et al., 2014) and in vitro during co-culture with tumor cells treated with a telomere-targeted drug (Mender et al., 2020). Our data suggests these discrepancies may relate to HMGB1 acting as a critical factor mediating the uptake of extracellular DNA, although the receptor and pathway mediating this uptake remain to be determined. HMGB1 may also play a role by mediating the movement of DNA into the cytosol and enhancing recognition by cGAS (Andreeva et al., 2017; Deng et al., 2018). Cumulatively, our data indicate that uptake of extracellular DNA by cDCs can promote an anti-tumor response – at least for the cDC1 population that does not express the P2X7R channel important for uptake of cGAMP (Zhou et al., 2020) – but that this pathway is normally inhibited by high expression of TIM-3.
Limitations of Study
This study focused on the role of TIM-3 in cDCs and did not delve into how TIM-3 or its ligands regulate the function of other tumor immune populations, particularly in cancers where TIM-3 is prominently expressed on infiltrating T cells. It is also possible that TIM-3 expression by cDCs will be irrelevant under conditions where 2’3’-cGAMP is produced at high levels by cancer cells, with type I IFN expression by other cell types bypassing a need for activation of the cGAS-STING pathway in cDCs. Although we showed that TIM-3 prevents uptake of extracellular DNA by human peripheral blood cDCs, the extent to which these findings apply to human tumors remains unclear. Finally, while an HMGB1 neutralizing antibody was able to prevent endocytosis of DNA, it is possible that other DNA-binding proteins are also involved, and additional work is required to characterize the endocytic process.
STAR Methods
Resource Availability
Lead Contact:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Ruffell (Brian.Ruffell@moffitt.org).
Materials Availability:
This study did not generate new unique reagents.
Data and Code Availability:
This study did not generate any new datasets or code.
Experimental Model and Subject Details
Human Studies:
De-identified peripheral blood mononuclear cells of unknown gender were purchased from OneBlood, with patient consent forms obtained at the time of tissue acquisition.
Animal Studies:
Animals were maintained in the University of South Florida Department of Comparative Medicine barrier facility, and the respective Institutional Animal Care and Use Committee approved all experiments. All mice were obtained from The Jackson Laboratory, with the exception of the Xcr1tm2(HBEGF/Venus)Ksho mice (Yamazaki et al., 2013) obtained directly from Matthew Krummel at UCSF. BM chimeric mice were generated by irradiating recipient mice with 2 doses of 500 rads, followed by a BM transfer from donor animals, with tumors implanted after 6 weeks of reconstitution. Implantation of orthotopic mammary tumors was performed in female mice (approximately 2–4 months of age) by using single-cell suspensions isolated from mammary tumors of MMTV-PyMT transgenic mice combined 1:1 with matrigel (Corning), and injecting 106 cells/100 μl into the right 2/3 mammary gland. Treatment schedules were initiated in non-blinded fashion when tumors reached an average approximate volume of 100 mm3, with mice randomized to treatment groups as indicated in the respective figures. In vivo monoclonal antibodies (IgG2a, clone 2A3; αTIM-3, clone RMT3–23) were obtained from BioXCell and were administered by intraperitoneal (i.p.) injection at 1.0 mg/mouse, with follow-up doses of 0.5 mg every 5 days. Clinical grade PTX (Alvogen) was administered intravenously every 5 days at 10 mg/kg.
Method Detail
Tumor cell debris:
Cell lines (PyMT-B6, MDA-MB-231) were cultured in a 75 mm2 flask until 70–80% confluence. Cells were then removed via trypsin-EDTA, resuspended in 10 ml of serum free RPMI, and subjected to heat shock (55°C for 1 hr with agitation every 15 min). Samples were then centrifuged at 300 g for 10 min, dispersed into aliquots, and stored at −80°C until use. To label DNA with EdU cells were incubated in 10 μM EdU for 24 hr before being subjected to heat shock.
Generating guideRNA-Cas9 plasmids:
DNA oligonucleotides were designed with the following flanking sequences containing the respective targeting guide RNA (gRNA; N1-N20) sequences (5’-aaaggacgaaacaccN1-N20gttttagagctagaa-3’) that allows cloning into BsmBI (Esp3l) digested lentiCRISPRv2 hygro vector from Addgene (#98291). Sense and antisense Oligo’s for each gRNA were reconstituted at 10 μM and annealed in a reaction using 10 μl of each DNA oligo, 10 μl NaCl (5M) and 10 μl ddH2O for 10 min at 95°C, followed by decreasing 1°C/min for 70 cycles on a thermocycler. Annealed dsOligo were then diluted 1/50 and 0.5 μl was used per 50 ng of digested vector in a cloning reaction using the In-Fusion® HD cloning plus kit from Takara Bio (Cat.No. 638909), per according to the manufacturer’s recommendations. The lentiCRISPRv2 hygro vector was digested with BsmBI (Esp3l; ThermoFisher #FD0454) according to the manufacturer’s recommendations and purified with Nucleospin® Gel/PCR clean up kit (ThermoFisher #740609.5) before being used in the cloning reaction. Ampicillin resistant clones were picked, cultured overnight in 3 ml LB media containing 50 μg/ml ampicillin at 37°C, purified using the Nucleospin® plasmid kit according to manufacturer’s recommendations, and sent for sequencing to GeneWiz. Positive plasmids were transformed back in Stellar™ competent cells and 50 ml of overnight bacteria culture was used to purify the plasmid using PureLink HiPure Plasmid Midiprep Kit (ThermoFisher K210004).
Generating stable gene knockout cell lines:
Lentivirus containing supernatant was produced by transfecting pVSV-G (1 μg), pSPAX2 (2 μg) and lentiCRISPRv2/gRNA (2.5 μg) per 10cm dish of 70–80% confluent LentiX-293T cells (TakaraBio). Transfection was performed by incubating plasmid mix (5.5 μg total) in 700 μl Opti-MEM with 16.5 μl polyethylenimine 25 kDa (PEI; Polysciences #23966–2) transfection reagent (1 μg/μl stock) for 15 min at room temperature before adding drop wise to the cells. Media was replaced 8 hr after transfection and supernatant was harvested 48 hr after and filtered using Target2 PVDF syringe filters (0.45μm; ThermoFisher F2500–5). Spinfection was then conducted at 150 g for 1 hr, followed by overnight incubation with 3 ml of lentiviral supernatant in a 6-well dish in the presence of Polybrene (8 μg/ml) to infect 2×105 target cells (PyMT-B6). Culture media was replaced without selective antibiotics for 24 hr, followed by the addition of Hygromycin B (600–800 μg/ml) for 5–10 days to select resistant cells. All cells were generated and passaged under equal conditions in parallel.
HMGB1 protein production:
pET28a-Flag-HMGB1–6xHIS constructs (Addgene #53561) were transformed into BL21(DE3) (ThermoFisherScientific: EC0114) according to manufacturer’s instructions. Selected clones were precultured overnight at 37°C and used to inoculate two containers of 800 mL LB medium supplemented with Kanamycin (34 μg/ml). The cell culture was grown at 37°C and induced by IPTG at 0.2 mM until the OD600 reached 0.7. Cell growth continued at 18°C for 20 hours before the cells were harvested by centrifugation at 43,600 g. The obtained cells were resuspended in lysis buffer (PBS, pH 7.2, 400 mM NaCl, 20 mM imidazole, 0.1 mg lysozyme/ml lysis buffer, 25 units DNase/mL lysis buffer) and lysed by a homogenizer (APV 2000, Invensys). The supernatant was loaded onto a Ni2+-NTA column (GE Healthcare Life Science) and the column was washed with 8 column volumes (CV) of buffer A (PBS, pH 7.2, 400 mM NaCl, 20 mM imidazole) followed by 14 CV buffer B (PBS, pH 7.2, 400 mM NaCl, 250 mM imidazole) with a flow rate of 0.8 mL/min. The fractions containing the His-tag protein were combined and concentrated by an Amicon Ultra Device (MWCO 10 000, Millipore). The buffer was changed to buffer C (20 mM K/Na phosphate, pH 7.2) using a HiPrep 26/10 Desalting column (GE Healthcare Life Science) before the protein sample was passed through a HisTrap SP HP column (GE Healthcare Life Science). The target protein was eluted out using a 0 to 1 M NaCl gradient over 20 CV buffer. Protein was further purified by size-exclusion chromatography using Superdex 75 26/60 column (GE Healthcare Life Science) in PBS buffer without visible contamination. The purified protein was analyzed by SDS-PAGE and the concentration was determined by NanoDrop™ One (ThermoScientific) at UV 280 nm with 260/280 ratio at 0.57.
Splenic cDC stimulation:
cDCs were enriched (~50% purity) from spleens by negative selection using biotinylated antibodies against Ly6C (10 μg), Ter119 (20 μg), CD49b (20 μg), Ly6G (30 μg), CD3e (40 μg), B220 (40 μg) in combination with MojoSort magnetic beads (BioLegend). Cells were plated at 1×106 per ml in serum free RPMI 1640 or suspended in supernatant containing tumor cell debris. Blocking antibodies against TIM-3 (clone RMT3–23), IFNRA1 (clone MAR1–5A3) HMGB1 (clone 3E8), or galectin-9 (clone RG9–1) were added to the supernatant at 10 μg/ml for the duration of the 2–6 hr stimulation. For studies employing intracellular staining with CXCL9 (clone MIG-2F5.5) the cells were incubated for the final 4 hr in the presence of 5 μg/ml brefeldin A and 2 μM monensin. For some experiments, cells were stimulated with IFNγ (10 ng/ml), IFNα (10 ng/ml), IFNβ1 (10 ng/ml), 2’3’-cGAMP (10 μg/ml) or 3’3-cGAMP (10 μg/ml).
Peripheral blood cDC stimulation:
Unfrozen peripheral blood buffy coats were enriched for mononuclear cells via Lympoprep (StemCell Technologies) density centrifugation, followed by enrichment for dendritic cells via negative selection using the EasySep Human Myeloid DC Enrichment Kit, both according to the manufacturer’s directions (StemCell Technologies). Cells were then plated at 1×106 per ml in serum free RPMI 1640 or suspended in supernatant containing EdU-labelled MDA-MB-231 cellular debris and blocking antibodies against TIM-3 (clone F38–2E2, TSR-022, TSR-A7) were added to the supernatant at 10 μg/ml. Intracellular detection of EdU was performed after 2 hr using Click-iT EdU Alexa Fluor 594 (Invitrogen) according to the manufacturer’s directions. Intracellular staining for CXCL9 (clone J1015E10) and CXCL10 (clone J034D6) was conducted after 24 hr, with cells incubated for the final 4 hr in the presence of 5 μg/ml brefeldin A and 2 μM monensin.
BMDC generation:
BM was harvested from C57BL6/J female mice and red blood cells lysed with 150 mM NH4Cl/10 mM NaHCO3/1 mM EDTA. Remaining cells were plated in RPMI 1640 containing 2.0 mM L-glutamine and 25 mM HEPES, supplemented with 100 U/ml penicillin/streptomycin, 55 μM β-ME, and 10% fetal calf serum (Life Technologies). To create FLT-3L BMDCs, cells were plated at 2×106/ml in 100 ng/ml recombinant human Flt-3 Ligand Immunoglobulin (Flt-3L-Ig; BioXCell) and then incubated untouched for 7 days. To generate iCD103+ BMDCs, cells were plated at 1.5×106/ml in 200 ng/ml Flt-3L-Ig and 5 ng/ml murine recombinant GM-CSF (Peprotech). A 50% volume of fresh medium was added to the culture on day 5, non-adherent cells were harvested and re-plated in fresh medium at 3×105/ml on day 9, and cells were used on day 15–16. Both protocols generated cultures with over 90% purity of CD11c+ cells. For stimulation the cells were harvested and resuspended at 106/ml in tumor cell debris for up to 24 hr. For DNA uptake, cells were either incubated for 2 hr with supernatant from EdU-labeled tumor cells or with 250 ng/ml Poly(dA:dT) rhodamine (synthetic B-DNA) mixed with recombinant murine HMGB1. In some experiments Ciliobrevin D (40 μM, 15 min) or Dynasore (80 μM, 30 min) were added to BMDCs prior to incubation with DNA. For efferocytosis, single cell suspensions of thymocytes were labelled with 5 μM CellTrace Violet per the manufacturer’s instructions, X-Ray irradiated at 20 Gy, and then incubated overnight. Thymocytes were then incubated with BMDCs pre-incubated with blocking antibodies for 30 min prior to washing, antibody staining, and flow cytometry.
Western blot:
To measure activation of the cGAS-STING pathway, 4.5×106 iCD103+ BMDCs were stimulated for 3 hr, and then divided in half to create either nuclear or total cell lysate. Total cell lysis buffer consisted of Triton X-100 (1X, Sigma Aldrich; T8787), Phosphatase inhibitor cocktail 2 (1X, Sigma Aldrich; P5726), Phosphatase inhibitor cocktail 3 (1X, Sigma Aldrich; P0044), Trypsin-chemotrypsin inhibitor (0.1 mg/ml, Sigma Aldrich T9777), Leupeptin (0.01 mg/ml, Roche 11017101001) and Aprotinin (0.01 mg/ml, Roche 10236624001). Nuclear lysate was generated using the NE-PER Nuclear and Cytoplasmic extraction kit (ThermoFisher), with the addition of Trypsin-chemotrypsin inhibitor, Leupeptin and Aprotinin. Equal protein amounts of cytolasmic and total cell lysates were then electrophoresed in 10% Tris-Glycine gels (Novex-Invitrogen), transferred to PVDF membranes by iBlot™ Gel Transfer Device (ThermoFisher), and blotted with the corresponding primary and secondary antibodies. Membrane-bound immune complexes were detected using the ChemiDoc™ Imaging System (Bio-Rad, #17001401). To verify the generation of gene knockout cell lines, whole cell lysate was generated using NP-40 lysis. Lysates were electrophoresed on a 4–15% gradient gel at 120V for 1.5–2 hr and transferred onto Immobilion-FL PVDF membrane (ThermoFisher) at 200 V for 1.5h at room temperature. Respective primary antibodies were used at 1/1000 dilution in 5% skim milk in TBS-Tween (0.1%) at 4°C overnight, and after washing, secondary fluorescently-labeled antibodies were incubated for 20–30 min at room temperature before membranes were imaged on a LI-COR Odyssey® Fc imaging system.
Flow/image cytometry:
Mice were cardiac-perfused with PBS containing 10 U/ml heparin to clear peripheral blood, and single cell suspensions were prepared by incubating minced tissue in 1 mg/ml collagenase (Roche) and 50 U/ml DNase I (Roche) at 35°C with agitation. Cells were used immediately or stored in 10% DMSO at −80°C. All incubations were performed for 30 min on ice. For staining of single cell suspensions, splenocytes, or BMDCs, cells were first incubated with Live/Dead Aqua (1/500, Invitrogen) or Zombie NIR (1/1000, BioLegend) diluted in PBS, then washed once and incubated with 10 μg/ml of anti-CD16/32 (clone 2.4G2) diluted in PBS, 1 mM EDTA, and 1 mg/ml BSA (FACS buffer). Cells were then incubated with a cocktail of antibodies (see Key Resource Table) in FACS buffer, washed once, and fixed with Cytofix (BD Biosciences). Intracellular staining for chemokines was performed in 1X Perm/Wash buffer (BD Biosciences), followed by a wash step and suspension in FACS buffer. Intracellular detection of EdU was performed using Click-iT EdU Alexa Fluor 594 (Invitrogen) according to the manufacturer’s directions. Intracellular staining of pIRF3 was performed after permeabilization with 90% methanol for 30 min, followed by staining for extracellular markers. Immune populations were identified with a previously described gating strategy (de Mingo Pulido et al., 2018; Ruffell et al., 2014). Data was collected with a LSRII flow cytometer (BD Bioscience) with analysis conducted using FlowJo version 9 or 10 (FlowJo LLC), or collected with an ImagestreamX Mark II with analysis conducted using the INSPIRE software (Amnis/Millipore Sigma).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse Ly6G clone 1A8 BUV395 | BD | Cat# 563978 RRID:AB_2716852 |
| Anti-mouse CD24 clone M1/69 BUV496 | BD | Cat# 564664 RRID:AB_2716853 |
| Anti-mouse CD19 clone 1D3 BUV737 | BD | Cat# 564296 RRID:AB_2716855 |
| Anti-mouse CD8 alpha clone 53.6–7 BUV800 | BD | Cat# 564920 RRID:AB_2716856 |
| Anti-mouse CD8 alpha clone 53.6–7 BB700 | BD | Cat# 566409 RRID:AB_2744467 |
| Anti-mouse MHCII M5/114.15.2 BV421 | BD | Cat# 562564 RRID:AB_2716857 |
| Anti-mouse CD11c clone N418 BV605 | BioLegend | Cat# 117334 RRID:AB_2562415 |
| Anti-mouse CD11c clone N418 APC | BioLegend | Cat# 117310 RRID:AB_313779 |
| Anti-mouse CD4 clone RM4–5 BV650 | BD | Cat# 563747 RRID:AB_2716859 |
| Anti-mouse/human CD11b M1/70 BV711 | BD | Cat# 563168 RRID:AB_2716860 |
| Anti-mouse/human CD11b M1/70 BB515 | BD | Cat# 564454 RRID:AB_2665392 |
| Anti-mouse CD45 30-F11 BV785 | BD | Cat# 564225 RRID:AB_2716861 |
| Anti-mouse CD69 H½F3 FITC | BioLegend | Cat# 104506 RRID:AB_313109 |
| Anti-mouse CD3 epsilon clone 17A2 PerCP-Cy5.5 | BD | Cat# 560527 RRID:AB_1727463 |
| Anti-mouse PDCA-1 clone 927 PE | BioLegend | Cat# 127010 RRID:AB_1953285 |
| Anti-mouse CD49b clone DX5 PE-Dazzle | BioLegend | Cat# 108924 RRID:AB_2565271 |
| Anti-mouse CD103 clone 2E7 PE-Cy7 | BioLegend | Cat# 121426 RRID:AB_2563691 |
| Anti-mouse F4/80 clone BM8 APC | BioLegend | Cat# 123116 RRID:AB_893481 |
| Anti-mouse Ly6C clone HK1.4 APC-Cy7 | BioLegend | Cat# 128026 RRID:AB_10640120 |
| Anti-mouse CXCL9 PE | BioLegend | Cat# 515604 RRID:AB_2245489 |
| Anti-mouse TIM-3 clone RMT3–23 PE | BioLegend | Cat# 119703 RRID:AB_345377 |
| Anti-mouse TIM-3 clone RMT3–23 FITC | ThermoFisher | Cat# 11–5870-82 RRID:AB_2688129 |
| Anti-mouse P2X7R clone 1F11 PE | BioLegend | Cat# 148703 RRID:AB_2650951 |
| Anti-mouse Galectin-9 clone 108A2 APC | BioLegend | Cat# 137912 RRID:AB_2750155 |
| Anti-mouse Galectin-9 clone RG9–35 PE | BioLegend | Cat# 136103 RRID:AB_1953306 |
| Anti-FLAG clone L5 PE | BioLegend | Cat# 637310 RRID:AB_2563148 |
| Anti-mouse CD16/CD32 clone 2.4G2 (Fc block) | BD | Cat# 553142 RRID:AB_394657 |
| Anti-mouse CD3 epsilon clone 145–2C11 biotin | BioLegend | Cat# 100304 RRID:AB_312669 |
| Anti-mouse/human B220 clone RA3–6B2 biotin | BioLegend | Cat# 103204 RRID:AB_312989 |
| Anti-mouse Ly6G clone 1A8 biotin | BioLegend | Cat# 127604 RRID:AB_1186108 |
| Anti-mouse CD49b clone DX5 biotin | BioLegend | Cat# 108904 RRID:AB_313411 |
| Anti-mouse Ter119 clone TER-119 biotin | BioLegend | Cat# 116204 RRID:AB_313705 |
| Anti-mouse TIM-3 clone RMT3–23 (LEAF) | BioLegend | Cat# 119708 RRID:AB_2564109 |
| Anti-mouse/human HMGB1 clone 3E8 (Ultra-LEAF) | BioLegend | Cat# 651414 RRID:AB_2728488 |
| Anti-human/mouse GAPDH (polyclonal) | ThermoFisher | Cat# PA1–16777 RRID:AB_568552 |
| Donkey anti-rabbit IgG (polyclonal) Alexa 488 | ThermoFisher | Cat# A21206 RRID:AB_2535792 |
| Anti-human/mouse phospho TBK1/NAK (Ser172) (D52C2) XP Rb mAb | Cell signaling | Cat# 5483 RRID:AB_10693472 |
| Anti-human/mouse TBK1/NAK (D1B4) Rb mAb | Cell signaling | Cat# 3504 RRID:AB_2255663 |
| Anti-human/mouse phospho IRF3 (Ser396) (D6O1M) Rb mAb | Cell signaling | Cat# 29047 RRID:AB_2773013 |
| Anti-human/mouse IRF3 (D83B9) Rb mAb | Cell signaling | Cat# 4302 RRID:AB_1904036 |
| Anti-mouse Cgas (D3O8O) Rb mAb | Cell signaling | Cat# 31659S RRID:AB_2799008 |
| Anti-human/mouse STING (D2P2F) Rb mAb | Cell signaling | Cat# 13647S RRID:AB_2732796 |
| Anti-mouse/human nuclear matrix protein p84 (5E10) | Abcam | Cat# Ab487 RRID:AB_304696 |
| Anti-mouse/human β-actin | Millipore-Sigma | Cat# A2228 RRID:AB_476697 |
| Anti-mouse/human Vinculin | Millipore-Sigma | Cat# V9131 RRID:AB_477629 |
| Anti-rabbit IgG HRP linked | Millipore-Sigma | Cat# NA934V RRID:AB_2722659 |
| Anti-mouse IgG HRP linked | BioLegend | Cat# 405306 RRID:AB_315009 |
| Anti-GAPDH mAb (GA1R) | ThermoFisher | Cat# MA5–15738 RRID:AB_10977387 |
| Goat anti-Rabbit IgG (H+L), DyLight 800 4X PEG | ThermoFisher | Cat# SA5–35571 RRID:AB_2556775 |
| Goat anti-Mouse IgG (H+L), DyLight 680 | ThermoFisher | Cat# 35519 RRID:AB_1965956 |
| Polyclonal goat anti-fluorescein, Alexa 488 | ThermoFisher | Cat# A11096 RRID:AB_221558 |
| Anti-mouse TIM-3 clone RMT3–23 | BioXCell | Cat# BE0115 RRID:AB_10949464 |
| Anti-mouse TIM-3 clone RMT3–23 (mouse IgG2a) | TESARO: A GSK Company | N/A |
| Anti-mouse TIM-3 clone RMT3–23 (mouse IgG1-D265A) | TESARO: A GSK Company | N/A |
| Anti-mouse Galectin-9 clone RG9–1 | BioXCell | Cat# BE0218 RRID:AB_2687702 |
| Anti-mouse IFNAR1 clone MAR1–5A3 | BioXCell | Cat# BE0241 RRID:AB_2687723 |
| Rat anti-HRPN Isotype Control (IgG1) | BioXCell | Cat# BE0088 RRID:AB_1107775 |
| Rat anti trinitrophenol Isotype Control (IgG2a) | BioXCell | Cat# BE0089 RRID:AB_1107769 |
| Anti-human CD45 clone H130 BV785 | BD | Cat# 563716 RRID:AB_2716864 |
| Anti-human HLA-DR clone L243 APC-Fire750 | BioLegend | Cat# 307658 RRID:AB_2572101 |
| Anti-human CD16 clone 3G8 BV421 | BD | Cat# 562874 RRID:AB_2716865 |
| Anti-human CD3 epsilon clone OKT3 PerCP710 | ThermoFisher (eBioscience) | Cat# 46–0037-42 RRID:AB_1834395 |
| Anti-human CD56 clone HCD56 BB700 | BD | Cat# 555518 RRID:AB_398601 |
| Anti-human CD19 clone SJ25C1 BB700 | BD | Cat# 566396 RRID:AB_2744310 |
| Anti-human CD11c clone 3.9 BV650 | BioLegend | Cat# 301638 RRID:AB_2563797 |
| Anti-human CD14 clone M5E2 BUV805 | BD | Cat# 565779 RRID:AB_2716868 |
| Anti-human CD11b clone ICRF44 BUV395 | BD | Cat# 563839 RRID:AB_2716869 |
| Anti-human BDCA1/CD1c clone F10/21A3 BB515 | BD | Cat# 565054 RRID:AB_2716870 |
| Anti-human BDCA3/CD141 clone M80 APC | BioLegend | Cat# 344106 RRID:AB_10899578 |
| Anti-human CD123 clone 6H6 BV650 | BioLegend | Cat# 306020 RRID:AB_2563827 |
| Anti-human CXCL9 clone J1015E10 | BioLegend | Cat# 357904 RRID:AB_2562009 |
| Anti-human CXCL10 clone J034D6 PE | BioLegend | Cat# 519504 RRID:AB_2561409 |
| Anti-human TIM-3 clone F38–2E2 (Ultra-LEAF) | BioLegend | Cat# 345009 RRID:AB_11150398 |
| Anti-human TIM-3 clone TSR-022 | TESARO: A GSK Company | N/A |
| Anti-human TIM-3 clone TSR-A7 | TESARO: A GSK Company | N/A |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| Adult peripheral blood mononuclear cells | OneBlood | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Paclitaxel | Alvogen | 47781–59307-0 |
| Human Flt-3L-Ig | BioXCell | Cat# BE0098 |
| Recombinant mouse IFNγ | Peprotech | Cat# 315–05 |
| Recombinant mouse GM-CSF | Peprotech | Cat# 315–03 |
| 2’3’-cGAMP | InvivoGen | Cat# tlrl-nacga23 |
| 3’3’-cGAMP | InvivoGen | Cat# tlrl-nacga |
| DMXAA | InvivoGen | Cat# tlrl-dmx |
| Poly(dA:dT) rhodamine (synthetic B-DNA analog) | InvivoGen | Cat# tlrl-patrh |
| Recombinant mouse IFNα | BioLegend | Cat# 751802 |
| Recombinant mouse IFNβ1 | BioLegend | Cat# 581302 |
| Recombinant mouse HMGB1 | BioLegend | Cat# 764004 |
| Zombie NIR Fixable Viability Kit | BioLegend | Cat# 423105 |
| TrueStain FcX Block | BioLegend | Cat# 422302 |
| Brefeldin A (1000x solution) | BioLegend | Cat# 420601 |
| Monensin (1000x solution) | BioLegend | Cat# 420701 |
| Recombinant mouse galectin-9 | R&D Systems | Cat# 3535-GA-050 |
| Recombinant mouse TIM-3-Fc | R&D Systems | Cat# 1529-TM-050 |
| D-Lactose monohydrate | Sigma-Aldrich | Cat# 61339 |
| Diphtheria Toxin | Sigma-Aldrich | Cat# D0564 |
| Polybrene infection/transfection reagent | Sigma-Aldrich | TR-1003-G |
| Collagenase A | Millipore Sigma | 11088793001 |
| DNAse I, grade II from bovine pancreas | Roche | 10104159001 |
| Matrigel GFR/LDEV-Free | ThermoFisher | Cat# CB-40230 |
| Live/Dead Fixable Aqua Dead Cell Stain | ThermoFisher | Cat# L34957 |
| Hygromycin B | ThermoFisher | Cat# 10–687-010 |
| FastDigest Esp3l | ThermoFisher | Cat# FD0454 |
| AccuCheck Counting Beads | ThermoFisher | Cat# PCB100 |
| polyethylenimine 25kDa (PEI) transfection reagent | Polysciences | Cat# 23966–2 |
| Dynasore, dynamin inhibitor I, CAS 30448–55-3 | Millipore-Sigma | Cat# 324410 |
| Ciliobrevin D, dynein inhibitor | Millipore-Sigma | Cat# 250401 |
| Critical Commercial Assays | ||
| Single Tube TaqMan Gene Expression Assays | ThermoFisher Scientific | Cat# 4331182 |
| nCounter Mouse Pan-Cancer Immune Panel | NanoString | XT-CSO-MIP1–12 |
| nCounter Human Pan-Cancer Immune Panel | NanoString | XT-CSO-HIP1–12 |
| Click-iT EdU Cell Proliferation Kit Alexa 594 | Invitrogen | Cat# C10339 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagent | ThermoFisher | Cat# 78833 |
| In-Fusion HD cloning kit | TakaraBio | Cat# 638909 |
| Nucleospin PCR/Gel purification kit | TakaraBio | Cat# 740609.5 |
| Nucleospin Plasmid miniprep | TakaraBio | Cat# 740588.5 |
| PureLink HiPure Plasmid Midiprep kit | ThermoFisher | Cat# K210004 |
| pHrodo Red Transferrin Conjugate | ThermoFisher | Cat# P35376 |
| pHrodo Deep Red E. coli BioParticles | ThermoFisher | Cat# P35360 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | Cat# HTB-26 RRID:CVCL_0062 |
| PyMT-B6 | David G. DeNardo, Washington University | Meyer et al. Nat. Commun. 2018 PMID: 29593283 |
| LentiX-239T | Takara Bio | Cat# 632180 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.FVB-Tg(MMTV-PyVT)634Mul/J | The Jackson Laboratory | RRID: IMSR_JAX:022974 |
| Mouse: B6.Cg-Tg(Itgax-cre)1–1Reiz/J | The Jackson Laboratory | RRID: IMSR_JAX:008068 |
| Mouse: BC(Cg)-Irf8tm1.1hm/J | The Jackson Laboratory | RRID: IMSR_JAX:014175 |
| Mouse: B6(Cg)-Xcr1tm2(HBEGF/Venus)Ksho (Xcr1-DTR) | Matthew Krummel, UCSF | RRID: IMSR_RBRC09485 |
| Mouse: B6(Cg)-Tmem173tm1.2Camb/J | The Jackson Laboratory | RRID: IMSR_JAX:025805 |
| B6.129P2(SJL)-Myd88tm1.1Defr/J | The Jackson Laboratory | RRID: IMSR_JAX:009088 |
| C57BL/6J-Ticam1Lps2/J | The Jackson Laboratory | RRID: IMSR_JAX:005037 |
| B6(C)-Cgastm1d(EUCOMM)Hmgu/J | The Jackson Laboratory | RRID: IMSR_JAX:026554 |
| B6;129-Mavstm1Zjc/J | The Jackson Laboratory | RRID: IMSR_JAX:008634 |
| Mouse: C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| Non-targeted (GGACATTACATATAAGACCA) | Integrated DNA Technology | N/A |
| Mb21d1 (CGGGCCGCAGCTTTCCGCGT) | Integrated DNA Technology | N/A |
| Tmem173 (CAGTAGTCCAAGTTCGTGCG) | Integrated DNA Technology | N/A |
| Recombinant DNA | ||
| lentiCRISPRv2 hygro | Addgene | Cat# 98291 RRID:Addgene_98291 |
| pCMV VSV-G | Addgene | Cat# 8454 RRID:Addgene_8454 |
| psPAX2 | Addgene | Cat# 12260 RRID:Addgene_12260 |
| pET28a-Flag-HMGB1–6xHIS | Addgene | Cat# 53561 RRID:Addgene_53561 |
| Software and Algorithms | ||
| FlowJo Version 9 and 10 | FlowJo LLC |
https://www.flowjo.com RRID:SCR_008520 |
| Prism Version 8 | GraphPad |
https://www.graphpad.com/scientific-software/prism/ RRID:SCR_002798 |
| Other | ||
| Mouse anti-human IgG (Fc) Coated Polystyrene Particles (10.0–14.0 μm) | Spherotech | Cat# HUAMP-100–4 |
| MojoSort Streptavidin Nanobeads | BioLegend | Cat# 480016 |
Bead Assay:
Mouse anti-human IgG (Fc) coated polystyrene beads (Spherotech) were coupled to 40 μg/ml recombinant mouse TIM-3-Fc (R&D Systems) by incubation at room temperature for 1 hr. Galectin-9 binding was measured by incubating 1 μg/ml recombinant mouse galectin 9 (R&D Systems) in the presence or absence of 10 μg/ml galectin-9 blocking antibody (RG9–1, BioXCell) or 100 mM lactose (Sigma-Aldrich) for 30 minutes. Galectin-9 bound to beads was then detected by incubation with 0.5 μg/ml of an APC conjugated anti-mouse galectin-9 antibody that recognizes the linker region (clone 108A2), with detection by flow cytometry. Binding of FLAG-tagged HMGB1ΔC was determined using an anti-FLAG-PE antibody. Binding of B-DNA/HMGB1ΔC complexes was detected by measuring rhodamine fluorescence by flow cytometry. Rat IgG2a isotype control or anti-HMGB1 (clone 3E8) were pre-incubated with HMGB1 at 10 μg/ml, prior to their addition to TIM-3-Fc-coated polystyrene beads.
Immunofluorescence microscopy:
For detection of EdU-labeled DNA, BMDCs were prepared as described above for staining with MHCII-BV421 or Click-iT EdU Alexa Fluor 594, with additional intracellular staining with rabbit antibodies against GAPDH (1:100, Invitrogen), and secondary detection using a 1:2000 dilution of donkey anti-rabbit Alexa 488 (Invitrogen). Cells were then adhered to glass slides using a Shandon Cytospin, and coverslips mounted using ProLong Glass Antifade (Invitrogen). Images were acquired with a Leica SP8 Confocal Microscope. ImageJ was used to segment cells by creating a mask in thresholded images with MHCII as a cell membrane reference, with the level of EdU expressed as the mean fluorescence intensity on a per cell basis. Colocalization between GAPDH and EdU-labeled DNA was determined within each image. For detection of TIM-3, BMDCs were treated with 2 μg/ml recombinant murine galectin-9 (R&D Systems) for 30 min, followed by incubation with 5 μg/ml of galectin-9 neutralizing antibody (clone RG9–1) for another 30 min at 37°C. Cells were then stained with TIM-3-FITC followed by goat anti-fluorescein Alexa 488 (1:800, Invitrogen), fixed with cytofix for 30 min, and adhered to glass slides using a Shandon Cytospin. Coverslips were mounted using Antifade Mounting Medium with DAPI (Vector Labs). Images were acquired with a Leica SP8 Confocal Microscope. Definiens Developer XD was used to quantify the number of TIM-3 clusters (area > 0.15 μm2) on a per cell basis (DAPI area < 25 μm2 and roundness > 1).
Gene Expression:
Fluorescent-activated cell sorting (FACS) was conducted on a FACSAriaII (BD Biosciences), with 2,000 to 50,000 sorted cells flash frozen in liquid nitrogen as a cell pellet. For real-time PCR analysis 20 μl of Ambion Cells-to-Ct buffer (Thermo Fisher Scientific) was added to a cell pellet and 10 μl was used to generate cDNA according to manufacturer’s directions. PCR was performed using individual TaqMan Assays following a preamplification step (Life Technologies). The comparative threshold cycle method was used to calculate fold change in gene expression, which was normalized to a single (Tbp) reference gene. For gene expression analysis by Nanostring nCounter, cell lysates were hybridized to the 770-gene Mouse Pancancer Immune Panel according to the manufacturer’s protocol (NanoString Technologies). Briefly, 20 μl of Ambion Cells-to-Ct buffer (Thermo Fisher Scientific) was added to a cell pellet and a 5.0 μl volume of lysate was hybridized to the NanoString reporter and capture probes in a thermal cycler for 16 hr at 65°C. Washing and cartridge immobilization were performed on the NanoString nCounter PrepStation, and the cartridge was scanned at 555 fields of view on the nCounter Digital Analyzer.
Quantification and statistical analysis
Nanostring:
Raw probe counts were captured by nCounter® Digital Analyzer and pre-processed by nSolver™ Analysis Software v2.5. Initial quality control (QC) was performed to inspect the quality of imaging, binding density, and positive controls following the nSolver user manual. Data normalization was then performed using the NanoStringNorm package (v1.2.1) (Waggott et al., 2012). Specifically, endogenous probes were normalized to the positive control probe counts using “geo.mean” method and to the housekeeping gene counts using the ‘housekeeping.geo.mean’ method. Differential expression analysis was performed on the log2-transformed normalized data using NanoStringNorm. Genes with false positive rate (FDR) adjusted p-value <0.05 and |fold-change|>2 were considered as significantly differentially expressed due to the presence of THS, and visualized by heatmaps and volcano plots. Pathway enrichment analysis of the differentially expressed genes was performed using MetaCore. The statistical significance of the enriched pathways was evaluated by hypergeometric distribution p-value adjusted for FDR.
Statistical analyses:
For growth curves significance was determined via 2-way ANOVA with Tukey’s multiple comparisons test, with significance shown for the final data point. A 2-way unpaired t-test or 2-way unpaired t-test with Welch’s correction was used for comparison between groups with equal or unequal variance, respectively. Comparisons between multiple groups were performed via 1-way ANOVA. Analyses were performed using Prism 8 or 9 (GraphPad). Significance is shown as *p<0.05, **p <0.01, ***p<0.001 as described in each figure legend.
Supplementary Material
Highlights.
TIM-3 blockade promotes endocytosis of extracellular DNA by dendritic cells
DNA uptake and CXCL9 expression by dendritic cells is HMGB1-dependent
Galectin-9 regulates TIM-3 cell surface clustering and inhibitory function
Anti-tumor efficacy of TIM-3 mAb and paclitaxel is dependent upon cGAS and STING
Acknowledgements
This work was supported by the Moffitt Cancer Center Flow Cytometry, Molecular Genomics, Analytic Microscopy, Chemical Biology, and Tissue Core Facilities, all comprehensive cancer center facilities designated by the National Cancer Institute (P30-CA076292). The authors would like to thank Matthew F. Krummel, Miranda Broz, Kathy Yang, Vivian Lee, Leenil Noel and Aysenur Keske for scientific/technical assistance, along with Melissa A. Meyer and David G. DeNardo for the PyMT-B6 cell line. K.H. was supported by a Postdoctoral Fellowship from the Swiss National Science Foundation. A.G. was supported by a NIH NRSA Predoctoral Fellowship (F31CA224963) and a NIH/NCI Predoctoral to Postdoctoral Fellow Transition Award (F99CA245807). Research reported herein was supported by the Florida Breast Cancer Foundation, the Shula Fund at Moffitt Cancer Center, the Florida Department of Health Bankhead-Coley Cancer Research Program (8BC02), the Department of Defense Breast Cancer Research Program (W81XWH-20–1-0012) and the NIH/NCI (R00CA185325, R01CA230610), all to B.R.
Declaration of Interests
This work was supported in part by a sponsored research agreement with TESARO: A GSK company. Johanna Kaufman is an employee of GSK and Kristen McEachern was an employee of TESARO: A GSK company. H.S. has received payments from Novartis International AG for consulting and advisory boards. B.R. has received payments from Merck & Co., Inc. and Roche Farma S.A. for consulting. H.S., V.L., P.C.R. and B.R. have courtesy faculty appointments at the University of South Florida, Tampa, FL 33620.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, Schmid-Burgk JL, Hemmerling I, Horvath GL, Schmidt T, Latz E, and Hornung V (2013). Cell intrinsic immunity spreads to bystander cells via the intercellular transfer of cGAMP. Nature 503, 530–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Xia T, Rabasa Capote A, Betancourt D, and Barber GN (2018). Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer cell 33, 862–873 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, Chandwaskar R, Karman J, Su EW, Hirashima M, et al. (2007). Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science 318, 1141–1143. [DOI] [PubMed] [Google Scholar]
- Anderson AC, Joller N, and Kuchroo VK (2016). Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44, 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L, Hiller B, Kostrewa D, Lassig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, and Hopfner KP (2017). cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo JA, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat Rev Immunol 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, and Reis e Sousa C (2018). NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 172, 1022–1037 e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinal J, Pan P, Dhupar R, Ross M, Nakao A, Lotze M, Billiar T, Geller D, and Tsung A (2009). Cisplatin prevents high mobility group box 1 release and is protective in a murine model of hepatic ischemia/reperfusion injury. Hepatology 50, 565–574. [DOI] [PubMed] [Google Scholar]
- Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, et al. (2012). Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nature immunology 13, 832–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral Activity of the CXCR3 Chemokine System Is Required for the Efficacy of Anti-PD-1 Therapy. Immunity 50, 1498–1512 e1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, et al. (2015). Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mingo Pulido A, Gardner A, Hiebler S, Soliman H, Rugo HS, Krummel MF, Coussens LM, and Ruffell B (2018). TIM-3 Regulates CD103(+) Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer cell 33, 60–74 e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria O, De Gassart A, Coso S, Gestermann N, Di Domizio J, Flatz L, Gaide O, Michielin O, Hwu P, Petrova TV, et al. (2015). STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proceedings of the National Academy of Sciences of the United States of America 112, 15408–15413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li XD, Mauceri H, Beckett M, Darga T, et al. (2014). STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. (2018). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753 e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JM, Vanpouille-Box C, Spada S, Rudqvist NP, Chapman JR, Ueberheide BM, Pilones KA, Sarfraz Y, Formenti SC, and Demaria S (2018). Exosomes Shuttle TREX1-Sensitive IFN-Stimulatory dsDNA from Irradiated Cancer Cells to DCs. Cancer Immunol Res 6, 910–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp O, Zitvogel L, and Kroemer G (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 17, 97–111. [DOI] [PubMed] [Google Scholar]
- Gardner A, de Mingo Pulido A, and Ruffell B (2020). Dendritic Cells and Their Role in Immunotherapy. Front Immunol 11, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, Engblom C, Pfirschke C, Siwicki M, Gungabeesoon J, et al. (2018). Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-gamma and IL-12. Immunity 49, 1148–1161 e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, Verneris MR, Cichocki F, Holman CJ, Panoskaltsis-Mortari A, et al. (2012). Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 119, 3064–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. (2016). Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7, 10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtulus S, Madi A, Escobar G, Klapholz M, Nyman J, Christian E, Pawlak M, Dionne D, Xia J, Rozenblatt-Rosen O, et al. (2019). Checkpoint Blockade Immunotherapy Induces Dynamic Changes in PD-1(−)CD8(+) Tumor-Infiltrating T Cells. Immunity 50, 181–194 e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, and Poltorak A (2017). Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. Journal of immunology 199, 397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, and Tan X (2016). Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep 6, 19049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luteijn RD, Zaver SA, Gowen BG, Wyman SK, Garelis NE, Onia L, McWhirter SM, Katibah GE, Corn JE, Woodward JJ, and Raulet DH (2019). SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer CT, Ghorbani P, Nandan A, Dudek M, Arnold-Schrauf C, Hesse C, Berod L, Stuve P, Puttur F, Merad M, and Sparwasser T (2014). Selective and efficient generation of functional Batf3-dependent CD103+ dendritic cells from mouse bone marrow. Blood 124, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mender I, Zhang A, Ren Z, Han C, Deng Y, Siteni S, Li H, Zhu J, Vemula A, Shay JW, and Fu YX (2020). Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, et al. (2002). Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M, Yagita H, and Okumura K (2009). Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830. [DOI] [PubMed] [Google Scholar]
- Ndhlovu LC, Lopez-Verges S, Barbour JD, Jones RB, Jha AR, Long BR, Schoeffler EC, Fujita T, Nixon DF, and Lanier LL (2012). Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 119, 3734–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, and Smyth MJ (2011). Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer research 71, 3540–3551. [DOI] [PubMed] [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, and Kane LP (2015). Tim-3 enhances FcepsilonRI-proximal signaling to modulate mast cell activation. The Journal of experimental medicine 212, 2289–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie C, Cordova AF, Hess GT, Bassik MC, and Li L (2019). SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Mol Cell 75, 372–381 e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, Kaisho T, Bogunovic D, Bhardwaj N, and Krummel MF (2016). Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer cell 30, 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer cell 26, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatos-Peyton CA, Nevin J, Brock A, Venable JD, Tan DJ, Kassam N, Xu F, Taraszka J, Wesemann L, Pertel T, et al. (2018). Blockade of Tim-3 binding to phosphatidylserine and CEACAM1 is a shared feature of anti-Tim-3 antibodies that have functional efficacy. Oncoimmunology 7, e1385690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. The Journal of experimental medicine 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlee M, and Hartmann G (2016). Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 16, 566–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesniere A, Schlemmer F, Boige V, Kepp O, Martins I, Ghiringhelli F, Aymeric L, Michaud M, Apetoh L, Barault L, et al. (2010). Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 29, 482–491. [DOI] [PubMed] [Google Scholar]
- Theisen DJ, Davidson J. T. t., Briseno CG, Gargaro M, Lauron EJ, Wang Q, Desai P, Durai V, Bagadia P, Brickner JR, et al. (2018). WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waggott D, Chu K, Yin S, Wouters BG, Liu F-F, and Boutros PC (2012). NanoStringNorm: an extensible R package for the pre-processing of NanoString mRNA and miRNA data. Bioinformatics 28, 1546–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hu S, Chen X, Shi H, Chen C, Sun L, and Chen ZJ (2017). cGAS is essential for the antitumor effect of immune checkpoint blockade. Proceedings of the National Academy of Sciences of the United States of America 114, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf Y, Anderson AC, and Kuchroo VK (2020). TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol 20, 173–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, et al. (2014). STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki C, Sugiyama M, Ohta T, Hemmi H, Hamada E, Sasaki I, Fukuda Y, Yano T, Nobuoka M, Hirashima T, et al. (2013). Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. Journal of immunology 190, 6071–6082. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wang Y, Wang W, Jia J, Li Y, Wang Q, Wu Y, and Tang J (2009). Generation of monoclonal antibodies against highly conserved antigens. PloS one 4, e6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Fei M, Zhang G, Liang WC, Lin W, Wu Y, Piskol R, Ridgway J, McNamara E, Huang H, et al. (2020). Blockade of the Phagocytic Receptor MerTK on Tumor-Associated Macrophages Enhances P2X7R-Dependent STING Activation by Tumor-Derived cGAMP. Immunity 52, 357–373 e359. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any new datasets or code.