

Summary
Intestinal Paneth cells modulate innate immunity and infection. In Crohn’s disease, genetic mutations together with environmental triggers can disable Paneth cell function. Here, we find that a Western diet (WD) similarly leads to Paneth cell dysfunction through mechanisms dependent on the microbiome and farnesoid X receptor (FXR) and type I interferon (IFN) signaling. Analysis of multiple human cohorts suggest obesity is associated with Paneth cell dysfunction. In mouse models, consumption of a WD for as little as four weeks led to Paneth cell dysfunction. WD consumption in conjunction with Clostridium spp. increased the secondary bile acid deoxycholic acid levels in the ileum, which in turn inhibited Paneth cell function. The process required excess signaling of both FXR and IFN within intestinal epithelial cells. Our findings provide a mechanistic link between poor diet and inhibition of gut innate immunity, and uncover an effect of FXR activation in gut inflammation.
Keywords: High fat diet, metabolism, cell-intrinsic, microbiota, transcriptomics
Graphical Abstract
eTOC blurb:
Small intestinal Paneth cells are gatekeepers of gut innate immunity. Liu et. al. identified a link between consumption of high fat, high sugar diet (Western diet; WD) and Paneth cell dysfunction. The mechanisms involve WD-associated microbiota conversion of bile acids, which activate both FXR and type I interferon pathways.
Introduction
Paneth cells are critical for gut innate immunity by producing and secreting antimicrobial peptides (Bevins and Salzman, 2011; Cederlund et al., 2011; Ouellette, 2010; Vaishnava et al., 2008) and providing a niche for intestinal stem cells (Sato et al., 2011). We and others have shown that Paneth cell defects/dysfunction could be the origin of gut inflammation in models of inflammatory bowel disease (IBD) (Kaser et al., 2008; Liu et al., 2017; Wehkamp et al., 2005). Paneth cell dysfunction leads to mucosal dysbiosis both in patients with Crohn’s disease (CD; a major subtype of IBD) and in mouse models (Gulati et al., 2012; Liu et al., 2016; Salzman et al., 2010). In addition to IBD and infection, Paneth cell homeostasis is also critical in the pathogenesis of graft-versus-host disease (Zhao et al., 2018), metabolism dysregulation (Larsen et al., 2019), and cirrhosis (Teltschik et al., 2012). Thus, optimal Paneth cell function is a critical anti-inflammatory mechanism in the intestine, and its dysfunction could lead to increased susceptibility to infection and IBD (Adolph et al., 2013; Burger et al., 2018).
While Paneth cell function can be indirectly evaluated by response to pathogen infections in mouse models (Bel et al., 2017; Burger et al., 2018), measuring this parameter in humans and laboratory mice is most easily accomplished by quantifying the distribution pattern of cytoplasmic antimicrobial proteins in Paneth cells on well-oriented tissue sections (Cadwell et al., 2008; Cadwell et al., 2010; Khaloian et al., 2020; Liu et al., 2016; Liu et al., 2018; Liu et al., 2017; VanDussen et al., 2014; Wu et al., 2015). Humans and mice with an elevated number of abnormal Paneth cells by this metric (abnormal Paneth cell phenotype; defined as <80% of the total Paneth cells contain normal cytoplasmic granule morphology) have reduced Paneth cell numbers, repressed mRNA expression for genes that control cell metabolism (Liu et al., 2018; VanDussen et al., 2014), and more aggressive disease course in CD (Khaloian et al., 2020; Liu et al., 2018; Liu et al., 2017; VanDussen et al., 2014).
An abnormal Paneth cell phenotype can be driven by either a genetic mutation or gene-environment interactions (Cadwell et al., 2008; Cadwell et al., 2010; Kaser et al., 2008; Liu et al., 2013; Liu et al., 2018). While we previously demonstrated that murine norovirus or cigarette smoking could trigger Paneth cell dysfunction in a host with Atg16l1 deficiency (Cadwell, 2010; Liu et al., 2018), the role of persistent viral infection in CD pathogenesis remains elusive (Norman et al., 2015), and abnormal Paneth cell phenotypes can also be found in CD patients lacking ATG16L1 T300A risk alleles or a cigarette smoking history (Liu et al., 2018). Our study of a pediatric CD cohort (of which none were active smokers) showed that an abnormal Paneth cell phenotype is present in ~50% of CD subjects (Liu et al., 2016), consistent with a previous study showing diminished α-defensins in pediatric CD (Perminow et al., 2010). Accordingly, we hypothesized that additional environmental factors could also trigger Paneth cell dysfunction. Previous studies suggest micronutrient candidates such as zinc, of which deficiency could lead to Paneth cell defects (Bohane et al., 1977; Elmes and Jones, 1980). However, no robust data exists to establish such connections in CD patients. Identifying additional environmental factors that could trigger Paneth cell defects could help develop intervention strategies.
Obesity has become an epidemic over the past 3 decades, with current prevalence of overweight/obese at 39% among adults and 18% among children in the USA (Hales et al., 2018). Given the link between obesity and the prevalence of IBD (Piovani et al., 2020), we hypothesized that diet-induced obesity is a potent environmental trigger for Paneth cell dysfunction. Herein we show that overweight and obese subjects are more prone to developing Paneth cell defects. Mice consuming an obesogenic diet (aka “western diet [WD]”) with high fat, high sugar content) (Naja et al., 2015) also develop Paneth cell defects via the secondary bile acid deoxycholic acid (DCA). Of note, the experimental diet used in this study included fat content (40%) closer to the average US adults (~34%) (Shan et al., 2019) then other preclinical studies. We show that WD-associated Paneth cell defects are dependent on enhanced signaling of two pathways with the intestinal epithelium: farnesoid X receptor (FXR) and type I interferon (IFN). Both pathways are required, as inhibition of either FXR or type I IFN signaling prevents WD-induced Paneth cell defects. Specifically, our findings have implications for how FXR activation may elicit previously unknown undesirable effects in the context of gut infection and inflammation.
Results
Overweight and obese individuals are prone to Paneth cell defects
While we can mechanistically explain the basis of abnormal Paneth cells in certain cohorts (i.e. ATG16L1 CD susceptibility alleles and smoking), for others (such as children with CD), the causal links, especially the environmental components, have remained elusive. To address this issue, we used our bank of Paneth cell-phenotyped individuals (now at 930 individuals from multiple disease states) to search for additional features that correlated with humans having abnormal Paneth cell phenotypes. For one cohort, North American adult non-IBD subjects (demographics described in Table S1), we found a correlation between the percentages of abnormal Paneth cells and elevated body mass index (BMI; ≥25) (Fig. 1A–B), and the number of Paneth cells per crypt also correlated with elevated BMI (Fig. S1A), in concordance with previous studies (Hodin et al., 2011; Tomas et al., 2016). Paneth cells appeared to be the primary intestinal epithelial cell type affected by elevated BMI, as the abundance of differentiated epithelial lineages, villous height and crypt depth did not correlate with BMI (Fig. 1C, Fig. S1B–S1E). The correlation of BMI to abnormal Paneth cell morphology also occurred in CD cohorts; CD subjects with BMI ≥25 had fewer normal Paneth cells (Fig. S2A). Taken together, we hypothesized that obesity can cause Paneth cell dysfunction.
Figure 1. Obesity is associated with Paneth cell defects in humans and mice.
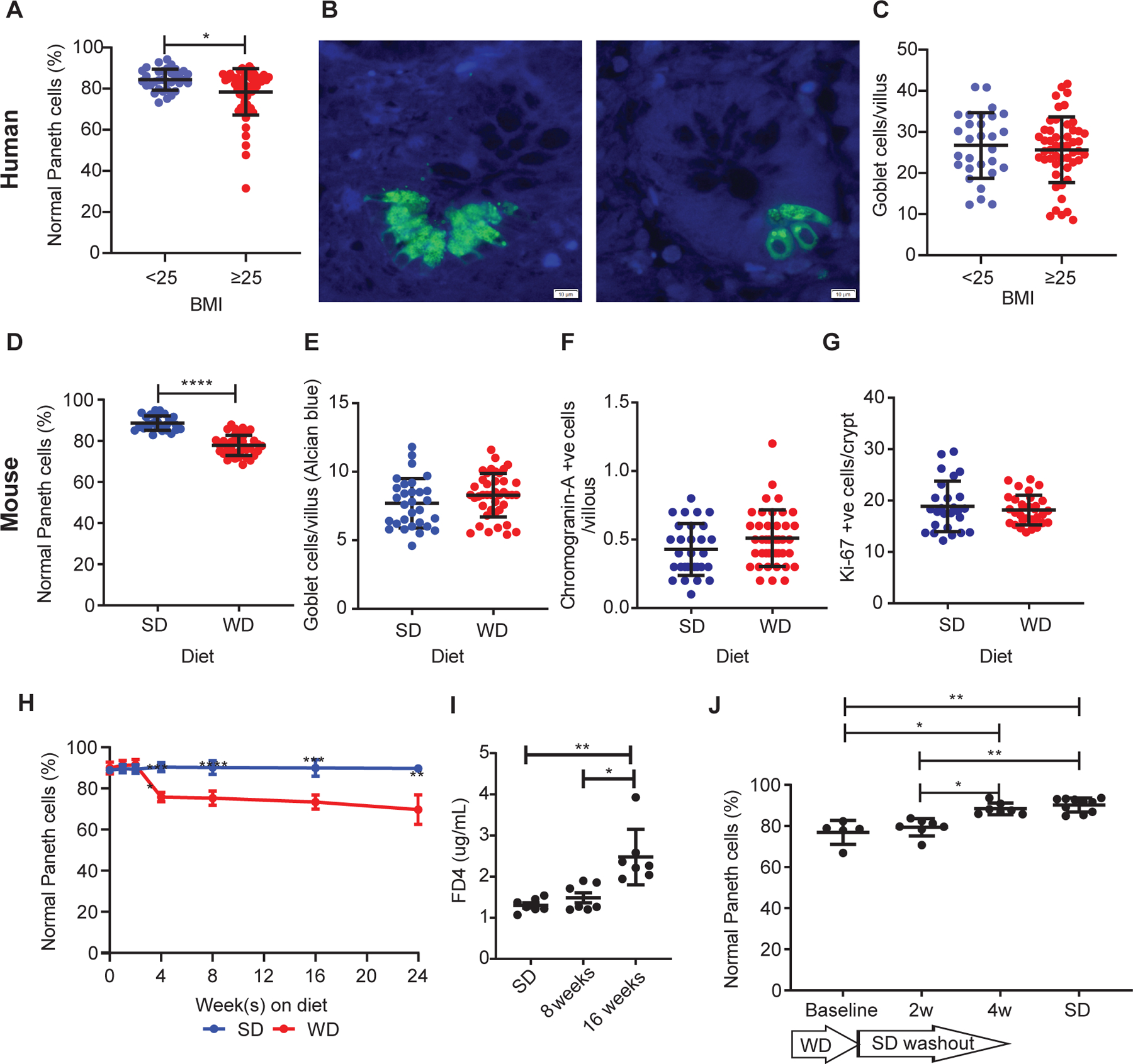
(A) In non-IBD patients, those with BMI≥25 (n=57) showed reduced percentage of normal Paneth cells compared to those with BMI<25 (n=34) (P=0.0129). Representative images of Paneth cells from patients with BMI<25 and BMI≥25 stained with HD5 immunofluorescence (green) are shown in (B). Scale bars: 10μm. (C) There was no difference in goblet cell density (P=0.6256) between the 2 groups. Compared to standard diet (SD)-fed mice (n=30), mice fed with Western diet (WD; n=41) for 8 weeks also resulted in (D) reduced percentage of normal Paneth cells (P<0.0001), and no significant changes in (E) goblet cell density/villus (P=0.1767), (F) neuroendocrine cells/villus (P=0.1193), or (G) crypt base proliferation (P=0.8982). (H) A time course study showed that 4 weeks of WD was sufficient to trigger Paneth cell defects (P<0.0001), whereas (I) WD only induced significant permeability change at 16 weeks (P=0.0022). (J) A 4-week washout period was sufficient to restore the percentage of normal Paneth cells (P=0.0198 compared to baseline). (H): n=3~10/group. (I): n=7/group. (J): baseline: n=5; 2 and 4wk: n=7; SD control: n=10. (A, C-G): Statistical analysis was performed by Mann-Whitney test. (H): Statistical analysis was performed by two-way ANOVA. (I, J): Statistical analysis was performed by Kruskal-Wallis test. *: P<0.05; **: P<0.01; ****: P<0.0001. Error bars represent standard deviations.
WD consumption results in Paneth cell defects in mice
We first tested if an abnormal Paneth cell phenotype could be induced by obesity generated by excess food consumption due to a genetic susceptibility. We found no significant changes in Paneth cell morphology or numbers per crypt in two well-characterized strains of genetically obese mice (ob/ob and db/db) fed with standard diet (SD) compared to their littermate controls (Fig. S3). In addition, CD patients harboring risk alleles in ATG16L1 T300A (Fig. S2B–C) and NOD2 (Fig. S2D–E) did not have increased probability of developing Paneth cell defects when BMI was above 25 compared to those without these risk alleles. These results suggest that host genetic susceptibility does not play a prominent role in Paneth cell defects associated with obese individuals.
We next tested if diet-induced obesity caused Paneth cell defects. Wild type (WT) C57BL/6 mice were fed with SD or WD for 8 weeks. We used a WD that contained 40% fat content, as this level is most similar to the diet of the average US adult (~34%) (Shan et al., 2019); this percentage of fat is lower than what was used in other published mouse studies (~60%) (Beyaz et al., 2016; Everard et al., 2019; Fu et al., 2019; He et al., 2019; Whitt et al., 2018). WD consumption in mice led to reduced numbers and percentages of morphologically normal Paneth cells (Fig. 1D, S1F, S1K, and S1L). There were no significant alterations in the abundance or distribution of other intestinal epithelial cell types (Fig. 1E–1G, Fig. S1G–S1J).
A critical functional consequence in mice with >6 months WD consumption is mucosal barrier dysfunction, which manifests as increased gut permeability (Genser et al., 2018; Guerville et al., 2017; Lam et al., 2012) and can lead to increased inflammation in the gastrointestinal tract (Li et al., 2008). To understand if Paneth cell defects may be a consequence of increased epithelial permeability, we determined the temporal relationship between WD-induced Paneth cell defects and permeability changes. A 4-week consumption of WD was sufficient to trigger Paneth cell defect (Fig. 1H), whereas increased gastrointestinal tract permeability required 16 weeks in our facility (Fig. 1I). Therefore, with WD consumption, the Paneth cell defects we observed are likely not secondary to barrier dysfunction.
Previous studies determining the effect of dietary changes on microbiome composition and metabolism suggested that certain community members and hepatic triglyceride metabolism are sensitive to dietary perturbation, and the resulting changes are irreversible after a washout period during which the diet was switched back to SD (Heden et al., 2014; Howe et al., 2016). Mice that were fed with WD for 8 weeks were switched back to SD for either 2 or 4 weeks. A 4-week washout was required for the Paneth cell defects to reverse (Fig 1J). Thus, Paneth cell defects associated with a WD consumption in mice are reversible.
Enhanced FXR and type I IFN signaling is associated with WD-induced Paneth cell defects
We used data from multiple OMICs approaches to determine the candidate pathways that mediate the WD-induced Paneth cell defects. First, we performed RNA-seq analysis on full thickness small intestine from mice fed with WD and SD. We used this approach because Paneth cells can receive signals from other cell types (Sehgal et al., 2018). We focused on metabolic pathways, as defective Paneth cells show profound alterations in metabolism (Adolph et al., 2013; Liu et al., 2018; Pentinmikko et al., 2019). Utilizing the Compbio analysis platform (Gehrig et al., 2019), we found that genes associated with Fxr activation were strongly associated with WD consumption (Fig. 2A, Fig. S4A). RT-qPCR of the well-established Fxr target gene, Fgf15 (Kim et al., 2007), showed highly enhanced expression in ileal samples from WD-fed mice than SD-fed mice (Fig. 2B). Furthermore, we found Fgf15 mRNA expression was increased in the crypt base compartment (enriched with Paneth cells (Fig. S4B) of WD fed mice. For a second approach, we performed bulk RNA-seq analysis on full-thickness ileal samples from non-IBD subjects. Subjects with BMI≥25 had elevated mRNA expression of genes associated with an activated FXR pathway (Fig. S4C). Collectively, these data show that activation of the FXR pathway was associated with Paneth cell defects in obese/overweight humans and a diet-induced obesity mouse model.
Figure 2. Activated FXR and type I IFN signaling in WD-associated Paneth cell defects in mice.
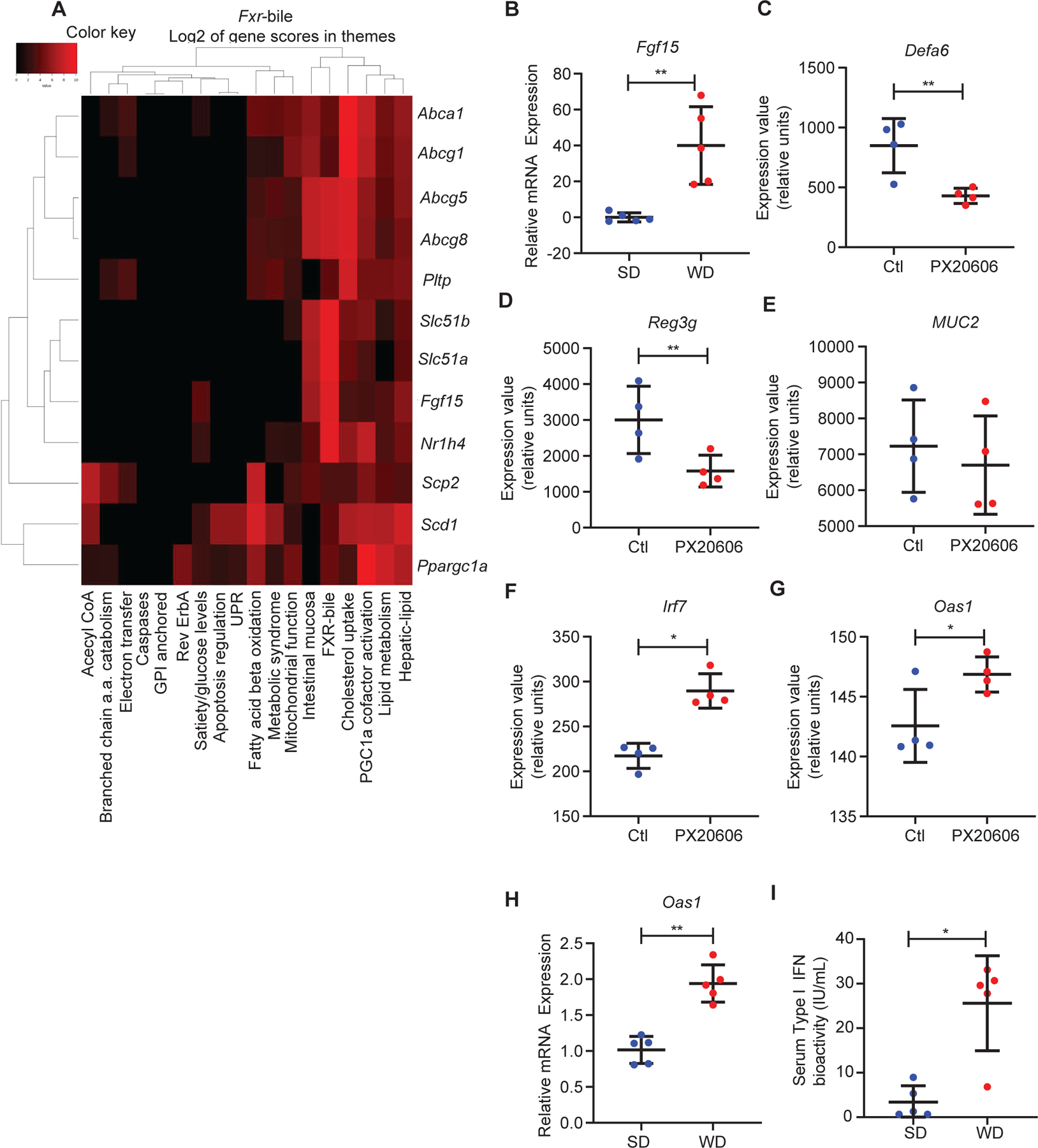
(A) A heat map showing upregulated Fxr-associated genes in full-thickness ileum from WD-fed mice and their level of contribution to specific themes in the data set. (B) RT-qPCR of Fxr target gene Fgf15 showed that WD treatment resulted in induction of Fgf15 in the ileum (n=5/group; P=0.0079). By mining publically-available dataset GSE74101 (FXR agonist PX20606 treatment in WT mice), we found that FXR activation reduced Paneth cell-associated (C) Defa6 (adjusted P=0.002) and (D) Reg3g (adjusted P=0.009) expression, without significantly altering the expression of (E) gob let cell-specific gene Muc2 (adjusted P=0.1143). PX20606 treatment also increased expression of (F) Irf7 (adjusted P=0.01) and (G) Oas1 (adjusted P=0.03). (H) Ileum from WD-fed mice showed enhanced expression of Oas1 (P=0.0079). (I) WD-fed mice showed higher serum type I IFN activity (P=0.0159). (C-G): Benjamini & Hochberg adjustment to the P value was applied (n=4/group). (B, H, I): Statistical analysis was performed by Mann-Whitney test (n=5/group). (B-H): Error bars represent standard deviations.
For a third approach, we mined a transcriptomic dataset (GSE 74101) from a published study where WT mice were administered vehicle or the FXR agonist PX20606 for 2 weeks (de Boer et al., 2017), and full-thickness small intestine tissue was collected for RNA-seq analysis. PX20606 significantly repressed the expression of Paneth cell-associated genes Defa6 (Fig. 2C) and Reg3g (which is also expressed in other intestinal epithelial cell types) (Fig. 2D), whereas expression of the goblet cell-specific gene, Muc2 was not altered (Fig. 2E), consistent with our findings with WD consumption. Therefore, excess FXR activation by a pharmacological approach specifically represses genes associated with Paneth cell function.
Previous studies by our group and others have shown that cytokines are important mediators of Paneth cell defects (Dannappel et al., 2014; Gunther et al., 2019; Liu et al., 2018). We queried the GSE 74101 dataset to determine if any signatures associated with cytokine activation were present. We found that PX20606 administration also led to the enhanced expression of the IFN-simulated genes Irf7 and Oas1 (Fig. 2F–G). We also found enhanced expression of IFN-stimulated gene Oas1 in the ileum of WD-fed mice (Fig. 2H). These results are consistent with elevated serum type I IFN bioactivity of WD-fed mice (Fig. 2I).
Secondary bile acid, deoxycholic acid, mediates WD-FXR-associated Paneth cell defects
The transcriptome analysis above suggests that excess FXR activation secondary to WD consumption causes Paneth cell defects. One consequence of defective Paneth cells is susceptibility to infections (Holly and Smith, 2018; Lassen et al., 2014; Ouellette, 2006; Wehkamp and Stange, 2006). Susceptibility to Salmonella typhimurium infection has been particularly shown to correlate with Paneth cell function (Bel et al., 2017; Holly and Smith, 2018; Lassen et al., 2014; Lu et al., 2020; Salzman et al., 2003). Therefore, we treated Salmonella-infected WT mice with vehicle or the FXR agonist GW4064. All mice received GW4064 upon infection, with two groups additionally getting pretreatment for 4 weeks or at the same time of Salmonella infection. Mice receiving the pretreatment of GW4064 showed increased mortality as compared to those treated with GW4064 only after Salmonella infection or with vehicle control (no treatment with GW4064) (Fig. 3A). This correlated with increased bacterial titers in the spleen and intestine of GW4064-pretreated mice (Fig. S4D, E). The detrimental effects on mortality with GW4064 pretreatment were reduced in Salmonella-infected Fxr−/− mice (Fig. 3B). These results show that excess FXR activation can lead to increased mortality with Salmonella infection, further suggesting the Paneth function may be a target of excess FXR signaling.
Figure 3. WD-DCA-mediated FXR activation induces Paneth cell defects.
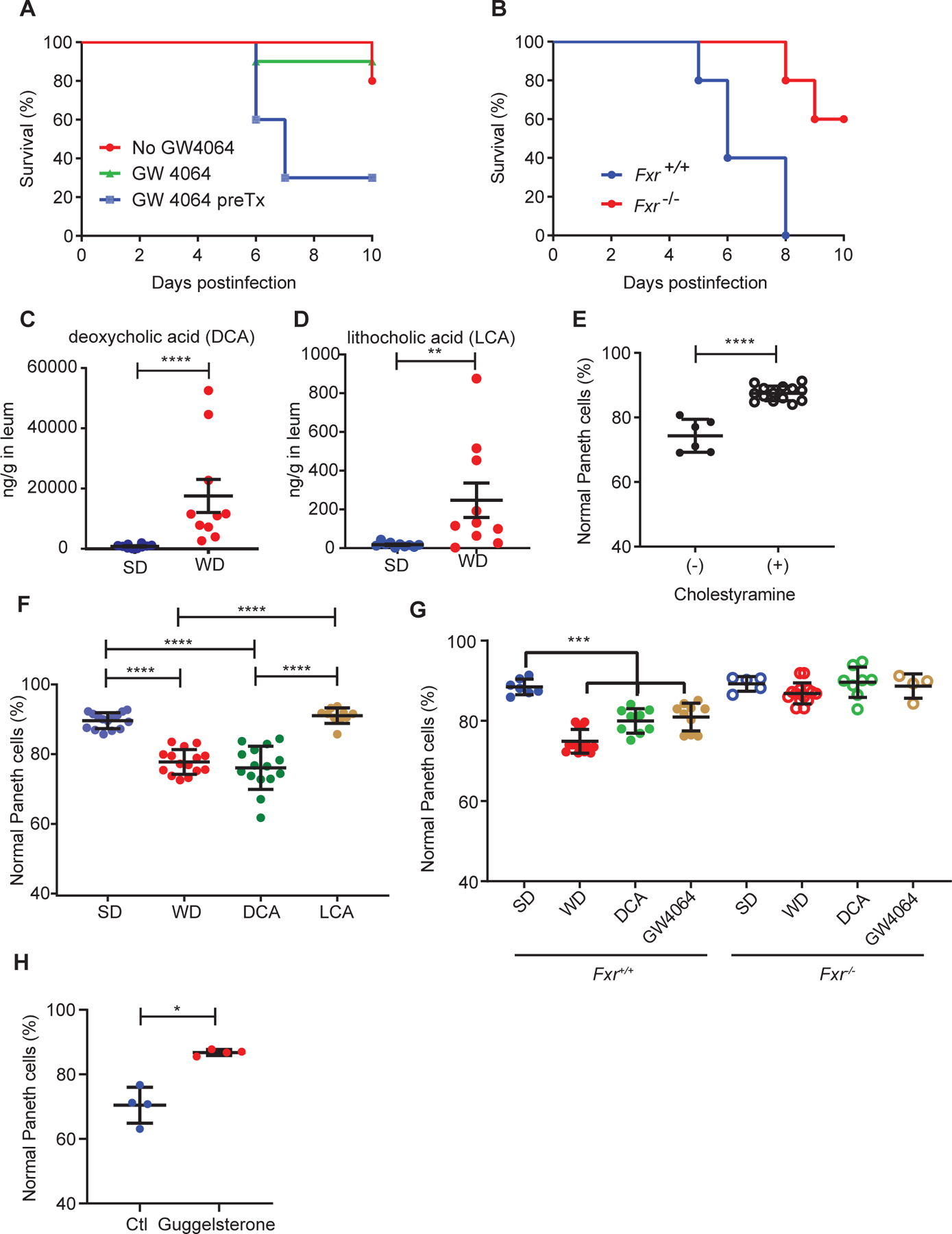
(A) WT mice pretreated with FXR agonist GW4064 showed worse mortality after Salmonella Typhimurium infection compared to non-GW4064 treated or treated with GW4064 at the onset of infection (n=10/group; P=0.0035). (B) GW4064 pretreatment increased the mortality of Fxr+/+ mice after Salmonella Typhimurium infection but not in Fxr−/− mice (n=5/group; P=0.0084). Distal ileum from WD-fed mice had higher levels of (C) DCA (P<0.0001) and (D) lithocholic acid (LCA; P=0.0021). n=10/group. (E) Reduction of DCA from WD-fed mice by bile acid sequestrant cholestyramine prevented Paneth cell defects (P<0.0001). Total n: no cholestyramine: n=6, with cholestyramine: n=14. (F) Administration of DCA but not LCA in SD-fed mice recapitulated WD-induced Paneth cell defects (P<0.0001). Total n: SD, WD, DCA: n=15/group, LCA: n=10. (G) Fxr−/− mice and littermates (Fxr+/+) were treated with SD, WD, SD+DCA, or SD+GW4064, and analyzed for Paneth cell morphology. While WD, DCA, and GW4064 all induced Paneth cell defects in the Fxr+/+ mice (P=0.0005), the Fxr−/− mice did not develop this phenotype (P=0.1257). n: 4–14/group. (H) WT, WD-fed mice treated with FXR antagonist guggelsterone did not develop Paneth cell defects (P=0.0286). n=4/group. (A, B): Kaplan-Meier curve analysis was performed with Logrank test. (C-H): Statistical analysis between 2 groups was performed by Mann-Whitney test. Statistical analysis between more than 2 groups was performed by Kruskal-Wallis test. *: P<0.05; **: P<0.01; ***: P<0.001. ****: P<0.0001. Error bars represent standard deviations.
The best characterized mechanism by which WD activates FXR pathway is through the induction of bile acids (Dermadi et al., 2017; Jena et al., 2018). Primary bile acids, the end product of cholesterol catabolism, are produced in the liver, and converted to secondary bile acids in the terminal ileum/cecum by Clostridium spp. and subsequently reabsorbed in the ileum as part of the enterohepatic circulation (Chiang, 2013). We measured the concentrations of primary bile acids cholic acid (CA), chenodeoxycholic acid (CDCA), muricholic acid (MCA) and secondary bile acids deoxycholic acid (DCA) and lithocholic acid (LCA) in the small intestine by mass spectrometry. These bile acids were analyzed because they are present in both humans and mice in significant quantity with the exception of MCA, which is predominantly present in mice (Chiang, 2013). Mice fed with WD had significantly increased DCA (13-fold) and LCA (7.6-fold) levels in the ileum (Fig. 3C–D), without changes to CA, CDCA, or MCA levels (Fig. S5). Therefore, WD exposure was associated with elevated secondary bile acids in the distal/terminal ileum.
To test if elevated WD-associated secondary bile acids caused Paneth cell defects, we performed the following experiments: (1) concomitant lumenal treatment with bile acid sequestrant cholestyramine to lower bile acid levels and (2) administration of secondary bile acids DCA and LCA in mice fed with SD. Co-administration of cholestyramine prevented Paneth cell defects in WD-fed mice (Fig. 3E), and reciprocally, DCA administration induced Paneth cell defects in SD-fed mice (Fig. 3F). The latter effect was specific for DCA, as LCA did not trigger Paneth cell defects. Therefore, increased levels of ileal DCA contribute to the WD-mediated Paneth cell defects in mice.
We tested the causality between WD-associated Fxr activation and Paneth cell defects in Fxr−/− mice and littermates. These mice were treated with SD, WD, SD+DCA, or SD+FXR agonist GW4064. As expected, Paneth cell defects were observed with WD, DCA, or GW4064 fed/treated WT littermates but not in Fxr−/− mice (Fig. 3G). WD-fed, WT mice additionally treated with FXR antagonist guggelsterone did not develop defective Paneth cells (Fig. 3H). These data further support the hypothesis that elevated activity of the FXR pathway in WD fed mice induces Paneth cell defects.
Excess FXR activation directly affects Paneth cells in the intestinal epithelium
FXR is active in a wide variety of cell types (Jiang et al., 2015a; Shih et al., 2001; Trabelsi et al., 2015). We first tested if DCA-mediated FXR activation in the intestinal epithelium was required for Paneth cell dysfunction. We determined the effect of WD/DCA on intestinal epithelium using intestinal epithelium-specific (Villin-Cre) Fxr conditional knockout mice (FxrΔIEC mice). In these experiments, Paneth cell defects were induced by WD, DCA, and GW4064 in the littermate control Fxrfl/fl mice but not in similarly treated FxrΔIEC mice (Fig. 4A). Similar results were obtained in vitro, by treating Fxr+/+ and Fxr−/− ileal organoids with DCA or GW4064. Excess FXR stimulation caused a reduction in Paneth cells in Fxr+/+ but not Fxr−/− organoids (Fig. 4B and Fig. S6A–E) without significantly affecting organoid sizes (Fig. S6F). Of note, the reduction of Paneth cells in vitro occurred using a DCA concentration comparable with the DCA concentration in the ileum of WD-fed mice (~45 μM; Fig. 3).
Figure 4. Fxr signaling in Paneth cells is essential for Western diet (WD)-mediated Paneth cell defects.
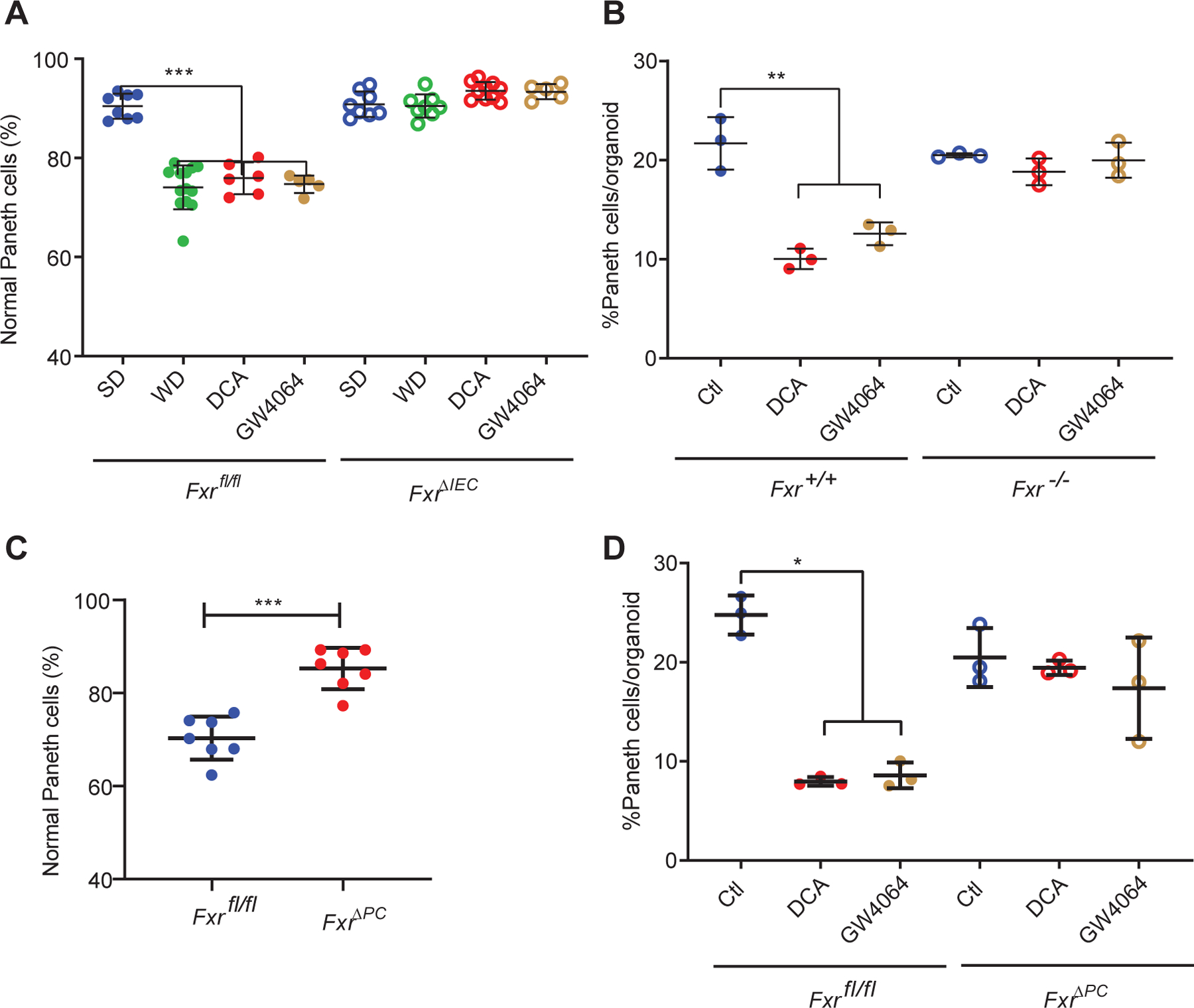
(A) WD, DCA, and GW4064 triggered Paneth cell defects in Fxrfl/fl mice, but the effects were abrogated in the FxrΔIEC mice (P=0.0005). Total n=5~13/group. (B) Ileal organoids derived from Fxr−/− and Fxr+/+ mice showed differential responses to DCA and GW4064. Whereas DCA and GW4064 both reduced the percentages of Paneth cells/organoid in the Fxr+/+ organoids, such an effect was abrogated in the Fxr−/− organoids (P=0.0036). (C) WD-mediated Paneth cell defects were abrogated in the FxrΔPC mice (P=0.0006). Total n=7/group. (D) Ileal organoids derived from the FxrΔPC mice did not show reduced percentages of Paneth cells/organoid when treated with DCA or GW4064. (B, D): Results were from 3 independent experiments, with each experiment containing 20 organoids/group. (A-D) Statistical analysis between 2 groups was performed by Mann-Whitney test. Statistical analysis between more than 2 groups was performed by Kruskal-Wallis test. *: P<0.05; **: P<0.01; ***: P<0.001; ****: P<0.0001. Error bars represent standard deviations.
To test if DCA acted directly on Paneth cells, we generated a Paneth cell-conditional knockout of FXR (α-defensin-4-IRES-Cre; herein termed FxrΔPC) (Liu et al., 2018) and showed that the FxrΔPC mice were protected from WD-driven Paneth cell defects (Fig. 4C). Similarly, ileal organoids from FxrΔPC mice did not show a reduction in Paneth cells with treated with either DCA or GW4064 (Fig. 4D). These data are consistent with a direct effect of DCA on Paneth cells through FXR.
The role of microbiota in WD-mediated Paneth cell defects
Dietary modulation of gut microbiome composition is well-established (Bisanz et al., 2019), and certain host phenotypes (including obesity) can be horizontally transmissible via microbiota transfer (Turnbaugh et al., 2009; Turnbaugh et al., 2006). WD-fed mice in our facility showed an increase in the abundance of Firmicutes and a relative reduction in abundance of Bacteroides, consistent with previous studies (Fig. S7A, 7B) (Ley et al., 2005; Turnbaugh et al., 2006). We next tested if WD-induced Paneth cell defects could be transmitted between mice. We first performed co-housing in a specific pathogen-free (SPF) facility where WT, microbiota-recipient mice were pretreated with broad-spectrum antibiotics to create a niche for donor microbiota. Microbial donors were mice fed for 4 weeks with WD or SD. The donors were replaced every other day to ensure fresh WD microbiota were provided (Fig. S7C), as microbial community composition changes rapidly as a function of diet (Turnbaugh et al., 2008). At the end of the co-housing period, the WD microbiota recipients showed no difference in the percentages of normal Paneth cells or Paneth cell density (Fig. S7D–E). In a second experiment, we performed microbiota transfer using cecal contents from SPF-housed WT mice fed either SD or WD into germ-free recipient mice, which were maintained on SD throughout the experiment. PBS-gavaged germ-free mice had lower percentages of normal Paneth cells (Fig. S7F), consistent with a prior study (Kernbauer et al., 2014). However, we observed no significant difference in Paneth cell phenotypes between mice gavaged with cecal contents from SD- and WD-fed SPF mice (Fig. S7F). Therefore, WD-associated Paneth cell defects were not induced by microbiome transfer alone.
The experiments above suggest that the primary role of the microbiome in WD-mediated Paneth cell defects is to facilitate the conversion of primary to secondary bile acid. Clostridium spp. harboring a BaiCD operon, are over-represented in mice fed with WD (Jena et al., 2018), and can mediate the conversion of primary to secondary bile acids (Jain et al., 2018). We found that the ileal mucosal microbiome in WD-fed, SPF-housed mice were indeed enriched with BaiCD expression (Fig. 5A). We next treated WD-fed, SPF-housed mice with or without vancomycin to deplete gram-positive bacteria, including Clostridium spp. Vancomycin-treated mice showed a reduction in BaiCD gene detection in the microbiome (Fig. 5B). As expected, vancomycin-treated mice were protected from WD-mediated Paneth cell defects (Fig. 5C). Furthermore, we gavaged WD-fed, germ-free mice with either PBS or Clostridium scindens (a well-characterized Clostridium sp. containing the BaiCD operon whose gene products convert primary to secondary bile acids) (Jain et al., 2018). Mice gavaged with C. scindens showed increased DCA levels in the ileum (Fig. S7G) and reduced percentages of normal Paneth cells (Fig. 5D). Thus, C. scindens promotes WD-induced Paneth cell defects. To determine if diet alone was sufficient to elicit Paneth cell defects in the absence of the microbiome, we also fed germ-free mice with either SD or WD for 4 weeks, without fecal transplantation. As expected, mice in each of these groups did not show increased abnormal Paneth cells (Fig. 5E).
Figure 5. The role of microbiota in WD-mediated Paneth cell defects.
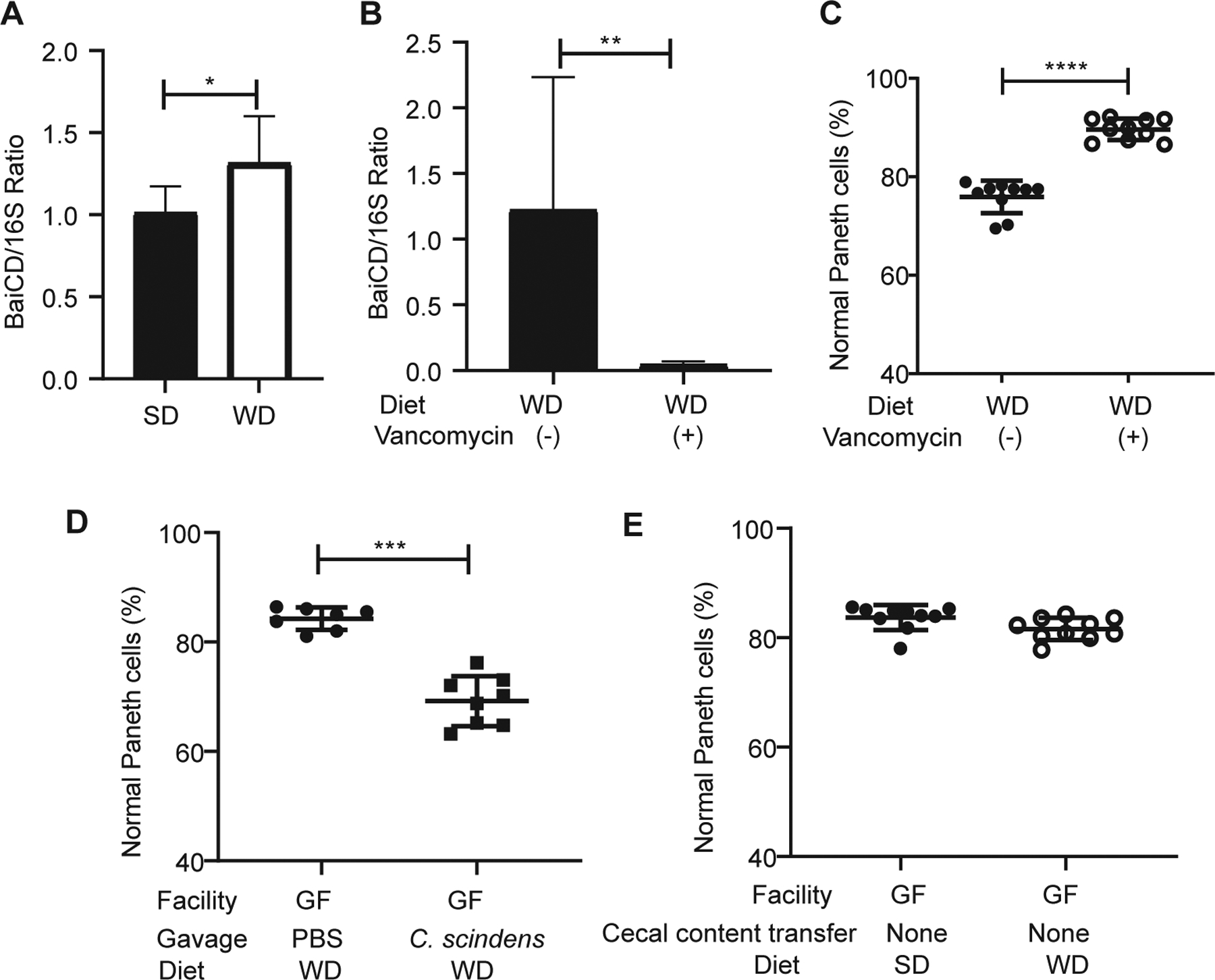
(A) Mucosal microbiome from WD-fed mice was enriched with BaiCD operon (n=8/group; P=0.0281). (B) The amount of BaiCD operon in the microbiota was diminished after vancomycin treatment (n=5/group; P=0.0079). (C) Vancomycin treatment prevented WD-mediated Paneth cell defects in SPF housed mice (n=10/group; P<0.0001). (D) Germ-free mice (GF) fed with WD and gavaged with C. scindens showed Paneth cell defects compared to those gavaged with PBS (P=0.0003). Total n: PBS: 7, C. scindens: 8. (E) GF mice fed with SD or WD without cecal content transfer did not develop Paneth cell defects (n=10/group; P=0.4309). Statistical analysis was performed by Mann-Whitney test. *: P<0.05; **: P<0.01; ***: P<0.001; ****: P<0.0001. Error bars represent standard deviations.
WD-FXR signaling activates Type I IFN and mediates Paneth cell defects
A second target of DCA-FXR signaling is type I IFNs that also have important roles in inhibition of Paneth cell function (Liu et al., 2013). FXR activation can lead to production and activation of type I IFN in the context of viral infection (Honke et al., 2017; Winkler et al., 2020). As noted above, we found evidence of elevated type I IFN activity in WD-fed mice (Fig. 2I) consistent with a previous study showing a direct correlation between systemic IFN-α and triglyceride levels (Tominaga et al., 2010). SD-fed mice administered with the FXR activator, GW4064 also showed increased serum type I IFN activity (Fig. 6A), as well as increased expression of Irf1/9 in the crypt base compartment (Fig. 6B, C). To test if elevated levels of type I IFN activity played a role in Paneth cell defects, we treated WD-fed mice with either a blocking monoclonal antibody to the type I IFN receptor (anti-Ifnar1, MAR1–5A3) or an isotype control antibody (GIR208). Remarkably, anti-Ifnar1 administration prevented WD-induced Paneth cell defects (Fig. 6D). Consistent with this result, Ifnar1−/− mice were also protected from WD-induced Paneth cell defects as compared to littermate genetic controls (Fig. 6E). Finally, Stat1ΔIECmice were protected from WD-induced Paneth cell defects (Fig. 6F), even though they maintained high levels of Fgf15 expression in the ileum (Fig. 6G), suggesting that type I IFN signaling in the intestinal epithelium was also critical in mediating Paneth cell defects.
Figure 6. Type I IFN mediates WD-associated Paneth cell defects.
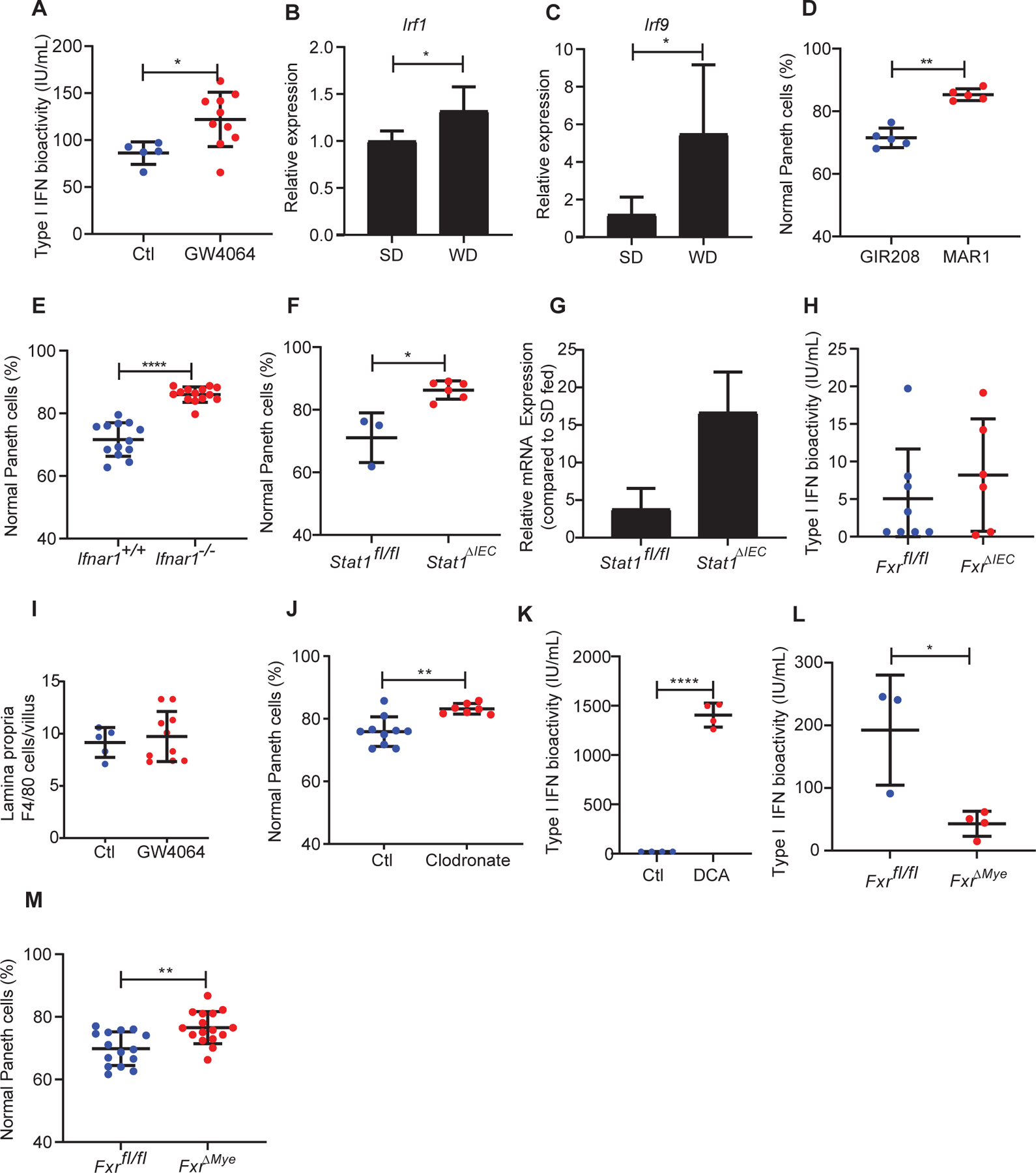
(A) Administration of GW4064 in WT, SD-fed mice showed induction of type I IFN (Ctl: n=5; GW4064: n=10; P=0.0193). This was associated with enhanced expression of type I IFN associated genes (B) Irf1 and (C) Irf9 in the crypt base compartment. (D) Treatment with anti-Ifnar1 (MAR1–5A3) but not isotype control antibody (GIR208) prevented WD-induced Paneth cell defects (n=5/group; P=0.0079). (E) Ifnar−/− mice were protected from WD-associated Paneth cell defects (Ifnar+/+: n=13, Ifnar−/−: n=14; P<0.0001). (F) Stat1ΔIEC mice fed with WD did not develop Paneth cell defects (P=0.0238). Total n: Stat1fl/fl: n=3; Stat1ΔIEC: n=6. (G) Stat1ΔIEC mice fed with WD maintained high Fgf15 expression compared to the Stat1fl/fl mice (P=0.1). (H) FxrΔIEC mice fed with WD did not show abrogation of type I IFN induction (P=0.6523). Total n: Fxrfl/fl: n=8; FxrΔIEC: n=6. (I) GW-4064-treated mice did not show increased lamina propria F4/80+ cells (P=0.6560). (J) WD-fed WT mice treated with clodronate were protected from Paneth cell defects (P=0.0031). Total n: Ct: n=10; clodronate: n=6. (K) DCA treatment induced type I IFN activity in macrophages in vitro (P<0.0001). n=4/group. (L) FxrΔMye mice fed with WD showed abrogation of type I IFN induction (P=0.0193). Total n: Fxrfl/fl: n=3; FxrΔMye: n=4. (M) FxrΔMye mice were protected from WD-mediated Paneth cell defects (P=0.0033). Total n: Fxrfl/fl: n=15; FxrΔMye: n=16. Statistical analysis for all panel was performed by Mann-Whitney test. *: P<0.05; **: P<0.01, ****: P<0.0001. Error bars represent standard deviations.
We found that a likely source of the type I IFN enhanced by FXR/WD diet was intestinal myeloid cells. FxrΔIEC mice showed similar levels of serum type I IFN activity as controls (Fig. 6H). We did not identify differences in the amount of F4/80+ cells infiltrating the lamina propria of GW4064-treated mice (Fig. 6I), suggesting the effect of FXR activation was not due to changes in macrophage cell numbers, but rather the function of these cells. WT mice fed with WD and treated with intraperitoneal clodronate to deplete intestinal myeloid cells (Weisser et al., 2012) were protected from WD-associated Paneth cell defects (Fig. 6J). Consistent with this result, DCA induced type I IFN bioactivity in macrophages in vitro (Fig. 6K). Finally, mice with Fxr conditionally knocked out in myeloid cells (FxrΔMye) when fed with WD, failed to show elevated serum type I IFN bioactivity compared to littermates (Fig. 6L), and these mice were protected from WD-mediated Paneth cell defects (Fig. 6M). These results support a role for elevated Type I IFNs in myeloid cells that was necessary for Paneth cell defects in vivo.
Discussion
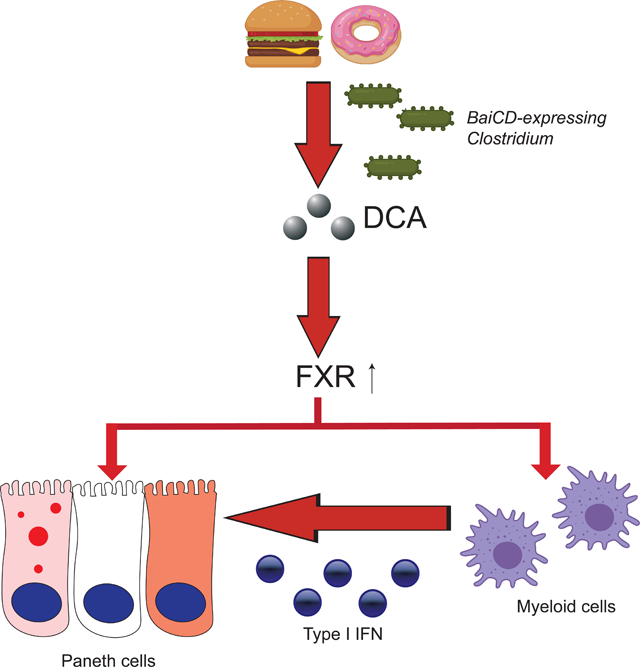
We previously showed that Paneth cell defects can be induced in the context of a gene-environment interaction (Liu et al., 2018). In this current study, we utilized our extensive collection of cohorts with Paneth cell phenotypes and observed a significant subset of overweight/obese human subjects had Paneth cell defects. Our studies in mice revealed that the obesity-associated Paneth cell dysfunction was not related to genetics but instead was likely related to WD consumption. Mechanistically, the defective Paneth cells driven by WD consumption was due to increased secondary bile acid (specifically DCA) in the ileum that were converted from primary bile acids by BaiCD-expressing Clostridium spp. This process was dependent on FXR signaling in two cell types: activation of FXR in Paneth cells and in myeloid cells; the latter augmented type I IFN production in response to FXR activation. Myeloid cells -derived type I IFNs in turn acted on the intestinal epithelium to produce abnormal Paneth cells. Both of these enhanced signaling activities are required to produce abnormal Paneth cells. (Fig. 7).
Figure 7. Proposed model of how WD triggers Paneth cell defects.
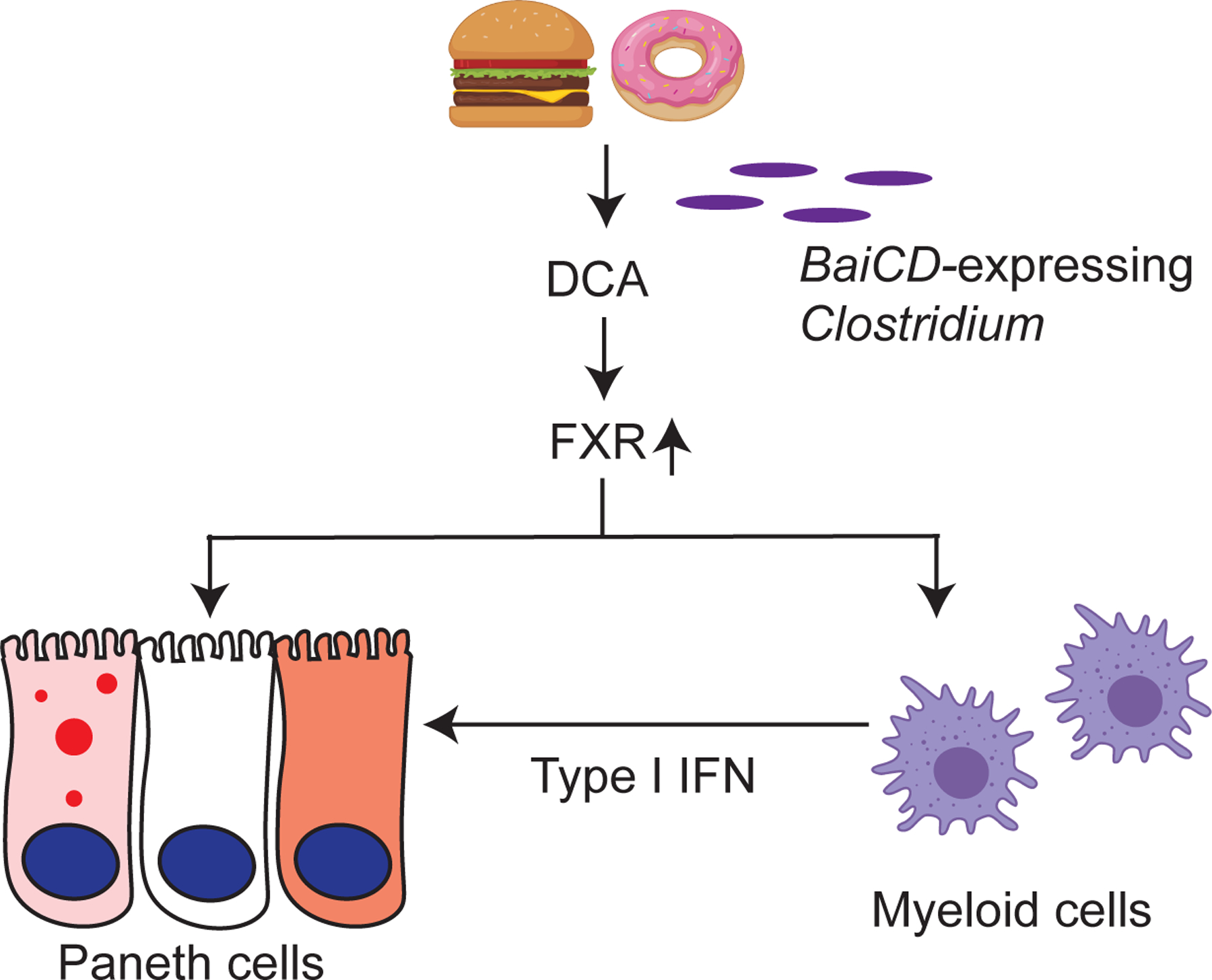
Our current study shows that potent environmental factors (such as WD consumption) could induce Paneth cell defects without host genetic susceptibility. WD induces FXR signaling that directly target Paneth cells, and also induces type I IFN production in myeloid cells such as macrophages. Both FXR and type I IFN signaling are required to trigger Paneth cell defects.
Paneth cell dysfunction indicates defects in metabolism that are driven by genetics and/or the environment. In this regard, mouse and human Paneth cells have shown remarkable concordance in several studies (Adolph et al., 2013; Cadwell et al., 2008; Cadwell et al., 2010; Jackson et al., 2020; Kaser et al., 2008; Liu et al., 2017; Lu et al., 2020), enabling reverse and forward translational approaches. Here we show that WD, which has been suspected to induce and exacerbate IBD (Ananthakrishnan, 2015; Ng et al., 2015), has a profound effect on Paneth cell function, which could lead to increased susceptibility and/or severity of IBD (Liu et al., 2018; Liu et al., 2017; VanDussen et al., 2014). This observation is of particular relevance, as the incidence of IBD is on the rise in historically low incidence areas which have now transitioned to a more western lifestyle (Garduno-Diaz and Khokhar, 2013; Ng et al., 2018; Siddiquee et al., 2015). While previous studies have shown that WD consumption affects nutrient absorption, metabolism, and cell cycle (Ait-Omar et al., 2011; de Wit et al., 2008; Newberry et al., 2009), how WD impacts low-abundance but functionally important cell types such as Paneth cells is relatively unknown. A key finding of our study is that Paneth cell dysfunction can be induced after 8 weeks of WD consumption, which in turn can lead to more aggressive disease in IBD. While we cannot exclude that intestinal stem cells or their niche is affected by bile acids, we did not observe alteration in lineage allocation in other secretory lineages and stem/progenitor proliferation was not altered. Prolonged WD consumption period may result in additional changes secondary to its effect on the stem cell niche, as suggested by a recent study (Beyaz et al., 2016). Nonetheless, our data suggest that, prior to permanent changes in intestinal stem cells, WD consumption is capable of inducing Paneth cell defects. Moreover, our study also suggests the potential reversibility of Paneth cell defect after cessation of short-term WD consumption. This has important implications in the management of IBD.
Using complementary models, we showed that WD has a direct effect on the intestinal epithelium, and that the nuclear receptor FXR is a central target. FXR signaling regulates metabolism in many cell types (Ryan et al., 2014; Sun et al., 2018), and appears to have this role in dysfunctional Paneth cells known to have altered metabolism (Cadwell et al., 2008; Liu et al., 2018). FXR widely affects many organs and may have different physiological effects in specific locations (Gonzalez et al., 2016; Jiang et al., 2015a; Rao et al., 2016; Trabelsi et al., 2015). In the gut, FXR has been implicated in improving microbiota composition, barrier function (Sorribas et al., 2019), and stem cell proliferation (Fu et al., 2019). Strategies of using FXR agonists are being developed for several diseases, including metabolic syndrome, insulin resistance, obesity, and nonalcoholic steatohepatitis (Fang et al., 2015; Jiang et al., 2015b; Mudaliar et al., 2013; Neuschwander-Tetri et al., 2015). Our study provides molecular and functional insight into how FXR activation may have unexpected and undesirable effects on gut innate immunity.
Our study also suggests that Paneth cell defects associated with WD consumption are mediated by commensal Clostridium spp. by affecting primary to secondary bile acid conversion, as suggested by a previous study (Jena et al., 2018). Because Paneth cell defects can trigger mucosal dysbiosis in CD patients (Liu et al., 2016), these data indicate that expansion of Clostridium spp., as part of WD-mediated dysbiosis, further affects bile acid-FXR-IFN signaling.
In addition to directly activating type I IFN by FXR under virus infection (Honke et al., 2017), previous studies have also shown that IFN regulatory transcription factor Irf7 can be recruited to the Fxr promotor (Renga et al., 2013) and that an Fxr deficiency results in repressed expression of type I IFN (Zheng et al., 2017). Our findings agree with previous reports showing that type I IFN signaling has a critical role in diet-induced obesity (Hannibal et al., 2017). Furthermore, Irgm knockout mice (with constitutively high levels of type I IFN) also possess Paneth cell defects (Liu et al., 2013; Sun et al., 2015). Studies using intestinal epithelium-specific Ifnar1-deficient mice showed that type I IFN signaling is critical in Paneth cell homeostasis (Tschurtschenthaler et al., 2014). While our in vitro data suggest that macrophages may be a source of type I IFN, it is possible that additional cell types targeted by the LysM-Cre (i.e., myeloid cells) are capable of producing type I IFN in this context. In addition, our study does not exclude the possibility that type I IFN activity can be induced by FXR-independent mechanisms. Together, with our data, an emerging theme is that dietary modulation of the FXR-type I IFN axis affects gut innate immunity and homeostasis. The involvement of type I IFN in modulating Paneth cell defects also raises the possibility that other disease states by which type I IFN is activated, such as chronic viral infections (Norman et al., 2015), could impact CD pathogenesis. In addition, our finding that WD-fed Stat1ΔIEC mice showed an even higher level of Fgf15 expression compared to WD-fed Stat1f/f mice suggests that while FXR activation is upstream of type I IFN activation, it is possible that a negative feedback loop is involved. Here, type I IFN could modulate the expression of FXR pathway-associated genes.
Paneth cells are important mediators for IBD and other disease processes (Wehkamp and Stange, 2020). Our study identifies specific signaling pathways by which WD consumption, a common environmental factor for these diseases, can trigger defective Paneth cells. These results have implications for the pathogenesis of IBD and for the potential utility for testing and monitoring the function of Paneth cells in IBD patients. Future experiments dissecting the different components of WD (e.g., fat and sugar) will provide additional insight into the pathogenesis of WD-associated Paneth cell defects.
STAR methods:
RESOURCE AVAILABILITY
Lead contact
Further information for resources and reagents should be directed to the Lead Contact, Thaddeus S. Stappenbeck (stappet@ccf.org).
Materials availability
The intestinal stem cells isolated for this study will be made available upon request and provided with completed Materials Transfer Agreement. The human material is part of routine clinical care and is under legal requirement to be maintained in the Pathology Departments of our institutions, and as a result will not be available.
Data and code availability
The full-thickness ileum transcriptomic dataset has been deposited to ArrayExpress, with accession number: E-MTAB-8332. The microbiome dataset has been deposited to ArrayExpress, with accession number: E-MTAB-8334.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human subjects
The study was approved by the Institutional Review Boards of Washington University School of Medicine (St. Louis) and Cedars-Sinai Medical Center (Los Angeles). CD or non-IBD subjects who underwent ileocolectomy between 2006–2013 at Washington University, St. Louis, or Cedars-Sinai Medical Center, Los Angeles, were previously described (Liu et al., 2018; Liu et al., 2017; VanDussen et al., 2014). De-identified tissue samples from ileal resection margins that were free of acute inflammation were used for Paneth cell phenotype analysis.
The following information was retrieved from the medical record: sex, body weight, height at time of operation (for body mass index [BMI] calculation). The genotypes of the patients were obtained using immunochip (VanDussen et al., 2014) or through Taqman genotyping assay (ThermoFisher) with genomic DNA extracted from formalin-fixed, paraffin-embedded tissue based on the vendor’s instructions (Liu et al., 2018; Liu et al., 2017). The average age, gender and their influences on the results are included in Table S1.
Mouse treatments
The animal studies were approved by the ethical committee at Washington University School of Medicine. Mice were either obtained from Jackson Laboratory or bred and maintained at Washington University School of Medicine. Mice were maintained under specific pathogen free (SPF) conditions unless otherwise specified. Only 4–6 week-old mice (post-weaning) in C57/BL6 background were used. Genotype information is included in Key Resources Table. Mice were exposed to the following diet: SD (Lab Supply Labdiet 5053; with calories provided by 62% carbohydrates, 13% fat, and 25% protein), WD (Research Diets D09100310; with calories provided by 40% carbohydrates, 40% fat, and 20% protein), and WD with 2% cholestyramine admixed (Research Diets D18030505; with calories provided by 40% carbohydrates, 40% fat, and 20% protein). Mice were exposed to different diets for the time indicated (4–24 weeks) and then sacrificed for tissue collection. For antibiotic treatment experiments, mice received vancomycin for the entire duration of diet exposure (Jain et al., 2018). For bile acid administration, sodium deoxycholate (0.2%; Sigma-Aldrich #30970) was added to the drinking water for 4 weeks. Lithocholic acid (20 mg/kg; Sigma-Aldrich #L6250) was administered via intraperitoneal (i.p.) route daily for 4 weeks. GW4064 (Tocris #2473) was administered i.p. at 10 mg/kg/day. Guggulsterone (Tocris #3570) was administered i.p. at 30 mg/kg/day. For type I IFN blockade, mouse anti-mouse monoclonal antibody specific for Ifnar1 (MAR1–5A3; Leinco) or isotype control antibody GIR208 (Leinco) was administered by using a loading dose of 2.5 mg/mouse, followed by a weekly dose of 0.5 mg/mouse (Pinto et al., 2011) per vendor instructions. To ensure that the bile acid levels did not fluctuate per fast/feed cycle change, all the mice were sacrificed in the hours between 10am – 3pm.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Lysozyme C Antibody (C-19) | Santa Cruz | Cat.# sc-27958 |
| Cleave caspase-3 Antibody | Cell signaling | Cat.# 9661 |
| Defensin alpha 5 Antibody (8C8) | Novus | Cat.# NB110–60002 |
| Mucin 2 Antibody (H-300) | Santa Cruz | Cat.# sc-15334 |
| F4/80 Antibody | Abcam | Cat.# 6640 |
| Lab Vision™ Ki-67, Rabbit Monoclonal Antibody | ThermoFisher | Cat.# RM9106 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat. # A-21206 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | ThermoFisher | Cat. # A-11058 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher | Cat. # A-21202 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Biotin-XX | ThermoFisher | Cat. # B-2770 |
| Chromogranin A antibody | DAKO | Cat. # A0430 |
| QIAamp DNA Stool Mini Kit | QIAGEN | Cat. # 51604 |
| Quant-iT™ RiboGreen® RNA Assay Kit | ThermoFisher | Cat. # R11490 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | Cat. #1708840 |
| TB Green Advantage qPCR Premix | Clontech | Cat. #639676 |
| RNeasy FFPE Kit | QIAGEN | Cat. # 73504 |
| RNeasy Mini Kit | QIAGEN | Cat. # 74104 |
| Bacterial and Virus Strains | ||
| Clostridium scindens | ATCC | Cat.# 35704 |
| Salmonella enterica serovar Typhimurium | ATCC | Cat.# 14028 |
| Biological Samples | ||
| Human ileum tissue blocks | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| GW4064 | Tocris | Cat. # 2473 |
| Sodium deoxycholate | Sigma-Aldrich | Cat. # 30970 |
| Lithocholic acid | Sigma-Aldrich | Cat. # L6250 |
| Z-Guggulsterone | Tocris | Cat. # 3570 |
| MAR1-5A3 | Leinco | Cat. # I-401 |
| GIR208 | Leinco | Cat. # I-443 |
| Vancomycin hydrochloride | Sigma-Aldrich | Cat. # V2002 |
| Fluorescein isothiocyanate-dextran | Sigma-Aldrich | Cat. # FD4 |
| Clodrosome,Clodronate liposome | Encapsula Nano Sciences | Cat. # CLD-8909 |
| Encapsome, Control liposome | Encapsula Nano Sciences | Cat. # CLD-8910 |
| Dulbacco’s modified Eagle’s medium (DMEM) | Life Technologies | Cat. # 11885–084 |
| Horse Serum | Sigma-Aldrich | Cat. # H-1207 |
| Fetal Bovine Serum | Sigma-Aldrich | Cat. # F-2442 |
| Penicillin-Streptomycin | Sigma-Aldrich | Cat. # P-4333 |
| HEPES | ThermoFisher | Cat. # 15630080 |
| L-glutamine | ThermoFisher | Cat. # 25030081 |
| Sodium Pyruvate | ThermoFisher | Cat. # 11360070 |
| Recombinant Mouse M-CSF (carrier-free) | Biolegend | Cat. # 576402 |
| Critical Commercial Assays | ||
| Custom TaqMan™ SNP Genotyping Assay | ThermoFisher | Cat. # 4332072 |
| Deposited Data | ||
| GSE 74101 | de Boer et al., 2017 | N/A |
| E-MTAB-8332 | This study | N/A |
| E-MTAB-8334 | This study | N/A |
| Experimental Models: Cell Lines | ||
| Small intestinal organoids from wild type mice | This study | N/A |
| Small intestinal organoids from Fxr−/− mice | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| SPF mice (C57BL/6J) | Jackson Laboratory | Cat.# 000664 |
| Fxr−/− | Jackson Laboratory | Cat.# 007214 |
| Stat1−/− | Jackson Laboratory | Cat.# 012606 |
| ob/ob | Jackson Laboratory | Cat.# 000632 |
| db/db | Jackson Laboratory | Cat.# 000697 |
| Villin-Cre | Jackson Laboratory | Cat. # 004586 |
| LysM-Cre | Jackson Laboratory | Cat. # 004781 |
| Fxrfl/fl | Gonzalez lab | N/A |
| α-defensin-4-IRES-Cre | Dempsey lab | N/A |
| Oligonucleotides | ||
| BaiCD Forward: GGWTTCAGCCCRCAGATGTTCTTTG | Wells et al., 2003 | N/A |
| BaiCD Reverse: GAATTCCGGGTTCATGAACATTCTKCKAAG | Wells et al., 2003 | N/A |
| Fgf15 Forward: AGTACCTGTACTCCGCTGGT | NM_008003.2 | N/A |
| Fgf15 Reverse: CAGCCCGTATATCTTGCCGT | NM_008003.2 | N/A |
| Oas1 Forward: CGCACTGGTACCAACTGTGT | Steed et al., 2017 | N/A |
| Oas1 Reverse: CTCCCATACTCCCAGGCATA | Steed et al., 2017 | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Graphpad Prism | Graphpad Software, Inc. | Version 9.0.0 |
| Olympus cellSens Dimension | Olympus | Version 1.17 |
| SAS | SAS Institute | Version 9.4 |
| Compbio | Washington University | N/A |
| Other | ||
| Standard diet | Lab Supply | Cat. # 5053 |
| Western diet | Research Diets | Cat. # D09100310 |
| Western diet (with 2% cholestyramine) | Research Diets | Cat. # D18030505 |
For co-housing experiments, 5 WT mice that had been and continued to be exposed to WD were designated as microbial donors. Additional WT mice exposed to SD designated as microbial recipients were exposed to an antibiotic cocktail of vancomycin, neomycin, ampicillin, and metronidazole for 2 weeks (Moon et al., 2015), followed by co-housing with microbial donors. The microbial donors and recipients were co-housed and all exposed to standard diet. A different microbial donor was used every other day to ensure the donor continue to provide microbes that are consistent with WD exposure. Co-housing lasted 2 weeks.
For germ-free mice experiment, mice were fed with irradiated SD or WD as indicated. Cecal contents from SD- or WD-fed, SPF-housed mice were used for fecal microbiota transplant every 2 weeks. For gavage of Clostridium scindens (ATCC #35704), mice were orally administered PBS or 7×108 bacteria per mouse every 2 weeks as described previously (Jain et al., 2018).
Salmonella infection model
Salmonella enterica serovar Typhimurium (ATCC # 14028) was grown overnight at 37°C in LB broth under mild aeration. Bacteria were washed twice in cold PBS and resuspended at 108 CFU/50 μL. Water and food were withdrawn 4 hours prior to treatment with 20 mg streptomycin in 75 μL; immediately following streptomycin treatment, water and food were again provided. Twenty hours later, water and food were again removed for 4 hours, after which mice were infected with 108 CFU S. Typhimurium per oral gavage. Water was resumed immediately and food was provided 2 hours post-infection.
METHOD DETAILS
Immunofluorescence and immunohistochemistry
For immunofluorescence, unstained slides were deparaffinized, followed by antigen retrieval using Trilogy (Cell Marque #920P-09; Rocklin, CA). The slides were blocked with 1% bovine serum albumin and 0.1% Triton X-100 in phosphate buffer saline for 1 hour, before applying primary antibody overnight. After overnight incubation, secondary antibody was applied for 45 minutes, and slides were subsequently washed and stained with Hoechst (1:20,000; Sigma-Aldrich #33258). The following primary antibodies were used: lysozyme (1:100; Santa Cruz# sc-27958), defensin 5 (1:2000; Novus #NB110–60002), chromogranin A (1:500; Dako # A0430), and MUC-2 (1:200; Santa Cruz# sc-15334). Secondary antibodies used include donkey anti-rabbit, donkey anti-goat, and donkey anti-mouse antibodies (1:500; Life Technologies). For Ki-67 immunohistochemistry, unstained slides were deparaffinized, followed by antigen retrieval using Diva Decloaker buffer (Biocare Medical). The slides were then quenched with 10% H2O2 in methanol and blocked as described above. The rabbit anti-human Ki-67 antibody was then applied (1:400; Lab Vision) overnight, followed by goat anti-rabbit biotinylated secondary antibody (1:200; Thermo Fisher Scientific). Signal detection was performed by using Vectastain Elite ABC kit (Vector) and DAB (Vector). The stains were reviewed using an Olympus BX53 microscope equipped with an Olympus DP73 camera and cellSens Dimension software for adjusting brightness and contrast and image cropping.
Paneth cell phenotype analysis
Lysozyme and defensin 5 immunofluorescence were interpreted as described previously (Cadwell et al., 2008; Cadwell et al., 2010; Liu et al., 2014; Liu et al., 2016; VanDussen et al., 2014). For both human and mouse samples, each Paneth cell was classified into normal or one of the 5 abnormal categories, including: disordered (abnormal distribution and size of the granules), diminished (≤10 granules), diffuse (smear of lysozyme or defensin within the cytoplasm with no recognizable granules), excluded (majority of the granules do not contain stainable material), and enlarged (rare, megagranules) (Liu et al., 2014; VanDussen et al., 2014). The last two categories were only observed in human samples.
BMDM stimulation
BMDMs were isolated as described previously (Steed et al., 2017). Briefly, cells from femurs and tibia of mice were isolated and plated in sterile dishes containing high glucose DMEM (Life Technologies) with 5% horse serum (Sigma-Aldrich), 10% fetal bovine serum (Sigma-Aldrich), Penicillin-Streptomycin (Sigma-Aldrich), 10mM HEPES (Corning), 1% L-glutamine, 1% sodium pyruvate and Macrophage Colony-Stimulating Factor (Biolegend) at 10 ng/ml for 7 days prior to experimental procedure.
For stimulation experiments, BMDMs were collected in DMEM by cell scraper and reseeded in 6 well plates containing DMEM supplemented with 10% fetal bovine serum, Penicillin-Streptomycin (Sigma-Aldrich), 10mM HEPES (Corning) and allowed to adhere. After 24 hours, cells were washed with sterile PBS, fresh media was added and cells were stimulated with DCA (10μM) or vehicle (PBS). 24 hours post-stimulation, supernatants were collected and assayed for type I IFN serum activity.
Laser capture microdissection
The distal ileum of the mice was prepared and fixed in methacarn as previously reported (Cadwell et al., 2010). The crypt base epithelial cells enriched for Paneth cells were captured and RNA isolated by previously described techniques (Cadwell et al., 2010; VanDussen et al., 2014).
Intestinal permeability assay
Food and water were removed from the mouse cage for 4 hours prior to performing permeability assays. FITC-dextran (average molecular weight 4,000; Sigma-Aldrich #FD4) solution (100 mg/ml) was gavaged into mice at 44 mg/100g. After 3 hours, serum was collected, and FITC-dextran concentration analyzed by fluorescence.
Clodronate treatment
Clodronate or control liposomes (40 μl from 23mg/ml stock; Encapsula Nano Sciences) were mixed with 160μL of molecular-grade PBS. Mice were then injected with clodronate or control liposomes intraperitoneally twice per week for 8 weeks.
In vitro studies
Small intestinal stem cells were isolated as described previously (VanDussen et al., 2015). Ileal crypts were cultured in Matrigel (BD Biosciences) in the presence of 50% L-WRN conditioned medium (contains WNT3A, R-SPONDIN 3 and NOGGIN) (Miyoshi et al., 2012; Miyoshi and Stappenbeck, 2013). Compared to other methods (Sato et al., 2009), this protocol allows for rapid expansion of intestinal epithelial spheroids enriched with stem cells. The spheroids were plated out at Day 0. From Day 1 to Day 7, the culture cells were cultured in ENR medium (Madison et al., 2015) which contains EGF, NOGGIN, and R-SPONDIN 1, to allow for Paneth cell differentiation. DCA and GW4064 were added to the ENR medium on Day 7. The organoids were harvested at Day 14 and processed for immunofluorescence.
Microbiota collection
Ileal mucosal microbiome was collected from the indicated groups, and microbial genomic DNA was isolated using the QIAamp DNA Stool Mini Kit (QIAGEN), followed by quantitative RT-PCR. The fecal microbiota was collected as described previously (Liu et al., 2018).
16s rRNA sequencing assay and data analysis
Fecal samples were processed for 16s rRNA sequencing as previously reported (Liu et al., 2018). Reads from the V4 region amplicon were used for downstream analyses. Analysis of alpha diversity, including Faith’s phylogenetic diversity (Faith and Baker, 2006) and Shannon diversity, was performed using alpha_diversity.py in QIIME on rarefied data. For microbiome studies, principal coordinate analysis was performed using ANOSIM with 999 permutations. Relative OTU abundance data were input into LEfSe to determine biomarkers with significant linear discriminant analysis effect size (Segata et al., 2011). The determination of sample size and data analysis for animal studies followed the general guideline of Festing and Altman (Festing and Altman, 2002). Based on the law of diminishing returns, Mead recommended that a degree of freedom (DF) of 10–20 associated with error term in an ANOVA will be adequate to estimate preliminary information (Mead, 1988).
Transcriptomic profiling and analysis
For mouse tissue, full-thickness small intestine tissue was preserved in RNAlater. RNA was isolated using Qiagen RNeasy Mini kit (#74104), according to the kit protocol. Total RNA was quantified using the Quant-iT™ RiboGreen® RNA Assay Kit (ThermoFisher; #R11490) and normalized to 4ng/μl. For human tissue, unstained slides from ileal resection margin were used to isolate RNA using RNeasy FFPE Kit (QIAGEN; # 73504) as described (VanDussen et al., 2018). Transcriptomic profiling was performed at the Genome Technology Access Center (GTAC) at Washington University, which has performed microarray (Liu et al., 2018) and RNA-seq before. RNA library preparation was performed with RiboZero and sequencing performed on an Illumina HiSeq 2500. RNA-seq reads were aligned to the GRCm38.76 assembly from Ensembl with STAR version 2.0.4 (Dobin et al., 2013). Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.5. Transcript counts were produced by Sailfish version 0.6.3. Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, genes and transcripts detected, ribosomal fraction and known junction saturation and read distribution over known gene models with RSeQC version 2.3. The results of the RNA-seq were further analyzed using the CompBio software package (PERCAYAI Inc., www.percayai.com/compbio). CompBio performs a literature analysis to identify relevant biological processes and pathways represented by the differentially expressed entities (genes, proteins, miRNA’s, or metabolites). This is accomplished with an automated Biological Knowledge Generation Engine that extracts all abstracts from PubMed that reference entities of interest (or their synonyms), using contextual language processing and a biological language dictionary that is not restricted to fixed pathway and ontology knowledge bases. Conditional probability analysis calculated the statistical enrichment of biological concepts (processes/pathways) over those that occur by random sampling. Related concepts built from the list of differentially expressed entities were further clustered into higher-level themes (e.g., biological pathways/processes, cell types and structures, etc.).
For analysis of the effect of FXR agonist on intestinal signaling, we used a publically-available transcriptomic dataset (GSE 74101), based on a published study testing FXR agonist PX20606 on intestine (de Boer et al., 2017). Expression of selected genes were retrieved and compared. Adjusted P values used in the original analysis were used.
RNA isolation and quantitative RT-PCR
To confirm the expression of selected genes, total RNA was purified from samples using the RNeasy Mini Kit (Qiagen #74104). cDNA was synthesized using the iScript™ Reverse Transcription Supermix (Bio-Rad #1708840). qRT-PCR was performed using TB Green Advantage qPCR Premix (Clontech #639676) with Applied Biosystems QuantStudio 3 and the primer sequences listed in STAR Methods. Expression levels of each target gene were normalized to Gapdh, which was similarly expressed in all samples, and analyzed using the ΔΔCT method.
Bile acid measurement
Tissues stored at −80°C were homogenized by sonication (Branson Sonifier 450, output 2.5, 50% duty cycle, 10– 20 sec) in 10 volumes of 50% MeOH in water. Homogenates were centrifuged (13,000 × g, 10 min, 4°C), and cleared supernatants were transferred to new tubes. One third volume of chloroform was added to the supernatant, mixed (vigorous shaking for 10 sec), and centrifuged (13,000 × g, 5 min, 4°C). The aqueous phase was transferred to a new tube and the chloroform addition was repeated and centrifuged again. After the second chloroform treatment, the clear aqueous solution (100 μl) was dried under vacuum at room temperature and stored in −80°C until analysis.
On the day of analysis, lyophilized samples were reconstituted with 50 μl of 50% MeOH in water, vortexed, and then centrifuged (13,000 × g, 5 min, 4°C). Clear supernatants were transferred to HPLC vials and 10 μl of each sample was used for the analysis. Bile acids were measured using LC-MS (Agilent 1290 LC and 6470 triple quadrupole). Samples and the series of deoxycholic acid and lithocholic acid dilutions (0 – 10 ng/ml in 50% MeOH in water, Sigma-Aldrich) were injected into HILIC HPLC column (Kinetex 2.6 μm HILIC, 150 × 2.1 mm) with mobile buffer (30% MeOH, 70% 5mM ammonium formate) and eluted by the MeOH gradient (0–2min 30% MeOH, 2–6min 30–50% MeOH, 6–8min 50–80% MeOH, 8–10min 80% MeOH, 10–15min 80–30% MeOH, and 15–25min 30% MeOH). Bile acids were detected using single ion monitoring mode (m/z = 391.3 for deoxycholic acid and m/z = 375.3 for lithocholic acid with fragmentation voltage 240V) and quantified using the standard curve.
Type I IFN bioactivity assay
Levels of biologically active type I IFN in serum were determined using an encephalomyocarditis virus (EMCV) L929 cell cytopathic effect bioassay as described previously (Sheehan et al., 2006; Sheehan et al., 2015). Briefly, serial dilutions of sera in DMEM containing 10% FBS were applied in duplicate to L929 cells (2 × 104 cells/well) in 96-well plates. Following incubation for 14 hours at 37°C, cells were infected with EMCV diluted in DMEM containing 2% FBS at a MOI of 7. At 8 hours post-infection, IFN-mediated protection was assayed using CellTiter 96 aqueous cell proliferation assay as per the manufacturer’s protocol (Promega). IFN levels were calculated from an IFN-β standard curve. To ensure protection from EMCV-induced death was due to type I IFN activity, L929 cells also were pretreated with anti-Ifnar1 mAb (MAR-1–5A3, 50 ug/mL) prior to the addition of serum samples.
QUANTIFICATION AND STATISTICAL ANALYSIS
For analysis between two groups, a nonparametric t test was used. For analysis between more than two groups, such as comparing different bile acids or different genotype and diet exposure combinations, Kruskal-Wallis tests followed by Dunn’s tests between groups was performed. Nonparametric statistical tests were selected based on the data distribution. All tests were two-tailed and a P value of <0.05 was considered significant. Data were plotted and analyzed using GraphPad Prism (version 9.0) and SAS version 9.4 (SAS Institute, Cary, North Carolina). All data represent mean ± SD. All statistical details of the experiments can be found in the figure legends, including the statistical tests used and exact value of n. n represents exact number of animals for in vivo studies and exact numbers of biological repeats for in vitro studies.
Supplementary Material
Highlights:
Diet-induced obesity results in Paneth cell dysfunction in humans and mice.
Consumption of Western diet leads to Clostridium-mediated deoxycholic acid conversion.
Deoxycholic acid activates both FXR and type I interferon (IFN) pathways.
Both FXR and type I IFN pathways are essential in triggering Paneth cell defects.
Acknowledgement
The study was supported by NIH grant U01DK062413 and a grant from Helmsley Charitable Trust (2014PGIBD010). T.C.L. was supported by NIH grants R01 DK125296 and R01 DK124274. K.L.V. was supported by NIH grant DK109081. M.S.D. was supported by NIH grants R01 AI143673, R01 AI127513, and R01 AI123348. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant# UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. The study was also supported in part by Digestive Disease Research Core Center at Washington University by grant P30DK052574 from the NIH. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. The authors also appreciate Drs. Ashly Steed and Megan Baldridge for sharing reagents.
Declaration of Interests
T.C. Liu received research funding from Pfizer on Paneth cell phenotype in inflammatory bowel disease, and advises Interline. R.D. Head and C. Storer may receive royalty income based on the CompBio technology developed by R.D. Head and licensed by Washington University to PercayAI. T. Stappenbeck advises Janssen, Boehringer Ingelheim, Kallyope, Takada, and Roche. All other authors declare no relevant competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. (2013). Paneth cells as a site of origin for intestinal inflammation. Nature 503, 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ait-Omar A, Monteiro-Sepulveda M, Poitou C, Le Gall M, Cotillard A, Gilet J, Garbin K, Houllier A, Chateau D, Lacombe A, et al. (2011). GLUT2 accumulation in enterocyte apical and intracellular membranes: a study in morbidly obese human subjects and ob/ob and high fat-fed mice. Diabetes 60, 2598–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthakrishnan AN (2015). Epidemiology and risk factors for IBD. Nature reviews Gastroenterology & hepatology 12, 205–217. [DOI] [PubMed] [Google Scholar]
- Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, and Hooper LV (2017). Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 357, 1047–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevins CL, and Salzman NH (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nature reviews Microbiology 9, 356–368. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Mana MD, Roper J, Kedrin D, Saadatpour A, Hong SJ, Bauer-Rowe KE, Xifaras ME, Akkad A, Arias E, et al. (2016). High-fat diet enhances stemness and tumorigenicity of intestinal progenitors. Nature 531, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz JE, Upadhyay V, Turnbaugh JA, Ly K, and Turnbaugh PJ (2019). Meta-Analysis Reveals Reproducible Gut Microbiome Alterations in Response to a High-Fat Diet. Cell host & microbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohane TD, Cutz E, Hamilton JR, and Gall DG (1977). Acrodermatitis enteropathica, zinc, and the Paneth cell. A case report with family studies. Gastroenterology 73, 587–592. [PubMed] [Google Scholar]
- Burger E, Araujo A, Lopez-Yglesias A, Rajala MW, Geng L, Levine B, Hooper LV, Burstein E, and Yarovinsky F (2018). Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell host & microbe 23, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K (2010). Crohn’s disease susceptibility gene interactions, a NOD to the newcomer ATG16L1. Gastroenterology 139, 1448–1450. [DOI] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. (2008). A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 456, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell K, Patel KK, Maloney NS, Liu TC, Ng AC, Storer CE, Head RD, Xavier R, Stappenbeck TS, and Virgin HW (2010). Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 141, 1135–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cederlund A, Gudmundsson GH, and Agerberth B (2011). Antimicrobial peptides important in innate immunity. The FEBS journal 278, 3942–3951. [DOI] [PubMed] [Google Scholar]
- Chiang JY (2013). Bile acid metabolism and signaling. Compr Physiol 3, 1191–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, et al. (2014). RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature 513, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer JF, Schonewille M, Boesjes M, Wolters H, Bloks VW, Bos T, van Dijk TH, Jurdzinski A, Boverhof R, Wolters JC, et al. (2017). Intestinal Farnesoid X Receptor Controls Transintestinal Cholesterol Excretion in Mice. Gastroenterology 152, 1126–1138.e1126. [DOI] [PubMed] [Google Scholar]
- de Wit NJ, Bosch-Vermeulen H, de Groot PJ, Hooiveld GJ, Bromhaar MM, Jansen J, Muller M, and van der Meer R (2008). The role of the small intestine in the development of dietary fat-induced obesity and insulin resistance in C57BL/6J mice. BMC medical genomics 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermadi D, Valo S, Ollila S, Soliymani R, Sipari N, Pussila M, Sarantaus L, Linden J, Baumann M, and Nystrom M (2017). Western Diet Deregulates Bile Acid Homeostasis, Cell Proliferation, and Tumorigenesis in Colon. Cancer research 77, 3352–3363. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmes ME, and Jones JG (1980). Ultrastructural changes in the small intestine of zinc deficient rats. The Journal of pathology 130, 37–43. [DOI] [PubMed] [Google Scholar]
- Everard A, Plovier H, Rastelli M, Van Hul M, de Wouters d’Oplinter A, Geurts L, Druart C, Robine S, Delzenne NM, Muccioli GG, et al. (2019). Intestinal epithelial N-acylphosphatidylethanolamine phospholipase D links dietary fat to metabolic adaptations in obesity and steatosis. Nature communications 10, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP, and Baker AM (2006). Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evolutionary bioinformatics online 2, 121–128. [PMC free article] [PubMed] [Google Scholar]
- Fang S, Suh JM, Reilly SM, Yu E, Osborn O, Lackey D, Yoshihara E, Perino A, Jacinto S, Lukasheva Y, et al. (2015). Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nature medicine 21, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Festing MF, and Altman DG (2002). Guidelines for the design and statistical analysis of experiments using laboratory animals. ILAR J 43, 244–258. [DOI] [PubMed] [Google Scholar]
- Fu T, Coulter S, Yoshihara E, Oh TG, Fang S, Cayabyab F, Zhu Q, Zhang T, Leblanc M, Liu S, et al. (2019). FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 176, 1098–1112.e1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garduno-Diaz SD, and Khokhar S (2013). South Asian dietary patterns and their association with risk factors for the metabolic syndrome. J Hum Nutr Diet 26, 145–155. [DOI] [PubMed] [Google Scholar]
- Gehrig JL, Venkatesh S, Chang HW, Hibberd MC, Kung VL, Cheng J, Chen RY, Subramanian S, Cowardin CA, Meier MF, et al. (2019). Effects of microbiota-directed foods in gnotobiotic animals and undernourished children. Science 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genser L, Aguanno D, Soula HA, Dong L, Trystram L, Assmann K, Salem JE, Vaillant JC, Oppert JM, Laugerette F, et al. (2018). Increased jejunal permeability in human obesity is revealed by a lipid challenge and is linked to inflammation and type 2 diabetes. The Journal of pathology 246, 217–230. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Jiang C, and Patterson AD (2016). An Intestinal Microbiota-Farnesoid X Receptor Axis Modulates Metabolic Disease. Gastroenterology 151, 845–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerville M, Leroy A, Sinquin A, Laugerette F, Michalski MC, and Boudry G (2017). Western-diet consumption induces alteration of barrier function mechanisms in the ileum that correlates with metabolic endotoxemia in rats. Am J Physiol Endocrinol Metab 313, E107–E120. [DOI] [PubMed] [Google Scholar]
- Gulati AS, Shanahan MT, Arthur JC, Grossniklaus E, von Furstenberg RJ, Kreuk L, Henning SJ, Jobin C, and Sartor RB (2012). Mouse background strain profoundly influences Paneth cell function and intestinal microbial composition. PloS one 7, e32403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther C, Ruder B, Stolzer I, Dorner H, He GW, Chiriac MT, Aden K, Strigli A, Bittel M, Zeissig S, et al. (2019). Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients With Crohn’s Disease. Gastroenterology 157, 1310–1322 e1313. [DOI] [PubMed] [Google Scholar]
- Hales CM, Fryar CD, Carroll MD, Freedman DS, and Ogden CL (2018). Trends in Obesity and Severe Obesity Prevalence in US Youth and Adults by Sex and Age, 2007–2008 to 2015–2016. JAMA 319, 1723–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannibal TD, Schmidt-Christensen A, Nilsson J, Fransen-Pettersson N, Hansen L, and Holmberg D (2017). Deficiency in plasmacytoid dendritic cells and type I interferon signalling prevents diet-induced obesity and insulin resistance in mice. Diabetologia 60, 2033–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Kahles F, Rattik S, Nairz M, McAlpine CS, Anzai A, Selgrade D, Fenn AM, Chan CT, Mindur JE, et al. (2019). Gut intraepithelial T cells calibrate metabolism and accelerate cardiovascular disease. Nature 566, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heden TD, Morris EM, Kearney ML, Liu TW, Park YM, Kanaley JA, and Thyfault JP (2014). Differential effects of low-fat and high-fat diets on fed-state hepatic triacylglycerol secretion, hepatic fatty acid profiles, and DGAT-1 protein expression in obese-prone Sprague-Dawley rats. Appl Physiol Nutr Metab 39, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodin CM, Verdam FJ, Grootjans J, Rensen SS, Verheyen FK, Dejong CH, Buurman WA, Greve JW, and Lenaerts K (2011). Reduced Paneth cell antimicrobial protein levels correlate with activation of the unfolded protein response in the gut of obese individuals. The Journal of pathology 225, 276–284. [DOI] [PubMed] [Google Scholar]
- Holly MK, and Smith JG (2018). Paneth Cells during Viral Infection and Pathogenesis. Viruses 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honke N, Shaabani N, Hardt C, Krings C, Haussinger D, Lang PA, Lang KS, and Keitel V (2017). Farnesoid X Receptor in Mice Prevents Severe Liver Immunopathology During Lymphocytic Choriomeningitis Virus Infection. Cell Physiol Biochem 41, 323–338. [DOI] [PubMed] [Google Scholar]
- Howe A, Ringus DL, Williams RJ, Choo ZN, Greenwald SM, Owens SM, Coleman ML, Meyer F, and Chang EB (2016). Divergent responses of viral and bacterial communities in the gut microbiome to dietary disturbances in mice. The ISME journal 10, 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson DN, Panopoulos M, Neumann WL, Turner K, Cantarel BL, Thompson-Snipes L, Dassopoulos T, Feagins LA, Souza RF, Mills JC, et al. (2020). Mitochondrial dysfunction during loss of prohibitin 1 triggers Paneth cell defects and ileitis. Gut 69, 1928–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain U, Lai CW, Xiong S, Goodwin VM, Lu Q, Muegge BD, Christophi GP, VanDussen KL, Cummings BP, Young E, et al. (2018). Temporal Regulation of the Bacterial Metabolite Deoxycholate during Colonic Repair Is Critical for Crypt Regeneration. Cell host & microbe 24, 353–363 e355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena PK, Sheng L, Di Lucente J, Jin LW, Maezawa I, and Wan YY (2018). Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 32, 2866–2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Li F, Zhang L, Nichols RG, Krausz KW, Cai J, Qi Y, Fang ZZ, Takahashi S, et al. (2015a). Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. The Journal of clinical investigation 125, 386–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, et al. (2015b). Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nature communications 6, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Lee AH, Franke A, Glickman JN, Zeissig S, Tilg H, Nieuwenhuis EE, Higgins DE, Schreiber S, Glimcher LH, et al. (2008). XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 134, 743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kernbauer E, Ding Y, and Cadwell K (2014). An enteric virus can replace the beneficial function of commensal bacteria. Nature 516, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaloian S, Rath E, Hammoudi N, Gleisinger E, Blutke A, Giesbertz P, Berger E, Metwaly A, Waldschmitt N, Allez M, et al. (2020). Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 69, 1939–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim I, Ahn SH, Inagaki T, Choi M, Ito S, Guo GL, Kliewer SA, and Gonzalez FJ (2007). Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. Journal of lipid research 48, 2664–2672. [DOI] [PubMed] [Google Scholar]
- Lam YY, Ha CW, Campbell CR, Mitchell AJ, Dinudom A, Oscarsson J, Cook DI, Hunt NH, Caterson ID, Holmes AJ, et al. (2012). Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PloS one 7, e34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen IS, Fritzen AM, Carl CS, Agerholm M, Damgaard MTF, Holm JB, Marette A, Nordkild P, Kiens B, Kristiansen K, et al. (2019). Human Paneth cell alpha-defensin-5 treatment reverses dyslipidemia and improves glucoregulatory capacity in diet-induced obese mice. Am J Physiol Endocrinol Metab 317, E42–E52. [DOI] [PubMed] [Google Scholar]
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ, et al. (2014). Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proceedings of the National Academy of Sciences of the United States of America 111, 7741–7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, and Gordon JI (2005). Obesity alters gut microbial ecology. Proceedings of the National Academy of Sciences of the United States of America 102, 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Lelliott C, Hakansson P, Ploj K, Tuneld A, Verolin-Johansson M, Benthem L, Carlsson B, Storlien L, and Michaelsson E (2008). Intestinal, adipose, and liver inflammation in diet-induced obese mice. Metabolism 57, 1704–1710. [DOI] [PubMed] [Google Scholar]
- Liu B, Gulati AS, Cantillana V, Henry SC, Schmidt EA, Daniell X, Grossniklaus E, Schoenborn AA, Sartor RB, and Taylor GA (2013). Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. American journal of physiology Gastrointestinal and liver physiology 305, G573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Gao F, McGovern DP, and Stappenbeck TS (2014). Spatial and temporal stability of paneth cell phenotypes in Crohn’s disease: implications for prognostic cellular biomarker development. Inflammatory bowel diseases 20, 646–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Gurram B, Baldridge M, Head R, Lam V, Luo C, Cao Y, Simpson P, Hayward M, Holtz M, et al. (2016). Paneth cell defects in Crohn’s disease promote dysbiosis. JCI Insight 1, e86907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Kern JT, VanDussen KL, Xiong S, Kaiko GE, Wilen CB, Rajala MW, Caruso R, Holtzman MJ, Gao F, et al. (2018). Interaction between smoking and ATG16L1T300A triggers Paneth cell defects in Crohn’s disease. The Journal of clinical investigation 128, 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Naito T, Liu Z, VanDussen KL, Haritunians T, Li D, Endo K, Kawai Y, Nagasaki M, Kinouchi Y, et al. (2017). LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight 2, e91917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Zhang Y, Xia Y, Zhang J, Kaser A, Blumberg R, and Sun J (2020). Paneth cell alertness to pathogens maintained by vitamin D receptors. Gastroenterology 160, 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison BB, Jeganathan AN, Mizuno R, Winslow MM, Castells A, Cuatrecasas M, and Rustgi AK (2015). Let-7 Represses Carcinogenesis and a Stem Cell Phenotype in the Intestine via Regulation of Hmga2. PLoS genetics 11, e1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead R (1988). The design of experiments. (Cambridge University Press; ). [Google Scholar]
- Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, and Stappenbeck TS (2012). Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science 338, 108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, and Stappenbeck TS (2013). In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat Protoc 8, 2471–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon C, Baldridge MT, Wallace MA, Burnham CA, Virgin HW, and Stappenbeck TS (2015). Vertically transmitted faecal IgA levels determine extra-chromosomal phenotypic variation. Nature 521, 90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, et al. (2013). Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582 e571. [DOI] [PubMed] [Google Scholar]
- Naja F, Hwalla N, Itani L, Karam S, Sibai AM, and Nasreddine L (2015). A Western dietary pattern is associated with overweight and obesity in a national sample of Lebanese adolescents (13–19 years): a cross-sectional study. The British journal of nutrition 114, 1909–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, et al. (2015). Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry EP, Kennedy SM, Xie Y, Luo J, and Davidson NO (2009). Diet-induced alterations in intestinal and extrahepatic lipid metabolism in liver fatty acid binding protein knockout mice. Mol Cell Biochem 326, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, Panaccione R, Ghosh S, Wu JCY, Chan FKL, et al. (2018). Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778. [DOI] [PubMed] [Google Scholar]
- Ng SC, Tang W, Leong RW, Chen M, Ko Y, Studd C, Niewiadomski O, Bell S, Kamm MA, de Silva HJ, et al. (2015). Environmental risk factors in inflammatory bowel disease: a population-based case-control study in Asia-Pacific. Gut 64, 1063–1071. [DOI] [PubMed] [Google Scholar]
- Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. (2015). Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 160, 447–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette AJ (2006). Paneth cell alpha-defensin synthesis and function. Current topics in microbiology and immunology 306, 1–25. [DOI] [PubMed] [Google Scholar]
- Ouellette AJ (2010). Paneth cells and innate mucosal immunity. Current opinion in gastroenterology 26, 547–553. [DOI] [PubMed] [Google Scholar]
- Pentinmikko N, Iqbal S, Mana M, Andersson S, Cognetta AB 3rd, Suciu RM, Roper J, Luopajarvi K, Markelin E, Gopalakrishnan S, et al. (2019). Notum produced by Paneth cells attenuates regeneration of aged intestinal epithelium. Nature 571, 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perminow G, Beisner J, Koslowski M, Lyckander LG, Stange E, Vatn MH, and Wehkamp J (2010). Defective paneth cell-mediated host defense in pediatric ileal Crohn’s disease. The American journal of gastroenterology 105, 452–459. [DOI] [PubMed] [Google Scholar]
- Pinto AK, Daffis S, Brien JD, Gainey MD, Yokoyama WM, Sheehan KC, Murphy KM, Schreiber RD, and Diamond MS (2011). A temporal role of type I interferon signaling in CD8+ T cell maturation during acute West Nile virus infection. PLoS pathogens 7, e1002407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovani D, Danese S, Peyrin-Biroulet L, and Bonovas S (2020). Environmental, Nutritional, and Socioeconomic Determinants of IBD Incidence: A Global Ecological Study. Journal of Crohn’s & colitis 14, 323–331. [DOI] [PubMed] [Google Scholar]
- Rao A, Kosters A, Mells JE, Zhang W, Setchell KD, Amanso AM, Wynn GM, Xu T, Keller BT, Yin H, et al. (2016). Inhibition of ileal bile acid uptake protects against nonalcoholic fatty liver disease in high-fat diet-fed mice. Science translational medicine 8, 357ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Cipriani S, D’Amore C, Carino A, Bruno A, Francisci D, Zampella A, Distrutti E, and Fiorucci S (2013). The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PloS one 8, e54472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F, et al. (2014). FXR is a molecular target for the effects of vertical sleeve gastrectomy. Nature 509, 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzman NH, Ghosh D, Huttner KM, Paterson Y, and Bevins CL (2003). Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 422, 522–526. [DOI] [PubMed] [Google Scholar]
- Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, et al. (2010). Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology 11, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, and Clevers H (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. (2009). Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome biology 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal A, Donaldson DS, Pridans C, Sauter KA, Hume DA, and Mabbott NA (2018). The role of CSF1R-dependent macrophages in control of the intestinal stem-cell niche. Nature communications 9, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Z, Rehm CD, Rogers G, Ruan M, Wang DD, Hu FB, Mozaffarian D, Zhang FF, and Bhupathiraju SN (2019). Trends in Dietary Carbohydrate, Protein, and Fat Intake and Diet Quality Among US Adults, 1999–2016. JAMA 322, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan KC, Lai KS, Dunn GP, Bruce AT, Diamond MS, Heutel JD, Dungo-Arthur C, Carrero JA, White JM, Hertzog PJ, et al. (2006). Blocking monoclonal antibodies specific for mouse IFN-alpha/beta receptor subunit 1 (IFNAR-1) from mice immunized by in vivo hydrodynamic transfection. J Interferon Cytokine Res 26, 804–819. [DOI] [PubMed] [Google Scholar]
- Sheehan KC, Lazear HM, Diamond MS, and Schreiber RD (2015). Selective Blockade of Interferon-alpha and -beta Reveals Their Non-Redundant Functions in a Mouse Model of West Nile Virus Infection. PloS one 10, e0128636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih DQ, Bussen M, Sehayek E, Ananthanarayanan M, Shneider BL, Suchy FJ, Shefer S, Bollileni JS, Gonzalez FJ, Breslow JL, et al. (2001). Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nature genetics 27, 375–382. [DOI] [PubMed] [Google Scholar]
- Siddiquee T, Bhowmik B, Da Vale Moreira NC, Mujumder A, Mahtab H, Khan AK, and Hussain A (2015). Prevalence of obesity in a rural Asian Indian (Bangladeshi) population and its determinants. BMC public health 15, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorribas M, Jakob MO, Yilmaz B, Li H, Stutz D, Noser Y, de Gottardi A, Moghadamrad S, Hassan M, Albillos A, et al. (2019). FxR-modulates the gut-vascular barrier by regulating the entry sites for bacterial translocation in experimental cirrhosis. Journal of hepatology 71, 1126–1140. [DOI] [PubMed] [Google Scholar]
- Steed AL, Christophi GP, Kaiko GE, Sun L, Goodwin VM, Jain U, Esaulova E, Artyomov MN, Morales DJ, Holtzman MJ, et al. (2017). The microbial metabolite desaminotyrosine protects from influenza through type I interferon. Science 357, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, Manieri NA, Fogel LA, French AR, Piwnica-Worms D, Piwnica-Worms H, et al. (2015). Type I interferons link viral infection to enhanced epithelial turnover and repair. Cell host & microbe 17, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, Liu J, Deng Y, Xia J, Chen B, et al. (2018). Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nature medicine 24, 1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teltschik Z, Wiest R, Beisner J, Nuding S, Hofmann C, Schoelmerich J, Bevins CL, Stange EF, and Wehkamp J (2012). Intestinal bacterial translocation in rats with cirrhosis is related to compromised Paneth cell antimicrobial host defense. Hepatology 55, 1154–1163. [DOI] [PubMed] [Google Scholar]
- Tomas J, Mulet C, Saffarian A, Cavin JB, Ducroc R, Regnault B, Kun Tan C, Duszka K, Burcelin R, Wahli W, et al. (2016). High-fat diet modifies the PPAR-gamma pathway leading to disruption of microbial and physiological ecosystem in murine small intestine. Proceedings of the National Academy of Sciences of the United States of America 113, E5934–E5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tominaga M, Uno K, Yagi K, Fukui M, Hasegawa G, Yoshikawa T, and Nakamura N (2010). Association between capacity of interferon-alpha production and metabolic parameters. J Interferon Cytokine Res 30, 451–454. [DOI] [PubMed] [Google Scholar]
- Trabelsi MS, Daoudi M, Prawitt J, Ducastel S, Touche V, Sayin SI, Perino A, Brighton CA, Sebti Y, Kluza J, et al. (2015). Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nature communications 6, 7629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschurtschenthaler M, Wang J, Fricke C, Fritz TM, Niederreiter L, Adolph TE, Sarcevic E, Kunzel S, Offner FA, Kalinke U, et al. (2014). Type I interferon signalling in the intestinal epithelium affects Paneth cells, microbial ecology and epithelial regeneration. Gut 63, 1921–1931. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Backhed F, Fulton L, and Gordon JI (2008). Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell host & microbe 3, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. (2009). A core gut microbiome in obese and lean twins. Nature 457, 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, and Gordon JI (2006). An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444, 1027–1031. [DOI] [PubMed] [Google Scholar]
- Vaishnava S, Behrendt CL, Ismail AS, Eckmann L, and Hooper LV (2008). Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proceedings of the National Academy of Sciences of the United States of America 105, 20858–20863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDussen KL, Liu TC, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. (2014). Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology 146, 200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDussen KL, Marinshaw JM, Shaikh N, Miyoshi H, Moon C, Tarr PI, Ciorba MA, and Stappenbeck TS (2015). Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 64, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanDussen KL, Stojmirovic A, Li K, Liu TC, Kimes PK, Muegge BD, Simpson KF, Ciorba MA, Perrigoue JG, Friedman JR, et al. (2018). Abnormal Small Intestinal Epithelial Microvilli in Patients With Crohn’s Disease. Gastroenterology 155, 815–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehkamp J, Salzman NH, Porter E, Nuding S, Weichenthal M, Petras RE, Shen B, Schaeffeler E, Schwab M, Linzmeier R, et al. (2005). Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proceedings of the National Academy of Sciences of the United States of America 102, 18129–18134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehkamp J, and Stange EF (2006). Paneth cells and the innate immune response. Current opinion in gastroenterology 22, 644–650. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, and Stange EF (2020). An Update Review on the Paneth Cell as Key to Ileal Crohn’s Disease. Front Immunol 11, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisser SB, van Rooijen N, and Sly LM (2012). Depletion and reconstitution of macrophages in mice. J Vis Exp, 4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt J, Woo V, Lee P, Moncivaiz J, Haberman Y, Denson L, Tso P, and Alenghat T (2018). Disruption of Epithelial HDAC3 in Intestine Prevents Diet-Induced Obesity in Mice. Gastroenterology 155, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler ES, Shrihari S, Hykes BL Jr., Handley SA, Andhey PS, Huang YS, Swain A, Droit L, Chebrolu KK, Mack M, et al. (2020). The Intestinal Microbiome Restricts Alphavirus Infection and Dissemination through a Bile Acid-Type I IFN Signaling Axis. Cell 182, 901–918 e918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Zhang YG, Lu R, Xia Y, Zhou D, Petrof EO, Claud EC, Chen D, Chang EB, Carmeliet G, et al. (2015). Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 64, 1082–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Kim YH, Jeong S, Greenson JK, Chaudhry MS, Hoepting M, Anderson ER, van den Brink MR, Peled JU, Gomes AL, et al. (2018). Survival signal REG3alpha prevents crypt apoptosis to control acute gastrointestinal graft-versus-host disease. The Journal of clinical investigation 128, 4970–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T, Kang JH, Sim JS, Kim JW, Koh JT, Shin CS, Lim H, and Yim M (2017). The farnesoid X receptor negatively regulates osteoclastogenesis in bone remodeling and pathological bone loss. Oncotarget 8, 76558–76573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The full-thickness ileum transcriptomic dataset has been deposited to ArrayExpress, with accession number: E-MTAB-8332. The microbiome dataset has been deposited to ArrayExpress, with accession number: E-MTAB-8334.