

Abstract
Asymmetric C(sp3)−H functionalization is a persistent challenge in organic synthesis. Here, we report an asymmetric benzylic C−H acylation of alkylarenes employing carboxylic acids as acyl surrogates for the synthesis of α-aryl ketones via nickel and photoredox dual catalysis. This mild yet straightforward protocol transforms a diverse array of feedstock carboxylic acids and simple alkyl benzenes into highly valuable α-aryl ketones with high enantioselectivities. The utility of this method is showcased in the gram-scale synthesis and late-stage modification of medicinally relevant molecules. Mechanistic studies suggest a photocatalytically generated bromine radical can perform benzylic C−H cleavage to activate alkylarenes as nucleophilic coupling partners which can then engage in a nickel-catalyzed asymmetric acyl cross-coupling reaction. This bromine-radical-mediated C−H activation strategy can be also applied to the enantioselective coupling of alkylarenes with chloroformate for the synthesis of chiral α-aryl esters.
Subject terms: Asymmetric catalysis, Homogeneous catalysis, Photocatalysis, Synthetic chemistry methodology
Chiral α-aryl ketones are versatile building blocks, and represent important pharmacophores existing in many drug molecules such as ibuprofen and naproxen. Here the authors for such ketones but using nickel and photoredox dual catalysis in asymmetric benzylic C−H acylation of alkylarenes and employing carboxylic acids as acyl surrogates.
Introduction
Chiral α-aryl ketones are versatile building blocks and represent important pharmacophores existing in many drug molecules such as ibuprofen and naproxen1,2. Although numerous enantioselective approaches for preparing quaternary α-aryl ketones have been reported3–5, asymmetric methods to access more commonly encountered tertiary variants remain limited presumably owing to the lability of tertiary stereocenters6. Nevertheless, transition-metal catalyzed asymmetric couplings of aryl organometallic reagents with α-bromo ketones7–9, benzylic zinc reagents with thioesters10, benzylic chlorides with acid chlorides under reductive conditons11, and aryl alkenes with activated carboxylic acids in the presence of a hydrosilane12,13 have been disclosed in seminal studies by Fu, Maulide, Reisman, and Buchwald, respectively (Fig. 1a). Despite this impressive progress, it remains highly desirable to develop complementary methods that use feedstock functional groups to avoid sensitive organometallic reagents, preformed organohalides, and stoichiometric reductants.
Fig. 1. Enantioselective metal-catalyzed approaches for the synthesis of α-aryl ketones.

a Previous approaches. b Dual nickel/photoredox catalyzed C(sp3)−H functionalization. c This work and its mechanistic hypothesis. O. A. oxidative addition, R. E. reductive elimination. Ir(III) = Ir[dF(CF3)ppy]2(dtbbpy)PF6.
In recent years, nickel and photoredox dual catalysis have emerged as a powerful tool for the direct C(sp3)−H functionalization of feedstock hydrocarbons by leveraging photoredox-mediated C−H activation and nickel’s unique ability in alkyl cross-couplings14–33 (Fig. 1b). This strategy allows the use of mild and robust conditions to perform C−H cleavage via a hydrogen atom transfer (HAT) or single-electron transfer (SET) pathway. These routes provide attractive strategic alternatives to challenging metal-catalyzed C(sp3)−H functionalization without the need for high reaction temperature and coordinating directing groups that are often encountered in traditional C−H activation reactions34. Pioneering works by MacMillan, Molander, and Doyle have led to numerous nonasymmetric methods for dual nickel/photoredox catalyzed C(sp3)−H coupling reactions17–33. In contrast, enantioselective approaches remain largely undeveloped35. Few successful examples in nickel and photoredox catalyzed asymmetric C(sp3)−H functionalization are only limited to the arylation of C(sp3)−H bonds with aryl bromides36,37.
Recently, our laboratory reported a direct enantioselective C(sp3)−H acylation of N-alkyl benzamides for the synthesis of α-amino ketones; wherein, a chiral nickel catalyst could engage photocatalytically generated α-amino radicals and in situ-activated carboxylic acids in acyl cross-couplings38. We reasoned that this strategy could be applied to the asymmetric benzylic C−H acylation of alkylarenes to address the challenges described above for the synthesis of α-aryl ketones via radical C(sp3)−H functionalization36–38. Despite that initial progress38, no examples of enantioselective benzylic C−H acylation have been reported. In addition, there is an increasing demand for the development of benzylic C−H functionalization reactions for the synthesis of high value-added molecules from simple alkylarenes39–50. In this work, we report an enantioselective benzylic C−H acylation of alkylarenes with in situ-activated carboxylic acids enabled by nickel and photoredox dual catalysis (Fig. 1c, top).
Results
Reaction design
The proposed catalytic cycle for this benzylic acylation is shown in the bottom of Fig. 1c. It has been reported that single-electron oxidation of bromide anion by photoexcited photocatalyst can generate bromine radical (E1/2[Ir(III*/II)] = +1.21 V vs SCE in CH3CN; E1/2ox [Br−/Br·] = +0.80 V vs SCE in DME)51–54. According to the literature precedent and our previous mechanistic experiments38,51–54, we hypothesize that the catalytic reaction is initiated by oxidative addition of Ni(0) catalyst I to an in situ-activated carboxylic acid to afford Ni(II) species II. Subsequent trapping of prochiral benzylic radicals generated from the bromine-radical-mediated HAT process provides Ni(III) complex III, which undergoes reductive elimination to yield the desired product and Ni(I) species IV. A recent computational study of nickel-catalyzed cross-coupling of photoredox-generated benzylic radicals suggested that reductive elimination is the stereochemistry-determining step55. Finally, SET between Ni(I) species IV and reduced photocatalyst regenerates the Ni(0) catalyst I and ground-state photocatalyst to close both catalytic cycles (E1/2red [Ir(II/III)] = −1.37 V vs SCE in CH3CN).
Reaction optimization
Our investigation began with an exploration of reaction conditions for the coupling of 4-ethylbiphenyl and 3-phenylpropanoic acid (Table 1). Based on previously reported elegant strategies and our recent conditions for carboxylic acid activation in ketone synthesis38,56–60, dimethyl dicarbonate (DMDC) was chosen as the activating agent to generate mixed anhydride in situ from carboxylic acids. After an extensive study of reaction parameters (also see Supplementary Table 1), we were delighted to find that a simple chiral nickel/bis(oxazoline) catalyst and a known Ir-photocatalyst could provide the acylation product in 85% yield and 94% ee (entry 1). An attractive feature of this transformation is that only commodity chemicals are involved in this reaction. From the standpoint of commercial availability, carboxylic acids are perhaps the most ubiquitous functional group. The reaction could be also performed at room temperature with similar efficiency (entry 2). The use of a nickel source free of bromide led to almost no product formation (entry 3). Interestingly, the addition of NaBr was found to restore the reaction with comparable outcome, showcasing the crucial role of bromide anion in the catalytic cycle (entry 4). The use of Boc2O instead of DMDC provided the desired product in 93% ee, but with poor yield (entry 5). Replacing the Ir-photocatalyst with a ketone triplet sensitizer, which has been employed in nickel/photoredox catalyzed C(sp3)−H functionalization reactions18,23,36 resulted in no product formation (entry 6). Running the reaction in the absence of NH4Cl, which has been previously employed to facilitate the formation of a mixed anhydride56,60, led to a significantly lower yield (entry 7). Control experiments revealed nickel, photocatalyst, and light are indispensable for product formation (entry 8). The use of other acyl surrogates in place of the in situ combination of carboxylic acid, DMDC, and NH4Cl did not provide improvements (entry 9). Other chiral ligands such as L1 and L2 delivered acylation products with similar enantioselectivities, albeit in diminished yields (entry 10).
Table 1.
Effect of reaction parameters.
 | |||
|---|---|---|---|
| Entry | Variation from standard conditions | Yield (%) | ee (%) |
| 1 | None | 85 | 94 |
| 2 | 25°C, instead of 10°C | 83 | 90 |
| 3 | Ni(acac)2, instead of NiBr2·glyme | 6 | - |
| 4 | as entry 3, but plus 1.5 equiv NaBr | 62 | 88 |
| 5 | Boc2O, instead of DMDC | 14 | 93 |
| 6 | DMBP, instead of PC | 0 | - |
| 7 | No NH4Cl | 54 | 93 |
| 8 | No Ni, or no PC, or no light | 0 | - |
| 9 | C1–C4, instead of (acid+DMDC + NH4Cl) | as shown below | |
| 10 | L1–L5, instead of (S)-L | as shown below | |
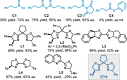 | |||
Reactions were conducted on a 0.1 mmol scale. The yields were determined through GC analysis using n-dodecane as an internal standard. DMDC dimethyl dicarbonate. PC Ir[dF(CF3)ppy]2(dtbbpy)PF6, DMBP 4,4’-dimethoxybenzophenone.
Evaluation of substrate scope
We next investigated the scope for cross-coupling of alkylarenes with carboxylic acids employing the optimized reaction conditions (Fig. 2). This transformation was compatible with many functional groups, such as chloride (7, 44), bromide (8, 43, 32, and 33), fluoride (9, 19, 24, and 31), ether (10, 14, and 42), nitrile (11), carbamate (12), ester (13, 40, and 41), olefin (15, 16), boronate ester (34), pyrazole (35), and heteroaromatic moieties (17, 36, and 46). Remarkably, the alkyl halide, aryl halide, aryl boronate ester, and terminal olefin can serve as versatile synthetic handles for further structural elaborations. Pyrazole- and thiophene-based heterocycles are commonly found in pharmaceutically relevant compounds. The coupling of 4-ethylbiphenyl with carboxylic acids bearing different steric properties furnished the corresponding α-aryl ketones in high yields and ee’s (2−5). Aromatic carboxylic acids were also suitable coupling partners to generate desired products, albeit with modest yields and enantioselectivities (18, 19). The corresponding methyl carboxylate was a significant side product for the cross-coupling of aromatic carboxylic acids. For the alkylarene component, acylation of para-substituted alkylarene bearing diverse electronic properties resulted in good yields and enantioselectivities (20−27). When the alkylarene featuring more than one benzylic C−H site was used, monoacylation products could be obtained in good yields and ee’s (28−30). The homobenzylic bulky substrate was a competent coupling partner (39). Acylation of indane provided 45 in good yield and slightly reduced enantioselectivity. Under the current reaction conditions, the sterically hindered coupling partners such as the α-branched carboxylic acids and ortho-substituted alkylarene (33) led to low efficiency or no product formation (also see Supplementary Table 1).
Fig. 2. Substrate scope of enantioselective acylation of benzylic C(sp3)−H bonds with carboxylic acids.

All data represent the average of two experiments. Unless otherwise stated, reactions were conducted on a 0.5 mmol scale under standard conditions. aIn place of the standard conditions, chiral ligand L3, 3.0 equiv DMDC, and 3.0 equiv K2HPO4 were used.
Late-stage functionalization
Given the particularly broad functional group tolerance of our method, we sought to demonstrate the utility of this operationally convenient method in the late-stage functionalization of medicinally relevant molecules (Fig. 3)61. Specifically, acylation of benzylic C−H bonds of drugs such as ibuprofen, fenoprofen, ketoprofen, and naproxen, provided corresponding drug analogs in good yields and enantioselectivities (47−51). Employing menthol and amino acid derivatives as alkylbenzene coupling partners led to good diastereoselectivities (52−55). With oxaprozin, stearic acid, oleic acid, 2,4-D, and lithocholic acid derivatives as acyl donors, the acylation proceeded with good stereoselectivity (56−61).
Fig. 3. Late-stage functionalization.

All data represent the average of two experiments. Unless otherwise stated, reactions were conducted on a 0.5 mmol scale under standard conditions. aIn place of the standard conditions, the reaction was conducted at 25 °C in dioxane. bIn place of the standard conditions, the reaction was conducted at 25 °C. cIn place of the standard conditions, 5.0 equiv of 4-ethylbiphenyl was used.
Gram-scale synthesis and parallel synthesis
To demonstrate the scalability of the present method, two 20.0 mmol scale reactions were performed in a common flask to produce 5.35 g of chiral ketone product 8, and 9.13 g of lithocholic acid derivative 60 with excellent stereoselectivity and good yield (Fig. 4a). To further demonstrate the synthetic utility, two types of drug analogs derived from (S)-flurbiprofen and artesunate were prepared in parallel with high yields and excellent stereoselectivities (Fig. 4b). More than 100 mg of product was obtained in all cases. It is noteworthy that the labile peroxide subunit in artesunate was tolerated particularly well under mild conditions. This powerful method enables the streamlined synthesis of drug analogs, providing attractive opportunities for the rapid exploration of structure-activity relationships in drug discovery62, as well as complementing the existing methods for the synthesis chiral α-aryl ketones7−13.
Fig. 4. Gram-scale synthesis and parallel synthesis.
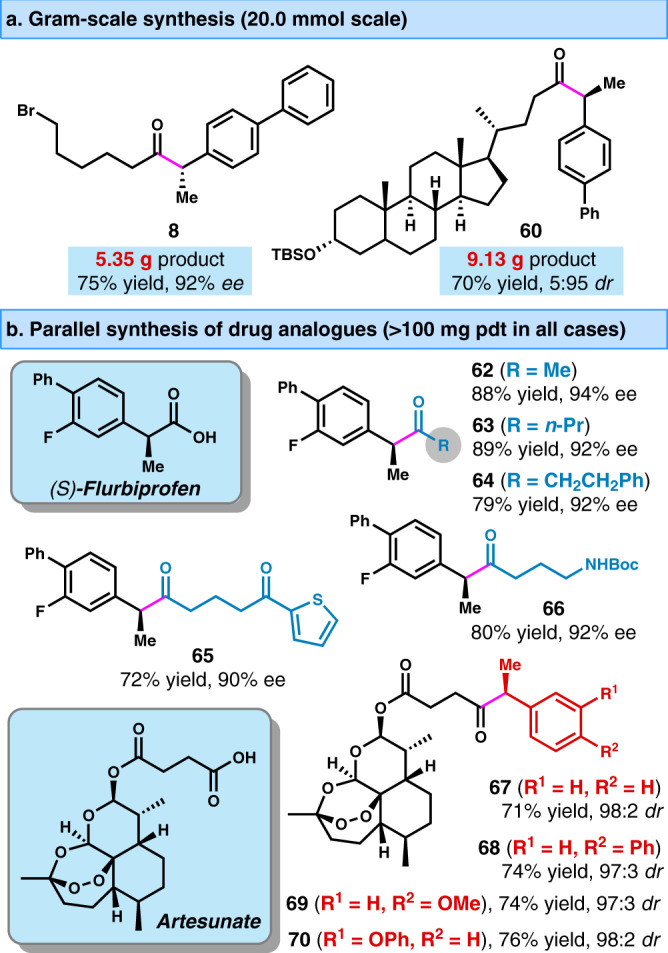
a Gram-scale synthesis. b Parallel synthesis of drug analogs.
Mechanistic observations
We next performed preliminary mechanistic studies for this newly developed method (Fig. 5). The primary kinetic isotope effect was observed in intermolecular parallel and competition experiments, which suggested that C−H cleavage significantly contributed to the rate-determining step (Fig. 5a). When the reaction was performed in the presence of an electron-deficient alkene, the benzylic acylation was completely inhibited, and a racemic adduct 71 was obtained in 58% yield (Fig. 5b, top). This observation supported the benzylic radical might be involved in the catalytic cycle. Moreover, in the absence of nickel catalyst and in situ-generated acyl electrophile (Fig. 5b, bottom), the addition of 1.5 equiv of NaBr to the coupling of 4-ethylbiphenyl with electron-deficient alkene led to the adduct 71 in 16% yield, which suggested photochemical oxidatively generated bromine radical was likely involved in the acylation reaction.
Fig. 5. Mechanistic observations.
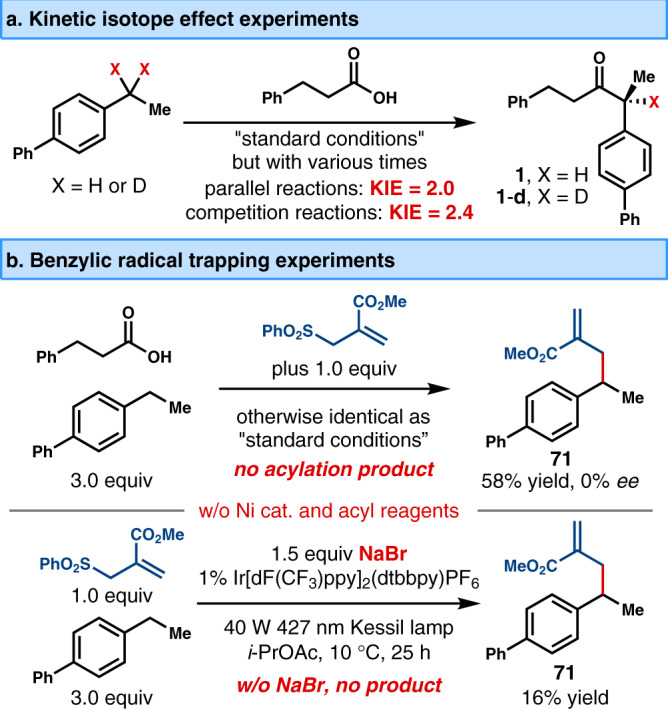
a Kinetic isotope effect experiments. b Benzylic alkyl radical trapping experiments.
Rational expansion
Finally, we questioned whether this bromine-radical-mediated C−H cleavage strategy could be applied to the synthesis of α-aryl esters rather than α-aryl ketones63–65. Indeed, replacing the in situ-generated mixed anhydride with commercially available phenyl chloroformate led to a number of α-aryl esters in good yields and selectivities under similar conditions (Fig. 6). Chiral ligand L2 proved to be optimal for this transformation.
Fig. 6. Rational expansion for the synthesis of α-aryl esters.
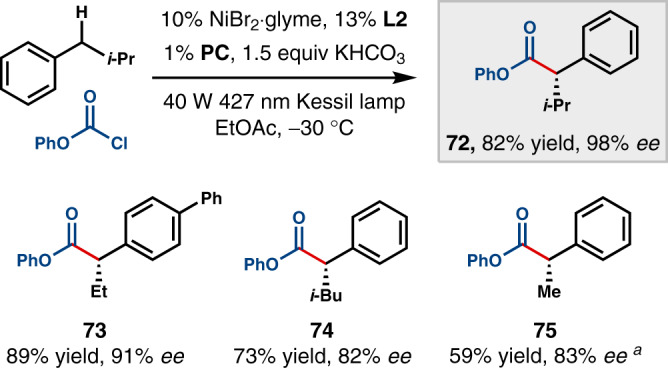
All data represent the average of two experiments. Unless otherwise noted, reactions were conducted on a 0.5 mmol scale under stated conditions. aIn place of the stated conditions, the reaction was conducted at −40°C with 5.0 equiv ethylbenzene.
Discussion
In summary, a direct enantioselective benzylic C(sp3)−H acylation for the synthesis of α-aryl ketones has been developed. Several attractive features are noteworthy. First, both coupling partners, carboxylic acids and alkylbenzenes, have broad commercial availability. Second, this operationally simple and scalable method has a broad substrate scope and excellent functional group tolerance. Third, this mild protocol can be applied to the late-stage modification of pharmaceutically relevant molecules. Finally, the asymmetric synthesis of α-aryl esters is also accessible based on a simply rational expansion. The development of enantioselective C(sp3)−H alkylation for the construction of C(sp3)−C(sp3) bonds is underway in our laboratory.
Methods
Representative procedure for the synthesis of α-aryl ketone 1
In a nitrogen-filled glovebox, Ir[dF(CF3)ppy]2(dtbbpy)PF6 (5.5 mg, 0.005 mmol), NiBr2·glyme (15.5 mg, 0.050 mmol), (S)-L (23.5 mg, 0.065 mmol), NH4Cl (26.5 mg, 0.50 mmol), Na2HPO4 (106.5 mg, 0.75 mmol), a Teflon stir bar, and anhydrous i-PrOAc (5.0 mL) were added sequentially to a 15 mL vial. The reaction mixture was stirred at room temperature for 30 min, after which it turned to a purple suspension. Next, 3-phenylpropanoic acid (75.0 mg, 0.50 mmol) was added as a solid, followed by addition of 4-ethylbiphenyl (0.75 mL, 2.0 M in i-PrOAc, 1.50 mmol) via a 1.0 mL syringe. The vial was then capped with a polytetrafluoroethylene septum cap, and DMDC (80.0 µL, 0.75 mmol) was added via a 100 µL syringe. The vial was next transferred out of the glovebox, and vacuum grease was applied to cover the entire top of the septum cap. Then, the reaction mixture was stirred at 10 °C in an EtOH bath for 5 min, followed by irradiation with a 40 W blue LED lamp (Kessil PR160L, 427 nm). The reaction was stirred at 10 °C under irradiation for 25 h. The reaction mixture was then passed through a short pad of silica gel, with Et2O as the eluent (~35 mL). The resulting mixture was concentrated, and the residue was purified by flash chromatography on silica gel, which provided the desired acylation product 1 in 82% yield and 94% ee as a white solid. All new compounds were fully characterized (See the Supplementary Methods).
Supplementary information
Acknowledgements
This work is dedicated to 100th anniversary of Xiamen University and its Chemistry. We are grateful for financial support from the NSFC (22071203) and the Xiamen University. We thank professor Hai-Chao Xu (Xiamen University) for assistance and helpful discussions. We acknowledge Wesley Harrison (UIUC) for manuscript revision.
Author contributions
L.H. and X.S. contributed equally to this work. L.H., X.S., W.Z., and D.Z. performed the experiments and analyzed the data. H.H. designed and directed the project and wrote the manuscript.
Data availability
The data that support the findings of this study are available within the article and its Supplementary Information files. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2058381 (59). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Leitao Huan, Xiaomin Shu.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-23887-2.
References
- 1.Harrington PJ, Lodewijk E. Twenty years of naproxen technology. Org. Process Res. Dev. 1997;1:72–76. doi: 10.1021/op960009e. [DOI] [Google Scholar]
- 2.Corey, E. J. & Kürti, L. Enantioselective Chemical Synthesis: Methods, Logic and Practice (Direct Book Publishing, 2010).
- 3.Bellina F, Rossi R. Transition metal-catalyzed direct arylation of substrates with activated sp3-hybridized C−H bonds and some of their synthetic equivalents with aryl halides and pseudohalides. Chem. Rev. 2010;110:1082–1146. doi: 10.1021/cr9000836. [DOI] [PubMed] [Google Scholar]
- 4.Johansson CCC, Colacot TJ. Metal-catalyzed α-arylation of carbonyl and related molecules: novel trends in C−C bond formation by C−H bond functionalization. Angew. Chem. Int. Ed. 2010;49:676–707. doi: 10.1002/anie.200903424. [DOI] [PubMed] [Google Scholar]
- 5.Kano T, Hayashi Y, Maruoka K. Construction of a chiral quaternary carbon center by catalytic asymmetric alkylation of 2-arylcyclohexanones under phase-transfer conditions. J. Am. Chem. Soc. 2013;135:7134–7137. doi: 10.1021/ja403340r. [DOI] [PubMed] [Google Scholar]
- 6.Bordwell FG, Zhang S, Zhang X-M, Liu W-Z. Homolytic bond dissociation enthalpies of the acidic H−A bonds caused by proximate substituents in sets of methyl ketones, carboxylic esters, and carboxamides related to changes in ground state energies. J. Am. Chem. Soc. 1995;117:7092–7096. doi: 10.1021/ja00132a008. [DOI] [Google Scholar]
- 7.Lundin PM, Esquivias J, Fu GC. Catalytic asymmetric cross-couplings of racemic α-bromoketones with arylzinc reagents. Angew. Chem. Int. Ed. 2009;48:154–156. doi: 10.1002/anie.200804888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lou S, Fu GC. Nickel/bis(oxazoline)-catalyzed asymmetric Kumada reactions of alkyl electrophiles: cross-couplings of racemic α-bromoketones. J. Am. Chem. Soc. 2010;132:1264–1266. doi: 10.1021/ja909689t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yin H, Fu GC. Mechanistic investigation of enantioconvergent Kumada reactions of racemic α-bromoketones catalyzed by a nickel/bis(oxazoline) complex. J. Am. Chem. Soc. 2019;141:15433–15440. doi: 10.1021/jacs.9b08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oost R, Misale A, Maulide N. Enantioconvergent Fukuyama cross-coupling of racemic benzylic organozinc reagents. Angew. Chem. Int. Ed. 2016;55:4587–4590. doi: 10.1002/anie.201600597. [DOI] [PubMed] [Google Scholar]
- 11.Cherney AH, Kadunce NT, Reisman SE. Catalytic asymmetric reductive acyl cross-coupling: synthesis of enantioenriched acyclic α,α-disubstituted ketones. J. Am. Chem. Soc. 2013;135:7442–7445. doi: 10.1021/ja402922w. [DOI] [PubMed] [Google Scholar]
- 12.Bandar JS, Ascic E, Buchwald SL. Enantioselective CuH-catalyzed reductive coupling of aryl alkenes and activated carboxylic acids. J. Am. Chem. Soc. 2016;138:5821–5824. doi: 10.1021/jacs.6b03086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Bandar JS, Buchwald SL. Enantioselective CuH-catalyzed hydroacylation employing unsaturated carboxylic acids as aldehyde surrogates. J. Am. Chem. Soc. 2017;139:8126–8129. doi: 10.1021/jacs.7b04937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Twilton J, et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017;1:0052. doi: 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- 15.Milligan JA, Phelan JP, Badir SO, Molander GA. Alkyl carbon–carbon bond formation by nickel/photoredox cross-coupling. Angew. Chem. Int. Ed. 2019;58:6152–6163. doi: 10.1002/anie.201809431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui JK, Lang SB, Heitz DR, Molander GA. Photoredox-mediated routes to radicals: the value of catalytic radical generation in synthetic methods development. ACS Catal. 2017;7:2563–2575. doi: 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Native functionality in triple catalytic cross-coupling: sp3 C–H bonds as latent nucleophiles. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heitz DR, Tellis JC, Molander GA. Photochemical nickel-catalyzed C–H arylation: synthetic scope and mechanistic investigations. J. Am. Chem.Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shields BJ, Doyle AG. Direct C(sp3)–H cross coupling enabled by catalytic generation of chlorine radicals. J. Am. Chem. Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ahneman DT, Doyle AG. C–H functionalization of amines with aryl halides by nickel-photoredox catalysis. Chem. Sci. 2016;7:7002–7006. doi: 10.1039/C6SC02815B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen MK, et al. Mild, redox-neutral formylation of aryl chlorides through the photocatalytic generation of chlorine radicals. Angew. Chem. Int. Ed. 2017;56:7191–7194. doi: 10.1002/anie.201702079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry IB, et al. Direct arylation of strong aliphatic C–H bonds. Nature. 2018;560:70–75. doi: 10.1038/s41586-018-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shen Y, Gu Y, Martin R. sp3 C–H arylation and alkylation enabled by the synergy of triplet excited ketones and nickel catalysts. J. Am. Chem. Soc. 2018;140:12200–12209. doi: 10.1021/jacs.8b07405. [DOI] [PubMed] [Google Scholar]
- 24.Dewanji A, Krach PE, Rueping M. The dual role of benzophenone in visible-light/nickel photoredox-catalyzed C−H arylations: hydrogen-atom transfer and energy transfer. Angew. Chem. Int. Ed. 2019;58:3566–3570. doi: 10.1002/anie.201901327. [DOI] [PubMed] [Google Scholar]
- 25.Das S, et al. Photocatalytic (het)arylation of C(sp3)–H bonds with carbon nitride. ACS Catal. 2021;11:1593–1603. doi: 10.1021/acscatal.0c05694. [DOI] [Google Scholar]
- 26.Deng H-P, Fan X-Z, Chen Z-H, Xu Q-H, Wu J. Photoinduced nickel-catalyzed chemo- and regioselective hydroalkylation of internal alkynes with ether and amide α-hetero C(sp3)–H bonds. J. Am. Chem. Soc. 2017;139:13579–13584. doi: 10.1021/jacs.7b08158. [DOI] [PubMed] [Google Scholar]
- 27.Le C, Liang Y, Evans RW, Li X, MacMillan DWC. Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature. 2017;547:79–83. doi: 10.1038/nature22813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thullen SM, Treacy SM, Rovis T. Regioselective alkylative cross-coupling of remote unactivated C(sp3)–H Bonds. J. Am. Chem. Soc. 2019;141:14062–14067. doi: 10.1021/jacs.9b07014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Joe CL, Doyle AG. Direct acylation of C(sp3)–H bonds enabled by nickel and photoredox catalysis. Angew. Chem. Int. Ed. 2016;55:4040–4043. doi: 10.1002/anie.201511438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ackerman LKG, Martinez Alvarado JI, Doyle AG. Direct C–C bond formation from alkanes using Ni-photoredox catalysis. J. Am. Chem. Soc. 2018;140:14059–14063. doi: 10.1021/jacs.8b09191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Z, Kumagai N, Shibasaki M. Photocatalytic α-acylation of ethers. Org. Lett. 2017;19:3727–3730. doi: 10.1021/acs.orglett.7b01552. [DOI] [PubMed] [Google Scholar]
- 32.Lee GS, Won J, Choi S, Baik M-H, Hong SH. Synergistic activation of amides and hydrocarbons for direct C(sp3)–H acylation enabled by metallaphotoredox catalysis. Angew. Chem. Int. Ed. 2020;59:16933–16942. doi: 10.1002/anie.202004441. [DOI] [PubMed] [Google Scholar]
- 33.Krach PE, Dewanji A, Yuan T, Rueping M. Synthesis of unsymmetrical ketones by applying visible-light benzophenone/nickel dual catalysis for direct benzylic acylation. Chem. Commun. 2020;56:6082–6085. doi: 10.1039/D0CC01480J. [DOI] [PubMed] [Google Scholar]
- 34.Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F, Yu J-Q. Enantioselective C(sp3)−H bond activation by chiral transition metal catalysts. Science. 2018;359:eaao4798. doi: 10.1126/science.aao4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipp A, Badir SO, Molander GA. Stereoinduction in metallaphotoredox catalysis. Angew. Chem. Int. Ed. 2020;60:1714–1726. doi: 10.1002/anie.202007668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng X, Lu H, Lu Z. Enantioselective benzylic C–H arylation via photoredox and nickel dual catalysis. Nat. Commun. 2019;10:3549. doi: 10.1038/s41467-019-11392-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rand AW, et al. Dual catalytic platform for enabling sp3 α C–H arylation and alkylation of benzamides. ACS Catal. 2020;10:4671–4676. doi: 10.1021/acscatal.0c01318. [DOI] [Google Scholar]
- 38.Shu X, Huan L, Huang Q, Huo H. Direct enantioselective C(sp3)–H acylation for the synthesis of α-amino ketones. J. Am. Chem. Soc. 2020;142:19058–19064. doi: 10.1021/jacs.0c10471. [DOI] [PubMed] [Google Scholar]
- 39.Nakafuku KM, et al. Enantioselective radical C–H amination for the synthesis of β-amino alcohols. Nat. Chem. 2020;12:697–704. doi: 10.1038/s41557-020-0482-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hu H, et al. Copper-catalysed benzylic C–H coupling with alcohols via radical relay enabled by redox buffering. Nat. Catal. 2020;3:358–367. doi: 10.1038/s41929-020-0425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang C-J, et al. Cu-catalysed intramolecular radical enantioconvergent tertiary β-C(sp3)–H amination of racemic ketones. Nat. Catal. 2020;3:539–546. doi: 10.1038/s41929-020-0460-y. [DOI] [Google Scholar]
- 42.Ye L, Tian Y, Meng X, Gu Q-S, Liu X-Y. Enantioselective copper(I)/chiral phosphoric acid catalyzed intramolecular amination of allylic and benzylic C−H bonds. Angew. Chem. Int. Ed. 2020;59:1129–1133. doi: 10.1002/anie.201911742. [DOI] [PubMed] [Google Scholar]
- 43.Clark JR, Feng K, Sookezian A, White MC. Manganese-catalysed benzylic C(sp3)–H amination for late-stage functionalization. Nat. Chem. 2018;10:583–591. doi: 10.1038/s41557-018-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang W, et al. Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science. 2016;353:1014–1018. doi: 10.1126/science.aaf7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang W, Wu L, Chen P, Liu G. Enantioselective arylation of benzylic C−H bonds by copper-catalyzed radical relay. Angew. Chem. Int. Ed. 2019;58:6425–6429. doi: 10.1002/anie.201902191. [DOI] [PubMed] [Google Scholar]
- 46.Ma J, et al. Visible-light-activated asymmetric β-C–H functionalization of acceptor-substituted ketones with 1,2-dicarbonyl compounds. J. Am. Chem. Soc. 2017;139:17245–17248. doi: 10.1021/jacs.7b09152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao X, Wang Q, Zhu J. Dual photoredox/copper catalysis for the remote C(sp3)–H functionalization of alcohols and alkyl halides by N-alkoxypyridinium salts. Angew. Chem. Int. Ed. 2019;58:2139–2143. doi: 10.1002/anie.201813356. [DOI] [PubMed] [Google Scholar]
- 48.Li Y, Lei M, Gong L. Photocatalytic regio- and stereoselective C(sp3)–H functionalization of benzylic and allylic hydrocarbons as well as unactivated alkanes. Nat. Catal. 2019;2:1016–1026. doi: 10.1038/s41929-019-0357-9. [DOI] [Google Scholar]
- 49.Tan Y, et al. Intramolecular C(sp3)–H bond oxygenation by transition-metal acylnitrenoids. Angew. Chem. Int. Ed. 2020;59:21706–21710. doi: 10.1002/anie.202009335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Z-H, et al. Copper-catalyzed enantioselective Sonogashira-type oxidative cross-coupling of unactivated C(sp3)−H bonds with alkynes. Nat. Commun. 2019;10:5689. doi: 10.1038/s41467-019-13705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang P, Le CC, MacMillan DWC. Silyl radical activation of alkyl halides in metallaphotoredox catalysis: a unique pathway for cross-electrophile coupling. J. Am. Chem. Soc. 2016;138:8084–8087. doi: 10.1021/jacs.6b04818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bacauanu V, et al. Metallaphotoredox difluoromethylation of aryl bromides. Angew. Chem. Int. Ed. 2018;57:12543–12548. doi: 10.1002/anie.201807629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kawasaki T, Ishida N, Murakami M. Dehydrogenative coupling of benzylic and aldehydic C–H bonds. J. Am. Chem. Soc. 2020;142:3366–3370. doi: 10.1021/jacs.9b13920. [DOI] [PubMed] [Google Scholar]
- 54.Kawasaki T, Ishida N, Murakami M. Photoinduced specific acylation of phenolic hydroxy groups with aldehydes. Angew. Chem. Int. Ed. 2020;59:18267–18271. doi: 10.1002/anie.202008897. [DOI] [PubMed] [Google Scholar]
- 55.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. Nickel-catalyzed cross-coupling of photoredox-generated radicals: uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J. Am. Chem. Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gooßen LJ, Ghosh K. Palladium-catalyzed synthesis of aryl ketones from boronic acids and carboxylic acids or anhydrides. Angew. Chem. Int. Ed. 2001;40:3458–3460. doi: 10.1002/1521-3773(20010917)40:18<3458::AID-ANIE3458>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 57.Gooßen LJ, Rodríguez N, Gooßen K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008;47:3100–3120. doi: 10.1002/anie.200704782. [DOI] [PubMed] [Google Scholar]
- 58.Badir SO, Dumoulin A, Matsui JK, Molander GA. Synthesis of reversed C-acyl glycosides through Ni/photoredox dual catalysis. Angew. Chem. Int. Ed. 2018;57:6610–6613. doi: 10.1002/anie.201800701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amani J, Molander GA. Direct conversion of carboxylic acids to alkyl ketones. Org. Lett. 2017;19:3612–3615. doi: 10.1021/acs.orglett.7b01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhao C, Jia X, Wang X, Gong H. Ni-catalyzed reductive coupling of alkyl acids with unactivated tertiary alkyl and glycosyl halides. J. Am. Chem. Soc. 2014;136:17645–17651. doi: 10.1021/ja510653n. [DOI] [PubMed] [Google Scholar]
- 61.Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016;45:546–576. doi: 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
- 62.Cannalire R, et al. Visible light photocatalysis in the late-stage functionalization of pharmaceutically relevant compounds. Chem. Soc. Rev. 2021;50:766–897. doi: 10.1039/D0CS00493F. [DOI] [PubMed] [Google Scholar]
- 63.Dai X, Strotman NA, Fu GC. Catalytic asymmetric Hiyama cross-couplings of racemic α-bromo esters. J. Am. Chem. Soc. 2008;130:3302–3303. doi: 10.1021/ja8009428. [DOI] [PubMed] [Google Scholar]
- 64.Mao J, et al. Cobalt–bisoxazoline-catalyzed asymmetric Kumada cross-coupling of racemic α-bromo esters with aryl Grignard reagents. J. Am. Chem. Soc. 2014;136:17662–17668. doi: 10.1021/ja5109084. [DOI] [PubMed] [Google Scholar]
- 65.DeLano, T. J. et al. Nickel-catalyzed asymmetric reductive cross-coupling of α-chloroesters with (hetero)aryl iodides. Chem. Sci. 10.1039/D1SC00822F (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available within the article and its Supplementary Information files. The X-ray crystallographic coordinates for structures reported in this article have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2058381 (59). The data can be obtained free of charge from The Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/data_request/cif.