

Abstract
Over the last 50 years, there has been a steady improvement in the treatment outcome of acute myeloid leukemia (AML). However, median survival in the elderly is still poor due to intolerance to intensive chemotherapy and higher numbers of patients with adverse cytogenetics. Fadraciclib (CYC065), a novel cyclin-dependent kinase (CDK) 2/9 inhibitor, has preclinical efficacy in AML. In AML cell lines, myeloid cell leukemia 1 (MCL-1) was downregulated following treatment with fadraciclib, resulting in a rapid induction of apoptosis. In addition, RNA polymerase II (RNAPII)-driven transcription was suppressed, rendering a global gene suppression. Rapid induction of apoptosis was observed in primary AML cells after treatment with fadraciclib for 6–8 h. Twenty-four hours continuous treatment further increased efficacy of fadraciclib. Although preliminary results showed that AML cell lines harboring KMT2A rearrangement (KMT2A-r) are more sensitive to fadraciclib, we found that the drug can induce apoptosis and decrease MCL-1 expression in primary AML cells, regardless of KMT2A status. Importantly, the diversity of genetic mutations observed in primary AML patient samples was associated with variable response to fadraciclib, confirming the need for patient stratification to enable a more effective and personalized treatment approach. Synergistic activity was demonstrated when fadraciclib was combined with the BCL-2 inhibitor venetoclax, or the conventional chemotherapy agents, cytarabine, or azacitidine, with the combination of fadraciclib and azacitidine having the most favorable therapeutic window. In summary, these results highlight the potential of fadraciclib as a novel therapeutic approach for AML.
Subject terms: Acute myeloid leukaemia, Preclinical research
Introduction
Acute myeloid leukemia (AML) is one of the most common hematologic malignancies, characterized by clonal, proliferative, and abnormally or poorly differentiated myeloid cells infiltrating the bone marrow, blood or extramedullary tissues1–3. Although treatment has progressed, in some AML patients, particularly the elderly1,4, outcome is dismal due to the remarkable genetic complexity5,6, epigenetic alterations7–9, and the dynamics of the disease10,11. Treatment-related morbidity/mortality and resistance to chemotherapy are major causes of treatment failure12. However, longer life expectancy and possibly a cure is achievable if complete remission (CR) is attained13,14. Therefore, there is substantial research to improve and personalize therapies, and lessen treatment toxicity15–18.
Currently, several novel agents acting through distinct molecular targets, identified in the pathophysiology of AML, are being investigated alone or combined with conventional chemotherapy, e.g., FLT3 inhibitors (midostaurin19, gilteritinib20), IDH1/2 inhibitors (ivosidenib21, enasidenib22,23), BCL-2 inhibitor (venetoclax24,25), and cyclin-dependent kinase (CDK) inhibitors (CDKi; alvocidib26–28, fadraciclib29), which cause cell cycle arrest and induce apoptosis.
The second generation CDKi fadraciclib is an orally available 2,6,9-trisubstituted purine analog designed to selectively inhibit CDK2 and CDK929–31. Direct inhibition of CDK2 results in cell cycle arrest of abnormally proliferating cells32. The blockade of CDK9 halts RNA polymerase II (RNAPII) transcriptional activity consequently inducing apoptosis33. Furthermore, CDK9 inhibition downregulates the anti-apoptotic protein myeloid cell leukemia 1 (MCL-1)34. Fadraciclib has already entered clinical trials in advanced solid tumors (NCT02552953), chronic lymphocytic leukemia (CLL) (NCT03739554) and AML (NCT04017546).
In this study, fadraciclib was assessed for efficacy against AML cell lines and primary AML samples in vitro either as a single agent or in combination with venetoclax (VEN), cytarabine (AraC), or azacitidine (AZA).
Results
Fadraciclib induces apoptosis of AML cell lines and reduces metabolic activity confirming anti-proliferative effect
IC50 of fadraciclib was established by resazurin reduction assay at 24, 48, and 72 h. The IC50 of fadraciclib was lower at 72 h, compared to 24 and 48 h, showing that longer exposure increased drug potency (Fig. 1A). At 72 h, the IC50 of fadraciclib in OCI-AML3, MOLM-13 and MV4–11 was approximately 0.44 ± 0.01 µM, 0.25 ± 0.01 µM and 0.52 ± 0.01 µM, respectively. As compared to the IC50, which is related to anti-proliferative activity, higher fadraciclib doses were required to induce apoptosis in 50% of cells, represented by the percentage of annexin V-positive cells (%annexinV). 0.75, 0.5, and 1 µM of fadraciclib was required to induce apoptosis in 50% of cells in OCI-AML3, MOLM-13 and MV4–11, respectively (Fig. 1B, C). This was confirmed by a significant concentration-dependent increase in the percentage of active caspase-3-positive cells (%active caspase-3; Fig. 1D). Cell cycle analysis at 72 h demonstrated a significant increase in the percentage of sub G0 (%subG0) population at concentrations of 0.75 μM, 0.5 μM, and 1 μM of fadraciclib for OCI-AML3, MOLM-13, and MV4–11, respectively, with no evidence of cell cycle arrest (Fig. 1E). However, at 4 h, G1 arrest occurred in OCI-AML3 and MV4–11, but not MOLM-13 (Supplementary Fig. S1).
Fig. 1. Fadraciclib reduces metabolic activity and causes apoptosis of AML cell lines.

A The IC50 of fadraciclib at 24, 48 and 72 h in AML cell lines using the rezasurin reduction assay. B Representative flow cytometry plots Annexin V/DAPI apoptosis assay for the OCI-AML3 cell line treated with fadraciclib for 72 h. C Bar charts based on flow cytometry data for the percentage of annexin V-positive cells in AML cell lines treated with fadraciclib for 72 h. D Representative flow cytometry plots and summary bar charts of the percentage of active caspase-3-positive cells in AML cell lines treated with fadraciclib for 72 h. E Representative flow cytometry plots and summary bar charts of cell cycle phase using PI staining of AML cell lines treated with fadraciclib for 72 h. n = 3.Graphs depict mean ± SD (****p < 0.0001). Data were compared using ANOVA.
Taken together, fadraciclib potently induces apoptosis, increases the subG0 population, and reduces proliferation as evidenced by reduced metabolic activity in AML cell lines.
Fadraciclib downregulates MCL-1, resulting in rapid induction of apoptosis in AML cell lines
AML cell lines were treated with 1 µM fadraciclib, for 4 and 24 h. An inhibitory effect on downstream targets was demonstrated by immunoblotting (Fig. 2A). Inhibition of CDK2 and CDK9 was evaluated by phosphorylation levels at serine 807/serine 811 of retinoblastoma (Rb) and serine 2 of RNAPII, respectively. Rb was not expressed in OCI-AML3. In MOLM-13, densitometry confirmed a 75% reduction in Rb levels by 4 h (p < 0.0001) and greater than 99% at 24 h, with a corresponding reduction in the phosphorylated form (p-Rb). In MV4–11, only a modest reduction in the levels of Rb and p-Rb at 4 h, with Rb level returning to baseline and a modest increase in p-Rb observed at 24 h (Fig. 2A, B). At 4 h, in all cell lines, the level of RNAPII decreased by approximately 60% with a corresponding decrease in phosphorylation (Fig. 2A, C); with similar results at 24 h.
Fig. 2. Fadraciclib downregulates MCL-1, resulting in rapid induction of apoptosis in lines treated with 1 µM of fadraciclib for 4 and 24 h. SH-PTP2 was used as an internal AML cell lines.

A Representative Western blot of OCI-AML3, MOLM-13, and MV4-11 cell protein loading control. B Densitometric analysis of serine 807/serine 811 (S807/S811) phosphorylated and total Rb. C Densitometric analysis of serine 2 (S2) phosphorylated RNAPII and total RNAPII. D Densitometric analysis of MCL-1. E Densitometric analysis of cleaved poly (ADP-ribose) polymerase-1 (PARP1). F Heatmap demonstrating fold changes relative to NDC of gene expression of OCI-AML3, MOLM-13, and MV4-11 cell lines treated with 0.75, 0.5, and 1 µM of fadraciclib, respectively, for 4 and 24 h. G Fold changes of CDK9, MCL1, PPP1R10, and E2F1 gene expression as compared with NDC. n = 3. Graphs depict mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Data were compared using the Student’s t-test.
A significant downregulation of MCL-1 occurred at 4 h in all cell lines and was maintained or further decreased at 24 h (Fig. 2A, D). This induced apoptosis as indicated by the accumulation of cleaved poly (ADP-ribose) polymerase-1 (Fig. 2A, E). The function of CDK1 was not perturbed as observed by an unchanged level of threonine 320 phosphorylated protein phosphatase 1 alpha (Supplementary Fig. S2A, B).
Owing to the impact of fadraciclib on MCL-1 levels, relevant signaling pathways, controlling MCL-1 were investigated. Threonine 180/tyrosine 182 phosphorylated p38 MAPK was markedly increased at 4 h in MOLM-13 (p < 0.0001) and MV4–11 (p < 0.001), and at 24 h in MOLM-13, but not in OCI-AML3 (Supplementary Fig. S2A, C). A decrease in threonine 202/tyrosine 204 phosphorylated Erk1/2 was also observed at 4 and 24 h (Supplementary Fig. S2A, D). By contrast, serine 473 phosphorylated Akt (Supplementary Fig. S2A, E) and serine 9 phosphorylated GSK3β (Supplementary Fig. S2A, F) were not significantly changed.
These results imply that stress response and apoptosis are potently induced in MOLM-13 and MV4–1135–38, and the observed downregulation of MCL-1 was unlikely to be via the proteasome-dependent degradation pathway modulated by GSK3β39.
Fadraciclib alters cell cycle regulatory gene expression to induce apoptosis
OCI-AML3, MOLM-13 and MV4–11 were treated with 0.75, 0.5, and 1 µM of fadraciclib, respectively, for 4 and 24 h and the effect on cell cycle regulatory gene expression investigated. The heatmap depicts fold change of gene expression as compared with NDC (Fig. 2F). Several target genes were downregulated following fadraciclib treatment, including; CDK family, transcription factors, key protein phosphatases, CDKI, DNA damage response regulators, anti-apoptotic related, and pro-apoptotic related genes (Supplementary Figs. S3–S5).
In particular, at 4 h, the expression of CDKs, especially CDK9, the transcription factor E2F1, and MCL1, were suppressed. PPP1R10 was also significantly downregulated (Fig. 2F, G). Effects on gene expression were less marked at 24 h. Conversely, the DNA damage response regulator gene CDKN1A, encoding p21Cip1, was upregulated at 24 h, indicating the intrinsic apoptosis pathway40 was activated following fadraciclib treatment (Supplementary Fig. S4B).
Fadraciclib induces apoptosis and decreases MCL-1 expression in primary AML cells, regardless of KMT2A-PTD status
Published results indicate AML cell lines bearing KMT2A rearrangements (KMT2A-r) are sensitive to fadraciclib29. Five primary AML samples harboring KMT2A-partial tandem duplication (PTD) and five primary AML samples with KMT2A wild-type (WT) were tested with fadraciclib as a single agent (Table. 1). All samples were treated with 0.5 and 1 µM fadraciclib for 24 h, then the drug was washed out and cells were cultured in fresh media for up to 72 h.
Table. 1.
Demographic data of AML patients with their cytogenetic abnormalities and genetic lesions classified by functions.
| Sample | Sex | Age (years) | Karyotype | Genetic lesions | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Signaling genes | DNA methylation-related genes | Chromatin-modifying genes | Nucleophosmin gene | Cohesin complex genes | Spliceosome complex genes | Transcription factor fusions | ||||
| An investigation of the effect of fadraciclib on primary AML cells harboring KMT2A-PTD or KMT2A WT | ||||||||||
| AML 22 | M | 68 | Normal | FLT3-ITD | DNMT3A, TET2 | KMT2A-PTD | SRSF2 | RUNX1 | ||
| AML 20B | M | 80 | NA | KMT2A-PTD | U2AF1 | |||||
| AML H002 | NA | NA | Normal | IDH2 | KMT2A-PTD, ASXL1 | |||||
| AML 12 | F | 65 | Normal | DNMT3A | KMT2A-PTD | |||||
| AML 17 | M | 72 | Trisomy 21 | NRAS | KMT2A-PTD, ASXL1 | NPM1 | U2F1 | |||
| AML 18 | M | 68 | Normal | NRAS | TET2 | ASXL1, EZH2 | ||||
| AML 13 | F | 43 | Normal | FLT3-ITD | NPM1 | |||||
| AML 44 | M | 79 | Monosomy 7 | FLT3-ITD | DNMT3A, IDH2 | |||||
| AML 5 | M | 46 | Trisomy 8 t(15;17) | TET2 | ASXL1 | SRSF2 | ||||
| AML 36 | M | 78 | Trisomy 11 | DNMT3A, IDH1 | STAG2 | SRSF2 | ||||
| Drug combination studies | ||||||||||
| AML H002 | NA | NA | Normal | IDH2 | KMT2A-PTD, ASXL1 | |||||
| AML 44 | M | 79 | Monosomy 7 | FLT3-ITD | DNMT3A, IDH2 | |||||
| AML 52 | M | 73 | Normal | FLT3-ITD | DNMT3 | NPM1 | RAD21 | |||
| AML 31 | F | 56 | Complex | KIT | NPM1 | |||||
| AML 36 | M | 78 | Trisomy 11 | DNMT3A, IDH1 | STAG2 | SRSF2 | ||||
| AML 22 | M | 68 | Normal | FLT3-ITD | DNMT3A TET2 | KMT2A-PTD | SRSF2 | RUNX1 | ||
At 24, 48, and 72 h time points, fadraciclib treatment resulted in an increase in apoptosis in both KMT2A-PTD and KMT2A WT patient samples (Fig. 3A, B). The proportion of apoptotic cells significantly increased over time. Specifically, at 72 h, a concentration-dependent maximal increase in %annexinV was observed in KMT2A-PTD patient samples following treatment with fadraciclib at 0.5 µM (20.5 ± 9.6%, p = 0.6051) and 1 µM (79.5 ± 12.9%, p < 0.0001), and in KMT2A WT patient samples following treatment with fadraciclib at 0.5 µM (33 ± 14.6%, p = 0.624) and 1 µM (74 ± 26%, p < 0.0001) compared with NDC (Fig. 3A). Responses were similar in KMT2A-PTD and KMT2A WT patient samples.
Fig. 3. Fadraciclib induces apoptosis and decreased MCL-1 expression in primary AML cells, regardless of KMT2A-PTD status.

All samples were treated with 0.5 µM or 1 µM of fadraciclib for 24 h, then the drug was washed out and cells were cultured in fresh media for up to 72 h. A Representative flow cytometry plots and summary bar charts of the percentage of annexin V-positive cells. n = 4–5. B Representative flow cytometry plots and summary bar charts of the percentage of active caspase-3-positive cells. n = 4–5. C Representative flow cytometry plots and summary bar charts of the percentage of the MCL-1 level. n = 3. Graphs depict mean ± SD (**p < 0.01, ****p < 0.0001). Data were compared using ANOVA.
A concentration-dependent increase in %active caspase-3 was observed in both KMT2A-PTD and KMT2A WT patient samples following fadraciclib treatment (Fig. 3B). However, due to genetic heterogeneity of AML samples (Table. 1), this difference did not reach statistical significance.
Treatment with fadraciclib resulted in a time- and concentration-dependent decrease in MCL-1 expression in both KMT2A-PTD and WT AML samples. Specifically, at 72 h, a decrease in MCL-1 in KMT2A-PTD patient samples at 0.5 µM (42.8 ± 20.3%, p = 0.9997) and 1 µM (5.4 ± 3.2%, p = 0.1852), and in KMT2A WT patient samples at 0.5 µM (31.2 ± 21.2%, p = 0.9463) and 1 µM (4 ± 1%, p = 0.1913) following fadraciclib treatment compared with NDC (Fig. 3C).
Pulsed dosing with fadraciclib in primary AML cells results in apoptosis and reduces cell viability, but is less effective than continuous dosing
To determine if pulsed dosing achieved an optimal response, primary AML cells were treated for up to 8 h with fadraciclib at a concentration of 1 μM. This resulted in a significant increase in %annexinV at both 6 and 8 h (p = 0.023 and p = 0.0411, respectively) and %active caspase-3 at 6 h (p = 0.0351) as compared with NDC (Supplementary Fig. S6A, B). As fadraciclib exerts a rapid effect, we investigated whether a 6-h pulse was as effective as 24-h continuous treatment at inducing apoptosis. Fadraciclib showed a higher %active caspase-3 after continuous treatment compared to the pulsed treatment (Supplementary Fig. S6C). MCL-1 levels were significantly reduced after 24-h continuous treatment compared to both the NDC and the 6-h pulsed treatment (p = 0.0167 and p = 0.0304, respectively; Supplementary Fig. S6D).
Increased apoptosis of primary human AML samples in combination studies of fadraciclib + VEN, fadraciclib + AraC, and fadraciclib + AZA
For combination studies, six primary AML samples were selected based on the presence of the most common favorable (e.g., NPM1) and unfavorable mutations (e.g., FLT3-internal tandem duplication (ITD) and KMT2A-PTD), in order to assess the efficacy of fadraciclib and its synergistic activity when combined with VEN, AraC, or AZA, towards these mutations.
Drug combination studies exploit opportunities for reduced drug resistance, decreased toxicity, and efficacy improvement41. Based on phase 1 clinical trial data, the maximum clinically achievable concentration of fadraciclib is 6–7 µM when administered as a single agent42. In this study, much lower fadraciclib concentrations of 0.25 µM (FAD1) and 0.5 µM (FAD2) in combination with VEN, AraC or AZA were assessed for efficacy against primary AML cells. The concentrations selected were based on previous studies: 0.025 µM (VEN1) and 0.5 µM43 (VEN2); 0.01 µM (AraC1) and 0.1 µM44 (AraC2); 0.5 µM45 (AZA1) and 2 µM46 (AZA2), aiming to optimize results for both sensitive and more therapy-resistant patient samples. All drug concentrations were within clinically achievable concentrations (1 µM VEN47, 1 µM AraC48, and 3–11 µM AZA49). Apoptosis, active caspase-3, cell cycle analyses, and proliferation assays were performed at 72 h.
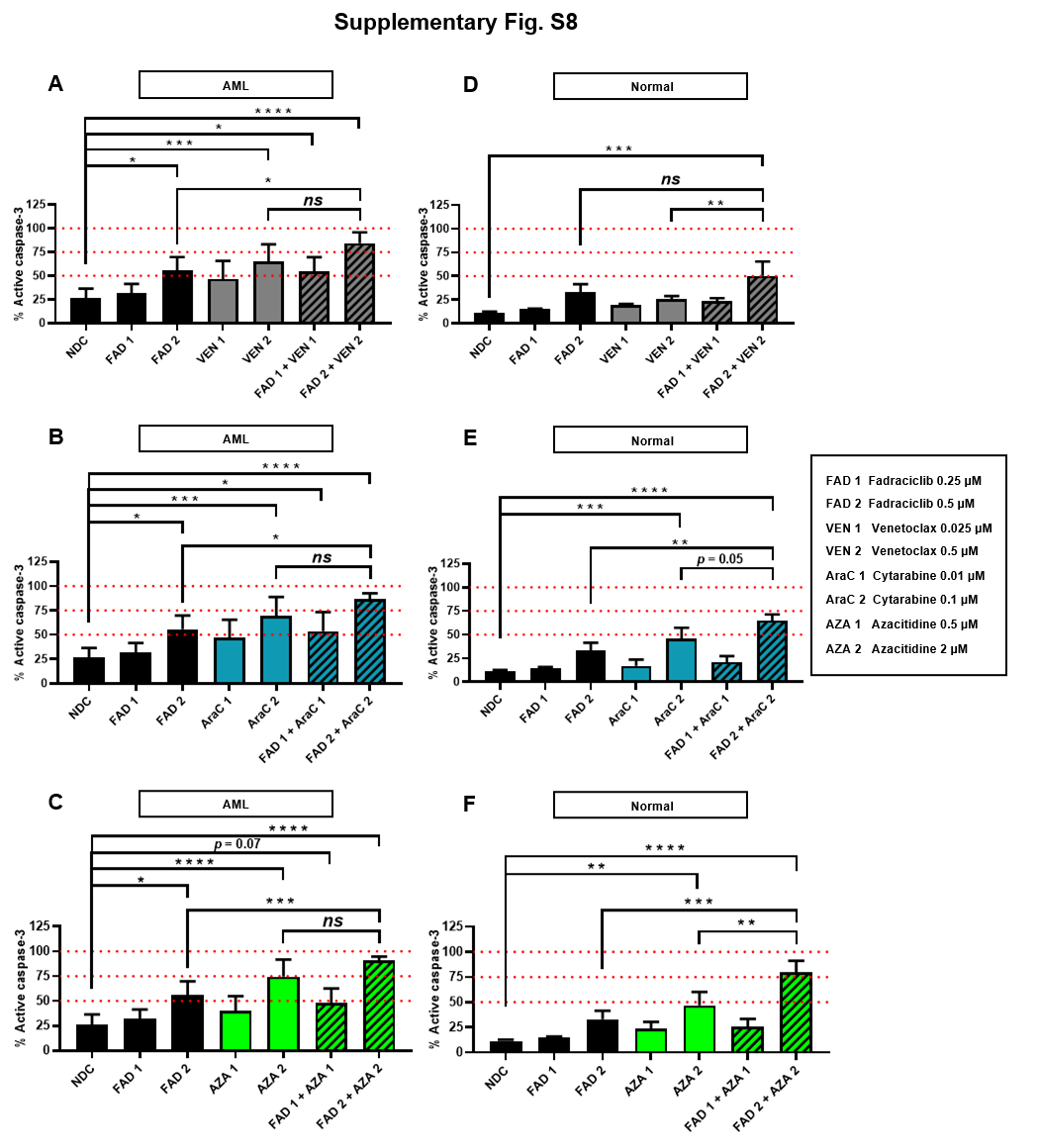
In primary AML samples, %annexinV and %active caspase-3 (Supplementary Figs. S7A–C and S8A–C, respectively) were not significantly different at low concentrations, with any drug combination pair tested. At high concentrations, a modest increase in %annexinV was observed; 82.2 ± 16.9% in FAD2 + VEN2 combination (as compared with 55.7 ± 14% in FAD2, p = 0.1059; and 64.6 ± 21.4% in VEN2 alone, p = 0.5166) (Supplementary Fig. S7A), 81.6 ± 13.4% in FAD2 + AraC2 combination (as compared with 55.7 ± 14% in FAD2, p = 0.1077; and 69.5 ± 22.5% in AraC2 alone, p = 0.8117) (Supplementary Fig. S7B), and 87.7 ± 12.3% in FAD2 + AZA2 combination (as compared with 55.7 ± 14% in FAD2, p = 0.0109; and 70.7 ± 22.8% in AZA2 alone, p = 0.437) (Supplementary Fig. S7C). Consistently, a modest increase in %active caspase-3 was observed at high concentrations; 84.3 ± 11.5% in FAD2 + VEN2 combination (as compared with 55.6 ± 14% in FAD2, p = 0.0217; and 65.2 ± 17.9% in VEN2 alone, p = 0.267) (Supplementary Fig. S8A), 86.8 ± 5.9% in FAD2 + AraC2 combination (as compared with 55.6 ± 14% in FAD2, p = 0.0143; and 69.3 ± 19.5% in AraC2 alone, p = 0.4081) (Supplementary Fig. S8B), and 90.7 ± 4% in FAD2 + AZA2 combination (as compared with 55.6 ± 14% in FAD2, p = 0.0006; and 74.4 ± 17.3% in AZA2 alone, p = 0.3197) (Supplementary Fig. S8C).
Cell cycle analysis revealed %subG0, but no other cell cycle phase, marginally increased in agreement with the increase in %annexinV and %active caspase-3 observed in FAD2 + VEN2, FAD2 + AraC2 and FAD2 + AZA2 combination (Supplementary Fig. S9A–C) as compared with each single drug treatment. Overall results indicate the more complex the molecular genetic lesions or complexity of karyotype, the less efficacious the combination therapy was (Supplementary Table S4).
Fadraciclib alone and low-dose combination therapies have limited cytotoxicity on normal hematopoietic cells
Fadraciclib alone and in combination with VEN, AraC and AZA was assessed in normal CD34 + hematopoietic samples to determine toxicity. Neither fadraciclib alone nor lower concentration drug combinations exerted a significant increase in %annexinV, %active caspase-3 or %subG0 (Supplementary Figs. S7D–F, S8D–F, and S9D–F, respectively) as compared to NDC. However, as expected, an increase in %annexinV was observed at high concentrations with a corresponding increase in %active caspase-3 (Supplementary Figs. S7D–F and S8D–F, respectively).
Fadraciclib + AZA combination therapy has a therapeutic window in in vitro cell proliferation assays
Finally, cell proliferation assays using CellTrace Violet staining were performed. Representative histograms for AML and normal CD34 + samples are shown (Supplementary Fig. S10). When considering the combination therapies, none of the low concentration drug combinations resulted in a significant anti-proliferative effect (increased percentage of undivided population, %Undivided %undivided) compared to either NDC or single drug therapies in either AML or normal CD34 + samples (Fig. 4A–F).
Fig. 4. A therapeutic window was observed in cell proliferation assays with the combination of fadraciclib and AZA.

A Summary bar charts of flow cytometry data of the percentage of undivided populations of primary AML and D normal hematopoietic patient samples treated with fadraciclib and/or VEN for 72 h. n = 6/3. B Summary bar charts of flow cytometry data of the percentage of undivided populations of primary AML and E normal hematopoietic patient samples treated with fadraciclib and/or AraC for 72 h. n = 6/3. C Summary bar charts of flow cytometry data of the percentage of undivided populations of primary AML and F normal hematopoietic patient samples treated with fadraciclib and/or AZA for 72 h. n = 6/3. Graphs depict mean ± SD (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Data were compared using ANOVA.
However, differences occurred with the high concentration drug combinations. Specifically, FAD2 + VEN2, had significant anti-proliferative effects when compared to VEN2, but might be explained by the effect of FAD2 (Fig. 4A). This combination had no anti-proliferative effect on normal CD34 + cells (Fig. 4D). Although FAD2 + AraC2, has significantly increased anti-proliferative effect as compared to FAD2 in both AML and normal CD34 + samples, this is fully accounted for by the known cytotoxic effects of AraC (Fig. 4B, E). For FAD2 + AZA2, the anti-proliferative effect in AML samples was significantly increased compared to both FAD2 and AZA2 (%Undivided %undivided of 68.7 ± 29.1% for FAD2 + AZA2 compared with 28.5 ± 10.2% in FAD2, p = 0.0004; and 33.5 ± 17% in AZA2, p = 0.0024), as well as NDC (Fig. 4C), indicating synergism of this combination with a combination index (CI) of 0.75. No anti-proliferative effect was demonstrated for this combination on normal CD34 + cells (Fig. 4F).
These findings indicate a therapeutic window and provide evidence for further exploration of combination treatment with fadraciclib and AZA as a promising treatment approach.
Discussion
Our study evaluated the therapeutic potential of fadraciclib in AML. AML cell lines harboring various genetic lesions were tested, these include OCI-AML3 (carries NPM1 and DNMT3AR882C mutations), MOLM-13 (carries FLT3-ITD and KMT2A-MLLT3), and MV4–11 (carries FLT3-ITD and KMT2A-AFF1), aiming to encompass stratification of major risk profiles according to the European LeukemiaNet recommendations50. Results indicate MV4–11, harboring adverse prognosis molecular abnormalities, was less sensitive with the highest IC50 (Fig. 1A) and a higher dose of fadraciclib required to induce apoptosis (Fig. 1C–E).
Inhibition of CDK2 and CDK9 was evaluated by phosphorylation of Rb51,52 and RNAPII53,54. We found the effects of fadraciclib on Rb were cell line specific (Fig. 2A, B), whereas, the endogenous level of RNAPII was decreased in all cell lines (Fig. 2A, C). A transcriptional downregulation of RNAPII is the most plausible explanation for the global gene suppression observed (Fig. 2F and Supplementary Figs. S3–S5). This is consistent with a previous study stating that a downregulation of POLR2A encoding the major subunit of RNAPII was observed in leukemic blasts following treatment with alvocidib, which potently inhibits CDK955. A significant decrease in short half-life MCL-1 at gene (Fig. 2G) and protein (Fig. 2A, D) levels highlights a potential target inhibited by fadraciclib.
A rapid decrease in MCL-1 level perturbs a balance in the BCL-2 family, activating the intrinsic apoptosis pathway (Fig. 5B). AML cells are more dependent on MCL-1 than normal hematopoietic cells56, highlighting the potential of fadraciclib as a therapeutic approach for AML. Downregulation of E2F1 (Fig. 2G) may explain the G1 cell cycle arrest seen in OCI-AML3 and MV4–11 following fadraciclib treatment (Supplementary Fig. S1). Interestingly, Erk1/2 were suppressed in all cell lines tested (Supplementary Fig. S2A, D), which potentially promotes a reduction in MCL-1 stability and half-life57. In various cancer cell lines, siRNA-mediated PPP1R10 knockdown results in tumor suppressor PTEN release from nuclear sequestration58. Hence, the significant decrease in PPP1R10 gene expression observed (Fig. 2G) potentially promotes an induction of apoptosis. Using leukemic blasts from adult patients with refractory AML in a phase 1 clinical trial (NCT00470197), treatment with the pan-CDKi alvocidib, resulted in a downregulation of various genes55. Among these, downregulation of POLR2A, encoding the major subunit of RNAPII, and E2F1 was seen. In addition, a decrease in phosphorylation of RNAPII and expression of MCL-1 was observed in leukemia blasts of some patients treated with alvocidib who achieved a CR59. Favorably, we found that the function of CDK1, which plays an important role in mitosis60,61, was not perturbed, suggesting better immune function with less impact on normal hematopoietic cells following fadraciclib treatment.
Fig. 5. A schematic diagram of the effects of fadraciclib at 4 h.
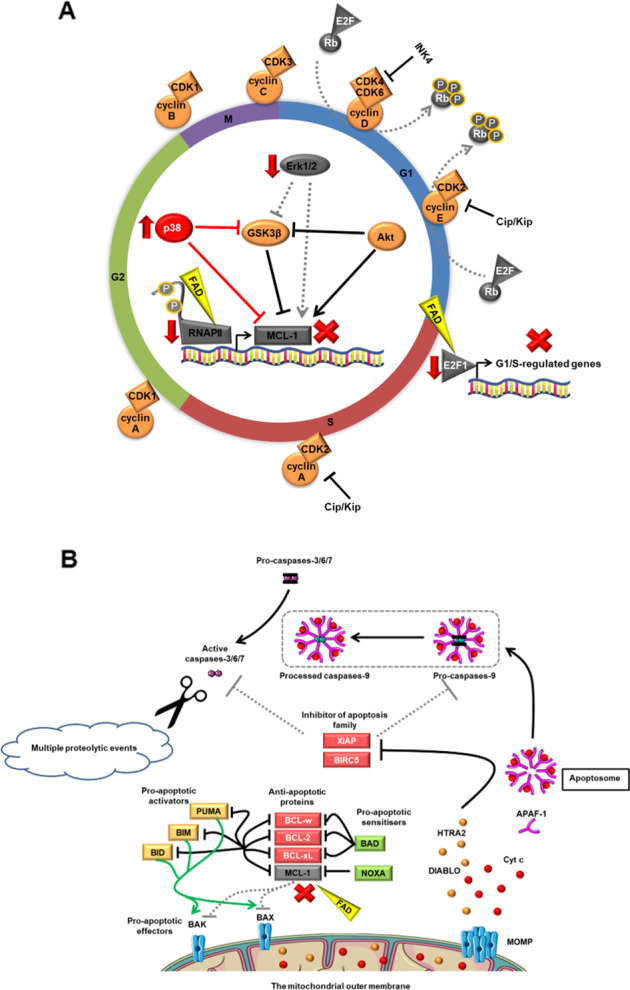
A Fadraciclib downregulates E2F1, resulting in cell cycle arrest at G1 phase. Downregulation of RNAPII is the most plausible explanation for the global gene suppression observed. A rapid decrease in short half-life MCL-1 level perturbs a balance in the BCL-2 family. A decrease in the Erk1/2 activity observed in all cell lines tested potentially promotes a reduction in MCL-1 stability and half-life. p38 MAPK is markedly increased in MOLM-13 cell line only, implying that the stress response and apoptosis are highly induced. B Fadraciclib downregulates MCL-1, which activates the intrinsic apoptosis pathway, followed by cell death. Indeed, a rapid decrease in MCL-1 level perturbs a balance in the BCL-2 family, which leads directly to activator BH3-only proteins-binding BAK and BAX, resulting in their homo-oligomerisation and MOMP. Following this, pro-apoptotic proteins within the mitochondrial intermembrane space, for example, Cyt c, DIABLO and HTRA2 are released. Cyt c binds to APAF-1 to form the apoptosome. Once formed, the apoptosome can then recruit and activate the inactive pro-caspase-9. Following this, caspases-3, 6, and 7 are activated and multiple proteolytic events occur. FAD, Fadraciclib; Cip/Kip, cyclin-dependent kinase interacting protein/kinase inhibitory protein; INK4, inhibitors of cyclin-dependent kinase 4; G1, Gap 1 phase; S, Synthesis phase; G2, Gap 2 phase; M, Mitotic phase; BCL-2, B-cell lymphoma 2; BCL-xL, B-cell lymphoma-extra large; BCL-w, B-cell lymphoma 2-like protein 2; BAD, B-cell lymphoma 2-associated death promoter; NOXA, phorbol-12-myristate-13-acetate-induced protein 1; BIM, B-cell lymphoma 2-like protein 11; BID, BH3 interacting-domain death agonist; PUMA, p53 upregulated modulator of apoptosis; BAK, B-cell lymphoma 2 homologous antagonist/killer; BAX, B-cell lymphoma 2-associated X protein; MOMP, mitochondrial outer membrane permeabilisation; Cyt c, cytochrome c; DIABLO, direct inhibitor of apoptosis proteins-binding protein with low pI; HTRA2, high-temperature requirement A serine peptidase 2; APAF-1, apoptotic protease activating factor-1; BIRC5, baculoviral inhibitor of apoptosis repeat-containing 5; XIAP, X-linked inhibitor of apoptosis protein.
Recently, a phase 1 clinical trial of fadraciclib in solid tumor patients was completed (NCT02552953)42. Dose-limiting toxicities were reversible and a biologically effective dose of 192 mg/m2/day, which corresponds to the drug concentration of 6–7 μM in vitro was established42.
Over the past few years, many targeted therapies for patients with AML have emerged with efficacy dependent on molecular subtype of AML62. This highlights the important of assessing the efficacy of fadraciclib on specific AML subtypes based on its mechanism of action of inhibiting CDK9. As part of the Super Elongation Complex (SEC), CDK9 is important for transcriptional elongation, in particular in KMT2A-r AML63, it is therefore rational to evaluate the efficacy of fadraciclib in this AML subtype. Preliminary results indicate KMT2A-r AML cell lines are more sensitive to fadraciclib29. Patient samples, however, display a high diversity of genetic mutations (Table. 1). We therefore observed a more variable fadraciclib response with KMT2A-PTD status not being a predictor for better treatment response. This is consistent with previous results showing that the CDK9-specific inhibitor atuveciclib inhibits the proliferation of seven AML cell lines, regardless of KMT2A-r status64. In addition, atuveciclib displayed potent in vitro activity in 80% of AML patient samples harboring KMT2A WT, including those with mutant NPM1 or FLT3-ITD.
Considering the combination studies, primary AML samples were selected based on the most common favorable (e.g., NPM1) and unfavorable mutations (e.g., FLT3-ITD and KMT2A-PTD). Initially, we hypothesized that the treatment response would be relevant to the risk profiles of the individual genetic lesions in primary AML samples. However, we found that complexity of the molecular genetic lesions and karyotypes have more influence. The more complex the molecular landscape, the less efficacious the combination therapy (Supplementary Table S4). The treatment response variability associated with the high diversity of genetic mutations observed reiterates the results of previous experiment showing the variable fadraciclib response, regardless of KMT2A-PTD status. Taken together, our results highlight the importance of genetic and molecular profiling prior to initiating therapy, to identify personalized therapeutic combinations most likely to benefit individual patient. Regarding the 2017 ELN risk stratification50, next generation sequencing (NGS), exome sequencing and genome wide assays65, are being developed to replace single gene assays. This will facilitate better prognostication and more personalized therapy in the future66. A short delay in commencing therapy in newly diagnosed AML had no effect on outcome after accounting for other prognostic covariates67, providing a potential window to perform NGS.
As a global effect, an increase in cell death in primary human AML cells was shown for apoptosis, active caspase-3 assays, and cell cycle analysis when fadraciclib was combined with VEN, AraC, or AZA. In primary AML cells, combining the MCL-1 inhibitor S63845 with VEN results in greater efficacy than either inhibitor alone, with a more potent activity against leukemic rather than normal hematopoietic progenitors68. In an AML patient-derived xenograft mouse model bearing FLT3-ITD, the CDK9 inhibitor A-1592668 combined with VEN provided a significant survival advantage over single treatments69. In a lymph node mimicking microenvironment, fadraciclib in combination with VEN for 24 h efficiently induced apoptosis of primary CLL cells70, a disease where MCL-1 plays a role in disease progression and fludarabine resistance71. Ongoing phase 1 clinical trials are evaluating the safety and tolerability of fadraciclib in combination with VEN in patients with relapsed/refractory AML or myelodysplastic syndromes (MDS) (NCT04017546); and relapsed/refractory CLL (NCT03739554).
When considering combining CDK9 inhibitors with AraC, a recent phase 2 study (NCT01349972), demonstrated a significantly higher efficacy of FLAM (alvocidib, cytarabine plus mitoxantrone) as compared with the standard “7 + 3” regimen in terms of a CR rate26,28. However, no increase in overall or event-free survival rates were observed. Potentially, this may be due to the higher toxicity of the combination regimen based on our preliminary data showing the impact of the fadraciclib + AraC on normal hematopoietic cells.
In preliminary experiments, pre-treatment with alvocidib reduced the IC50 of AZA in MV4–11 cell line72. In the same manner seen in fadraciclib + AZA combination presented here, alvocidib + AZA also increased %active caspase-3 as compared with single treatments. In a MOLM-13 xenograft model, the combination showed greater tumor growth inhibition compared to single agent treatments. Our results, demonstrated a potential therapeutic window with the FAD2 + AZA2 combination, as measured by %Undivided %undivided of AML cells in cell proliferation assay (Fig. 4C). These interesting results identify an attractive therapeutic strategy warranting consideration of clinical development in AML.
In summary, our studies indicate the preclinical efficacy of the CDK2/9 inhibitor fadraciclib as a single agent or in conjunction with frontline AML chemotherapeutics, highlighting its potential as a novel therapeutic approach. Our work implicates the importance of molecular genetic profiling as a critical step prior to initiating therapy for AML, highlighting the need for a more personalized medicine approach to improve outcomes for patients.
Materials and methods
Cell lines and primary AML samples cell culture
OCI-AML3, MOLM-13, and MV4–11 (DSMZ) were cultured at 37 °C in a 5% CO2 atmosphere in RPMI 1640 media (Sigma-Aldrich) with 20% or 10% fetal bovine serum (Gibco), supplemented with 1 mM glutamine and 1% penicillin-streptomycin (Invitrogen).
Healthy donor and diagnostic primary AML samples were taken in accordance with the Declaration of Helsinki, and Ethics Committee approval (15/WS/0077). Demographic data, cytogenetic abnormalities and genetic lesions of AML patients are shown in Table. 1. Primary samples were thawed and cultured overnight in serum-free medium II (Stem Cell Technologies) supplemented with hIL-3, hIL-6, hSCF, and hFLT3L (all at 10 ng/mL) (Stem Cell Technologies).
Inhibitors
Ten micromolar stock solutions of fadraciclib (Cyclacel), AZA (Stratech), and VEN (Stratech) were prepared in DMSO. 10 mM AraC (Sigma-Aldrich) was prepared in water. All stocks were stored at −20 °C and dilutions freshly prepared in cell culture media.
Rezasurin reduction assay
Half-maximal inhibitory concentration (IC50) of AML cell lines treated with fadraciclib for up to 72 h was established using 7-Hydroxy-3H-phenoxazin-3-one-10-oxide sodium salt resazurin assay (Sigma-Aldrich) according to manufacturer’s instructions. IC50 were calculated using GraphPad Prism 8 software (GraphPad Software).
Gene expression analysis
RNA was extracted using RNA Easy Micro kits (Qiagen) and converted to cDNA using high-capacity cDNA reverse transcription kit (Applied Biosystems). Quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed using Fluidigm Biomark technology and data were collected as per manufacturer’s instructions. Data was analyzed using the 2−ΔΔCT compared to no drug control (NDC) as previously described73. Internal sample control was ensured by subtracting the average of six housekeeping genes ATP5F1B, B2M, CYC1, RNF20, TYW1, and UBE2D2 from the Ct value of each gene of interest. Mean and standard deviation of fold change in expression were calculated. For Primer sequences see Supplementary Table S1.
Protein extraction and quantification
In all, 1–5 × 106 cells were harvested and washed twice in ice-cold PBS and pelleted at 300 × g for 5 min. Then, the pellets were lyzed in ice-cold solubilization buffer containing phosphatase and protease inhibitors at a concentration of 1 × 106 cells per 10 μl solubilization buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Nonidet P40, 10% Glycerol, cOmplet ULTRA 1 tablet and PhosSTOP phosphatase inhibitor 1 tablet per 10 mL of solubilization buffer). This suspension was left on ice and vortexed every 15 min for 1 h, then centrifuged at 14,000 × g for 10 min to pellet all cell debris. The supernatant was transferred to a fresh eppendorf and stored at −80 o C until required.
Protein concentration was quantified using the Quick Start Bradford Dye reagent and compared to a standard curve prepared using Quick StartTM BSA protein. The colorimetric protein assay was measured by visual absorbance on the Spectramax M5 plate reader at 595 nm at using SoftMax Pro software.
Protein analysis
Cell lysates were prepared and 30 µg of protein was resolved. Western blotting was performed using the NuPAGE electrophoresis system (Invitrogen) as per manufacturer’s instructions, and protein detection via Odyssey Fc Imaging System. Densitometry was performed using Image Studio Lite version 5.2. Antibodies and experimental conditions are outlined in Supplementary Table S2.
Flow cytometry
For proliferation analysis, primary AML and normal CD34+ cells were stained with CellTrace Violet (Thermo Fisher Scientific) as per manufacturer’s protocol. For establishing a maximum point of fluorescence staining, cells were cultured with Colcemid (100 ng/mL, Sigma-Aldrich) to determine non-dividing cells. All primary cells were treated with fadraciclib, VEN, AraC, AZA or fadraciclib in combination with VEN, AraC or AZA in physiological growth factor conditions. Annexin V/7-aminoactinomycin D (7-AAD) or DAPI (4’,6-diamidino-2-phenylindole) (BD Biosciences) staining to assess apoptosis by flow cytometry using 1 × 105 cells per condition. Propidium iodide (PI) staining buffer to assess cell cycle progression as per manufacturer’s protocol. Fixation/Permeabilization Solution Kit (BD Biosciences) was used prior to MCL-1 or active caspase-3 staining. For staining reagents used, see Supplementary Table S3.
Combination studies
CompuSyn software (ComboSyn, Inc) was used to investigate the synergism in the cell proliferation assays. The software was based on the Chou-Talalay method for drug combination based on the median-effect equation derived from the mass-action law principle74. The CI provides a quantitative definition for additive effect (CI = 1), synergism (CI < 1) or antagonism (CI > 1) in drug combinations.
Statistics
Average responses from at least three independent experiments are shown (mean ± SD). Statistical analysis used GraphPad Prism 8 for Student’s t-test and one-way ANOVA (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
The authors thank Alan Hair for processing patient samples, Jennifer Cassels for preparing culture media, Joana Silvestre for creating the heatmap, Chinmay Munje and Caroline Busch for technical support.
Author contributions
Contribution: W.C. designed and performed experiments, collected and analyzed data, and wrote the manuscript; Y.-C.H. performed experiments, collected and analyzed data; D.Z. and S.F. provided reagents, discussed experiments and edited the manuscript; H.W. supervised the project, designed experiments, and edited the manuscript; and M.C. supervised the project, designed experiments, and edited the manuscript.
Funding
This research was funded by a Ph.D. studentship from the Royal Thai Government. Additional funding was provided by Cyclacel Ltd. This study was supported by the Glasgow Experimental Cancer Medicine Center, which is funded by Cancer Research UK (C58789/A251741) and the Chief Scientist’s Office, Scotland. Cell-sorting facilities were funded by the Kay Kendall Leukemia Fund (KKL501) and the Howat Foundation.
Conflict of interest
M.C.: research funding (Cyclacel Ltd, Novartis, BMS, Incyte), advisory board member (BMS, Novartis, Incyte, Pfizer, Daiichi Sankyo), and honoraria (Astellas, BMS, Novartis, Incyte, Pfizer, Takeda, Celgene). D.Z. and S.F. are employees of Cyclacel Ltd. The remaining authors declare no competing interests.
Ethics statement
Healthy donor and diagnostic primary AML samples were taken in accordance with the Declaration of Helsinki, and Ethics Committee approval (15/WS/0077).
Footnotes
Edited by Carlo Ganini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
7/6/2021
A Correction to this paper has been published: 10.1038/s41420-021-00558-1
Supplementary information
The online version contains supplementary material available at 10.1038/s41420-021-00496-y.
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N. Engl. J. Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.De Kouchkovsky I, Abdul-Hay M. ‘Acute myeloid leukemia: a comprehensive review and 2016 update’. Blood Cancer J. 2016;6:e441. doi: 10.1038/bcj.2016.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pelcovits A, Niroula R. Acute myeloid leukemia: a review. R. I. Med. J. (2013) 2020;103:38–40. [PubMed] [Google Scholar]
- 4.Rangoonwala HI, Gootee J, Silberstein PT. Epidemiology and survival outcomes in elderly acute myeloid leukemia patients: an NCDB study. Abstracts of the 75th Annual Meeting of the American Society of Hematology. Blood. 2019;134:3840. [Google Scholar]
- 5.Rowe JM. The increasing genomic complexity of acute myeloid leukemia. Best. Pr. Res. Clin. Haematol. 2014;27:209–213. doi: 10.1016/j.beha.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Hou HA, Tien HF. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J. Biomed. Sci. 2020;27:81–94. doi: 10.1186/s12929-020-00674-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldman SL, et al. Epigenetic modifications in acute myeloid leukemia: prognosis, treatment, and heterogeneity. Front. Genet. 2019;10:133–148. doi: 10.3389/fgene.2019.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gambacorta V, Gnani D, Vago L, Di Micco R. Epigenetic therapies for acute myeloid leukemia and their immune-related effects. Front. Cell Dev. Biol. 2019;7:207–226. doi: 10.3389/fcell.2019.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plass C, Oakes C, Blum W, Marcucci G. Epigenetics in acute myeloid leukemia. Semin. Oncol. 2008;35:378–387. doi: 10.1053/j.seminoncol.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akinduro O, et al. Proliferation dynamics of acute myeloid leukaemia and haematopoietic progenitors competing for bone marrow space. Nat. Commun. 2018;9:519–531. doi: 10.1038/s41467-017-02376-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tzoran I, Rebibo-Sabbah A, Brenner B, Aharon A. Disease dynamics in patients with acute myeloid leukemia: new biomarkers. Exp. Hematol. 2015;43:936–943. doi: 10.1016/j.exphem.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 12.Walter RB, et al. Prediction of early death after induction therapy for newly diagnosed acute myeloid leukemia with pretreatment risk scores: a novel paradigm for treatment assignment. J. Clin. Oncol. 2011;29:4417–4423. doi: 10.1200/JCO.2011.35.7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eleni LD, Nicholas ZC, Alexandros S. Challenges in treating older patients with acute myeloid leukemia. J. Oncol. 2010;2010:943823. doi: 10.1155/2010/943823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burnett AK, et al. A comparison of low-dose cytarabine and hydroxyurea with or without all-trans retinoic acid for acute myeloid leukemia and high-risk myelodysplastic syndrome in patients not considered fit for intensive treatment. Cancer. 2007;109:1114–1124. doi: 10.1002/cncr.22496. [DOI] [PubMed] [Google Scholar]
- 15.Quiroga-Campano AL, et al. Personalized and optimized low-dose and intensive chemotherapy treatments for patients with acute myeloid leukemia (AML). Abstracts of the 75th Annual Meeting of the American Society of Hematology. Blood. 2018;132:3500. [Google Scholar]
- 16.Collignon A, et al. A chemogenomic approach to identify personalized therapy for patients with relapse or refractory acute myeloid leukemia: results of a prospective feasibility study. Blood Cancer J. 2020;10:64–75. doi: 10.1038/s41408-020-0330-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Illangeswaran RSS, Das S, Paul DZ, Mathews V, Balasubramanian P. A personalized approach to acute myeloid leukemia therapy: current options. Pharmgenomics Pers. Med. 2019;12:167–179. doi: 10.2147/PGPM.S168267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hourigan CS, Karp JE. Personalized therapy for acute myeloid leukemia. Cancer Discov. 2013;3:1336–1338. doi: 10.1158/2159-8290.CD-13-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone RM, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 Mutation. N. Engl. J. Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perl AE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N. Engl. J. Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 21.DiNardo CD, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N. Engl. J. Med. 2018;378:2386–2398. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 22.Stein EM, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein EM. Enasidenib, a targeted inhibitor of mutant IDH2 proteins for treatment of relapsed or refractory acute myeloid leukemia. Future Oncol. 2018;14:23–40. doi: 10.2217/fon-2017-0392. [DOI] [PubMed] [Google Scholar]
- 24.DiNardo CD, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wei AH, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J. Clin. Oncol. 2019;37:1277–1284. doi: 10.1200/JCO.18.01600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeidner JF, et al. Final results of a randomized multicenter phase II study of alvocidib, cytarabine, and mitoxantrone versus cytarabine and daunorubicin (7 + 3) in newly diagnosed high-risk acute myeloid leukemia (AML) Leuk. Res. 2018;72:92–95. doi: 10.1016/j.leukres.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeidner JF, Karp JE. Clinical activity of alvocidib (flavopiridol) in acute myeloid leukemia. Leuk. Res. 2015;39:1312–1318. doi: 10.1016/j.leukres.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Zeidner JF, et al. Randomized multicenter phase II study of flavopiridol (alvocidib), cytarabine, and mitoxantrone (FLAM) versus cytarabine/daunorubicin (7+3) in newly diagnosed acute myeloid leukemia. Haematologica. 2015;100:1172–1179. doi: 10.3324/haematol.2015.125849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frame S, et al. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS ONE. 2020;15:e0234103. doi: 10.1371/journal.pone.0234103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawakami M, et al. Next-generation CDK2/9 inhibitors and anaphase catastrophe in lung cancer. J. Natl Cancer Inst. 2017;109:6–17. doi: 10.1093/jnci/djw297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borthakur GM, et al. Combining CDK2/9 Inhibitor CYC065 with Venetoclax, a BCL2 Inhibitor, to Treat Patients with Relapsed or Refractory AML or MDS. Abstracts of the 75th Annual Meeting of the American Society of Hematology. Blood. 2019;134:1379. [Google Scholar]
- 32.Pellerano M, et al. Targeting conformational activation of CDK2 kinase. Biotechnol. J. 2017;12:1600531. doi: 10.1002/biot.201600531. [DOI] [PubMed] [Google Scholar]
- 33.Morales F, Giordano A. Overview of CDK9 as a target in cancer research. Cell Cycle. 2016;15:519–527. doi: 10.1080/15384101.2016.1138186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polier G, et al. Wogonin and related natural flavones are inhibitors of CDK9 that induce apoptosis in cancer cells by transcriptional suppression of Mcl-1. Cell Death Dis. 2011;2:e182. doi: 10.1038/cddis.2011.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cai B, Chang SH, Becker EBE, Bonni A, Xia Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006;281:25215–25222. doi: 10.1074/jbc.M512627200. [DOI] [PubMed] [Google Scholar]
- 36.Kralova J, Dvorak M, Koc M, Kral V. p38 MAPK plays an essential role in apoptosis induced by photoactivation of a novel ethylene glycol porphyrin derivative. Oncogene. 2008;27:3010–3020. doi: 10.1038/sj.onc.1210960. [DOI] [PubMed] [Google Scholar]
- 37.Deacon K, Mistry P, Chernoff J, Blank JL, Patel R. p38 Mitogen-activated protein kinase mediates cell death and p21-activated kinase mediates cell survival during chemotherapeutic drug-induced mitotic arrest. Mol. Biol. Cell. 2003;14:2071–2087. doi: 10.1091/mbc.E02-10-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eriksson M, et al. Agonistic targeting of TLR1/TLR2 induces p38 MAPK-dependent apoptosis and NFκB-dependent differentiation of AML cells. Blood Adv. 2017;1:2046–2057. doi: 10.1182/bloodadvances.2017006148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamaguchi H, Hsu JL, Hung MC. Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front. Oncol. 2012;2:15–24. doi: 10.3389/fonc.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hernandez AM, et al. Upregulation of p21 activates the intrinsic apoptotic pathway in β-cells. Am. J. Physiol. Endocrinol. Metab. 2013;304:E1281–E1290. doi: 10.1152/ajpendo.00663.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Foucquier J, Guedj M. Analysis of drug combinations: current methodological landscape. Pharm. Res. Perspect. 2015;3:e00149. doi: 10.1002/prp2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Do KT, et al. Abstract CT037: Phase I safety, pharmacokinetic and pharmacodynamic study of CYC065, a cyclin dependent kinase inhibitor, in patients with advanced cancers ( NCT02552953). Abstracts of the AACR 109th Annual Meeting. Cancer Res. 2018;78:CT037. [Google Scholar]
- 43.Teh TC, et al. Enhancing venetoclax activity in acute myeloid leukemia by co-targeting MCL1. Leukemia. 2018;32:303–312. doi: 10.1038/leu.2017.243. [DOI] [PubMed] [Google Scholar]
- 44.Leonard SM, Perry T, Woodman CB, Kearns P. Sequential treatment with cytarabine and decitabine has an increased anti-leukemia effect compared to cytarabine alone in xenograft models of childhood acute myeloid leukemia. PLoS ONE. 2014;9:e87475. doi: 10.1371/journal.pone.0087475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adachi Y, Ishikawa Y, Kiyoi H. Identification of volasertib-resistant mechanism and evaluation of combination effects with volasertib and other agents on acute myeloid leukemia. Oncotarget. 2017;8:78452–78465. doi: 10.18632/oncotarget.19632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Min C, et al. Selective inhibitors of histone deacetylases 1 and 2 synergize with azacitidine in acute myeloid leukemia. PLoS ONE. 2017;12:e0169128. doi: 10.1371/journal.pone.0169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson MA, et al. The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood. 2016;127:3215–3224. doi: 10.1182/blood-2016-01-688796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heasman SA, Zaitseva L, Bowles KM, Rushworth SA, MacEwan DJ. Protection of acute myeloid leukaemia cells from apoptosis induced by front-line chemotherapeutics is mediated by haem oxygenase-1. Oncotarget. 2011;2:658–668. doi: 10.18632/oncotarget.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hollenbach PW, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS ONE. 2010;5:e9001. doi: 10.1371/journal.pone.0009001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Döhner H, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tetsu O, McCormick F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell. 2003;3:233–245. doi: 10.1016/s1535-6108(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 52.Chytil A, et al. Construction of a cyclin D1-Cdk2 fusion protein to model the biological functions of cyclin D1-Cdk2 complexes. J. Biol. Chem. 2004;279:47688–47698. doi: 10.1074/jbc.M405938200. [DOI] [PubMed] [Google Scholar]
- 53.Santo L, Siu KT, Raje N. Targeting cyclin-dependent kinases and cell cycle progression in human cancers. Semin. Oncol. 2015;42:788–800. doi: 10.1053/j.seminoncol.2015.09.024. [DOI] [PubMed] [Google Scholar]
- 54.Bacon CW, D’Orso I. CDK9: a signaling hub for transcriptional control. Transcription. 2019;10:57–75. doi: 10.1080/21541264.2018.1523668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nelson DM, et al. Flavopiridol induces BCL-2 expression and represses oncogenic transcription factors in leukemic blasts from adults with refractory acute myeloid leukemia. Leuk. Lymphoma. 2011;52:1999–2006. doi: 10.3109/10428194.2011.591012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glaser SP, et al. Anti-apoptotic Mcl-1 is essential for the development and sustained growth of acute myeloid leukemia. Genes Dev. 2012;26:120–125. doi: 10.1101/gad.182980.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hermanson DL, Das SG, Li Y, Xing C. Overexpression of Mcl-1 confers multidrug resistance, whereas topoisomerase IIβ downregulation introduces mitoxantrone-specific drug resistance in acute myeloid leukemia. Mol. Pharm. 2013;84:236–243. doi: 10.1124/mol.113.086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kavela S, et al. PNUTS functions as a proto-oncogene by sequestering PTEN. Cancer Res. 2013;73:205–214. doi: 10.1158/0008-5472.CAN-12-1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karp JE, et al. Phase I and pharmacokinetic study of flavopiridol followed by 1-beta-D-arabinofuranosylcytosine and mitoxantrone in relapsed and refractory adult acute leukemias. Clin. Cancer Res. 2005;11:8403–8412. doi: 10.1158/1078-0432.CCR-05-1201. [DOI] [PubMed] [Google Scholar]
- 60.Hao S, Chen C, Cheng T. Cell cycle regulation of hematopoietic stem or progenitor cells. Int. J. Hematol. 2016;103:487–497. doi: 10.1007/s12185-016-1984-4. [DOI] [PubMed] [Google Scholar]
- 61.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 62.Richard-Carpentier G, DiNardo CD. Single-agent and combination biologics in acute myeloid leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2019;2019:548–556. doi: 10.1182/hematology.2019000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.He N, et al. Human polymerase-associated factor complex (PAFc) connects the super elongation complex (SEC) to RNA polymerase II on chromatin. Proc. Natl Acad. Sci. USA. 2011;108:E636–E645. doi: 10.1073/pnas.1107107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Scholz A, et al. Abstract 3022: BAY 1143572, a first-in-class, highly selective, potent and orally available inhibitor of PTEFb/CDK9 currently in Phase I, shows convincing antitumor activity in preclinical models of acute myeloid leukemia (AML). Abstracts of the AACR 107th Annual Meeting. Cancer Res. 2016;76:3022. [Google Scholar]
- 65.Kuo FC, Mar BG, Lindsley RC, Lindeman NI. The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood. 2017;130:433–439. doi: 10.1182/blood-2017-03-734533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Estey EH. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am. J. Hematol. 2018;93:1267–1291. doi: 10.1002/ajh.25214. [DOI] [PubMed] [Google Scholar]
- 67.Bertoli S, et al. Time from diagnosis to intensive chemotherapy initiation does not adversely impact the outcome of patients with acute myeloid leukemia. Blood. 2013;121:2618–2626. doi: 10.1182/blood-2012-09-454553. [DOI] [PubMed] [Google Scholar]
- 68.Moujalled DM, et al. Combining BH3-mimetics to target both BCL-2 and MCL1 has potent activity in pre-clinical models of acute myeloid leukemia. Leukemia. 2019;33:905–917. doi: 10.1038/s41375-018-0261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Phillips DC, et al. A novel CDK9 inhibitor increases the efficacy of venetoclax (ABT-199) in multiple models of hematologic malignancies. Leukemia. 2019;34:1646–1657. doi: 10.1038/s41375-019-0652-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen R, et al. Abstract 3905: Strategic combination of the cyclin-dependent kinase inhibitor CYC065 with venetoclax to target anti-apoptotic proteins in chronic lymphocytic leukemia. Abstracts of the AACR 109th Annual Meeting. Cancer Res. 2018;78:3905. [Google Scholar]
- 71.Pepper C, et al. Mcl-1 expression has in vitro and in vivo significance in chronic lymphocytic leukemia and is associated with other poor prognostic markers. Blood. 2008;112:3807–3817. doi: 10.1182/blood-2008-05-157131. [DOI] [PubMed] [Google Scholar]
- 72.Kim W, et al. The CDK9 inhibitor, alvocidib, potentiates the non-clinical activity of azacytidine or decitabine in an MCL-1-dependent fashion, supporting clinical exploration of a decitabine and alvocidib combination. Abstracts of the 74th Annual Meeting of the American Society of Hematology. Blood. 2018;132:4355. [Google Scholar]
- 73.Rao X, Huang X, Zhou Z, Lin X. An improvement of the 2(−ΔΔCT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinforma. Biomath. 2013;3:71–85. [PMC free article] [PubMed] [Google Scholar]
- 74.Chao T. C. & Martin N. CompuSyn for Drug Combinations: PC Software and User’s Guide: A Computer Program for Quantitation of Synergism and Antagonism in Drug Combinations, and the Determination of IC50 and ED50 and LD50 Values (ComboSyn Inc, 2005).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.