

Summary
Metabolic reprogramming in cancer cells can create metabolic liabilities. KEAP1-mutant lung cancer is refractory to most current therapies. Here we show that KEAP1 deficiency promotes glucose dependency in lung cancer cells, and KEAP1-mutant/deficient lung cancer cells are more vulnerable to glucose deprivation than their WT counterparts. Mechanistically, KEAP1 inactivation in lung cancer cells induces constitutive activation of NRF2 transcription factor and aberrant expression of NRF2 target cystine transporter SLC7A11; under glucose limitation, high cystine uptake in KEAP1-inactivated lung cancer cells stimulates toxic intracellular disulfide buildup, NADPH depletion, and cell death, which can be rescued by genetic ablation of NRF2-SLC7A11 axis or treatments inhibiting disulfide accumulation. Finally, we show that KEAP1-inactivated lung cancer cells or xenograft tumors are sensitive to glucose transporter inhibitor. Together, our results reveal that KEAP1 deficiency induces glucose dependency in lung cancer cells and uncover a therapeutically relevant metabolic liability.
Subject areas: physiology, cell biology, cancer
Graphical abstract
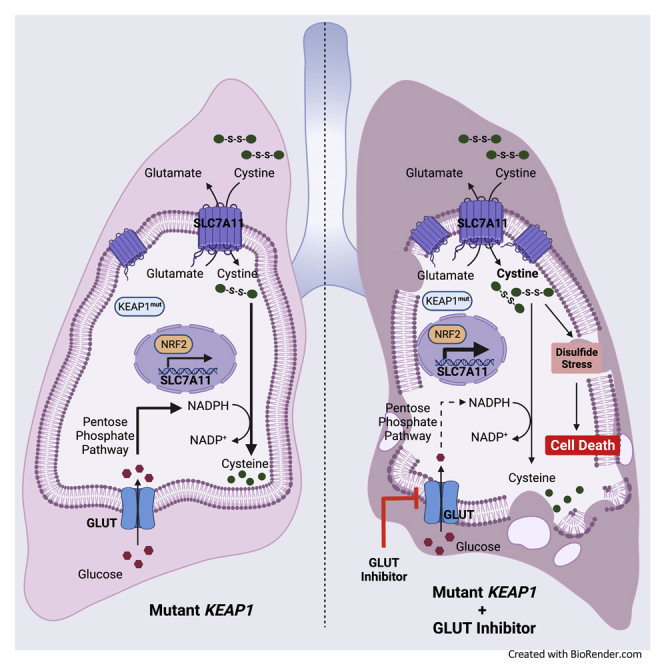
Highlights
-
•
Mutant KEAP1 exposes a metabolic liability provoked by high cystine uptake
-
•
KEAP1-NRF2-SLC7A11 axis drives glucose dependency
-
•
KEAP1 mutant NSCLC tumors are sensitive to GLUT inhibitor
Physiology; Cell biology; Cancer
Introduction
Nutrient flexibility is defined as the ability of cells to adapt to changing metabolic environment to maintain cellular homeostasis encompassing survival and proliferation. Specific genetic alterations in cancer cells can reprogram their metabolic networks to render them dependent on particular nutrients for survival or growth, thereby limiting their nutrient flexibility, which is commonly known as nutrient dependency (Pavlova and Thompson, 2016). Mechanistic understanding of nutrient dependency in cancer cells may offer attractive therapeutic strategies in cancer treatment, as it suggests that drugs that impair nutrient uptake or metabolism may be effective in killing or arresting tumor cells that are particularly dependent on corresponding nutrients while sparring normal cells (Wolpaw and Dang, 2018). A widely recognized therapeutic regimen that targets nutrient dependency is the use of asparaginase to treat patients with acute lymphoblastic leukemia (ALL) (Narta et al., 2007). Unlike normal cells, ALL cells cannot synthesize asparagine because they lack the corresponding catalytic enzyme asparagine synthetase and thus are reliant on extracellular asparagine for proliferation. However, to date there has been limited success in targeting nutrient dependency in solid tumors.
Metabolic reprogramming in cancer cells also increases oxidative stress (Hayes et al., 2020). To maintain the redox balance, cancer cells upregulate their antioxidant programs through a diverse array of mechanisms, prominent among which is the activation of the transcription factor NRF2 (Chio and Tuveson, 2017; Hayes et al., 2020; Lee et al., 2017). Under basal unstressed conditions, NRF2 is targeted for KEAP1-Cullin3 ubiquitin ligase-mediated polyubiquitination and proteasomal degradation (Baird and Yamamoto, 2020; Kensler et al., 2007; Sykiotis and Bohmann, 2010). Oxidative stress impairs NRF2 degradation by the KEAP1-Cullin3 ubiquitin ligase complex. The stabilized NRF2 subsequently translocates into the nucleus, binds to antioxidant response elements in gene promoter regions, and regulates the transcription of a host of target genes involved in the antioxidant defense and cellular redox maintenance (Kensler et al., 2007; Rojo de la Vega et al., 2018; Sykiotis and Bohmann, 2010). One critical transcriptional target of NRF2 in cellular antioxidant defense is solute carrier family 7 member 11 (SLC7A11, also called xCT), a cystine-glutamate antiporter (Koppula et al., 2018; Sato et al., 1999). Cancer cells obtain cysteine mainly through the uptake of extracellular cystine—an oxidized cysteine dimer—via SLC7A11 (Sato et al., 1999). Cysteine then serves as a critical precursor for glutathione (GSH), as well as other metabolites with antioxidant functions, such as hydrogen sulfide and taurine (Combs and DeNicola, 2019). SLC7A11 is overexpressed in many cancers, including KEAP1-mutant lung cancer (Koppula et al., 2020). Pancancer analysis shows that KEAP1 is mutated in about 3.93% of all cancers, with the highest frequency of mutation in NSCLC at 15.8%. Another mode of alterations in KEAP1 expression in cancers is its epigenetic control. KEAP1 promoter methylation has also been reported in a number of cancers including NSCLC, malignant gliomas, primary breast cancer, colorectal cancer and clear cell renal cancer (DeNicola et al., 2011; Huang et al., 2015; Krall et al., 2017; Wang et al., 2017; Yamadori et al., 2012). Several of these tumor tissues also exhibit a concomitant increase in NRF1 protein expression. KEAP1 is highly mutated in non-small cell lung cancer (NSCLC), including up to 20% of lung adenocarcinoma (Cancer Genome Atlas Research Network, 2014) and 12% of lung squamous cell carcinoma (Cancer Genome Atlas Research Network, 2012). Patients with lung cancer with KEAP1 mutations have shorter overall survival, and KEAP1-mutant lung cancers are resistant to most current therapies, such as radiotherapy and immunotherapy (Arbor et al., 2018; Jeong et al., 2017), underscoring the urgent need to develop effective targeted therapies to treat patients with this type of lung cancer.
In this study, we show that KEAP1 mutation or deficiency in lung cancer cells promotes glucose dependency by promoting NRF2-dependent SLC7A11 expression and inducing disulfide stress–associated cellular toxicity under glucose limiting conditions. We further show that KEAP1-mutant or -deficient lung cancer cells or tumors are sensitive to GLUT inhibitor KL-11743, which represents a series of compounds currently progressing toward clinical development. Our study therefore identifies a targetable metabolic liability in KEAP1-mutant lung cancer which can be potentially exploited in cancer therapy for treating this type of largely incurable disease.
Results
KEAP1 deficiency promotes glucose dependency in lung cancer cells at least partly through SLC7A11
To study the potential role of KEAP1 in regulating nutrient dependency, we compared the survival rates of a panel of KEAP1-wild-type (WT) and -mutant lung cancer cells in medium deprived of specific nutrients. As expected, KEAP1-mutant lung cancer cells exhibited markedly increased NRF2 levels compared with KEAP1-WT lung cancer cells (Figure 1A). We found that KEAP1-mutant lung cancer cells (H2126, H460, and A549 cells) were more dependent on glucose (i.e., more sensitive to glucose starvation-induced cell death) than KEAP1-WT counterparts (H1299, H23, and H1703 cells) (Figure 1B). Importantly, the glucose dependency phenotype in these lung cancer cell lines correlated with the mutation status of KEAP1 but not of other lung cancer genes (Table S1).
Figure 1.

KEAP1 deficiency promotes glucose dependency in lung cancer cells mainly through SLC7A11
(A) Protein levels of KEAP1, NRF2, and SLC7A11 in different NSCLC cell lines were determined by Western blotting. Vinculin is used as a loading control.
(B) Cell death upon glucose (Glc) starvation for 0–24 hr in different NSCLC cell lines was analyzed by PI staining.
(C) Western blotting analysis of KEAP1, NRF2, and SLC7A11 protein levels in the control (sgC) and KEAP1 knockout (sg1/2/3) H1299 cells.
(D) Cell death in KEAP1 knockout H1299 cells upon glucose withdrawal was analyzed by PI staining.
(E) Western blotting analysis of KEAP1, NRF2, and SLC7A11 protein levels in KEAP1-overexpressing H460 cells. EV, empty vector, OE, KEAP1 overexpressing.
(F) Cell death in KEAP1-overexpressing H460 cells upon glucose withdrawal was analyzed by PI staining.
(G) Western blotting analysis of KEAP1, NRF2, and SLC7A11 protein levels in NRF2-KEAP1 double knockout H1299 cells.
(H) Cell death in NRF2-KEAP1 double knockout H1299 cells upon glucose withdrawal was analyzed by PI staining.
(I) Correlation analysis of SLC7A11 expression in KEAP1 WT vs mutant LUAD tumors from TCGA.
(J) Correlation analysis of SLC7A11 expression with KEAP1 mutation status of NSCLC in CCLE database.
(K) Western blotting analysis of KEAP1, NRF2, and SLC7A11 protein levels in SLC7A11-KEAP1 double knockout H1299 cells.
(L) Cell death in SLC7A11-KEAP1 double knockout H1299 cells upon glucose withdrawal was analyzed by PI staining.
(M) Cell death in SLC7A11-KEAP1 double knockout H1299 cells upon glucose withdrawal supplemented with erastin was analyzed by PI staining.
(N) Western blotting analysis of SLC7A11 protein levels in SLC7A11 knockdown (sh1/2) H460 cells.
(O) Cell death upon glucose withdrawal was analyzed by PI staining in SLC7A11 knockdown H460 cells.
(P) SLC7A11 protein levels in SLC7A11-overexpressing H1299 cells. EV, empty vector, OE, SLC7A11 overexpressing.
(Q) Cell death in SLC7A11-overexpressing H1299 cells upon glucose withdrawal was analyzed by PI staining.
Data are represented as mean ± SD; n = 3. ∗, p < 0.05, ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
To study whether KEAP1 plays a causal role in regulating glucose dependency in lung cancer cells, we generated KEAP1-knockout (KO) H1299 cells (a KEAP1-WT lung cancer cell line) using CRISPR-Cas9 technology. KEAP1 deficiency in H1299 cells markedly increased NRF2 levels (Figure 1C) and rendered these cells more dependent on glucose (Figure 1D). We made similar observations by deleting KEAP1 in H23 cells (Figures S1A and S1B; of note, H23 cells harbor a KEAP1 silent mutation and therefore still express functional KEAP1 protein (Singh et al., 2006), which was confirmed by NRF2 upregulation caused by KEAP1 deletion in this cell line.) Conversely, overexpression of WT KEAP1 in KEAP1-mutant H460 cells (to the level slightly lower than that of endogenous mutant KEAP1) decreased NRF2 levels and attenuated glucose dependency (i.e., rendered cells less sensitive to glucose starvation-induced cell death) (Figures 1E and 1F). Together, our results suggest that KEAP1 deficiency promotes glucose dependency in lung cancer cells.
We next sought to understand the mechanisms by which KEAP1 regulates glucose dependency in lung cancer cells. Considering that NRF2 is a major effector of KEAP1 (Lee et al., 2017; Rojo de la Vega et al., 2018), we examined the potential relevance of NRF2 to KEAP1 regulation of glucose dependency. As shown in Figures 1G and 1H, NRF2 deletion reversed the enhanced glucose dependency in KEAP1-KO H1299 cells, suggesting that KEAP1 deficiency promotes glucose dependency mainly through NRF2 in lung cancer cells. This further raised the question of which NRF2 transcriptional target(s) might mediate glucose dependency in KEAP1-deficient lung cancer cells. One obvious NRF2 target linking the KEAP1-NRF2 signaling to glucose metabolism is glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the pentose phosphate pathway (PPP) (Mitsuishi et al., 2012). We confirmed that KEAP1 deletion increased G6PD levels; however, G6PD deficiency in KEAP1-KO cells further promoted glucose starvation–induced cell death (Figures S1Cand S1D), rather than attenuated it as did NRF2 deficiency (see Figures 1G and 1H). These data are consistent with the known role of the PPP in maintaining cell survival (Liu et al., 2020b) but does not support a role of G6PD as the NRF2 target to mediate glucose dependency in KEAP1-deficient lung cancer cells.
We and others previously reported that cystine transporter SLC7A11 (also known as xCT) promotes glucose dependency in cancer cells (Goji et al., 2017; Joly et al., 2020; Koppula et al., 2017; Liu et al., 2020b; Shin et al., 2017). SLC7A11 is a well-established NRF2 transcriptional target (Rojo de la Vega et al., 2018; Sasaki et al., 2002). Consistent with this, analyses of data sets from The Cancer Genome Atlas (TCGA) or Cancer Cell Line Encyclopedia (CCLE) revealed that KEAP1-mutant lung tumors (or lung cancer cell lines) exhibit significantly higher expression of SLC7A11 than do KEAP1-WT counterparts (Figures 1I and 1J and Table S2). We confirmed that KEAP1-mutant or -KO lung cancer cells used in our study exhibited markedly increased SLC7A11 levels (see Figures 1A and 1C, and NRF2 deficiency reversed the increased SLC7A11 expression in KEAP1-KO H1299 cells (Figure 1G). Importantly, similar to NRF2 deficiency (Figure 1H), deleting SLC7A11 in KEAP1-KO H1299 or H23 cells largely reversed the increased glucose dependency caused by KEAP1 deficiency (Figures 1K, 1L, S1E, and S1F). Treatment with SLC7A11 inhibitor erastin in KEAP1-KO H1299 cells had similar effects (Figure 1M; of note, as expected, KEAP1 deficiency rendered cells to be more resistant to erastin-induced ferroptosis). Likewise, SLC7A11 knockdown in KEAP1-mutant H460 cells decreased glucose starvation-induced cell death (Figures 1N and 1O). Conversely, SLC7A11 overexpression in H1299 or H23 cells (which express KEAP1 WT protein and exhibit low SLC7A11 expression) promoted glucose dependency (Figures 1P, 1Q, S1G, and S1H). Collectively, our data suggest that KEAP1 deficiency promotes lung cancer cell dependency on glucose at least partly through SLC7A11.
Glucose dependency in KEAP1-deficient lung cancer cells is partly caused by increased NADPH consumption associated with SLC7A11-mediated cystine uptake
We recently showed that SLC7A11-mediated cystine uptake and its subsequent reduction to cysteine presents a substantial drain on cellular NADPH reservoir, leading to increased dependency of cancer cells with high SLC7A11 expression (SLC7A11high) on the glucose-PPP route for NADPH supply; glucose starvation causes NADPH depletion and marked accumulation of cystine and other disulfide molecules, resulting in rapid cell death in SLC7A11high cancer cells (Liu et al., 2020b). This raised the question of whether the glucose dependency phenotype observed in KEAP1-deficient lung cancer cells can be attributed to similar mechanisms as revealed in SLC7A11high cancer cells (of note, the SLC7A11high cancer cells used in our previous study do not harbor KEAP1 or NRF2 mutation). It is important to note that, although KEAP1-deficient lung cancer cells generally exhibit high SLC7A11 expression, KEAP1 deficiency is not equivalent to SLC7A11 overexpression on regulating NADPH homeostasis, because KEAP1 deficiency also increases the expression of several NADPH-generating enzymes, such as G6PD, 6-phosphogluconate dehydrogenase (PGD), malic enzyme 1 (ME1), and isocitrate dehydrogenase 1 (IDH1), all of which are NRF2 targets and presumably would buffer the increased NADPH consumption caused by cystine reduction upon SLC7A11 overexpression (Lee et al., 2017).
We first confirmed that KEAP1-mutant lung cancer cells exhibited significantly increased cystine uptake than KEAP1-WT lung cancer cells (Figure 2A). Likewise, KEAP1 deletion in lung cancer cells increased cystine uptake (Figures 2B and S2A); interestingly, glucose starvation also increased cystine uptake, resulting in a further increase of cystine uptake in KEAP1-KO H1299 cells under glucose starvation (Figure 2B), which correlated with a KEAP1-independent effect of glucose starvation on inducing NRF2 and SLC7A11 levels (Figures 2C and 2D). Conversely, KEAP1 restoration in KEAP1-mutant H460 cells decreased cystine uptake (Figure S2B). Furthermore, deleting NRF2 or SLC7A11 largely abolished the increased cystine uptake in KEAP1-KO H1299 cells (Figures 2E and 2F), suggesting that KEAP1 deficiency promotes cystine uptake mainly through NRF2-SLC7A11.
Figure 2.

Glucose dependency of KEAP1 deficient cells is partly due to SLC7A11 mediated increased cystine uptake mediated NADPH consumption
(A) Cystine uptake levels in different NSCLC cells. DPM, disintegrations per min.
(B) Cystine uptake levels in KEAP1 knockout H1299 cells upon control (Glc(+)) and glucose starvation (Glc(−)) conditions.
(C) Western blotting analysis of KEAP1, NRF2, SLC7A11 protein levels in KEAP1 knockout H1299 cells.
(D) qRT-PCR analysis of SLC7A11 in KEAP1 knockout H1299 cells upon glucose starvation.
(E and F) Cystine uptake in NRF2 - KEAP1 double knockout H1299 (E) or SLC7A11-KEAP1 double knockout cells (F).
(G, H, and J), NADP+/NADPH ratios in different NSCLC cells (G), KEAP1 knockout H1299 cell (H), and SLC7A11-KEAP1 double knockout H1299 cells (J) cultured with glucose (Glc(+)) or without glucose (Glc(−)).
(I) Correlation analysis of NADP+ metabolite abundance with KEAP1 mutation status of NSCLC cell lines in the CCLE database.
(K and L) NADP+/NADPH ratios (K) and cell death analysis (L) of H1299 KEAP1 knockout cells cultured in complete medium or glucose-free medium with and without 2 mM 2-deoxy glucose (2DG) for 24 hr.
(M and N), NADP+/NADPH ratios (M) and cell death analysis (N) of H1299 KEAP1 knockout cells cultured in complete medium or glucose-free medium with and without 1 mM tris(2-carboxyethyl)phosphine (TCEP) for 24 hr.
Data are represented as mean ± SD; n = 3. ∗, p < 0.05, ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
As expected, glucose starvation increased cellular NADP+/NADPH ratio (which corresponds to NADPH depletion); importantly, under glucose starvation, KEAP1-mutant or -deficient lung cancer cells exhibited much more dramatic NADPH depletion than KEAP1-WT cells (Figures 2G, 2H, and S2C). Analysis of metabolomic datasets from CCLE (Li et al., 2019) also revealed higher levels of NADP+ in KEAP1-mutant cancer cells than in KEAP1-WT ones (Figure 2I). Further, SLC7A11 deletion in KEAP1-deficient H1299 cells largely restored NADPH levels under glucose starvation (Figure 2J). Conversely, KEAP1 overexpression in KEAP1-mutant H460 cells alleviate NAPDH depletion by glucose starvation (Figure S2D). These data suggest that, while KEAP1 deficiency leads to the increased expression of genes involved in both NADPH generation (such as PPP enzymes and ME1) and consumption (such as SLC7A11), it seems that NADPH consumption caused by SLC7A11-associated cystine metabolism exceeds NADPH generation in KEAP1-deficent cells, resulting in more NADPH depletion under glucose limiting conditions.
Next, to establish the causal role of NADPH depletion in glucose dependency in KEAP1-KO lung cancer cells, we tested whether supplying NADPH by 2-deoxyglucose (2DG) treatment can rescue glucose starvation-induced cell death in KEAP1-KO lung cancer cells. 2DG is a glucose analog, with the 2-hydroxyl group in glucose replaced by a hydrogen in 2DG. Similar to glucose, 2DG can still be shunted to the G6PD reaction in the PPP to generate NADPH (Liu et al., 2020b), but unlike glucose, it cannot be further oxidized by the PGD reaction in the PPP to support nucleic acid biosynthesis or into the glycolysis pathway downstream of the phosphoglucose isomerase reaction; thus, 2DG uncouples NADPH production by the oxidative PPP from energy generation by glycolysis. We showed that 2DG treatment rescued NADPH levels and correspondingly restored cell survival in KEAP1-KO cells under glucose starvation (Figures 2K and 2L). We also tested the potential rescuing effect of adding reducing agent tris(2-carboxyethyl)phosphine (TCEP) into the medium, which enables cells to obtain cysteine from medium independent of SLC7A11-mediated cystine transport (and consequently, there is no need to increase NADPH supply for intracellular cystine reduction). In support of our hypothesis, we found that TCEP treatment also rescued NADPH depletion and cell death in KEAP1-KO cells under glucose starvation (Figures 2M and 2N). Further analyses confirmed that TCEP treatment did not affect cyst(e)ine (cystine and cysteine) uptake in control and KEAP1-KO cells (Figure S2E). Taken together, our data suggest that high SLC7A11 expression and its associated cystine metabolism have a significant demand for NADPH supply, resulting in glucose dependency in KEAP1-deficient lung cancer cells. It should be noted that there was no significant difference in growth rates between KEAP1-deficient/-mutant lung cancer cells and their WT counterparts (Figures S2F and S2G), thereby ruling out the possibility that the increased glucose dependency observed in KEAP1- deficient/-mutant lung cancer cells is simply caused by their high growth rates.
Glucose starvation leads to accumulation of disulfide molecules but not cystine in KEAP1-deficient lung cancer cells
We recently showed that NADPH depletion in SLC7A11high cancer cells under glucose starvation is accompanied with redox system collapse characterized by depletion of reduced GSH but drastic accumulation of cystine and other disulfide molecules including γ-glutamylcystine, glutathionylcysteine, and oxidized glutathione (GSSG) (Liu et al., 2020b, 2020c). We conducted similar thiol analyses in KEAP1 deficient lung cancer cells. As expected, KEAP1 deficiency in H1299 cells dramatically increased GSH levels under glucose replete conditions (Figure 3A). Consistent with this, KEAP1-mutant cancer cells exhibited higher GSH levels than KEAP1-WT cancer cells from CCLE (Figure S3A). Glucose starvation depleted GSH but markedly increased the levels of GSSG, γ-glutamylcystine, and glutathionylcysteine in KEAP1-KO cells (Figures 3A–3D). As expected, KEAP1 deficiency significantly increased intracellular cysteine levels under either glucose replete or depletion condition (Figure 3E). Surprisingly, cystine levels did not apparently accumulate in KEAP1-KO cells under glucose starvation (Figure 3F).
Figure 3.

KEAP1 deletion in lung cancer cells leads to buildup of disulfide molecules but not cystine upon glucose starvation
(A–F) The concentrations or relative fold changes of glutathione (GSH; (A), oxidized glutathione (GSSG; (B), γ-glutamylcystine (C), glutathionylcysteine (D), Cysteine (E), and Cystine (F) in KEAP1 KO H1299 cells upon glucose starvation.
(G–L) The concentrations or relative fold changes of GSSG (G), γ-glutamylcystine (H), glutathionylcysteine (I), GSH (J), cysteine (K), and cystine (L) in KEAP1 KO H1299 cells upon glucose starvation supplemented with either 2-DG or TCEP.
Data are represented as mean ± SD; n = 3. ∗, p < 0.05, ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
We also conducted thiol analyses in KEAP1-mutant H460 cells with KEAP1 restoration, which revealed that glucose starvation markedly depleted GSH and resulted in drastic accumulation of disulfides in H460 cells; KEAP1 restoration attenuated glucose starvation-induced disulfide accumulation of cystine, γ-glutamylcystine, and glutathionylcysteine (Figures S3B–S3G). The relative moderate phenotype from KEAP1-restored cells likely relates to the low level of ectopically expressed KEAP1 in H460 cells; correspondingly, KEAP1 restoration only moderated decreased NRF2 and SLC7A11 in H460 cells (see Figure 1E).
Since our data showed that 2DG or TCEP treatment rescued NADPH depletion and cell death in KEAP1-KO H1299 cells under glucose starvation (Figures 2K–2N), we further characterized the effect of 2DG or TCEP treatment on thiol profiles in KEAP1-KO cells under glucose starvation. As shown in Figures 3G–3I, 2DG or TCEP treatment totally rescued the accumulation of GSSG, γ-glutamylcystine, or glutathionylcysteine in KEAP1-KO cells under glucose starvation. 2DG also largely restored GSH levels, whereas TCEP only moderately increased GSH levels in KEAP1-KO cells under glucose starvation (Figure 3J). Consistent with the reducing effect of TCEP, we observed that TCEP treatment dramatically increased intracellular cysteine levels but depleted intracellular cystine (Figures 3K and 3L). Together, our data show that glucose starvation depletes GSH and results in accumulation of disulfides molecules in KEAP1-deficient lung cancer cells, and further suggest that the disulfide accumulation plays a causal role in inducing cell death in KEAP1-deficient lung cancer cells under glucose starvation.
Our results also reveal that, under glucose starvation, the redox system change in KEAP1-KO H1299 cells largely phenocopied that of SLC7A11 high cancer cells (with intact KEAP1 status) (Liu et al., 2020b) with one major difference, namely the lack of cystine accumulation in KEAP1-KO H1299 cells under glucose starvation. We reasoned that this discrepancy can potentially be explained by upregulation of additional NRF2 targets in KEAP1-deficient lung cancer cells (Lee et al., 2017): (1) upregulation of glutamate-cysteine ligase (GCL, the rate-limiting enzyme in GSH biosynthesis which consists of GCLM and GCLC) would promote cystine flux to γ-glutamylcystine and glutathionylcysteine under glucose starvation; Figure S4A), thereby preventing cystine accumulation in KEAP1-deficient lung cancer cells; (2) upregulation of presumed cystine reductases glutathione reductase (GSR) and thioredoxin reductase 1 (TXNRD1) would facilitate cystine reduction to cysteine and mitigate cystine accumulation under glucose starvation in KEAP1-deficient lung cancer cells. Indeed, KEAP1-mutant lung tumors or KEAP1-deficient H1299 cells exhibited increased expression of these NRF2 target genes (Figures S4B and S4C). Furthermore, treatment with buthionine sulfoximine (BSO; a GCL inhibitor), 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU; a GSR inhibitor), or auranofin (a TXNRD1 inhibitor) further promoted cell death in KEAP1-KO H1299 cells under glucose starvation (Figures S4D–S4F). However, these treatments did not increase cystine levels in KEAP1-KO cells under glucose starvation (Figure S4G), suggesting that the lack of cystine accumulation in KEAP1-KO H1299 cells is less likely caused by the increased expression of GCL, GSR, or TXNRD1. Another potential reason for the lack of cystine accumulation in KEAP1-KO H1299 cells is the more moderate upregulation of SLC7A11 in this cell line compared with that in other SLC7A11high cancer cells which exhibit drastic cystine accumulation under glucose starvation, such as UMRC6 cells (Liu et al., 2020b) (Figure S4H). Consistent with this, KEAP1 deletion in H1299 cells resulted in less potent cell death than that in UMRC6 cells under glucose starvation (Figure S4I).
KEAP1 deficiency sensitizes lung cancer cells or lung tumors to GLUT inhibition
Our aforementioned data prompted us to further test whether KEAP1-mutant lung cancer cells are sensitive to pharmacologic inhibition of glucose transporter (GLUT)-mediated glucose uptake. In our study, we tested KL-11743, a potent pan-GLUT1 and GLUT3 inhibitor developed by Kadmon (Liu et al., 2020a) (note that KL-11743 was named as compound 8 in this study), considering that both GLUT1 and GLUT3 are often co-expressed in tumors or cancer cell lines. We found that, compared to KEAP1-WT lung cancer cells, KEAP1-mutant lung cancer cells were more sensitive to KL-11743 (Figure 4A), which correlated with more NADPH depletion under KL-11743 treatment in KEAP1-mutant lung cancer cells (Figure 4B). Extending this correlative observation, we further showed that KEAP1 deletion sensitized H1299 cells to GLUT inhibition and that GLUT inhibition induced more NADPH depletion in KEAP1-KO H1299 cells than in their WT counterparts (Figures 4C and 4D). We made similar observations in H23 cells with KEAP1 deletion (Figures S5A and S5B). Conversely, KEAP1 restoration in KEAP1-mutant H460 cells alleviated KL-11743-induced cell death and NADPH depletion (Figures S5C and S5D). Unlike NRF2 target genes, GLUT1 or GLUT3 (also called SLC2A1 or SLC2A3, respectively) did not exhibit increased expression in KEAP1-mutant lung cancers (or lung cancer cells) compared with KEAP1-WT counterparts (Figures S5E–S5H). Consistent with this, KEAP1 deletion in H1299 cells did not significantly affect GLUT1 or GLUT3 levels (Figure S5I) or glucose uptake with or without KL-11743 treatment (Figure S5J), suggesting that the differential sensitivity to GLUT inhibition cannot be attributed to the differential GLUT expression or glucose uptake between KEAP1-WT and -KO cells. Using the same double KO cells we generated earlier, we further showed that NRF2 or SLC7A11 deletion largely abolished the increased GLUT inhibition-induced cell death or NADPH depletion in KEAP1-KO lung cancer cells (Figures 4F–4I and S5K). Likewise, treatment with SLC7A11 inhibitor erastin had similar effects (Figure S5L). Therefore, mirroring our data from glucose starvation, our results show that KEAP1 deficiency sensitizes lung cancer cells to GLUT inhibition largely through the NRF2-SLC7A11 axis.
Figure 4.

KEAP1 deficiency sensitizes lung cancer cells or lung tumors to GLUT inhibition
(A) Cell viability in different NSCLC cell lines upon KL-11743 treatment was analyzed by CCK8.
(B) NADP+/NADPH ratios in different NSCLC cell lines upon KL-11743 treatment.
(C and D) Cell viability in KEAP1 KO H1299 cell lines upon KL-11743 treatment was analyzed by CCK8 (C) or PI staining (D).
(E) NADP+/NADPH ratios in control and KEAP1 KO H1299 cell lines upon KL-11743 treatment.
(F and G) Cell death in NRF2-KEAP1 double knockout H1299 cells (F) or SLC7A11-KEAP1 double knockout H1299 cells (G) upon KL-11743 treatment was analyzed by PI staining.
(H and I), NADP+/NADPH ratios in NRF2-KEAP1 double knockout H1299 cells (H) and SLC7A11-KEAP1 double knockout H1299 cells (I) treated with KL-11743.
(J) Tumor volumes of control (sgC) or KEAP1-knockout (KEAP1 sg2) H1299 xenograft tumors at different times after treatment with KL-11743 or vehicle. Data are mean +/− SD; n = 7 independent repeats.
(K and L), Endpoint weights of sgC (K) or KEAP1 sg2 (L) H1299 xenograft tumors treated with KL-11743 or vehicle. Error bars are mean ± s.d., n = 7 independent repeats.
(M) Representative images of hematoxylin and eosin and cleaved caspase-3 or Ki67 immunohistochemical staining of sgC and KEAP1 sg2 H1299 xenograft tumors treated with KL-11743 or vehicle. The experiment was repeated twice, independently, with similar results. Scale bars, 20 μm.
(N and O) Immunochemistry scoring of cleaved caspase-3 (N) and Ki67 (O) staining. Error bars are means +/− SD, n = 8 randomly selected magnification fields.
Data are presented as (if mentioned otherwise) mean ± SD; n = 3. ∗, p < 0.05, ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Our aforementioned data suggest that KEAP1 deficiency sensitizes lung cancer cells to pharmacological inhibition of GLUTs. To further interrogate these concepts in vivo, we tested KL-11743 in xenograft models established from control and KEAP1-KO H1299 cells (sgC and KEAP1 sg2 tumors). KEAP1 deletion moderately promoted H1299 xenograft tumor development (Figure 4J; compare vehicle-treated groups between sgC and KEAP1 sg2 tumors), which is consistent with the tumor suppression function of KEAP1 in lung cancer (Romero et al., 2017). Analysis of xenograft tumor samples confirmed significantly increased SLC7A11 levels in KEAP1 KO xenograft tumors (Figure S5M). Importantly, while KL-11743 had negligible effects on the growth of control tumors with no obvious impact on end point tumor weight (Figures 4J and 4K), KL-11743 significantly suppressed the growth of KEAP1 KO tumors, resulting in dramatic reduction in tumor weight at endpoint (Figures 4J, 4L, and S5N). Immunohistochemical (IHC) analyses in these tumor samples revealed more cleaved caspase-3 staining in KEAP1-KO tumors than in control tumors treated with KL-11743, indicating that KL-11743 treatment induced more cell death in KEAP1-KO tumors than in control tumors (Figures 4M and 4N). While KEAP1-KO tumors exhibited increased cell proliferation compared with control tumors as measured by Ki-67 staining, KL-11743 treatment did not significantly affect tumor cell proliferation (Figures 4M and 4O). Finally, we showed that KL-11743 treatment did not have obvious adverse effects on animal weight throughout the treatment (Figures S5O and S5P), suggesting that the drug is well tolerated in animals.
Recently we showed that KL-11743 treatment selectively suppressed SLC7A11high tumor growth in lung cancer patient-derived xenografts (PDXs) (Liu et al., 2020b). As shown in Table S3, gene mutation analyses in these PDXs revealed that the two SLC7A11low PDXs that are insensitive to KL-11743 (TC393 and TC551) are KEAP1 WT; among the three SLC7A11high PDXs that are sensitive to KL-11743, TC333 and TC494 are KEAP1 mutant, whereas TC453 is KEAP1 WT (as well as NFE2L2 WT; NFE2L2 gene encodes NRF2). It is possible that the high expression of SLC7A11 in TC453 is driven by other mechanisms independent of the KEAP1-NRF2 signaling axis. It should be noted that KL-11743 sensitivity in these five PDX lines does not correlate with mutation status of other commonly mutated lung cancer genes, such as TP53, KRAS, or PIK3CA, except that there is somewhat correlation with STK11 mutation (Table S3), which is not surprising, considering that KEAP1 and STK11 are significantly co-mutated in lung adenocarcinoma (Skoulidis et al., 2015). Together, our results show that KEAP1 deficiency sensitizes lung cancer cells or tumors to GLUT inhibition and suggest to use GLUT inhibitors for the treatment of KEAP1 mutant lung cancer.
Discussion
NRF2-mediated antioxidant programs require cysteine to provide the key precursor for synthesizing GSH and other antioxidants. Owing to the high demand for antioxidant defense, cancer cells mainly rely on obtaining cysteine from extracellular environment through nutrient transporters. However, because of the highly oxidizing extracellular environment, extracellular cysteine is highly unstable, with only ~30-min half-life in culture medium (Ishii and Bannai, 1985), and quickly oxidized to cystine; consequently, cystine concentration is much higher than that of cysteine in extracellular space or culture medium. Therefore, cancer cells obtain cysteine mainly through SLC7A11-mediated cystine uptake. Consistent with this, SLC7A11 is a strong NRF2 transcriptional target (Sasaki et al., 2002). However, SLC7A11high cancer cells also have to endure a significant cost resulting from SLC7A11-mediated cystine uptake: the buildup of intracellular cystine and its derived disulfide molecules can potentially cause disulfide stress and be highly toxic to cells. This forces cells to quickly reduce cystine to cysteine (or disulfide molecules to their corresponding reduced metabolites) through NADPH-consuming reduction reactions. Correspondingly, NRF2 also upregulates the transcription of multiple NADPH-generating enzymes (such as G6PD, PGD, and malic enzymes) and reductases (such as GSR and TXNRD1) to buffer NADPH consumption caused by disulfide reduction (Lee et al., 2017).
Cytosolic NADPH is mainly supplied from glucose via the oxidative PPP (which generates NADPH through G6PD and PGD). Consequently, glucose starvation significantly limits NADPH supply in KEAP1-deficient cancer cells, despite that these cells express high levels of NADPH-generating enzymes and reductases; combining glucose starvation with high cystine uptake in KEAP1-deficient lung cancer cells leads to NADPH depletion, drastic accumulation of disulfide molecules (including GSSG, γ-glutamylcystine, and glutathionylcysteine), and ultimately cell death. While previous studies showed that glucose starvation induces massive accumulation of intracellular cystine (as well as these other disulfides mentioned above) in SLC7A11high cancer cells (which are non-lung cancer cells and do not harbor KEAP1 mutation) (Liu et al., 2020b; Yamaguchi et al., 2020), surprisingly, in this study, we found that glucose starvation does not induce obvious cystine accumulation in KEAP1-KO H1299 cells. The lack of cystine accumulation in this cell line is particularly striking given that glucose starvation actually increases cystine uptake in these cells (Figure 2B).
Apparently, the resulting redox defects caused by NADPH depletion and accumulation of other disulfide molecules can still cause substantial cell death in KEAP1-KO H1299 cells under glucose starvation, although the cell death is somewhat less potent than that in other SLC7A11high cancer cells which exhibit dramatic cystine accumulation under glucose starvation (Liu et al., 2020b) (Figure S4I). These observations suggest that (i) different reductase enzymes, potentially with different Km values for NADPH, likely govern the reduction of cystine and other disulfides, such that with the extent of NADPH depletion in KEAP1-KO H1299 cells under glucose starvation, cystine reductase(s) can still reduce cystine to cysteine whereas the reductases for other disulfides cannot function appropriately, resulting in selective accumulation of these other disulfides but not cystine in this context; (ii) other SLC7A11high cancer cells presumably would experience more severe “NADPH bankruptcy” under glucose starvation than do KEAP1-KO H1299 cells used in this study, likely because these other SLC7A11high cancer cells express higher SLC7A11 (see Figure S4H) and/or because the upregulation of other NADPH-generating enzymes (such as malic enzymes) by NRF2 activation helps alleviates the “NADPH bankruptcy” in KEAP1-KO H1299 cells under glucose deprivation. It should also be noted that the disulfide profiles in KEAP1-overexpressing H460 cells and KEAP1 KO H1299 cells are somewhat different: under glucose starvation, KEAP1-overexpressing H460 cells showed KEAP1-dependent changes in cystine, γ-glutamylcystine, and glutathionylcysteine but not GSSG, whereas KEAP1 KO H1299 cells exhibited KEAP1-dependent changes in γ-glutamylcystine, glutathionylcysteine, and GSSG but not cystine, suggesting that KEAP1 regulation of disulfide stress under glucose starvation could be context (cell line) dependent. Further studies will be directed to further address these important questions.
Previous studies also showed that KEAP1 deficiency promotes glutamine dependency at least partly through SLC7A11-mediated glutamate export, and KEAP1-mutant lung cancer cells or tumors are sensitive to glutaminase inhibitors (Galan-Cobo et al., 2019; Muir et al., 2017; Romero et al., 2017; Sayin et al., 2017). Consistent with this, we found that KEAP1 deficiency in H1299 or H23 cells decreased intracellular glutamate levels under basal, glucose starvation or GLUT inhbition condition (Figures S6A and S6B); importantly, SLC7A11 deletion reversed the decreased intracellular glutamate levels in KEAP1 KO H1299 cells (Figure S6C). Further, SLC7A11 overexpression, similar to KEAP1 deficiency, decreased intracellular glutamate levels in H1299 or H23 cells (Figures S6D and S6E). These data could suggest a model that, with decreased intracellular glutamate levels, more glucose is shunted through the glycolysis pathway to support the TCA cycle, leading to glucose dependency in KEAP1-deficient cells, which would further predict that KEAP1-deficient cells should be sensitive to 2DG (a glycolysis inhibitor), much like these cells are sensitive to GLUT inhibition. However, our data showed that 2DG actually completely rescued glucose starvation-induced cell death in KEAP1-deficient cells (see Figure 2L). Therefore, our current data does not appear to support this model.
While the exact contribution of SLC7A11-mediated glutamate export in determining glucose dependency in KEAP1-deficient/-mutant cells remains to be further studied, currently we favor a model that the metabolic underpinnings underlying the glucose or glutamine dependency phenotype in KEAP1-deficient lung cancer cells are different: while KEAP1 deficiency-induced glutamine dependency is primarily driven by the cellular need for maintaining anaplerosis (because SLC7A11high cancer cells export a large amount of intracellular glutamate), KEAP1 deficiency-induced glucose dependency mainly relates to redox maintenance resulting from cystine uptake (Koppula et al., 2020). Consequently, glucose starvation (or GLUT inhibition) mainly induces cell death in KEAP1-deficient lung cancer cells, whereas the major cellular defect in KEAP1-deficient lung cancer cells with glutamine starvation (or glutaminase inhibition) is cell growth arrest (Galan-Cobo et al., 2019; Romero et al., 2017; Sayin et al., 2017). Because of the different underlying mechanisms and resulting cellular phenotypes of these two nutrient dependencies in KEAP1-mutant lung cancer, it will be interesting to test whether combining GLUT and glutaminase inhibitors might achieve synergistic therapeutic effects for treating KEAP1-mutant lung cancer (as well as whether this combinatorial treatment will also cause more toxicities). In future preclinical studies, GLUT inhibitors should also be further combined with other standard of cares, such as immunotherapy and radiotherapy, for treating KEAP1-mutant lung cancer.
Limitations of study
We acknowledge the limitation of the NADPH measurement method used in our study, as discussed in previous studies (Lu et al., 2018). However, while this technique limitation could present a particular problem for quantitative flux modeling of NADPH metabolism, it should not change the direction and approximate magnitude of large and qualitative changes as shown in this study, and therefore should not affect our conclusion that KEAP1 deficiency promotes NADPH depletion under glucose starvation or GLUT inhibition condition. We also acknowledge the limited number of xenograft models tested in this study. Future work with additional preclinical models is needed to further explore therapeutically targeting glucose dependency in KEAP1-mutant lung cancer.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| KEAP1 | Santa Cruz Biotechnology | Cat# sc-365626, RRID:AB_10844829 |
| G6PD | Cell Signaling Technology | Cat# 12263 S, RRID:AB_2797861 |
| Vinculin | Sigma | Cat# V4505, RRID:AB_477617 |
| Myc | Cell Signaling Technology | Cat# 2276 S, RRID:AB_331783 |
| SLC7A11 | Cell Signaling Technology | Cat# 12691 S, RRID:AB_2687474 |
| GLUT1 | Thermo Fisher | Cat# PA5-16793, RRID:AB_10986893 |
| GLUT3 | Santa Cruz Biotechnology | Cat# sc-74497, RRID:AB_1124974 |
| NRF2 | Cell Signaling Technology | Cat# 12721 S, RRID:AB_2715528 |
| anti-Ki-67 (D2H10) | Cell Signaling Technology | Cat# 9027, RRID:AB_2636984 |
| anti-cleaved caspase-3 | Cell Signaling Technology | Cat# 9661, RRID:AB_2341188 |
| Biological samples | ||
| PDX TC393 | University of Texas MD Anderson Cancer Center | N/A |
| PDX TC551 | University of Texas MD Anderson Cancer Center | N/A |
| PDX TC333 | University of Texas MD Anderson Cancer Center | N/A |
| PDX TC494 | University of Texas MD Anderson Cancer Center | N/A |
| PDX TC453 | University of Texas MD Anderson Cancer Center | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Deoxy-D-glucose, 2-[1-14C]- | Perkin Elmer | Cat# NEC495A050UC |
| L-[1,2,1ʹ,2ʹ-14C]-cystine | Perkin Elmer | Cat# NEC854010UC |
| Buthionine Sulfoximine (BSO) | Cayman Chemicals | Cat# 14484 |
| 2DG | Sigma | Cat# D8375 |
| TCEP | Sigma | Cat# C4706 |
| Methylcellulose | Sigma | Cat# M0512-1 |
| TWEEN-80 | Sigma | Cat# P1754 |
| BCNU | Sigma | Cat# C0400 |
| Auranofin | Sigma | Cat# A6733 |
| Erastin | Cayman Chemicals | Cat# 571203-78-6 |
| Dialyzed Fetal Bovine Serum | Sigma | Cat# F0392 |
| KL-11743 | Kadmon Corp. | N/A |
| Propidium Iodide | Roche | Cat# 11348639001 |
| G6PD enzyme | Sigma | Cat# G4134 |
| DMEM without glucose | Thermo Fisher | Cat# 11966 |
| RPMI-1640 | Sigma | Cat# R8758 |
| Penicillin-Streptomycin | Life Technologies | Cat# 15140-122 |
| DMEM | Sigma | Cat# D6429 |
| DPBS | Sigma | Cat# D8537 |
| NADPH Tetrasodium Salt | Roche | Cat# 10107824001 |
| Critical commercial assays | ||
| cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 43-688-14 |
| QuantiTect SYBR Green PCR Kit | Qiagen | Cat# 204143 |
| TaqMan™ Universal PCR Master Mix | Applied Biosystems | Cat# 4305719 |
| CCK8 | Dojindo | Cat# CK0413 |
| Experimental models: cell lines | ||
| H1299 | Laboratory of Bingliang Fang | N/A |
| H23 | Laboratory of Bingliang Fang | N/A |
| H1703 | Laboratory of Bingliang Fang | N/A |
| H2126 | ATCC | Cat# CRL-5925, RRID:CVCL_1532 |
| H460 | Laboratory of Bingliang Fang | N/A |
| A549 | ATCC | Cat# CRL-7909, RRID:CVCL_0023 |
| HEK 293T | ATCC | CRL-11268; RRID: CVCL_1926 |
| UMRC6 | Laboratory of W. G. Kaelin | N/A |
| H1299 KEAP1 knockout cell line | This paper | N/A |
| H23 KEAP1 knockout cell line | This paper | N/A |
| H460 KEAP1 overexpressing cell line | This paper | N/A |
| H1299 KEAP1 & NRF2 double knockout cell line | This paper | N/A |
| H1299 KEAP1 & SLC7A11 double knockout cell line | This paper | N/A |
| H23 KEAP1 & SLC7A11 double knockout cell line | This paper | N/A |
| H1299 KEAP1 knockout G6PD knockdown cell line | This paper | N/A |
| H460 SLC7A11 knockdown cell line | This paper | N/A |
| H1299 SLC7A11 overexpressing cell line | This paper | N/A |
| H23 SLC7A11 overexpressing cell line | This paper | N/A |
| Experimental models: organisms/strains | ||
| Mouse: athymic nude mice (female, 4-6 weeks old, homozygous for Foxn1 nu) | Experimental Radiation Oncology Breeding Core Facility at MD Anderson Cancer Center | N/A |
| Oligonucleotides | ||
| RT-PCR primers (Table S4) | This paper | N/A |
| SLC7A11-sg3 5’-CACCGATGAGCTTGATCGCAAGTTC-3’ | This paper | N/A |
| SLC7A11-sg4 5’-CACCGAAGTATTACGCGGTTGCCAC-3’ | This paper | N/A |
| NRF2-sg2 5’-CACCGTTACAACTAGATGAAGAGAC-3’ | This paper | N/A |
| NRF2-sg3 5’-CACCGCAGATCCACTGGTTTCTGAC-3’ | This paper | N/A |
| sgC 5’-CACCGGCACTACCAGAGCTAACTCA-3’ | This paper | N/A |
| KEAP1-sg1 5’-CACCGCTTGTGGGCCATGAACTGGG-3’ | This paper | N/A |
| KEAP1-sg2 5’-CACCGTGTGTCCTCCACGTCATGAA-3’ | This paper | N/A |
| KEAP1-sg3 5’-CACCGGAGGACACACTTCTCGCCCA-3’ | This paper | N/A |
| Recombinant DNA | ||
| LentiCRISPR-V2 | Addgene | Cat# 52961 |
| pGIPZ-shSLC7A11-1 | MD Anderson Cancer Center shRNA and ORFeome Core Facility | Cat# V2LHS_204910 |
| pGIPZ-shSLC7A11-2 | MD Anderson Cancer Center shRNA and ORFeome Core Facility | Cat# V2LHS_251161 |
| pGIPZ-shG6PD-1 | MD Anderson Cancer Center shRNA and ORFeome Core Facility | Cat# V2LHS_92826 |
| pGIPZ-shG6PD-2 | MD Anderson Cancer Center shRNA and ORFeome Core Facility | Cat# V3LHS_315154 |
| pLVX-M-puro | Zhang et al., 2018 | Plasmid# 125839, RRID:Addgene_125839 |
| pLVX-M-blast SLC7A11 | Liu et al., 2020b, 2020c | N/A |
| pLVX-M-puro KEAP1 | This paper | N/A |
| Software and algorithms | ||
| GraphPad | GraphPad | https://www.graphpad.com, RRID:SCR_002798 |
| ImageJ | (Schindelin et al., 2012) | https://imagej.net/Fiji/, RRID:SCR_003070 |
| Cancer Dependency Map Portal | Cancer Dependency Map Portal | https://depmap.org/portal/, RRID:SCR_017655 |
| FlowJo | FlowJo |
https://www.flowjo.com/solutions/flowjo, RRID:SCR_008520 |
| BD Accuri C6 Plus | BDBiosciences | https://www.bdbiosciences.com/us/instruments/research/cell-analyzers/bd-accuri/m/1294932/features/software, RRID:SCR_014422 |
| UCSC Xena | UCSC Xena | http://xena.ucsc.edu/, RRID:SCR_018938 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Boyi Gan (bgan@mdanderson.org)
Materials availability
All unique reagents generated in this study are available from the lead contact with a completed Material Transfer Agreement.
Data and code availability
This study did not generate such unique reagents.
Experimental model and subject details
H1299, H23, H460, H1703, A549 and H2126 were cultured in a 37°C incubator in a 5% CO2 atmosphere. A549 and H2126 cells were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum and 10,000 U/mL of penicillin-streptomycin. H460, H1299, H1703, HCC-193 and H23 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum and 10,000 U/mL of penicillin-streptomycin. For glucose- or cystine-deprivation experiments, cells were cultured in glucose and/or cystine-free DMEM with dialyzed FBS as previously described (Chauhan et al., 2019; Lee et al., 2020; Lin et al., 2014b). Glucose-free DMEM was obtained from Life Technologies (no. 11966-025).
Method details
Constructs and generation of overexpression, knockdown, or knockout cell lines
CRISPR-mediated knockout plasmids containing guide RNAs targeting KEAP1, NRF2 and SLC7A11 were generated in LentiCRISPR-V2 (Addgene, #52961) according to the standard protocol. The sequences of G6PD shRNAs and guide RNAs are listed in Table S4. The expression vectors containing SLC7A11 cDNA are described in our previous publication (Koppula et al., 2017). The KEAP1 cDNA was a kind gift from Dr. Bing Xia at Rutgers Cancer Institute of New Jersey. The KEAP1 cDNA was subsequently cloned into the lentivirus expression vector pLVX-Puromycin or Blasticidin stable cell lines were generated as described previously (Lei et al., 2020; Zhang et al., 2020).
Real-time PCR
Real-time PCR was performed as described previously (Dai et al., 2017; Lee et al., 2016). Briefly, total RNA was extracted from cells using RNeasy (Qiagen), and first-strand cDNA was prepared with a high-capacity cDNA reverse transcription kit (Applied Biosystems, ABI). Real-time PCR was subsequently performed using the QuantiTect SYBR Green PCR kit (Qiagen) or TaqMan Universal PCR Master Mix (ABI) and was run on Stratagene MX3000P. For quantification of gene expression, the 2-ΔΔCt method was used. β-Actin expression was used for normalization. The sequences of all the primers are listed in Table S4.
Cell death and viability assays
To measure cell death or cell viability, cells were seeded into 12 well plates or 96 well plates 1 day before treatment. Cell death was measured by PI staining followed by FACS analysis. Following appropriate treatment, cells were trypsinized and resuspended in 100 μl of PBS containg 2μg/ml of PI. The cells were incubated for 30 min at room temperature and analyzed with an LSRII flow cytometer (BD Biosciences). Cell viability was measured using the CCK8 kit. Following appropriate treatment, media was replaced with media containing CCK8 reagent. The plate was incubated for 1 hr at 37°C in the Co2 incubator. Cell viability was measured by microplate reader (FLUOstar Omega, BMG Labtech).
NADP+ and NADPH measurement
The intracellular levels of NADPH and total NADP (NADPH + NADP+) were measured as previously described (Liu et al., 2020b; Wagner and Scott, 1994). Briefly, cells were cultured in six-well plates overnight. On the next day, the cells were lysed in 300 μl extraction buffer (20 mM nicotinamide, 20 mM NaHCO3 and 100 mM Na2CO3), centrifuged, and supernatant was split into two 150 μl aliquots. For total NADP measurement, 20 μl cell supernatant was added from one 150 μl aliquot into a 96-well plate and mixed with 80 μl of NADP-cycling buffer (100 mM Tris-HCl pH8.0, 0.5 mM thiazolyl blue, 2 mM phenazine ethosulfate and 5 mM EDTA) containing 0.75 U of G6PD enzyme (Sigma, no. G4134). After 1 min incubation in the dark at 30 °C, 20 μl of 10 mM glucose-6-phosphate was added to the mixture, and the change in absorbance at 570 nm was measured every 1 min for 6 min at 30 °C with a microplate reader. For NADPH measurement, the other 150 μl supernatant was incubated at 60 °C for 30 min (to destroy NADP+ without affecting NADPH), followed by the same procedures in parallel as for total NADP measurement. Eventually, the concentration of NADP+ was calculated by subtracting [NADPH] from [total NADP].
Cystine and glucose uptake assays
Cystine uptake using [14C]cystine (Perkin Elmer) was conducted as described previously (Zhang et al., 2018, 2021). Glucose uptake was conducted similar to cystine uptake. In brief, cells were washed with PBS once and replaced with glucose-free medium containing 0.1 μCi 2-[1-14C]deoxy-D-glucose (Perkin Elmer) with or without indicated drugs. After incubation for 2 h at 37 °C, cells were washed twice with cold PBS and lysed in 0.1 mM NaOH. Radioactivity was measured using a Tri-Carb Liquid Scintillation Analyzer (PerKinElmer, model 4810TR) with a quench curve. All experiments were carried out in triplicate.
Western blotting
Western blotting was conducted as previously described (Liu and Gan, 2016; Liu et al., 2016). Tumor tissues were ground to powder in a liquid nitrogen-cooled mortar and pestle and then lysed in ten volumes of NP40 buffer as previously described (Lin et al., 2014a). The primary antibodies and concentrations used for Western blotting were following: KEAP1 (Santa Cruz, sc-365626, 1:1000 dilution), G6PD (1:1,000, Cell Signaling Technology, no. 12263 S), vinculin (1:5,000, Sigma, no. V4505), Myc tag (1:2,000, Cell Signaling Technology, no. 2276 S), SLC7A11 (1:2,000, Cell Signaling Technology, no. 12691 S), GLUT1 (1:1,000, Thermo Fisher, no. PA5-16793), GLUT3 (1:1,000, Santa Cruz, no. sc-74497), NRF2 (1:1,000, Cell Signaling Technology, no. 12721 S).
Xenograft experiments
All the xenograft experiments were performed as previously described (Lei et al., 2021; Xiao et al., 2017) and in accordance with a protocol approved by the Institutional Animal Care and Use Committee and Institutional Review Board at The University of Texas MD Anderson Cancer Center. The study is compliant with all relevant ethical regulations regarding animal research. Female 4- to 6- week-old athymic nude mice (Foxn1nu/Foxn1nu) were purchased from the Experimental Radiation Oncology Breeding Core Facility at MD Anderson Cancer Center and housed in the Animal Care Facility at the Department of Veterinary Medicine and Surgery at MD Anderson. Cancer cell lines were resuspended in FBS-free RPMI and the same number of cells were injected into mice subcutaneously. Tumor progression was monitored by bi-dimensional tumor measurements twice a week until the endpoint. The tumor volume was calculated according to the equation volume = 0.5 × length × width2. When the tumors had grown to around 50–100 mm3 in volume, the mice were assigned randomly into 2 groups (n > 5 per group) and treated with 100 mg kg−1 KL-11743 or vehicle (0.5% methylcellulose, 0.25% Tween-80 in water) by intraperitoneal administration every 2 d.
Histology and immunohistochemistry
Xenograft tumor samples were collected and fixed in 10% neutral-buffered formalin (ThermoFisher Scientific) overnight. Tumors were washed with PBS and then transferred to 70% ethanol followed by embedding, sectioning, and hematoxylin and eosin staining. For immunohistochemical staining, tissue sections were processed according to methods previously described (Gan et al., 2010; Gan et al., 2006). The primary antibodies used for immunohistochemistry were anti-Ki-67 (D2H10) (1:500), Cell Signaling Technology, 9027 S) and anti-cleaved caspase-3 (1:500), Cell Signaling Technology, 9661 s). Images were obtained at 400× magnification on an Olympus BX43 microscope.
Thiol metabolite extraction & analysis
Intracellular levels of labile thiol species were quantified through a modified extraction and sample derivatization procedure optimized to prevent oxidation prior to analysis as previously described (Liu et al., 2020b). Briefly, cells cultured in appropriate treatment conditions were quickly washed in 1ml ice-cold PBS and then extracted once in 500 μl of ice cold extraction buffer consisting of 40% methanol, 40% acetonitrile and 20% water containing 100mM formic acid and 1mM EDTA. The levels of glutathione and cysteine were maintained for at least 48h under these conditions. These extracts were further derivatized with benzyl chloroformate with the following standard mixture: [U-13C-15N-cysteine, 1 μgml−1; U-13C-15N-cystine, 10 μgml−1; 2×13C-1×15N-glutathione (labeled on the glycine carbons and nitrogen), 50 μgml−1 ; 4×13C-2×15N-glutathione disulfide. Analyte concentrations were quantified by comparison to standard curves prepared by the same method. To determine intracellular concentrations, the total cell volume on replicate plates was determined using packed cell volume tubes.
Gene expression and metabolomic correlation analysis
Data from the cancer dependency map (Tsherniak et al., 2017) was downloaded from the depmap data portal (https://depmap.org/portal/). Cell lines were selected based on their NSCLC classification. The cell lines were further classified based on their KEAP1 mutation status. Data from the TCGA LUAD cohort were downloaded from the UCSC Xena browser and analyzed. The generation of data is described in detail on the UCSC Xena website (Goldman et al., 2020).
Quantification and statistical analysis
Results of cell culture experiments were obtained from at least 3 independents repeats. Data are represented as means ± standard deviation (SD). Statistical significance (p values) were calculated using upaired Student’s t-tests, 2-way ANOVA analysis by GraphPad Prism 8.0 (GraphPad Software, Inc.). ∗∗, p < 0.01; ∗∗∗, p < 0.001; ∗∗∗∗, p < 0.0001.
Acknowledgments
This research was supported by Institutional Research Fund and Bridge Fund from The University of Texas MD Anderson Cancer Center, Emerson Collective Cancer Research Fund, and R01CA181196 and R01CA244144 from the National Institutes of Health (to B.G.). B.G. was an Andrew Sabin Family Fellow. P.K. is supported by CPRIT Research Training Grant (RP170067) and Dr. John J. Kopchick Research Award from The University of Texas MD Anderson Cancer Center UTHealth Graduate School of Biomedical Sciences. PDX generation and annotation were supported by the University of Texas MD Anderson Cancer Center Moon Shots Program, and Specialized Program of Research Excellence (SPORE) grant CA070907. This research has also been supported by the National Institutes of Health Cancer Center Support Grant P30CA016672 to The University of Texas MD Anderson Cancer Center.
Author contributions
P.K. performed most of the experiments with assistance from Y.Z., X.L., G.L., M.D.; B.G. supervised the project; K.O. and L.K. conducted thiol metabolite analyses; K.O. and M.V.P. provided KL-11743; B. F. analyzed gene expression and mutation in PDX models; B.G. and P.K. designed the experiments and wrote the manuscript; all authors commented on the manuscript.
Declaration of interests
K.O., L.K., and M.V.P. are full-time employees of Kadmon Corporation. B.G. is an inventor on patent applications involving targeting ferroptosis in cancer therapy. The other authors declare no competing interests.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102649.
Supplemental information
References
- Arbour K.C., Jordan E., Kim H.R., Dienstag J., Yu H.A., Sanchez-Vega F., Lito P., Berger M., Solit D.B., Hellmann M. Effects of Co-occurring genomic alterations on outcomes in patients with KRAS-mutant non-small cell lung cancer. Clin. Cancer Res. 2018;24:334–340. doi: 10.1158/1078-0432.CCR-17-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird L., Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol. Cell. Biol. 2020;40 doi: 10.1128/MCB.00099-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A.S., Liu X., Jing J., Lee H., Yadav R.K., Liu J., Zhou Y., Gan B. STIM2 interacts with AMPK and regulates calcium-induced AMPK activation. FASEB J. 2019;33:2957–2970. doi: 10.1096/fj.201801225R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chio I.I.C., Tuveson D.A. ROS in cancer: the burning question. Trends Mol. Med. 2017;23:411–429. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs J.A., DeNicola G.M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers (Basel) 2019;11 doi: 10.3390/cancers11050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai F., Lee H., Zhang Y., Zhuang L., Yao H., Xi Y., Xiao Z.D., You M.J., Li W., Su X., Gan B. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc. Natl. Acad. Sci. U S A. 2017;114:3192–3197. doi: 10.1073/pnas.1619588114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola G.M., Karreth F.A., Humpton T.J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K.H., Yeo C.J., Calhoun E.S. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan-Cobo A., Sitthideatphaiboon P., Qu X., Poteete A., Pisegna M.A., Tong P., Chen P.H., Boroughs L.K., Rodriguez M.L.M., Zhang W. LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 2019;79:3251–3267. doi: 10.1158/0008-5472.CAN-18-3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B., Lim C., Chu G., Hua S., Ding Z., Collins M., Hu J., Jiang S., Fletcher-Sananikone E., Zhuang L. FoxOs enforce a progression checkpoint to constrain mTORC1-activated renal tumorigenesis. Cancer Cell. 2010;18:472–484. doi: 10.1016/j.ccr.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan B., Peng X., Nagy T., Alcaraz A., Gu H., Guan J.L. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J. Cell Biol. 2006;175:121–133. doi: 10.1083/jcb.200604129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goji T., Takahara K., Negishi M., Katoh H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J. Biol. Chem. 2017;292:19721–19732. doi: 10.1074/jbc.M117.814392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M.J., Craft B., Hastie M., Repecka K., McDade F., Kamath A., Banerjee A., Luo Y., Rogers D., Brooks A.N. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020;38:675–678. doi: 10.1038/s41587-020-0546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes J.D., Dinkova-Kostova A.T., Tew K.D. Oxidative stress in cancer. Cancer Cell. 2020;38:167–197. doi: 10.1016/j.ccell.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Li W., Su Z.Y., Kong A.N. The complexity of the Nrf2 pathway: beyond the antioxidant response. J. Nutr. Biochem. 2015;26:1401–1413. doi: 10.1016/j.jnutbio.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii T., Bannai S. The synergistic action of the copper chelator bathocuproine sulphonate and cysteine in enhancing growth of L1210 cells in vitro. J. Cell Physiol. 1985;125:151–155. doi: 10.1002/jcp.1041250119. [DOI] [PubMed] [Google Scholar]
- Jeong Y., Hoang N.T., Lovejoy A., Stehr H., Newman A.M., Gentles A.J., Kong W., Truong D., Martin S., Chaudhuri A. Role of KEAP1/NRF2 and TP53 mutations in lung squamous cell carcinoma development and radiation resistance. Cancer Discov. 2017;7:86–101. doi: 10.1158/2159-8290.CD-16-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly J.H., Delfarah A., Phung P.S., Parrish S., Graham N.A. A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose. J. Biol. Chem. 2020;295:1350–1365. doi: 10.1074/jbc.RA119.011471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler T.W., Wakabayashi N., Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- Koppula P., Zhang Y., Shi J., Li W., Gan B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017;292:14240–14249. doi: 10.1074/jbc.M117.798405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P., Zhang Y., Zhuang L., Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018;38:12. doi: 10.1186/s40880-018-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P., Zhuang L., Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2020 doi: 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall E.B., Wang B., Munoz D.M., Ilic N., Raghavan S., Niederst M.J., Yu K., Ruddy D.A., Aguirre A.J., Kim J.W. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife. 2017;6 doi: 10.7554/eLife.18970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Dai F., Zhuang L., Xiao Z.D., Kim J., Zhang Y., Ma L., You M.J., Wang Z., Gan B. BAF180 regulates cellular senescence and hematopoietic stem cell homeostasis through p21. Oncotarget. 2016;7:19134–19146. doi: 10.18632/oncotarget.8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Zandkarimi F., Zhang Y., Meena J.K., Kim J., Zhuang L., Tyagi S., Ma L., Westbrook T.F., Steinberg G.R. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020;22:225–234. doi: 10.1038/s41556-020-0461-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.B., Sellers B.N., DeNicola G.M. The regulation of NRF2 by nutrient-responsive signaling and its role in anabolic cancer metabolism. Antioxid. Redox Signal. 2017 doi: 10.1089/ars.2017.7356. [DOI] [PubMed] [Google Scholar]
- Lei G., Zhang Y., Hong T., Zhang X., Liu X., Mao C., Yan Y., Koppula P., Cheng W., Sood A.K. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021 doi: 10.1038/s41388-021-01790-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei G., Zhang Y., Koppula P., Liu X., Zhang J., Lin S.H., Ajani J.A., Xiao Q., Liao Z., Wang H., Gan B. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–162. doi: 10.1038/s41422-019-0263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Ning S., Ghandi M., Kryukov G.V., Gopal S., Deik A., Souza A., Pierce K., Keskula P., Hernandez D. The landscape of cancer cell line metabolism. Nat. Med. 2019;25:850–860. doi: 10.1038/s41591-019-0404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A., Piao H.L., Zhuang L., Sarbassov dos D., Ma L., Gan B. FoxO transcription factors promote AKT Ser473 phosphorylation and renal tumor growth in response to pharmacologic inhibition of the PI3K-AKT pathway. Cancer Res. 2014;74:1682–1693. doi: 10.1158/0008-5472.CAN-13-1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin A., Yao J., Zhuang L., Wang D., Han J., Lam E.W., Network T.R., Gan B. The FoxO-BNIP3 axis exerts a unique regulation of mTORC1 and cell survival under energy stress. Oncogene. 2014;33:3183–3194. doi: 10.1038/onc.2013.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.G., Kim J.I., Olszewski K., Barsotti A.M., Morris K., Lamarque C., Yu X., Gaffney J., Feng X.J., Patel J.P., Poyurovsky M.V. Discovery and optimization of glucose uptake inhibitors. J. Med. Chem. 2020;63:5201–5211. doi: 10.1021/acs.jmedchem.9b02153. [DOI] [PubMed] [Google Scholar]
- Liu X., Gan B. lncRNA NBR2 modulates cancer cell sensitivity to phenformin through GLUT1. Cell Cycle. 2016;15:3471–3481. doi: 10.1080/15384101.2016.1249545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Olszewski K., Zhang Y., Lim E.W., Shi J., Zhang X., Zhang J., Lee H., Koppula P., Lei G. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat. Cell Biol. 2020;22:476–486. doi: 10.1038/s41556-020-0496-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Xiao Z.D., Han L., Zhang J., Lee S.W., Wang W., Lee H., Zhuang L., Chen J., Lin H.K. LncRNA NBR2 engages a metabolic checkpoint by regulating AMPK under energy stress. Nat. Cell Biol. 2016;18:431–442. doi: 10.1038/ncb3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Zhang Y., Zhuang L., Olszewski K., Gan B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edge sword in cellular redox regulation. Genes Dis. 2020 doi: 10.1016/j.gendis.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W., Wang L., Chen L., Hui S., Rabinowitz J.D. Extraction and quantitation of nicotinamide adenine Dinucleotide redox cofactors. Antioxid. Redox Signal. 2018;28:167–179. doi: 10.1089/ars.2017.7014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Muir A., Danai L.V., Gui D.Y., Waingarten C.Y., Lewis C.A., Vander Heiden M.G. Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife. 2017;6 doi: 10.7554/eLife.27713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narta U.K., Kanwar S.S., Azmi W. Pharmacological and clinical evaluation of L-asparaginase in the treatment of leukemia. Crit. Rev. Oncol. Hematol. 2007;61:208–221. doi: 10.1016/j.critrevonc.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo de la Vega M., Chapman E., Zhang D.D. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R., Sayin V.I., Davidson S.M., Bauer M.R., Singh S.X., LeBoeuf S.E., Karakousi T.R., Ellis D.C., Bhutkar A., Sanchez-Rivera F.J. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017;23:1362–1368. doi: 10.1038/nm.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H., Sato H., Kuriyama-Matsumura K., Sato K., Maebara K., Wang H., Tamba M., Itoh K., Yamamoto M., Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- Sato H., Tamba M., Ishii T., Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sayin V.I., LeBoeuf S.E., Singh S.X., Davidson S.M., Biancur D., Guzelhan B.S., Alvarez S.W., Wu W.L., Karakousi T.R., Zavitsanou A.M. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife. 2017;6 doi: 10.7554/eLife.28083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S. Biological imaging software tools. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C.S., Mishra P., Watrous J.D., Carelli V., D'Aurelio M., Jain M., Chan D.C. The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat. Commun. 2017;8:15074. doi: 10.1038/ncomms15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A., Misra V., Thimmulappa R.K., Lee H., Ames S., Hoque M.O., Herman J.G., Baylin S.B., Sidransky D., Gabrielson E. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoulidis F., Byers L.A., Diao L., Papadimitrakopoulou V.A., Tong P., Izzo J., Behrens C., Kadara H., Parra E.R., Canales J.R. Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 2015;5:860–877. doi: 10.1158/2159-8290.CD-14-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykiotis G.P., Bohmann D. Stress-activated cap'n'collar transcription factors in aging and human disease. Sci. Signal. 2010;3:re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A., Vazquez F., Montgomery P.G., Weir B.A., Kryukov G., Cowley G.S., Gill S., Harrington W.F., Pantel S., Krill-Burger J.M. Defining a cancer dependency map. Cell. 2017;170:564–576 e516. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner T.C., Scott M.D. Single extraction method for the Spectrophotometric quantification of oxidized and reduced pyridine nucletides in erythrocytes. Anal. Biochem. 1994;222:417–426. doi: 10.1006/abio.1994.1511. [DOI] [PubMed] [Google Scholar]
- Wang Q., Ma J., Lu Y., Zhang S., Huang J., Chen J., Bei J.X., Yang K., Wu G., Huang K. CDK20 interacts with KEAP1 to activate NRF2 and promotes radiochemoresistance in lung cancer cells. Oncogene. 2017;36:5321–5330. doi: 10.1038/onc.2017.161. [DOI] [PubMed] [Google Scholar]
- Wolpaw A.J., Dang C.V. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell. Biol. 2018;28:201–212. doi: 10.1016/j.tcb.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z.D., Han L., Lee H., Zhuang L., Zhang Y., Baddour J., Nagrath D., Wood C.G., Gu J., Wu X. Energy stress-induced lncRNA FILNC1 represses c-Myc-mediated energy metabolism and inhibits renal tumor development. Nat. Commun. 2017;8:783. doi: 10.1038/s41467-017-00902-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamadori T., Ishii Y., Homma S., Morishima Y., Kurishima K., Itoh K., Yamamoto M., Minami Y., Noguchi M., Hizawa N. Molecular mechanisms for the regulation of Nrf2-mediated cell proliferation in non-small-cell lung cancers. Oncogene. 2012;31:4768–4777. doi: 10.1038/onc.2011.628. [DOI] [PubMed] [Google Scholar]
- Yamaguchi I., Yoshimura S.H., Katoh H. High cell density increases glioblastoma cell viability under glucose deprivation via degradation of the cystine/glutamate transporter xCT (SLC7A11) J. Biol. Chem. 2020;295:6936–6945. doi: 10.1074/jbc.RA119.012213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Shi J., Liu X., Feng L., Gong Z., Koppula P., Sirohi K., Li X., Wei Y., Lee H. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018;20:1181–1192. doi: 10.1038/s41556-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Shi J., Liu X., Xiao Z., Lei G., Lee H., Koppula P., Cheng W., Mao C., Zhuang L. H2A Monoubiquitination links glucose availability to epigenetic regulation of the endoplasmic reticulum stress response and cancer cell death. Cancer Res. 2020;80:2243–2256. doi: 10.1158/0008-5472.CAN-19-3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Swanda R.V., Nie L., Liu X., Wang C., Lee H., Lei G., Mao C., Koppula P., Cheng W. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat. Commun. 2021;12:1589. doi: 10.1038/s41467-021-21841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate such unique reagents.