

Abstract
A key tool for metabolic engineering is the ability to express heterologous genes. One obstacle to gene expression in non-model organisms, and especially in relatively uncharacterized bacteria, is the lack of well-characterized promoters. Here we test 17 promoter regions for their ability to drive expression of the reporter genes β-galactosidase (lacZ) and NADPH-alcohol dehydrogenase (adhB) in Clostridium thermocellum, an important bacterium for the production of cellulosic biofuels. Only three promoters have been commonly used for gene expression in C. thermocellum, gapDH, cbp and eno. Of the new promoters tested, 2638, 2926, 966 and 815 showed reliable expression. The 2638 promoter showed relatively higher activity when driving adhB (compared to lacZ), and the 815 promoter showed relatively higher activity when driving lacZ (compared to adhB).
Keywords: Promoter, Copy number, Structural instability, Rolling circle replication, lacZ, adhB
Highlights
-
•
Tested 17 different putative promoter sequences with 2 different reporter genes.
-
•
The eno, cbp, cbp_2, 815, 966, 2638 and 2926 promoters resulted in high enzymatic activity.
-
•
The gapDH, gapDH_2, 1194 and 3011_2 promoters apparently exacerbate problems with plasmid instability.
-
•
The 307, 544, 2463, lac and lacUV5 promoters did not result in reporter gene activity.
-
•
Increasing antibiotic selection pressure did not increase expression of the reporter gene.
1. Introduction
Clostridium thermocellum is a promising candidate for the conversion of biomass to ethanol due to its native ability to use cellulose and produce ethanol (Olson et al., 2012). Recently there has been significant progress engineering C. thermocellum to improve ethanol production (Argyros et al., 2011, Deng et al., 2013, Van der Veen et al., 2013), however ethanol yield needs further improvement for commercial viability.
To date, most metabolic engineering of C. thermocellum has focused on gene deletion but many metabolic engineering strategies require increased or heterologous gene expression, in addition to gene deletion. Only a few genes have been successfully expressed in C. thermocellum, including the antibiotic resistance markers cat and neo; the counter-selectable markers pyrF, hpt and tdk (Olson and Lynd, 2012a); the cellulosome scaffoldin cipA (Olson et al., 2013) and the metabolic enzyme pyruvate kinase (Deng et al., 2013).
The most commonly used promoters are from the regions upstream of the gapDH (Clo1313_2095), cbp (Clo1313_1954) and eno (Clo1313_2090) genes in C. thermocellum. The gapDH gene encodes the glyceraldehyde-3-phosphate dehydrogenase enzyme, which is one of the most highly expressed proteins in the C. thermocellum proteome (Olson et al., 2013, Olson et al., 2010, Rydzak et al., 2012). While none of these promoters have been extensively characterized, the cbp promoter has been reliably used to drive expression of the pyrF (Tripathi et al., 2010) and tdk (Argyros et al., 2011) selectable markers. The eno promoter has been used to drive expression of an exogenous pyruvate kinase from Thermoanaerobacterium saccharolyticum (Deng et al., 2013). The transcription start sites have not been determined nor is anything known about which sigma factor is responsible for promoter recognition.
In the absence of detailed analysis, it is still useful to search for suitable promoters for expression by scanning those available. It is important to appreciate that any such study based on the assay of a reporter gene activity will also depend on mRNA stability and enzyme efficiency. An ideal promoter for metabolic engineering would have the following characteristics:
-
1.
Low expression of the gene of interest in cloning strains of Escherichia coli.
-
2.
Consistently high expression in C. thermocellum, independent of genetic context.
-
3.
Low homology to the chromosome (in the case of native promoters, shorter is better).
The goal of this work is to identify new promoters for gene expression in C. thermocellum, and compare them with ones currently in use.
2. Materials and methods
2.1. Plasmid construction
Plasmids were constructed using standard molecular biology techniques (Sambrook and Russell, 2001) and isothermal DNA assembly (Gibson, 2011). Plasmid sequence was confirmed by Sanger sequencing of the promoter region and reporter gene. Plasmids were transformed into C. thermocellum DSM 1313 using standard techniques (Olson and Lynd, 2012a). Plasmids were also transformed in to E. coli C2566 (New England Biolabs) using standard techniques (Sambrook and Russell, 2001). The thermostable lacZ gene was a gift from James Liao (Lin et al., 2014). A list of the promoters and reporter genes in each plasmid is given in Table 1.
Table 1.
Plasmids used in this study.
| Plasmid | Promoter | Putative SigA/RpoD motif | Predicted conventional RBS Sequence | Predicted TIE strength(arbitrary units)a | Reporter gene |
|---|---|---|---|---|---|
| pDGO-66 | gapDH | TTGA(A)A–N17–TA(A)AAT | AGGAGG | – | none |
| pDGO80 | none | 369 | adhB | ||
| pDGO81 | 0544 | GGAGG | 5190 | adhB | |
| pDGO83 | cbp | TTGA(A)(T)–N17–TATAAT | AGGAGG | 244,912 | adhB |
| pDGO84 | eno | TTGA(A)A–N18–(C)AT(T)AT | GGAG | 8763 | adhB |
| pDGO86 | 1194 | TTG(T)(T)(T)–N15–TATAAT | AGG(G)GG | 19,141 | adhB |
| pDGO87 | 0966 | TTG(C)(A)(T)–N15–TGNTATAAT | AGGA | 5587 | adhB |
| pDGO88 | 2638 | TT(A)A(A)A–N15–TATAAT | AGGAGG | 42,645 | adhB |
| pDGO89 | 0815 | TT(T)A(A)A–N12–TGNTAT(T)AT | GAGG | 19,843 | adhB |
| pDGO90 | 2926 | AGGAGG | 15,731 | adhB | |
| pDGO92 | 0307 | GGAG | 2764 | adhB | |
| pDGO95 | gapDH | TTGA(A)A–N17–TA(A)AAT | AGGAGG | 7372 | lacZ |
| pDGO98 | none | – | lacZ | ||
| pDGO99 | 0544 | GGAGG | 716 | lacZ | |
| pDGO100 | gapDH_2 | TTGA(A)A–N17–TA(A)AAT | AGGAGG | 1154 | lacZ |
| pDGO102 | eno | TTGA(A)A–N18–(C)AT(T)AT | GGAG | 32,320 | lacZ |
| pDGO104 | 1194 | TTG(T)(T)(T)–N15–TATAAT | AGG(G)GG | 18,381 | lacZ |
| pDGO105 | 0966 | TTG(C)(A)(T)–N15–TGNTATAAT | AGGA | 1734 | lacZ |
| pDGO106 | 2638 | TT(A)A(A)A–N15–TATAAT | AGGAGG | 6441 | lacZ |
| pDGO107 | 0815 | TT(T)A(A)A–N12–TGNTAT(T)AT | GAGG | 2091 | lacZ |
| pDGO108 | 2926 | TGNTA(A)(T)AT | AGGAGG | 7656 | lacZ |
| pDGO109 | 2463 | AGGAGG | 8067 | lacZ | |
| pDGO110 | 0307 | GGAG | 858 | lacZ | |
| pDGO111 | lac | TT(T)ACA–N18–TAT(G)(T)T | AGGA | 390 | lacZ |
| pDGO112 | lacUV5 | TT(T)ACA–N18–TATAAT | AGGA | 390 | lacZ |
| pDGO117 | cbp_2 | TTGA(A)(T)–N17–TATAAT | AGGAGG | 50,011 | lacZ |
| pDGO118 | 3011_2 | TTGAC(T)–N17–TATAAT | AGGAGG | 5978 | lacZ |
See Section 2.10 for details about calculation of translation initiation efficiency (TIE).
2.2. Choice of promoter regions
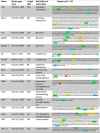
Promoter regions were chosen based on comparison of several sets of published gene expression data. These sets include Riederer et al. (2011), Raman et al. (2011), Gowen and Fong (2010) and Van der Veen et al. (2013). Note that for the last reference, strain characterization is described in the referred paper, but the transcription data is available from the GEO database, accession GSE27046. Promoters were selected on the basis of showing moderate to high expression of the genes they control across several of the data sets. For a given gene, the region upstream of the translation start site of the gene was selected. In general this region was 100–500 bp in length (Table 2). For promoters that are listed twice (i.e. cbp and cbp_2), the first version of the promoter caused problems with cloning, so a slightly different region was selected.
Table 2.
Description of promoter sequences. Predicted SigA/RpoD binding sites are highlighted: “-35 boxes” in green, “-10 boxes” in yellow and “extended-10 boxes” in pink. Predicted ribosome binding sites are highlighted in blue. Start codons at the end of each promoter are indicated by bold font.
 |
2.3. Making cell-free extracts
To prepare cell-free extracts (CFE), C. thermocellum cells were grown to mid-log phase. 10 ml of cells was centrifuged and the pellet was resuspended in B-PER buffer (Thermo Scientific, IL). The cells were lysed by addition of Lysonase according to the manufacturer׳s directions (EMD Millipore, MA). After centrifugation for 1 min at 15,000g to remove cell debris, the resulting supernatant was used for protein and enzyme assays. E. coli cells were also lysed using B-PER buffer and Lysonase enzyme. Protein concentration of the CFE was measured using the Coomassie Plus Bradford Assay (Thermo Scientific, IL) using bovine serum albumin as the standard. Absorbance was measured at 595 nm and 450 nm and the ratio of the two was used to determine concentration.
2.4. LacZ activity assay
The activity of the LacZ enzyme was determined by measuring the formation of the yellow cleavage product of o-nitrophenyl-β-d-galactoside (ONPG) as described by Miller (1972) with modifications as described below. The 0.2 ml assay solution contained 100 mM sodium phosphate pH 7.5, 1 mM magnesium chloride, 50 mM beta mercaptoethanol, 0.655 mg/ml ONPG and varying amounts of CFE. The assay solution was incubated at 37 °C for 4 h and the rate of increase in absorbance at 420 nm was measured in a 96-well plate with a BioRad Powerwave XS spectrophotometer. Purified LacZ enzyme (G4155 from Sigma) was used as a standard. Several 2-fold dilutions of CFE were added, and activity was determined in the linear range.
2.5. AdhB activity assay
AdhB is a bifunctional secondary alcohol and aldehyde dehydrogenase from Thermoanaerobacter pseudethanolicus ATCC 33223 (Teth39_0218) that is NADPH-dependent (Burdette et al., 1997). The activity of the AdhB enzyme was determined by measuring the disappearance of NADPH by measuring changes in absorbance at 340 nm. The 0.2 ml assay solution contained 100 mM Tris–HCl pH 7.6, 0.4 mM NADPH, 4 mM acetaldehyde and varying amounts of CFE. The assay solution was incubated at 55 °C for 1 h and the rate of decrease in absorbance at 340 nm was measured. Measurements were performed in a BioRad Powerwave XS spectrophotometer. The path length was 0.52 cm. Several 2-fold dilutions of CFE were added, and activity was determined in the linear range.
2.6. Plasmid copy number
Plasmid copy number was determined by quantitative PCR (qPCR). Copies of the plasmid genes chloramphenicol (cat) and B-galactosidase (lacZ) were normalized to levels of the celS gene (Clo1313_2747) levels, which is present at single copy on the C. thermocellum genome (Olson et al., 2010). Primer sequences are given in Supporting information Table S4. Reactions were performed in triplicate using 2 µl of bacterial cultures as template and SsoFast EvaGreen Supermix (BioRad) following the manufacturer’s recommendation and an annealing temperature of 55 °C. Standard curves for cat and celS were generated using the linearized plasmid pDGO-28 (Olson and Lynd, 2012b), which contains a single copy of both genes. Standard curves for lacZ were generated using linearized plasmid pDGO98, which contains a single copy of the lacZ gene. All data was normalized using a 529 bp synthetic gBlock (IDT) containing the cat, celS and lacZ amplicons.
2.7. RNA isolation
1 ml of bacterial culture was pelleted and lysed by digestion with lysozyme (15 mg/ml) and proteinase K (20 mg/ml). RNA was isolated with an RNAeasy minikit (Qiagen) and digested with TURBO DNase (Applied Biosystems) to remove contaminating DNA. cDNA was synthesized from 500 ng of RNA using the iScript cDNA synthesis kit (BioRad).
2.8. Gene expression
qPCR was performed in triplicate using cDNA with SsoFast EvaGreen Supermix (BioRad) at an annealing temperature of 55 °C to determine RNA levels of the gene of interest, which were normalized to recA RNA levels. In order to confirm removal of contaminating DNA from RNA samples, for a subset of samples, a control cDNA synthesis reaction was performed in the absence of reverse transcriptase enzyme. Standard curves were generated using a 1904 bp synthetic gBlock (IDT) containing all of the amplicons.
2.9. PCR for plasmid structural stability analysis
To determine plasmid stability primers were designed that generated an approximately 6000 bp PCR product (the exact amplicon size depended upon the promoter inserted) spanning the repB, cat, amp and lacZ genes of plasmids pDGO95 through pDGO118. The PCR product was generated using 1 µl of bacterial culture, previously transformed with the plasmid, and Quick Load 2× Taq (NEB). Primers were annealed at 60 °C (forward: 5′-AAGGTGCGTTGAAGTGTTGGTATGT-3′, reverse: 5′-GGCTTCTGTACGCCTGACCCTATTA-3′). PCR products were analyzed by electrophoresis on 1% agarose gels.
2.10. Predicted translation initiation efficiency (TIE) analysis
The translation initiation efficiency was predicted based on a biophysical model of translation initiation that considers the hybridization of the 16s rRNA to the 50 bp region around the translation start site. For this model, the 16s sequence from C. thermocellum was used (Salis et al., 2009). The calculations were performed using a freely available calculator on the Salis lab website, https://salis.psu.edu/software/reverse. The predicted relative rate of translation initiation is expressed in arbitrary units.
2.11. Ribosome binding site prediction and SigA promoter prediction
Conventional ribosome binding sites (RBS) were identified using the Prodigal Gene Prediction algorithm (Hyatt et al., 2010) since that is an integral part of the algorithm and part of the output. Predicted SigA binding sites were identified manually and are based on the known Bacillus subtilis SigA and E. coli RpoD (http://dbtbs.hgc.jp/) consensus sequence TTGACA–N16-19–TATAAT.
3. Results and discussion
Sequences of DNA upstream from genes in C. thermocellum were screened for promoter sequences. First these sequences were cloned 5′ of a thermostable beta galactosidase gene (lacZ). For sequences that gave promising results with lacZ, we built a second set of constructs using the NADPH-linked secondary alcohol dehydrogenase from Thermoanaerobacterium ethanolicus (adhB). In total, 27 plasmid constructs were made (Table 1), using 17 promoter regions (Table 2).
3.1. E. coli vs. C. thermocellum activity
For our purposes we were interested in finding promoters with low activity in E. coli and high activity in C. thermocellum, so we measured LacZ activity in cell-free extracts of both organisms (Fig. 1, Supporting information Table S1).
Fig. 1.

Comparison of LacZ activity between E. coli and C. thermocellum in units of activity per mg of cell-free extract (CFE). Error bars represent one standard deviation, n≥3.
It can be seen that there is a low correlation between promoters that give good activity in C. thermocellum vs. E. coli. The promoters 2638, cbp_2, gapDH and 3011_2 have the largest differential. It is interesting to note the difference between gapDH and gapDH_2, since these promoters only differ by 46 bp. To understand the cause of the substantial variability observed, we performed a second round of experiments.
Colonies from the first round were inoculated into media with different amounts of thiamphenicol (6, 24 or 96 µg/ml). For each culture, we measured 6 parameters including: DNA copy number of the lacZ reporter gene, DNA copy number of the cat antibiotic resistance gene, mRNA expression of the lacZ reporter gene, mRNA expression of the native gene (i.e. the gene whose promoter was used in the construct) and specific activity of the LacZ protein (Fig. 2, Supporting information Table S2).
Fig. 2.

Comparison of LacZ activity for different promoters and different thiamphenicol concentrations. The thick line represents the median, the box encloses the 25th–75th percentile of data. Individual data points are plotted.
To understand the effect of antibiotic concentration on plasmid copy number, we grew plasmid-containing strains of C. thermocellum on various concentrations of thiamphenicol. Although we did see a slight correlation between antibiotic strength and the copy number of the cat gene (Supporting information Table S2), this did not result in higher LacZ activity. In general, the highest activity was found when cells were grown in the presence of 6 µg/ml thiamphenicol (Fig. 2). Promoters 307, 544, 2463, lac and lacUV5 did not show activity above the negative control.
After these experiments had been performed, these promoters were analyzed for the presence of SigA binding sites (Table 2, green boxes). For sequences 307, 544, 2463 and 2926, these sites were not found, suggesting either that the sequence does not contain a promoter or that the sequence contains a promoter that is recognized by a sigma factor other than SigA. In addition, the four genes downstream of these sequences appear to be internal genes in operons. The 2926 sequence is interesting since it contains three potential -10 boxes (yellow), no -35 boxes (green), and a weak extended -10 box (magenta–TGNTA(A)(T)AT), is expressed well in E. coli but, not at all in C. thermocellum. We speculate that it might contain a binding site for an E. coli positive regulator that overcomes the lack of a good -35 box. Furthermore, this is the only sequence of the four that showed LacZ activity above the negative control.
The lac and lacUV5 promoters are derived from E. coli and require the cAMP receptor protein (CRP). Apparently C. thermocellum does not have a CRP that interacts with the lac or lacUV5 promoters.
Constructs with sequences 1194 and 3011_2 were difficult to transform into C. thermocellum, transformation efficiency was lower than expected. For 3011_2, despite numerous attempts, we only had one successful transformation and that transformation yielded only two colonies. We speculate that these promoters may have contributed to plasmid instability for the inherently unstable plasmid.
3.2. Expression of lacZ vs. native gene
To test whether lacZ expression was similar to expression of the native gene, we measured both by RT-PCR (Fig. 3). Since the lacZ gene is present on a plasmid that can be present in multiple copies, lacZ gene expression was normalized to lacZ DNA copy number. In general, lacZ expression was higher than that of the native gene. Since these are translational fusions, we speculate that there may be differences in mRNA stability or the activity of the gene product.
Fig. 3.

Comparison of mRNA expression between lacZ and native gene. Thick bar represents the median. The box encompasses the 25th–75th percentile of data. Individual data points are plotted.
To investigate whether the results obtained with lacZ are applicable to another reporter gene, we measured the expression of adhB driven by some of these promoters (Fig. 4, Supporting information Table S3). In general, there was a good correlation between constructs driving the lacZ or adhB reporter genes. Two notable exceptions are promoters 2638 and 815. For promoter 2638, the AdhB activity is about 8-fold higher than what would be expected based on the LacZ activity. For promoter 815, the AdhB activity is about 16-fold lower than what would be expected based on the LacZ activity.
Fig. 4.

Comparison of activity levels for two different reporter genes with the same promoter. Error bars represent one standard deviation, n≥3. For the cbp promoter, the LacZ activity was measured with the cbp_2 promoter and the AdhB activity was measured with the cbp promoter. The plasmid with cbp driving lacZ could not be cloned in E. coli, and the plasmid with cbp_2 driving adhB could not be transformed into C. thermocellum.
We do not have a convincing explanation for these differences, however we speculate that translation initiation may play a role. For a given promoter, the RBS sequence upstream of lacZ or adhB is identical. However differences in mRNA secondary structure near the RBS due to the different sequences of the lacZ and adhB genes can affect translation initiation (Salis et al., 2009) and mRNA stability. For each promoter and reporter gene pair, the translation initiation efficiency was predicted (Table 2). In the case of promoter 2638, the predicted RBS strength is calculated to be about 7-fold higher for the adhB construct than the lacZ construct. This explanation does not, however, explain the lower adhB expression with promoter 815, as the predicted RBS strength of the adhB construct was also higher than the lacZ construct (10-fold).
Another possible explanation for the differences in gene expression between lacZ and adhB with promoters 0815 and 2638 is that these promoters are affected by redox levels. Since adhB encodes an alcohol dehydrogenase that could affect redox levels in the cell, that would create a feedback loop and explain the differences between the reporter genes.
3.3. Plasmid instability
Although not the primary focus of this work, we observed several indications that plasmid instability is a serious problem. One indication is the variability of the DNA copy numbers of the lacZ and cat genes. Since they are both present in a single copy on the plasmid, it was expected that their copy number would remain in a 1:1 ratio. In fact, we found an excess of the cat gene relative to the lacZ gene in about a quarter of the colonies we analyzed (Supporting information Table S2). Another indication of structural instability was results seen in PCR across the region containing the repB, cat, amp and lacZ genes. Although we would have expected a ~6 kb amplicon, in about 40% of the colonies we analyzed, we found either no amplicon or an amplicon that was much shorter or longer. Plasmid instability is commonly observed in rolling-circle-replicating plasmids (Bron et al., 1991), and since the cat gene is upstream of the lacZ gene, premature termination of plasmid replication could result in a truncated plasmid with the cat gene able to provide thiamphenicol resistance but lacking the lacZ gene.
Another interesting observation was that some promoter sequences caused problems with recovery of frozen cultures. For all of the colonies tested, about a third could not be recovered after freezing. However the problems with recovery did not seem to be distributed randomly. Rather, the sequences that resulted in high lacZ activity also tended to have more problems with strain recovery (data not shown). The worst results were observed with the gapDH promoters, where 7 of 8 colonies initially frozen could not be recovered. Interestingly, this problem only occurred when both the gapDH promoter and the lacZ reporter gene were present. In control constructs missing either the gapDH promoter sequence (pDGO98) or the lacZ reporter gene (pDGO-66), this effect was not observed, and all frozen colonies (4 of 4 in both cases) were recovered. One possible explanation is that the presence of these sequences resulted in homologous recombination and integration of this unstable plasmid at the gapDH locus. This integration could be toxic to cells because it disrupts expression of the native gapDH gene which is essential for glycolysis.
4. Conclusions
Promoters 815, 966, 2638, 2926, eno, cbp and cbp_2 all show activity and seem to be more generally useful than the gapDH and gapDH_2 promoters. Several of these promoter regions are less than 250 bp in length, which suggests that this length of promoter region is sufficient.
An important direction for future work will be understanding and eliminating the causes of plasmid structural instability observed here.
Acknowledgments
We thank Professor Jan Westpheling for critical review of the manuscript and useful discussions. The BioEnergy Science Center is a U.S. Department of Energy Bioenergy Research Center supported by the Office of Biological and Environmental Research in the DOE Office of Science subcontract number 4000115284. The DOE Office of Science did not play any role in study design, writing the report or the decision to submit the manuscript for publication.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.meteno.2015.03.002.
Contributor Information
Daniel G. Olson, Email: dan268@gmail.com.
Lee R. Lynd, Email: Lee.R.Lynd@dartmouth.edu.
Appendix A. Supporting information
Supplementary material
References
- Argyros D.A., Tripathi S.A., Barrett T.F., Rogers S.R., Feinberg L.F., Olson D.G., Foden J.M., Miller B.B., Lynd L.R., Hogsett D.A., Caiazza N.C. High ethanol titers from cellulose by using metabolically engineered thermophilic, anaerobic microbes. Appl. Environ. Microbiol. 2011;77:8288–8294. doi: 10.1128/AEM.00646-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bron S., Meijer W., Holsappel S., Haima P. Plasmid instability and molecular cloning in Bacillus subtilis. Res. Microbiol. 1991;142:875–883. doi: 10.1016/0923-2508(91)90068-l. [DOI] [PubMed] [Google Scholar]
- Burdette D.S., Secundo F., Phillips R.S., Dong J., Scott R. a, Zeikus J.G. Biophysical and mutagenic analysis of Thermoanaerobacter ethanolicus secondary-alcohol dehydrogenase activity and specificity. Biochem. J. 1997;326(Pt 3):717–724. doi: 10.1042/bj3260717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y., Olson D.G., Zhou J., Herring C.D., Joe Shaw A., Lynd L.R. Redirecting carbon flux through exogenous pyruvate kinase to achieve high ethanol yields in Clostridium thermocellum. Metab. Eng. 2013;15:151–158. doi: 10.1016/j.ymben.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Gibson D.G. Chapter fifteen - Enzymatic Assembly of Overlapping DNA Fragments. In: Voigt C.A., editor. Methods in Enzymology. Academic Press; Waltham, MA: 2011. pp. 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen C.M., Fong S.S. Genome-scale metabolic model integrated with RNAseq data to identify metabolic states of Clostridium thermocellum. Biotechnol. J. 2010;5:759–767. doi: 10.1002/biot.201000084. [DOI] [PubMed] [Google Scholar]
- Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P.P., Rabe K.S., Takasumi J.L., Kadisch M., Arnold F.H., Liao J.C. Isobutanol production at elevated temperatures in thermophilic Geobacillus thermoglucosidasius. Metab. Eng. 2014;24:1–8. doi: 10.1016/j.ymben.2014.03.006. [DOI] [PubMed] [Google Scholar]
- Miller J.H. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1972. Experiments in Molecular Genetics. [Google Scholar]
- Olson D.G., Giannone R.J., Hettich R.L., Lynd L.R. Role of the CipA scaffoldin protein in cellulose solubilization, as determined by targeted gene deletion and complementation in Clostridium thermocellum. J. Bacteriol. 2013;195:733–739. doi: 10.1128/jb.02014-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson D.G., Lynd L.R. Chapter seventeen – transformation of Clostridium thermocellum by electroporation. In: Gilbert H.J., editor. Methods in Enzymology. Academic Press; Waltham, MA: 2012. pp. 317–330. [DOI] [PubMed] [Google Scholar]
- Olson D.G., Lynd L.R. Computational design and characterization of a temperature sensitive plasmid replicon for gram positive thermophiles. J. Biol. Eng. 2012;6:5. doi: 10.1186/1754-1611-6-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson D.G., McBride J.E., Shaw A.J., Lynd L.R. Recent progress in consolidated bioprocessing. Curr. Opin. Biotechnol. 2012;23:396–405. doi: 10.1016/j.copbio.2011.11.026. [DOI] [PubMed] [Google Scholar]
- Olson D.G., Tripathi S.A., Giannone R.J., Lo J., Caiazza N.C., Hogsett D.A., Hettich R.L., Guss A.M., Dubrovsky G., Lynd L.R. Deletion of the Cel48S cellulase from Clostridium thermocellum. Proc. Natl. Acad. Sci. U. S. A. 2010;107:17727–17732. doi: 10.1073/pnas.1003584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman B., McKeown C.K., Jr., Brown M.R., Mielenz S.D., Rodriguez J.R., Rodriguez M., Jr. Transcriptomic analysis of Clostridium thermocellum ATCC 27405 cellulose fermentation. BMC Microbiol. 2011;11:134. doi: 10.1186/1471-2180-11-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riederer A., Takasuka T.E., Makino S., Stevenson D.M., Bukhman Y. V, Elsen N.L., Fox B.G. Global gene expression patterns in Clostridium thermocellum as determined by microarray analysis of chemostat cultures on cellulose or cellobiose. Appl. Environ. Microbiol. 2011;77:1243–1253. doi: 10.1128/aem.02008-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydzak T., McQueen P.D., Krokhin O. V, Spicer V., Ezzati P., Dwivedi R.C., Shamshurin D., Levin D.B., Wilkins J.A., Sparling R. Proteomic analysis of Clostridium thermocellum core metabolism: relative protein expression profiles and growth phase-dependent changes in protein expression. BMC Microbiol. 2012;12:214. doi: 10.1186/1471-2180-12-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis H.M., Mirsky E.A., Voigt C.A. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Russell D.W. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2001. Molecular Cloning: A Laboratory Manual. [Google Scholar]
- Tripathi S.A., Olson D.G., Argyros D.A., Miller B.B., Barrett T.F., Murphy D.M., McCool J.D., Warner A.K., Rajgarhia V.B., Lynd L.R., Hogsett D.A., Caiazza N.C. Development of pyrF-based genetic system for targeted gene deletion in Clostridium thermocellum and creation of a pta mutant. Appl. Environ. Microbiol. 2010;76:6591–6599. doi: 10.1128/AEM.01484-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Veen D., Lo J., Brown S.D., Johnson C.M., Tschaplinski T.J., Martin M., Engle N.L., van den Berg R.A., Argyros A.D., Caiazza N.C., Guss A.M., Lynd L.R. Characterization of Clostridium thermocellum strains with disrupted fermentation end-product pathways. J. Ind. Microbiol. Biotechnol. 2013;40:725–734. doi: 10.1007/s10295-013-1275-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material