

Abstract
The aim of the review was to describe a complex microstructure and biomechanical properties of the articular cartilage as well as a current review of its pathologies encountered in veterinary practice. The articular cartilage with its unique features: complex microarchitecture, significant mechanical durability and elasticity, lacking blood, lymphatic vessels, and innervation, seems to stand in contradiction to the laws of biology. It can be involved in a vast majority of diseases, from osteoarthrosis as a result of natural aging process to more complex in nature like osteochondromatosis. The primary role of articular cartilage is to provide the surface for movement in any single joint in the body. Therefore, its diseases lead to physical impairment and deterioration of the quality of life. Treatment of articular cartilage poses a formidable challenge in both modern human and animal medicine.
Keywords: Articular cartilage, cartilage pathologies, osteoarthrosis, polyarthritis rheumatoidea, hypervitaminosis A, chondromatosis, review
The articular cartilage (cartilago articularis) belongs to the group of chondral tissues (textus cartilagineus) or more broadly supporting connective tissues originating from the mesenchymal tissue (1). It is a type of hyaline cartilage (cartilago hyalina), however, what sets it apart from other tissues of this kind is the lack of perichondrium – a connective fibrous membrane serving nutritional and regenerative functions (2).
The articular cartilage is a type of tissue characterised by complex histochemistry, morphology and architecture, significant mechanical durability and elasticity, while at the same time lacking any blood vessels, which seems to stand in contradiction to the laws of biology (3).
In an adult organism, the articular cartilage covers the contacting surfaces of epiphyses constituting parts of synovial articulations – diarthrodial joints (juncturae synoviales; articulationes) (4-6). Its function is to transfer and absorb loads, and facilitate effective, friction-free action of adjacent elements of the skeletal system (2).
It is primarily composed of chondrocytes and the extracellular matrix (ECM). A characteristic trait of the articular cartilage is its small number of cells relative to the volume of extracellular matrix – actual chondrocytes comprise only 1-2% (up to 5% in some reports) of its total volume, while the ECM takes up the remaining 98-99% (1,4,7). It has been calculated that the mean number of chondrocytes in 1 cm3 of mature human cartilage is approximately 100•106 (5), individually scattered or clustered in isogenic groups known as chondrons. These constitute the primary architectonic unit of cartilaginous tissue. Within the said groups, each chondrocyte is surrounded by a thin layer of extracellular matrix that is structurally different from that located further from the cells, hence it is usually referred to as the PCM – pericellular matrix (8). The number and distribution of cells within the isogenic groups depends on the type and location of the particular cartilage. Notably, what distinguishes articular cartilage is its absence of intercellular connections and ability to function in low-oxygen environments (1).
Between 60 and 80% wet weight of the extracellular matrix consists of water, with the remaining part comprising collagen fibres (15-22% wet weight) and ground substance composed of non-collagenous proteins and polysaccharides (glycosaminoglycans) and their connections – proteoglycans and glycoproteins (4-7% wet weight). In healthy cartilage, chondroitin sulphate constitutes 65-90% of total glycosaminoglycans, with the remainder composed of keratan sulphate (5-25%) and hyaluronic acid (2-10%) (9,10). The most characteristic proteoglycan found in cartilage is aggrecan. It constitutes 10% of the cartilage dry weight and is composed of 87% chondroitin sulphate and 6% keratan sulphate. Its name refers to its capacity for aggregation, i.e. the ability to form noncovalent bonds with hyaluronic acid, the only glycosaminoglycan not forming covalence bonds with protein molecules. Approx. 200 aggrecan molecules can bind with a single hyaluronic acid molecule, thus forming multimolecular complexes known as proteoglycan aggregates. These are primarily responsible for maintaining high osmotic pressure and compressive strength (compressibility) of articular cartilage. Approx. 85% of proteoglycans present in cartilage are in the form of aggregates. The glycoproteins found in cartilage include link protein responsible for stabilising proteoglycan connections, chondronectin, chondrocalcin, fibronectin, and cartilage oligomeric matrix protein (COMP). Apart from the above, the cartilaginous matrix also contains many other structural proteins (C- and N-terminal peptides of collagen type II, fibronectin, tenascin-C, GLA proteins (vitamin K-dependent, containing gamma-carboxyglutamic acid) and functional proteins. The latter include enzymes responsible for degradation of the matrix components, inhibitors thereof, and some cytokines (7,11). The collagen fibres present in articular cartilage are primarily type II collagen – approx. 90-95% of all cartilaginous collagen. The remaining 5% include: collagen type VI, IX, X, XI, and XII (7). The presence of other types (I, III, V) is negligible, mainly detectable in immature articular cartilage.
The arrangement of collagen fibres is ordered and reflects the directions of forces acting on the cartilage. The arcade-like distribution of collagen fibres facilitates reversible deformations of the cartilage within specific physiological limits and facilitates better tensile strength and elasticity.
The most characteristic trait of articular cartilage, responsible for its unique properties, is the absence of blood and lymphatic vessels, and innervation. The provision of nutrients and removal of unneeded metabolites from chondrocytes takes place by way of diffusion, via synovial membrane synoviocytes (11). Due to this method of chondrocyte nutrition and their relatively small number, the partial pressure of oxygen in cartilaginous tissue is very low. Hence, glucose metabolism in chondrocytes takes place primarily in the form of anaerobic glycolysis (2). The distance that the diffusing nutrients from the synovial liquid can travel is limited, which consequently restricts the possible thickness of cartilaginous tissue. Based on the above, Maroudas established in 1972 that human cartilage can reach up to 4-6 mm in height (7). This physiological nutritional limit is also the cause of local cartilaginous necrosis observed in many pathological conditions in humans and animals alike.
Articular cartilage is deprived of nerve endings. Pain stimuli and proprioceptive sensibility are mediated by numerous nociceptors and proprioceptors present in other joint structures: capsule, ligament, muscle, and bone (11,12). The receptors can be stimulated mechanically (by trauma, pressure, stretching) or chemically by substances such as bradykinin, histamine, or prostaglandin E2 (11).
Under a light microscope, one can distinguish respective layers that differ in terms of both the shape and spatial arrangement of their structural elements.
Subchondral bone takes the form of a thin layer of high-density osseous tissue located between the articular cartilage and the cancellous bone of the epiphysis. It contains a large quantity of thickly woven collagen fibres and ground substance saturated with calcium and phosphorus minerals. Its task is to transfer and distribute loads between the flexible articular cartilage and stiff cancellous bone (13).
Cartilage is characterised by a very low coefficient of friction, both static (0.01-0.02) and dynamic (0.003). The same is inversely proportional to increasing load and decreases if the cartilage is coated in synovial fluid (14). Under physiological loads, articular cartilage may be compressed by up to 40% of its resting height (7). This is possible due to its high elasticity and compressibility – traits facilitated by the collagen fibre network as well as proteoglycans and their capacity to bind water molecules and cations, which allows them to maintain high osmotic pressure within the articular cartilage (15). The physiological range of loads and the resulting mechanical stimuli are necessary for the correct development of cartilage (9). Biomechanical studies conducted on cartilage cell cultures revealed that intermittent, cyclical joint loads within the physiological range condition the maintenance of homeostasis in the extracellular matrix (16). Cyclical loads on cartilaginous tissues facilitates correct circulation of articular fluid and consequently supply of oxygen and nutrients to chondrocytes. In turn, continuous loads or intermittent loads exceeding the physiological range shift the metabolic equilibrium towards catabolism (16). This results in degenerative changes to the cartilage. Conversely, immobilisation and lack of loads also weakens the cartilage and exacerbates katabolic processes (17). The application of regular loads with roughly the same strength on the articular cartilage contributes to increasing the density of proteoglycans and cells meshwork, thus increasing its durability (9). This means that similarly to bone, cartilage behaves in accordance with the commonly accepted Wolff’s law of biomechanics stating that the correct development of living organism tissues requires the application of evenly distributed pressure stretching forces (17). The maintenance of join homeostasis, including in the articular cartilage, is the responsibility of chondrocytes and synovial membrane cells, synoviocytes. They produce the components of the matrix as well as the enzymes that degrade the same (3). Cartilage contains cysteine proteases (cathepsin), serine proteases (plasminogen activator), and metalloproteases: collagenase (MMP-1), gelatinase A and B (MMP-2 and MMP-9), as well as stromelysin (MMP-3), responsible for the degradation of collagen and proteoglycans (18). This dual function is strictly regulated by cytokines and growth factors whose receptors are present on chondrocyte surfaces. The formation of cartilage is also affected by certain hormones and vitamins. The equilibrium between synthesis and degradation processes is pivotal to the correct function of the tissue.
Until recently, cartilage was believed to be a poorly immunogenic tissue. It has since been discovered, however, that collagen type II (unlike collagen type I or III), aggrecan core protein, and CH65 protein in chondrocytes contain autoantigenic determinants capable of inducting inflammatory response in experimental systems. Chondrocytes can be recognised and destroyed by cytotoxic NK (natural killer) cells. Irrespective of the same, chondrocyte surfaces show the expression of the major histocompatibility complex antigens class I and II (MHC I, II), which means that they are themselves capable of presenting antigens to T cells. This allows chondrocytes to not only act as targets of immunological response but also actively participate in the induction of the same in the course of autoimmune and degenerative diseases of the cartilaginous tissue (19).
The above qualities of the cartilage condition its functioning under the conditions of physiological homeostasis both in terms of loads and metabolic processes (3,8). Hence, the nature of cartilage diseases lies in the destabilisation of the processes of synthesis and decomposition taking place within it. Such destabilisation can result from a variety of biomechanical, autoimmune, and genetic factors (20). The most common reasons for local defects of articular cartilages include: mechanical trauma, degenerative joint conditions, osteoarthritis, and cancer (21).
Pathologies of Articular Cartilage
Osteoarthrosis. The cartilage pathology of the greatest clinical significance in both human and veterinary medicine is osteoarthrosis, also referred to as degenerative joint disease (OA/DJD). It has been estimated that OA currently affects over 25% of the adult population and is the main cause of physical impairment in patients over 40 years of age (6). It is prognosed that by 2030, approximately 20% of west European and American population will suffer from the osteoarthrosis (22). The disease entails broadly understood cartilage degeneration resulting from upset processes of its catabolism and de novo synthesis (7,23). Clinically, it is diagnosed as functional impairment of articular cartilages. It is noteworthy that the name does not refer to a single disease entity but rather a group of diseases whose underlying causes may stem from a variety of pathophysiological mechanisms. Osteoarthritis is, therefore, a syndrome with a range of possible causes that jointly contribute to a specific pathomorphological and clinical output (16,21,24,25). Moreover, osteoarthritis is a condition that affects an entire joint as a functional entity, i.e. not only the cartilage but also all the accompanying intra and extraarticular structures, including the particular muscle, nervous, and bone groups (8,25,26). The character of OA is usually primary (idiopathic) with an undefined underlying cause. It mainly affects older patients and can extend to one or more joints. Mankin et al. observed that natural ageing of an articular cartilage is not synonymous to OA as the lesions observed in the tissue are not the same (2). Secondary osteoarthritis can emerge as a result of trauma, obesity, inflammation, genetic factors, or systemic diseases (metabolic or endocrine disorders) (6). In clinical terms, the disease manifests itself through chronic pain and swelling of joints, limited joint mobility, crepitation of exposed bone surfaces, and limb deformation. The progressing process leads to significant functional impairment and deterioration of the quality of life (6,22,23,27-30). A number of biomechanisms have been suggested as the underlying causes of OA but none have been conclusively and comprehensively studied and confirmed as such, consequently an effective method of inhibiting the process of cartilage degradation and its restoration to full functional capacity has yet to be discovered (6).
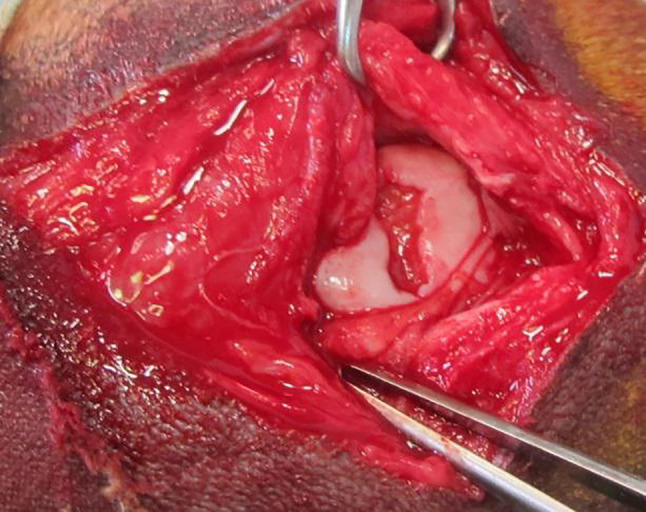
In humans, what acts as a trigger for the development of degenerative changes is a focal articular cartilage defect which is traumatic in origin (31). In domestic animals, however, apart from osteochondrosis, there are no available statistics regarding specifically trauma-induced cartilage defects. Instead, they are only observed incidentally during joint surgery or arthroscopy, even though they accompany numerous orthopaedic conditions and injuries such as cruciate ligament tear, intra-articular fracture, and other conditions leading to non-physiological load distribution (Figure 1). The resulting mechanical damage to the cartilage can occur either due to long-term effects of relatively low forces (“wear&tear”) or short-term application of a force exceeding the physiological range (3,16). The is exacerbated by: long bone misalignment, varus and valgus angulation, ligamentous instability, obesity, or meniscus damage.
Figure 1. Intraoperative view of accidentally found focal cartilage defect in the femoral trochlea of the dog.
In response to such mechanical factors, the cartilage and synovial membrane cells release inflammatory mediators such as tumour necrosis factor alpha (TNF-α), transforming growth factor-β (TGF-β) and interleukins IL1-IL-7. These, in turn, stimulate the production of metalloproteinases (MMPs) capable of degrading all the components of the ECM (21). The primary role is played by MMP1 produced by synoviocytes and MMP 13 produced by chondrocytes (6,22). Metalloproteinases MMP-2, MMP-3, and MMP-9 are in turn primarily responsible for the decomposition of non-collagenic proteins (22). A significant role in the genesis of OA is played by the characteristic cartilage collagen type II which has the ability to induce autoantibodies. In mice with a mutation of the encoding gene, the protein triggered spontaneous development of OA (6). Genetic factors include disturbance of the biochemical pathways of: TGF-β and fibroblast growth factor (FGF) as well as Indian Hedgehog (IHH) protein (6).
In experimental OA models, the first histologically observable differences included changes to the extracellular matrix and pericellular matrix: reduced concentration of proteoglycans (lowered stainability with toluidine blue), increased water content, separation and disorganisation of the surface network of collagen fibres (8,9). The first degenerative symptom observed macroscopically is the surface fibrillation of the cartilage, i.e. chondromalacia. It is caused by disarrangement of the network of collagen fibres and fibrils, separation of their chains, which in turn leads to increased intake of water molecules and formation of vertical cracks between chondrocyte columns (22). The impaired integrity of surface layers causes proliferation of the pathogenic process to deeper layers of the cartilage, forcing them to withstand excessive loads. This further reduces the content of glycosaminoglycans and triggers destructive lesions in the networks of collagen type II fibres resulting from the influence of metalloproteinases. Simultaneous changes also occur in the calcified layer of the cartilage and subchondral bone which are intensively reformed and thickened (24,26).
In orthopaedics, such lesions are referred to as subchondral bone eburnation, which in macroscopic view forms surfaces reminiscent of polished ivory or marble (2,24,26,32). The process deprives the bone of its cushioning capacity, which renders the articular cartilage more susceptible to damage (16). Jointly, the fibrillation of the cartilage and progressing eburnation of the subchondral bone are considered to constitute the irreversible stage of degenerative joint disease (9). Additionally, foci of aseptic bone necrosis and subchondral cysts are formed. At such places the articular cartilage loses the support of subchondral bone, which forms fissures through which synovia can penetrate to reach cancellous bone and induce chondrometaplastic and fibrotic changes in the mesenchymal precursor cells. As a result, macroscopically visible, nodular or cauliflower, fibrochondral thickening occurs on the surface of the cartilage in late stages of the disease. On joint surfaces opposite to those affected by subchondral bone exposure, in a way by mirror image, erosion can occur due to excessive pressure. Literature refers to the same as “kissing lesions” (2).
On the other end of the spectrum there are proliferative lesions of osteochondral tissue. These are small, initially chondral forms emerging at the border of periosteum and articular cartilage, at the attachment of the articular capsule – osteophytes, and at the attachment of tendons and ligaments to the bone – enthesophytes (16,26,33). Their shape and size are determined by the direction and intensity of forces acting on the given area of the joint. With the progressing disease process, the forms undergo secondary ossification due to excessive local mechanical loads (26) McDevitt et al. demonstrated the presence of osteophytes already on the seventh day after trauma (2). They may also appear outside the joint area, at the attachment of tendons to bone. Such exostoses are referred to as enthesophytes. The reasons for their formation and their role in the pathogenesis of the degenerative disease remain unknown (16). As the disease progresses, the articular capsule thickens, both in its fibrous and synovial membrane (2). The reformation of subchondral bone leads to active hyperaemia of the epiphysis and increase in local blood pressure. This results in the formation of fibrovascular channels in deeper layers of the cartilage and infiltration of blood vessels (neoangiogenesis) and sensory nerve endings, which is the main cause of pain experienced in the course of the degenerative disease (16).
The disease process also affects the morphology and behaviour of chondrocytes which become pleomorphic. The balance between cell death and proliferation is upset as a consequence of impaired gene expression. In regions affected by the disease process, chondrocytes become heterogenous with a different phenotype and form characteristic clusters that are symptoms of excessive proliferation and the tissue’s response to the pathological process. Paradoxically, such cellular conglomerates are incapable of rebuilding the degenerated matrix as they are anabolically inactive. Moreover, they may in fact cause the emergence of gaps, which further destabilises the cartilage (34).
In veterinary orthopaedics, one can refer to the Collins system classifying the subsequent histopathological stages in the development osteoarticular inflammation (2,32): a) damage to surface layers of the cartilage; b) chaotic increase in the number of chondrocytes; c) decrease in proteoglycan content (poorer metachromatic staining); d) blood vessels invading the layer of calcified cartilage; e) surface cracks in the cartilage; f) chondromalacia (fibrillation); g) further decrease in proteoglycan content; h) formation of chondrocyte clusters; i) exposure of subchondral bone affected by eburnation; j) formation of subchondral bone cysts.
Presently, there is yet to be a definitive determination as to the initial location of OA lesions. Some authors posit that it is the subchondral bone that initiates the degenerative process (24,26,35). Repeated joint overload leads to microfractures at early stages of the disease, whose reconstruction leads to pathological thickening, increased stiffness, and consequently weakening of the bone (16,26,35). As follows from this hypothesis, it is the subchondral bone’s loss of shock absorption capacity that triggers secondary degenerative lesions in the articular cartilage (13). This sequence of OA stages has been confirmed in a series of experimental studies on rabbits, Guineapigs, and dogs (26).
In veterinary clinical practice, OA is most commonly diagnosed in dogs, less frequently cats. Unlike humans, animal OA is usually of secondary character. It typically develops as a consequence of trauma (e.g. injury to the crucial ligament, meniscus, sprains, intraarticular fractures), congenital and developmental defects (dysplasia, osteochondrosis, Legg-Calve-Perthes disease, premature epiphyseal closure), or incorrect limb alignment (valgity, varus deformity). Additional significant factors include obesity, advanced age, and excessive physical exertion. OA is listed as the primary cause of chronic pain in dogs. In a 2009 study, between 10 and 12 million cases of degenerative diseases in dogs were reported in the USA. It is estimated that a typical veterinary practitioner in the USA will encounter approximately 45 dogs suffering from OA every month, of which 21% will be severe cases (36).
Osteoarthritis (polyarthritis rheumatoidea). Osteoarthritis (polyarthritis rheumatoidea) entails the development of chronic and progressing inflammatory lesions within various structures of the connective tissue proper, usually auto-immunologically triggered. The inflammatory process originates from the synovial membrane and its secondary progression extends to the articular cartilage, although the associated micro- and macroscopic lesions are largely similar to those observed in primary degenerative disease (37-39). It is commonly accepted in literature that due to the absence of blood vessels in the articular cartilage, inflammatory conditions do not originate therefrom. The disease foci are always located in adjacent, periarticular tissues and damage to the cartilage is secondary by nature (38). The primary role in the induction of rheumatoid inflammation is played by T cells accumulating in the synovial membrane and adhere to connective tissue fibrils. Those aggregates further attract macrophages and B cells, which results in the formation of so-called rheumatoid granulation. The cascade of immune response is triggered leading to disorders in the synthesis and deposition of proteoglycans and collagen, release of proinflammatory cytokines, prostaglandins, and cartilage-degrading enzymes. Plasmacytes produced in the granulation release the rheumatoid factor (IgG antibodies) which binds the complement system. The influx of chemotactic factors exacerbates the process of phagocytosis, liposomal enzyme activation, and release of free radicals. The increased permeability of synovial membrane vessels results in exudation which allows globulins, erythrocytes, fibroblasts, fibrinogen, and thrombocytes to infiltrate the articular cavity. The rheumatoid granulation and fibrinous proteins are consolidated to form a new tissue that can be penetrated by blood vessels. This leads to the development of articular pannus which confirms the rheumatism diagnosis (38). Clinically, polyarticular osteoarthritis can be classified as erosive or nonerosive depending on the emergence of local bone damage foci in the epiphyseal and metaphyseal region. Additionally, periosteal response can also be observed in the form of new bone formation (39,40). Clinically, particularly in the early stages, this is often misdiagnosed as degenerative joint disease. In human medicine there are a number of criteria facilitating the diagnosis of rheumatism, e.g. the 7-point symptom scale developed by the American Rheumatism Association, where the diagnosis is condition by the recognition of at least four of the same in a given patient (2). However, the same cannot be applied to canine or feline treatment as some of the symptoms do not emerge in the animals: e.g. there are no reports on the formation of subcutaneous rheumatoid nodules (2). Clinical symptoms observed in veterinary practice include: difficulty and unwillingness to move, lameness, polyarticular swelling, pain on palpation. Accompanying systemic symptoms may include fever, loss of appetite, apathy, skin inflammation, and haemolytic anaemia (39,40). Rheumatoid inflammation is relatively rare in cats and dogs, it is mostly diagnosed in the Siamese breed (40). Diagnosis is based on clinical symptoms, radiological imaging, analysis of the synovial fluid, histopathological analysis of the synovial membrane, as well as through elimination of other possible disease entities (8). Coupled with clinical symptoms, the diagnosis can be further corroborated by testing for the presence of the rheumatoid factor in blood serum, but a negative result thereof does not exclude the existence of the disease (2,40,41). Polyarticular inflammation can affect dogs suffering from systemic lupus erythematosus, in cases of Ehrlichia sp. or Borrelia burgdorferi infections, while in some breeds genetic factors have also been mentioned as the cause of immunological complexes’ accumulation in periarticular tissues and the development of sterile synovitis (Akita Inu, Boxer, Weimaraner, Bernese Mountain Dog) (39). In one paper, the author describes the diagnosis and treatment of osteoarthritis in 12 cats meeting all Bennet’s criteria relevant to this disease entity, including: subchondral bone erosion, articular capsule thickening, watery synovial fluid with high leukocyte content, positive result of rheumatoid factor test in 10 animals, limited joint mobility and pain (40,42). No correlations have been observed with viral diseases such as FELV, FIV, or FIP (40). Contrary to earlier reports, the author demonstrated clinical improvement and regression of lesions after treatment with methotrexate (a cytostatic drug, folic acid antagonist) and leflunomide (an immunosuppressant) (40).
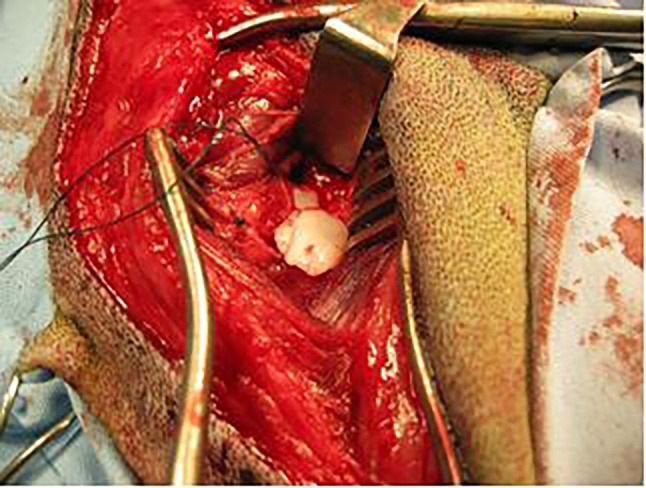
Osteochondrosis. Osteochondrosis (OCD) is a disease with significant incidence in veterinary medicine. Its therapy is the object of efforts aimed at developing potential techniques of chondral repair. The disease affects a variety of domestic animal species (dogs, pigs, horses, cattle, cats, rats) as well as humans (43,44). Osteochondrosis is often diagnosed on large and giant dog breeds. The early clinical symptoms develop in animals aged between 4 and 8 months. It can affect a number of joints: humeral, elbow, tarsal, knee, and spinal (2). The exact disease background remains unknown; hence it is treated as polyetiological. Its probable causes include: genetic factors, rapid skeleton growth relative to the muscle mass, anatomical joint characteristics, trauma, and incorrectly balanced diet (2,43,44). OCD primarily entails a local disorder in cell differentiation in the process of endochondral ossification and local subchondral bone necrosis. This leads to thickening of the cartilage and its necrosis due to malnutrition. In the last stage, the affected fragment of the cartilage is detached from its bone bed and displaced to the articular cavity (so-called articular mouse), which is accompanied by inflammatory lesions (Figure 2). This stage of the disease is described as osteochondritis dissecans – OCD (2,43). If untreated, it leads to degenerative lesions in the affected joint, increased synovial fluid production, and subchondral bone eburnation (45).
Figure 2. Intraoperative view of a detached cartilage flap during surgery of OCD of the humeral head in the dog.
Five distinct disease variants were identified in the context of the elbow joint: ununited anconeal process (UAP), fragmented medial coronoid process (FMCP), osteochondritis dissecans of the medial humeral condyle, OCD, ununited medial epicondyle (UME) joint incongruity (2). The generally accepted method of treating osteochondral defects involves removal of the detached cartilage fragment, surgical clearance of joint tissues, and drilling into the subchondral bone. The purpose of the latter is to stimulate the process of healing by triggering the production of fibrochondral tissue. Clinical improvement is observed in most patients although lameness and progressing degenerative lesions persist in some cases, which justifies efforts made in the search for more effective therapeutic methods (45).
Another condition affecting articular cartilage is cartilage calcification (chondrocalcinosis), a metabolically mediated disease. It is caused by the build-up of calcium pyrophosphate crystals in the articular cartilage and its accompanying structures – articular capsule, meniscus, ligaments, and tendons. In most cases its course is subclinical, only in advanced stages does it lead to degenerative lesions and inflammation. It bears some resemblance to a gout - a form of inflammatory arthritis, but unlike chondrocalcinosis, gout rarely extends to articular cartilage (34).
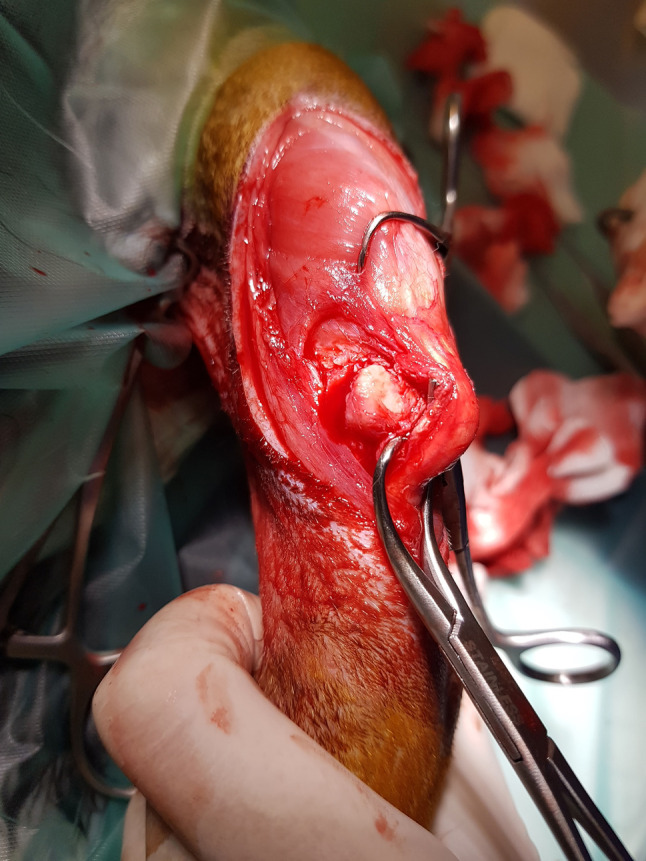
Chondromatosis. Chondromatosis is a rare disease entailing non-malignant hyperplasia of cartilaginous tissue from the synovial membrane of the articular capsule, tendon sheaths, and bursas (46-48). It may affect one or a number of joints at a time – usually knees and elbows. It is most often diagnosed in cats and tends to be accompanied by epicondylitis (49). In dogs, osteochondral hyperplasia tends to be located in the region of the epiphyseal plate and is related to endochondral ossification. Unlike in cats, it is usually self-limiting – the growth ceases once skeletal maturity is reached. There has been one confirmed case of an extraskeletal osteochondral exostosis in a dog’s trachea (50). The pathogenesis and causes of the disease remain unknown. One of the possible causes of cartilagineous exostoses proposed in human medicine is genetic – mutations of genes encoding Exostosin-1 (EXT1) and Exostosin-2 (EXT2) proteins (46,47). The possible causes of periarticular exostoses in cats proposed in some reports include nutrition, environmental factors, degenerative joint disease, obesity, as well as viral infections – feline leukaemia virus FeLV (47,50). Migrating chondrocytes colonise empty spaces between synoviocytes and, through metaplasia, form numerous chondromas – chaotically distributed clusters of chondral tissue. Over time, this may be followed by neovascularisation and gradual ossification (osteochondromatosis). Osteochodrotic lesions may detach from the stroma and migrate, as bodies, to the articular cavity (48). This causes pain, lameness, reduced joint mobility, and secondary degenerative disease. As the disease progresses, numerous fibrochondromas which cause deformation of and pressure on periarticular structures such as ligaments and nerves, which in turn leads to joint destabilisation, secondary sprains and limb paralysis (Figure 3) (49,50). Pressure and irritation may result in local erosion of healthy cartilage (so-called “kissing lesions”) (45) and development of secondary degenerative joint disease (48). It has been estimated that approximately 20% of osteochondral exostoses in cats have a tendency towards malignant transformation into chondrosarcoma and osteosarcoma (50,51) as well as fibrosarcoma (47). The treatment of osteochondromatosis entails surgical removal of chondral formations, where possible together with the fragments of metaplastically altered synovial membrane (48).
Figure 3. Intraoperative view of a large fibrocartilage tissue lying under the patellar ligament in the cat during the course of osteochondromatosis.
Hypervitaminosis A. Another characteristic feline joint disease is hypervitaminosis A. It is caused by excessive vitamin A supply in the diet, typically affecting cats consuming large amounts of liver or receiving vitamin A diet supplements (52,53). The disease has been first described as metabolic osteopathy in the 1950s in cats fed exclusively with milk and raw liver (beef, poultry, pork) (52). It is most commonly diagnosed in cats aged between 2 and 9 years, without significant predilections in terms of sex or breed (52). For reasons yet to be discovered, it triggers periosteal response and formation of individual or multiple osteochondral outgrowths (exostoses, osteophytes, enthesophytes). They usually emerge in spinal, mainly cervical and thoracic, joints and cause spondylopathies (52-54), but can also form around large joints such as knees or elbows (e.g. in the intrapatellar fat body). Symptoms depend on the location and are similar to those observed in osteochondromatosis, i.e. pain, erosion of the articular cartilage, progressing joint stiffness (ankylosis), and secondary degenerative lesions (52,53). The newly emerging osteochondral lesions can press against or expand to periarticular structures such as nerves, nerve plexuses and muscles, causing chronic pain, lameness and paralysis, paresis, as well as other neurological symptoms such as anisocoria and Horner’s syndrome (52). Due to the immobilisation of spinal joints, cats suffering from the disease tend to assume the so-called marsupial-like posture, with the spine bent, head lowered, and thoracic limbs raised. In cases where the proliferative lesions on ventral surfaces of cervical vertebra press on the oesophagus and larynx, symptoms including voice alteration, swallowing disorders and regurgitation (dysphagia) have been reported. A case of a cat suffering from hypervitaminosis A has been described where incarceration of the shoulder plexus in the osteochondral mass was observed (52). Correlations between long-term oversupply of vitamin A in the diet and hepatotoxicity, nephrotoxicity, and hypercalcaemia have been mentioned in literature. A currently unknown mechanism leads to fibrosis of the hepatic and renal parenchyma, thickening of organ vessels, local cell necrosis, and accumulation of calcium deposits in parenchymal organs. Said mechanism has been confirmed in an experiment conducted on rats (55), a case of hepatocyte lipidosis and fibrosis has also been described in a cat in relation to excessive consumption of vitamin A (55). Prognoses tend to be doubtful to unfavourable given the fact that the massive osteochondral formations prevent full functional recovery of the affected joints. However, cases of spontaneous remission after diet correction have also been reported (52).
Diseases related to articular cartilage pathologies continue to be subject to scientific scrutiny. Effective treatment is yet to be discovered, as is the exact aetiology of many of the conditions. At the same time, their incidence on both humans and animals is growing due to increasing life expectancy as well as factors related to physical activity and environmental pollution.
Conflicts of Interest
The Authors declare that they have no conflict of interest in regard to this study.
Authors’ Contributions
B. Żylińska (first author): Study design, literature research, data analysis, editing and writing of the article. A. Sobczyńska-Rak (corresponding author): Literature research, editing, writing and proofreading of the article. U. Lisiecka: Co-editing, proofreading of the article. E. Stodolak-Zych: Data analysis and proofreading of the article. Ł. Jarosz: Literature research and proofread the article. T. Szponder: literature research and proofreading of the article. All Authors made pertinent contributions to the article, and proofread and approved the final article before submission.
References
- 1.Bhosale AM, Richardson JB. Articular cartilage: Structure, injuries and review of management. Br Med Bull. 2008;87:77–95. doi: 10.1093/bmb/ldn025. [DOI] [PubMed] [Google Scholar]
- 2.DeCamp CE. Saunders Elsevier LTD, 5th edition, Oxford. 2016. Brinker, Piermattei and Flo’s Handbook of small animal orthopedics and fracture repair. [Google Scholar]
- 3.Marczyński W. Pathology of articular cartilage – dynamics of changes, prevention. Wiad Lek. 2007;60(1-2):53–59. [PubMed] [Google Scholar]
- 4.Ge Z, Li C, Heng BC, Cao G, Yang Z. Functional biomaterials for cartilage regeneration. J Biomed Mater Res A. 2012;100(9):2526–2536. doi: 10.1002/jbm.a.34147. [DOI] [PubMed] [Google Scholar]
- 5.Chiang H, Jiang CC. Repair of articular cartilage defects: Review and perspectives. J Formos Med Assoc. 2009;108(2):87–101. doi: 10.1016/S0929-6646(09)60039-5. [DOI] [PubMed] [Google Scholar]
- 6.Chen D, Shen J, Zhao W, Wang T, Han L, Hamilton JL, Im HJ. Osteoarthritis: Toward a comprehensive understanding of pathological mechanism. Bone Res. 2017;5:16044. doi: 10.1038/boneres.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mollenhauer JA. Perspectives on articular cartilage biology and osteoarthritis. Injury. 2008;39(Suppl 1):S5–12. doi: 10.1016/j.injury.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 8.Guilak F, Nims RJ, Dicks A, Wu CL, Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. 2018;71-72:40–50. doi: 10.1016/j.matbio.2018.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arokoski JP, Jurvelin JS, Väätäinen U, Helminen HJ. Normal and pathological adaptations of articular cartilage to joint loading. Scand J Med Sci Sports. 2000;10(4):186–198. doi: 10.1034/j.1600-0838.2000.010004186.x. [DOI] [PubMed] [Google Scholar]
- 10.Zeyland J, Lipiński D, Juzwa W, Pławski A, Słomski R. Structure and application of select glycosaminoglycans. Med Wet. 2006;62(2):139–144. [Google Scholar]
- 11.Tyyni A, Karlsson J. Biological treatment of joint cartilage damage. Scand J Med Sci Sports. 2000;10(5):249–265. doi: 10.1034/j.1600-0838.2000.010005249.x. [DOI] [PubMed] [Google Scholar]
- 12.Kalson NS, Gikas PD, Briggs TW. Current strategies for knee cartilage repair. Int J Clin Pract. 2010;64(10):1444–1452. doi: 10.1111/j.1742-1241.2010.02420.x. [DOI] [PubMed] [Google Scholar]
- 13.Swieszkowski W, Tuan BH, Kurzydlowski KJ, Hutmacher DW. Repair and regeneration of osteochondral defects in the articular joints. Biomol Eng. 2007;24(5):489–495. doi: 10.1016/j.bioeng.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 14.Krishnan R, Mariner EN, Ateshian GA. Effect of dynamic loading on the frictional response of bovine articular cartilage. J Biomech. 2005;38(8):1665–1673. doi: 10.1016/j.jbiomech.2004.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han EH, Chen SS, Klisch SM, Sah RL. Contribution of proteoglycan osmotic swelling pressure to the compressive properties of articular cartilage. Biophys J. 2011;101(4):916–924. doi: 10.1016/j.bpj.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Łapaj Ł, Markuszewski J, Wierusz-Kozłowska M. Current views on the pathogenesis of osteoarthritis. Chir Narzadow Ruchu Ortop Pol. 2010;75(4):248–260. [PubMed] [Google Scholar]
- 17.Trzaskacz T. The influence of degeneration of synovial joints on his tribological properties. Doctoral dissertation. Department of machine design and research, Wroclaw University of Technology, Faculty of Mechanical Engineering 15/2005. Available at: https://www.dbc.wroc.pl/dlibra/publication/1133/edition/1356/content?&action=ChangeMetaLangAction&lang=pl [Last accessed on March 3, 2021]
- 18.Ito A, Nose T, Takahashi S, Mori Y. Cyclooxygenase inhibitors augment the production of pro-matrix metalloproteinase 9 (progelatinase B) in rabbit articular chondrocytes. FEBS Lett. 1995;360(1):75–79. doi: 10.1016/0014-5793(95)00085-n. [DOI] [PubMed] [Google Scholar]
- 19.Lance EM, Kimura LH, Manibog CN. The expression of major histocompatibility antigens on human articular chondrocytes. Clin Orthop Relat Res. 1993;(291):266–282. [PubMed] [Google Scholar]
- 20.Widuchowski W, Tomaszewski W, Widuchowski J, Czamara A. Contemporary possibilities of treatment of cartilage lesions with special regard to the knee joint. Ortopedia Traumatologia Rehabilitacja. 2017;13(4):327–341. doi: 10.5604/15093492.955721. [DOI] [PubMed] [Google Scholar]
- 21.Cai X, Lin Y, Ou G, Luo E, Man Y, Yuan Q, Gong P. Ectopic osteogenesis and chondrogenesis of bone marrow stromal stem cells in alginate system. Cell Biol Int. 2007;31(8):776–783. doi: 10.1016/j.cellbi.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 22.Mehana EE, Khafaga AF, El-Blehi SS. The role of matrix metalloproteinases in osteoarthritis pathogenesis: An updated review. Life Sci. 2019;234:116786. doi: 10.1016/j.lfs.2019.116786. [DOI] [PubMed] [Google Scholar]
- 23.Kubomura D, Ueno T, Yamada M, Nagaoka I. Evaluation of the chondroprotective action of N-acetylglucosamine in a rat experimental osteoarthritis model. Exp Ther Med. 2017;14(4):3137–3144. doi: 10.3892/etm.2017.4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldring MB, Goldring SR. Articular cartilage and subchondral bone in the pathogenesis of osteoarthritis. Ann NY Acad Sci. 2010;1192:230–237. doi: 10.1111/j.1749-6632.2009.05240.x. [DOI] [PubMed] [Google Scholar]
- 25.Hardingham TE. Fell-Muir lecture: Cartilage 2010 - the known unknowns. Int J Exp Pathol. 2010;91(3):203–209. doi: 10.1111/j.1365-2613.2010.00719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Felson DT, Neogi T. Osteoarthritis: Is it a disease of cartilage or of bone. Arthritis Rheum. 2004;50(2):341–344. doi: 10.1002/art.20051. [DOI] [PubMed] [Google Scholar]
- 27.Baltzer AW, Moser C, Jansen SA, Krauspe R. Autologous conditioned serum (Orthokine) is an effective treatment for knee osteoarthritis. Osteoarthritis Cartilage. 2009;17(2):152–160. doi: 10.1016/j.joca.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Haasper C, Zeichen J, Meister R, Krettek C, Jagodzinski M. Tissue engineering of osteochondral constructs in vitro using bioreactors. Injury. 2008;39 Suppl 1:S66–S76. doi: 10.1016/j.injury.2008.01.037. [DOI] [PubMed] [Google Scholar]
- 29.Shah MR, Kaplan KM, Meislin RJ, Bosco JA 3rd. Articular cartilage restoration of the knee. Bull NYU Hosp Jt Dis. 2007;65(1):51–60. [PubMed] [Google Scholar]
- 30.Euppayo T, Siengdee P, Buddhachat K, Pradit W, Chomdej S, Ongchai S, Nganvongpanit K. In vitro effects of triamcinolone acetonide and in combination with hyaluronan on canine normal and spontaneous osteoarthritis articular cartilage. In Vitro Cell Dev Biol Anim. 2016;52(7):723–735. doi: 10.1007/s11626-016-0022-4. [DOI] [PubMed] [Google Scholar]
- 31.Chu CR, Szczodry M, Bruno S. Animal models for cartilage regeneration and repair. Tissue Eng Part B Rev. 2010;16(1):105–115. doi: 10.1089/ten.TEB.2009.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pritzker KP, Gay S, Jimenez SA, Ostergaard K, Pelletier JP, Revell PA, Salter D, van den Berg WB. Osteoarthritis cartilage histopathology: Grading and staging. Osteoarthritis Cartilage. 2006;14(1):13–29. doi: 10.1016/j.joca.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 33.Benjamin M, Toumi H, Ralphs JR, Bydder G, Best TM, Milz S. Where tendons and ligaments meet bone: attachment sites (‘entheses’) in relation to exercise and/or mechanical load. J Anat. 2006;208(4):471–490. doi: 10.1111/j.1469-7580.2006.00540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Archer C, Ralphs J. Woodhead Publishing Limited, Cambridge. 2010. Regenerative medicine and biomaterials for the repair of connective tissues; p. pp. 83. [Google Scholar]
- 35.Westacott C. Interactions between subchondral bone and cartilage in OA. Cells from osteoarthritic bone can alter cartilage metabolism. J Musculoskelet Neuronal Interact. 2002;2(6):507–509. [PubMed] [Google Scholar]
- 36.Fox S.M, Millis D. London, UK. 2010. Multimodal management of canine osteoarthritis. Manson Publishing/The Veterinary Press (eds.) p. pp. 41. [Google Scholar]
- 37.van Holten J, Plater-Zyberk C, Tak PP. Interferon-beta for treatment of rheumatoid arthritis. Arthritis Res. 2002;4(6):346–352. doi: 10.1186/ar598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sudoł-Szopińska I, Zaniewicz-Kaniewska K, Warczyńska A, Matuszewska G, Saied F, Kunisz W. The pathogenesis of rheumatoid arthritis in radiological studies. Part II: Imaging studies in rheumatoid arthritis. J Ultrason. 2012;12(50):319–328. doi: 10.15557/JoU.2012.0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stull JW, Evason M, Carr AP, Waldner C. Canine immune-mediated polyarthritis: Clinical and laboratory findings in 83 cases in western Canada (1991-2001) Can Vet J. 2008;49(12):1195–1203. [PMC free article] [PubMed] [Google Scholar]
- 40.Hanna FY. Disease modifying treatment for feline rheumatoid arthritis. Vet Comp Orthop Traumatol. 2005;18(2):94–99. [PubMed] [Google Scholar]
- 41.Bennett D, Kirkham D. The laboratory identification of serum rheumatoid factor in the dog. J Comp Pathol. 1987;97(5):541–550. doi: 10.1016/0021-9975(87)90004-1. [DOI] [PubMed] [Google Scholar]
- 42.DIAGNOSTIC criteria for rheumatoid arthritis. 1958 revision by a committee of the American Rheumatism Association. Ann Rheum Dis. 1959;18(1):49–51; French transl 51-2. Spanish transl 52-3. [PMC free article] [PubMed] [Google Scholar]
- 43.Ytrehus B, Carlson CS, Ekman S. Etiology and pathogenesis of osteochondrosis. Vet Pathol. 2007;44(4):429–448. doi: 10.1354/vp.44-4-429. [DOI] [PubMed] [Google Scholar]
- 44.Nečas A, Dvořák M, Zatloukal J. Incidence of osteochondrosis in dogs and its late diagnosis. Acta Veterinaria Brno. 2018;68(2):131–139. doi: 10.2754/avb199968020131. [DOI] [Google Scholar]
- 45.Fitzpatrick N, Yeadon R, Smith TJ. Early clinical experience with osteochondral autograft transfer for treatment of osteochondritis dissecans of the medial humeral condyle in dogs. Vet Surg. 2009;38(2):246–260. doi: 10.1111/j.1532-950X.2008.00492.x. [DOI] [PubMed] [Google Scholar]
- 46.Hameetman L, Bovée JV, Taminiau AH, Kroon HM, Hogendoorn PC. Multiple osteochondromas: Clinicopathological and genetic spectrum and suggestions for clinical management. Hered Cancer Clin Pract. 2004;2(4):161–173. doi: 10.1186/1897-4287-2-4-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laeta M, Kompanje EJO, Watson A, Souza SMFM, Dittmar K, Cuenca SC, Hassel LB, Siciliano S. Osteochondromatosis (multiple cartilaginous exostoses) in an immature killer whale Orcinus orca. Dis Aquat Organ. 2019;134(3):209–213. doi: 10.3354/dao03372. [DOI] [PubMed] [Google Scholar]
- 48.Ryan RS, Harris AC, O’Connell JX, Munk PL. Synovial osteochondromatosis: The spectrum of imaging findings. Australas Radiol. 2005;49(2):95–100. doi: 10.1111/j.1440-1673.2005.01416.x. [DOI] [PubMed] [Google Scholar]
- 49.Perry KL. Surgical management of medial humeral epicondylitis, cubital synovial osteochondromatosis and humeroradial subluxation in a cat. JFMS Open Rep. 2017;3(1):2055116917695053. doi: 10.1177/2055116917695053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosa C, Kirberger RM. Extraskeletal osteochondroma on a cat´s elbow. J S Afr Vet Assoc. 2012;83(1):104. doi: 10.4102/jsava.v83i1.104. [DOI] [PubMed] [Google Scholar]
- 51.Ranade S, Pacchiana P. What Is Your Diagnosis. Journal of the American Veterinary Medical Association. 2011;238(10):1243–1244. doi: 10.2460/javma.238.10.1243. [DOI] [PubMed] [Google Scholar]
- 52.Polizopoulou ZS, Kazakos G, Patsikas MN, Roubies N. Hypervitaminosis A in the cat: A case report and review of the literature. J Feline Med Surg. 2005;7(6):363–368. doi: 10.1016/j.jfms.2005.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guerra JM, Daniel AG, Aloia TP, de Siqueira A, Fukushima AR, Simões DM, Reche-Júior A, Cogliati B. Hypervitaminosis A-induced hepatic fibrosis in a cat. J Feline Med Surg. 2014;16(3):243–248. doi: 10.1177/1098612X13516121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Allevi G, Serafini F. Polyostotic cortical hyperostosis in an 8-week-old cat with a 3-year follow-up. J Small Anim Pract. 2021;62(1):59–64. doi: 10.1111/jsap.13013. [DOI] [PubMed] [Google Scholar]
- 55.Ahmed RA. Histological assessment and quantification of hypervitaminosis A-induced fibrosis in liver, kidney and testis of albino rats. World J Pharm Sci. 2017;5(8):209–220. [Google Scholar]