

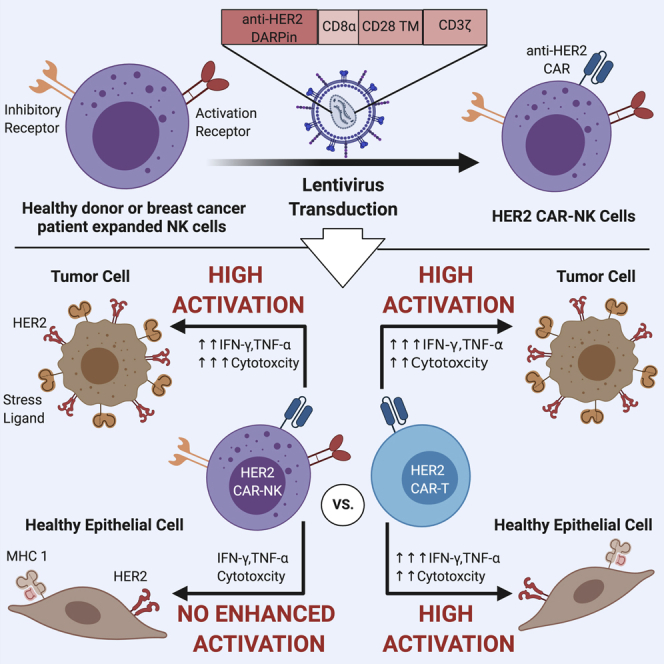
Summary
Despite the remarkable success of chimeric antigen receptor (CAR)-T cells against hematologic malignancies, severe off-tumor effects have constrained their use against solid tumors. Recently, CAR-engineered natural killer (NK) cells have emerged as an effective and safe alternative. Here, we demonstrate that HER2 CAR-expression in NK cells from healthy donors and patients with breast cancer potently enhances their anti-tumor functions against various HER2-expressing cancer cells, regardless of MHC class I expression. Moreover, HER2 CAR-NK cells exert higher cytotoxicity than donor-matched HER2 CAR-T cells against tumor targets. Importantly, unlike CAR-T cells, HER2 CAR-NK cells do not elicit enhanced cytotoxicity or inflammatory cytokine production against non-malignant human lung epithelial cells with basal HER2 expression. Further, HER2 CAR-NK cells maintain high cytotoxic function in the presence of immunosuppressive factors enriched in solid tumors. These results show that CAR-NK cells may be a highly potent and safe source of immunotherapy in the context of solid tumors.
Subject areas: Immunology, Cancer
Graphical abstract
Highlights
-
•
Primary HER2 CAR-NK cells from patients with cancer have potent anti-tumor functions
-
•
HER2 CAR-NK cells have a higher tumor killing capacity than HER2 CAR-T cells
-
•
HER2 CAR-NK cells are not overly activated against HER2+ lung epithelial cells
-
•
CAR-NK cells can overcome inhibition by the immunosuppressive factors TGF-β and PGE2
Immunology; Cancer
Introduction
Natural killer (NK) cells play a critical role in cancer immunosurveillance through their innate ability to recognize and kill malignant cells without prior antigen sensitization (Vivier et al., 2008). Hence, NK cell-based cancer immunotherapy has been of recent interest due to their selective and potent anti-tumor activity. The adoptive transfer of highly activated, feeder cell-expanded NK cells has shown high clinical efficacy against hematologic malignancies (Ciurea et al., 2017). However, the use of expanded NK cells against solid tumors has been limited thus far, largely due to the harsh immunosuppressive tumor microenvironment (TME). The TME consists of a variety of different cell types including tumor cells, stromal cells, and tumor-induced immunosuppressive immune cells that can release immunosuppressive cytokines and factors such as transforming growth factor (TGF)-β and prostaglandin E2 (PGE2) (Binnewies et al., 2018). Studies using human cytokine-activated NK cells show that TGF-β and PGE2 directly inhibit NK cell-mediated cytotoxicity through a downregulation of activation receptor expression and reduce interferon-γ (IFN-γ) cytokine production (Joshi et al., 2001; Lee et al., 2004; Martinet et al., 2010; Park et al., 2011).
Immune effector cells can be specifically redirected and activated against tumor cells through the expression of recombinant chimeric antigen receptors (CARs). CARs consist of an extracellular binding domain which recognizes a tumor-specific antigen and intracellular signaling domains which mediate cell activation leading to effector function. CAR-engineered T cells have shown remarkable clinical efficacy against hematological malignancies, leading to U.S. Food and Drug Administration (FDA) approval of three anti-CD19 CAR-T cell products for B-cell lymphomas and leukemia (Maude et al., 2014; Neelapu et al., 2017; Wang et al., 2020). Despite these successes, the use of CAR-T cells for solid tumors has proven challenging due to the uncontrolled nature of CAR-T cell activity upon engagement with the target antigen. In fact, previous treatment with HER2 CAR-T cells in metastatic colon cancer has caused life-threatening adverse effects, likely due to the recognition and attack of normal lung tissue which expresses basal levels of HER2 (Morgan et al., 2010). Indeed high-grade toxicities, namely cytokine release syndrome (CRS) and neurotoxicity are severe and at times, lethal complications from CAR-T cell therapy (Bonifant et al., 2016). CRS is mediated by the extensive release of inflammatory cytokines such as IFN-γ, TNF-α, and IL-6, resulting from uncontrolled immune cell activation following target-dependent CAR-T cell expansion and activation (Bonifant et al., 2016; Giavridis et al., 2018; Titov et al., 2018).
Given the inherent ability of NK cells to discriminate between healthy and malignant cells, NK cells are being considered as a safe alternative for CAR-based therapies. NK cells express germ-line encoded activation and inhibitory receptors which orchestrate their activation through the balance of activating and inhibitory signaling (Vivier et al., 2008). NK cell activation receptors recognize stress-induced ligands on the surface of malignant target cells, while their inhibitory receptors constrain their killing toward normal cells expressing self-major histocompatibility complex (MHC) class I molecules (Vivier et al., 2008). Previously, adoptive transfer of allogeneic NK cells has shown to be safe in clinical settings, with low incidence of graft-versus-host disease (GVHD) or other toxicities (Lee et al., 2016; Miller et al., 2005). Recently, umbilical cord blood derived anti-CD19 CAR-NK cells have shown impressive clinical efficacy leading to a 73% objective response rate in B-cell lymphoid malignancies (Liu et al., 2020). Notably, a high safety prolife was observed in which none of the treated patients developed CRS or neurotoxicity-related symptoms (Liu et al., 2020). Yet despite these current advances in CAR-NK cell therapy, their efficacy and safety against solid tumors have been less explored.
In this study, we evaluated the efficacy and safety of human epidermal growth factor receptor 2 (HER2)-specific CAR-NK cells generated from ex vivo expanded NK cells derived from healthy donors and patients with breast cancer. We chose HER2 as the target antigen in this study as a proof of concept, as HER2 is expressed in low levels in some normal tissues and overexpressed in various carcinomas of epithelial origin including breast, ovarian, and gastric cancer (Press et al., 1990; Yan et al., 2015). In breast cancer, HER2 overexpression accounts for 15–30% of cases and is a predictor of poor clinical outcome and reduced survival rates (Freudenberg et al., 2009). We hypothesized that expanded NK cells engineered to express the HER2 CAR will be specific against HER2-expressing cancer cells, exert high cytotoxicity in the presence of immunosuppressive factors, and have minimal to no toxicity against non-malignant cells. We first assessed HER2 CAR-NK cell anti-tumor functions against various cancer cell lines with varying levels of HER2 and MHC I expression. To investigate off-tumor effects, we tested HER2 CAR-NK cell killing against non-malignant human bronchial epithelial cells expressing low levels of target antigen. Additionally, we tested whether HER2 CAR-NK cells can overcome the immunosuppressive effects of factors present in the TME. We demonstrate that HER2 CAR-expression enhances the cytotoxicity of highly activated expanded NK cells against breast cancer cells, and unlike HER2 CAR-T cells, they exhibit minimal activation toward normal cells.
Results
HER2 CAR expression enhances the anti-tumor functions of primary expanded NK cells from healthy donors and patients with breast cancer
Our group has previously demonstrated that K562-mb-IL21 expanded NK cells from healthy donors and patients with cancer exert potent anti-tumor effects in pre-clinical human ovarian and breast cancer models (Nham et al., 2018; Poznanski et al., 2018; Shenouda et al., 2017). We first assessed the cytotoxicity and IFN-γ production of unmodified expanded NK cells compared to the human NK-92 cell line, as CAR-engineered NK-92 cells have been shown to exert robust cytotoxicity against a variety of target tumor cells in pre-clinical studies and can be continuously expanded in culture (Zhang et al., 2017; Zhang et al., 2019). We generated HER2 CAR-NK-92 cells and control NK-92 cells (Figures S1A and S1B) by transduction with a lentivirus construct containing the HER2 CAR or a control vector containing only NGFR which lacks an intracellular signaling domain (Figure 1A). We found that unmodified expanded NK cells exhibited significantly higher cytotoxicity and IFN-γ release (Figures S1C and S1D) against the HER2-overexpressing SKBR3 breast cancer cell line compared to HER2 CAR-NK-92 cells. Further, the unmodified expanded NK cells exhibited a trend toward higher killing and IFN-γ release in response to triple-negative MDA-MD-231 breast cancer cells in comparison to parental NK-92 cells (Figures S1E and S1F). These findings suggest that K562-mb-IL21 expanded NK cells may be a potent source for CAR-based immunotherapy.
Figure 1.

Primary NK cells from healthy donors and patients with breast cancer engineered with a HER2-specific CAR have strong anti-tumor functions against HER2+ breast cancer cells
(A) Schematic representation of the lentiviral vector containing the HER2 CAR and truncated NGFR as a transduction control.
(B) Representative histograms of the transgene expression on HER2 CAR or control vector transduced NK cells, compared to non-transduced controls.
(C–J) (C and G) HER2 CAR and NGFR expression on NK cells was assessed four days following transduction. (D and H) Cell-mediated cytotoxicity of HER2 CAR-, control vector transduced-, and non-transduced NK cells from healthy donors or breast cancer patients against SKBR3 cells. Graphs show percent-specific lysis for NK cells from two representative donors. (E and I) Relative change in specific lysis for HER2 CAR- and control vector transduced-NK cells compared to the non-transduced control. (F and J) Relative change in IFN-γ release was calculated for HER2 CAR- and control vector transduced-NK cells compared to non-transduced controls. Data represent mean ± SEM of seven to fourteen biological replicates per condition.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (C and G, two-sided t tests; E, F, I, and J, one-way ANOVA with Tukey's post hoc tests). ns, no significant difference.
See also Figures S1 and S2.
We next tested whether the additional activating signal provided by a CAR would enhance the anti-tumor functions of the already highly activated expanded NK cells. We transduced healthy donor expanded NK cells with the HER2 CAR or the control vector lentiviral construct, obtaining a mean transduction efficiency of 41.4% (range: 21.4–66.4%) and 35.2% (range: 19–52.3%), respectively (Figures 1B and 1C). To evaluate the functionality of HER2 CAR-NK cells, we tested their cytotoxicity and IFN-γ production against the HER2-overexpressing SKBR3 cells. Due to the high sensitivity of SKBR3 cells to expanded NK cell-mediated killing, low effector-to-target (E:T) ratios were chosen to better assess differences in killing between the HER2 CAR- and the control vector transduced- or non-transduced NK cells. HER2 CAR-NK cells killed significantly higher than control vector transduced- and non-transduced controls, with a 1.7-fold mean increase in cytotoxicity relative to the non-transduced-NK cells (Figures 1D and 1E). Additionally, HER2 CAR-NK cells released significantly greater amounts of IFN-γ after 5 hr incubation with HER2-positive breast cancer cells, with a 1.6-fold mean increase in IFN-γ production relative to the non-transduced controls (Figure 1F). There was no significant difference in cytotoxicity and IFN-γ release between the control vector transduced- and non-transduced NK cells.
Given the high transduction efficiency and enhanced function of healthy donor HER2 CAR-NK cells, we assessed whether the same effect would be observed in CAR-NK cells generated from six breast cancer patients (BCP). Expanded BCP-NK cells had similar transduction efficiencies compared to healthy donors (Figure 1C), with a mean 50% (range: 27.3–68.4%) HER2 CAR and mean 33.5% (range: 23.4–43.3%) NGFR transgene expression (Figure 1G). HER2 CAR expression increased expanded BCP-NK cell-mediated cytotoxicity at different E:T ratios, with a significant 1.5-fold mean increase in killing relative to non-transduced controls (Figures 1H and 1I). BCP HER2 CAR-NK cells also produced significantly greater amounts of IFN-γ than non-transduced NK cells (Figure 1J). Of note, there was some level of non-specific activation by the lentiviral transduction as BCP-NK cells transduced with the control vector exhibited a trend for increased killing and IFN-γ release relative to non-transduced NK cells.
To investigate the non-specific enhancement in anti-tumor function observed, we measured the expression of various NK cell activation and inhibitory receptors in expanded NK cells after lentiviral transduction with the control vector. We found no apparent differences in the expression of CD16, NKG2D, NKp30, NKp44, NKp46 and CD69 activation markers (Figures S2A and S2B) or NKG2A, CD158a, CD158b and CD158e1 inhibitory markers (Figures S2C and S2D) in control vector transduced-NK cells (NGFR-positive) compared to the untransduced population (NGFR-negative) and non-modified NK cells. We also assessed the expression of the Glut1, CD71, and CD98 cell surface nutrient receptors as it is becoming increasingly clear that the metabolism of NK cells is an important determinant of their anti-tumor function (Poznanski and Ashkar, 2019). Additionally, our lab has demonstrated that higher expression of these nutrient receptors in expanded NK cells compared to cytokine-activated NK cells contributes to their potent cytotoxic function (Poznanski et al., 2021a). Interestingly, we observed higher expression of the nutrient receptors CD71 and CD98 specifically in the control vector transduced, NGFR-positive NK cell population (Figures S2E and S2F). This suggests that lentiviral transduction of expanded NK cells may increase their metabolic function, consequently leading to some enhancement in killing and cytokine production.
Altogether, these results show that HER2 CAR expression heightens the anti-tumor functions of already highly cytotoxic expanded NK cells, in both healthy donors and patients with breast cancer.
HER2 CAR expression enhances expanded NK cell cytotoxicity against various HER2-expressing cancer cell lines
We next assessed the killing capacity of HER2 CAR-NK cells against other breast cancer and ovarian cancer cell lines with varying levels of HER2 (Figure 2A) and MHC class I (Figure S3) expression. We generated HER2 CAR-NK cells from healthy donor expanded NK cells with a minimum transduction efficiency of 15.6% for the HER2 CAR construct (Figures 2B and 2E) and assessed their cytotoxicity at different E:T ratios. There was no significant difference in killing between HER2 CAR-NK cells and non-transduced controls against the triple-negative MDA-MB-231 breast cancer cell line which lowly expresses HER2 (Figures 2C and 2D). Against the highly NK cell-resistant and HER2-expressing BT-474 breast cancer cell line, HER2 CAR-NK cells exhibited a significant 1.9-fold mean increase in killing relative to non-transduced expanded NK cells (Figures 2F and 2G). We further wanted to test the cell-mediated killing of HER2 CAR-NK cells against a cancer cell line, which does not downregulate MHC class I. We chose the SKOV-3 ovarian cancer cell line which overexpresses HER2 and expresses MHC class I significantly higher than SKBR3 breast cancer cells (Figure S3B). Against this cell line, HER2 CAR-NK cells demonstrated a significant 1.5-fold mean increase in killing compared to non-transduced controls (Figures 2H and 2I). These results demonstrate that HER2 CAR-NK cells can efficiently target both breast and ovarian cancer cells expressing the HER2 target antigen irrespective of MHC class I expression.
Figure 2.

HER2 CAR-NK cell killing against HER2-low and HER2-overexpressing breast cancer and ovarian cancer cell lines
(A) Histograms of HER2 expression on SKBR3 breast cancer, triple-negative MDA-MB-231 breast cancer, BT-474 breast cancer, and SK-OV-3 ovarian cancer cells.
(B–I) (B and E) HER2 CAR and NGFR transgene expression on healthy donor expanded NK cells. (C, F, and H) Cell-mediated cytotoxicity of HER2 CAR-, control vector transduced-, and non-transduced controls against MDA-MB-231, BT-474, or SK-OV-3 cancer cell lines. Graphs show percent-specific lysis for NK cells from one representative donor. (D, G, and I) Relative change in specific lysis was calculated for HER2 CAR- and control vector transduced-NK cells compared to non-transduced controls at a 1:1 (E)T ratio for each cell line. Data represent mean ± SEM of three to four biological replicates per condition.
∗p < 0.05 (B and E, two-sided t tests; D, G, and I, one-way ANOVA with Tukey's post hoc tests). ns, no significant difference.
See also Figure S3.
HER2 CAR-NK cells show greater cytotoxic function than HER2 CAR-T cells
CAR-T cell therapy has revolutionized the field of cancer immunotherapy, leading to remarkable clinical success against hematologic malignancies. Despite lower efficacy in solid tumors, HER2-redirected CAR-T cells have shown to be highly specific and demonstrate robust killing and cytokine production in response to activation by target cells in vitro and in vivo (Hammill et al., 2015; Helsen et al., 2018). To evaluate the activation potential of human HER2 CAR-NK cells, we generated donor-matched HER2 CAR-NK and -T cells and compared killing and cytokine production against HER2-positive SKBR3 breast cancer cells. HER2 CAR transgene expression was similar in NK cells compared to CD4+ and CD8+ T cell populations in the matched donors, although the fluorescence intensity of the HER2 CAR transgene was lower in NK cells compared to T cells (Figures 3A and 3B). Despite the difference in HER2 CAR expression, HER2 CAR-NK cells exhibited significantly higher cell-mediated killing of the SKBR3 target cells compared to donor-matched HER2 CAR-T cells at all E:T ratios tested (Figure 3C). This suggests that HER2 CAR-NK cells have a lower activation threshold. While HER2 CAR-NK cells exhibited potent tumor cell killing, IFN-γ and TNF-α production remained low (Figures 3D and 3E). This was in contrast to HER2 CAR-T cells which released extensive amounts of IFN-γ (mean: 4,400 pg/mL) and TNF-α (mean: 2,000 pg/mL), despite having low cytotoxicity. While inflammatory cytokine production is necessary for tumor immune surveillance, sustained exposure to these inflammatory cytokines can lead to severe CRS. These results show that HER2 CAR-NK cells have a high capacity to respond to HER2-positive target cells, leading to a high degree of specific lysis, but elicit a more controlled cytokine response.
Figure 3.

HER2 CAR-NK cells have greater killing against HER2-positive breast cancer target cells compared to HER2 CAR-T cells
(A) Representative histograms of HER2 CAR and NGFR surface expression in donor-matched NK cells and T cells.
(B) Transgene expression of HER2 CAR and NGFR on NK cells, CD4+ and CD8+ T cells.
(C) Cell-mediated cytotoxicity against SKBR3 cells was measured and relative change in specific lysis for HER2 CAR- and control vector transduced-NK cells was compared to HER2 CAR-T cells.
(D and E) IFN-γ (D) and TNF-α (E) release by HER2 CAR-, control vector transduced-, and non-transduced NK cells or T cells in response to SKBR3 target cells. Data represent mean ± SEM from two independent experiments with four donors per condition.
∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (B, two-way ANOVA with Sidak's post hoc tests; C, one-way ANOVA with Dunnet's post hoc tests; D and E, two-way ANOVA with Tukey's post hoc tests). ns, no significant difference.
HER2 CAR-NK cells are not further activated against healthy human bronchial epithelial cells
The potential risk of severe on-target off-tumor effects has been a major limiting factor in extending CAR-T cell therapy for solid tumors. This can result from uncontrolled CAR-T cell activation against healthy tissues that express basal levels of the target antigen. Expanded non-transduced NK cells have recently proven to be safe, with no adverse toxicities in patients with hematologic malignancies (Ciurea et al., 2017). However, given the augmented cytotoxicity exerted by HER2 CAR-NK cells against HER2-positive breast cancer cells, we asked whether expression of the CAR would override the inhibitory signaling in NK cells and cause off-tumor effects. Since normal lung tissues expressing low levels of HER2 has shown to be an off-tumor target site for HER2 CAR-T cells, we used a non-malignant human bronchial epithelial cell line (HBEC-6KT) as a translational model to assess potential off-tumor effects of CAR-NK cells. We first verified HER2 expression in HBEC-6KT cells compared to primary HBECs isolated from healthy donor bronchial brushings and HER2-overexpressing SKBR3 tumor cells via flow cytometry (Figure 4A). HER2 mean fluorescence intensity in HBEC-6KT cells was much lower compared to SKBR3 tumor cells (Figure 4B). We also confirmed MHC class I expression in HBEC-6KT cells and found that HBEC-6KT cells express MHC class I significantly higher than SKBR3 cells (Figures S3A and S3B). To further characterize the HBEC-6KT cells we measured their expression of stress-induced ligands typically upregulated on malignant cells (Figure S4). Compared to the various tumor cell lines used in this study, HBEC-6KT cells significantly expressed lower levels of all stress-induced ligands tested except for CD155 which is known to promote tumor immunosuppression through binding to the NK cell inhibitory receptor TIGIT (Li et al., 2014). To assess off-tumor effects, we measured cell-mediated killing of HBEC-6KT cells by HER2 CAR-NK cells and donor-matched HER2 CAR-T cells at a 5:1 E:T ratio using the same donors as the experiments performed with SKBR3 target cells. HER2 CAR expression in expanded NK cells did not lead to enhanced killing of non-malignant HBEC-6KT cells, relative to the control vector transduced-NK cells (Figure 4C). In contrast, donor-matched HER2 CAR-T cells became highly activated and exhibited a significant 7.8-fold mean increase in killing against the low HER2-expressing HBEC-6KT cells. Further, HER2 CAR-T cells killed SKBR3 breast cancer targets less efficiently than the HBEC-6KT cells (Figure S5A) unlike HER2 CAR-NK cells which exhibited significantly higher cytotoxicity toward the tumor cells only (Figure S5B). In response to stimulation by non-malignant HBEC-6KT cells, HER2 CAR-T cells produced significantly higher amounts of IFN-γ (mean: 6,000 pg/mL) and TNF-α (mean: 2,400 pg/mL) than the control vector transduced-T cells (Figures 4D and 4E). However, HER2 CAR-NK cells did not exhibit enhanced IFN-γ or TNF-α release compared to control vector transduced-NK cells. These findings demonstrate that unlike CAR-T cells, HER2 CAR-NK cells remain discriminatory against healthy cells that express basal levels of target antigen through recognition of MHC class I and lower levels of stress ligand expression.
Figure 4.

HER2 CAR NK cells discriminate healthy from malignant HER2-expressing cells
(A) Representative histograms of HER2 expression and (B) quantified mean fluorescence intensity (MFI) of HER2 expression on HBEC-6KT, primary HBEC, and SKBR3 breast cancer cells.
(C) Relative change in specific lysis was calculated for HER2 CAR-NK cells and T cells compared to their control vector transduced controls.
(D and E) IFN-γ (D) and TNF-α (E) production following 5 hr incubation of HER2 CAR-, and control vector transduced NK cells or T cells with HBEC-6KT cells. Data represent mean ± SEM of two independent experiments and three to four replicates per condition.
∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 (B, two-sided t test; C-E, two-way ANOVA with Sidak's post hoc tests). ns, no significant difference.
See also Figures S3, S4, and S5.
HER2 CAR-NK cells maintain cytotoxic function in the presence of the immunosuppressive factors TGF-β and PGE2
The TME of solid tumors is enriched with immunosuppressive cytokines and soluble factors, such as TGF-β and PGE2, that can directly inhibit NK cell function (Vitale et al., 2014). Here we evaluated the anti-tumor functions of HER2 CAR-NK cells after sustained exposure to both TGF-β and PGE2. We first assessed the inhibitory effect of the immunosuppressive factors on expanded NK cell-mediated killing and inflammatory cytokine production against SKBR3 breast cancer target cells. Expanded NK cell cytotoxicity and IFN-γ release were both significantly inhibited after stimulation with TGF-β and PGE2 for up to 48 hr (Figures S6A and S6B). We then generated HER2 CAR-NK cells and tested whether exposure to TGF-β and PGE2 would lead to the same immunosuppressive effects. We found that in comparison to non-transduced NK cells, which were significantly inhibited in the presence of TGF-β and PGE2, HER2 CAR-NK cells did not have a significant reduction in cytotoxic function (Figure 5A). In fact, HER2 CAR-NK cells exhibited significantly higher cytotoxicity than non-transduced NK cells within the same inhibitory conditions. However, TGF-β and PGE2 completely abrogated IFN-γ production in both non-transduced- and HER2 CAR-NK cells (Figure 5B). Assessment of HER2 CAR expression showed that there was no significant downregulation of the CAR after exposure to the inhibitors (Figure 5C). These results demonstrate that with sustained CAR expression, HER2 CAR-NK cells may maintain their high cytotoxic function in the immunosuppressive TME compared to unmodified expanded NK cells.
Figure 5.

HER2 CAR-NK cells remain cytotoxic in the presence of immunosuppressive factors
(A) Cytotoxicity of non-transduced- and HER2 CAR-NK cells against SKBR3 target cells was compared between NK cells cultured with TGF-β and PGE2 for 24 hr or media only controls.
(B) IFN-γ release was measured after 5 hr incubation of TGF-β and PGE2 pre-stimulated NK cells with target cells and compared to media only controls.
(C) Representative histogram and quantified median fluorescence intensity (MFI) of HER2 CAR after culture in TGF-β and PGE2 or media only. Data represent mean ± SEM of two independent experiments and three to six biological replicates per condition.
∗p < 0.05, ∗∗p < 0.001 (A and B, two way ANOVA with Sidak's post hoc tests; C, two-sided t test). ns, no significant difference.
See also Figure S6.
Discussion
While CAR-T cells have been successful in treating CD19-expressing B-cell malignancies, extending this therapy against other cancer types has been challenging due to difficulties in identifying a suitable antigen expressed exclusively on malignant cells (Vigneron, 2015). This has limited CAR-T cell implementation particularly against solid tumors, as antigen recognition on normal tissues can lead to adverse on-target, off-tumor effects (Morgan et al., 2010). CAR-engineered NK cells have been shown to be safe against hematologic malignancies; however their potential off-tumor effects in the context of solid tumors has been less explored (Liu et al., 2020). Here we engineered expanded NK cells derived from healthy donors and patients with breast cancer with a HER2-specific CAR to redirect their killing against HER2-expressing cancer cells and assess safety against non-malignant cells.
We demonstrate that HER2 CAR-NK cells from both healthy donors and patients with breast cancer exhibited enhanced cytotoxicity and IFN-γ production against HER2-expressing breast and ovarian cancer cells in vitro. This is in line with other studies showing that primary NK cells can be efficiently redirected and highly activated against various target antigens with CARs containing CD28 co-stimulatory and CD3ξ intracellular signaling domains (Kruschinski et al., 2008; Liu et al., 2018; Yu et al., 2018). While in our study the enhancement of NK cell anti-tumor functions through the CAR is modest, our group has previously shown that K562-mb-IL21 expansion converts human NK cells to a unique and highly activated CD56superbright NK cell subset (Poznanski et al., 2018). Indeed, we have shown that these expanded NK cells already exert highly potent cytotoxic function against a variety of primary tumor cells and cancer cell lines independent of tumor antigen expression (Nham et al., 2018; Poznanski et al., 2021b; Shenouda et al., 2017). This work demonstrates that CAR-expression can specifically redirect and further increase the activation potential of highly activated expanded NK cells toward tumor cells. Interestingly, we did observe a slight enhancement in NK cell anti-tumor functions due to the lentiviral transduction alone. Similarly to previous reports, we found no change in NK cell activation or inhibitory receptor expression as a result of the transduction (Colamartino et al., 2019). We did however see an increase in the expression of nutrient receptors CD71 and CD98 in the control vector transduced-NK cell population compared to untransduced and non-modified NK cells. Our recent work has shown that higher expression of the amino acid transporter CD98 and CD71 transferrin receptor does correlate with enhanced NK cell anti-tumor function (Poznanski et al., 2021a). This suggests that NK cells transduced with the control vector may have heightened nutrient uptake capacity leading to some level of non-specific activation.
Further, we found that non-transduced expanded NK cells killed breast cancer cell lines to a higher degree than both parental and HER2 CAR-expressing NK-92 cells. The human NK-92 cell line has been widely used in CAR-based preclinical studies, with some clinical trials underway, due to their high cytotoxic ability and expansion without the need of feeder cells (Schönfeld et al., 2015; Zhang et al., 2017). However, CAR-NK-92 cells must be irradiated prior to infusion which may impede their persistence and expansion in vivo, limiting their clinical efficacy (Tang et al., 2018). The ability to feasibly generate highly functional CAR-NK cells from both healthy donor and patients with cancer makes expanded NK cells a more suitable candidate for CAR-based therapy.
In contrast to hematologic malignancies, the efficacy of adoptive cell therapies against solid tumors has been limited by the suppression of immune effector function by the hostile TME. The soluble factors TGF-β and PGE2 are abundantly present in solid tumors and have each been implicated in the downregulation of NK cell anti-tumor functions (Holt et al., 2011; Lee et al., 2004; Nakanishi and Rosenberg, 2013; Narai et al., 2002; Otegbeye et al., 2018). We found that unmodified expanded NK cells had a marked reduction in cytotoxicity and IFN-γ production after exposure to TGF-β and PGE2 in combination. In comparison, HER2 CAR-NK cells maintained high cytotoxic function due to sustained HER2 CAR transgene expression after TGF-β and PGE2 treatment. Studies with IL-15 activated NK cells showed that PGE2 leads to suppression of IFN-γ production at both mRNA and protein levels (Joshi et al., 2001). Here we saw that CAR expression and signaling did not rescue the inhibition of IFN-γ release in expanded NK cells. It has been shown that CD28 co-stimulation in CAR-T cells can overcome TGF-β inhibitory signaling through the LCK-activating motif and IL-2 stimulation (Golumba-Nagy et al., 2018). However, PGE2 can activate protein kinase A type 1 which downregulates LCK activation inhibiting activation and cytotoxicity of NK cells (Martinet et al., 2010). Since the CD28/CD3ξ based CAR construct relies on protein tyrosine kinase-dependent pathways for signaling, this may account for the slight, albeit non-significant, reduction in cytotoxicity observed with the HER2 CAR-NK cells. Nonetheless, we demonstrate that HER2 CAR-NK cells are a potent source of immunotherapy for solid tumors through their ability to retain high cytotoxic function in the presence of highly immunosuppressive factors.
Difficulties in homing and persistence in the solid tumor is still an important factor influencing the clinical application of CAR-NK cell therapies against solid tumors. While we were unable to test these factors within our system, the first CAR-NK cell trial demonstrated that CD19 CAR-NK cells expressing human IL-15 were detected in the blood of patients by flow cytometry up to three weeks following CAR-NK infusion (Liu et al., 2020). Additionally, NK cells can be engineered to express chemokine receptors specific for chemokine ligands expressed by the target tumor to improve NK cell trafficking (Kremer et al., 2017). Hence, additional strategies such as dual expression of exogenous cytokines which support NK cell proliferation in vivo or chemokine receptors may be necessary to ensure high CAR-NK cell homing, infiltration, and persistence within solid tumor sites.
The heterogeneity of solid tumors is one major limiting factor of CAR-T cell efficacy, since CAR-T cell activation and subsequent effector function rely solely on antigen expression by target cells. In this study, we surprisingly observed that HER2 CAR-NK cells exerted superior cell-cell dependent cytotoxicity against breast cancer targets compared to donor-matched HER2 CAR-T cells. This was despite the lower fluorescence intensity of the HER2 CAR construct in expanded NK cells compared to the donor-matched T cells. It has been shown that NK cells typically have lower transduction efficiencies than T cells due to NK cells resistance to viral transduction and higher sensitivity to the insertion of genetic material (Schmidt et al., 2021; Sutlu et al., 2012). Thus, the resistance to transduction and large CAR vector used in our study could account for these differences in HER2 CAR expression. Regardless, we found that HER2 CAR-NK cells were highly efficient at killing HER2-expressing target cells in vitro. The ability of CAR-NK cells to retain their native array of activation receptors allows them to exert both antigen-independent and CAR-dependent activation which may account for the higher cytotoxic function observed. This unique ability of NK cells to recognize tumor cells in a non-antigen specific manner can overcome tumor escape problems observed in CAR-T cell therapy, in which CAR-T cell-driven downregulation of the CAR-specific antigen and epitope loss can lead to relapse post-treatment (Orlando et al., 2018; Sotillo et al., 2015).
Further, the safety of CAR-T cells has been a persisting concern in the field given the high incidence of observed toxicities with CD19 CAR-T cell therapy in clinical trials (Fitzgerald et al., 2017; Gardner et al., 2017; Maude et al., 2018). These CAR-T cell associated toxicities have been unfortunately coupled with anti-tumor efficacy, and most commonly manifest as CRS and neurotoxicity. Additionally, serious off-tumor effects can occur from CAR-T cell recognition of target antigen expressed on normal cells. In fact, B-cell aplasia has been a common adverse effect in B-cell malignancies due to CD19 CAR-T cell targeting of healthy CD19-expressing B cells (Brudno and Kochenderfer, 2016; Kansagra et al., 2019). While B-cell aplasia can be successfully treated with infusion of immunoglobulin, off-tumor activation against important normal tissues such as the lung can lead to fatal effects (Brudno and Kochenderfer, 2016; Morgan et al., 2010). In contrast to T cells, the expression of germline-encoded inhibitory receptors that recognize MHC class I molecules restrict NK cell killing against non-malignant cells. Expanded NK cells have proven to be safe in clinical settings against both hematologic malignancies and patients with HER2-positive solid tumors, leading to no CRS or neurotoxicity (Lee et al., 2020; Liu et al., 2020). Seeing that in our study HER2 CAR-NK cells exhibited very potent cytotoxicity against HER2-expressing breast cancer cells, it was important to assess whether CAR expression could override MHC class I inhibition. We found that HER2 CAR-NK cells did not exhibit increased activation toward non-malignant human bronchial epithelial cells that express low levels of both, HER2 and stress-induced ligands overexpressed by tumor cells, with no enhancement in cytotoxicity or IFN-γ and TNF-α production. This was in stark contrast to donor-matched HER2 CAR-T cells which elicited a significant increase in cytotoxicity and robust inflammatory cytokine production. The uncontrolled activation exhibited by HER2 CAR-T cells against normal epithelial cells expressing very low levels of target antigen has led to serious adverse effects. In vivo studies show that these HER2 CAR-T cells have the capacity to infiltrate into the lungs and heart, leading to cross-reactivity and activation causing lethal toxicities in tumor xenograft mice (Hammill et al., 2020). Since MHC class I molecules are constitutively expressed on normal cells while also often downregulated on tumors, our findings suggest that CAR-NK cells will overcome the current on-target, off-tumor toxicity concerns with CAR-T cells redirected against solid tumors without compromising anti-tumor function. Altogether, our results provide evidence that CAR-NK cells can uncouple high anti-tumor efficacy with toxicity, which may allow their safe clinical implementation for solid tumor malignancies.
Using HER2 CAR-NK cells as a model, we have demonstrated that CAR-engineered NK cells have the capacity to overcome major obstacles that adoptive cell therapies have faced in achieving therapeutic efficacy against solid tumors, including suppression by the TME, tumor heterogeneity, and off-tumor toxicity. This strategy could be used as a personalized treatment option for patients with current unmet therapeutic need.
Limitations of the study
Here we show the potent anti-tumor efficacy and limited off-tumor activation of CAR-modified expanded NK cells generated from healthy donor and patients with breast cancer against HER2-expressing target cells in vitro. Further work is needed to evaluate the persistence and homing of HER2 CAR-NK cells within the solid tumor, and whether their higher anti-tumor functions are maintained in vivo.
STAR★methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE Mouse Anti-Human IgG Fc | BioLegend | Cat#409304; Clone HP6017; RRID:AB_10895907 |
| FITC Mouse Anti-Human IgG Fc | BioLegend | Cat#409310; Clone HP6017; RRID:AB_2561855 |
| APC Mouse Anti-Human CD271 (NGFR) | BioLegend | Cat#345108; Clone ME20.4; RRID:AB_10645515 |
| VioBright FITC Mouse Anti-Human CD271 (LNGFR) | Miltenyi Biotec | Cat#130-113-423; Clone ME20.4-1.H4; RRID:AB_2734064 |
| BV421 Mouse Anti-Human CD56 | BD Biosciences | Cat#562751; Clone NCAM16.2; RRID:AB_2732054 |
| PerCP-Cy5.5 Mouse Anti-Human CD3 | BioLegend | Cat#300430; Clone UCHT1; RRID:AB_893299 |
| APC-H7 Mouse Anti-Human CD3 | BD Biosciences | Cat#560275; Clone SK7; RRID:AB_1645475 |
| AlexaFluor 700 Mouse Anti-Human CD4 | BD Biosciences | Cat#557922; Clone RPA-T4; RRID:AB_396943 |
| PerCP-Cy5.5 Mouse Anti-Human CD8 | BD Biosciences | Cat#565310; Clone SK1; RRID:AB_2687497 |
| PerCP-Cy5.5 Mouse Anti-Human CD340 (erbB2/HER2) | BioLegend | Cat#324415; Clone 24D2; RRID:AB_2562999 |
| PE Mouse Anti-Human HLA-A,B,C | BioLegend | Cat#311406; Clone W6/32; RRID:AB_314875 |
| PE Mouse Anti-Human MICA/MICB | BioLegend | Cat#320906; Clone 6D4; RRID:AB_493193 |
| PE Mouse Anti-Human ULBP-2/5/6 | R&D Systems | Cat#FAB1298P; Clone 165903; RRID:AB_2214693 |
| PE Mouse Anti-Human CD155 | BioLegend | Cat#337610; Clone SKII.4; RRID:AB_2174019 |
| PE Mouse Anti-Human CD112 | BioLegend | Cat#337410; Clone TX31; RRID:AB_2269088 |
| PerCP-Cy5.5 Mouse Anti-Human NKG2D | BioLegend | Cat#320818; Clone 1D11; RRID:AB_2562792 |
| AlexaFluor 700 Mouse Anti-Human CD16 | BioLegend | Cat#302026; Clone 3G8; RRID:AB_2278418 |
| APC Mouse Anti-Human NKp30 | BioLegend | Cat#325210; Clone P30-15; RRID:AB_2149449 |
| PE Mouse Anti-Human NKp44 | BioLegend | Cat#325108; Clone P44-8; RRID:AB_756100 |
| BV786 Mouse Anti-Human NKp46 | BD Biosciences | Cat#563329; Clone 9E2/NKp46; RRID:AB_2738139 |
| PE-CF594 Mouse Anti-Human CD69 | BD Biosciences | Cat#562617; Clone FN50; RRID:AB_2737680 |
| PE-Vio770 Anti-Human NKG2A | Miltenyi Biotec | Cat#130-105-647; Clone REA110; RRID:AB_2655388 |
| APC Mouse Anti-Human CD158a | BD Biosciences | Cat#564319; Clone HP-3E4; RRID:AB_2738742 |
| PE Mouse Anti-Human CD158b | BioLegend | Cat#312606; Clone DX27; RRID:AB_2130554 |
| AlexaFluor 700 Mouse Anti-Human CD158e1 | BioLegend | Cat#312712; Clone DX9; RRID:AB_2130824 |
| PE Mouse Anti-Human CD98 | BD Biosciences | Cat#556077; Clone UM7F8; RRID: AB_396344 |
| BV786 Mouse Anti-Human CD71 | BD Biosciences | Cat#563768; Clone M-A712; RRID:AB_2738414 |
| Human Glut1Fluorescein-conjugated Antibody | R&D Systems | Cat#FAB1418F; Clone 202915; RRID: AB_2191041 |
| Bacterial and virus strains | ||
| Third Generation Self-Inactivating Lentivirus | Generated in this study | NA |
| Biological samples | ||
| Healthy adult PBMCs | Isolated from peripheral blood of healthy volunteers | NA |
| Cancer patient PBMCs | Isolated from peripheral blood of breast cancer patients | NA |
| Bronchial Epithelial Cells | Isolated from bronchial brushings of subject undergoing bronchoscopy | NA |
| Chemicals, peptides, and recombinant proteins | ||
| Lymphoprep | Stemcell Technologies | Cat#07861 |
| Recombinant Human IL-2 | Peprotech | Cat#200-02; Accession# P60568 |
| Recombinant Human IL-15 | Peprotech | Cat#200-15; Accession# P40933 |
| Recombinant Human TGF-β | R&D Systems | Cat#240-B-002; Accession# P01137 |
| Recombinant Human ErbB2/Her2 Fc Chimera Protein | R&D Systems | Cat#1129-ER-050; Accession# NP_004439 |
| Prostaglandin E2 | Sigma-Aldrich | Cat#P0409; CAS: 363-24-6 |
| Recombinant Human IL-7 | Peprotech | Cat#200-07; Accession# P13232 |
| Hexadimethrine bromide (Polybrene) | Sigma-Aldrich | Ca# H9268; CAS: 28728-55-4 |
| Dynabeads Human T-Activator CD3/CD28 | Thermo Fisher | Cat#11161D |
| 2-Mercaptoethanol | Thermo Fisher | Cat#31350010 |
| 5(6)-Carboxyfluorescein diacetate N-succinimidyl ester (CFSE) | Sigma-Aldrich | Cat#21888; CAS 150347-59-4 |
| eBioscience Fixable Viaility Dye eFluor 780 | Thermo Fisher | Cat#65-0865-18 |
| Fixable Viability Stain 510 | BD Biosciences | Cat#564406; RRID: AB_2869572 |
| Lipofectamine 2000 | Thermo Fisher | Cat#11668019 |
| Sodium butyrate | Sigma-Aldrich | Cat#B5887; CAS: 156-54-7 |
| Keratinocyte Serum Free Media (KSFM) | Thermo Fisher | Cat#17005042 |
| Opti-MEM | Thermo Fisher | Cat#31985070 |
| Critical commercial assays | ||
| Human IFN-gamma DuoSet ELISA | R&D Systems | Cat#DY285B |
| Human TNF-alpha DuoSet ELISA | R&D Systems | Cat#DY210 |
| Experimental models: cell lines | ||
| Human: K562-mb-IL21 (Clone 9) cells | Laboratory of Dean A. Lee | Denman et al. (2012) |
| Human: SKBR-3 cells | Laboratory of Karen Mossman | RRID: CVCL_0033 |
| Human: MDA-MB-231 cells | Laboratory of Karen Mossman | RRID: CVCL_0062 |
| Human: BT-474 cells | Laboratory of Karen Mossman | RRID: CVCL_0179 |
| Human: SK-OV-3 cells | Laboratory of Karen Mossman | RRID: CVCL_0532 |
| Human: HBEC-6KT cells | Laboratory of Jeremy A. Hirota | Hirota et al. (2015) |
| Human: NK-92 cells | ATCC | RRID: CVCL_2142 |
| Human: HEK 293T cells | Laboratory of Jonathan Bramson | RRID: CVCL_0063 |
| Recombinant DNA | ||
| Plasmid: pCCL-Darpin-hCD8a NGFR | Laboratory of Jonathan L. Bramson | Hammill et al., 2015 |
| Plasmid: pCCL-CMV-NGFR | Laboratory of Jonathan L. Bramson | Hammill et al., 2015 |
| Plasmid: pRSV-REV | Laboratory of Jonathan L. Bramson | NA |
| Plasmid: pMD2.G | Laboratory of Jonathan L. Bramson | NA |
| Plasmid: pMDLg-pRRE | Laboratory of Jonathan L. Bramson | NA |
| Software and algorithms | ||
| FACSDIVA Software | BD Biosciences | https://www.bdbiosciences.com/ |
| FlowJo Software | BD Biosciences | https://www.flowjo.com/ |
| Prism Software (version 7.0) | GraphPad | https://www.graphpad.com/ |
| Other | ||
| LSRFortessa Flow Cytometer | BD Biosciences | NA |
| LSRII Flow Cytometer | BD Biosciences | NA |
| SpectraMax i3 | Molecular Devices | NA |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ali A. Ashkar (ashkara@mcmaster.ca).
Materials availability
The study did not generate new unique reagents.
Data and code availability
This study did not generate/analyze any datasets/code.
Experimental models and subject details
Human samples
All research conducted was approved by the Hamilton Integrated Research Ethics Board in Hamilton, Ontario. Peripheral blood was obtained from healthy donors with written informed consent at McMaster University in Hamilton, Ontario (11 participants aged between 20 and 60 years; Gender M:8 F:3). Cryopreserved peripheral blood mononuclear cells (PBMCs) were used from peripheral blood obtained from breast cancer patients with written informed consent (6 participants; Gender F). Two bronchial brushings from a consented subject undergoing bronchoscopy as part of routine clinical care were collected at St. Joseph's Healthcare in Hamilton, Ontario (1 participant aged 79; Gender F). Human bronchial epithelial cells (HBEC) were released from the cytology brush by gentle vortex and washing with EMEM media.
Cell lines and reagents
K562 myelogenous leukemia feeder cells (ATCC CCL-243, female) expressing membrane-bound IL-21(K562mb-IL21, Clone 9) were kindly provided by Dr. Dean A. Lee (Nationwide Children's Hospital, Ohio State University Comprehensive Cancer Center, USA). K562 mb-IL21 cells were cultured at 0.5 × 106 cells/mL in RPMI 1640 media containing 10% heat-inactivated fetal bovine serum (FBS). Human SKBR3 (RRID: CVCL_0033, female), MDA-MB-231 (RRID:CVCL_0062, female), BT-474 (RRID:CVCL_0179, female), SK-OV-3 (RRID:CVCL_0532, female) and HEK 293T (RRID:CVCL_0063, fetal) cells were cultured in DMEM media supplemented with 10% FBS until 70–80% confluency. The human NK-92 cell line (RRID:CVCL_2142, male) was cultured at 0.3 × 106 cells/mL in RMPI 1640 media supplemented with 12.5% heat-inactivated horse bovine serum, 12.5% FBS, and 55 μM 2-Mercaptoethanol (Thermo Fisher). The minimally immortalized human bronchial epithelial cell line, HBEC-6KT (RRID:CVCL_ER01, male) was generated by expressing the human telomerase reverse transcriptase and cyclin dependent kinase 4, as previously described (Hirota et al., 2015; Ramirez et al., 2004). HBEC-6KT cells were cultured in Keratinocyte Serum Free Media (Thermo Fisher) supplemented with 50 μg/mL bovine pituitary extract and 0.4 ng/mL epidermal growth factor until 80% confluency. All culture media except HBEC-6KT cell media contained 2 mM L-glutamine, 10 mM HEPES, 100 U/mL penicillin, and 100 ug/mL streptomycin. All cells were grown at 37°C and 5% CO2.
Method details
NK cell expansion and stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated from the whole blood of healthy donors or breast cancer patients using density gradient centrifugation with Lymphoprep (Stemcell Technologies) density gradient. NK cells from freshly isolated or cryopreserved PBMCs were expanded using irradiated K562 mb-IL-21 cells at a ratio of 1:2, as previously described (Denman et al., 2012; Somanchi et al., 2010). Cells were cultured in NK cell media (RPMI 1640 media supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 100 U/mL penicillin, 100 ug/mL streptomycin) and 100 U/mL rhIL-2 at 37°C and 5% CO2. Co-cultures were replenished with irradiated K562 mb-IL21 feeder cells on a weekly basis. For inhibition experiments, control expanded NK cells or HER2 CAR-NK cells were co-cultured in the presence of low-dose rhIL-15 (10 ng/mL) with TGF-β (5 ng/mL) and PGE2 (10 μg/mL) or media control (low dose rhIL-15 only) for the indicated durations (24 hr–72 hr) at 37°C and 5% CO2. Prior to all in vitro cytotoxicity assays, NK cells were washed and resuspended in fresh NK cell media to remove continued rhIL-15, TGF-β, or PGE2 exposure.
CAR-encoding vector and lentivirus production
The second generation HER2 CAR and mock lentiviral constructs have been described previously (Hammill et al., 2015; Helsen et al., 2018). The lentiviral vector encoding the anti-HER2 CAR consists of a human DARPin28z, a CD8 hinge region, a transmembrane and cytoplasmic domain of CD28, and the cytoplasmic region of CD3zeta (pCCL-Darpin-hCD8a NGFR) under the control of the human EF-1α promoter and truncated NGFR under the control of a human cytomegalovirus (hCMV) promoter as a transduction marker. The CAR negative control vector only includes the truncated NGFR gene under the hCMV promoter (pCCL-CMV-NGFR).
Third generation, self-inactivating and non-replicative lentivirus was produced as previously described (Hammill et al., 2016; Helsen et al., 2018). Briefly, 9 × 106 HEK 293T cells were first cultured on 15 cm diameter tissue culture-treated plates (NUNC; Thermo Fisher) until 70% confluency. The HEK 293T cells were then transfected with the packaging plasmids pRSV-REV (6.25 μg), pMD2.G (9 μg), pMDLg-pRRE (12.5 μg), and the transfer plasmid pCCL containing the transgene (32 μg) using Opti-MEM (Thermo Fisher) and Lipofectamine 2000 (Thermo Fisher). Twelve to sixteen hours after transfection, media was removed and supplemented with fresh HEK 293T media containing sodium butyrate (1mM; Sigma-Aldrich Canada Co.). After 36–48 hr post media replenishment, media containing lentivirus particles was collected, filtered with 0.45 μm filters and concentrated by ultracentrifugation (4°C, 1 hr 40min, 1.3 × 10 rcf). Viral pellet was resuspended in PBS (phosphate buffered saline) and stocks were stored at −80°C. Viral titer in TU/mL was determined by serial dilution and transduction of HEK 293T cells with thawed virus aliquots. Titer was determined by measuring the %NGFR positive population by flow cytometry using a VioBrightFITC-conjugated anti-NGFR antibody (Miltenyi Biotec).
Transduction of expanded NK cells and NK-92 cells
NK cells from healthy donors or breast cancer patients were expanded for a minimum of 2 weeks prior to use in experiments. 1 × 105 expanded NK cells were replenished at a 1:1 ratio with irradiated K562 mb-IL21 feeder cells, supplemented with 100U/mL rhIL-2 and seeded in a U-bottom 96-well plate. Alternatively, 1 × 105 human NK-92 cells were plated in a U-bottom 96-well plate with NK-92 media containing 100 U/mL rh-IL2. Twenty-four hours after plating, expanded NK cells and NK-92 cells were resuspended in transduction medium containing polybrene (8 μg/mL; Sigma-Aldrich Canada Co.) and 500U/mL rhIL-2 or 200 U/mL rh-IL2, respectively. Cells were transduced with thawed lentivirus at MOIs between 2 and 8. The plates were immediately centrifuged at 1000 × g for 45 min at 32 C° as a spinfection step and further incubated at 37 C°, 5% CO2 overnight. The transduced cells were maintained in NK or NK-92 cell media containing 100 U/mL rhIL-2 at 37°C and 5% CO2 prior to use in functional assays. For NK cell gating strategy see Figure S6.
Transduction of human T cells
Engineered T cells were generated from cryopreserved PBMCs of healthy donors as previously described (Hammill et al., 2016). Briefly, 1 × 105 human T cells were activated from cryopreserved PBMCs using anti-CD3/CD28 beads at a 0.8:1 bead-to-cell ratio (Dynabeads, Thermo Fisher) in a U-bottom 96-well plate with T cell media (RPMI media supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1× non-essential amino acids, 55 μM 2-Mercaptoethanol, and 100 U/mL penicillin +100 ug/mL streptomycin) containing 1.5 ng/mL rhIL-2 and 10 ng/mL rhIL-7 at 37°C. Twenty-four hours after activation T cells were transduced with thawed lentivirus at a MOI of 5. Transduced T cells were expanded in T cell media containing 1.5 ng/mL rhIL-2 and 10 ng/mL rhIL-7 and cultured for a period of 14 days at 37°C and 5% CO2 prior to use in functional assays. For gating strategy see Figure S7.
In vitro cytotoxicity assays
The SKBR3, MDA-MB-231, BT-474, SK-OV-3 or the HBEC-6KT target cells were first labeled with carboxyfluorescein succinimidyl ester (CFSE; Sigma-Aldrich Canada Co.) according to manufacturer's instructions. To evaluate cytotoxicity of CAR-NK cells, CAR-T cells and CAR-NK-92 cells, CFSE-labeled SKBR3, MDA-MD-231, BT-474, SK-OV-3 or HEBC-6KT cells were co-cultured with the HER2 CAR-engineered effector cells or corresponding control cells in a U-bottom 96-well culture plate at indicated effector:target (E:T) ratios for 5 hr at 37°C and 5% CO2. 100, 000–200, 000 target cells were used per duplicate wells for experiments. After 5 hr incubation, the cells were spun down and supernatants were collected for cytokine analysis. Cells were then washed in PBS and stained with a Fixable Viability Dye, eFluor 780 (Thermo Fisher) at 4°C for 30 min. Cells were gated on CFSE-positive events and cell death was calculated by gating on the Fixable Viability Dye eFluor 780 (APC-Cy7) positive gate. Percent specific cell lysis was calculated as: ((% tumor cell death – % basal tumor cell death)/(100- % basal tumor cell death)) x 100.
Cytokine quantification
To examine cytokine production of effector cells, supernatants were collected directly from the in vitro cytotoxicity assays for the CAR-NK cells, CAR-T cells, CAR-NK-92s, or control cells and frozen at −20°C. Samples were thawed on ice and cytokine release was analyzed using an IFN-γ (R&D Systems) and TNF-α (R&D Systems) specific enzyme-linked immunoabsorbent assay kit according to the manufacturer's instructions. Measurements that fall below 9.36 pg/mL (IFN-γ) and 15.6 pg/mL (TNF-α) are below the detection range of the assays. Absorbance was measured using the SpectraMax i3 plate reader (Molecular Devices).
Cell staining and flow cytometry
Cell viability was determined using Fixable Viability Dye eFluor 780 or Fixable Viability Stain 510 (BD Biosciences) according to the manufacturer's instructions. Cells were then stained with anti-human fluorescently labeled extracellular antibodies for the indicated extracellular markers in FACS buffer (0.2% BSA in PBS) at 4°C for 30 min. Detection of the HER2 CAR construct on the surface of human expanded CAR-NK cells, CAR-NK-92 cells, and CAR-T cells was determined by incubation with 2.5 μg of HER2 Fc chimeric protein (R&D Systems) in FACS buffer for 30 min at room temperature, followed by staining with a PE- or FITC-conjugated anti-human IgG Fc antibody (BioLegend). Transduced cells were also stained with APC-conjugated anti-NGFR (CD271) antibody (BioLegend) or VioBrightFITC-conjugated anti-NGFR antibody. All stained samples were fixed for 1 hr with 1% paraformaldehyde. All flow cytometry was conducted on a BD LSRFortessa or BDLSRII cytometer (BD Biosciences) and analyzed using FlowJo software (FlowJo, LLC, Ashland, OR).
Quantification and statistical analysis
All the data are reported as mean ± Standard Error of the Mean (SEM). Graphs were generated and all statistical analysis was conducted using GraphPad Prism (version 7.0). A two-sided Student's t-test was used to compare two groups. A one-way ANOVA test was used to compare three or more groups, and a two-way ANOVA was used to compare groups with two independent variables. The post hoc tests used for multiple comparisons are specified in the corresponding figure legends. The significance is shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, p > 0.05 was considered not significant (ns). The number of replicates used per condition are provided within the corresponding figure legends.
Acknowledgments
This work was supported by a grant from the Canadian Institutes of Health Research (20009360) awarded to A.A.A. A.A.A is a holder of a Tier 1 Canada Research Chair in Natural Immunity and NK Cell Function. The graphical abstract and schematics were created using bio-render.com
Author contributions
A.A.A. conceived the study. A.L.P., R.H., S.M.P., and A.A.A. contributed to the experimental design of the study. A.L.P. and R.H. performed experiments. E.A.R., N.J.C., M.V.C., M.M.S., and T.M.R. contributed to performing experiments. A.L.P. and A.A.A. analyzed the data, interpreted the data, and wrote the manuscript. S.M.P., E.A.R., and T.M.R. edited the manuscript. J.A.H. and J.L.B. provided HEK 293T cells, all lentiviral constructs used and experimental input for CAR-T cell experiments. Q.T.C. and J.A.H. provided the primary HBEC cells and HBEC-6KT cell line used for in vitro killing assays. S.D.-T. provided clinical samples from patients with breast cancer.
Declaration of interests
The authors declare no potential conflicts of interest.
Published: June 25, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102619.
Supplemental information
References
- Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24:541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifant C.L., Jackson H.J., Brentjens R.J., Curran K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. - Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brudno J.N., Kochenderfer J.N. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciurea S.O., Schafer J.R., Bassett R., Denman C.J., Cao K., Willis D., Rondon G., Chen J., Soebbing D., Kaur I. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–1868. doi: 10.1182/blood-2017-05-785659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colamartino A.B.L., Lemieux W., Bifsha P., Nicoletti S., Chakravarti N., Sanz J., Roméro H., Selleri S., Béland K., Guiot M. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front. Immunol. 2019;10:2873. doi: 10.3389/fimmu.2019.02873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denman C.J., Senyukov V.V., Somanchi S.S., Phatarpekar P.V., Kopp L.M., Johnson J.L., Singh H., Hurton L., Maiti S.N., Huls M.H. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald J.C., Weiss S.L., Maude S.L., Barrett D.M., Lacey S.F., Melenhorst J.J., Shaw P., Berg R.A., June C.H., Porter D.L. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit. Care Med. 2017;45:e124–e125. doi: 10.1097/CCM.0000000000002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg J.A., Wang Q., Katsumata M., Drebin J., Nagatomo I., Greene M.I. The role of HER2 in early breast cancer metastasis and the origins of resistance to HER2-targeted therapies. Exp. Mol. Pathol. 2009;87:1–11. doi: 10.1016/j.yexmp.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R.A., Finney O., Annesley C., Brakke H., Summers C., Leger K., Bleakley M., Brown C., Mgebroff S., Kelly-Spratt K.S. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129:3322–3331. doi: 10.1182/blood-2017-02-769208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giavridis T., Van Der Stegen S.J.C., Eyquem J., Hamieh M., Piersigilli A., Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade letter. Nat. Med. 2018;24:731–738. doi: 10.1038/s41591-018-0041-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golumba-Nagy V., Kuehle J., Hombach A.A., Abken H. CD28-ζ CAR T cells resist TGF-β repression through IL-2 signaling, which can Be mimicked by an engineered IL-7 autocrine loop. Mol. Ther. 2018;26:2218–2230. doi: 10.1016/j.ymthe.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammill J.A., Afsahi A., Bramson J.L., Helsen C.W. Methods in Molecular Biology. Humana Press Inc.; New York: 2016. Viral engineering of chimeric antigen receptor expression on murine and human T lymphocytes; pp. 137–157. [DOI] [PubMed] [Google Scholar]
- Hammill J.A., Kwiecien J.M., Dvorkin-Gheva A., Lau V.W.C., Baker C., Wu Y., Bezverbnaya K., Aarts C., Heslen C.W., Denisova G.F. A cross-reactive small protein binding domain provides a model to study off-tumor CAR-T cell toxicity. Mol. Ther. Oncolytics. 2020;17:278–292. doi: 10.1016/j.omto.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammill J.A., VanSeggelen H., Helsen C.W., Denisova G.F., Evelegh C., Tantalo D.G.M., Bassett J.D., Bramson J.L. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J. Immunother. Cancer. 2015;3:55. doi: 10.1186/s40425-015-0099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsen C.W., Hammill J.A., Lau V.W.C., Mwawasi K.A., Afsahi A., Bezverbnaya K., Newhook L., Hayes D.L., Aarts C., Bojovic B. The chimeric TAC receptor co-opts the T cell receptor yielding robust anti-tumor activity without toxicity. Nat. Commun. 2018;9:1–13. doi: 10.1038/s41467-018-05395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota J.A., Gold M.J., Hiebert P.R., Parkinson L.G., Wee T., Smith D., Hansbro P.M., Carlsten C., VanEeden S., Sin D.D. The nucleotide-binding domain, leucine-rich repeat protein 3 inflammasome/IL-1 receptor I axis mediates innate, but not adaptive, immune responses after exposure to particulate matter under 10 μm. Am. J. Respir. Cell Mol. Biol. 2015;52:96–105. doi: 10.1165/rcmb.2014-0158OC. [DOI] [PubMed] [Google Scholar]
- Holt D., Ma X., Kundu N., Fulton A. Prostaglandin E 2 (PGE 2) suppresses natural killer cell function primarily through the PGE 2 receptor EP4. Cancer Immunol. Immunother. 2011;60:1577–1586. doi: 10.1007/s00262-011-1064-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi P.C., Zhou X., Cuchens M., Jones Q. Prostaglandin E 2 suppressed IL-15-mediated human NK cell function through down-regulation of common γ-chain. J. Immunol. 2001;166:885–891. doi: 10.4049/jimmunol.166.2.885. [DOI] [PubMed] [Google Scholar]
- Kansagra A.J., Frey N.V., Bar M., Laetsch T.W., Carpenter P.A., Savani B.N., Heslop H.E., Bollard C.M., Komanduri K.V., Gastineau D.A. Clinical utilization of chimeric antigen receptor T-cells (CAR-T) in B-cell acute lymphoblastic leukemia (ALL)–an expert opinion from the European Society for Blood and Marrow Transplantation (EBMT) and the American Society for Blood and Marrow Transplantation (ASBMT) Bone Marrow Transpl. 2019;54:1868–1880. doi: 10.1038/s41409-019-0451-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer V., Ligtenberg M.A., Zendehdel R., Seitz C., Duivenvoorden A., Wennerberg E., Colón E., Scherman-Plogell A.-H., Lundqvist A. Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J. Immunother. Cancer. 2017;5:73. doi: 10.1186/s40425-017-0275-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruschinski A., Moosmann A., Poschke I., Norell H., Chmielewski M., Seliger B., Kiessling R., Blankenstein T., Abken H., Charo J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. U. S. A. 2008;105:17481–17486. doi: 10.1073/pnas.0804788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D.A., Denman C.J., Rondon G., Woodworth G., Chen J., Fisher T., Kaur I., Fernandez-Vina M., Cao K., Ciurea S. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: a phase I trial. Biol. Blood Marrow Transpl. 2016;22:1290–1298. doi: 10.1016/j.bbmt.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-C., Lee K.-M., Kim D.-W., Heo D.S. Elevated TGF-β1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J. Immunol. 2004;172:7335–7340. doi: 10.4049/jimmunol.172.12.7335. [DOI] [PubMed] [Google Scholar]
- Lee S.C., Shimasaki N., Lim J.S.J., Wong A., Yadav K., Yong W.P., Tan L.K., Koh L.P., Poon M.L.M., Tan S.H. Phase I trial of expanded, activated autologous NK-cell infusions with trastuzumab in patients with HER2-positive cancers. Clin. Cancer Res. 2020;26:4494–4502. doi: 10.1158/1078-0432.CCR-20-0768. [DOI] [PubMed] [Google Scholar]
- Li M., Xia P., Du Y., Liu S., Huang G., Chen J., Zhang H., Hou N., Cheng X., Zhou L. T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J. Biol. Chem. 2014;289:17647–17657. doi: 10.1074/jbc.M114.572420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E., Marin D., Banerjee P., MacApinlac H.A., Thompson P., Basar R., Kerbauy L.N., Overman B., Thall P., Kaplan M. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinet L., Jean C., Dietrich G., Fournié J.J., Poupot R. PGE2 inhibits natural killer and γδ T cell cytotoxicity triggered by NKR and TCR through a cAMP-mediated PKA type I-dependent signaling. Biochem. Pharmacol. 2010;80:838–845. doi: 10.1016/j.bcp.2010.05.002. [DOI] [PubMed] [Google Scholar]
- Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.S., Soignier Y., Panoskaltsis-Mortari A., McNearney S.A., Yun G.H., Fautsch S.K., McKenna D., Le C., Defor T.E., Burns L.J. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of t cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M., Rosenberg D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013;35:123–137. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narai S., Watanabe M., Hasegawa H., Nishibori H., Endo T., Kubota T., Kitajima M. Significance of transforming growth factor β1 as a new tumor marker for colorectal cancer. Int. J. Cancer. 2002;97:508–511. doi: 10.1002/ijc.1631. [DOI] [PubMed] [Google Scholar]
- Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nham T., Poznanski S.M., Fan I.Y., Shenouda M.M., Chew M.V., Lee A.J., Vahedi F., Karimi Y., Butcher M., Lee D.A. Ex vivo-expanded NK cells from blood and ascites of ovarian cancer patients are cytotoxic against autologous primary ovarian cancer cells. Cancer Immunol. Immunother. 2018;67:575–587. doi: 10.1007/s00262-017-2112-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando E.J., Han X., Tribouley C., Wood P.A., Leary R.J., Riester M., Levine J.E., Qayed M., Grupp S.A., Boyer M. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018;24:1504–1506. doi: 10.1038/s41591-018-0146-z. [DOI] [PubMed] [Google Scholar]
- Otegbeye F., Ojo E., Moreton S., Mackowski N., Lee D.A., De Lima M., Wald D.N. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PLoS One. 2018;13:e0191358. doi: 10.1371/journal.pone.0191358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y.P., Choi S.C., Kiesler P., Gil-Krzewska A., Borrego F., Weck J., Krzewski K., Coligan J.E. Complex regulation of human NKG2D-DAP10 cell surface expression: opposing roles of the γ c cytokines and TGF-β1. Blood. 2011;118:3019–3027. doi: 10.1182/blood-2011-04-346825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznanski S.M., Ashkar A.A. What defines NK cell functional fate: phenotype or metabolism? Front. Immunol. 2019;10:1414. doi: 10.3389/fimmu.2019.01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poznanski S.M., Nham T., Chew M.V., Lee A.J., Hammill J.A., Fan I.Y., Butcher M., Bramson J.L., Lee D.A., Hirte H.W. Expanded CD56superbrightCD16+ NK cells from ovarian cancer patients are cytotoxic against autologous tumor in a patient-derived xenograft murine model. Cancer Immunol. Res. 2018;6:1174–1185. doi: 10.1158/2326-6066.CIR-18-0144. [DOI] [PubMed] [Google Scholar]
- Poznanski S.M., Singh K., Ritchie T.M., Aguiar J.A., Fan I.Y., Portillo A.L., Rojas E.A., Vahedi F., El-Sayes A., Xing S. Metabolic flexibility determines human NK cell functional fate in the tumor microenvironment. Cell Metab. 2021;33 doi: 10.1016/j.cmet.2021.03.023. 1205-1220.e5. [DOI] [PubMed] [Google Scholar]
- Poznanski S.M., Ritchie T.M., Fan I.Y., El-Sayes A., Portillo A.L., Ben-Avi R., Rojas E.A., Chew M.V., Shargall Y., Ashkar A.A. Expanded human NK cells from lung cancer patients sensitize patients’ PDL1−negative tumors to PD1-blockade therapy. J. Immunother. Cancer. 2021;9:1933. doi: 10.1136/jitc-2020-001933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Press M.F., Cordon-Cardo C., Slamon D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene. 1990;5:953–962. [PubMed] [Google Scholar]
- Ramirez R.D., Sheridan S., Girard L., Sato M., Kim Y., Pollack J., Peyton M., Zou Y., Kurie J.M., DiMaio J.M. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–9034. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- Schmidt P., Raftery M.J., Pecher G. Engineering NK cells for CAR therapy—recent advances in gene transfer methodology. Front. Immunol. 2021;11:3404. doi: 10.3389/fimmu.2020.611163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönfeld K., Sahm C., Zhang C., Naundorf S., Brendel C., Odendahl M., Nowakowska P., Bönig H., Köhl U., Kloess S. Selective inhibition of tumor growth by clonal NK cells expressing an ErbB2/HER2-specific chimeric antigen receptor. Mol. Ther. 2015;23:330–338. doi: 10.1038/mt.2014.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenouda M.M., Gillgrass A., Nham T., Hogg R., Lee A.J., Chew M.V., Shafaei M., Aarts C., Lee D.A., Hassell J. Ex vivo expanded natural killer cells from breast cancer patients and healthy donors are highly cytotoxic against breast cancer cell lines and patient-derived tumours. Breast Cancer Res. 2017;19:76. doi: 10.1186/s13058-017-0867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somanchi S.S., Senyukov V.V., Denman C.J., Lee D.A. Expansion, purification, and functional assessment of human peripheral blood NK cells. J. Vis. Exp. 2010 doi: 10.3791/2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E., Barrett D.M., Black K.L., Bagashev A., Oldridge D., Wu G., Sussman R., Lanauze C., Ruella M., Gazzara M.R. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutlu T., Nyström S., Gilljam M., Stellan B., Applequist S.E., Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum. Gene Ther. 2012;23:1090. doi: 10.1089/hum.2012.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083. [PMC free article] [PubMed] [Google Scholar]
- Titov A., Petukhov A., Staliarova A., Motorin D., Bulatov E., Shuvalov O., Soond S.M., Piacentini M., Melino G., Zaritskey A. The biological basis and clinical symptoms of CAR-T therapy-associated toxicites. Cell Death Dis. 2018;9:1–15. doi: 10.1038/s41419-018-0918-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron N. Human tumor antigens and cancer immunotherapy. Biomed. Res. Int. 2015;2015:948501. doi: 10.1155/2015/948501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale M., Cantoni C., Pietra G., Mingari M.C., Moretta L. Effect of tumor cells and tumor microenvironment on NK-cell function. Eur. J. Immunol. 2014;44:1582–1592. doi: 10.1002/eji.201344272. [DOI] [PubMed] [Google Scholar]
- Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. Functions of natural killer cells. Nat. Immunol. 2008;9:503–510. doi: 10.1038/ni1582. [DOI] [PubMed] [Google Scholar]
- Wang M., Munoz J., Goy A., Locke F.L., Jacobson C.A., Hill B.T., Timmerman J.M., Holmes H., Jaglowski S., Flinn I.W. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N. Engl. J. Med. 2020;382:1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M., Schwaederle M., Arguello D., Millis S.Z., Gatalica Z., Kurzrock R. HER2 expression status in diverse cancers: review of results from 37,992 patients. Cancer Metastasis Rev. 2015;34:157–164. doi: 10.1007/s10555-015-9552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Luo H., Fan M., Wu X., Shi B., Di S., Liu Y., Pan Z., Jiang H., Li Z. Development of GPC3-specific chimeric antigen receptor-engineered natural killer cells for the treatment of hepatocellular carcinoma. Mol. Ther. 2018;26:366–378. doi: 10.1016/j.ymthe.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C., Oberoi P., Oelsner S., Waldmann A., Lindner A., Tonn T., Wels W.S. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017;8:533. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Zheng H., Diao Y. Natural killer cells and current applications of chimeric antigen receptor-modified NK-92 cells in tumor immunotherapy. Int. J. Mol. Sci. 2019;20:317. doi: 10.3390/ijms20020317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze any datasets/code.