

Abstract
The only medication available currently to prevent and treat opioid overdose (naloxone) was approved by the US Food and Drug Administration (FDA) nearly 50 years ago. Because of its pharmacokinetic and pharmacodynamic properties, naloxone has limited utility under some conditions and would not be effective to counteract mass casualties involving large-scale deployment of weaponized synthetic opioids. To address shortcomings of current medical countermeasures for opioid toxicity, a trans-agency scientific meeting was convened by the US National Institute of Allergy and Infectious Diseases/National Institutes of Health (NIAID/NIH) on August 6 and 7, 2019, to explore emerging alternative approaches for treating opioid overdose in the event of weaponization of synthetic opioids. The meeting was initiated by the Chemical Countermeasures Research Program (CCRP), was organized by NIAID, and was a collaboration with the National Institute on Drug Abuse/NIH (NIDA/NIH), the FDA, the Defense Threat Reduction Agency (DTRA), and the Biomedical Advanced Research and Development Authority (BARDA). This paper provides an overview of several presentations at that meeting that discussed emerging new approaches for treating opioid overdose, including the following: (1) intranasal nalmefene, a competitive, reversible opioid receptor antagonist with a longer duration of action than naloxone; (2) methocinnamox, a novel opioid receptor antagonist; (3) covalent naloxone nanoparticles; (4) serotonin (5-HT)1A receptor agonists; (5) fentanyl-binding cyclodextrin scaffolds; (6) detoxifying biomimetic “nanosponge” decoy receptors; and (7) antibody-based strategies. These approaches could also be applied to treat opioid use disorder.
Opioid receptor agonists have been used for medicinal purposes for centuries and remain the first-line treatment for some types of moderate-to-severe pain; however, the ongoing opioid epidemic has prompted a re-evaluation of the widespread clinical use of opioids. In addition to their well-established therapeutic effects, this class of drugs has significant adverse effects as evidenced by the large number of opioid-related overdose deaths in the United States and elsewhere, most recently fueled by the availability of fentanyl and related synthetic opioids. In fact, synthetic opioids are currently linked to over two-thirds of all opioid fatalities.1 The high potency, rapid onset of action (due, in part, to very high lipophilicity relative to opioids like morphine), and relatively long half-life of fentanyl and related synthetic opioids pose particular challenges for rescue by naloxone (e.g., Narcan), a competitive, reversible opioid receptor antagonist and currently the only US Food and Drug Administration (FDA)-approved medication for treating opioid overdose. Multiple reports have now emerged2,3 indicating that rescue of a victim who has overdosed on a synthetic opioid often requires larger or repeated doses of naloxone, compared with the standard (4 mg intranasal and 2 mg intramuscular) unit doses currently available in the commercial products and used by first responders, medical personnel, and others to reverse overdose caused by other opioids, such as heroin.
The very high potency, ease of synthesis, and widespread availability of synthetic opioids pose a significant public health risk not only to civilians, but also to law enforcement personnel, first responders, and the military because these chemicals, in particular ultra-potent fentanyl analogs, such as carfentanil, have the potential to be weaponized, which could lead to mass casualties.4 Moreover, there is evidence that high potency synthetic opioids have, in fact, been weaponized.5 In 2002, the Russian military released an aerosolized mixture of carfentanil and remifentanil into a ventilation system to immobilize Chechen terrorists who had stormed the Dubrovka Opera House in Moscow. More than 120 hostages died in the operation, and 650 of the survivors required hospitalization. The current defense against weaponized opioids is naloxone; however, given its short duration of action (typically not more than 2 hours), naloxone would be of limited value in a mass casualty event involving opioid poisoning, particularly with ultra-potent long-acting synthetic opioids. Thus, the need for a potent, long-acting antidote for opioid receptor agonist poisoning as a medical countermeasure for use in a mass casualty event now intersects its use as a traditional rescue medication. This situation has prompted leadership at the National Institutes of Health (NIH) to call for the development of “…stronger, longer-acting formulations of antagonists…”6 To address this vulnerability and a major shortcoming in medical countermeasures for the weaponization of synthetic opioids, a trans-agency scientific meeting was convened by the US National Institute of Allergy and Infectious Diseases/NIH (NIAID/NIH) on August 6 and 7, 2019, to explore new countermeasures for opioid toxicity. The meeting was initiated by the Chemical Countermeasures Research Program (CCRP), was organized by NIAID, and was a collaboration with the National Institute on Drug Abuse/NIH (NIDA/NIH), the FDA, the Defense Threat Reduction Agency (DTRA), and the Biomedical Advanced Research and Development Authority (BARDA). An overview of the meeting, attended by nearly 200 subject-matter experts from industry, academia, and government, was published in an Executive Summary.7 This paper provides a more detailed overview of several presentations at that meeting that discussed emerging new approaches for treating opioid overdose, including the following: (1) intranasal nalmefene, a competitive, reversible opioid receptor antagonist with a longer duration of action than naloxone; (2) methocinnamox (MCAM), a novel opioid receptor antagonist; (3) covalent naloxone nanoparticles; (4) serotonin (5-HT)1A receptor agonists; (5) fentanyl-binding cyclodextrin scaffolds; (6) detoxifying biomimetic “nanosponge” decoy receptors; and (7) antibody-based strategies.
NOVEL TREATMENTS: INTRANASAL NALMEFENE
An opioid receptor antagonist with higher affinity, more rapid onset, and longer duration of action than naloxone could significantly improve the treatment of opioid overdose. Based on these criteria, the pharmacokinetic and pharmacodynamic properties of the opioid receptor antagonist nalmefene are particularly well-suited to treat opioid overdose, including overdose caused by synthetic opioids. Both radioligand binding and functional assays have demonstrated that the potency of nalmefene is more than fivefold higher than naloxone at mu opioid receptors.8 Moreover, its reported plasma terminal half-life (t1/2) following parenteral dosing (t1/2~ 8 hours)9 is significantly longer than naloxone (t1/2~ 2 hours), reducing the likelihood of renarcotization if a victim has taken a long-acting opioid receptor agonist like fentanyl (t1/2 ~ 7–8 hours).10,11 Nalmefene injection was approved to treat opioid overdose in 1995, but withdrawn from the market by its sponsor for commercial reasons with no safety or efficacy concerns.
A pilot study8 was conducted in healthy male and female volunteers to determine the feasibility of developing an intranasal nalmefene product to treat opioid overdose. This study compared the pharmacokinetic properties of intranasal nalmefene in the presence and absence of a nasal absorption enhancer (dodecyl maltoside (DDM), an alkylsaccharide that enhances transmucosal absorption12; Intravail) with those of an intramuscular injection. Nalmefene (1.5 and 3 mg delivered in a volume of 0.1 ml) was slowly absorbed following intranasal administration, with a time to peak plasma concentration (Tmax) of 2 hours. Because a rapid onset of action must be considered a cardinal feature of an effective opioid overdose reversal agent, DDM was studied for its effects on the pharmacokinetic properties of nalmefene. Addition of 0.25% DDM to the 3-mg dose of nalmefene markedly reduced Tmax from 2 hours to 0.25 hours and increased peak plasma concentration (Cmax) by ~ 2.2-fold. Alkylsaccharides, such as DDM, have previously been shown to enhance the nasal absorption of both peptides and proteins as well as low molecular weight compounds, including the structurally related opioid receptor antagonist naltrexone, the 5-HT receptor agonist sumatriptan, and the benzodiazepine diazepam.12 The concentration of DDM selected for this study was based on a concentration range (0.063–0.5%) reported to enhance the effects of peptides, proteins, and small molecules. Although its effects on the pharmacokinetic profile of nalmefene were dramatic, the concentration of DDM used in this pilot study might not be optimum.
The pharmacokinetic characteristics of a 3-mg dose of intranasal nalmefene containing 0.25% DDM are consistent with an effective rescue medication: its onset of action is as fast as and possibly faster than an intramuscular dose (Tmax = 0.33 hours) of nalmefene (1.5 mg) that was previously approved to treat opioid overdose. Furthermore, the Cmax following intranasal administration is ~ 3-fold higher than the Cmax following intramuscular administration and is comparable to previously reported plasma concentrations of nalmefene observed 5 minutes after a 1-mg i.v. dose. At present, naloxone is the only opioid receptor antagonist approved for the treatment of suspected or confirmed opioid overdose. If Tmax can be used to approximate onset of action between structurally similar compounds acting at the same molecular target, then the Tmax of a 3-mg dose of intranasal nalmefene (0.25 hours) predicts a more rapid onset than the Tmax of a 4-mg dose of intranasal naloxone (0.5 hours).8 In addition, the high affinity (5-fold higher than naloxone) and long half-life (at least 7 hours compared with not more than 2 hours for naloxone) of nalmefene8 present distinct advantages as a rescue medication, making it particularly well-suited as a rescue agent against long-lasting synthetic opioids, such as fentanyl. A 3-mg nasal nalmefene product (containing DDM) is currently in development. A new drug application for this product candidate is planned for submission in 2021.
NOVEL TREATMENTS: METHOCINNAMOX
A drug that reverses as well as protects against opioid overdose in a manner that is not surmounted by taking more opioid, particularly potent synthetic opioids like fentanyl, could be a significant improvement over naloxone. MCAM is structurally similar to buprenorphine and was described first in 2000 by Broadbear and colleagues13 who reported MCAM to be a novel opioid receptor antagonist with a long duration of action. Although MCAM has affinity for all three types of opioid receptors, it interacts with mu receptors, but not kappa or delta receptors, in a functionally irreversible manner (i.e., pseudo-irreversible). Unlike the long-acting mu opioid receptor antagonist beta-funaltrexamine that also has agonist properties at kappa opioid receptors,14 there is no evidence that MCAM has intrinsic activity at any opioid receptor in vivo or in vitro13; rather, it blocks agonist binding to opioid receptors15 and antagonizes many effects of mu opioid receptor agonists in vivo. For example, in mice, MCAM antagonizes the antinociceptive effects of morphine.13 In rats, MCAM blocks the antinociceptive effects of morphine and fentanyl as well as morphine-induced inhibition of gastrointestinal motility15,16; it also precipitates withdrawal in morphine-dependent rats, producing withdrawal signs that are similar to those that emerge when withdrawal is precipitated by naloxone.16 In nonhuman primates, MCAM both reverses and prevents the ventilatory depressant effects of heroin17 and attenuates intravenous self-administration of heroin and remifentanil, but not self-administration of cocaine.18
Broadbear and colleagues reported that the antinociceptive effects of morphine are antagonized 48 hours after a single injection of MCAM.13 In fact, the antagonist properties of an acute injection of MCAM persist for 2 weeks or longer in rats (blocking antinociceptive effects16) and for a week or longer in nonhuman primates (blocking ventilatory depression and self-administration17–19). A dose of 3.2 mg/kg MCAM, which is 10 times larger than the dose that antagonizes the positive reinforcing and ventilatory depressant effects of mu opioid receptor agonists for a week or longer, does not affect responding for food and has no meaningful effect on heart rate, blood pressure, body weight, or activity in nonhuman primates.18 MCAM does not affect performance in monkeys responding under a delayed matching-to-sample (memory) task at doses that antagonize the response-rate decreasing effects of morphine in the same monkeys for a week or longer.20 In vitro studies conducted under a NIDA contract (unpublished) found no effect of MCAM in the AMES (mutagenicity) assay or a bacterial cytotoxicity assay, no effect in the hERG (cardiac toxicity) assay, no effect on any of 6 isozymes in a cytochrome p450 inhibition assay, and, with the exception of opioid receptors, no significant affinity for 66 different receptors and enzymes, up to a concentration of 100 nM.
After the initial characterization of MCAM in 200013 with a second publication appearing in 2005,15 investigation of MCAM stopped until recent studies that were prompted by the ongoing opioid epidemic and the potential of MCAM for treating opioid use disorder (OUD). Because MCAM has no known agonist properties, it would not be expected to result in detrimental interactions when combined with other drugs, such as benzodiazepines or alcohol, as can be the case with methadone and buprenorphine, further increasing the safety profile of MCAM. Moreover, if a single administration of MCAM blocks insurmountably the effects of mu opioid receptor agonists for a week or longer in humans, it could help to reduce the poor medication compliance that is common among patients being treated for OUD. Based on studies with the irreversible opioid receptor ligands clocinnamox21 and beta-funaltrexamine,22 there does not appear to be any adverse consequence from long-term occupancy of mu opioid receptors. In a recent study,19 an effective dose of MCAM (0.32 mg/kg) was administered to rhesus monkeys every 12 days for 5 total injections. Monkeys could self-administer fentanyl daily with the exception of every fourth day when cocaine was available for self-administration. For more than 10 weeks, MCAM blocked fentanyl self-administration without affecting cocaine self-administration. Importantly, recovery of responding for (sensitivity to) fentanyl after the last of 5 injections was not significantly different from recovery of responding after a single injection of the same dose of MCAM.
MCAM might also be useful for treating opioid overdose and could have advantages over naloxone. The sustained antagonist properties of MCAM could mitigate the high risk of another overdose in patients rescued with naloxone.23 MCAM would block the re-emergence of toxic effects of the opioid(s) that necessitated rescue with naloxone (i.e., renarconization) and block the effects of any subsequently administered opioid(s) for a week or longer. Based on studies in rats,16 MCAM, like naloxone, would be expected to precipitate withdrawal in opioid-dependent patients, although withdrawal signs did not last longer in rats receiving MCAM compared with rats receiving naloxone, despite sustained antagonism by MCAM of other effects of mu opioid receptor agonists.
Finally, and most relevant to this paper, there are situations in which MCAM might be particularly effective in protecting against (prophylaxis) opioid poisoning. For example, MCAM would protect law enforcement, first responders, and military personnel who are exposed to opioids in the line of duty, accidentally or intentionally. A single prophylactic dose of MCAM could protect against opioid toxicity for up to a week and, if needed and based on studies in nonhuman primates,19 repeated dosing would provide extended protection for as long as necessary. A more challenging situation requiring highly effective opioid antagonists would occur in the event of deployment of weaponized opioids when potentially large numbers of individuals need to be treated.4 One justification for the meeting on which this paper is based is the current vulnerability of US civilians and military to the weaponization of opioids. It was clear from the meeting that opioids, particularly highly potent fentanyl analogs, such as carfentanil, pose a significant risk to public health not only because of the ongoing opioid epidemic, but also because of the toxicity and potential mass lethality that could occur from nefarious use of these opioids.5 It is difficult to imagine how the only drug currently available to treat opioid overdose, naloxone, could be deployed effectively in a mass casualty situation, particularly after exposure to large doses of fentanyl or its analogs.2 A rapid-acting, potent, and safe opioid receptor antagonist that provides extended (hours or days) protection after a single administration could be a much needed medication for defending civilian and military populations.
NOVEL TREATMENTS: COVALENT NALOXONE NANOPARTICLES AS LONG-ACTING OPIOID ANTAGONISTS
The short half-life and low metabolic stability24–28 of naloxone, compared with many opioid receptor agonists (e.g., fentanyl), can require the delivery of relatively large doses of naloxone to reverse overdose from a synthetic opioid and prevent renarcotization,28–31 even for relatively short-acting opioid agonists.32 Naloxone would also be of limited value in the face of the weaponization and deployment of a synthetic opioid resulting in mass casualties, which would likely require re-administration throughout the decontamination and recovery process.33 Furthermore, the administration of large acute doses of mu opioid receptor antagonists to overdose patients can precipitate withdrawal signs and symptoms (i.e., acute opioid dependence and acute withdrawal34), even in patients who are not otherwise taking opioids.3,35,36
A long-acting antagonist that has linear “low and slow” delivery might be an especially useful tool for reversing overdose from synthetic opioids and providing sustained protection from renarcotization, particularly in the context of mass exposure to opioids. The low and slow antidote approach would avoid the need for re-administration of the antidote while avoiding adverse effects that can occur with administration of large doses of naloxone, including the precipitation of acute opioid withdrawal.34 A low and slow antidote approach would be ideal from both a patient and provider perspective in a mass casualty incident because protection from overdose is ensured without risk of precipitating withdrawal or renarcotization.
To design this next generation countermeasure for synthetic opioid toxicity, a drug delivery platform was used that is based on polymer nanoparticle technology. Common matrices for polymer nanoparticles include polylactic acid (PLA), polyglycolic acid (PGA), and their copolymer poly(lactic-co-glycolic acid (PLGA)). PLA/PGA/PLGA polymeric nanoparticles have been administered successfully using systemic, oral, pulmonary, and transdermal routes for various medical conditions.37–39 Because these polymers are safe and already constituents in many FDA-approved drugs, the time required to move a new formulation to the clinic will be comparatively short. In previous studies with PLA/PGA/PLGA nanoparticles, drugs were loaded into the matrix via coprecipitation of the polymer and drug into an aqueous solution. This approach leads to a nano/microparticle matrix of polymer with drugs noncovalently distributed throughout. The entrapped drugs are released in vivo by diffusion and by action of endogenous esterases on the polymer backbone. Limitations to the noncovalent system include the following: (1) burst release of drugs from the particles due to rapid desorption of drugs from the surface of the particles; (2) challenging batch-to-batch control over drug loading; and (3) overall lower drug loadings.40–43 For example, Vivitrol, a well-known extended-release preparation of naltrexone, has naltrexone noncovalently loaded into PLGA, leading to burst release. Vivitrol reportedly can precipitate severe withdrawal possibly due to this release property.44
A covalent loading approach was used to address the issue of burst release. In this approach, available hydroxyl groups in a small-molecule drug were used as initiators in the ring opening polymerization of lactide (PLA backbone) to form a drug-polymer hybrid. This drug-polymer hybrid is formulated into a nanoparticle using the one-stage emulsion approach. Particles with controlled drug loading, near uniform size, and no burst release are achieved due to covalent drug loading.45
Nanoparticles with naloxone covalently bound to polymer (cNP-Nal) were prepared with ~ 7% naloxone per particle.45 Particles have a diameter of ~ 250 nm and a narrow monomodal size distribution with polydispersity of 0.15, as determined using dynamic light scattering. The cNP-Nal were biocompatible with no burst release and no in vitro cell toxicity. The particles were evaluated in several in vivo models to determine their ability to attenuate the effects of the mu opioid receptor agonist morphine. In a neuropathic pain model, a single injection of cNP-Nal (1 mg/ml at 7% w/w and 300 μl volume = ~ 0.75 mg/kg) blocked the antinociceptive effects of morphine for several days, with complete antagonism of morphine observed both 24 and 48 hours after cNP-Nal administration.45 Antagonism of morphine by cNP-Nal was diminished but still significant 98 hours after administration. In contrast, free naloxone (10 mg/kg) antagonized morphine only on the day of administration. Importantly, the effective naloxone dose used in cNP-Nal was, on average, only 0.023 mg/mouse compared with 0.3 mg/mouse for the free naloxone. These data suggest that controlled naloxone release allows for sustained systemic dosing while avoiding the quick metabolic inactivation that occurs with free naloxone. Delivery of a small dose of naloxone via the cNP-Nal might prevent agonist activity (e.g., toxicity), while also avoiding the precipitation of withdrawal.
Recently, cNP-Nal was shown to produce significantly fewer withdrawal signs in opioid-dependent mice, compared with withdrawal precipitated by free naloxone.46 In morphine-dependent mice, significant behavioral signs of withdrawal were observed after the administration of 8 mg/kg of free naloxone. In contrast, the behavior of morphine-dependent mice that received cNP-Nal (0.75 or 7.5 mg/kg) was not different from behavior of morphine-dependent mice that received control injections (saline or cNP-Empty). Both 0.75 and 7.5 mg/kg of cNP-Nal reversed the agonist effects of morphine. These results, coupled with the ease of synthesizing and manufacturing cNP-Nal, are encouraging and prompting further study of this drug platform as a countermeasure to synthetic opioid poisoning.
The relative advantages of the low and slow approach for naloxone delivery include the following: (1) use of a known, FDA-approved medication with a broad therapeutic window and well-established low toxicity; (2) potential to reverse synthetic opioid overdose; and (3) avoiding the precipitation of withdrawal. The biocompatible PLA backbone has proven clinical safety but has never been applied to the delivery of overdose rescue agents. Collectively, this formulation is very low risk (i.e., the safety records of PLA and naloxone) and highly innovative (i.e., covalent-loaded PLA for overdose treatment). Moreover, compared with alternative biological-based sequestration strategies, cNP-Nal is not specific for synthetic opioids and provides a shelf-stable formulation that is simple to manufacture. Importantly, the cNP-Nal formulation is not expected to interfere with and thus could conceivably be co-administered with a biological sequestration mechanism of action. Potential drawbacks of the cNP-Nal approach include the possible extended off-target effects. Although the existing data suggest reduced precipitation of withdrawal in nonhuman subjects, it is possible that some patients might experience some withdrawal signs and symptoms. Such adverse effects might persist longer with cNP-Nal, compared with free naloxone, because the active pharmaceutical ingredient would be available for up to 4 days. At this point, the cNP-Nal strategy has been evaluated only in the context of morphine antinociception and physical dependence. Ongoing studies include the evaluation of cNP-Nal in fentanyl-induced antinociception and, critically, in fentanyl-induced overdose. Overall, the cNP-Nal strategy could be applied to reverse synthetic opioid poisoning in a mass casualty incident or to treat overdose in patents with OUD, particularly in patients suffering from overdose with a long-lasting synthetic opioid.
NOVEL TREATMENTS: SEROTONIN (5-HT)1A RECEPTOR AGONISTS FOR TREATING OPIOID-INDUCED RESPIRATORY DEPRESSION
The neurotransmitter 5-HT plays an important role in respiration47; 5-HT neurons of the medullary raphe together with those in the parapyramidal region of the ventrolateral medulla innervate almost all of the interconnected brainstem respiratory nuclei including the preBötzinger complex (preBötC), a major group of neurons responsible for inspiratory drive.47 These 5-HT neurons are potential CO2 sensors and their activation stimulates respiration.48 Although respiratory nuclei express several 5-HT receptor subtypes, there is considerable evidence supporting a primary regulatory role for the 5-HT1A receptor.49,50 Buspirone, a drug with 5-HT1A receptor agonist properties that is used to treat depression, stimulates respiration.51,52 Moreover, microinjection of 8-hydroxy-n,n-dipropylaminotetralin (8-OH-DPAT, a compound with 5-HT1A-7 receptor agonist properties) into the preBötC counteracts hypoxia-induced apneusis (dysregulated breathing53). Conversely, blockade of 5-HT1A receptors has been reported to impair respiratory rhythm.54
The above observations suggest that 5-HT1A receptor agonists have utility as generalized respiratory stimulants, and there could be important advantages of using 5-HT1A receptor agonists to treat opioid-induced respiratory depression. In animal models, it has been shown that 5-HT1A receptor agonists can reverse respiratory depression induced by morphine or fentanyl.55–57 Collectively, these data support further investigation of 5-HT1A receptor agonists as a novel class of drugs for treating or preventing the respiratory depression triggered by opioids, including synthetic opioids, or other chemical toxins. In this context, the primary advantage of drugs in this class is that they do not act at opioid receptors. Although naloxone is useful for reversing opioid-induced respiratory depression, it has significant limitations.28 Indeed, the longer-acting opioid receptor antagonist nalmefene has been proposed to circumvent the pharmacokinetic limitations of naloxone (vide supra). The potential weaponization of “super” opioids that are significantly more potent than carfentanil, which has at least 20 times greater affinity for mu opioid receptors compared with naloxone, necessitates the development of antidote medications that act independently of opioid receptors.
Another therapeutic advantage of generalized respiratory stimulants is that they offer the potential for broad protection against an array of respiratory poisons. For example, neither naloxone nor nalmefene would be effective against a weaponized respiratory toxin that does not act at opioid receptors. Similarly, overdose in opioid abusers often involves a cocktail of substances that depress respiration, including ethanol and benzodiazepines,58 and a 5-HT receptor agonist might have therapeutic utility in treating overdose in patients that received multiple drugs from different classes. Furthermore, because they stimulate respiration through distinct pathways, combined treatment with a 5-HT1A receptor agonist and naloxone might produce a superior therapeutic outcome for opioid overdose, extending therapeutic potency, effectiveness, and duration compared with administering either compound alone.
There are a number of factors to consider regarding the development of 5-HT1A receptor agonists targeting respiration. For example, 5-HT1A receptors couple to diverse downstream cellular signaling pathways (cAMP, MAP kinase, and Ca2+) and many ligands exhibit a bias to one or more of these cellular responses.59 Moreover, several agonists selectively activate and desensitize pre-synaptic vs. post-synaptic 5-HT1A receptors. Thus, a key step in developing 5-HT1A receptor agonists would be to identify specific signaling pathways and ligands that are optimal for activating respiratory neurons. Potentially, this could involve medicinal chemistry approaches and the development of novel compounds. On the other hand, there are currently a number of selective 5-HT1A receptor agonists either in clinical or experimental use (intended as anxiolytic/neurologic agents) that could be readily repurposed as respiratory stimulant drugs.
Other important considerations in developing 5-HT1A receptor agonists for treating respiratory depression are pharmacokinetics and dosage. Although 5-HT1A receptor agonists have proven efficacy in reversing opioid-induced respiratory depression in nonhuman subjects, that effect was not observed in humans.60 The discrepancy between humans and nonhumans is likely due to pharmacokinetic factors and inadequate oral dosing of 5-HT1A receptor agonists in humans (e.g., ~ 2% of orally administered buspirone reaches the brain due to extensive first-pass metabolism61). In summary, 5-HT1A receptor agonists represent a novel drug class that could be developed, either alone or in combination with an opioid receptor antagonist, to reverse opioid overdose, including overdose caused by synthetic opioids; because drugs from this pharmacological class stimulate respiration, they would also have the distinct advantage of blocking the effects of a broad range of non-opioid respiratory toxins and could be especially useful when the toxin is not known.
NOVEL TREATMENTS: FENTANYL-BINDING CYCLODEXTRIN SCAFFOLDS
Cyclodextrins (CDs) are cyclic oligosaccharides composed of glucose units joined by α-1,4-glycosidic linkages. The type of linkage that holds these units together gives rise to a well-defined, rigid, 3D structure resembling that of a frustum with a hydrophobic interior and a hydrophilic exterior.62 Thus, most organic molecules (i.e., hydrophobic) tend to seek the interior of CDs forming host:guest complexes. Work at the Lawrence Livermore National Laboratory has taken advantage of the uniqueness of a centralized, collaborative effort involving computational chemistry,63,64 biochemistry,63 and organic synthesis65 in a highly iterative fashion with the goal of identifying CD candidates that can serve as capturing hosts for fentanyl and related synthetic opioids. Molecular dynamic simulations are used along with Molecular Mechanics-Generalized Born Surface Area calculations to assess CD scaffolds in silico for their binding affinities toward fentanyl. Lead CD candidates that are identified by the computational component are synthesized for their subsequent in vitro toxicological assessment, binding affinity measurements by isothermal titration calorimetry, and CD:fentanyl structure determination by nuclear magnetic resonance (NMR) spectroscopy.63,66 Initial modeling studies using β-CD, experimentally supported by detailed NMR spectroscopy, revealed that fentanyl can reside in the interior of the CD in two possible orientations, denoted up or down (vide infra63). Furthermore, these initial studies demonstrated that β-CD is the most optimal CD for harboring the synthetic opioid in its interior, allowing further investigation focusing solely on analogs based on this CD. Various CDs have been evaluated that include β-CD, heptakis-6-deoxy-6-amino-β-CD, 6-deoxy-6-amino-β-CD, 2-hydroxypropyl-β-CD, and heptakis-6-(3-O-propyl-1-sulfonic acid)-β-CD.
Molecular dynamic simulations involving fentanyl and β-CD were stable, although it was not uncommon for fentanyl to diffuse out of the cavity. When that occurred, the most common molecular complex obtained after allowing the simulation to continue was the one involving the down conformation (ΔG = −19.5 kcal/mole; Figure 1; unpublished data). Simulations on the modified, more elaborate β-CDs provided lower ΔG values indicating stronger CD:fentanyl inclusion complexes (Figure 1). Calculations found 2-hydroxypropyl-β-CD (HPCD) and heptakis-6-deoxy-6-(3-O-propyl-l-sulfonic acid)-β-CD to form the strongest inclusion complexes with fentanyl featuring free energy values of ΔG = −33.1 kcal/mole (up) and ΔG = −33.5 kcal/mole (up), respectively. Binding of fentanyl to the mono-substituted 6-deoxy-6-amino-β-CD appears to be more favorable than binding to its fully substituted counterpart, heptakis-6-deoxy-6-amino-β-CD, (ΔG = −26.9 kcal/mol (up) vs. −19.0 kcal/mol (up)).
Figure 1.
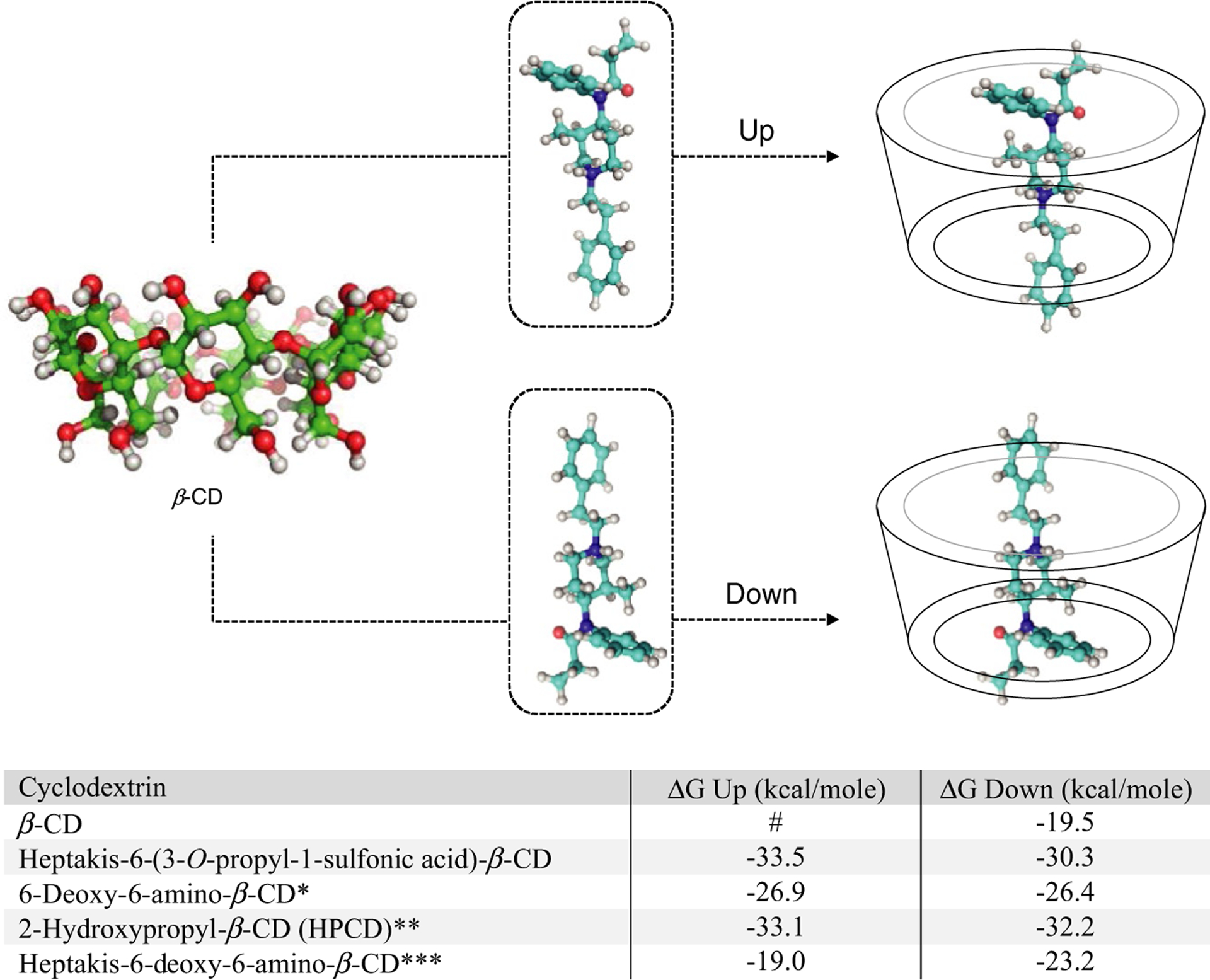
Modes of binding of fentanyl to beta-cyclodextrin (β-CD; up vs. down conformation) and free energy (ΔG) values calculated using Molecular Mechanics-Generalized Born Surface Area (MM/GBSA) energy calculations for both orientations (unpublished data): *protonated form of amine used; **only 1° rim hydroxypropyl modification used in calculations; ***protonated form of amines used; #fentanyl in the up conformation in β-CD tended to diffuse out of the cavity.
The cytotoxic effects associated with β-CDs are thought to arise from their host:guest complexation with cholesterol and membrane lipids.67,68 Cytotoxicity assays were conducted using human brain endothelial cells using the well-established methylthiazol tetrazolium assay. There was significant toxicity at 10 and 20 mM for β-CD (Figure 2a; unpublished data) and heptakis-6-(3-O-propyl-1-sulfonic acid)-β-CD (Figure 2b), respectively. Other CDs did not demonstrate toxicity until much higher concentrations: 50–100 mM for 6-deoxy-6-amino-β-CD (Figure 2c) and 75 and 100 mM for 2-hydroxypropyl-β-CD (Figure 2d), and no significant toxicity was noted for heptakis-6-deoxy-6-amino-β-CD (Figure 2e) at any concentration; however, there was greater variability between replicates for this CD. Hemolytic activity was also evaluated for CDs at 10 minutes and 4 hours after incubation with rat erythrocytes. At 10 minutes (Figure 2f), hemolysis varied depending on CD and concentration (~ 0.5–130%). Low levels of hemolysis (< 13%) were noted for all CDs, with the exception of β-CD at 10 mg/ml (complete hemolysis). At 4 hours (Figure 2g), hemolytic activity generally increased compared with the respective 10-minute time point. For β-CD, hemolytic activity for 0.1 and 1 mg/ml increased by 2.3-fold (5.87% ± 0.32) and 8.7-fold (5.33% ± 0.13), respectively. A small increase was also noted for 10 mg/ml β-CD at the 4-hour time point (~ 1.1-fold). For heptakis-6-deoxy-6-amino-β-CD, increases in hemolytic activity were observed for all three concentrations evaluated (1.3-fold to 3.6-fold increases). In contrast, for 6-deoxy-6-amino-β-CD, an increase was observed only for the highest concentration (10 mg/ml, 7.8-fold increase, ~ 98%); minimal hemolytic activity was noted at the lower concentrations (0–0.8%). Taken together, these data demonstrate that whereas cytotoxicity for CDs is evident at high concentrations (i.e., 10–100 mM), much lower concentrations of CDs cause significant hemolysis in blood (< 10 μM).
Figure 2.
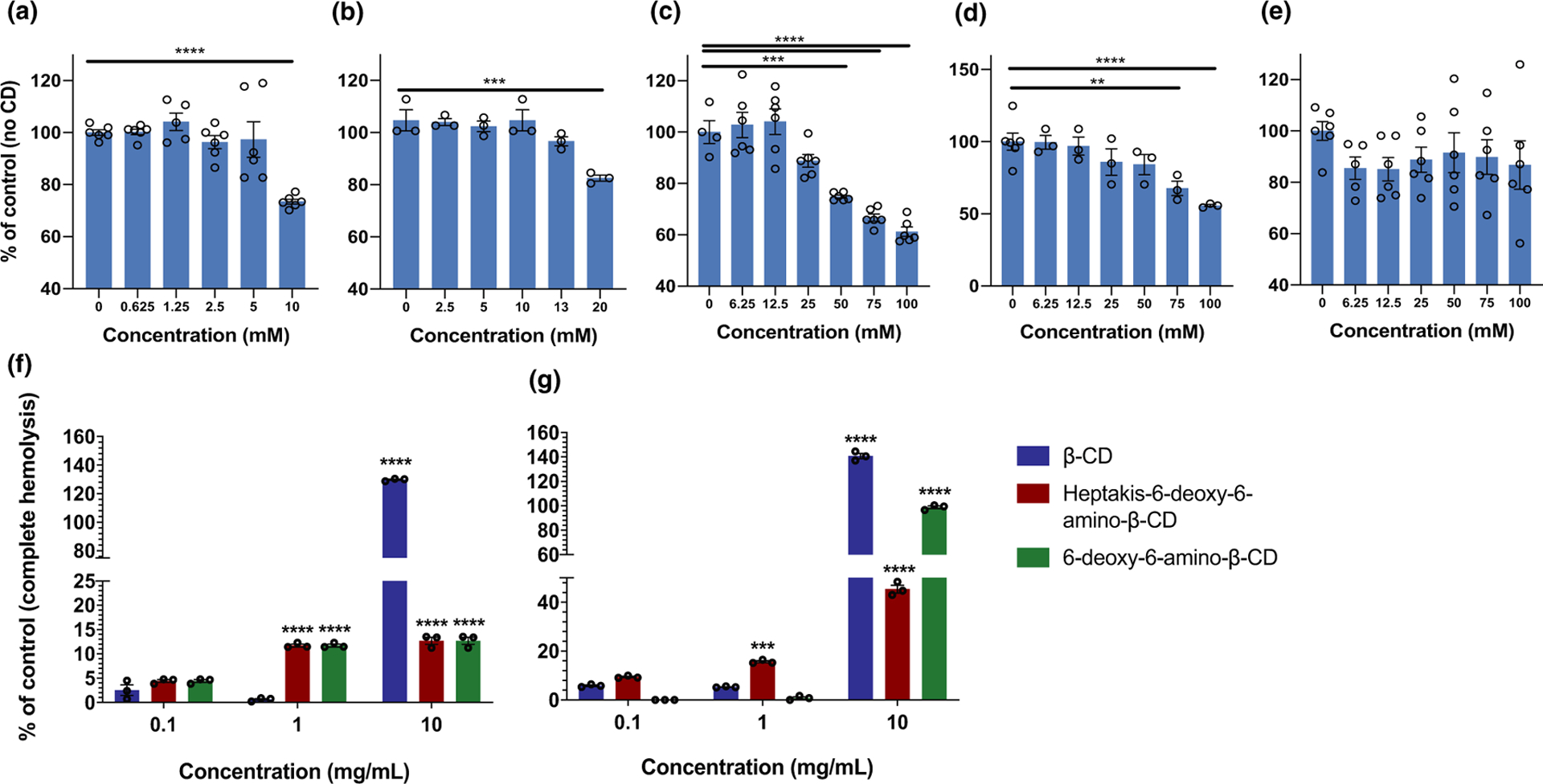
Comparison of induced cytotoxicity on human brain endothelial cells by five cyclodextrins as follows: (a) beta-cyclodextrin (β-CD); (b) heptakis-6-(3-O-propyl-1-sulfonic acid)-β-CD sodium salt; (c) 6-deoxy-6-amino-β-CD; (d) 2-hydroxypropyl-β-CD; and (e) heptakis-6-deoxy-6-amino-β-CD. Data (unpublished) are expressed as a percentage relative to the negative control (no CD), (f) and (g) hemolytic activity on β-CD, heptakis-6-deoxy-6-amino-β-CD and 6-deoxy-6-amino-β-CD. Data are expressed as a percentage relative to the positive control (complete hemolysis). For all experiments, data are presented as the mean ± the standard error of the mean (SEM). Individual replicates are shown and indicated by circular data points on each bar. A two-way analysis of variance (ANOVA) with a Tukey’s post hoc test was used to determine statistical significance. Significance relative to the 0.1 mg/mL concentration is shown: ***P < 0.001, ****P < 0.0001.
CDs have become important platforms on which to build medical countermeasures against fentanyl and other synthetic opioids with the ultimate goal of providing suitable protection to law enforcement, first responders, and military personnel. Thus, their ability to form inclusion complexes with fentanyl and analogs would serve as a protective layer (prophylaxis) prior to exposure to a synthetic opioid. Investigators at the Lawrence Livermore National Laboratory are conducting highly collaborative research spearheading this emerging application of CDs. From the ability to chemically modify CDs, to a plethora of in vitro assays to assess their toxicological profile, to the complete structural resolution of their complexes along with their binding affinities using NMR and isothermal titration calorimetry, to powerful computational modeling, these investigators have systematically characterized the host:guest complexes of numerous CD members with fentanyl and its analogs. Current work involves evaluating more complex, chemically modified CDs that have been initially identified by Molecular Mechanics-Generalized Born Surface Area methods using fentanyl as the primary target.
NOVEL TREATMENTS: DETOXIFYING BIOMIMETIC “NANOSPONGE” DECOY RECEPTORS
The detoxifying treatment NarcoBond is effective against any opioid that binds to the mu opioid receptor, the cellular target that mediates the respiratory depressant effects of opioids, including fentanyl and its analogs. NarcoBond is a proteolipid biomimetic “nanosponge,” 100 nm diameter nanospheres, assembled from a mixture of cholesterol and synthetic choline-based phospholipids that mimic the lipid bilayer of cell membranes). Its multifunctional surface displays an optimized milieu of human membrane proteins originally isolated from the following cellular sources: (1) human erythrocytes to increase t1/2 in peripheral blood and circulation; (2) human neurons to increase opioid receptor functionality; and (3) recombinant human mu opioid receptors to bind circulating opioids. The repertoire of native opioid receptors in NarcoBond bind and sequester a comprehensive array of opioids thereby causing an antagonist pharmacological effect by rapidly reducing the free concentration of opioids. NarcoBond directly captures opioids in liquid tissues (e.g., blood and lymph) while it indirectly extracts opioids from other tissues (e.g., fat) via the diffusion of opioids to the plasma and the subsequent sequestration in the nanosponge (Le Chatelier’s principle). In competitive receptor binding studies with (3H)-DAMGO, NarcoBond showed affinities for opioids consistent with those reported for opioid binding to cells expressing opioid receptors69,70 and matching those measured in control experiments using human induced pluripotent stem cell-derived neurons and Chinese hamster ovary (CHO) cells expressing mu opioid receptors (unpublished data). For example, NarcoBond bound buprenorphine, fentanyl, and oxycodone with Ki values of 0.28, 1.45, and 529 nM, respectively, mimicking the cell membrane environment that maintains the functional form of mu receptors. The ability of NarcoBond to bind any class of opioid could be a significant advantage over some other treatments because the particular opioid(s) taken by a patient needing rescue from overdose is typically unknown.
In contrast to the short duration of action of naloxone, NarcoBond is relatively stable in vivo being found in blood, lungs, kidneys, spleen, and liver 24 hours after injection. Importantly, NarcoBond is safely excluded from the central nervous system by not crossing the blood brain barrier. Nonetheless, NarcoBond traps centrally acting opioids, presumably using equilibrium processes. After 48 hours, it is cleared from tissues and undergoes digestion in the liver. Although more studies are needed, this pattern of clearance indicates that NarcoBond and trapped opioids are gradually processed in the liver and excreted in urine, a metabolic fate seen with other nanosponges.71 Preliminary safety testing in mice showed 90–100% survival after i.v. injection of NarcoBond from 0.8 mg/kg (effective dosage) to 160 mg/kg (200 times larger than the effective dose).
The effect of NarcoBond on distribution and antinociception of opioids was studied in rats. NarcoBond sequesters opioids in the plasma and blocks the effects of fentanyl, heroin, and oxycodone. Rats challenged with a single dose of fentanyl (n = 12, 0.05 mg/kg, i.v., T = 0), or heroin (n = 6, 1 mg/kg, s.c., T = 0), or oxycodone (n = 6, 2.25 mg/kg, s.c., T = 0) were treated with NarcoBond (0.8 mg/kg, i.v., T = 5 minutes). Animals were tested for opioid-induced antinociception with a hot plate test (T = 30 minutes), serum and brain were isolated (fentanyl rats only), and fentanyl was extracted and quantitated (gas chromatography/mass spectroscopy). These studies found that NarcoBond attenuated antinociceptive effects of fentanyl, oxycodone, and hydrocodone, and sequestered fentanyl in serum away from the brain.
Rats were challenged with cumulative doses of fentanyl (s.c. at successive intervals: 12.5, 25, 50, and 100 μg/kg); 15 minutes after each dose, antinociception, oxygen saturation, and heart rate were measured. Animals then received 0.8 mg/kg NarcoBond (i.v.) immediately after the final antinociception and oxygen saturation measurements. NarcoBond reversed fentanyl-induced antinociception (vs. vehicle; Figure 3a; unpublished data), fentanyl-induced respiratory depression (Figure 3b), and fentanyl-induced bradycardia (Figure 3c). Furthermore, NarcoBond sequestered > 70% of fentanyl in the peripheral blood and away from the brain (Figure 3d). Taken together, these experiments demonstrate the ability of NarcoBond to attenuate opioid-induced antinociception, respiratory depression, and bradycardia, as well as sequester opioids in peripheral blood.
Figure 3.
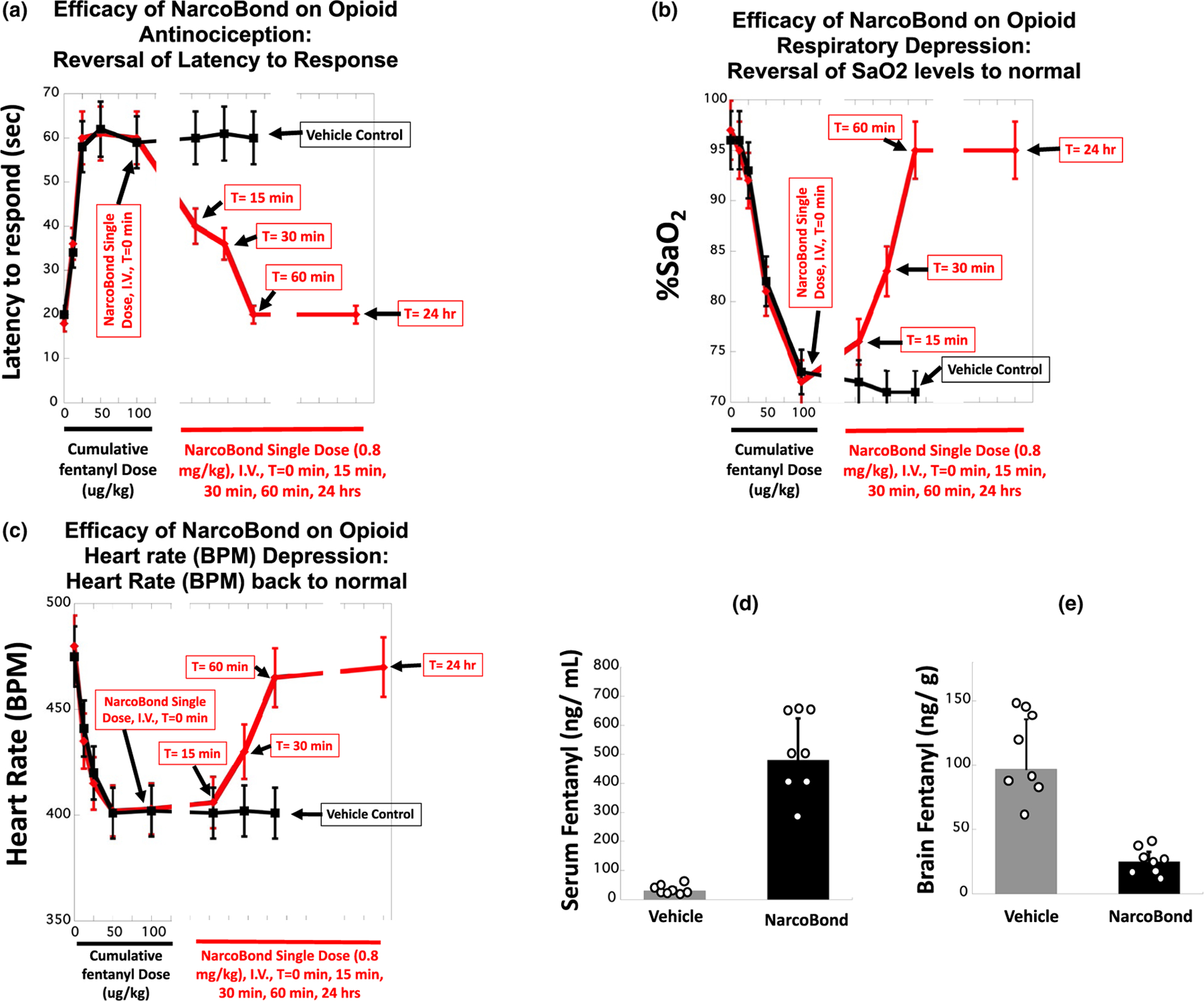
Effects of NarcoBond (unpublished data) on opioid antinociception, respiratory depression, heart rate, and distribution in rats (n = 6–12) challenged with repeated cumulative doses of fentanyl (100 μg/kg). Acute injection of NarcoBond (i.v., 0.8 mg/kg at T = 0) reversed fentanyl-induced antinociception (a) and respiratory depression (b) and heart rate (c), observed at T = 15 minutes and returning to normal by T = 60 minutes after treatment (compared with vehicle). NarcoBond sequestered > 70% of fentanyl in circulation (d) away from brain (e). a–c: Mean ± SEM. d: Mean ± SD. BPM, beats per minute.
Naloxone should also bind to mu opioid receptors on NarcoBond. Unlike naloxone, NarcoBond does not cross the blood brain barrier and remains in the periphery creating an opioid buffer zone in circulation (i.e., a partial functional and compartmental separation between the activity of naloxone (mostly in the brain) and NarcoBond (only in the periphery)). A recent study compared the effects of fentanyl in rats treated with NarcoBond alone, naloxone alone, or both NarcoBond and naloxone. Activity of naloxone was not diminished in rats treated with the mixture of NarcoBond and naloxone (similar to unpublished data shown in Figure 3). In fact, the mixture of naloxone and NarcoBond protected rats from much larger doses of fentanyl (up to 20 times larger than doses used in Figure 3) compared with protection from naloxone alone (reversing antinociception and respiratory depression). Naloxone alone did not rescue animals from these larger doses of fentanyl, whereas NarcoBond was effective under the same conditions. Thus, NarcoBond does not appear to interfere with the antagonist properties of naloxone. NarcoBond represents a novel addition to the current arsenal of opioid overdose treatments that can be used alone or in combination with naloxone by first responders and emergency department personnsel72 to reverse acute respiratory depression and to subsequently clear opioids from the blood, thereby preventing renarcotization.
To date, preclinical studies have not shown any adverse allergic reactions to NarcoBond. To minimize immunogenicity, NarcoBond is formulated with only humanized and defined proteins that are overexpressed in cell free systems, eliminating the possibility of immunogenicity from contaminates derived from cell expression systems. Moreover, NarcoBond contains integrin associated protein (CD47), the red blood cell membrane protein that is a universal “marker-of-self,” inhibiting phagocytosis and conferring anti-inflammatory properties. Animals treated with NarcoBond have not shown any acute or delayed anaphylactic responses (suggesting no sensitization by or to endogenous mu opioid receptors). It is envisioned that NarcoBond will be used in the clinic as a single or a titrated dose short-term therapy (in conjunction with or following naloxone), minimizing the possibility of any allergic and immune reactions.
To enable efficient production of clinical grade material, a simplified formulation of NarcoBond was devised using iterative rounds of screening and optimization. The amount of mu opioid receptor protein added per NarcoBond particle can be varied systematically, with initial data showing that adding more protein per liposomal particle results in a corresponding increase in opioid binding capacity. The goal is to decrease adverse effects by minimizing the number of NarcoBond particles while maintaining a high capacity to bind opioids. This simple yet efficacious formulation enables scale-up of Good Laboratory Practice-grade NarcoBond, which is being used to complete preclinical and clinical development. Nanoparticle nanosponges are used in nearly two dozen clinically approved products.73 Furthermore, scores of liposomal products are in clinical testing for diverse areas, including the delivery of anticancer drugs, antifungal drugs, antibiotics, oligonucleotides, anesthetics, imaging agents, and anti-inflammatory drugs.73
The “nanosponge” decoy receptor mechanism used for NarcoBond is readily adaptable to other drugs of abuse (e.g., methamphetamines), toxins (e.g., nerve gas), or contagions (e.g., severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2)) using specific protein/receptor targets, providing practical, safe, and efficacious countermeasures. Currently, this approach is being adapted to mitigate SARS-COV-2 (proteolipid nanosponge called: ViruClear). ViruClear uses human angiotensin-converting enzyme 2 receptor, the human receptor targeted by the spike protein of the SARS-CoV-2 virus74 as well as other cellular membrane proteins vital for viral attachment and cellular penetration (e.g., transmembrane protease, serine 2). Preliminary in vitro experiments show that ViruClear captures the virus with high affinity and mitigates viral infection in human cell culture models of SARS-CoV-2 infection. Of paramount significance is the synthetic nature of the designed proteolipid nanosponges, which readily permit multiplexing of different toxins, agents, and receptors, providing for an ideal tool for developing countermeasures against a vast number of the current list of chemical and biological threats, and contagions of interest.75
NOVEL TREATMENTS: ANTIBODY-BASED STRATEGIES
Vaccines and monoclonal antibodies (mAbs) are being developed as medical countermeasures to reduce the prevalence of OUD and the incidence of opioid-induced overdose.76,77 Active immunization with vaccines, consisting of opioid-based small molecule haptens conjugated to larger immunogenic carrier proteins or particles, stimulates innate and adaptive immunity to generate polyclonal antibodies that bind and sequester the target opioid in serum.77 Because antibody-drug complexes do not cross the blood brain barrier, vaccine-induced polyclonal antibodies prevent the distribution of the free (unbound) drug to the brain and reduce drug-induced behavioral and pharmacological effects.77 Although anti-opioid mAbs display in vitro and in vivo properties comparable to vaccine-induced polyclonal antibodies, passive immunization with mAbs has the advantage (over active immunization) of providing almost immediate protection upon administration while minimizing concerns of individual variability because mAbs do not rely on the patient’s immune system.77
Because of their selectivity for the target opioid and its metabolites or related structural analogs, both vaccines and mAbs are not expected to interfere with current FDA-approved medications for treating OUD (e.g., naltrexone) or opioid-induced toxicity and overdose (i.e., naloxone). Moreover, vaccines and mAbs do not bind endogenous opioid neuropeptides or off-target drugs used in pain management and critical care (e.g., anesthetics). In clinical settings, vaccines and mAbs could be used in combination with standard care to provide additional prophylactic and therapeutic options to patients diagnosed with OUD or other substance use disorders. Beyond OUDs and other substance use disorders, vaccines and mAbs could also be administered to individuals who are at higher risk of opioid poisoning and overdose through accidental or deliberate (e.g., weaponization) exposure, including airport or customs personnel, law enforcement, first responders, and military personnel. Although vaccines provide safe, long-lasting, and cost-effective interventions suitable for long-term treatment, mAbs may offer a viable medical countermeasure in acute clinical scenarios. For instance, anti-opioid mAbs have a longer half-life compared with the opioid receptor antagonist naloxone, and mAbs could be co-administered with naloxone or other opioid receptors antagonists to prevent renarcotization. On the other hand, mAb deployment could be limited by a higher cost per dose and the potential for immune-related adverse effects (e.g., auto-antibodies). Ultimately, the target product profile of anti-opioid vaccines and mAbs will be based upon evaluation of their clinical safety and efficacy, along with other commercial or marketing considerations.
Preclinical studies have shown that anti-opioid vaccines effectively reduce behavioral effects of opioids, such as antinociception and motor activity, as well as acquisition and maintenance of intravenous self-administration in mice, rats, and nonhuman primates challenged with heroin, oxycodone, hydrocodone, fentanyl, fentanyl analogs, and opioid mixtures.76 Anti-opioid vaccines have been shown to reduce opioid-induced respiratory depression and bradycardia in rats challenged with either oxycodone or fentanyl.78,79 The ability of naloxone to reverse effects of opioid receptor agonists was preserved in vaccinated rats,78,79 providing further evidence that anti-opioid vaccines will not interfere with life-saving medications, such as naloxone (Narcan). For instance, co-administration of an oxycodone vaccine with an extended-release naltrexone depot formulation (Vivitrol) offered greater protection against oxycodone-induced respiratory depression compared with either treatment alone.80 As these preclinical data support translation and clinical evaluation, one candidate vaccine targeting oxycodone is currently moving toward phase I clinical trials.81 To accelerate implementation of anti-opioid vaccines in clinical management of OUD, biomarkers predictive of vaccine efficacy will be useful tools to guide patient stratification in clinical trials and individualized therapy. In this context, it has been shown that the frequency of opioid-specific B cell lymphocyte population subsets in blood prior to immunization is predictive of vaccine efficacy against opioids in mice.82 Efforts in the laboratory of Dr. Pravetoni are currently underway to test whether B cell-based biomarkers are likely to predict vaccine efficacy against opioids in humans. Promising preclinical data also support use of anti-opioid mAbs to counteract acute respiratory depression and bradycardia in mice and rats challenged with heroin, oxycodone, or fentanyl.83 Furthermore, it has been shown that anti-opioid mAbs can increase survival from opioid overdose in mice and that anti-opioid mAbs can be administered in postexposure scenarios to reduce opioid-induced behavioral effects.84 These data provide encouraging evidence to support translation of anti-opioid mAbs, but more advanced studies are needed to fully characterize their safety and efficacy. Specifically, efforts should focus on generating and characterizing humanized or fully human anti-opioid mAbs to minimize concerns for immune-related adverse effects upon repeated dosing. Future efforts will be directed toward exploring the clinical potential of vaccines and mAbs for treating OUD and preventing opioid-induced overdose.
SUMMARY AND CONCLUSIONS
On the one hand, opioid receptor agonists are still the most effective drugs available for treating certain types of moderate-to-severe pain. On the other hand, the adverse effects of opioid receptor agonists continue to be a major public health challenge, both in terms of OUD and opioid overdose and death. Despite medications for treating OUD being available for more than 70 years (methadone since 1947, buprenorphine since 1981, and naltrexone since 1984), and the fact that these medications are effective in many patients, the opioid epidemic continues and, in fact, appears to have worsened during the current pandemic.85 Methadone and buprenorphine replace abused opioids, including fentanyl and other synthetic opioids and, thus, help mitigate risks associated with illicit drug use. However, methadone and buprenorphine have agonist properties at mu opioid receptors and, therefore, share many adverse effects of abused opioids (e.g., abuse and overdose). Because of its antagonist properties at mu opioid receptors, naltrexone avoids the adverse effects that can occur with methadone and buprenorphine, although there are limitations to the clinical use of naltrexone. Because it binds to receptors in a competitive, reversible manner, the effects of naltrexone can be surmounted by taking more opioids. Moreover, the utility of naltrexone for treating OUD is limited by its relatively short duration of action, compared with the longer duration of action of some abused opioids. A sustained-release formulation (Vivitrol) extends the duration of action of naltrexone, although it is not clear how effective this formulation will be for treating OUD and surmountability of the antagonist properties of Vivitrol remains a concern.86 Collectively, a large body of epidemiological evidence underscores the need for more and better treatments for OUD.
The only medication available currently to treat opioid overdose (naloxone) was approved by the FDA nearly 50 years ago, yet more than 70,000 Americans died from a drug overdose in 2019, most from opioids, and many from synthetic opioids.87 Further compounding this public health challenge is the fact that, despite the effectiveness of naloxone in rescuing individuals from opioid overdose, the risk of overdose and death remains very high after rescue with naloxone.23 Thus, a similarly large body of evidence underscores the need for more and better treatments for opioid overdose. That need is magnified significantly by the possible weaponization and deployment of synthetic opioids in a manner that could lead to mass casualties. Currently available formulations of naloxone would be of limited value in a mass casualty scenario, primarily because of its short duration of action. This paper describes emerging alternative approaches for treating opioid overdose, particularly in the event of weaponization of synthetic opioids, and these strategies could also be applied to treating OUD. First, an intranasal formulation of nalmefene would be a significant improvement over naloxone by providing an equally effective and much longer duration of protection after rescue from overdose. Second, MCAM blocks the effects of opioids for a week or longer and could be particularly useful as a prophylaxis to protect law enforcement and military personnel against possible opioid poisoning. Third, covalent naloxone nanoparticles have the advantage of using an FDA-approved medication in a formulation that would reverse and protect against the toxic effects of opioids while avoiding the precipitation of withdrawal. Fourth, because 5-HT1A receptor agonists can stimulate respiration generally, they could attenuate respiratory depression by a variety of chemical agents, including synthetic opioids; this approach would be especially useful in situations where the chemical toxin is not known. Fifth, the physical and chemical properties of cyclodextrin scaffolds allow for specific inactivation of targeted molecules, including fentanyl. Sixth, biomimetic “nanosponge” decoy receptors, such as NarcoBond (a liposome encapsulated human opioid receptor), can deactivate and sequester any molecule that has affinity for opioid receptors. Finally, by targeting specific molecular features of opioids, antibody-based strategies can rapidly deactivate and prevent penetration to the central nervous system or limit distribution of opioids to other organs, thereby reducing opioid toxicity. The magnitude of the problem facing the nation, both in terms of the continuing opioid epidemic and our vulnerability to the weaponization of opioids, demands a multifaceted aggressive approach to develop new medications for opioid overdose. This paper reviews some of the innovative approaches that academic, government, and industry scientists are advancing to address this pressing matter.
FUNDING
Nalmefene (Skolnick): the clinical study described was conducted under a Clinical Trial Agreement supported in part by Opiant Pharmaceuticals and the National Institute on Drug Abuse (NIDA), National Institutes of Health (NIH) contracts N01DA-128905, N01DA-13-8920, and N01DA-14-8914. Methocinnamox (France, Gerak, Maguire, Disney, Husbands): supported by NIDA/NIH grants R01DA005018 (France), R01DA048417 (France), and R01DA007315 (Husbands), and the Welch Foundation grant AQ-0039 (France). Pravetoni: supported by NIDA/NIH awards U01DA038876, UG3DA048386, UG3DA047711, UG3DA048775, R01DA041730, and a CounterACT Administrative Supplement to U01DA038876, and U01DA051658. Valdez: supported by the Defense Threat Reduction Agency (DTRA) grant DTRA13081-32915.
CONFLICT OF INTEREST
The authors make the following disclosures: C.P.F. is co-holder of a US provisional patent for methocinnamox. M.P. is inventor of patents related to vaccines and antibodies against fentanyl and other opioids. P.S. is an employee of Opiant Pharmaceuticals, Inc. B.E.-A. is the Chief Executive Office and has ownership interest in CellCure (CiBots, Inc.) and he is the inventor of patents related to NarcoBond. G.M. and A.F. are employees of CellCure (CiBots, Inc.). J.Z. receives research support from and is a consultant and advisory board member for CellCure (CiBots, Inc.). All other authors declared no competing interests for this work.
Footnotes
DISCLOSURE
From presentations at a meeting (NIAID/NIH, August 6 and 7, 2019) entitled “Developing Medical Countermeasures to Rescue Opioid-Induced Respiratory Depression (A Trans-Agency Scientific Meeting).” This meeting brought together academic, industry, and government scientists to discuss medical countermeasures and other approaches for protecting against opioid poisoning, particularly poisoning caused by synthetic opioids such as fentanyl.
References
- 1.Centers for Disease Control and Prevention (CDC). Vital statistic rapid release: Provisional drug overdose death counts <https://www.cdc.gov/nchs/nvss/vsrr/drug-overdose-data.htm> (2020) Accessed August 17, 2020.
- 2.Sutter ME et al. Fatal fentanyl: one pill can kill. Acad. Emerg. Med 24, 106–113 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Ross RB & Carlo DJ Higher doses of naloxone are needed in the synthetic opioid era. Subst. Abuse Treat. Prev. Policy 14, 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shafer SL Carfentanil: a weapon of mass destruction. Can. J. Anaesth 66, 351–355 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Wax P, Becker C & Curry S Unexpected, “gas” casualties in Moscow: a medical toxicology perspective. Ann. Emerg. Med 41, 700–705 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Volkow N & Collins F The role of science in addressing the opioid crisis. N. Engl. J. Med 377, 391–394 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Yeung DT, Bough KJ, Harper JR & Platoff GE Jr. National Institutes of Health (NIH) Executive Meeting Summary: developing medical countermeasures to rescue opioid-induced respiratory depression (a trans-agency scientific meeting) – August 6/7 2019. J. Med. Toxicol 16, 87–105 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krieter P et al. Fighting fire with fire: development of intranasal to treat synthetic opioid overdose. J. Pharmacol. Exp. Ther 371, 409–415 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon R et al. Nalmefene: intravenous safety and kinetics of a new opioid antagonist. Clin. Pharmacol. Ther 39, 49–53 (1986). [DOI] [PubMed] [Google Scholar]
- 10.Ahonen J et al. Comparison of alfentanil, fentanyl and sufentanil for total intravenous anesthesia with Propofol in patients undergoing coronary artery bypass surgery. Br. J. Anesthes 85, 533–540 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Kharasch E Opioid half-lives and hemlines: the long and short of fashion. Anesthesiology 122, 969–970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krieter P et al. Enhanced intranasal absorption of naltrexone by dodecyl maltopyranoside: implications for the treatment of opioid overdose. J. Clin. Pharmacol 59, 947–957 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broadbear JH et al. Methocinnamox is a potent, long-lasting, and selective antagonist of morphine-mediated antinociception in the mouse: comparison with clocinnamox, β-funaltrexamine, and β-chlornaltrexamine. J. Pharmacol. Exp. Ther 294, 933–940 (2000). [PubMed] [Google Scholar]
- 14.Qi JA, Heyman JS, Sheldon RJ, Koslo RJ & Porreca F Mu antagonist and kappa agonist properties of beta-funaltrexamine (beta-FNA) in vivo: long-lasting spinal analgesia in mice. J. Pharmacol. Exp. Ther 252, 1006–1011 (1990). [PubMed] [Google Scholar]
- 15.Peckham EM, Barkley LM, Divin MF, Cicero TJ & Traynor JR Comparison of the antinociceptive effects of acute morphine in female and male Sprague-Dawley rats using the long-lasting mu-antagonist methocinnamox. Brain Res. 1058, 137–147 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Gerak LR et al. Methocinnamox (MCAM) produces long-lasting antagonism of the behavioral effects of μ opioid receptor agonists but not prolonged precipitated withdrawal in rats. J. Pharmacol. Exp. Ther 371, 507–516 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gerak LR, Maguire DR, Woods JH, Husbands SM, Disney A & France CP Reversal and prevention of the respiratory-depressant effects of heroin by the novel μ-opioid receptor antagonist methocinnamox in rhesus monkeys. J. Pharmacol. Exp. Ther 368, 229–236 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maguire DR, Gerak LR, Woods JH, Husbands SM, Disney A & France CP Long-lasting effects of methocinnamox on opioid self-administration in rhesus monkeys. J. Pharmacol. Exp. Ther 368, 88–99 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maguire DR et al. Effects of acute and repeated treatment with methocinnamox, a mu opioid receptor antagonist, on fentanyl self-administration in rhesus monkeys. Neuropsychopharmacology. 45, 1986–1993 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minervini V, Disney A, Husbands SM & France CP Methocinnamox (MCAM) antagonizes the behavioral suppressant effects of morphine without impairing delayed matching-to-sample accuracy in rhesus monkeys. Psychopharmacology 237, 3057–3065 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zernig G, Butelman ER, Lewis JW, Walker EA & Woods JH In vivo determination of mu opioid receptor turning in rhesus monkeys after irreversible blockade with clocinnamox. J. Pharmacol. Exp. Ther 269, 57–65 (1994). [PubMed] [Google Scholar]
- 22.Gmerek DE & Woods JH Effects of beta-funaltrexamine in normal and morphine-dependent monkeys: observational studies. J. Pharmacol. Exp. Ther 235, 296–301 (1985). [PubMed] [Google Scholar]
- 23.Weiner SG, Baker O, Bernson D & Schuur JD One-year mortality of patients after emergency department treatment for nonfatal opioid overdose. Ann. Emerg. Med 75, 13–17 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comer SD & Cahill CM Fentanyl: receptor pharmacology, abuse potential, and implications for treatment. Neurosci. Biobehav. Rev 106, 49–57 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cone EJ Recent discoveries in pharmacokinetics of drugs of abuse. Toxicol. Lett 102–103, 97–101 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Skopp G, Ganssmann B, Cone EJ & Aderjan R Plasma concentrations of heroin and morphine-related metabolites after intranasal and intramuscular administration. J. Anal. Toxicol 21, 105–111 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Boyer EW Management of opioid analgesic overdose. N. Engl. J. Med 367, 146–155 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rzasa Lynn R & Galinkin J.l. Naloxone dosage for opioid reversal: current evidence and clinical implications. Ther. Adv. Drug Saf 9, 63–88 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneir AB et al. Massive OxyContin ingestion refractory to naloxone therapy. Ann. Emerg. Med 40, 425–428 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Lindsay CD, Riches JR, Roughley N & Timperley CM Chemical warfare toxicology. In: Chemical Warfare Toxicology Volume 2: Management of poisoning. Ch. 8 (eds. Worek F, Jenner J, Thiermann H) 259–313 (Royal Society of Chemistry, Cambridge, UK, 2016). [Google Scholar]
- 31.Uddayasankar U, Lee C, Oleschuk C, Eschun G & Ariano RE The pharmacokinetics and pharmacodynamics of carfentanil after recreational exposure: a case report. Pharmacotherapy 38, e41–e45 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Evans JM, Hogg MI, Lunn JN & Rosen M Degree and duration of reversal by naloxone of effects of morphine in conscious subjects. Br. Med. J 2, 589–591 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lindsay CD, Riches JR, Roughley N & Timperley CM Chemical Defence Against Fentanyls. In: Chemical Warfare Toxicology Volume 2: Management of poisoning. Ch. 8 (eds. Worek F, Jenner J, Thiermann H) 259–313 (Royal Society of Chemistry, Cambridge, UK, 2016). [Google Scholar]
- 34.Harris AC & Gewirtz JC Acute opioid dependence: characterizing the early adaptations underlying drug withdrawal. Psychopharmacology 178, 353–366 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Loimer N, Hofmann P & Chaudhry HR Nasal administration of naloxone for detection of opiate dependence. J. Psychiatr. Res 26, 39–43 (1992). [DOI] [PubMed] [Google Scholar]
- 36.Clarke SF, Dargan PI & Jones AL Naloxone in opioid poisoning: walking the tightrope. Emerg. Med. J 22, 612–616 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahoo SK, Panyam J, Prabha S & Labhasetwar V Residual polyvinyl alcohol associated with poly (D, L-lactide-co-glycolide) nanoparticles affects their physical properties and cellular uptake. J. Control. Release 82, 105–114 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Görner T et al. Lidocaine-loaded biodegradable nanospheres. I. Optimization of the drug incorporation into the polymer matrix. J. Control. Release 57, 259–268 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Tyler B, Gullotti D, Mangraviti A, Utsuki T & Brem H Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug. Deliv. Rev 107, 163–175 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Vanbever R Transdermal delivery of fentanyl: rapid onset of analgesia using skin electroporation. J. Control. Release 50, 225–235 (1998). [DOI] [PubMed] [Google Scholar]
- 41.Diaz del Consuelo I, Falson F, Guy RH & Jacques Y Ex vivo evaluation of bioadhesive films for buccal delivery of fentanyl. J. Control. Release 122, 135–140 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Anselmo AC & Mitragotri S An overview of clinical and commercial impact of drug delivery systems. J. Control. Release 190, 15–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liaw J & Lin Y-C Evaluation of poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide) (PEO–PPO–PEO) gels as a release vehicle for percutaneous fentanyl. J. Control. Release 68, 273–282 (2000). [DOI] [PubMed] [Google Scholar]
- 44.Wightman RS, Nelson LS, Lee JD, Fox LM & Smith SW Severe opioid withdrawal precipitated by Vivitrol(R). Am. J. Emerg. Med 36, 1128 e1121–1128 e1122 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Kassick AJ et al. Covalent poly(lactic acid) nanoparticles for the sustained delivery of naloxone. ACS Applied Bio. Materials 2, 3418–3428 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lewter L, Johnson MC, Kassick AJ, Averick S & Kolber B Slow-sustained delivery of naloxone reduces typical naloxone-induced precipitated opioid withdrawal effects in male morphine-dependent mice. J. Neurosci 10.1002/jnr.24627. [e-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hodges MR & Richerson GB The role of medullary serotonin (5-HT) neurons in respiratory control: contributions to eupneic ventilation, CO2 chemoreception, and thermoregulation. J. Appl. Physiol 1985, 1425–1432 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guyenet PG & Bayliss DA Neural control of breathing and CO2 homeostasis. Neuron 87, 946–961 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilken B et al. Treatment of apneustic respiratory disturbance with a serotonin-receptor agonist. J. Pediatr 130, 89–94 (1997). [DOI] [PubMed] [Google Scholar]
- 50.Lalley PM, Bischoff AM & Richter DW Serotonin 1A-receptor activation suppresses respiratory apneusis in the cat. Neurosci. Lett 172, 59–62 (1994). [DOI] [PubMed] [Google Scholar]
- 51.Mendelson WB, Martin JV & Rapoport DM Effects of buspirone on sleep and respiration. Am. Rev. Respir. Dis 141, 1527–1530 (1990). [DOI] [PubMed] [Google Scholar]
- 52.Garner SJ, Eldridge FL, Wagner PG & Dowell RT Buspirone, an anxiolytic drug that stimulates respiration. Am. Rev. Respir. Dis 139, 946–950 (1989). [DOI] [PubMed] [Google Scholar]
- 53.Pierrefiche O, Schwarzacher SW, Bischoff AM & Richter DW Blockade of synaptic inhibition within the pre-Botzinger complex in the cat suppresses respiratory rhythm generation in vivo. J. Physiol 509(Pt 1), 245–254 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhingra RR, Dutschmann M & Dick TE Blockade of dorsolateral pontine 5HT1A receptors destabilizes the respiratory rhythm in C57BL6/J wild-type mice. Respir. Physiol. Neurobiol 226, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sahibzada N, Ferreira M, Wasserman AM, Taveira-DaSilva AM & Gillis RA Reversal of morphine-induced apnea in the anesthetized rat by drugs that activate 5-hydroxytryptamine(1A) receptors. J. Pharmacol. Exp. Ther 292, 704–713 (2000). [PubMed] [Google Scholar]
- 56.Ren J, Ding X & Greer JJ 5-HT1A receptor agonist Befiradol reduces fentanyl-induced respiratory depression, analgesia, and sedation in rats. Anesthesiology 122, 424–434 (2015). [DOI] [PubMed] [Google Scholar]
- 57.Guenther U et al. Repinotan, a selective 5-HT1A-R-agonist, antagonizes morphine-induced ventilatory depression in anesthetized rats. Anesth. Analg 111, 901–907 (2010). [DOI] [PubMed] [Google Scholar]
- 58.White JM & Irvine RJ Mechanisms of fatal opioid overdose. Addiction 94, 961–972 (1999). [PubMed] [Google Scholar]
- 59.Sniecikowska J, Newman-Tancredi A & Kolaczkowski M From receptor selectivity to functional selectivity: the rise of biased agonism in 5-HT1A receptor drug discovery. Curr. Top. Med. Chem 19, 2393–2420 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Oertel BG et al. The partial 5-hydroxytryptamine1A receptor agonist buspirone does not antagonize morphine-induced respiratory depression in humans. Clin. Pharmacol. Ther 81, 59–68 (2007). [DOI] [PubMed] [Google Scholar]
- 61.Khan S, Patil K, Yeole P & Gaikwad R Brain targeting studies on buspirone hydrochloride after intranasal administration of mucoadhesive formulation in rats. J. Pharm. Pharmacol 61, 669–675 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Szejtli J Introduction and general overview of cyclodextrin chemistry. Chem. Rev 98, 1743–1754 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Mayer BP, Kennedy DJ, Lau EY & Valdez CA Solution-state structure and affinities of cyclodextrin:fentanyl complexes by nuclear magnetic resonance spectroscopy and molecular dynamics simulation. J. Phys. Chem. B 120, 2423–2433 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Bennion BJ et al. Predicting a drug’s membrane permeability: a computational model validated with in vitro permeability assay data. J. Phys. Chem. B 121, 5228–5237 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Valdez CA, Leif RN & Mayer BP An efficient, optimized synthesis of fentanyl and related analogs. PLoS One 9, e108250 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mayer BP, Albo RL, Hok S & Valdez CA NMR spectroscopic investigation of inclusion complexes between cyclodextrins and the neurotoxin tetramethylenedisulfotetramine. Magn. Reason. Chem 50, 229–235 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Motoyama K et al. Involvement of PI3K-Akt-Bad pathway in apoptosis induced by 2,6-di-O-methyl-β-cyclodextrin, not 2–6-di-O-methyl-α-cyclodextrin, through cholesterol depletion from lipid rafts on plasma membranes in cells. Eur. J. Pharm. Sci 38, 249–261 (2009). [DOI] [PubMed] [Google Scholar]
- 68.Rothblat HG et al. Cell cholesterol efflux: integration of old and new observations provides new insights. J. Lipid Res 40, 781–796 (1999). [PubMed] [Google Scholar]
- 69.Olson KM, Duron DI, Womer D, Fell R & Streicher JM Comprehensive molecular pharmacology screening reveals potential new receptor interactions for clinically relevant opioids. PLoS One 14, e0217371 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pasternak GW & Pan Y-X Mu opioids and their receptors: evolution of a concept. Pharmacol. Rev 65, 1257–1317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Bars D, Gozariu M & Cadden SW Animal models of nociception. Pharm. Rev 53, 597–652 (2001). [PubMed] [Google Scholar]
- 72.George S, Boulay S & Begley D “I saved a life”: a heroin addict’s reflections on managing an overdose using “take home naloxone”. BMJ Case Rep. 2010, BCR0520102986 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anselmo AC & Mitragotri S Nanoparticles in the clinic: an update. Bioeng. Transl. Med 4, e10143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoffmann M et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.US Drug Enforcement Administration. Drug Enforcement Administration Counterfeit Prescription Pills Containing Fentanyls: A Global Threat (US Drug Enforcement Administration, Springfield, VA, 2016). [Google Scholar]
- 76.Pravetoni M & Comer S Development of vaccines to treat opioid use disorders and reduce incidence of overdose. Rev. Neuropharmacol 158, 107662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pravetoni M Biologics to treat substance use disorders: current status and new directions. Hum. VaccIN. Immunother 12, 3005–3019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Raleigh MD et al. Safety and efficacy of an oxycodone vaccine: addressing some of the unique considerations posed by opioid abuse. PLoS One 12, e0184876 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Raleigh MD et al. Vaccination reduces fentanyl distribution to the brain and fentanyl-induced toxicity in mice and rats: a potential role for a prophylactic vaccine against fentanyl-induced overdose. J. Pharmacol. Exp. Ther 10.1124/jpet.118.253674. [e-pub ahead of print] [DOI] [Google Scholar]
- 80.Raleigh MD, Accetturo C & Pravetoni M Combining a candidate vaccine for opioid use disorders with extended-release naltrexone increases protection against oxycodone-induced behavioral effects and toxicity. J. Pharmacol. Exp. Ther 10.1124/jpet.120.000014. [e-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baruffaldi F et al. Formulation and characterization of conjugate vaccines to reduce opioid use disorders suitable for pharmaceutical manufacturing and clinical evaluation. ACS Mol. Pharma 10.1021/acs.molpharmaceut.8b01296. [e-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Laudenbach M et al. The frequency of naïve and early-activated hapten-specific B cell subsets dictates the efficacy of a therapeutic vaccine against prescription opioid abuse. J. Immunol 194, 5926–5936 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baehr C et al. Monoclonal antibodies counteract opioid-induced behavioral and toxic effects in mice and rats. J. Pharmacol. Exp. Ther 10.1124/jpet.120.000124. [e-pub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smith LC et al. Monoclonal antibodies for combating synthetic opioid intoxication. J. Am. Chem. Soc 141, 10489–10503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Becker WC & Fiellin DA When epidemics collide: coronavirus disease 2019 (COVID-19) and the opioid crisis. Ann. Intern. Med 173, 59–60 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morgan JR, Schackman BR, Weinstein ZM, Walley AY & Linas BP Overdose following initiation of naltrexone and buprenorphine medication treatment for opioid use disorder in a United States commercially insured cohort. Drug Alcohol Depend. 200, 34–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Centers for Disease Control and Prevention (CDC). CDC Wonder <https://wonder.cdc.gov/> (2020). Accessed July 2, 2020