

Abstract
It is known that the humanin (HN) peptide binding to amyloid-β (Aβ) protects against its cytotoxic effects, while acetylcholinesterase (AChE) binding to Aβ increases its aggregation and cytotoxicity. HN is also known to bind the insulin-like growth factor binding protein-3 (IGFBP-3). Here, we examined the regulation of Aβ conformations by HN, AChE, and IGFBP-3 both in vitro and in the conditioned media from A549 and H1299 lung cancer cells. Our in vitro results showed the following: IGFBP-3 binds HN and blocks it from binding Aβ in the absence or presence of AChE; HN and AChE can simultaneously bind Aβ but not when in the presence of IGFBP-3; HN is unable to reduce the aggregation of Aβ in the presence of IGFBP-3; and HN abolishes the aggregation of Aβ induced by the addition of AChE in the absence of IGFBP-3. In the media, AChE and HN can simultaneously bind Aβ. While both AChE and HN are detected when using 6E10 Aβ antibodies, only AChE is detected when using the Aβ 17–24 antibody 4G8, the anti-oligomer A11, and the anti-amyloid fibril LOC antibodies. No signal was observed for IGFBP-3 with any of the anti-amyloid antibodies used. Exogenously added IGFBP-3 reduced the amount of HN found in a complex when using 6E10 antibodies and correlated with a concomitant increase in the amyloid oligomers. Immunodepletion of HN from the media of the A549 and H1299 cells increased the relative abundance of the oligomer vs the total amount of Aβ, the A11-positive prefibrillar oligomers, and to a lesser extent the LOC-positive fibrillar oligomers, and was also correlated with diminished cell viability and increased apoptosis.
Amyloid-β (Aβ), produced by almost all types of cells, is well recognized for its importance in the different stages of the development and progression of Alzheimer’s disease (AD).1-5 The approximately 4 kDa Aβ peptide is generated when the higher molecular weight amyloid precursor protein is sequentially processed by two membrane-bound endoproteases, β- and γ-secretase.1,6 Processing by γ-secretase yields different C-terminal Aβ heterogeneities, where Aβ40 and Aβ42 represent ~90 and 10% of the isoforms, respectively.1,6 The Aβ40 and Aβ42 peptides, which are thought to self-assemble into amyloid fibrils, are associated with and linked to the pathology of more than 20 devastating and serious human diseases, including AD and other neurodegenerative disorders.3-5,7-10 Aβ40, which has a lower tendency to form oligomers, co-localizes in plaques with Aβ42, which has a high tendency to aggregate into oligomers, is more fibrillogenic due to an additional two hydrophobic amino acids (Ile and Ala) at the C-terminus, and is the main form deposited in the brains of people with AD.11 Two different types of Aβ plaques are found in the AD brain, vascular amyloid and Aβ plaques, which are mainly composed of Aβ40 and Aβ42, respectively.12 The amino-terminal region of Aβ is relatively hydrophilic, while the carboxyl-terminal region is highly hydrophobic; this has been proposed to account for its propensity to aggregate at neutral pH.13
The mechanisms by which the primary sequence of Aβ is converted into functional entities and dysfunctional assemblies are largely obscure.14 Complementary approaches15 using molecular dynamics simulations along with experimental methodologies have been widely used in order to provide structural details of the various Aβ assemblies, which range from monomers16-19 to oligomers3,20 to protofibrils21 and fibrils,22 and the aggregation inhibitors of different Aβ species.23,24
More recently, multiple studies have shown that patients with AD might have a reduced risk of and some protection against cancer, and that an inverse relationship between cancer and AD25-30 exists such that patients with AD generally had a significantly reduced rate of developing cancer with time while the rate of developing AD was reduced in cancer patients. The incidence of AD was reduced not only with glioblastoma but also with other types of cancer, including lung cancer.30 Aβ was shown to be protective against certain types of cancer and is capable of inhibiting the growth of tumor cells.31,32 Treatment of cancer cell lines with conditioned media that contain Aβ reduced the proliferation of human breast adenocarcinoma, melanoma, and glioblastoma,32 while Aβ suppressed tumor growth in mice upon direct injection into human lung adenocarcinoma xenografts.31 In all cancer patients, the levels of plasma Aβ40 and Aβ42 were reported to be higher than the levels of the normal controls.33 In this study, we used two human non-small-cell lung carcinoma (NSCLC) cell lines34 and A549 (p53-positive) and H1299 (p53-null) cells35 in order to gain mechanistic insights into the Aβ regulation in lung cancer cells.
Humanin (HN) is a secreted mitochondrially derived peptide discovered initially by the Nishimoto laboratory.36,37 When translated in the cytoplasm, it is composed of 24 amino acids; when translated in the mitochondria, it is composed of 21 amino acids.38 Certain residues in HN have been implicated in different activities, including binding to Aβ.38,39 Multiple lines of evidence suggest that HN possesses broad cyto- and neuroprotective activities against different types of stress and a wide range of disease models.38,40,41
HN was previously identified as a binding partner to Aβ, likely counteracting its deleterious and damaging effects.39,40,42 HN was shown to alter the morphology of Aβ40 from fibrillary to amorphous,43 providing protective functions against the cytotoxic effects induced by Aβ. Circular dichroism and NMR studies were previously employed44 in order to show that HN is unstructured and flexible in aqueous solutions. However, in a less polar environment it takes up a helical structure (Gly5–Leu18), suggesting that these conformational changes allow the peptide, in its unstructured form, to interact with different receptors while enabling it in its helical conformation to pass through membranes and form more specific interactions.44
Numerous attempts have been put forth in order to design molecules that modify the kinetics of fibril formation in order to prevent or delay the self-assembly of monomeric Aβ into its oligomeric forms.2,10,45 Minimal information is currently available regarding the three-dimensional structures of the monomers and oligomers of both the Aβ40 and Aβ42 peptides in an aqueous solution.3 HN has been shown to directly interact with Aβ oligomers.46 Because HN binds directly to Aβ, and due to its known cytoprotective functions, it may play a role as a natural protective peptide-based tool with the potential to interfere with the formation and properties of toxic Aβ assemblies. Whether HN is able to interact with homogeneous unaggregated, oligomeric, and fibrillar Aβ peptide fragments is largely unknown.
The amino acid residues that form direct interactions between HN and Aβ40 were identified earlier by molecular modeling.47 HN5-15 and Aβ17-28 were further identified by proteolytic epitope excision and extraction as well as by affinity mass spectrometric data analysis as the specific epitopes at the binding interface between HN and Aβ40.47 The binding of HN to Aβ17-28 was proposed to block Aβ from interacting with its receptors.39 Inhibition of the 17–28 region of Aβ was found earlier to decrease the aggregation of the neurotoxic amyloid fibrils and the related cytotoxicity in SH-SY5Y, a human neuroblastoma cell line.5 More recently, HN was found to bind directly to Aβ42 and exhibited anti-oligomeric activity.46 We also found that the Leu11 of HN is important for its interactions with Aβ40.48 Using nuclear magnetic resonance (NMR) in an alcohol/water solution, HN with a d-isomerized Ser14 was found to bind Aβ40 with higher affinity than either wild-type HN or HNS14G and had potent inhibitory effects against Aβ40 fibrillation.49 Interestingly, the d-isomerization of the Ser14 residue resulted in a drastic conformational change in HN that might provide a molecular mechanism for its cytoprotective activity.49
AChE is an enzyme that breaks down the neurotransmitter acetylcholine at the synaptic cleft.50 Most of the cortical AChE activity present in the Alzheimer’s brain is known to be predominantly associated with the amyloid core of senile plaques.51-55 AChE forms a stable toxic complex with the Aβ peptide during its assembly into filaments, increasing the aggregation and neurotoxicity of the Aβ fibrils.55,56 AChE is known to increase Aβ42 oligomeric formation57 and is associated with the amyloid plaque accumulation of abnormally folded Aβ40, a main component of the amyloid plaques found in the brains of AD patients.50-56 The addition of AChE significantly increased the aggregation of Aβ40.51,53-56 It has been shown earlier that the enzyme may have non-catalytic functions, since the catalytic active center of AChE was not required for Aβ40 amyloid fibril formation.58 The peripheral anionic site of the enzyme was found to be the site where Aβ interacts, accelerating the formation of the amyloid fibrils and resulting in a highly toxic complex.59 The toxicity of the AChE–amyloid complexes was found to be higher than that of the Aβ aggregates alone.55 Binding assays indicated54 that AChE binds to Aβ (12–28) and the Aβ (1–16) peptide (Figure 1) and is able to directly promote Aβ40 aggregation and its assembly into amyloid fibrils. Thus, since the binding sites of AChE and HN on Aβ overlap, HN and AChE may serve to regulate the central domain of Aβ (residues 17–24) that is flanked by Lys16 and Lys28, which is known to be a critical structural element in fibrillar Aβ aggregates.60-62
Figure 1.

Experimental methods show that HN and AChE bind a shared sequence on Aβ42. (A) The amino acid sequence of Aβ40 and Aβ42. (B) AChE binds to regions Aβ 1–16 as well as Aβ 12–28. HN binds to region 17–28.
IGFBP-3 is a member of a family of six insulin-like growth factor (IGF) binding proteins that have highly conserved structures.63-68 Among the IGFBPs, IGFBP-3 is the most abundant and is a primary carrier of IGF-I in circulation. It also employs its antiproliferative effects by binding IGF-1, by blocking IGF/IGF-IR interactions,63,66,68 or via mechanisms independent of the IGF/IGF-IR axis.68-71 The expression of IGFBP-3 is decreased in lung cancer34 and is associated with tumor metastasis and a poor diagnosis in stage I NSCLC patients.72-76 An inverse relationship has been reported to exist between the plasma or serum levels of IGFBP-3 and lung cancer risk.63,68,77 The expression of IGFBP-3 resulted in the inhibition of MAPK signaling, increased cleaved caspase-3, and corresponded with a decreased human lung cancer cell survival.78
Previously, we found that HN binds IGFBP-3 and interferes with the interaction of importin-β1 with IGFBP-3 in vitro, providing a probable functional role for HN as a regulator of the nuclear translocation of IGFBP-3.79 We also found that amino acid residues 215–232 in the C-terminal region of IGFBP-3 bind hyaluronan, blocking it from interacting with its receptor CD44 and reducing A549 human lung cancer cell viability;80 HN binding to IGFBP-3 blocked the protein from exerting these effects. More recently, we found that IGFBP-3 can block hyaluronan-CD44 signaling by a mechanism that depends on both acetylcholinesterase and p53.81
Here, we show that while the binding of AChE to Aβ is reduced in the presence of HN it is not abolished, and that the aggregation of Aβ in the presence of both HN and AChE is greatly reduced. We also show that the interaction of IGFBP-3 with HN restores AChE’s ability to increase Aβ aggregation. Moreover, the immunodepletion of HN from A549 and H1299 lung cancer cell-conditioned media increased the relative amount of the Aβ oligomer vs the total amount of Aβ, decreasing cell viability and increasing apoptosis.
EXPERIMENTAL PROCEDURES
Materials.
Most of the materials used were purchased as we previously reported.80 Nitrocellulose membranes, phosphate buffer saline (PBS), recombinant human AChE (C1682, UniProt C9JD78), oligomer anti-amyloid fibril (LOC, rabbit, AB2287) antibodies, streptavidin-conjugated horseradish peroxidase (HRP) conjugate, Ponceau S solution, and phenyl-methylsulfonyl fluoride (PMSF) were purchased from Sigma-Aldrich. Recombinant human IGFBP-3 protein (YCP1009, UniProt P17936) was purchased from Speed BioSystems. Mouse IgG isotype control (mIgG), ultra 3,3′,5,5′-tetrame-thylbenzidine (TMB)-ELISA substrate solution, annexin V human ELISA kits (BMS252), halt protease and phosphatase inhibitor cocktail, humanin polyclonal antibodies (rabbit, PA1-41610), IGFBP-3 polyclonal antibodies (goat, PA5-18791), A11 polyclonal antibodies (rabbit, AHB0052), and Nunc MaxiSorpTM 96-well flat bottom plates were from ThermoFisher. Goat anti-AChE antibodies (ab31276) and rabbit anti-goat IgG H&L (HRP, ab6741) were from Abcam. Mouse monoclonal amyloid-β antibodies (sc-53822), IGFBP-3 mouse monoclonal antibodies (sc-374365), and goat anti-rabbit IgG-HRP (sc-2004) were from Santa Cruz Biotechnology. Super signal west pico luminol (chemiluminescence) reagent and BCA protein assay kits were from Pierce. Aβ40-HFIP (AS-64128-05), Aβ42-HFIP (AS-64129-05), biotin-Aβ40 (AS-23512-01), and biotin-Aβ42 (AS-23523-05) were purchased from AnaSpec. Humanin (018-26) and biotin-humanin (B-018--26, UniProt Q8IVG9) were purchased from Phoenix Pharmaceuticals. Anti-Aβ (6E10, 1–16, mouse), anti-Aβ antibody (4G8, 17–24, mouse), anti-Aβ42 antibody (mouse), and biotin anti-Aβ antibody (4G8, 17–24) were from BioLegend.
Cell Culture.
Human NSCLC cell lines A549 (ATCC CCL-185) and H1299 (ATCC CRL-5803) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were seeded as we reported earlier80 in 5 mL of HyClone Dulbecco’s modified Eagles medium/nutrient mixture F-12 (DMEM/F12) (GE Healthcare Life Sciences, Pittsburgh, PA); supplemented with 10% Fetalgro bovine growth serum (FBS, RMBIO, Missoula, MT), 50 U/mL penicillin, and 50 U/mL streptomycin (Invitrogen Life Technologies, Carlsbad, CA) in 25 cm2 tissue culture flasks; and allowed to grow overnight in an incubator at 37 °C, 95% humidity, and 5% CO2. The cells were counted after trypan blue staining with a hemocytometer
ELISA.
ELISAs were conducted as we reported previously.48,80,81 Nunc MaxiSorp 96-well flat bottom plate wells were coated with the samples as indicated. The plates were incubated overnight at 4 °C on a shaker in order to allow binding to the plate wells. After the incubation, the wells were washed four times with TBST, filled with 400 μL of blocking buffer (110 mM KCl, 5 mM NaHCO3, 5 mM MgCl2, 1 mM EGTA, 0.1 mM CaCl2, 20 mM HEPES, and 1% BSA, pH 7.4), and incubated overnight at 4 °C on a shaker. The wells were then washed four times with TBST, and 100 μL of the sample at the desired concentration was added to each well before incubating overnight at 4 °C on a shaker. TBST was then used to wash the wells four times before proceeding in one of the following two ways: (1) biotinylated samples were analyzed by adding 100 μL of streptavidin-HRP conjugate in TBST (1:2500 dilution) to the samples before incubating for 3 h at rt on a shaker, or (2) samples without biotin were analyzed by adding 100 μL of TBST containing the primary antibody at the manufacturer’s recommendation and incubated for 3 h at rt on a shaker before washing four times with TBST. The secondary antibody in 100 μL of TBST was then added to the samples following the manufacturer’s recommendation and incubated for 1 h at rt on a shaker. Plates containing either the biotinylated or non-biotinylated samples were then washed five times with TBST, followed by the addition of 100 μL of TMB that resulted in a blue color change. The reaction was stopped with 100 μL of 2 M H2SO4 after incubating at rt for 0.5–15 min, which resulted in a yellow color change that was measured by absorbance spectroscopy at 450 nm. In order to monitor the nonspecific binding, the negative control wells on the plates included, for example, bound Aβ peptide. Then, all the components were added, i.e., streptavidin-horseradish peroxidase and TMB, but without the addition of biotin-HN. Some wells were coated with 2.5, 10, 50, 100, 500, and 5000 nM biotin-HN or Aβ in order to allow the conversion of the OD measurements to concentrations of the bound material. Before analysis, the OD measurements from the data were corrected for nonspecific binding by subtracting the mean background absorbance for the negative controls. Typically, in control wells incubated on each plate the background binding was about 10–15% of the maximum binding seen with the addition of biotin-peptides or antibodies. Statistical analysis was performed using GraphPad Prism 8.3.1. Data were expressed as the mean ± the standard deviation (SD). Three to five independent experiments were carried out in triplicate for each assay condition.
Quantitation of Aβ.
Aβ ELISAs were carried out according to previous protocols82,83 for determining the oligomeric and monomeric concentrations of Aβ. Briefly, the total amount of Aβ (monomers + oligomers) was measured by two-site binding ELISAs using the capture 6E10 monoclonal antibody and 4G8 conjugated to biotin as the detection antibody, which recognizes a distinct epitope, and then quantitated using streptavidin-horseradish peroxidase.
Using the same samples, oligomerized Aβ was measured by a single-site ELISA in which antibodies targeting the same primary sequence epitope were used for both capture (4G8) and detection (4G8-biotin). Only oligomers are detected with this approach, since the 4G8-biotin antibody cannot bind to the captured monomer because the epitope is blocked by the 4G8 capture antibody. Therefore, only oligomeric or multimeric Aβ containing additional exposed 4G8 epitopes not engaged by the capture antibody is reported by the streptavidin-horseradish peroxidase. The amount of the monomer was then estimated as the difference between the concentration of total Aβ and the concentration of the oligomer.
Dot Blotting.
Dot blots were done following our previously published procedures.80,81 Cells were grown in a 10% FBS-supplemented medium overnight in 25 cm2 flasks (ThermoFisher). The medium was collected and analyzed following the incubation of the cell monolayers in a serum-free medium for 24, 48, and 72 h in the presence of Halt protease, a phosphatase inhibitor cocktail, and 1 mM PMSF. After the protein concentrations were determined using the BCA protein assay kit, 3 μL of the 600 μg/mL total protein of the conditioned media was spotted onto a nitrocellulose membrane and allowed to dry. Nonspecific sites were blocked by soaking the blot for 1 h at rt in a 10 cm Petri dish containing TBST with 5% BSA. The blot was then incubated with the primary antibodies in the BSA/TBST overnight at rt following the manufacturer’s recommendation. After washing the membrane with TBST (3 × 5 min), the secondary antibodies conjugated with HRP were added according to the manufacturer’s recommendation. The membrane was incubated for 30 min at rt and then washed for 3 × 5 min with TBST and once with TBS for 5 min. Super signal west pico luminol (chemiluminescence) reagent was added in order to detect the amount of peptide or protein on the membrane, which was then imaged using a Bio-Rad molecular imager and quantitated with ImageJ ver. 1.47 software. Distilled water was used as a negative control, while the purified protein or peptide was used as a positive control. The aggregation of Aβ was monitored via dot blotting by methods described previously84 using oligomer or fibrillar and sequence specific antibodies.
Thioflavin T (ThT) Aβ Aggregation Assay.
The ThT assay was carried out as we reported previously.48 The effects of HN, IGFBP-3, and AChE on the aggregation kinetics of Aβ were investigated using the SensoLyte Thioflavin T (ThT) Aβ aggregation kits (Anaspec, AS-72213 and AS-72214). AggreSure Aβ40 peptide (Anaspec, AS-72215) and AggreSure Aβ42 peptide (Anaspec, AS-72216) were purchased pretreated in order to ensure that they were in a high-percent monomeric state. The fluorescence signal of ThT (Ex/Em = 440/484 nm) increases upon binding to amyloid fibrils and aggregated Aβ peptides.85 Aβ (2 μM) in 50 mM Tris or 150 mM NaCl (pH 7.2 containing 20 μM ThT) was incubated without or with 2 μM AChE or IGFBP-3 (2 or 6 μM). Samples (3 × 100 μL) were incubated at 37 °C in 96-microplate wells (ThermoFisher), and the ThT fluorescence intensity was monitored using the plate reader for 335 min at 37 °C every 2.5 min, with 15 s of shaking between reads. In order to correct for the fluorescence in the absence of Aβ, control experiments were conducted by incubating the assay buffer and 2 μM HN or IGFBP-3 with 20 μM ThT. The data were normalized by plotting the change in the relative fluorescence units (RFU) in relation to the first measurement at t0. The data were plotted using GraphPad Prism 8.3.1.
Immunodepletion.
The conditioned media were immunodepleted according to methods previously described.86 Briefly, specific antibodies were bound to the ELISA wells overnight. The wells were then blocked and washed, and then the media were incubated with the antibodies bound to the ELISA wells for 24 h. The immunodepleted media were then carefully removed and analyzed for the presence of the target protein or peptide by ELISA. Significant depletion (95–100%) was observed upon using each of the antibodies employed in this study.
MTT Assay.
The MTT reduction assay (Sigma-Aldrich), which is used to measure cell viability, was used as we reported earlier.80,81,87 Cells were seeded in 96-well plates as indicated in 200 μL of a 10% FBS-supplemented medium per well and maintained overnight at 95% humidity and 5% CO2. After overnight incubation, the medium was replaced with 200 μL of a serum-free medium, and the cells were then allowed to incubate for a further 24, 48, or 72 h. The final concentration of DMSO in each well never exceeded 0.1%. Following treatment, the cells were incubated for 4 h with MTT (0.5 mg/mL) in the dark. The medium was carefully removed, and DMSO (100 μL) was added in order to dissolve the formazan crystals. The absorbance was measured at 570 nm in a plate reader. Untreated cells or wells containing only DMSO and media were used as positive and negative controls, respectively. Statistical analysis was conducted using GraphPad Prism ver. 8.3.1 software for Windows. Significant values were considered at p < 0.05 and more significant values at p < 0.01, as compared to the control.
Apoptosis Assays.
The cells were grown as described above, and then apoptosis was measured using the annexin V human ELISA kit (Thermofisher) as we reported earlier.81 A matched antibody pair was used in order to detect annexin88 in the cell culture medium and quantitated using a human Annexin V standard curve according to the manufacturer’s instructions. The plates were washed following the addition of HRP conjugates and incubation for 30 min at rt. Next, the TMB substrate was added to each well and incubated in the dark at rt. After 15 min, the TMB stop solution was typically added in order to terminate the reaction when the blue color was apparent. Within 30 min after the reaction was stopped, the absorbance was measured at 450 nm. Cells that were treated with a 0.1% DMSO vehicle control and contained all the reagents except the primary antibodies were used as a control. The average of all replicate nonspecific background signal controls was subtracted, and then the average absorbance at 450 nm was calculated.
Statistical Analysis.
Each experiment in this study was performed in triplicate and repeated a minimum of three times. Statistical values are expressed as the mean ± SD. In order to evaluate the statistical differences, Mann–Whitney or Kruskal–Wallis (ANOVA) tests were used. All the statistical tests were two-sided, and a p-value of <0.05 was considered statistically significant in all cases. GraphPad Prism (GraphPad Software ver. 8.3.1) was used for the statistical analysis.
RESULTS
IGFBP-3 Blocks the Binding of HN to Aβ in the Absence or Presence of AChE.
HN has been previously reported to bind Aβ.39,40,46-48,89 In order to examine the binding of HN to either Aβ40 or Aβ42, Aβ (100 nM) was bound to the plate wells. Increasing concentrations of biotinylated HN were then added to the wells and processed as described in the Experimental Procedures (Figure 2A). The optical density measurements (450 nm) were normalized for both curves by expressing each point in relation to the best-fit Emax value (set to 100%). The data were then plotted as a function of the increasing biotinylated HN concentrations and fit to a single binding site model with a nonlinear regression curve fitting approach using GraphPad Prism 8.3.1. No significant difference was observed between the binding of HN to either Aβ.
Figure 2.
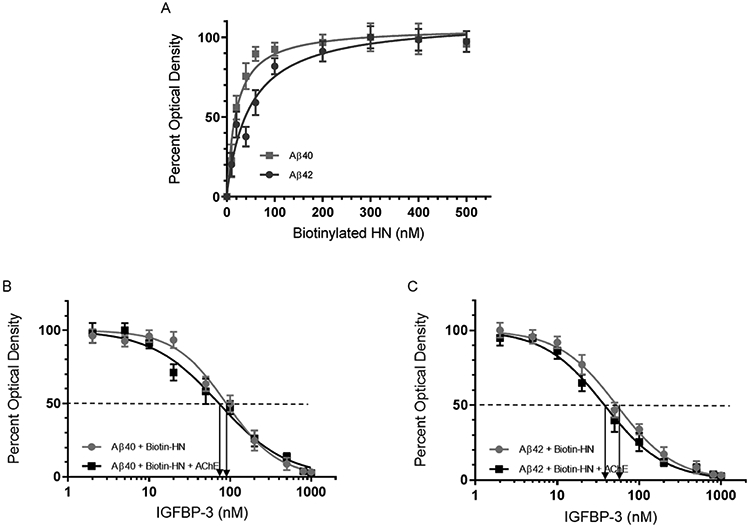
Increasing concentrations of IGFBP-3 bind HN and abolish its binding to Aβ in the absence or presence of AChE. (A) Interaction of Aβ40 (■) or Aβ42 (●) with biotinylated HN. Aβ (100 nM) was bound to the wells. Increasing concentrations of biotinylated HN were then added and processed as described in the experimental procedures. Optical density measurements (450 nm) were normalized by expressing each point in relation to the best-fit Emax value (set to 100%). The data were then plotted as a function of the increasing biotinylated HN concentrations. Using GraphPad Prism 8.3.1, the data were fit to a single binding site model with a nonlinear regression curve fitting approach and plotted as the mean ± SD of three independent trials, each of which was run in triplicate. (B and C) Competition of IGFBP-3 for the complex between HN and Aβ40 or Aβ42. Aβ (100 nM) was bound to the plate wells. Then, a single concentration of biotinylated HN (100 nM, ●) or a combination of HN and AChE [biotinylated HN (100 nM) + AChE (100 nM)] (■) was incubated for 1 h with or without increasing concentrations of IGFBP-3 and loaded into the Aβ coated wells. The signal was then processed as described in the Experimental Procedures. Arrows on the x-axis indicate the IGFBP-3 concentration corresponding to 50% inhibition for each of the following curves: Aβ40 (●, 93 ± 16 nM), Aβ40 (■, 74 ± 14 nM), Aβ42 (●, 58 ± 11 nM), and Aβ42 (■, 39 ± 7 nM). The dashed line indicates 50% of the maximum binding. Prior to data analysis, the OD was corrected for nonspecific binding by subtracting the mean background absorbance for the negative controls prepared with all components except biotinylated HN. Data were then normalized by plotting the mean absorbances for each concentration as a fraction of the maximal binding (set to 100%) and as a function of the IGFBP-3 concentrations. Using GraphPad Prism 8.3.1, the data were analyzed with a nonlinear regression curve fitting approach then expressed as the mean ± SD of three independent experiments, each of which was carried out in triplicate.
Certain amino acids of HN are known to bind Aβ, and F6 in particular is known to bind Aβ or IGFBP-3.39,90 We therefore tested whether IGFBP-3 competes with Aβ for binding HN or whether it can be found in a complex with Aβ and HN. Since AChE is known to bind Aβ,7,51,53,54,59 we also examined this competition in the presence of AChE. Aβ (100 nM) was bound to the plate wells (Figure 2B and C). A single concentration of biotinylated HN (100 nM) or a combination of HN and AChE [biotinylated HN (100 nM) + AChE (100 nM)], was incubated for 1 h without or with increasing concentrations of IGFBP-3 before loading into the Aβ coated wells, and the signal was processed as described in the Experimental Procedures. Before the analysis of the data, the OD was corrected for nonspecific binding by subtracting the mean background absorbance for the negative controls that contained all the components except biotinylated HN. The data were then normalized by plotting the mean absorbances for each concentration as a fraction of the maximal binding (set to 100%) and a function of the IGFBP-3 concentrations. The data were analyzed using GraphPad Prism 8.3.1 with a nonlinear regression curve fitting approach. The IGFBP-3 concentration that corresponded to 50% inhibition for Aβ40 in the presence of HN was found to be 93 ± 16 nM, while that for Aβ40 in the presence of both HN and AChE was found to be 74 ± 14 nM. Similar effects were observed for Aβ42. In the presence of Aβ42 and HN, the IGFBP-3 concentration that corresponded to 50% inhibition was found to be 58 ± 11 nM while that upon the addition of AChE was 39 ± 7 nM. These results show that HN binds with comparable affinity to either Aβ40 or Aβ42 but that IGFBP-3 is able to compete with Aβ for this binding. Moreover, the addition of AChE did not appear to modulate the effects of the added IGFBP-3.
Both HN and AChE Can Simultaneously Bind Aβ, But Not in the Presence of IGFBP-3.
The binding of Aβ to AChE is well documented.51,53,54,59 In order to test the binding of Aβ to AChE, the enzyme (100 nM) was bound to ELISA plate wells. Then, increasing concentrations of biotinylated Aβ were added to the wells and processed as described in the Experimental Procedures (Figure 3A). The optical density measurements (450 nm) were normalized for both curves by expressing each point in relation to the best fitted Emax value (set to 100%). The data were then plotted as a function of the increasing concentrations of biotinylated Aβ and fit using GraphPad Prism 8.3.1 to a single binding site model with a nonlinear regression curve fitting approach. Data were expressed as the mean ± SD of three independent experiments, each of which was carried out in triplicate. No significant difference was observed in the binding of AChE to either Aβ40 or Aβ42 (Figure 3A). These results were not surprising, since previous reports54 showed that AChE binds to Aβ12-28 as well as to the Aβ1-16 peptide (Figure 1).
Figure 3.
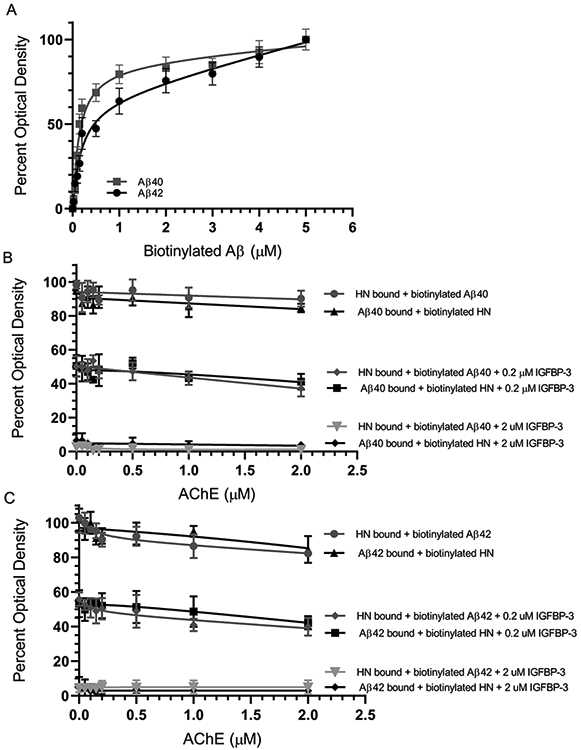
IGFBP-3, but not AChE, blocks the binding of HN and Aβ. (A) Interaction of AChE with Aβ40 (■) or Aβ42 (●). AChE (100 nM) was bound to the plate wells. Increasing concentrations of biotinylated Aβ were then added to the wells and processed as described in the Experimental Procedures. Optical densities (450 nm) were normalized for both curves by expressing each point relative to the best fitted Emax value (set to 100%). The data were then plotted as a function of the increasing biotinylated Aβ concentrations and, using GraphPad Prism 8.3.1, fit to a single binding site model with a nonlinear regression curve fitting approach. Data were expressed as the mean ± SD of three independent experiments, each of which was carried out in triplicate. (B and C) HN or Aβ (0.2 μM) was bound to the wells. Biotinylated Aβ (0.2 μM) or biotinylated HN (0.2 μM) was then pre-incubated with increasing concentrations of AChE for 1 h at rt and then added to the wells in the absence or presence of IGFBP-3 (0.2 μM or 2 μM). For samples without IGFBP-3, the optical density measurements were normalized by plotting the mean absorbances for each concentration as a fraction of the maximal binding (set to 100%), followed by plotting the data as a function of AChE concentrations. The negative control had the same concentrations of HN or Aβ and AChE, but water was substituted in place of biotinylated Aβ or biotinylated HN. For samples with IGFBP-3, the optical density was expressed as percent of the corresponding sample without IGFBP-3. Data were presented as the mean ± SD of three independent assays.
HN was shown previously to bind amino acid residues 17–28 of Aβ40.47 Since the binding sites of AChE and HN on Aβ overlap, we expected an antagonistic binding between HN and AChE on Aβ. In order to test this hypothesis, HN or Aβ (0.2 μM) were bound to ELISA plate wells. Biotinylated Aβ (0.2 μM) or biotinylated HN (0.2 μM) was pre-incubated with increasing concentrations of AChE for 1 h at rt and then added to the wells in the absence or presence of IGFBP-3 (0.2 or 2 μM). Data were expressed as the mean ± SD of three independent experiments. The optical density was normalized by plotting the mean absorbance values for each concentration as a fraction of the maximal binding in the absence of the added IGFBP-3 (set to 100%). The data were then plotted as a function of the AChE concentrations (Figure 3B and C).
We were surprised to find that the increasing concentrations of AChE had little effect on the ability of HN to bind Aβ. At equimolar concentrations of AChE (0.2 μM), only about a 10–15% reduction was observed using either biotinylated Aβ with HN or biotinylated HN with Aβ (Figure 3B and C). Moreover, there was only a 14–16% inhibition using 10-fold higher concentrations of AChE. These results clearly show that both HN and AChE can bind Aβ simultaneously despite each being capable of directly binding amino acid residues 17–28 of the Aβ peptide (Figure 1).
The addition of an equimolar concentration (0.2 μM) of IGFBP-3 and biotinylated Aβ to the HN bound to the wells resulted in an approximatly 50% decrease in the signal relative to samples without added IGFBP-3 (Figure 3B and C). Similarly, when Aβ was first bound to the wells the addition of equimolar concentrations of IGFBP-3 and biotinylated HN also resulted in a comparable decrease in signal relative to the control without IGFBP-3. Increasing the concentration of added IGFBP-3 from 0.2 to 2 μM completely abolished the signal. These results might suggest that the addition of equimolar concentrations of IGFBP-3 competes with Aβ for binding HN; however, the addition of a 10-fold higher concentration of IGFBP-3 is able to completely sequester HN, blocking its binding to Aβ. Moreover, no change in the signal was observed with increasing concentrations of AChE, suggesting that the reduction of the signal observed upon the addition of either 0.2 or 2 μM IGFBP-3 was due only to IGFBP-3 and not to AChE.
HN Is Unable to Reduce the Aggregation of Aβ in the Presence of IGFBP-3 In Vitro.
It is known that while both Aβ peptides can form toxic oligomers and rapidly aggregate, Aβ42 aggregates faster and to a significantly greater extent than Aβ40.91 HN is known to reduce the aggregation of Aβ.38,39,47,48 Here, we examined the effect of added IGFBP-3 on the ability of HN to reduce Aβ aggregation in vitro. Pretreated monomeric Aβ (2 μM) was incubated with 2 μM HN in the absence or presence of 2 or 6 μM IGFBP-3 (Figure 4). Thioflavin fluorescence (Ex 440 nm, Em 484 nm) was monitored at 37 °C for 335 min every 2.5 min, with 15 s of shaking between reads. Assay buffer alone, IGFBP-3, or HN was used as a blank. Each measurement was corrected for the fluorescence obtained without Aβ. The change in the relative fluorescence units (RFU) to the first measurement at t0 is shown. Not surprisingly, Aβ42 (Figure 4B) exhibited faster aggregation kinetics compared to those of Aβ40 (Figure 4A). For both Aβs, the addition of 2 μM IGFBP-3 reduced the ability of HN to block aggregation of Aβ, but only partially. These results might show that the addition of equimolar concentrations of IGFBP-3 to HN and Aβ allows IGFBP-3 to bind a portion of HN, sequestering it away from Aβ and increasing its aggregation. In support of this hypothesis, the addition of 6 μM IGFBP-3 to HN and Aβ, each added at 2 μM, completely abolished the ability of HN to block the aggregation of Aβ.
Figure 4.

HN reduced Aβ aggregation, an effect reversed upon the addition of IGFBP-3. Pretreated monomeric Aβ (2 μM) was incubated with 2 μM HN in the absence or presence of 2 or 6 μM IGFBP-3 ((A) Aβ40, AS-72215 and (B) Aβ42, AS-72216). Thioflavin fluorescence (Ex 440 nm, Em 484 nm) was monitored at 37 °C for 335 min every 2.5 min, with 15 s of shaking between the readings. Assay buffer alone, IGFBP-3, or HN was used as a blank. Each measurement was corrected for the baseline fluorescence in the absence of Aβ. The change in fluorescence units (RFU) relative to the first measurement at t0 is shown. The data were normalized to the maximum ThT level and plotted using GraphPad Prism 8.3.1.
HN Abolishes the Aggregation of Aβ Induced by the Addition of AChE in the Absence of IGFBP-3.
Since AChE does not appear to compete with HN for binding Aβ (Figure 3), and since HN and AChE are known to have opposing effects on the oligomerization of Aβ, 36,37,43,46-48,51,53-55,59 we questioned whether HN retains its ability to reduce Aβ aggregation (Figure 4) in the presence of AChE. Pretreated monomeric Aβ (2 μM, Aβ40, AS-72215) or Aβ42 (AS-72216) (Figure 5) was incubated with 2 μM AChE, 2 μM HN, or both in the absence or presence of 2 or 6 μM IGFBP-3. HN, IGFBP-3, or assay buffer alone was used as a blank. Thioflavin fluorescence (Ex 440 nm, Em 484 nm) was monitored at 37 °C for 335 min every 2.5 min, with 15 s of shaking between reads. Each measurement was corrected for the fluorescence measurement obtained without Aβ. As expected, the addition of AChE to either Aβ40 or Aβ42 resulted in increased aggregation with changes in both the lag phase and the final ThT fluorescence value (Figure 5). The addition of an equimolar concentration of HN and AChE, however, reduced the aggregation of both Aβ40 (Figure 5A) and Aβ42 (Figure 5B) to almost the same level induced by HN alone. These results suggest that HN binding to Aβ reduces its aggregation despite the binding of AChE.
Figure 5.

HN blocks the aggregation of Aβ induced by the addition of AChE, an effect abolished upon the addition of IGFBP-3. Pretreated monomeric Aβ (2 μM) was incubated with 2 μM AChE, 2 μM HN, or both in the absence or presence of 2 or 6 μM IGFBP-3 ((A) Aβ40, AS-72215 and (B) Aβ42, AS-72216). Assay buffer alone, HN, or IGFBP-3 was used as a blank. Thioflavin fluorescence (Ex 440 nm, Em 484 nm) was monitored at 37 °C for 335 min every 2.5 min, with 15 s of shaking between readings. Each measurement was corrected for the baseline fluorescence in the absence of Aβ. The change of the RFU in relation to the first measurement at t0 is shown. The data were normalized to the maximum ThT level and plotted using GraphPad Prism 8.3.1.
Incubation of an equimolar concentration of Aβ, AChE, HN, and IGFBP-3 (Figure 5) partially diminished the ability of HN to reduce the aggregation of Aβ. This is likely due to the ability of IGFBP-3 to compete with Aβ for binding HN. In support of this possibility, the addition of higher concentrations of IGFBP-3 (6 μM) to 2 μM Aβ, AChE, and HN resulted in aggregation kinetics comparable to those of Aβ incubated with only AChE. These results suggest that at higher concentrations IGFBP-3 is able to bind most HN and can better compete with Aβ for the interaction with HN.
AChE and HN Can Simultaneously Bind Aβ from the Media of A549 Cells.
Our in vitro data (Figure 3) show that HN and AChE can be found together in a complex with either Aβ40 or Aβ42. We then wondered whether such a complex can be found in the conditioned media of two human lung carcinoma NSCLC cell lines34 (A549 (p53-positive) or H1299 cells with a p53-null genotype due to a biallelic deletion of the TP53 gene).35 Anti-Aβ specific antibodies (6E10) were added (1:1000 dilution) to the ELISA wells (Figure 6). These antibodies are known to react with the monomers, oligomers, and fibrils of Aβ92,93 and recognize the N-terminal hydrophilic sequence amino acids 1–16 of Aβ. This epitope, previously shown to be exposed in Aβ aggregates,92 is believed to be residues 4–10.94 The wells were blocked, and then 300 μL of the conditioned medium (0.5 μg/μL of A549 cells or H1299 cells, 72 h after serum starvation) was added. The amount of HN and AChE bound was detected using the corresponding specific primary antibodies. The fold change values relative to the controls that included all components but without the primary antibodies were calculated and fit with a nonlinear regression curve using GraphPad Prism 8.3.1. Barely detectable levels of AChE in the H1299 conditioned medium were found to bind Aβ (Figure 6). This is not surprising, since we recently showed81 that there are minimal levels of AChE in the conditioned medium of the p53-null cell line, H1299, compared to that of the medium of the p53-positive cell line, A549. Consistent with this finding, detectable levels of AChE in the A549 cell medium were found to bind Aβ that is bound to its antibody, 6E10, in the wells (Figure 6). In the conditioned media of both cell lines, HN was found bound to Aβ as well. These results, however, do not indicate whether HN and AChE are found in the same complex with Aβ. In order to examine whether HN is able to compete off the binding of AChE to Aβ bound to the immobilized 6E10 antibodies in the wells, we incubated the wells with increasing concentrations of the HN peptide, followed by washing the unbound material as described in the Experimental Procedures. The addition of exogenous HN resulted in the increased binding of HN to Aβ from both A549 and H1299 media bound to its immobilized antibody, 6E10. In both cases, the signal increased and then began to level off around 50 nM (Figure 6). At the highest concentrations of HN added (400 nM), the fold increase relative to samples with no exogenous HN added was ~2.5-fold. This concentration of HN is much higher than the concentrations of HN (~50 nM) we measured previously80 in the A549 medium. Despite this increase in the HN signal indicative of binding to Aβ, there was only a ~1.5-fold decrease in AChE binding to Aβ using the A549 medium, suggesting that while HN blocks AChE binding to Aβ it is unable to completely inhibit this interaction at the concentrations used.
Figure 6.

Both AChE and HN were found in a complex with Aβ from the medium of A549 cells using 6E10 antibodies; however, the addition of an exogenous HN peptide reduced but did not abolish the binding of AChE to Aβ. Anti-Aβ specific antibodies (6E10) were added (1:1000 dilution) to the ELISA wells. The wells were blocked, and 300 μL of the conditioned medium (0.5 μg/μL of A549 cells or H1299 cells, 72 h after serum starvation) was added. The amount of HN and AChE bound was detected using the corresponding specific primary antibodies and then processed as described in the Experimental Procedures. The fold change relative to the controls incubated with all components except the primary antibodies was calculated and fit with a nonlinear regression curve using GraphPad Prism 8.3.1. The data represent the mean ± SD of three separate experiments, each of which was performed in triplicate.
Both AChE and HN Were Detected upon Using 6E10 Aβ Antibodies; However, Only AChE Was Detected with 4G8, A11, and LOC Antibodies, and No Signal Is Observed for IGFBP-3 with Any of the Antibodies Used.
Anti-Aβ antibodies 6E10 and 4G8 react with the monomers, oligomers, and fibrils of Aβ.92,93 The 6E10 antibody recognizes the N-terminal hydrophilic amino acid sequence 1–16 of Aβ95 while 4G8 binds to a central hydrophobic sequence (residues 17–24).94 Recent studies using a high-resolution mapping approach showed that 6E10 more specifically maps to amino acid residues 4–10, while the 4G8 epitope more specifically lies within residues 18–22 (VFFAE) of Aβ.96 Both epitopes have been previously shown to be exposed in Aβ aggregates.92 In addition to the sequence specific 6E10 and 4G8 antibodies that recognize all species of Aβ without regard to conformation, anticonformational antibodies were also used. The anti-oligomer specific A11 polyclonal antibody, which recognizes all types of prefibrillar oligomers but not monomers or fibrils, was used in order to distinguish the oligomer among the different conformations. The anti-amyloid fibrils LOC antibody, which recognizes epitopes common to amyloid fibrils and fibrillar oligomers but not monomers or prefibrillar oligomers, was used in order to detect the amyloid fibrils.
Our results show that HN is able to reduce the aggregation of Aβ in vitro (Figure 4), which is consistent with our previous findings.48 Since HN was previously found to reduce the amount of toxic oligomers in vivo,46 we tested whether HN is detected in a complex using 6E10, 4G8, A11, and LOC antibodies bound to ELISA plate wells. HN was detected upon using the anti-6E10 antibodies (Figure 7), which is not surprising since the direct binding of HN to Aβ is well documented.47,48,89,97 No signal was detected, however, upon the incubation of the media with the 4G8 antibody (Figure 7). Since this antibody recognizes amino acid residues 17–24 as the epitope on Aβ,94,96 it is likely that it acts to block the ability of HN to bind residues 17–28. Minimal detection of HN was observed upon using either A11 or LOC antibodies, suggesting that HN is likely not found in a complex with the prefibrillar oligomers detected by A11 or the anti-amyloid fibrils and Aβ fibrillar oligomers detected by LOC (Figure 7).
Figure 7.

HN was detected upon using Aβ 6E10 antibodies, while different levels of AChE were detected using all the amyloid antibodies. Cells (0.2 × 105 cells per well) were seeded in 96-well plates in a 10% FBS-supplemented medium. The next day, the cells were incubated in a serum-free medium for 72 h. Specific antibodies were added (1:1000 dilution) to the ELISA wells. After blocking the wells, 300 μL of the A549 cell conditioned medium (0.5 μg/μL, 72 h post serum starvation) was added. The proteins and peptides were detected using their corresponding primary antibodies and then processed as described in the Experimental Procedures. Each column represents the mean ± SD of three independent and separate experiments, each of which was performed in triplicate. Data processing was carried out using the GraphPad 8.3.1 software. Asterisks (*) indicate a statistically significant difference between each treatment relative to the mIgG controls. The absence of asterisks indicates no significance (Mann–Whitney test, *p < 0.05, **p < 0.01).
It is known that AChE directly promotes the aggregation of Aβ into amyloid fibrils.51,53-56 It was therefore not surprising to find AChE was bound upon using not only 6E10 and 4G8 but also A11 and LOC antibodies (Figure 7). Less enzyme was found to be bound upon using the 4G8 antibody (Figure 7). A likely explanation might be that while 4G8 may weaken binding of AChE to Aβ it is unable to completely block its binding to the peptide. This observation is similar to that found upon using HN to examine the binding of AChE to Aβ (Figure 6) and suggests that the binding of HN to residues 17–28 or 4G8 to residues 17–24 weakens but does not completely block the interaction of AChE with Aβ. The fraction of AChE bound to the anti-oligomer antibody (A11) or the anti-amyloid fibrils antibody (LOC) must not contain HN, since no signal above the background was obtained using the HN antibodies (Figure 7). No IGFBP-3 was detected upon using any of the antibodies tested, suggesting that the protein is not found in complexes bound to these antibodies (Figure 7).
Exogenously Added IGFBP-3 Results in a Reduction of HN Found in a Complex Using 6E10 Antibodies and Correlates with a Concomitant Increase in Amyloid Oligomers.
Our results (Figure 7) suggest that IGFBP-3 is not detected in a complex upon using 6E10, 4G8, A11, or LOC antibodies, and that HN is detected in a complex with Aβ only upon using anti-6E10 antibodies. We therefore examined whether exogenously added IGFBP-3 is able to sequester HN, resulting in changes in the conformation of Aβ. Anti-Aβ specific antibodies (6E10) were added to the ELISA wells. The wells were blocked, and then 300 μL of the conditioned medium (0.5 μg/μL of A549 cells or H1299 cells, 72 h post serum starvation) was added in the absence or presence of increasing IGFBP-3 concentrations (Figure 8). IGFBP-3, HN, amyloid oligomers, and fibrils were detected using the corresponding specific primary antibodies. The fold change relative to controls in the absence of added IGFBP-3 (Figure 8A) or controls using 300 μL of the medium not incubated with cells (Figure 8B and C) was calculated. The addition of IGFBP-3 concentrations (400 nM) (Figure 8A-C) that far exceed those we calculated previously for IGFBP-3 in the A549 conditioned medium80 of ~45–50 nM did not result in detection of IGFBP-3 in the complex obtained using 6E10 antibodies incubated with either the A549 or the H1299 medium. As expected, and consistent with the results reported in Figure 6, HN was detected in a complex using the 6E10 antibodies (Figure 8). The concentration of HN, however, was diminished upon the addition of 50 nM IGFBP-3 (Figure 8A) and completely depleted upon the addition of 100 nM IGFBP-3. No further decrease was observed upon the addition of higher concentrations of the protein, suggesting that 100 nM IGFBP-3 is sufficient in order to completely sequester the HN bound in a complex using the anti-6E10 antibodies (Figure 8A). In an inverse correlation with the HN levels, the addition of 50–100 nM IGFBP-3 resulted in increased signal detection of the amyloid oligomer by the anti-A11 antibody. That signal remained relatively constant upon the addition of increasing concentrations of the protein (Figure 8). While similar trends were observed upon using the anti-LOC antibody that detects the amyloid fibrils, the signal was increased only ~1.7-fold compared to ~3.8-fold using anti-A11 antibodies relative to the control (Figure 8A). While the levels of HN bound to the complex using anti-6E10 antibodies were comparable when using the A549 or H1299 medium (~4-fold relative to the control) (Figure 8B and C), the addition of IGFBP-3 resulted in a more modest increase in the signal when using the anti-A11 antibodies in H1299 at ~2.5-fold (Figure 8C) relative to the control with no IGFBP-3 compared to that detected under the same conditions using the A549 medium (~3.9-fold relative to the control without IGFBP-3) (Figure 8B). Moreover, there was a ~1.6-fold increase upon using the anti-LOC antibodies in the A549 medium relative to the control with no IGFBP-3, while no signal was observed under the same conditions using the medium from the H1299 cells (Figure 8C). These observations might be a result of a higher expression of AChE in A549 as compared to H1299 (Figure 6), which might allow AChE access to Aβ and thus increase its aggregation in the A549 medium but not in the H1299 medium.
Figure 8.

The concentration of HN found in a complex using 6E10 antibodies is reduced upon the addition of IGFBP-3 and correlates with an increase in the amount of amyloid oligomers. Anti-Aβ specific antibodies (6E10) were added (1:1000 dilution) to the ELISA wells. The wells were blocked, and then 300 μL of the conditioned medium (0.5 μg/μL of A549 cells or H1299 cells, 72 h after serum starvation) was added in the absence or presence of increasing IGFBP-3 concentrations. IGFBP-3, HN, amyloid oligomers, and fibrils were detected using the corresponding specific primary antibodies and then processed as described in the Experimental Procedures. The fold change relative to the controls with no IGFBP-3 (A) or using 300 μL of the medium not incubated with cells (B and C) was calculated. The curves (A) were fit with a nonlinear regression curve using GraphPad Prism 8.3.1. The data represent the mean ± SD of three separate assays, each of which was performed in triplicate. Asterisks (*) indicate a statistically significant difference from the corresponding cell line control, *p < 0.05 and **p < 0.01, of each cell line. The absence of asterisks indicates no significance (Mann–Whitney test).
Immunodepletion of HN Increases the Relative Abundance of A11-Positive Prefibrillar Oligomers and to a Lesser Extent the LOC-Positive Fibrillar Oligomers in A549 and H1299 Media.
Synthetic Aβ peptides are known to oligomerize and aggregate when added to growth media.25,32,33 While the different forms of Aβ are always in equilibrium, a linkage has been shown to exist between the small soluble oligomers of Aβ, neuronal toxicity, and failure of the synapse.98 Solubles Aβ oligomers were found to be more neurotoxic with a stronger impact on synaptic loss and cognitive impairment than fibrils and insoluble larger aggregates.8,32,98,99 Since the addition of synthetic Aβ peptides to the growth media is known to form heterogeneous Aβ assemblies due to the oligomerization and formation of peptide aggregates,8,32,84 numerous studies have focused instead on examining the cell toxicity of naturally synthesized cultured cell-derived Aβ peptides. For example, in vivo inhibition of hippocampal long-term potentiation was specifically attributed to oligomeric assemblies and not to monomers or fibrils of the naturally secreted human Aβ.98 Inhibition of tumor cell proliferation positively correlated with increased naturally secreted Aβ concentrations in the conditioned media enriched in Aβ that was added to the following three tumor cell lines: human glioblastoma multiforme, breast cancer, and mouse melanoma cells.32
In order to examine the effects of HN, AChE, or IGFBP-3 on amyloid conformations, we used specific primary antibodies in order to immunodeplete the medium of each component and then tested for the abundance of oligomeric and fibrillar Aβ conformations. Cells (0.2 × 105) were grown in a 10% FBS-supplemented medium for 24 h. The medium was then replaced with a serum-free medium, and the cells were allowed to incubate for 72 h. Subsequent to the 72 h incubation, the medium was collected. The specific antibodies for the immunodepletion (ID) were added to the ELISA wells (Figure 9). The wells were blocked, and then 300 μL of the conditioned medium (0.5 μg/μL, 72 h post serum starvation) was added. After the overnight incubation, the immunodepleted medium was removed. The same amount of protein (3 μL of 600 μg/mL total protein) of each sample was then spotted onto a nitrocellulose membrane (Figure 9). Since H1299 does not express IGFBP-334,72,100 and since the IGFBP-3 protein is not detected using any of the antibodies used here (6E10, 4G8, A11, LOC, Figures 7 and 8), anti-IGFBP-3 antibodies were used as a negative control (Figure 9). The blots were either stained with Ponceau (Figure 9A) or incubated with anti-6E10 (Figure 9B), anti-A11 (Figure 9C), and anti-LOC (Figure 9D) antibodies, and the signal on the membrane was detected using the super signal west pico luminol (chemiluminescence) reagent. The membrane was imaged using a Bio-Rad molecular imager, and the signal was quantitated using ImageJ software (Experimental Procedures). The dots from five independent experiments, each of which was carried out in triplicate, were quantitated, averaged, normalized, and expressed as a fold change relative to the control cells that were immunodepleted using the anti-IGFBP-3 antibodies (Figure 9E-G).
Figure 9.

The relative abundance of A11-positive prefibrillar oligomers, and to a lesser extent the LOC-positive fibrillar oligomers, is increased in the A549 and H1299 media immunodepleted of HN. Cells (0.2 × 105) were grown in a 10% FBS-supplemented medium for 24 h. The cells were then incubated in a serum-free medium for 72 h, and the medium was collected. The indicated antibodies for the immunodepletion (ID) were added (1:1000 dilution) to the ELISA wells. After blocking the wells, 300 μL of the conditioned medium (0.5 μg/μL, 72 h post serum starvation) was added. Subsequent to incubation overnight, the immunodepleted medium was removed, and the same amount of protein (3 μL of 600 μg/mL total protein) of each sample was spotted onto a nitrocellulose membrane. The blots were either stained with Ponceau (A) or incubated with anti-6E10 (B), anti-A11 (C), or anti-LOC (D) antibodies, and the signal on the membrane was detected using the super signal west pico luminol (chemiluminescence) reagent. The membranes were imaged with a Bio-Rad molecular imager and quantitated with ImageJ software. The dots from five independent assays, each of which was carried out in triplicate, were quantitated, averaged, normalized, and expressed as a fold change relative to the control medium (IGFBP-3). The graphs (E–G) were prepared using the GraphPad 8.3.1 software and summarize the results expressed as the mean ± SD. Asterisks (*) indicate a statistically significant difference from the corresponding cell line control, *p < 0.05 and **p < 0.01, of each cell line. The absence of asterisks indicates no statistical significance (Mann–Whitney test).
Immunodepletion of AChE (Figure 9BE) decreased the amount of Aβ detected using anti-6E10 antibodies in the A549 medium, while no change was observed in the medium of the H1299 cells compared to the control. These results support our previous findings81 that show that there is minimal expression of AChE in H1299, likely due to its deficiency in p53. The decreased levels of Aβ in the A549 medium depleted of AChE might reflect the elimination of the Aβ fraction normally bound to AChE. Immunodepletion of HN from both the A549 and H1299 media resulted in a comparable decrease in the levels of detected Aβ using the anti-6E10 antibodies (Figure 9B and E). These results suggest that Aβ is normally bound to a fraction of HN in the media.
No difference in the signal obtained using the anti-oligomer A11 antibody was observed between the A549 medium immunodepleted of either AChE or IGFBP-3 compared to that of H1299, which had minimal expression and no expression of AChE and IGFBP-3, respectively (Figure 9C and F). These results are not surprising, since the removal of AChE, which is known to increase Aβ oligomer formation,51,54,55 is expected to reduce detection by the A11 antibody. While the immunodepletion of IGFBP-3 is also expected to deplete a fraction of HN associated with it, the lack of a signal above that obtained by using H1299 (Figure 9C and F), which does not express IGFBP-3, suggests that the protein is unable to affect the fraction of HN bound to Aβ in the A549 medium. This possibility might be supported by the finding (Figure 8) that HN is found in a complex bound to the 6E10 antibodies, and that addition of ~50–100 nM exogenous IGFBP-3 is needed in order to block this binding in either the A549 or H1299 medium. These results might also support the in vitro data (Figures 4 and 5), which show that while IGFBP-3 is able to sequester HN by increasing the aggregation of Aβ in the absence or presence of AChE, higher concentrations are needed in order to exhibit the full effect. The immunodepletion of HN resulted in an increased oligomer signal detection by the anti-A11 antibodies in the media of both the A549 and H1299 cells (Figure 9C and F). This signal was higher in the A549 medium compared to that of H1299. The relatively high expression of AChE in A549 compared to that in H1299 might explain the increased oligomer signal in the A549 medium, since the immunodepletion of HN might lead to the increased aggregation of Aβ caused by AChE. Similar but much more modest results were obtained upon blotting for the amyloid fibrils using anti-LOC antibodies (Figure 9D and G), suggesting that HN serves to bind Aβ and largely reduces oligomer formation.
Since A11 antibodies are known to recognize the oligomeric species of other polypeptides that are amyloidogenic, such as human insulin, prion, and α-synuclein,101 and, similarly, since LOC antibodies recognize generic epitopes present in several amyloid fibrils and fibrillar oligomers,101 we tested whether the increased signal obtained with the A11 (Figure 9C and F) or LOC (Figure 9D and G) antibodies upon the immunodepletion of HN corresponded to that of Aβ. Immunodepletion with both HN and 6E10 completely blocked the A11 and LOC signals in the media of both A549 and H1299 cells (Figure 9F and G). These results suggest that HN is important in reducing amyloid oligomer formation, which is consistent with previous reports showing that HN reduces the aggregation of Aβ.37,46,48,89 We next tested whether exogenously added IGFBP-3 is able to bind HN and alter the Aβ conformational states. The addition of 100 nM IGFBP-3 resulted in a ~4- and ~2.5-fold increased signal using the anti-A11 antibodies in A549 and H1299, respectively, while a ~1.7-fold increase was detected using anti-LOC antibodies only in the A549 medium (Figure 10). The addition of 400 nM IGFBP-3 did not have any further effect, suggesting that a 100 nM concentration of IGFBP-3 is sufficient in order to bind HN in the media. These results support those obtained in Figure 8.
Figure 10.

Exogenously added IGFBP-3 increases the relative abundance of A11-positive prefibrillar oligomers and to a lesser extent the LOC-positive fibrillar oligomers in both A549 and H1299 media. Cells (0.2 × 105) were grown in a 10% FBS-supplemented medium for 24 h. The cells were then incubated in a serum-free medium for 72 h, and the medium was collected. IGFBP-3 was added exogenously to 10 μL of the conditioned medium (0.5 μg/μL, 72 h post serum starvation) and allowed to incubate for 2 h. Following the incubation, 3 μL portions of the samples were spotted onto a nitrocellulose membrane. The blots were stained with either A11 or LOC antibodies, and the signal on the membrane was detected using the super signal west pico luminol (chemiluminescence) reagent. The blots were then imaged with a Bio-Rad molecular imager and quantitated with ImageJ software. The dots from five independent assays, each of which was performed in triplicate, were quantitated, averaged, normalized, and expressed as fold change relative to the control with no added IGFBP-3. GraphPad 8.3.1 software was used in order to prepare the graphs, which summarize the results and are expressed as mean ± SD. Asterisks (*) indicate a statistically significant difference from the corresponding cell line control, *p < 0.05 and **p < 0.01, of each cell line. The absence of asterisks indicates no significance (Mann–Whitney test).
The Relative Amount of Oligomer vs the Total Amount of Aβ Increases upon the Immunodepletion of HN from the A549 or H1299 Cell-Conditioned Medium and Correlates with Diminished Cell Viability and Increased Apoptosis.
Both epitopes recognized by the 6E10 and 4G8 antibodies have been previously shown to be exposed in Aβ aggregates.92,102 The 4G8:6E10 ratio was suggested to be a marker for the relative amount of aggregated vs monomeric Aβ.102 In order to determine the effect of the immunodepletion of AChE, IGFBP-3, or HN on this ratio as well as whether there was a correlation with cell viability or apoptosis, 0.2 × 105 cells per well were seeded in 96-well plates in a 10% FBS-supplemented medium. The next day, the cells were incubated in a serum-free medium for 72 h and then immunodepleted of AChE, HN, or IGFBP-3 as described in the Experimental Procedures. The antibodies 6E10 or 4G8 were bound (1:1000 dilution) to the ELISA wells. The wells were blocked and then incubated with 300 μL of the immunodepleted medium (0.5 μg/μL). Biotin-4G8 was then added, and the signal was processed as described in the Experimental Procedures. The fold change relative to the controls using anti-6E10 or anti-4G8 antibodies incubated with 300 μL of the medium not incubated with the cells was calculated. For the cell viability and apoptosis assays, cells were seeded in 96-well plates at 0.2 × 105 cells per well in 200 μL of the 10% FBS-supplemented medium. The next day, the cells were incubated in a serum-free medium for 12 h and then treated with the immunodepleted medium for 48 h. The medium containing the specific components in the different treatments was replaced every 12 h.
Compared to the total amount of Aβ, there was more oligomer in the A549 medium (~63%) compared to that found in the H1299 (~48%) medium (Figure 11A and B). The immunodepletion of AChE from the A549 medium decreased the total amount of Aβ in the media to ~57%, results that are consistent with those obtained in Figure 9B and E where ~62% remained in the media. The amount of the oligomer was ~47% of the total amount of Aβ (Figure 11A), suggesting that removing AChE from the A549 medium reduces the amount of the oligomer relative to undepleted medium. No change upon the immunodepletion of AChE was observed in the H1299 medium, which is not surprising since the expression of AChE is minimal in this cell line.81 The immunodepletion of HN reduced the total amount of Aβ in the media to ~63% in A549 and ~58% in H1299 in accord with the results obtained in Figure 9B and E (~63% in A549 and ~57% in H1299). Compared to the total amount of Aβ that remained after HN immunodepletion, there was relatively more oligomer (~86% of total) in the A549 medium compared to in H1299 (~69%). Those results are consistent with those in Figure 9 and might suggest that the depletion of HN increases the ability of AChE to result in increased Aβ oligomer formation in the A549 medium. No effects were observed on either the total amount of Aβ or the oligomer formation upon the immunodepletion of IGFBP-3.
Figure 11.
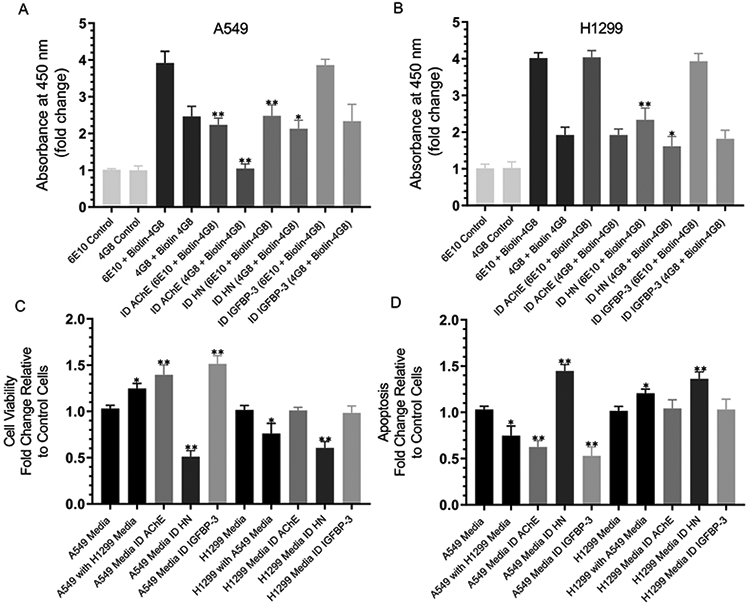
Immunodepletion of HN from an A549 or H1299 cell-conditioned medium increases the relative amount of oligomer vs the total amount of Aβ, decreasing cell viability and increasing apoptosis. (A and B) Cells (0.2 × 105 cells per well) were seeded in 96-well plates in a 10% FBS-supplemented medium. The next day, the cells were incubated in a serum-free medium for 72 h and then immunodepleted (ID) of AChE, HN, or IGFBP-3 as described in the Experimental Procedures and in the Figure 9 legend. The antibodies 6E10 or 4G8 were bound (1:1000 dilution) to the ELISA wells. The wells were blocked and then incubated with 300 μL of the ID medium (0.5 μg/μL). Biotin-4G8 was then added, and the signal was processed as described in the Experimental Procedures. The fold change relative to the controls using anti-6E10 and anti-4G8 antibodies and 300 μL of the medium not incubated with cells was calculated. Viability (C) or apoptosis (D) of the A549 or H1299 cells was assessed by the MTT assay or the annexin V method, respectively. Cells were seeded in 96-well plates at 0.2 × 105 cells per well in 200 μL of a 10% FBS-supplemented medium. The next day, the cell monolayers were incubated in a serum-free medium for 12 h and then treated with the immunodepleted medium for 48 h, with the medium containing the specific components in the various treatments replaced every 12 h. Data were processed using the GraphPad 8.3.1 software. The graphs summarize the results expressed as the mean ± SD of three separate experiments, each of which was performed in triplicate. Asterisks (*) indicate a statistically significant difference relative to the control. The absence of asterisks indicates no significance (Mann–Whitney test, *p < 0.05 and **p < 0.01).
Since H1299 does not express IGFBP-334 with minimal expression of AChE,81 we asked whether swapping the media affects either the cell viability or apoptosis. Incubation of A549 cells with a H1299 medium increased cell viability (Figure 11C) and correlated with diminished apoptosis (Figure 11D). This observation suggests that the H1299 media might either lack or contain additional components compared to that of A549, leading to an increased cell viability and reduced apoptosis. The immunodepletion of AChE or IGFBP-3 increased the A549 cell viability and reduced apoptosis but had no effect on H1299 (Figure 11C and D). This was not surprising as H1299 does not express IGFBP-3 with minimal expression of AChE. In both cell lines, the immunodepletion of HN reduced the cell viability (Figure 11C and D) and correlated with increased apoptosis, suggesting that HN plays an important role in regulating these cellular processes.
DISCUSSION
Earlier reports showed that HN binds Aβ.39,40,46-48,89 HN was found to bind amino acid residues 17–28 of Aβ40,47 decreasing the amount of toxic oligomers in vivo,46 while AChE binds amino acid residues 1–16 and 12–28 of Aβ (Figure 1),54 directly promoting Aβ40 aggregation and its assembly into amyloid fibrils. As AChE and HN can both bind residues 17–28 of Aβ, they may serve to regulate the central domain (residues 17–24) flanked by Lys16 and Lys28, which is known to be important in the formation of fibrillar Aβ aggregates.60-62
No significant difference was observed between the binding of HN to either Aβ40 or Aβ42 (Figure 2A). As amino acid F6 of HN is known to bind IGFBP-3 or Aβ,39,90 we tested whether IGFBP-3 was able to compete with Aβ for binding HN. Moreover, since AChE is known to bind Aβ,7,51,53,54,59 we tested this competition in the presence of AChE. We found that HN binds to either Aβ40 or Aβ42 with comparable affinity, and that IGFBP-3 is able to compete with Aβ for this binding. For both Aβ40 and Aβ42, no difference was observed upon the addition of AChE on the competition of IGFBP-3 with Aβ for binding HN (Figure 2B and C), suggesting that IGFBP-3 can block the binding of HN to Aβ in the absence or presence of AChE.
Binding of AChE to Aβ has been previously documented.51,53,54,59 Both Aβ40 and Aβ42 bound AChE (Figure 3A), which is not surprising since AChE was shown previously54 to bind Aβ (12–28) as well as the Aβ (1–16) peptide (Figure 1). Since HN was found earlier to bind amino acid residues 17–28 of Aβ40,47 and since the HN and AChE binding sites on Aβ are overlapping, we expected an antagonistic binding between HN and AChE on Aβ. We were surprised, however, to find that the addition of increasing concentrations of AChE had a minimal effect on the ability of HN to bind Aβ. Equimolar concentrations of AChE resulted in only about a 10–13% reduction in the binding of either Aβ40 or Aβ42 to HN (Figure 3B and C). The addition of 10-fold higher concentrations of AChE only resulted in a 14–16% inhibition of Aβ binding to HN. These results suggest that both HN and AChE can bind Aβ simultaneously despite each being able to directly bind amino acid residues 17–28 of the peptide (Figure 1). This binding of HN and AChE to Aβ was blocked by the addition of IGFBP-3 (Figure 3B and C). Equimolar concentrations of IGFBP-3 resulted in an approximately 50% decrease in the binding of Aβ to HN while increasing the concentration of added IGFBP-3 10-fold, which completely abolished the signal relative to samples without added IGFBP-3 (Figure 3B and C). These results might indicate that at equimolar concentrations IGFBP-3 competes with Aβ for binding HN but that at higher concentrations IGFBP-3 is able to completely sequester HN, thereby blocking its binding to Aβ (Figure 3B and C).
Both Aβ peptides are known to form toxic oligomers and rapidly aggregate, with Aβ42 aggregating faster and to a greater extent than Aβ40,91 while HN is known to decrease the aggregation of Aβ.38,39,47,48 Consistent with these reports, Aβ42 (Figure 4B) exhibited faster aggregation kinetics compared to those of Aβ40 (Figure 4A), and HN was able to reduce the aggregation of both Aβ40 and Aβ42. For both Aβs, the addition of equimolar concentrations of IGFBP-3 partially reduced the ability of HN to block the aggregation of Aβ, while the addition of 3-fold higher concentrations of the protein completely blocked the HN effects. These results might indicate that equimolar concentrations of IGFBP-3 bind a portion of the HN, sequestering it away from Aβ and resulting in increased aggregation; however, higher concentrations of IGFBP-3 are necessary in order to bind HN, leading to aggregation kinetics similar to those using Aβ alone (Figure 4).
Since the central domain of Aβ is critical for fibrillar aggregation, AChE may bind this region, inducing or enhancing Aβ aggregation. Meanwhile, HN may act to bind Aβ and shield this hydrophobic region, thereby blocking the binding of other Aβ monomers to this region; this would effectively arrest any further growth of the Aβ aggregates. Residues 5–15 of HN were found earlier to bind residues 17–28 of Aβ40,47 a region essential for Aβ oligomerization and fibril formation. Recently, we showed that HN (5–15) was a more effective inhibitor of Aβ40-HN interactions than Aβ (17–28).48 The peptide HN (5–15, L11S), where Ser replaced the conserved Leu11, reduced the ability of HN (5–15) to compete with the Aβ40-HN interactions. This might highlight the importance of the hydrophobic amino acid residues in regulating the binding and formation of the HN-Aβ complex. Amino acids 22 and 23 of Aβ were shown previously103,104 to form a β-turn, which allows for intermolecular parallel β-sheet formation between segments 3–21 and 24–42 and results in aggregation. It was also found earlier that the substitution of two phenylalanine residues, F19 and F20, with alanine105-107 or proline99 reduced fibril formation. These aromatic residues, located within the 17–21 self-recognition element (SRE) of Aβ, are known to promote Aβ aggregation into toxic oligomers or fibrils.108-111 Substitution of the l-form of F19 or F20 or both F19 and F20 with their d-enantiomers112 in an Aβ (14–23) peptide construct abolished the formation of ThT-positive aggregates. It is thus likely that the binding of HN hinders Aβ aggregation due to binding to region 17–28 of Aβ.
Our results suggest that AChE does not compete with the binding of HN to Aβ (Figure 3). However, since HN and AChE are known to have opposing effects on Aβ oligomerization,36,37,43,46-48,51,53-55,59, we tested whether HN can reduce Aβ aggregation (Figure 4) in the presence of AChE (Figure 5). An increased aggregation of Aβ40 or Aβ42 was observed upon the addition of AChE, with changes in both the lag phase and the final ThT fluorescence (Figure 5). The addition of equimolar concentrations of HN and AChE, however, reduced the aggregation of both Aβ40 and Aβ42 almost to the same levels as those resulting from the addition of only HN, suggesting that HN is able to reduce the aggregation of Aβ despite the presence of AChE. The ability of HN to reduce the aggregation of Aβ was partially diminished upon the incubation of equimolar concentrations of Aβ, AChE, HN, and IGFBP-3 (Figure 5) and was completely blocked upon the addition of 3-fold higher IGFBP-3 concentrations. Comparable to the results obtained in Figure 4, these findings indicate that at higher concentrations IGFBP-3 can bind and sequester most HN and better compete with Aβ for interactions with HN, leading to aggregation kinetics comparable to those of Aβ incubated with only AChE (Figure 5).
Since our in vitro data (Figure 3) suggest that HN and AChE can be found together in a complex with Aβ, we wondered whether such a complex can be detected in the conditioned media of two human lung carcinoma NSCLC cell lines34 (A549 (p53-positive) or H1299 cells with a p53-null genotype due to a biallelic deletion of the TP53 gene).35 Anti-Aβ specific antibodies (6E10) (Figure 6) that recognize the N-terminal hydrophilic sequence 1–16 of Aβ were shown to be exposed in Aβ aggregates92 and are known to react with monomers, oligomers, and fibrils92,93 bound to the ELISA wells, followed by an incubation with the conditioned medium from A549 or H1299 cells 72 h post serum starvation. AChE was detected in the medium of the A549 cells, while barely detectable levels of AChE were found in the H1299-conditioned medium (Figure 6). These results are not surprising, since we recently showed81 that there are minimal levels of AChE in the conditioned medium of the p53-null cell line, H1299, as compared to that of the p53-positive cell line, A549. In both cell lines, HN from the conditioned media was found bound to Aβ (Figure 6). These results, however, provide no information on whether HN and AChE are found in the same complex with Aβ. In order to test whether HN can compete off the binding of AChE with Aβ bound to the immobilized 6E10 antibodies in the wells, increasing concentrations of HN (Figure 6) were added. Using the media from both cell lines, the addition of exogenous HN resulted in increased binding of HN to Aβ bound to its immobilized antibody 6E10. In both cases, there was an increased signal that began to level off around 50 nM HN, with a ~2.5-fold increase with the highest concentration of HN added (400 nM) relative to samples with no exogenous HN added. However, despite this increase in the HN signal indicative of binding to Aβ, only a ~1.5-fold decrease in AChE binding was found (Figure 6) using the A549 medium, suggesting that while HN blocks the binding of AChE to Aβ it is unable to completely inhibit this interaction.
We next tested whether HN is detected in a complex by using 6E10 and 4G8 antibodies known to react with the monomers, oligomers, and fibrils of Aβ92,93 with anti-oligomer specific A11 antibodies that recognize all types of prefibrillar oligomers but not monomers or fibrils, and with anti-amyloid fibril LOC antibodies that recognize epitopes common to amyloid fibrils and fibrillar oligomers but not monomers or prefibrillar oligomers. Since direct binding of HN to Aβ is well documented,47,48,89,97 HN was not surprisingly detected upon using 6E10 antibodies (Figure 7). No signal was detected, however, upon the incubation of the media with the 4G8 antibody. As this antibody recognizes the central hydrophobic sequence of amino acid residues 17–2494 with its epitope located within residues 18–22 (VFFAE) of Aβ,94,96 it is plausible that it binds this region and blocks the ability of HN to bind to residues 17–28. Minimal signal detection of HN was found upon using either A11 or LOC antibodies, indicating that HN is likely not found in a complex with prefibrillar oligomers detected by the anti-A11 or anti-amyloid fibrils and the Aβ fibrillar oligomers detected by the anti-LOC antibodies. Since AChE has been reported previously to directly promote the aggregation of Aβ into amyloid fibrils,51,53-56 we expected to find the protein bound in a complex using not only 6E10 and 4G8 but also A11 and LOC. However, less enzyme was found to be bound upon using the 4G8 antibodies. A likely explanation might be that while the addition of 4G8 does not block the binding of AChE to Aβ, it reduces its binding by competing for binding residues 17–24 of Aβ (Figure 1 and Figure 7). These finding are comparable to those found upon using HN in order to examine the binding of AChE to Aβ (Figure 6) and suggest that the binding of HN to residues 17–28 or binding of 4G8 to residues 17–24 diminishes but does not completely block the interaction of AChE with Aβ. Moreover, the fraction of AChE bound to the anti-oligomer antibody, A11, or the anti-amyloid fibril antibody, LOC, must not contain HN, since no signal above the background was observed when using the HN antibodies. IGFBP-3 was not detected upon using any of the antibodies tested, suggesting that the protein is not found in complexes bound to these antibodies.
Since we did not detect IGFBP-3 in a complex using the 6E10, 4G8, A11, or LOC antibodies (Figure 7), we tested whether exogenously added IGFBP-3 can bind and sequester HN and result in changes in the conformation of Aβ. The addition of 400 nM IGFBP-3, which was far above those we measured previously for IGFBP-3 in A549-conditioned media80 of ~45–50 nM, did not result in binding of IGFBP-3 in the complex bound to 6E10 antibodies incubated with either the A549 or H1299 medium (Figure 8A-C). The HN concentration detected in a complex using 6E10 antibodies was diminished upon the addition of 50 nM IGFBP-3 and completely depleted upon the addition of 100 nM IGFBP-3 (Figure 8A), with no further decrease observed upon the addition of higher concentrations of the protein; this suggests that 100 nM IGFBP-3 is sufficient in order to completely sequester the HN bound in a complex using the anti-6E10 antibodies. A concomitant increase in amyloid oligomer formation detected using anti-A11 antibodies was found upon the addition of 50–100 nM IGFBP-3 and was not further changed upon the addition of increasing concentrations of the protein (Figure 8A). While similar results were obtained using the anti-LOC antibody that detects amyloid fibrils, the signal was increased only ~1.7-fold relative to the control compared to ~3.9-fold when using the anti-A11 antibodies. In the absence of added IGFBP-3, the levels of HN in the conditioned media bound to the complex using anti-6E10 antibodies were comparable when using either the A549 or H1299 cell medium (~4-fold relative to control). The incubation of anti-6E10 antibodies with the A549 cell medium supplemented with exogenously added IGFBP-3 resulted in a ~3.9-fold increase in the signal detection by the anti-A11 antibodies compared to a ~2.5-fold signal increase detected under the same conditions when using the H1299 medium (Figure 8B and C). Using the anti-LOC antibodies in the presence of media with exogenously added IGFBP-3 showed a ~1.59-fold increase in signal using the A549 medium and no increase from the H1299 medium under the same conditions (Figure 8B and C). These results might be a consequence of the presence of AChE in the A549 medium and not the H1299 medium (Figure 6), enhancing oligomerization of Aβ when HN is sequestered by IGFBP-3.
When added to the growth media, synthetic Aβ peptides are known to oligomerize and aggregate.25,32,33 Soluble oligomers of Aβ have been shown to be associated with more neuronal toxicity and failure of the synapse compared to those of fibrils and insoluble larger aggregates.8,32,98,99 On the other hand, Aβ monomers from the conditioned media were previously found to be neuroprotective, devoid of toxicity, and able to activate the IGF-IR in neuronal cells, leading to activation of the PI3–K/AKT pathway and regulating the function of cyclic adenosine monophosphate response element binding protein (CREB).113 Since synthetic Aβ peptides added to the cell growth media are known to form heterogeneous Aβ assemblies due to the oligomerization and formation of peptide aggregates,8,32,84 we have focused instead on examining Aβ conformations and the function of naturally synthesized cultured cell-derived Aβ peptides. The immunodepletion of AChE (Figure 9B and E) decreased the amount of Aβ in the A549 medium, which might reflect the elimination of the Aβ fraction normally bound to AChE. In support of our previous findings81 that showed that there is a minimal expression of AChE in H1299, likely due to its deficiency in p53, no change in the Aβ levels was observed in the medium of the H1299 cells immunodepleted of AChE as compared to the control (Figure 9B and E). A comparable decrease in the levels of detected Aβ was found upon the immunodepletion of HN from both the A549 and H1299 media, indicating that Aβ is normally bound to a fraction of HN in the media of both cell lines (Figure 9B and E).
Since AChE is known to increase Aβ oligomer formation,51,54,55 the immunodepletion of AChE was predicted to reduce detection by the A11 antibody. In support of this prediction, no difference in the A11 signal was observed between the A549 media immunodepleted of either AChE or IGFBP-3 as compared to that of H1299, which does not express IGFBP-334 with minimal expression of AChE.81 In the media of both the A549 and H1299 cells (Figure 9C and F), the immunodepletion of HN resulted in increased oligomer signal detection using the anti-A11 antibodies. This signal was higher when using the A549 cell medium compared to that of the medium from the H1299 cells. Similar but much more modest effects were obtained upon blotting for amyloid fibrils using anti-LOC antibodies (Figure 9D and G). These results might indicate that HN serves to bind Aβ, mainly leading to reduced oligomer formation. Since A11 antibodies have been shown to detect oligomeric species of other amyloidogenic polypeptides, such as α-synuclein, human insulin, and prion,101 and similarly since LOC antibodies can recognize generic epitopes present in several amyloid fibrils and fibrillar oligomers, we examined whether the increased signal obtained with anti-A11 (Figure 9C and F) or anti-LOC (Figure 9D and G) antibodies upon HN immunodepletion corresponded to that of Aβ. Immunodepletion of the media using both 6E10 and HN antibodies completely blocked the A11 and LOC signal in the media of both the A549 and H1299 cells (Figure 9F and G). These findings suggest that HN is important for reducing amyloid oligomer formation, results that are consistent with previous reports showing that HN reduces Aβ aggregation.37,46,48,89 The increased oligomer signal in the A549 medium relative to that of H1299 might be due to the relatively high expression of AChE in A549 compared to that of H1299. Immunodepletion of HN might, therefore, increase the aggregation of Aβ caused by AChE in the absence of HN.
We next tested whether the exogenous addition of IGFBP-3 to the conditioned media can bind and sequester HN, increasing the signals obtained using the A11 and LOC antibodies (Figure 10). The addition of 100 nM IGFBP-3 to the media of either cell line resulted in an increased signal using the A11 antibodies (Figure 10), while the signal detected upon using the LOC antibodies was only observed when using the medium from the A549 cells. No further increase in signal was detected upon the addition of 400 nM IGFBP-3, suggesting that 100 nM concentrations of IGFBP-3 are sufficient in order to bind HN in the media; these results support those obtained in Figure 8.
Both epitopes recognized by the the 6E10 and 4G8 antibodies have been reported to be exposed in Aβ aggregates.83,93 The 4G8:6E10 ratio was proposed as a marker for the relative amount of aggregated vs monomeric Aβ.102 The physiological concentration of Aβ42 was reported previously in a range between 1 and 20 nM.82 The plasma levels of the total amount of Aβ (Aβ40 or Aβ42) were not found to be significantly different in patients with lung cancer.33 More oligomer, compared to the total amount of Aβ, was detected in the A549 medium compared to that found in the H1299 medium (Figure 11A and B). The immunodepletion of AChE from the A549 medium reduced both the total amount of Aβ and the amount of the oligomer relative to the control medium, while no change compared to control was found upon AChE immunodepletion from the H1299 medium (Figure 11A and B). This is not surprising, as H1299 cells express minimal levels of AChE.81 HN immunodepletion decreased the total amount of Aβ in the media from both A549 and H1299 cells; however, a relatively higher percent of oligomer of the total amount of Aβ was found in the A549 medium compared to that from the H1299 cells (Figure 11A and B). Those results support those obtained in Figure 9 and might suggest that the depletion of HN increases the ability of AChE to promote Aβ oligomer formation. The immunodepletion of IGFBP-3 had no effect (Figure 11A and B) on either the total amount of Aβ or the oligomer formation when using the medium from either cell line.
The incubation of A549 cells with the H1299 medium resulted in increased cell viability (Figure 11C) and correlated with diminished apoptosis (Figure 11D). Conversely, cell viability decreased with a corresponding increase in apoptosis upon the incubation of H1299 cells with the A549 cell medium. This observation might be due to the lack of IGFBP-3 expression34 in H1299 cells and a minimal expression of AChE.81 In support of this hypothesis, the immunodepletion of AChE or IGFBP-3 increased A549 cell viability and decreased apoptosis with no effect on the H1299 cells. The immunodepletion of HN diminished cell viability, which correlated with increased apoptosis in both cell lines, suggesting that HN has an important role in these cellular processes.
Our results shed light on a mechanism whereby IGFBP-3 binds HN, blocking it from binding Aβ. This then results in increased Aβ aggregation, an effect further exaggerated in the presence of AChE, increasing apoptosis and decreasing cell viability (Figure 12). Findings from this work might allow a rational design of smaller peptides as tools in order to investigate the basic principles of protein/peptide interactions aimed at inhibiting amyloid formation and increasing our fundamental understanding of the molecular underpinnings governing the interlinkages between biomolecular interactions and amyloid assembly.
Figure 12.

A graphic representation of the key findings of the current investigation. Binding of HN to Aβ reduces the prefibrillar and fibrillar oligomers in the absence or presence of AChE, reducing apoptosis and increasing cell survival. IGFBP-3 binds HN and blocks its interaction with Aβ, resulting in increased prefibrillar and fibrillar oligomer formation and leading to increased apoptosis and decreased cell survival.
ACKNOWLEDGMENTS
The authors gratefully thank Katarina Evans for help with the statistical analysis.
Funding
The research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award no. R15GM131222 to H.G.E. This work was also supported by an Eastern Michigan University Provost Research Support Award/Chemistry Seller’s Fund and an EMU Faculty Research Fellowship.
ABBREVIATIONS
- Aβ
amyloid-β
- IGFBP-3
insulin-like growth factor binding protein-3
- AChE
acetylcholinesterase
- HN
humanin
- ThT
thioflavin T dye
- AD
Alzheimer’s disease
- RFU
relative fluorescence units
- ID
immunodepletion
Footnotes
REFERENCES
- (1).Murphy MP, and LeVine H (2010) Alzheimer’s Disease and the β-Amyloid Peptide. J. Alzheimer's Dis. 19, 311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Villegas S (2015) [Alzheimer’s disease: New therapeutic strategies]. Med. Clin. (Barcelona) 145, 76–83. [DOI] [PubMed] [Google Scholar]
- (3).Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete N-V, Coté S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, and Derreumaux P (2015) Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev 115, 3518–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lukiw WJ (2008) Emerging amyloid beta (Ab) peptide modulators for the treatment of Alzheimer’s disease (AD). Expert Opin. Emerging Drugs 13, 255–271. [DOI] [PubMed] [Google Scholar]
- (5).Kawasumi M, Hashimoto Y, Chiba T, Kanekura K, Yamagishi Y, Ishizaka M, Tajima H, Niikura T, and Nishimoto I (2002) Molecular mechanisms for neuronal cell death by Alzheimer’s amyloid precursor protein-relevant insults. Neurosignals 11, 236–250. [DOI] [PubMed] [Google Scholar]
- (6).Alzheimer’s Association, Thies W, and Bleiler L (2013) 2013 Alzheimer’s disease facts and figures. Alzheimer's Dementia 9, 208–245. [DOI] [PubMed] [Google Scholar]
- (7).Lukiw WJ (2012) Amyloid beta (Aβ) peptide modulators and other current treatment strategies for Alzheimer’s disease (AD). Expert Opin. Emerging Drugs 17, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Sakono M, and Zako T (2010) Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 277, 1348–1358. [DOI] [PubMed] [Google Scholar]
- (9).LaFerla FM, Green KN, and Oddo S (2007) Intracellular amyloid-beta in Alzheimer’s disease. Nat. Rev. Neurosci 8, 499–509. [DOI] [PubMed] [Google Scholar]
- (10).Benilova I, Karran E, and De Strooper B (2012) The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci 15, 349–357. [DOI] [PubMed] [Google Scholar]
- (11).Schmidt M, Sachse C, Richter W, Xu C, Fändrich M, and Grigorieff N (2009) Comparison of Alzheimer Aβ(1–40) and Aβ(1–42) amyloid fibrils reveals similar protofilament structures. Proc. Natl. Acad. Sci. U. S. A 106, 19813–19818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Goure WF, Krafft GA, Jerecic J, and Hefti F (2014) Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimer's Res. Ther 6, 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Barrow CJ, and Zagorski MG (1991) Solution structures of beta peptide and its constituent fragments: relation to amyloid deposition. Science 253, 179–182. [DOI] [PubMed] [Google Scholar]
- (14).Eichner T, and Radford SE (2011) A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 43, 8–18. [DOI] [PubMed] [Google Scholar]
- (15).Ma B, and Nussinov R (2006) Simulations as analytical tools to understand protein aggregation and predict amyloid conformation. Curr. Opin. Chem. Biol 10, 445–452. [DOI] [PubMed] [Google Scholar]
- (16).Kirshenbaum K, and Daggett V (1995) pH-dependent conformations of the amyloid beta(1–28) peptide fragment explored using molecular dynamics. Biochemistry 34, 7629–7639. [DOI] [PubMed] [Google Scholar]
- (17).Rojas AV, Liwo A, and Scheraga HA (2011) A Study of the α-Helical Intermediate Preceding the Aggregation of the Amino-Terminal Fragment of the β Amyloid Peptide (Aβ1–28). J. Phys. Chem. B 115, 12978–12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ball KA, Phillips AH, Nerenberg PS, Fawzi NL, Wemmer DE, and Head-Gordon T (2011) Homogeneous and heterogeneous tertiary structure ensembles of amyloid-β peptides. Biochemistry 50, 7612–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Flöck D, Colacino S, Colombo G, and Di Nola A (2006) Misfolding of the amyloid beta-protein: a molecular dynamics study. Proteins: Struct., Funct., Genet 62, 183–192. [DOI] [PubMed] [Google Scholar]
- (20).Zhu X, Bora RP, Barman A, Singh R, and Prabhakar R (2012) Dimerization of the Full-Length Alzheimer Amyloid β-Peptide (Aβ42) in Explicit Aqueous Solution: A Molecular Dynamics Study. J. Phys. Chem. B 116, 4405–4416. [DOI] [PubMed] [Google Scholar]
- (21).Fawzi NL, Kohlstedt KL, Okabe Y, and Head-Gordon T (2008) Protofibril assemblies of the arctic, Dutch, and Flemish mutants of the Alzheimer’s Abeta1–40 peptide. Biophys. J 94, 2007–2016, DOI: 10.1529/biophysj.107.121467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Buchete N-V, Tycko R, and Hummer G (2005) Molecular dynamics simulations of Alzheimer’s beta-amyloid protofilaments. J. Mol. Biol 353, 804–821. [DOI] [PubMed] [Google Scholar]
- (23).Convertino M, Pellarin R, Catto M, Carotti A, and Caflisch A (2009) 9,10-Anthraquinone hinders beta-aggregation: how does a small molecule interfere with Abeta-peptide amyloid fibrillation? Protein Sci. 18, 792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chebaro Y, Jiang P, Zang T, Mu Y, Nguyen PH, Mousseau N, and Derreumaux P (2012) Structures of Aβ17–42 trimers in isolation and with five small-molecule drugs using a hierarchical computational procedure. J. Phys. Chem. B 116, 8412–8422. [DOI] [PubMed] [Google Scholar]
- (25).Behrens MI, Lendon C, and Roe CM (2009) A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res 6, 196–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhang Q, Guo S, Zhang X, Tang S, Shao W, Han X, Wang L, and Du Y (2015) Inverse relationship between cancer and Alzheimer’s disease: a systemic review meta-analysis. Neurol. Sci 36, 1987–1994. [DOI] [PubMed] [Google Scholar]
- (27).Okereke OI, and Meadows M-E (2019) More Evidence of an Inverse Association Between Cancer and Alzheimer Disease. JAMA Netw Open 2, e196167–e196167. [DOI] [PubMed] [Google Scholar]
- (28).Nixon DW (2017) The Inverse Relationship Between Cancer and Alzheimer’s Disease: A Possible Mechanism. Curr. Alzheimer Res 14, 883–893. [DOI] [PubMed] [Google Scholar]
- (29).Catalá-López F, Crespo-Facorro B, Vieta E, Valderas JM, Valencia A, and Tabarés-Seisdedos R (2014) Alzheimer’s Disease and Cancer: Current Epidemiological Evidence for a Mutual Protection. NED 42, 121–122. [DOI] [PubMed] [Google Scholar]
- (30).Majd S, Power J, and Majd Z (2019) Alzheimer’s Disease and Cancer: When Two Monsters Cannot Be Together. Front. Neurosci, DOI: 10.3389/fnins.2019.00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Brothers HM, Gosztyla ML, and Robinson SR (2018) The Physiological Roles of Amyloid-β Peptide Hint at New Ways to Treat Alzheimer’s Disease. Front. Aging Neurosci, DOI: 10.3389/fnagi.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Zhao H, Zhu J, Cui K, Xu X, O’Brien M, Wong KK, Kesari S, Xia W, and Wong STC (2009) Bioluminescence imaging reveals inhibition of tumor cell proliferation by Alzheimer’s amyloid beta protein. Cancer Cell Int. 9, 15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jin W-S, Bu X-L, Liu Y-H, Shen L-L, Zhuang Z-Q, Jiao S-S, Zhu C, Wang Q-H, Zhou H-D, Zhang T, and Wang Y-J (2017) Plasma Amyloid-Beta Levels in Patients with Different Types of Cancer. Neurotoxic. Res 31, 283–288. [DOI] [PubMed] [Google Scholar]
- (34).Lee H-Y, Moon H, Chun K-H, Chang Y-S, Hassan K, Ji L, Lotan R, Khuri FR, and Hong WK (2004) Effects of Insulin-like Growth Factor Binding Protein-3 and Farnesyltransferase Inhibitor SCH66336 on Akt Expression and Apoptosis in Non–Small-Cell Lung Cancer Cells. J. Natl. Cancer Inst 96, 1536–1548. [DOI] [PubMed] [Google Scholar]
- (35).Leroy B, Girard L, Hollestelle A, Minna JD, Gazdar AF, and Soussi T (2014) Analysis of TP53 Mutation Status in Human Cancer Cell Lines: A Reassessment. Hum. Mutat 35, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, Abe Y, Kita Y, and Nishimoto I (2001) Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer’s disease-relevant insults. J. Neurosci 21, 9235–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K, and Nishimoto I (2001) A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer’s disease genes and Abeta. Proc. Natl. Acad. Sci. U. S. A 98, 6336–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yen K, Lee C, Mehta H, and Cohen P (2013) The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J. Mol. Endocrinol 50, R11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Gong Z, Tas E, and Muzumdar R (2014) Humanin and Age-Related Diseases: A New Link? Front. Endocrinol. (Lausanne, Switz.), DOI: 10.3389/fendo.2014.00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lee C, Yen K, and Cohen P (2013) Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol. Metab 24, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zuccato CF, Asad AS, Nicola Candia AJ, Gottardo MF, Moreno Ayala MA, Theas MS, Seilicovich A, and Candolfi M (2019) Mitochondrial-derived peptide humanin as therapeutic target in cancer and degenerative diseases. Expert Opin. Ther. Targets 23, 117–126. [DOI] [PubMed] [Google Scholar]
- (42).Yen K, Wan J, Mehta HH, Miller B, Christensen A, Levine ME, Salomon MP, Brandhorst S, Xiao J, Kim S-J, Navarrete G, Campo D, Harry GJ, Longo V, Pike CJ, Mack WJ, Hodis HN, Crimmins EM, and Cohen P (2018) Humanin Prevents Age-Related Cognitive Decline in Mice and is Associated with Improved Cognitive Age in Humans. Sci. Rep, DOI: 10.1038/s41598-018-32616-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zou P, Ding Y, Sha Y, Hu B, and Nie S (2003) Humanin peptides block calcium influx of rat hippocampal neurons by altering fibrogenesis of Abeta(1–40). Peptides 24, 679–685. [DOI] [PubMed] [Google Scholar]
- (44).Benaki D, Zikos C, Evangelou A, Livaniou E, Vlassi M, Mikros E, and Pelecanou M (2005) Solution structure of humanin, a peptide against Alzheimer’s disease-related neurotoxicity. Biochem. Biophys. Res. Commun 329, 152–160. [DOI] [PubMed] [Google Scholar]
- (45).Becker RE, and Greig NH (2008) Alzheimer’s disease drug development: old problems require new priorities. CNS Neurol. Disord.: Drug Targets 7, 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Romeo M, Stravalaci M, Beeg M, Rossi A, Fiordaliso F, Corbelli A, Salmona M, Gobbi M, Cagnotto A, and Diomede L (2017) Humanin Specifically Interacts with Amyloid-β Oligomers and Counteracts Their in vivo Toxicity. J. Alzheimer's Dis 57, 857–871. [DOI] [PubMed] [Google Scholar]
- (47).Maftei M, Tian X, Manea M, Exner TE, Schwanzar D, Arnim CAF, and Przybylski M (2012) Interaction structure of the complex between neuroprotective factor humanin and Alzheimer’s β-amyloid peptide revealed by affinity mass spectrometry and molecular modeling: HUMANIN-í-AMYLOID PEPTIDE INTERACTION. J. Pept. Sci 18, 373–382. [DOI] [PubMed] [Google Scholar]
- (48).Heyl DL, Iwaniec B, Esckilsen D, Price D, Guttikonda P, Cooper J, Lombardi J, Milletti M, and Evans HG (2019) Using Small Peptide Segments of Amyloid-β and Humanin to Examine their Physical Interactions. Protein Pept. Lett 26, 502–511. [DOI] [PubMed] [Google Scholar]
- (49).Alsanousi N, Sugiki T, Furuita K, So M, Lee Y-H, Fujiwara T, and Kojima C (2016) Solution NMR structure and inhibitory effect against amyloid-β fibrillation of Humanin containing a d-isomerized serine residue. Biochem. Biophys. Res. Commun 477, 647–653. [DOI] [PubMed] [Google Scholar]
- (50).Soreq H, and Seidman S (2001) Acetylcholinesterase — new roles for an old actor. Nat. Rev. Neurosci 2, 294–302. [DOI] [PubMed] [Google Scholar]
- (51).Reyes AE, Chacón MA, Dinamarca MC, Cerpa W, Morgan C, and Inestrosa NC (2004) Acetylcholinesterase-Abeta complexes are more toxic than Abeta fibrils in rat hippocampus: effect on rat beta-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am. J. Pathol 164, 2163–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Inestrosa NC, Alvarez A, Godoy J, Reyes A, and De Ferrari GV (2000) Acetylcholinesterase–amyloid-β-peptide interaction and Wnt signaling involvement in Aβ neurotoxicity. Acta Neurol. Scand 102, 53–59. [DOI] [PubMed] [Google Scholar]
- (53).Inestrosa NC, Sagal JP, and Colombres M (2005) Acetylcholinesterase interaction with Alzheimer amyloid beta. Subcell. Biochem 38, 299–317. [DOI] [PubMed] [Google Scholar]
- (54).Alvarez A, Opazo C, Alarcón R, Garrido J, and Inestrosa NC (1997) Acetylcholinesterase promotes the aggregation of amyloid-beta-peptide fragments by forming a complex with the growing fibrils. J. Mol. Biol 272, 348–361. [DOI] [PubMed] [Google Scholar]
- (55).Bartolini M, Bertucci C, Cavrini V, and Andrisano V (2003) beta-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies. Biochem. Pharmacol 65, 407–416. [DOI] [PubMed] [Google Scholar]
- (56).De Ferrari GV, Canales MA, Shin I, Weiner LM, Silman I, and Inestrosa NC (2001) A Structural Motif of Acetylcholinesterase That Promotes Amyloid β-Peptide Fibril Formation. Biochemistry 40, 10447–10457. [DOI] [PubMed] [Google Scholar]
- (57).Inestrosa NC, Dinamarca MC, and Alvarez A (2008) Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 275, 625–632. [DOI] [PubMed] [Google Scholar]
- (58).Inestrosa NC, Alvarez A, Pérez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, and Garrido J (1996) Acetylcholinesterase Accelerates Assembly of Amyloid-β-Peptides into Alzheimer’s Fibrils: Possible Role of the Peripheral Site of the Enzyme. Neuron 16, 881–891. [DOI] [PubMed] [Google Scholar]
- (59).Inestrosa NC, Dinamarca MC, and Alvarez A (2008) Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 275, 625–632. [DOI] [PubMed] [Google Scholar]
- (60).Tjernberg LO, Näslund J, Lindqvist F, Johansson J, Karlström AR, Thyberg J, Terenius L, and Nordstedt C (1996) Arrest of beta-amyloid fibril formation by a pentapeptide ligand. J. Biol. Chem 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- (61).Williams AD, Portelius E, Kheterpal I, Guo J, Cook KD, Xu Y, and Wetzel R (2004) Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J. Mol. Biol 335, 833–842. [DOI] [PubMed] [Google Scholar]
- (62).Kheterpal I, Chen M, Cook KD, and Wetzel R (2006) Structural differences in Abeta amyloid protofibrils and fibrils mapped by hydrogen exchange–mass spectrometry with on-line proteolytic fragmentation. J. Mol. Biol 361, 785–795. [DOI] [PubMed] [Google Scholar]
- (63).Firth SM, and Baxter RC (2002) Cellular actions of the insulin-like growth factor binding proteins. Endocr. Rev 23, 824–854. [DOI] [PubMed] [Google Scholar]
- (64).Baxter RC (2014) IGF binding proteins in cancer: mechanistic and clinical insights. Nat. Rev. Cancer 14, 329–341.24722429 [Google Scholar]
- (65).Allard JB, and Duan C (2018) IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. (Lausanne, Switz.) 9, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Butt AJ, and Williams AC (2001) IGFBP-3 and apoptosis—a licence to kill? Apoptosis 6, 199–205. [DOI] [PubMed] [Google Scholar]
- (67).Forbes BE, McCarthy P, and Norton RS (2012) Insulin-Like Growth Factor Binding Proteins: A Structural Perspective. Front. Endocrinol. (Lausanne, Switz.), DOI: 10.3389/fendo.2012.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Jogie-Brahim S, Feldman D, and Oh Y (2009) Unraveling insulin-like growth factor binding protein-3 actions in human disease. Endocr. Rev 30, 417–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Martin JL, Coverley JA, Pattison ST, and Baxter RC (1995) Insulin-like growth factor-binding protein-3 production by MCF-7 breast cancer cells: stimulation by retinoic acid and cyclic adenosine monophosphate and differential effects of estradiol. Endocrinology 136, 1219–1226. [DOI] [PubMed] [Google Scholar]
- (70).Rajah R, Valentinis B, and Cohen P (1997) Insulin-like growth factor (IGF)-binding protein-3 induces apoptosis and mediates the effects of transforming growth factor-beta1 on programmed cell death through a p53- and IGF-independent mechanism. J. Biol. Chem 272, 12181–12188. [DOI] [PubMed] [Google Scholar]
- (71).Liu B, Lee HY, Weinzimer SA, Powell DR, Clifford JL, Kurie JM, and Cohen P (2000) Direct functional interactions between insulin-like growth factor-binding protein-3 and retinoid X receptor-alpha regulate transcriptional signaling and apoptosis. J. Biol. Chem 275, 33607–33613. [DOI] [PubMed] [Google Scholar]
- (72).Chang YS, Wang L, Liu D, Mao L, Hong WK, Khuri FR, and Lee H-Y (2002) Correlation between Insulin-like Growth Factor-binding Protein-3 Promoter Methylation and Prognosis of Patients with Stage I Non-Small Cell Lung Cancer. Clin. Cancer Res 8 (12), 3669–3675. [PubMed] [Google Scholar]
- (73).Põld M, Krysan K, Põld A, Dohadwala M, Heuze-Vourc’h N, Mao JT, Riedl KL, Sharma S, and Dubinett SM (2004) Cyclooxygenase-2 Modulates the Insulin-Like Growth Factor Axis in Non–Small-Cell Lung Cancer. Cancer Res. 64, 6549–6555. [DOI] [PubMed] [Google Scholar]
- (74).Ho GYF, Zheng SL, Cushman M, Perez-Soler R, Kim M, Xue X, Wang T, Schlecht NF, Tinker L, Rohan TE, Wassertheil-Smoller S, Wallace R, Chen C, Xu J, and Yu H (2016) Associations of Insulin and IGFBP-3 with Lung Cancer Susceptibility in Current Smokers. J. Natl. Cancer Inst 108 (7), djw012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).McCarthy K, Laban C, McVittie CJ, Ogunkolade W, Khalaf S, Bustin S, Carpenter R, and Jenkins PJ (2009) The expression and function of IGFBP-3 in normal and malignant breast tissue. Anticancer Res. 29 (10), 3785–3790. [PubMed] [Google Scholar]
- (76).Marzec KA, Baxter RC, and Martin JL (2015) Targeting Insulin-Like Growth Factor Binding Protein-3 Signaling in Triple-Negative Breast Cancer. BioMed Res. Int 2015, 638526–638533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Lee HY, Chun KH, Liu B, Wiehle SA, Cristiano RJ, Hong WK, Cohen P, and Kurie JM (2002) Insulin-like growth factor binding protein-3 inhibits the growth of non-small cell lung cancer. Cancer Res. 62 (12), 3530–3537. [PubMed] [Google Scholar]
- (78).Wang YA, Sun Y, Palmer J, Solomides C, Huang L-C, Shyr Y, Dicker AP, and Lu B (2017) IGFBP3Modulates Lung Tumorigenesis and Cell Growth through IGF1 Signaling. Mol. Cancer Res 15, 896–904. [DOI] [PubMed] [Google Scholar]
- (79).Njomen E, Evans HG, Gedara SH, and Heyl DL (2015) Humanin Peptide Binds to Insulin-Like Growth Factor-Binding Protein 3 (IGFBP3) and Regulates Its Interaction with Importin-β. Protein Pept. Lett 22, 869–876. [DOI] [PubMed] [Google Scholar]
- (80).Muterspaugh R, Price D, Esckilsen D, McEachern S, Guthrie J, Heyl D, and Evans HG (2018) Interaction of Insulin-Like Growth Factor-Binding Protein 3 With Hyaluronan and Its Regulation by Humanin and CD44. Biochemistry 57, 5726–5737. [DOI] [PubMed] [Google Scholar]
- (81).Price D, Muterspaugh R, Clegg B, Williams A, Stephens A, Guthrie J, Heyl D, and Evans HG (2020) IGFBP-3 Blocks Hyaluronan-CD44 Signaling, Leading to Increased Acetylcholinesterase Levels in A549 Cell Media and Apoptosis in a p53-Dependent Manner. Sci. Rep 10 (1), 5083–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).LeVine H (2004) Alzheimer’s beta-peptide oligomer formation at physiologic concentrations. Anal. Biochem 335, 81–90. [DOI] [PubMed] [Google Scholar]
- (83).Izzo NJ, Staniszewski A, To L, Fa M, Teich AF, Saeed F, Wostein H, Walko T, Vaswani A, Wardius M, Syed Z, Ravenscroft J, Mozzoni K, Silky C, Rehak C, Yurko R, Finn P, Look G, Rishton G, Safferstein H, Miller M, Johanson C, Stopa E, Windisch M, Hutter-Paier B, Shamloo M, Arancio O, LeVine H, and Catalano SM (2014) Alzheimer’s Therapeutics Targeting Amyloid Beta 1–42 Oligomers I: Abeta 42 Oligomer Binding to Specific Neuronal Receptors Is Displaced by Drug Candidates That Improve Cognitive Deficits. PLoS One 9 (11), e111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Pryor NE, Moss MA, and Hestekin CN (2012) Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. Int. J. Mol. Sci 13, 3038–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).LeVine H (1993) Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: detection of amyloid aggregation in solution. Protein Sci. 2, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Patel BB, Barrero CA, Braverman A, Kim PD, Jones KA, Chen DE, Bowler RP, Merali S, Kelsen SG, and Yeung AT (2012) Assessment of Two Immunodepletion Methods: Off-Target Effects and Variations in Immunodepletion Efficiency May Confound Plasma Proteomics. J. Proteome Res 11, 5947–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Evans HG, Guthrie JW, Jujjavarapu M, Hendrickson N, Eitel A, Park Y, Garvey J, Newman R, Esckilsen D, and Heyl DL (2017) D-Amino Acid Analogues of the Antimicrobial Peptide CDT Exhibit Anti- Cancer Properties in A549, a Human Lung Adenocarcinoma Cell Line. Protein Pept. Lett 24 (7), 590–598. [DOI] [PubMed] [Google Scholar]
- (88).Shojaie M, Sotoodah A, Roozmeh S, Kholoosi E, and Dana S (2009) Annexin V and anti-Annexin V antibodies: two interesting aspects in acute myocardial infarction. Thromb. J 7, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Yamagishi Y, Hashimoto Y, Niikura T, and Nishimoto I (2003) Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer’s disease-relevant insults. Peptides 24, 585–595. [DOI] [PubMed] [Google Scholar]
- (90).Ikonen M, Liu B, Hashimoto Y, Ma L, Lee K-W, Niikura T, Nishimoto I, and Cohen P (2003) Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. U. S. A 100, 13042–13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Garai K, and Frieden C (2013) Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc. Natl. Acad. Sci. U. S. A 110, 3321–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Necula M, Kayed R, Milton S, and Glabe CG (2007) Small molecule inhibitors of aggregation indicate that amyloid beta oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem 282, 10311–10324. [DOI] [PubMed] [Google Scholar]
- (93).Ramakrishnan M, Kandimalla KK, Wengenack TM, Howell KG, and Poduslo JF (2009) Surface plasmon resonance binding kinetics of Alzheimer’s disease amyloid beta peptide-capturing and plaque-binding monoclonal antibodies. Biochemistry 48, 10405–10415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Thakker DR, Weatherspoon MR, Harrison J, Keene TE, Lane DS, Kaemmerer WF, Stewart GR, and Shafer LL (2009) Intracerebroventricular amyloid-beta antibodies reduce cerebral amyloid angiopathy and associated micro-hemorrhages in aged Tg2576 mice. Proc. Natl. Acad. Sci. U. S. A 106, 4501–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Perdivara I, Deterding LJ, Cozma C, Tomer KB, and Przybylski M (2009) Glycosylation profiles of epitope-specific anti-beta-amyloid antibodies revealed by liquid chromatography-mass spectrometry. Glycobiology 19, 958–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Baghallab I, Reyes-Ruiz JM, Abulnaja K, Huwait E, and Glabe C (2018) Epitomic Characterization of the Specificity of the Anti-Amyloid Aβ Monoclonal Antibodies 6E10 and 4G8. J. Alzheimers Dis 66 (3), 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu Z-X, Le Y, Cui Y, and Wang JM (2004) Humanin, a newly identified neuroprotective factor, uses the G protein-coupled formylpeptide receptor-like-1 as a functional receptor. J. Immunol 172, 7078–7085. [DOI] [PubMed] [Google Scholar]
- (98).Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- (99).Bernstein SL, Wyttenbach T, Baumketner A, Shea J-E, Bitan G, Teplow DB, and Bowers MT (2005) Amyloid beta-protein: monomer structure and early aggregation states of Abeta42 and its Pro19 alloform. J. Am. Chem. Soc 127, 2075–2084. [DOI] [PubMed] [Google Scholar]
- (100).Chang YS, Wang L, Suh Y-A, Mao L, Karpen SJ, Khuri FR, Hong WK, and Lee H-Y (2004) Mechanisms underlying lack of insulin-like growth factor-binding protein-3 expression in non-small-cell lung cancer. Oncogene 23, 6569–6580. [DOI] [PubMed] [Google Scholar]
- (101).Kayed R, Head E, Sarsoza F, Saing T, Cotman CW, Necula M, Margol L, Wu J, Breydo L, Thompson JL, Rasool S, Gurlo T, Butler P, and Glabe CG (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener 2, 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Du J, and Murphy RM (2010) Characterization of the interaction of β-amyloid with transthyretin monomers and tetramers. Biochemistry 49, 8276–8289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Morimoto A, Irie K, Murakami K, Ohigashi H, Shindo M, Nagao M, Shimizu T, and Shirasawa T (2002) Aggregation and neurotoxicity of mutant amyloid beta (A beta) peptides with proline replacement: importance of turn formation at positions 22 and 23. Biochem. Biophys. Res. Commun 295, 306–311. [DOI] [PubMed] [Google Scholar]
- (104).Murakami K, Irie K, Morimoto A, Ohigashi H, Shindo M, Nagao M, Shimizu T, and Shirasawa T (2002) Synthesis, aggregation, neurotoxicity, and secondary structure of various A beta 1–42 mutants of familial Alzheimer’s disease at positions 21–23. Biochem. Biophys. Res. Commun 294, 5–10. [DOI] [PubMed] [Google Scholar]
- (105).Hilbich C, Kisters-Woike B, Reed J, Masters CL, and Beyreuther K (1991) Aggregation and secondary structure of synthetic amyloid beta A4 peptides of Alzheimer’s disease. J. Mol. Biol 218, 149–163. [DOI] [PubMed] [Google Scholar]
- (106).Chen Y-R, Huang H, Lo C-J, Wang C-C, Su C-L, Liu H-T, Shiao M-S, Lin T-H, and Chen Y-C (2011) Aβ40(L17A/F19A) mutant diminishes the aggregation and neurotoxicity of Aβ40. Biochem. Biophys. Res. Commun 405, 91–95. [DOI] [PubMed] [Google Scholar]
- (107).Päiviö A, Nordling E, Kallberg Y, Thyberg J, and Johansson J (2004) Stabilization of discordant helices in amyloid fibril-forming proteins. Protein Sci. 13, 1251–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, and Dobson CM (2005) Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol 350, 379–392. [DOI] [PubMed] [Google Scholar]
- (109).Tartaglia GG, Cavalli A, Pellarin R, and Caflisch A (2004) The role of aromaticity, exposed surface, and dipole moment in determining protein aggregation rates. Protein Sci. 13, 1939–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Inouye H, Gleason KA, Zhang D, Decatur SM, and Kirschner DA (2010) Differential effects of Phe19 and Phe20 on fibril formation by amyloidogenic peptide A beta 16–22 (Ac-KLVFFAE-NH2). Proteins: Struct. Funct., Genet 78, 2306–2321. [DOI] [PubMed] [Google Scholar]
- (111).Capone R, Jang H, Kotler SA, Kagan BL, Nussinov R, and Lal R (2012) Probing structural features of Alzheimer’s amyloid-β pores in bilayers using site-specific amino acid substitutions. Biochemistry 51, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Kumar J, Namsechi R, and Sim VL (2015) Structure-Based Peptide Design to Modulate Amyloid Beta Aggregation and Reduce Cytotoxicity. PLoS One 10, No. e0129087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Zimbone S, Monaco I, Gianí F, Pandini G, Copani AG, Giuffrida ML, and Rizzarelli E (2018) Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell 17 (1), e12684. [DOI] [PMC free article] [PubMed] [Google Scholar]