

Abstract
Adoptive cell therapy (ACT) for cancer shows tremendous potential, however, several challenges preclude its widespread use. These include poor T cell function in hostile tumor microenvironments, a lack of tumor-specific target antigens, and the high cost and poor scalability of cell therapy manufacturing. Creative genome-editing strategies are beginning to emerge to address each of these limitations, which has initiated the next generation of cell therapy products now entering clinical trials. CRISPR is at the forefront of this revolution, offering a simple and versatile platform for genetic engineering. This article provides a comprehensive overview of CRISPR applications that have advanced ACT.
Keywords: CRISPR, genetic engineering, cancer, adoptive cell therapy, immunotherapy
INTRODUCTION
The promise of adoptive cell therapy (ACT) for cancer has long been recognized with clinical trials in the 1980’s providing some of the first evidence that administration of tumor-reactive immune cells can produce profound tumor regression in some patients (1–4). Building upon this groundwork, Rosenberg and colleagues pioneered the development ACT using T cells that were isolated directly from the tumor site (tumor-infiltrating lymphocytes (TILs)). Their method of TIL ACT yielded robust response rates of 40–60% in metastatic melanoma patients (5), a result that has now been replicated across several centers (6–9). More recently, emerging data has demonstrated the efficacy of TIL ACT for other solid tumors, including cervical and head and neck cancers (10,11).
In the 1990’s and early 2000’s, the field of ACT moved towards generating T-cell therapies with defined and highly specific tumor reactivities, contrary to the polyclonal and often undefined specifies of TILs. Three dominant approaches emerged that involved (1) isolating and expanding endogenous T cells with defined antigen specificities, (2) redirecting T-cell specificity via introduction of transgenic T-cell receptors (tgTCRs), and (3) engineering novel T-cell specificities via transduction of chimeric antigen receptors (CARs).
Philip Greenberg and colleagues demonstrated that high avidity, tumor specific T cells can be isolated and expanded from the peripheral blood of cancer patients (12), and initial clinical use of such cells for ACT elicited antigen-specific tumor regression in melanoma patients (13). Alternatively, bulk peripheral blood lymphocytes can be genetically engineered to express a tgTCR that recognizes a tumor antigen. Rosenberg and colleagues were the first to demonstrate tumor regression in cancer patients treated with such TCR-transduced T cells. They reported that 2 of 15 advanced melanoma patients experienced durable tumor regression following treatment with T cells engineered to recognize a melanocyte differentiation antigen (14). Research is currently ongoing to explore the clinical potential of ACT using TCR-transduced T cells targeting a variety of antigens and cancer types (15).
CAR-engineered T cells have proven to be particularly impactful for the treatment of select hematologic malignancies. CARs, as originally developed by Eshhar and colleagues (16–18), are fusion proteins that combine the antigen binding capability of the antibody variable region with T-cell signaling motifs. Thus, CARs are able to recognize cell surface antigens independent of peptide presentation by major histocompatibility complexes. CAR T cells engineered to recognize the CD19 surface antigen have demonstrated tremendous clinical success for the treatment of CD19+ B-cell malignancies, as described by the groups of Carl June (19), Steven Rosenberg (20), Michel Sadelain (21) among others. This success ultimately led to U.S. Food and Drug Administration approval of axicabtagene ciloleucel (Yescarta) (Package Insert available at: https://www.fda.gov/media/108377/download) and tisagenlecleucel (Kymriah) (Package Insert available at: https://www.novartis.us/sites/www.novartis.us/files/kymriah.pdf) in 2017.
Despite these successes, hurdles remain at every step along the pipeline of developing safe, effective cellular therapies for cancer. A dearth of safe target antigens has hampered the development of CAR T cells for broader therapeutic indications beyond CD19+ lymphomas and leukemias. Achieving durable responses of solid tumors to ACT is met by the additional challenges of poor T-cell function in immunosuppressive tumor microenvironments, heterogeneity of antigen expression, and the need for human leukocyte antigen matching in engineered TCR approaches. At the other end of the development pipeline, the high cost and poor scalability of clinical-grade cell therapy manufacturing limits widespread access to these potentially life-saving treatments.
As genome-editing technologies have become increasingly accessible, their use in cellular therapy has expanded greatly. This has initiated the next generation of cellular products that harbor creative genetic manipulations to improve T cells’ potency and safety profiles and that can mitigate immunosuppressive triggers in the tumor microenvironment. Furthermore, genetic engineering approaches that enable use of allogeneic cell sources hold the potential to substantially improve the scalability of cell therapy manufacturing. CRISPR genome-editing techniques are at the forefront of this revolution, offering a versatile platform for applications ranging from multiplexed gene knockout to site-specific transgene insertion to genome-wide genetic screening. The purpose of this review is to detail recent applications of CRISPR that have advanced the field of cellular therapy, beginning with a discussion of specific genetic manipulations that hold the potential to enhance T-cell potency, safety, and scalability and ending with a review of CRISPR screening efforts that have unveiled novel targets for cell therapy enhancement. Emerging applications of CRISPR in non-T-cell-based ACTs are also described.
CRISPR: Why and How
CRISPR technology has transformed the landscape of genome editing with its simplicity and versatility (22). Genome editing via CRISPR is achieved using a two-part system comprising a guide RNA (gRNA) and a Cas9 nuclease. The 5’ end of the gRNA directs site specificity via Watson-Crick base pairing, whereas the Cas9 nuclease binds to the 3’ end of the gRNA, positioning it to induce a double-strand break in the target genomic DNA (22). The double-strand break then undergoes repair via the cell’s native machinery (nonhomologous end joining and homology-directed repair [HDR]), which often results in small insertions or deletions that disrupt the protein-coding sequence to produce a functional knockout. Furthermore, HDR can be exploited for targeted gene insertion at the CRISPR cut site via inclusion of an HDR template (23).
CRISPR offers a number of notable advantages over earlier genome editing technologies, such as zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs). ZFNs and TALENs rely on protein-DNA binding to guide site-specific DNA cleavage. As a result, new ZFN or TALEN proteins must be engineered for each new target site and design restrictions (particularly for ZFNs) limit the number of sites that can be targeted efficiently (24). ZFNs and TALENs have limited potential for multiplexed genome editing and are not amenable to the generation of large-scale libraries (25). In contrast, the CRISPR system can be designed to target virtually any genomic sequence simply by altering the gRNA sequence. This enables (1) rapid design and implementation of new CRISPR gene knockout/knock-in strategies, (2) efficient multiplexing via incorporation of multiple gRNAs, and (3) application of whole-genome CRISPR libraries for high throughput screening. Finally, the efficiency and specificity of genome editing via CRISPR is generally superior to that of ZFNs and TALENs (25,26). Specificity is a key safety consideration for clinical applications as off-target mutagenesis poses a risk of oncogenic transformation in genetically engineered cells. Recent technological advances have substantially reduced the risk of off-target mutagenesis with CRISPR, as discussed later in this review. For an in-depth comparison of CRISPR, ZFNs, and TALENs, readers are referred to a review by Khan (25).
Despite the aforementioned strengths of CRISPR genome editing, fully implementing this technology in primary T cells was a challenge for many years. Standard viral transduction approaches to introducing Cas9 and gRNA expression constructs into primary T cells is inefficient (27,28), likely due to the large size of the Cas9 gene and relative resistance of primary T cells to transduction. Furthermore, stable expression of Cas9 and gRNA following viral integration increases the risk of off-target editing and may result in Cas9 immunogenicity upon adoptive transfer of the edited cells into immunocompetent hosts. DNA nucleofection has been attempted with varying degrees of success but often results in low transfer efficiency and/or high toxicity in T cells, limiting the broad applicability of this method (29–31).
These challenges were overcome with the development of methods to transfect primary T cells with Cas9 ribonucleoprotein (RNP) complexes comprising recombinant Cas9 protein bound to a gRNA (28). Electrotransfer of RNP complexes into primary T cells can be accomplished efficiently without sacrificing cell viability, resulting in highly efficient gene knockout that is observable within about 48 hours. Cas9 delivered via transfection is degraded within 48–72 hours, which limits off-target editing and limits concerns about downstream Cas9 immunogenicity (32). Importantly, applying this technique is feasible within the constraints and timelines of clinical-grade cell therapy manufacturing in compliance with Good Manufacturing Practices, and incorporating a CRISPR gene editing step is not expected to increase overall production time (33). For the generation for CAR and TCR T-cell therapies, RNP electrotransfer can simply be applied about 1 day prior to standard viral transduction of CAR and TCR transgenes.
Given these benefits, Cas9 RNP electrotransfer has become the primary method of CRISPR editing in T cells and other primary immune cells. This system has proven to be quite versatile, with success seen in 1) single-gene knockout using one or more gRNA sequences (Fig. 1A) (28,34), 2) multiplexed gene knockout (Fig. 1B) (35,36), and 3) nonviral site-specific gene insertion via inclusion of homologous repair templates in the form of linear DNA oligonucleotides (Fig. 1C) (23,37,38). Furthermore, CRISPR editing via Cas9 RNP electrotransfer has been successfully applied to additional primary immune cells, including natural killer (NK) cells (39), B cells (40,41), and hematopoietic stem and progenitor cells (42–45). Oh et al. (46) and Naeimi Kararoudi et al. (39) reported detailed protocols describing Cas9 RNP electrotransfer in T and NK cells, respectively.
Figure 1: Electrotransfer of Cas9 RNP Complexes is a Versatile and Efficient Method of CRISPR Genome Editing in Primary T Cells.
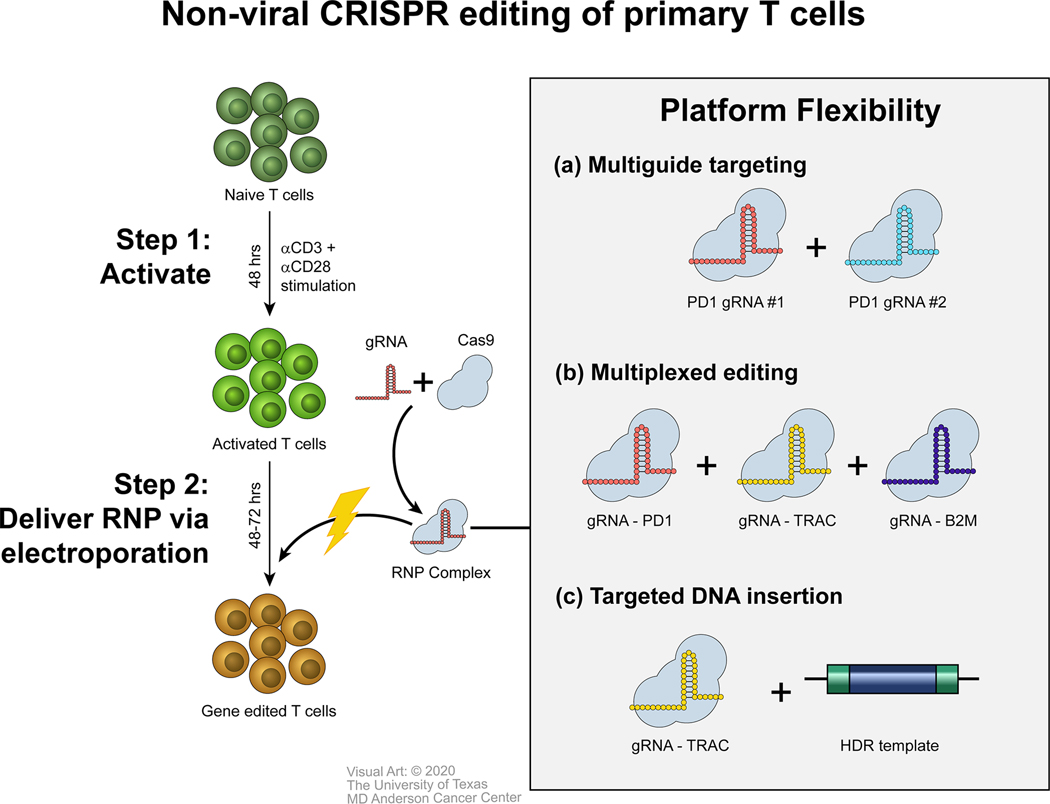
Cas9 RNP complexes comprising Cas9 protein and a gRNA are delivered into activated T cells via electroporation. Genomic editing is detectable about 48 hours after electroporation. (A) Multiple RNP complexes targeting the same gene can be delivered simultaneously to increase knockout efficiency. (B) Multiple RNP complexes targeting different genes can be delivered simultaneously to achieve multiplexed gene editing. (C) RNP complexes can be combined with HDR templates for targeted DNA insertion into the CRISPR cut site.
APPLICATIONS OF CRISPR TO IMPROVE THE POTENCY, SAFETY, AND SCALABILITY OF CELLULAR THERAPIES
Eliminating the “Brakes” to Create More Potent Cellular Therapies
Resistance of cancer to ACTs can develop through T-cell-extrinsic and T-cell-intrinsic mechanisms. Extrinsic resistance involves changes in tumor cells that render them resistant to T-cell killing, such as loss of the target antigen (47,48). Conversely, intrinsic resistance develops when the transferred T cells fail to persist in the patient or when they become hyporesponsive and unable to kill target tumor cells. The latter is particularly relevant in the treatment of solid tumors, where the tumor microenvironment, with its characteristically low pH, lack of nutrients, hypoxia, and abundance of immunosuppressive molecules, presents a major obstacle to T-cell function (49). Furthermore, tumors often upregulate inhibitory proteins such as programmed death-ligand 1 that engage with co-inhibitory receptors on the T-cell surface (e.g., programmed cell death protein 1 [PDCD1 or PD-1]) to suppress their antitumor activity (50).
CRISPR can be used to engineer T cells to overcome key pathways of immunosuppression that lead to T-cell intrinsic resistance of cancer to ACTs. The most thoroughly explored example of this is using CRISPR to knock out PD-1, which allows adoptively transferred T cells to resist programmed death-ligand 1-mediated co-inhibition (Fig. 2A) (29–31,34,51–57). Preclinical data demonstrate the therapeutic potential of this approach, with improved tumor control reported following treatment with PD-1–knockout T cells in models of gastric cancer (53), leukemia (52,54), hepatocellular carcinoma (55), triple-negative breast cancer (34), and glioma (31). The first two reports of the use of CRISPR-edited T cells to treat cancer were published earlier this year (2020) and involved PD-1 knockout (ClinicalTrials.gov identifiers: NCT03399448 and NCT02793856) (Table 1) (29) (35). Although these early clinical studies were not powered to draw conclusions regarding the therapeutic impact of PD-1 knockout, they did provide initial evidence suggesting that CRISPR gene editing is feasible and safe clinically.
Figure 2: Applications of CRISPR to Improve the Potency, Safety, and Scalability of T-cell Therapies.

(A) T-cell therapies are made more potent by knocking out inhibitory receptors (e.g., PD-1) that dampen T cell function upon ligation with cognate ligands in the tumor microenvironment. (B) The safety of TCR T-cell therapies can be improved via CRISPR knockout of the endogenous TCR (eTCR), which prevents formation of novel TCR mismatches with unpredictable neoreactivity. (C) With CRISPR gene knockout, it is feasible to generate CAR T-cell therapies from allogeneic T cells. Knockout of the eTCR reduces risk of GvHD while knockout of major histocompatibility complex (MHC) class I genes prevents rejection of the allogeneic cells upon adoptive transfer. Use of allogeneic cells for CAR T-cell therapy holds the potential to increase the scalability of CAR T-cell manufacturing as many doses can be generated from cells isolated from a single healthy donor. Furthermore, creation of allogeneic T-cell therapies may enable treatment of cancers that are otherwise difficult to treat with autologous T cells. (D) CRISPR can be used to knockout target antigens in off-target cells. For example, knocking out CD7 in CD7-directed CAR T cells prevents fratricide of the CAR T cells during manufacturing, enabling CAR T-cell therapy for T cell-based cancers.
Table 1: Ongoing Clinical Trials Involving CRISPR-Edited ACT for Cancer.
Information was gathered through Clinicaltrials.gov unless otherwise noted.
| Citation | ClinicalTrials.gov identifier | Company/Institution | CRISPR gene targets | Cell therapy details | Summary/Status |
|---|---|---|---|---|---|
| Trials for which reports are available | |||||
| Stadtmauer et al. (35) | NCT03399448 | University of Pennsylvania | PDCD1, TRAC, TRBC | NY-ESO-1 TCR T cells | Results demonstrate that multiplexed CRISPR/Cas9 genome editing is feasible at clinical scale and suggest acceptable safety |
| Xinxin Wang et al.a | NCT04264078 | Xinqiao Hospital of Chongqing | TRAC, CD7 | CD7 CAR T cells | Results demonstrate deep clinical responses and an acceptable safety profile; GvHD was not observed in any patients treated with allogeneic CAR T cells |
| Lu et al. (29) | NCT02793856 | Sichuan University | PDCD1 | Peripheral blood lymphocytes | Results for patients given CRISPR/Cas9 PD-1-edited T cells suggest that CRISPR editing in adoptively transferred T cells is feasible in a clinical setting and generally safe |
| Trials for which reports are not yet available | |||||
| NCT03545815 | Chinese PLA General Hospital | TCR, PDCD1 | Mesothelin CAR T cells | Recruiting | |
| NCT04035434 | CRISPR Therapeutics AG | B2M, TRAC | CD19 T cells | Recruiting | |
| NCT03747965 | Chinese PLA General Hospital | PDCD1 | Mesothelin CAR T cells | Recruiting | |
| NCT03081715 | Hangzhou Cancer Hospital | PDCD1 | Peripheral blood lymphocytes | Completed | |
| NCT04037566 | Xijing Hospital | HPK1 | CD19 CAR T cells | Recruiting | |
| NCT03690011 | Baylor College of Medicine | CD7 | CD7 CAR T cells | Not yet recruiting | |
| NCT04244656 | CRISPR Therapeutics AG | ? | B-cell maturation antigen T cells | Recruiting | |
| NCT03044743 | The Affiliated Nanjing Drum Tower Hospital of Nanjing University Medical School | PDCD1 | Epstein-Barr virus-specific T cells | Recruiting | |
| NCT03398967 | Chinese PLA General Hospital | ? | CD19 and CD20 or CD22 CAR T cells | Recruiting | |
| NCT03166878 | Chinese PLA General Hospital | TCR, B2M | CD19 CAR T cells | Recruiting | |
111th Annual Meeting of the American Association for Cancer Research (abstract available at: https://www.abstractsonline.com/pp8/#!/9045/presentation/10760)
Using CRISPR to knock out PD-1 (or other co-inhibitory receptors) in T cells is an alternative to combining ACT with systemic checkpoint-blocking antibodies (e.g., anti-PD-1/programmed death-ligand 1, anti-cytotoxic T-lymphocyte-associated protein 4) and offers attractive benefits. First, checkpoint pathways are engaged throughout the body to promote peripheral tolerance and prevent autoimmunity. Therefore, systemic blockade often results in autoimmune-related side effects (e.g., colitis, pneumonitis) (58,59). Using CRISPR to eliminate PD-1 specifically in transferred cells limits the risk of breaking peripheral tolerance, especially when any self-reactive TCRs are eliminated from the infusion product (see below). Second, cellular therapies can persist in patients and provide tumor surveillance for many years. CRISPR knockout of a checkpoint receptor protects the transferred cells from PD-1–mediated inhibition for their entire lifetimes and the lifetimes of their progeny. In contrast, checkpoint-blocking antibodies have half-lives of 15–25 days and would require prolonged repeat dosing to achieve the same benefit.
Permanent checkpoint elimination via CRISPR knockout is not without limitations. Emerging evidence from studies of patients with cancer treated with PD-1–knockout TCR T cells suggested that cells deficient in PD-1 are less persistent than their PD-1–proficient counterparts, which is consistent with a defect in memory formation (35). In this context, administering a mixed population of T cells may be advantageous, as the PD-1–knockout population may provide potent effector activity in the short term, whereas those with intact PD-1 may persist and provide longer anti-tumor protection. Alternatively, advanced CRISPR approaches are being explored to provide reversible genetic control of PD-1 expression. In 2019, Qi et al. described a system whereby transcriptional repression of PD-1 was transiently induced in CAR T cells upon antigen engagement (Abstract presented in: SITC Adoptive Cellular Therapies Workshop 2019 https://www.sitcancer.org/education/actworkshop/schedule). This nuanced control of PD-1 may enhance effector function while maintaining memory differentiation and long-term persistence of the edited cells in vivo. Given the complexity of most checkpoint signaling pathways, the multidimensional consequences of CRISPR knockout should be carefully evaluated for each gene target and therapeutic application.
Going beyond elimination of canonical checkpoint receptors, CRISPR offers the potential to inhibit the activity of metabolic regulators, transcription factors, and signaling molecules that are otherwise challenging to block using conventional antibodies or small molecules. Tang et al. (60) used CRISPR to knock out the transforming growth factor (TGF)-β receptor (TGFBR2) in CAR T cells, which rendered these cells unresponsive to high levels of exogenous TGF-β in vitro and improved their antitumor activity in vivo. This is an attractive approach given the pleiotropic roles of TGF-β throughout the body, which present challenges and safety concerns for the development of systemic TGF-β inhibitors (61). Also, Jung et al. (62) knocked out the metabolic enzyme diacylglycerol kinase in CAR T cells, which enhanced their effector function and antitumor activity via increased TCR signaling and resistance to immunosuppressive factors. The specificity of CRISPR knockout is also attractive in this context given the roles of diacylglycerol kinases in other cell types and a lack of specific inhibitors targeting this kinase (63).
These methods of enhancing T-cell potency via CRISPR gene editing may ultimately decrease the minimum effective doses of cellular therapies and lessen the burden of manufacturing high T-cell numbers. However, eliminating inherent checks and balances that restrain T-cell responses may exacerbate the risk of cytokine release syndrome (a common and serious side effect of CAR T-cell therapy (64)) and/or cause uncontrolled lymphoproliferation in vivo. These risks should be carefully evaluated for any target in longitudinal preclinical safety studies prior to clinical translation.
Disrupting the Endogenous TCR to Enhance the Safety and Efficacy of T cell Therapies
Generating engineered TCR T-cell therapies typically involves viral transfer of a tumor-reactive tgTCR into peripheral T cells. Sustained expression of endogenous TCRs in the final T-cell product presents safety and efficacy challenges. First, α and β chains from the endogenous TCR and the tgTCR can form mixed dimers with unpredictable and dangerous neoreactiviy (65). Furthermore, mixed dimer formation limits surface expression of the transgenic αβ pair (33). Competition for CD3 binding by the endogenous TCR and tgTCR further limits surface expression of tgTCR complexes (66). Ultimately, reduced tgTCR expression dampens the antigen sensitivity and therapeutic efficacy of the final TCR T-cell product.
CRISPR can be used to efficiently knock out endogenous αβTCR by targeting the genes that encode for the α chain (TRAC) and β chain (TRBC) constant regions (Fig. 2B). Whereas single knockout of TRAC or TRBC is sufficient to increase tgTCR surface expression and antigen sensitivity in some contexts (33,67), double knockout of both genes is necessary to eliminate the risk of endogenous TCR and tgTCR mispairing and to ensure optimal expression of weakly competitive tgTCRs (33).
Researchers have proposed two methods of endogenous TCR disruption in TCR T cells. First, efficient TRAC/TRBC double knockout can be achieved via single-step transfection with TRAC- and TRBC-specific Cas9 RNP complexes (33,57). This method is compatible with clinical TCR T-cell manufacturing pipelines, as it can be simply applied about 1 day prior to standard viral transduction of the tgTCR (33). The clinical feasibility of this approach was confirmed in a recent report by Stadtmauer et al. (35) describing the use of NY-ESO-1–directed TCR T cells that had undergone TRAC, TRBC, and PDCD1 knockout (ClinicalTrials.gov identifier: NCT03399448) (Table 1).
Second, Cas9 RNP complexes can be used to nonvirally guide the integration of tgTCR α- and β-expressing DNA templates into endogenous TRAC and/or TRBC loci (37). These complexes are delivered into cells via electroporation and create double-strand breaks at specific locations within the TRAC and/or TRBC gene. Subsequently, tgTCR templates bearing homology arms (co-delivered via electroporation) are integrated into double-strand break sites via HDR. This approach disrupts endogenous TCR expression while putting the transgenes under the control of the endogenous TCR promoters. A strength of this approach is that it does not involve viral transduction for tgTCR integration. Avoiding the lengthy and expensive process of generating clinical-grade viral vectors has the potential to accelerate the development of new TCR T-cell therapies and decrease T-cell therapy production costs. Furthermore, avoiding viral transduction mitigates the risk of oncogenic mutagenesis via random viral integration.
CRISPR also has been used to insert CAR constructs into the TRAC locus (68). Placing the CAR under the transcriptional control of the TRAC gene promotes more consistent and moderate baseline expression of the CAR when compared with random viral integration. This limits tonic signaling at baseline and promotes optimal receptor internalization and recycling upon antigen exposure. Ultimately, these features enhance CAR T-cell potency and delay terminal differentiation and exhaustion of the CAR T cells. An added benefit is that integrating the CAR into the TRAC locus abolishes surface expression of the endogenous TCR. Although disrupting endogenous TCR expression in CAR T cells does not inherently increase CAR T-cell potency, it does limit the risk of undesired autoreactivity and alloreactivity.
Creating Off-the-Shelf Allogeneic Therapies to Broaden Their Disease Applications and Improve Scalability
The majority of TCR and CAR T cells in the clinic today are engineered ex vivo from patients’ own (i.e., autologous) T cells. Not only does the use of autologous cells result in costly, time-consuming manufacturing, it also limits the variety of malignancies that can be treated with TCR and CAR T cells. For instance, patients with some of the most aggressive malignancies (e.g., glioblastoma multiforme) often do not have time to wait for an autologous therapy to be manufactured before rapid tumor progression renders them ineligible for treatment (64). For treating T-cell-based malignancies, using autologous T cells for CAR or TCR T-cell manufacturing is met with the critical challenge of ensuring that the final T-cell product is not contaminated with malignant T cells that express many of the same surface markers as healthy T cells (69).
Creating off-the-shelf allogeneic cell therapies holds the potential to overcome these challenges. T cells from a single healthy donor could be used to generate many doses of TCR or CAR T-cell products, which would increase the scalability, cost-effectiveness, and standardization of cell therapy manufacturing. Furthermore, manufacturing of allogeneic products could be completed well in advance of patient enrollment, decreasing time to treatment for the patient. However, using allogeneic cells has its own complications, including the risk of life-threatening graft vs. host disease (GvHD) and alloreactivity resulting in rapid rejection of the transferred cells. Recent advances demonstrate the potential of CRISPR to overcome these challenges and enable the development of allogeneic T-cell therapies that are safe and effective (Fig. 2C).
As described above, allogeneic CAR T cells are particularly attractive for treating T-cell malignancies. Cooper et al. (70) developed an off-the-shelf CAR T cell targeting the CD7 antigen expressed by many T-cell malignancies. The authors rendered their CAR T cells “universal” by using CRISPR to knock out the endogenous TCR, thereby limiting the risk of GvHD. Importantly, they also used CRISPR to knock out CD7 and prevent fratricide of the CAR T cells (see below). A similar approach was recently taken to the clinic by Gracell (ClinicalTrials.gov identifier: NCT04264078) (Table 1), with their initial clinical results presented at the 111th Annual Meeting of the American Association for Cancer Research (2020) (Abstract available at: https://www.abstractsonline.com/pp8/#!/9045/presentation/10760). The authors gave treatment to an initial five T-cell leukemia patients with allogeneic CD7-directed CAR T cells that had undergone CRISPR knockout of CD7 (to prevent fratricide of the CAR T cells) and TRAC (to prevent GvHD). All five patents experienced complete remission, although one quickly had a relapse, and four had minimal residual disease-negative complete responses at 1 month. Encouragingly, none of these patients experienced acute GvHD. The impact of alloreactivity on the long-term persistence of these CAR T cells remains to be determined.
CRISPR can be used to limit rejection of allogeneic T-cell therapies by knocking out major histocompatibility complex class I expression, which renders the engineered cells invisible to the recipient’s CD8+ T cells. Proof-of-principle studies by Liu et al. (51) and Choi et al. (36) demonstrated the technical feasibility of this approach using CD19- and EGFRvIII-directed CAR T cells, respectively. The authors achieved efficient TCR knockout by targeting the constant region of the TCRα chain (TRAC) and major histocompatibility complex class I knockout by targeting the β2-microglobulin gene (B2M), which encodes for a necessary component of the major histocompatibility complex class I complex. Whereas major histocompatibility complex class I expression prevents elimination of allogeneic cells by the host’s CD8+ T cells, it may lead to downstream activation of NK cells. Additional genetic manipulation, such as via enforced expression of nonclassical human leukocyte antigen molecules (71), could be explored if this challenge comes to fruition.
The initial reports reviewed herein demonstrate the promise of using CRISPR to generate off-the-shelf allogeneic T-cell therapies for cancer. These advances hold the potential to greatly expand the reach of cellular therapies by making production more scalable and cost-effective and by opening doors to the treatment of malignancies such as glioblastoma multiforme and T-cell cancers that are otherwise challenging to treat with autologous products.
Overcoming On-Target Off-Tumor Challenges to Broaden Disease Applications for T cell Therapies
The power of CAR and TCR T-cell therapy is rooted in the ability of these cells to kill tumors in an antigen-specific manner. However, these T cells cannot distinguish between malignant and heathy cells expressing the same antigen, resulting in so-called on-target off-tumor side effects. This is readily observed in patients given CD19-directed CAR T cells, as tumor regression is associated with long-term B-cell aplasia (72,73). Nevertheless, patients can tolerate B-cell aplasia, and immunoglobulin replacement therapy can be given to treat hypogammaglobulinemia (72–74). Identifying antigens with similarly acceptable safety profiles has been a major challenge preventing widespread adoption of CAR T-cell therapy for hematologic malignancies.
CRISPR can be used to ameliorate on-target off-tumor challenges by knocking out the target antigen in off-target cells. Candidate acute myeloid leukemia antigens such as CD33 are shared by normal and malignant myeloid cells, so targeting these antigens would result in persistent myeloablation and serious morbidity and/or mortality in the treated leukemia patients. This can be overcome by combining CD33-directed CAR T-cell therapy with hematopoietic stem cell transplantation using stem cells that have undergone CRISPR deletion of CD33 (43–45). This facilitates eradication of CD33+ acute myeloid leukemia cells with simultaneous reconstitution of the hematopoietic system with CD33-null cells that can function normally while resisting death induced by CAR T cells. This synthetic approach to creating tumor-specific antigens holds the potential to widen the therapeutic window of CAR T-cell therapy and broaden the cancer types for which this therapy is feasible.
Treating T-cell malignancies with CAR T-cells is met with an additional on-target off-tumor obstacle: fratricide. Candidate antigens expressed by T-cell tumors, such as CD7 and CD5, are also expressed by CAR T cells themselves, resulting in self-activation and self-killing of CAR T cells during manufacture of the cellular therapy. CRISPR can be used to overcome this challenge by simply knocking out the target antigen in T cells prior to CAR transduction (Fig. 2D). Researchers have successfully applied this approach to generating fratricide-resistant CAR T cells targeting CD7 (70,75) and CD5 (76), and several such approaches are currently entering clinical trials (Table 1).
Fratricide-resistant CAR T cells can kill not only malignant T cells but also healthy endogenous T cells expressing the same antigens. This makes patients susceptible to life-threatening immunodeficiency via T-cell aplasia. Gomes-Silva et al. (75) demonstrated that their fratricide-resistant CD7-directed CAR T cells were responsive to viral peptides in vitro due to the cells’ native TCRs. This suggests that autologous CD7-directed CAR T cells can provide patients with T-cell malignancies with some level of protection from pathogens despite elimination of native T cells. On the other hand, Cooper et al. (70) argued for the use of allogeneic CAR T cells for the treatment of T-cell malignancies given the risk of malignant contamination of autologous T-cell products and the poor quantity and/or quality of T cells isolated from this heavily pretreated patient population. As discussed above, the use of allogeneic CAR T cells requires TCR knockout to prevent GvHD, which inadvertently eliminates any protection that CAR T cells provide against pathogens via their TCRs.
As an alternative, CAR T-cell therapy for T-cell malignancies can be viewed as a “bridge” to transplantation. Following tumor eradication via treatment with autologous CAR T cells, a patient can receive an allogeneic stem cell transplant, which would terminate CAR T-cell activity (due to alloreactivity/rejection) and reconstitute the hematopoietic system. Alternatively, the target antigen (e.g., CD7) may be knocked out in stem cells (as described previously for CD33 (43–45)) to enable long-term co-existence of antitumor CAR T cells and healthy CD7- T cells if the same allogeneic donor is used for CAR T-cell generation and stem cell transplantation.
Applications of CRISPR for ACT Using Cell Types Other than T cells
Whereas T-cell-based approaches to ACT are by far the most prevalent and mature, the potential of other immune cell subsets in ACT should not be overlooked, and applications of CRISPR in this context are beginning to emerge.
Like T cells, NK cells can directly kill cancer, making them attractive for ACT. NK cells can do so through several mechanisms, including degranulation and antibody-dependent cellular cytotoxicity initiated by receptor (e.g., CD16a [FCGR3A]) binding to the Fc portion of IgG-opsonized target cells (77). In contrast with T cells, NK cell-mediated killing is not antigen-specific, and NK cells do not induce GvHD in the allogeneic setting, making them ideal as off-the-shelf cellular therapy products (77,78). However, the function of NK cells can be suppressed via exposure to inhibitory factors in the tumor microenvironment and via NK cells’ intrinsic checks and balances. Thus, the potency of adoptively transferred NK cells may be enhanced via CRISPR editing.
Pomeroy et al. (79) demonstrated that PD-1 knockout increased the functional capacity of primary NK cells in vitro and enhanced their antitumor efficacy in an ovarian cancer model. Furthermore, they demonstrated that CRISPR can be used to enhance antibody-dependent cellular cytotoxicity by preventing CD16a shedding from the NK cell surface via either knockout of ADAM17 (a protease that cleaves CD16a) or CRISPR knock-in of a noncleavable CD16a variant (Fig. 3A) (79). Zhu et al. (80) and Daher et al. (81) knocked out an important cytokine checkpoint (cytokine-inducible SH2-containing protein [CISH or CIS]) to enhance the metabolic fitness of induced pluripotent stem cell-derived NK cells and IL-15–secreting CAR NK cells, respectively. In both reports, CIS knockout improved NK cell effector function in vitro and tumor control upon adoptive transfer in vivo.
Figure 3: Applications of CRISPR for ACT Using Cell Types Other than T cells.

(A) Killing of target cells by NK cells via antibody-dependent cellular cytotoxicity can be enhanced by CRISPR knockout of ADAM17, a protease that cleaves CD16a from the NK cell surface. (B) Cancer cells often upregulate CD47, which transmits a “don’t eat me” signal to macrophages via binding to signal regulatory protein (SIRP)-α on the macrophage cell surface. Thus, SIRP-α knockout in macrophages increases their ability to phagocytose tumor cells.
Investigators have also applied CRISPR to macrophage-based immunotherapies. Macrophages can engulf cancer cells, and they play a role in tumor control. However, many cancer cells have surface overexpression of CD47, which engages with signal regulatory protein-α (SIRPA) to deliver a “don’t eat me” signal to macrophages. Ray et al. (82) demonstrated that CRISPR knockout of signal regulatory protein-α in macrophages increased their ability to phagocytose tumor cells in vitro (Fig. 3B). We anticipate that the use of CRISPR to genetically engineer non-T-cell-based cellular therapies for cancer will continue to grow in the coming years as these therapies continue to mature.
Emerging Technologies to Enhance the Safety of CRISPR-Engineered Adoptive Cell Therapies
The first two reports of CRISPR-modified T cells being used to treat human cancer were published earlier this year (29,35), with additional clinical trials now underway (Table 1). As CRISPR-modified cellular therapies continue to advance into clinical use, it is critical to ensure their safety. A primary concern regarding CRISPR-modified ACTs is that induction of double strand breaks at off-target sites may result in pathogenic cellular modifications and genotoxicity. In the context of multiplexed CRISPR editing, the risk of chromosomal translocations between double strand break sites presents additional safety concerns. Therefore, prior to clinical use, CRISPR-modified cellular products must be comprehensively evaluated for off-target mutations using an unbiased assay based on whole genome sequencing. Chromosomal translocations must also be evaluated when multiplexed editing is performed.
Efforts to limit the off-target effects of CRISPR have yielded exciting technological advances in recent years, holding the potential to enhance the safety profile of CRISPR-modified cellular therapies and ease clinical translation. One particular area of advancement is the generation of high specificity Cas9 variants characterized by demonstrably lower off-target cleavage compared to wild type Cas9. These engineered proteins include the so-called high fidelity Cas9s (spCas9-HF1 (83), and HiFi Cas9 (84)), enhanced specificity Cas9 (eSpCas9(1.1)) (85), evolved Cas9 (evoCas9) (86), and hyper accurate Cas9 (HypaCas9) (87). The HiFi Cas9 generated by Vakulskas et al. is particularly relevant as it retains high on-target editing efficiency when delivered into cells transiently as an RNP complex (84). Furthermore, HiFi Cas9 is now commercially available as a Good Manufacturing Practice-grade protein applicable for clinical cell therapy manufacturing.
Recent innovations in CRISPR-based methodology, such as the development of base editors and prime editors, offer the potential to further increase the safety of CRISPR genome editing by eliminating double strand break formation all together. Base editors, which are fusions of a catalytically deficient or nicking Cas9 variant and a deaminase, induce nucleotide modifications within a short DNA window (~5 base pair) directed by gRNA binding (88,89). Cytosine (90,91) and adenine (92) base editors have been developed to induce site-specific C to T or A to G mutations, respectively.
Base editors can be used to (1) alter gene function by introducing a key point mutation (92) or (2) disrupt gene expression by inducing a premature stop codon (93,94) or disrupting a splice site (95). A key advantage of this system is that the induced genomic changes occur with single-nucleotide-level precision, resulting in an altered genotype that is homogenous and predictable. This contrasts with the heterogenous mix of insertions and deletions formed via conventional CRISPR gene knockout. Furthermore, since base editors do not induce double strand breaks, they are associated with a low risk of off-target insertions and deletions and a low risk of chromosomal translocation in the context of multiplexed editing (95). Encouragingly, Webber et al. recently optimized a base editing method for efficient multiplexed gene disruption in primary T cells, demonstrating the power of this technology in the context of ACT (95).
Despite the attractive benefits of CRISPR base editors, this technology is prone to its own characteristic off-target mutations that should be monitored and reduced for any clinical application. Base editors can induce bystander deamination if multiple cytosines or adenines are present within the editing window (88,91). Furthermore, base editors have been shown to induce single nucleotide mutations at off-target DNA or even RNA sites (88). While recent advances in base editor design have reduces these risks (89), off-target mutations should be comprehensively analyzed prior to their clinical use.
CRISPR prime editing offers a novel technique to introduce a wide range of base substitutions, insertions, and deletions without inducing double strand breaks (96). This technology relies on the fusion of a nicking Cas9 variant and a reverse transcriptase. A modified gRNA directs site specific editing and serves as a primer for the transcription of a modified DNA sequence at the target site. Prime editing offers a genetic engineering platform that is more versatile than base editing and more efficient than genetic modification via CRISPR-mediated insertion of HDR templates (96). Furthermore, prime editing is characterized by lower off-target modification compared to conventional CRISPR/Cas9 methods (96). Despite the tremendous potential of CRISPR prime editing, further research is required to establish the efficiency of this technique in primary immune cells and to fully characterize its safety with respect to off-target mutations. With further development, we believe that CRISPR base editors and prime editors will allow genetic engineering of adoptive cell therapies with unprecedented precision and flexibility while enhancing safety through the reduction of off-target mutations.
HIGH-THROUGHPUT CRISPR SCREENS TO IDENTIFY NEW THERAPEUTIC TARGETS
Preclinical Discovery
As detailed in the first half of this review, CRISPR genome editing is a powerful platform for enhancing the efficacy of cellular therapies for cancer. However, determining which genetic alterations among the endless possibilities will be the most therapeutically impactful is difficult. CRISPR-based genetic screening is an unbiased, high-throughput method of testing dozens to tens of thousands of genetic alterations in a single experiment. Genetic screens can be designed in 1) an arrayed format, where individual gRNAs are introduced into cells in the wells of a culture plate, or 2) a pooled format, where barcoded gRNA libraries are combined and simultaneously introduced into single populations of cells. Here we focus on pooled CRISPR screens, as they are generally advantageous for high-throughput analyses, and this technique recently yielded fruitful discoveries for immunotherapy advancement.
In its simplest form, a pooled CRISPR screen involves transducing a cell type of interest with a barcoded library of gRNAs targeting any number of genes (97). The cell line is typically pre-engineered to stably express Cas9, or the Cas9 gene is incorporated into the gRNA library constructs. Following library transduction, edited cells are subjected to a screening assay that involves application of a selective pressure or stimulus (e.g., drug-based treatment, TCR activation) followed by next-generation sequencing to identify enriched and depleted gRNA barcodes in surviving and/or phenotypically isolated cell populations (97). This approach is generally referred to as a loss-of-function screen, as it reveals the consequences of gene loss through CRISPR-mediated knockout. Alternative CRISPR screening techniques rely on CRISPR activation or CRISPR interference to transiently induce or repress gene expression, respectively (97), or CRISPR-mediated gene knock-in to explore the consequences of targeted DNA insertion (23) (98).
Performing pooled CRISPR screens with primary T cells has been a major challenge. Primary T cells are only viable in culture for a short period of time, making the lengthy process of Cas9 transduction, selection, expansion, and subsequent library transduction impractical. Furthermore, T cells are notoriously difficult to transduce, and they do not accept large constructs for simultaneous expression of Cas9 and gRNA.
To circumvent these challenges, researchers performed the first loss-of-function CRISPR screens with T cells using immortalized murine (99) and human (100) cell lines amenable to multiple rounds of transduction and selection (Table 2). They transduced T cell lines with Cas9 before lentiviral transduction of pooled gRNA libraries spanning the entire genome (for a detailed protocol, see Shang et al. (101)). Using this methodology, Okada et al. (99) revealed a novel regulatory mechanism of PD-1 expression on the surface of T cells (core fucosylation) that could be targeted pharmacologically to improve the antitumor efficacy of adoptively transferred T cells. Shang et al. (100) identified previously unknown regulators of T-cell activation, including FAM49B (CYRIB), enhancing our understanding of TCR signaling and its regulation. Whereas these initial reports demonstrate the potential of genome-wide CRISPR screens with T cells, discoveries made using CRISPR screening with immortalized T-cell lines may not always translate to primary cultures or ACT applications.
Table 2: Summary of CRISPR Genetic Screens with T Cells.
Reports are listed in chronological order of publication.
| Citation | Cell type | In vitro/In vivo | Screening details | Screening assay | Technological advance and/or discovery |
|---|---|---|---|---|---|
| Okada et al. (99) | Immortalized mouse T-cell line (68–41) | In vitro | Genome-wide loss-of-function screen to identify genes involved in PD-1 expression induction/maintenance | Identified gRNA enriched in sorted PD-1low cells | Discovery: knocking out genes associated with core fucosylation decreases surface expression of PD-1 on T cells; treating antigen-specific murine T cells with a core fucosylation inhibitor decreases PD-1 expression and improves antigen-specific tumor control (B16-Ovalbumin) in vivo |
| Shang et al. (100) | Immortalized human T-cell line (Jurkat) | In vitro | Genome-wide loss-of-function screen to identify genes that regulate T-cell activation | gRNA abundance compared in CD69low and CD69high cells | Discovery: identified FAM49B as a negative regulator of TCR activation, which is involved in actin cytoskeleton remodeling |
| Ting et al. (102) | Primary human CD4+ T cells | In vitro | Proof-of-principle loss-of-function screen with ~13,000 gRNAs; developed Guide Swap method | Proof-of-principle report demonstrated feasibility of this screening methodology | Technological advance: development of Guide Swap to enable large-scale pooled screening with primary human cells (including T cells) that cannot be transduced with Cas9-encoding vectors |
| Shifrut et al. (103) | Primary human CD8+ T cells | In vitro | Genome-wide loss-of-function screens to identify genes that regulate T-cell stimulation and suppression; developed SLICE method | gRNA abundance compared in CFSEhigh and CFSElow T cells after TCR stimulation |
Technological advance: development of SLICE screening method to enable large-scale pooled screening in primary human T cells Discovery: identified genes involved in T-cell suppression that, upon knockout, result in improved antigen-specific killing of tumor cells |
| Dong et al. (104) | Antigen-specific primary mouse CD8+ T cells | In vivo and in vitro | Genome-wide loss-of-function screen to identify genes that modulate CD8+ T cell effector function in vitro and in vivo | Identify gRNA associated with increased infiltration in tumors (in vivo); identify gRNA associated with increased T-cell degranulation (in vitro) | Discovery: complementary in vivo and in vitro screening reveals genes that can be targeted to improve T-cell function in the context of immunotherapy; authors validated that Dxh37 knockout in CD8+ T cells improves effector function in vitro and tumor control following adoptive transfer in vivo |
| Ye et al. (105) | Primary mouse CD8+ T cells | In vivo | Loss-of-function screen focused on genes encoding for membrane-bound proteins | Identify gRNA associated with enhanced T-cell infiltration in glioblastoma tumors following adoptive transfer | Discovery: identified multiple gene targets (novel and known) that, upon knockout, can increase the antitumor efficacy of adoptively transferred T cells; validated the potential of PDIA3 as an immunotherapy target using multiple mouse models with mechanistic characterization |
| Wei et al. (106) | Antigen-specific primary mouse CD8+ T cells | In vivo | Loss-of-function screen focused on metabolism-associated genes aimed at identifying factors to improve adoptive cell therapy for cancer | Identify gRNA associated with enhanced T-cell infiltration in B16 melanomas following adoptive transfer | Discovery: loss of REGNASE-1 improves CD8+ T-cell accumulation and persistence in the tumor microenvironment, improving antitumor efficacy. Follow-up genome-wide loss-of-function screen identified mechanistic targets of REGNASE-1 and candidates for combination therapy |
| Roth et al. (23) | Primary human T cells | In vitro and in vivo | Pooled knock-in screen to identify synthetic constructs that enhance T-cell fitness and antitumor activity; nonviral HDR template library delivered via Cas9 RNP electroporation | Identify constructs that enhance T-cell expansion in various contexts (in vitro) and that enhance T-cell infiltration and an anti-tumor phenotype in tumors (in vivo) |
Major technologic advance: developed methodology to screen large numbers of knock-in constructs in vivo and in vitro using primary T cells Discovery: identified a novel switch receptor (TGFBR2–41BB) that promotes solid tumor clearance by tgTCR T cells in vivo |
Ting et al. (102) and Shifrut et al. (103) made breakthroughs in the application of CRISPR screens to primary human T cells by circumventing the need for viral Cas9 transduction (Table 2) (102,103). Both reports described a method that involves transduction of primary T cells with lentiviral gRNA libraries followed by electroporation-based delivery of Cas9 protein either alone (SLICE method) (103) or complexed with a nontargeting gRNA (Guide Swap method) (Fig. 4) (102). In two independent genome-wide screens, Shifrut and colleagues validated the use of this approach to identify regulators of TCR activation and gene deletions that confer resistance of T cells to immunosuppressive adenosine signaling. The techniques developed in these two studies represent a major milestone in the application of CRISPR screening to primary T cells and other immune cells that are difficult to transduce.
Figure 4: High-Throughput CRISPR Screening in Primary T cells.

Novel gene targets to enhance ACT can be identified in an unbiased fashion using pooled CRISPR screens. First, T cells are transduced with a lentiviral library of barcoded gRNA constructs. Second, Cas9 is delivered via electroporation either alone (SLICE method) or in complex with a non-targeting gRNA (Guide Swap method). Finally, the pool of T cells harboring unique CRISPR gene knockouts is subjected to a screening assay and enriched and/or depleted gRNA barcodes are detected via next generation sequencing (NGS).
CRISPR screens with human T cells tend to be restricted to in vitro screening assays, in which gRNA enrichment and/or depletion is assessed after application of a stimulus (e.g., TCR stimulation) and/or fluorescence-activated cell sorting of distinct populations (e.g., nonproliferating CFSEhigh and proliferating CFSElow cells). Conversely, CRISPR screening with primary murine T cells can be paired with in vivo assays to reveal gene knockouts that promote T-cell infiltration and persistence in complex and immunosuppressive tumor microenvironments. This approach has been used to identify therapeutic targets to improve ACT in models of breast cancer (104), glioblastoma (105), and melanoma (106) as summarized in Table 2.
For CRISPR screening with primary murine T cells, Cas9–knock-in mice are used as sources of T cells that constitutively express Cas9, enabling one-step transduction with a viral library of gRNAs. In the context of cancer immunotherapy, Cas9 mice are typically crossed with a TCR transgenic strain to enable screening with antigen-specific, Cas9-expressing T cells (104,106). This approach has two limitations. First, genetically crossing mice is a lengthy process that requires a great deal of technical expertise. This adds significant developmental lead time to screening projects and limits the accessibility of this technique. Second, for in vivo screening assays, Cas9+ T cells are typically transferred into wild-type recipient mice that do not express Cas9. Cas9 is an immunogenic bacterial protein whose expression may result in premature clearance of the transferred cells, biasing screening results.
Adapting the aforementioned methodology developed by Ting et al. (102) and Shifrut et al. (103) to screening of mouse T cells holds the potential to overcome these two limitations. Transient Cas9 delivery via electroporation would prevent the need for crossing TCR transgenic and Cas9–knock-in mice, decreasing project lead time and increasing accessibility of this technique. Furthermore, Cas9 delivered via electroporation would be present in the T cells transiently, reducing the risk of Cas9 immunogenicity upon adoptive transfer.
All T-cell screens described thus far have been loss-of-function screens based on CRISPR gene knockout. Building on this groundwork, more advanced CRISPR-based screening applications are now possible. For example, Roth et al. (23) (98) recently reported on the first pooled CRISPR knock-in screen with primary human T cells (Table 2). The authors designed a library of 36 HDR templates encoding for synthetic constructs hypothesized to enhance T-cell fitness. These constructs included dominant-negative receptors, stimulatory switch receptors, transcription factors, and metabolic regulators. The researchers exposed library-modified cells to in vitro and in vivo screening assays to identify the constructs that most reliably increased T-cell fitness and antitumor function. They ultimately identified and validated that a novel switch receptor with the extracellular domain of the TGF-β receptor (TGFBR2) and the intracellular signaling domain of 4–1BB (TNFRSF9) increased T-cell fitness in vitro and enhanced tumor clearance by adoptively transferred T cells in vivo.
The CRISPR screening methods discussed herein are expected to have a substantial positive impact on the development of more potent ACTs. Genome-wide loss-of-function screens will continue to reveal novel targets that can be manipulated to enhance T-cell function and overcome key pathways of immunosuppression. Meanwhile, focused screens of 10–50 genetic alterations will enable researchers to systematically and efficiently evaluate the therapeutic benefit of numerous candidate alterations, accelerating the translational development of genetic engineering approaches that are expected to have the most profound clinical impact.
Lentiviral Insertional Mutagenesis as a Genetic Screening Platform Applicable to Cancer Patients
Cancer patients are the most relevant subjects for genetic screening assays aimed at uncovering mechanisms to improve the efficacy of cellular immunotherapy in a given patient population. Whereas the aforementioned CRISPR screens cannot be applied to human subjects directly, analyzing the consequences of lentiviral insertional mutagenesis from a screening perspective can reveal gene disruptions that enhance the antitumor efficacy and persistence of cellular therapies in the clinic. This provides a platform for reverse translational research that is expected to yield therapeutic advances with direct relevance to specific patient populations.
Fraietta et al. (107) made the serendipitous observation that lentiviral integration of a CD19-directed CAR construct in the TET2 gene was associated with remission in a patient with chronic lymphocytic leukemia. Despite infusing a polyclonal pool of cells harboring unique lentiviral integration sites, more than 90% of the persisting CAR T cells observed during peak response originated from a single clone with TET2 integration. In-depth analysis demonstrated that TET2 disruption was associated with profound CAR T-cell expansion in vivo accompanied by differentiation into a central memory phenotype, prolonged persistence, and ultimately robust antitumor efficacy. This highlights the therapeutic potential of disrupting TET2 (e.g., via CRISPR knockout) to enhance the therapeutic performance of CAR T cells in chronic lymphocytic leukemia patients.
Building upon this initial observation, Nobles et al. (108) set out to correlate lentiviral integration sites with clonal expansion and clinical outcomes in an additional 39 chronic lymphocytic leukemia patients given treatment with CD19-directed CAR T cells. Indeed, the authors identified a number of genes at integration sites that were associated with enhanced proliferation and long-term persistence of CAR T cells. These genes may prove to be valuable targets for improving the therapeutic efficacy of CAR T cells in chronic lymphocytic leukemia patients. This powerful approach can be expanded to identify therapeutic targets in additional patient populations already being given virally engineered T-cell therapies. Taking any gene targets identified using this approach back to the laboratory to rigorously test their safety and therapeutic tractability will be important. This approach of reverse translational research holds tremendous potential for the development of therapeutic strategies that have direct clinical relevance.
CONCLUSIONS
The development of methods to apply CRISPR genome editing to primary T cells has opened the door to numerous advances aimed at improving the potency, safety, and scalability of ACTs. First, CRISPR knockout of inhibitory factors that restrain T-cell function increases the antitumor efficacy of ACT in preclinical studies. This offers the potential for treating cancer with lower doses of highly potent gene-modified T cells than those doses used currently, which would lessen the burden of manufacturing massive cell numbers for clinical infusion. Second, novel techniques of guiding TCR and CAR transgene integration into the native TCR locus offers an alternative to laborious and costly viral transduction with the added benefit of improving safety via elimination of endogenous TCR expression. Third, CRISPR knockout of the TCRα gene paved the way for the first clinical use of allogeneic CAR T cells with no evidence of acute GvHD. Use of allogeneic T cells for CAR T-cell generation is expected to dramatically reduce the cost of T-cell therapy manufacturing, as cells from a single healthy donor can be used to manufacture many CAR T-cell doses. Furthermore, access to off-the-shelf T-cell products is expected to be particularly impactful for patients with rapidly progressing cancer who do not have time to wait for generation of an autologous therapy. Fourth, creative methods of overcoming on-target off-tumor toxicity offer the potential to extend the success of CAR T-cell therapy to indications beyond CD19+ B-cell malignancies. About a dozen open clinical trials involve CRISPR-edited T-cell therapies, and we anticipate the number to grow continually as the many preclinical approaches described herein continue to mature. Furthermore, recent breakthroughs have resulted in a spike in CRISPR genetic screening with primary T cells. These screens have yielded fruitful discoveries and novel targets for enhancing the efficacy of ACT. Continued use of CRISPR screens will accelerate preclinical discovery and enable systematic prioritization of genetic edits that have the greatest potential therapeutic impact.
Significance.
The clinical impact of ACT for cancer can be expanded by implementing specific genetic modifications that enhance the potency, safety, and scalability of cellular products. Here we provide a detailed description of such genetic modifications, highlighting avenues to enhance the therapeutic efficacy and accessibility of ACT for cancer. Furthermore, we review high-throughput CRISPR genetic screens that have unveiled novel targets for cell therapy enhancement.
Acknowledgments
The authors thank Rachel Hicklen and Donald Norwood of MD Anderson Scientific Publications, Research Medical Library for literature searching and editing of the manuscript, respectively. The authors thank David Aten of MD Anderson Department of Creative Communications for figure illustrations.
Financial Support: Authors wish to acknowledge generous support from the Dunwoody-Edwards Ovarian Cancer Philanthropic Fund and the Miriam and Jim Mulva Research Fund. SMF was supported by the CPRIT Research Training Program (RP170067), the Center for Clinical and Translational Sciences TL1 Training Program (NIH Grant No. TL1 TR003169), and an NIH/NCI National Research Service Award (F32CA253968). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Disclosure of Potential Conflicts of Interest: AAJ has no known conflicts of interest. PH reports being a consultant and Scientific Advisory Board member for Immatics, Dragonfly, GlaxoSmithKline and Sanofi. SMF reports grants from National Institutes of Health and grants from Cancer Prevention and Research Institute of Texas during preparation of the article. SMF and PH have a patent for Engineered T Cells and Tumor-Infiltrating Lymphocytes to Overcome Immunosuppression in the Tumor Microenvironment pending.
REFERENCES
- 1.Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the Systemic Administration of Autologous Lymphokine-Activated Killer Cells and Recombinant Interleukin-2 to Patients with Metastatic Cancer. N Engl J Med [Internet]. N Engl J Med; 1985. [cited 2020 Jul 16];313:1485–92. Available from: https://pubmed.ncbi.nlm.nih.gov/3903508/ [DOI] [PubMed] [Google Scholar]
- 2.Thompson JA, Shulman KL, Benyunes MC, Lindgren CG, Collins C, Lange PH, et al. Prolonged continuous intravenous infusion interleukin-2 and lymphokine-activated killer-cell therapy for metastatic renal cell carcinoma. J Clin Oncol [Internet]. Lippincott Williams and Wilkins; 1992. [cited 2020 Jul 16];10:960–8. Available from: https://pubmed.ncbi.nlm.nih.gov/1588376/ [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A Progress Report on the Treatment of 157 Patients with Advanced Cancer Using Lymphokine-Activated Killer Cells and Interleukin-2 or High-Dose Interleukin-2 Alone. N Engl J Med [Internet]. N Engl J Med; 1987. [cited 2020 Jul 16];316:889–97. Available from: https://pubmed.ncbi.nlm.nih.gov/3493432/ [DOI] [PubMed] [Google Scholar]
- 4.Thompson JA, Lee DJ, Lindgren CG, Benz LA, Collins C, Shuman WP, et al. Influence of schedule of interleukin 2 administration on therapy with interleukin 2 and lymphokine activated killer cells. Cancer Res. United States; 1989;49:235–40. [PubMed] [Google Scholar]
- 5.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. United States; 1988;319:1676–80. [DOI] [PubMed] [Google Scholar]
- 6.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012;18:6758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Besser MJ, Shapira-Frommer R, Itzhaki O, Treves AJ, Zippel DB, Levy D, et al. Adoptive transfer of tumor-infiltrating lymphocytes in patients with metastatic melanoma: intent-to-treat analysis and efficacy after failure to prior immunotherapies. Clin Cancer Res. United States; 2013;19:4792–800. [DOI] [PubMed] [Google Scholar]
- 8.Andersen R, Donia M, Ellebaek E, Borch TH, Kongsted P, Iversen TZ, et al. Long-Lasting Complete Responses in Patients with Metastatic Melanoma after Adoptive Cell Therapy with Tumor-Infiltrating Lymphocytes and an Attenuated IL2 Regimen. Clin Cancer Res. United States; 2016;22:3734–45. [DOI] [PubMed] [Google Scholar]
- 9.Goff SL, Dudley ME, Citrin DE, Somerville RP, Wunderlich JR, Danforth DN, et al. Randomized, Prospective Evaluation Comparing Intensity of Lymphodepletion Before Adoptive Transfer of Tumor-Infiltrating Lymphocytes for Patients With Metastatic Melanoma. J Clin Oncol. 2016;34:2389–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jazaeri AA, Zsiros E, Amaria RN, Artz AS, Edwards RP, Wenham RM, et al. Safety and efficacy of adoptive cell transfer using autologous tumor infiltrating lymphocytes (LN-145) for treatment of recurrent, metastatic, or persistent cervical carcinoma. J Clin Oncol [Internet]. American Society of Clinical Oncology; 2019;37:2538. Available from: 10.1200/JCO.2019.37.15_suppl.2538 [DOI] [Google Scholar]
- 11.Chesney JA, Lutzky J, Thomas SS, Nieva JJ, Munoz Couselo E, Martin-Liberal J, et al. A phase II study of autologous tumor infiltrating lymphocytes (TIL, LN-144/LN-145) in patients with solid tumors. J Clin Oncol [Internet]. American Society of Clinical Oncology; 2019;37:TPS2648–TPS2648. Available from: 10.1200/JCO.2019.37.15_suppl.TPS2648 [DOI] [Google Scholar]
- 12.Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD. Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol. United States; 1999;162:2227–34. [PubMed] [Google Scholar]
- 13.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A [Internet]. Proc Natl Acad Sci U S A; 2002. [cited 2020 Jul 17];99:16168–73. Available from: https://pubmed.ncbi.nlm.nih.gov/12427970/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (80- ). 2006;314:126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang J, Wang L. The emerging world of TCR-T cell trials against cancer: A systematic review [Internet]. Technol. Cancer Res. Treat SAGE Publications Inc.; 2019. [cited 2020 Jul 18]. Available from: /pmc/articles/PMC6391541/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A [Internet]. Proc Natl Acad Sci U S A; 1989. [cited 2020 Jul 16];86:10024–8. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/2513569/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the γ or ζ subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A [Internet]. Proc Natl Acad Sci U S A; 1993. [cited 2020 Jul 16];90:720–4. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/8421711/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor γ chain. J Exp Med [Internet]. J Exp Med; 1993. [cited 2020 Jul 18];178:361–6. Available from: https://pubmed.ncbi.nlm.nih.gov/8315392/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med [Internet]. Sci Transl Med; 2011. [cited 2020 Jul 14];3. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/21832238/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood [Internet]. Blood; 2010. [cited 2020 Jul 14];116:4099–102. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/20668228/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med [Internet]. Sci Transl Med; 2013. [cited 2020 Jul 14];5. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/23515080/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. United States; 2014;346:1258096. [DOI] [PubMed] [Google Scholar]
- 23.Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell. 2020;181:728–744.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.LaFountaine JS, Fathe K, Smyth HDC. Delivery and therapeutic applications of gene editing technologies ZFNs, TALENs, and CRISPR/Cas9. Int. J. Pharm Elsevier B.V.; 2015. page 180–94. [DOI] [PubMed] [Google Scholar]
- 25.Khan SH. Genome-Editing Technologies: Concept, Pros, and Cons of Various Genome-Editing Techniques and Bioethical Concerns for Clinical Application. Mol. Ther. - Nucleic Acids Cell Press; 2019. page 326–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, et al. Evaluation of TCR gene editing achieved by TALENs, CRISPR/Cas9, and megaTAL nucleases. Mol Ther [Internet]. 2016;24:570–81. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med13&AN=26502778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang W, Ye C, Liu J, Zhang D, Kimata JT, Zhou P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One. 2014;9:e115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seki A, Rutz S. Optimized RNP transfection for highly efficient CRI SPR/Cas9-mediated gene knockout in primary T cells. J Exp Med [Internet]. 2018;215:985–97. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29436394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat Med. United States; 2020;26:732–40. [DOI] [PubMed] [Google Scholar]
- 30.Su S, Hu B, Shao J, Shen B, Du J, Du Y, et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci Rep [Internet]. 2016;6:20070. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med13&AN=26818188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu B, Zou Y, Zhang L, Tang J, Niedermann G, Firat E, et al. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum Gene Ther [Internet]. 2019;30:446–58. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medl&AN=29706119 [DOI] [PubMed] [Google Scholar]
- 32.Liang X, Potter J, Kumar S, Zou Y, Quintanilla R, Sridharan M, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. Netherlands; 2015;208:44–53. [DOI] [PubMed] [Google Scholar]
- 33.Morton LT, Reijmers RM, Wouters AK, Kweekel C, Remst DFG, Pothast CR, et al. Simultaneous Deletion of Endogenous TCRalphabeta for TCR Gene Therapy Creates an Improved and Safe Cellular Therapeutic. Mol Ther [Internet]. 2020;28:64–74. Available from: https://www.journals.elsevier.com/molecular-therapy [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother [Internet]. 2019;68:365–77. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85058058256&doi=10.1007%2Fs00262-018-2281-2&partnerID=40&md5=8dce36572192d13c2e738dbd814632ab [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (80- ) [Internet]. 2020;367 (6481). Available from: https://science.sciencemag.org/content/367/6481/eaba7365/tab-pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer [Internet]. 2019;7. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85075114384&doi=10.1186%2Fs40425-019-0806-7&partnerID=40&md5=58bdf36b33225616ed65a9789911513f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. England; 2018;559:405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, Gate RE, et al. Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci U S A [Internet]. 2015;112:10437–42. Available from: http://www.pnas.org/content/112/33/10437.full.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naeimi Kararoudi M, Dolatshad H, Trikha P, Hussain S-RA, Elmas E, Foltz JA, et al. Generation of Knock-out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J Vis Exp. 2018;58237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hung KL, Meitlis I, Hale M, Chen CY, Singh S, Jackson SW, et al. Engineering Protein-Secreting Plasma Cells by Homology-Directed Repair in Primary Human B Cells. Mol Ther [Internet]. 2018;26:456–67. Available from: https://www.journals.elsevier.com/molecular-therapy [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu C-AM, Roth TL, Baglaenko Y, Ferri DM, Brauer P, Zuniga-Pflucker JC, et al. Genetic engineering in primary human B cells with CRISPR-Cas9 ribonucleoproteins. J Immunol Methods. 2018;457:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu L, Wang J, Liu Y, Xie L, Su B, Mou D, et al. CRISPR-Edited Stem Cells in a Patient with HIV and Acute Lymphocytic Leukemia. N Engl J Med [Internet]. 2019;381:1240–7. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=medc&AN=31509667 [DOI] [PubMed] [Google Scholar]
- 43.Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell [Internet]. 2018;173:1439–1453.e19. Available from: http://www.cell.com [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Humbert O, Laszlo GS, Sichel S, Ironside C, Haworth KG, Bates OM, et al. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia [Internet]. 2019;33:762–808. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85054480766&doi=10.1038%2Fs41375-018-0277-8&partnerID=40&md5=cff0b9891772f18d16c0b27b38a96e56 [DOI] [PubMed] [Google Scholar]
- 45.Borot F, Wang H, Ma Y, Jafarov T, Raza A, Ali AM, et al. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc Natl Acad Sci U S A [Internet]. 2019;116:11978–87. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=prem&AN=31138698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh SA, Seki A, Rutz S. Ribonucleoprotein Transfection for CRISPR/Cas9-Mediated Gene Knockout in Primary T Cells. Curr Protoc Immunol [Internet]. 2019;124. Available from: http://onlinelibrary.wiley.com/book/10.1002/0471142735 [DOI] [PubMed] [Google Scholar]
- 47.Shah NN, Fry TJ. Mechanisms of resistance to CAR T cell therapy. Nat Rev Clin Oncol. England; 2019;16:372–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng J, Zhao L, Zhang Y, Qin Y, Guan Y, Zhang T, et al. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies [Internet]. Front. Oncol. 2019. page 1237. Available from: https://www.frontiersin.org/article/10.3389/fonc.2019.01237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen G, Huang AC, Zhang W, Zhang G, Wu M, Xu W, et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature. 2018;560:382–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res [Internet]. 2017;27:154–7. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85001073672&doi=10.1038%2Fcr.2016.142&partnerID=40&md5=9829c5ebdf8400fc48236cfee9f15423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res. United States; 2017;23:2255–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Su S, Zou Z, Chen F, Ding N, Du J, Shao J, et al. CRISPR-cas9-mediated disruption of PD-1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology [Internet]. 2017;6. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85007398518&doi=10.1080%2F2162402X.2016.1249558&partnerID=40&md5=c67e77fe64b48165d84f69ba40e2b49b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rupp LJ, Schumann K, Roybal KT, Gate RE, Ye CJ, Lim WA, et al. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep [Internet]. 2017;7:737. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=emed18&AN=623731738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo X, Jiang H, Shi B, Zhou M, Zhang H, Shi Z, et al. Disruption of PD-1 Enhanced the Anti-tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front Pharmacol [Internet]. 2018;9:1118. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=prem2&AN=30327605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang C, Peng Y, Hublitz P, Zhang H, Dong T. Genetic abrogation of immune checkpoints in antigen-specific cytotoxic T-lymphocyte as a potential alternative to blockade immunotherapy. Sci Rep [Internet]. 2018;8:5549. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med15&AN=29615718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ouchi Y, Patil A, Tamura Y, Nishimasu H, Negishi A, Paul SK, et al. Generation of tumor antigen-specific murine CD8 + T cells with enhanced anti-tumor activity via highly efficient CRISPR/Cas9 genome editing. Int Immunol [Internet]. 2018;30:141–54. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85045459533&doi=10.1093%2Fintimm%2Fdxy006&partnerID=40&md5=c6b6014dde210ce29ea7ca5d92ec21db [DOI] [PubMed] [Google Scholar]
- 58.Chan KK, Bass AR. Autoimmune complications of immunotherapy: pathophysiology and management. BMJ. England; 2020;369:m736. [DOI] [PubMed] [Google Scholar]
- 59.Xing P, Zhang F, Wang G, Xu Y, Li C, Wang S, et al. Incidence rates of immune-related adverse events and their correlation with response in advanced solid tumours treated with NIVO or NIVO+IPI: a systematic review and meta-analysis. J Immunother Cancer [Internet]. 2019;7:341. Available from: 10.1186/s40425-019-0779-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020/January/31. 2020;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smith AL, Robin TP, Ford HL. Molecular Pathways: Targeting the TGF-β Pathway for Cancer Therapy. Clin Cancer Res [Internet]. 2012;18:4514 LP – 4521. Available from: http://clincancerres.aacrjournals.org/content/18/17/4514.abstract [DOI] [PubMed] [Google Scholar]
- 62.Jung IY, Kim YY, Yu HS, Lee M, Kim S, Lee J. CRISPR/Cas9-Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res [Internet]. 2018;78:4692–703. Available from: https://www.scopus.com/inward/record.uri?eid=2-s2.0-85051470972&doi=10.1158%2F0008-5472.CAN-18-0030&partnerID=40&md5=8b8c902eff86ddb4977720830ed4b2cf [DOI] [PubMed] [Google Scholar]
- 63.Purow B. Molecular Pathways: Targeting Diacylglycerol Kinase Alpha in Cancer. Clin Cancer Res [Internet]. 2015;21:5008 LP – 5012. Available from: http://clincancerres.aacrjournals.org/content/21/22/5008.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Santomasso B, Bachier C, Westin J, Rezvani K, Shpall EJ. The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden. Am Soc Clin Oncol Educ B [Internet]. American Society of Clinical Oncology; 2019;433–44. Available from: 10.1200/EDBK_238691 [DOI] [PubMed] [Google Scholar]
- 65.van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, et al. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc Natl Acad Sci U S A. United States; 2010;107:10972–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ahmadi M, King JW, Xue S-A, Voisine C, Holler A, Wright GP, et al. CD3 limits the efficacy of TCR gene therapy in vivo. Blood. United States; 2011;118:3528–37. [DOI] [PubMed] [Google Scholar]
- 67.Legut M, Dolton G, Mian AA, Ottmann OG, Sewell AK. CRISPR-mediated TCR replacement generates superior anticancer transgenic t cells. Blood [Internet]. 2018;131:311–22. Available from: http://www.bloodjournal.org/content/bloodjournal/131/3/311.full.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eyquem J, Mansilla-Soto J, Giavridis T, Van Der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature [Internet]. 2017;543:113–7. Available from: http://www.nature.com/nature/index.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alcantara M, Tesio M, June CH, Houot R. CAR T-cells for T-cell malignancies: challenges in distinguishing between therapeutic, normal, and neoplastic T-cells. Leukemia. England; 2018;32:2307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cooper ML, Choi J, Staser K, Ritchey JK, Devenport JM, Eckardt K, et al. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. England; 2018;32:1970–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Torikai H, Reik A, Soldner F, Warren EH, Yuen C, Zhou Y, et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood. 2013;122:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Porter DL, Hwang W-T, Frey N V, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hill JA, Giralt S, Torgerson TR, Lazarus HM. CAR-T - and a side order of IgG, to go? - Immunoglobulin replacement in patients receiving CAR-T cell therapy. Blood Rev. 2019;38:100596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gomes-Silva D, Srinivasan M, Sharma S, Lee CM, Wagner DL, Davis TH, et al. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. United States; 2017;130:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raikar SS, Fleischer LC, Moot R, Fedanov A, Paik NY, Knight KA, et al. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. Oncoimmunology [Internet]. 2018;7:e1407898. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=prem2&AN=29399409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Curr Opin Immunol. 2018;51:146–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pomeroy EJ, Hunzeker JT, Kluesner MG, Lahr WS, Smeester BA, Crosby MR, et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol Ther J Am Soc Gene Ther [Internet]. 2020;28:52–63. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=prem&AN=31704085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell. Cell Press; 2020;27:224–237.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Daher M, Basar R, Gokdemir E, Baran N, Uprety N, Nunez Cortes AK, et al. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood. Blood; 2020;doi: 10.1182/blood.2020007748. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ray M, Lee YW, Hardie J, Mout R, Yesilbag Tonga G, Farkas ME, et al. CRISPRed Macrophages for Cell-Based Cancer Immunotherapy. Bioconjug Chem [Internet]. 2018;29:445–50. Available from: https://ovidsp.ovid.com/ovidweb.cgi?T=JS&CSC=Y&NEWS=N&PAGE=fulltext&D=med15&AN=29298051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature [Internet]. Nature Publishing Group; 2016. [cited 2020 Sep 10];529:490–5. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/26735016/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vakulskas CA, Dever DP, Rettig GR, Turk R, Jacobi AM, Collingwood MA, et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat Med [Internet]. 2018;24:1216–24. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30082871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science (80- ) [Internet]. American Association for the Advancement of Science; 2016 [cited 2020 Sep 10];351:84–8. Available from: http://biorxiv.org/content/early/2015/06/12/020834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Casini A, Olivieri M, Petris G, Montagna C, Reginato G, Maule G, et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol [Internet]. Nature Publishing Group; 2018. [cited 2020 Sep 10];36:265–71. Available from: https://www.nature.com/articles/nbt.4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen JS, Dagdas YS, Kleinstiver BP, Welch MM, Sousa AA, Harrington LB, et al. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature [Internet]. Nature Publishing Group; 2017. [cited 2020 Sep 10];550:407–10. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/28931002/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Broeders M, Herrero-Hernandez P, Ernst MPT, van der Ploeg AT, Pijnappel WWMP. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience. Elsevier Inc.; 2020;23:100789. doi: 10.1016/j.isci.2019.100789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kantor A, McClements M, MacLaren R. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int J Mol Sci [Internet]. NLM (Medline); 2020 [cited 2020 Sep 11];21:6240. Available from: https://www.mdpi.com/1422-0067/21/17/6240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature [Internet]. Nature Publishing Group; 2016. [cited 2020 Sep 12];533:420–4. Available from: https://www.nature.com/articles/nature17946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nishida K, Arazoe T, Yachie N, Banno S, Kakimoto M, Tabata M, et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science (80- ) [Internet]. American Association for the Advancement of Science; 2016. [cited 2020 Sep 12];353. Available from: https://science.sciencemag.org/content/353/6305/aaf8729 [DOI] [PubMed] [Google Scholar]
- 92.Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature [Internet]. 2017;551:464–71. Available from: 10.1038/nature24644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kuscu C, Parlak M, Tufan T, Yang J, Szlachta K, Wei X, et al. CRISPR-STOP: Gene silencing through base-editing-induced nonsense mutations. Nat Methods [Internet]. Nature Publishing Group; 2017. [cited 2020 Sep 12];14:710–2. Available from: https://www.nature.com/articles/nmeth.4327 [DOI] [PubMed] [Google Scholar]
- 94.Billon P, Bryant EE, Joseph SA, Nambiar TS, Hayward SB, Rothstein R, et al. CRISPR-Mediated Base Editing Enables Efficient Disruption of Eukaryotic Genes through Induction of STOP Codons. Mol Cell. Cell Press; 2017;67:1068–1079.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Webber BR, lin Lonetree C, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun. 2019/November/21. 2019;10:5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Anzalone A V, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature [Internet]. Nature Research; 2019. [cited 2020 Sep 11];576:149–57. Available from: https://pubmed-ncbi-nlm-nih-gov.elibrary.mdanderson.org/31634902/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schuster A, Erasimus H, Fritah S, Nazarov P V, van Dyck E, Niclou SP, et al. RNAi/CRISPR Screens: from a Pool to a Valid Hit. Trends Biotechnol [Internet]. 2019;37:38–55. Available from: http://www.sciencedirect.com/science/article/pii/S0167779918302300 [DOI] [PubMed] [Google Scholar]
- 98.Roth TL, Li PJ, Blaeschke F, Nies JF, Apathy R, Mowery C, et al. Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell. United States; 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Okada M, Chikuma S, Kondo T, Hibino S, Machiyama H, Yokosuka T, et al. Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Rep. United States; 2017;20:1017–28. [DOI] [PubMed] [Google Scholar]
- 100.Shang W, Jiang Y, Boettcher M, Ding K, Mollenauer M, Liu Z, et al. Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proc Natl Acad Sci U S A [Internet]. 2018;115:E4051–60. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29632189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shang W, Wang F, Zhu Q, Wang L, Wang H. CRISPR/Cas9-Based Genetic Screening to Study T-Cell Function. Methods Mol Biol. 2020/January/15. 2020;2111:59–70. [DOI] [PubMed] [Google Scholar]
- 102.Ting PY, Parker AE, Lee JS, Trussell C, Sharif O, Luna F, et al. Guide Swap enables genome-scale pooled CRISPR-Cas9 screening in human primary cells. Nat Methods. United States; 2018;15:941–6. [DOI] [PubMed] [Google Scholar]
- 103.Shifrut E, Carnevale J, Tobin V, Roth TL, Woo JM, Bui CT, et al. Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell [Internet]. 2018;175:1958–1971 e15. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30449619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell [Internet]. 2019;178:1189–1204 e23. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31442407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.. Ye L, Park JJ, Dong MB, Yang Q, Chow RD, Peng L, et al. In vivo CRISPR screening in CD8 T cells with AAV-Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nat Biotechnol. 2019/09/25. 2019;37:1302–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature. England; 2019;576:471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature [Internet]. 2018;558:307–12. Available from: https://www.ncbi.nlm.nih.gov/pubmed/29849141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Invest. United States; 2020;130:673–85. [DOI] [PMC free article] [PubMed] [Google Scholar]