

Abstract
Development of effective therapeutics for neurological disorders has historically been challenging partly due to lack of accurate model systems in which to investigate disease etiology and test new therapeutics at the pre-clinical stage. Human stem cells, particularly patient-derived induced pluripotent stem cells (iPSC) upon differentiation, have ability to recapitulate aspects of disease pathophysiology, and are increasingly recognized as robust scalable systems for drug discovery. We review advances in deriving cellular models of human central nervous system (CNS) disorders using iPSCs along with strategies for investigating disease-relevant phenotypes, translatable biomarkers and therapeutic targets. Given their potential to identify novel therapeutic targets and leads, we focus on phenotype-based, small-molecule screens employing human stem cell-derived models. Integrated efforts to assemble patient iPSC-derived cell models with deeply annotated clinicopathological data, along with molecular and drug-response signatures, may aid in stratification of patients, diagnostics and clinical trials success, shifting translational science and precision medicine approaches. A number of remaining challenges, including optimization of cost-effective, large-scale culture of iPSC-derived cell types, incorporation of aging into neuronal models, as well as robustness and automation of phenotypic assays to support quantitative drug efficacy, toxicity and metabolism testing workflows are covered. Continued advancement of the field is expected to help fully ‘humanize’ the process of CNS drug discovery.
Keywords: human induced pluripotent stem cells, neuroscience, psychiatric disorders, neurodegenerative disorders, screening, drug discovery
Introduction
Central nervous system (CNS) diseases represent one of the most challenging areas for drug development and successful clinical trials.1–3 Parallel to a general increase in human lifespan, the economic burden of aging-associated neurological diseases is on the rise, with more than 44 million people affected by dementia worldwide, including 5.4 million Americans affected by Alzheimer’s disease (AD).4–5 On the other end of the spectrum, epidemiological data on pediatric neurological diseases is more scarce, but high numbers are attributed to encephalopathy, epilepsy, cerebral palsy, autism spectrum disorders (ASD) and intellectual disability. Collectively, these diseases represents a significant proportion of global disease burden, contributing to early mortality and years lived with disability dependent on geographic location and socioeconomical factors.6 The heterogeneity of symptoms, age range and less than optimal diagnostic tools make CNS disorders difficult to study, diagnose and treat. While significant progress has been made for diseases like multiple sclerosis, stroke and cancer, the failure rate in neurodegenerative and psychiatric clinical trials is high, making the need for new research and innovation in drug discovery urgent.2–4, 7–8
Reliable disease biomarkers, which require understanding of disease-specific molecular profiles, can be used for diagnosis and can report on patient’s response to experimental therapeutics and successful drug development, but are not necessarily the same.1 However, the landscape of biomarkers in neurology, specifically in neurodegeneration, is modest due to an incomplete understanding of disease molecular mechanisms and how it relates to clinical symptomology and pathology. Also, for brain diseases, access to disease-affected tissue and samples is severely limited.1, 3 AD serves as one example of how biomarkers (e.g. positron emission tomography (PET) and cerebrospinal fluid (CSF) levels of amyloid-β and tau) have contributed to a better understanding of disease onset and progress. Here, collaborative, worldwide consortia (e.g. Alzheimer’s Disease Neuroimaging Initiative, ADNI), are shaping diagnostics as well as drug development.1, 9–10 Amyotrophic lateral sclerosis (ALS) serves as another example for which a range of measurable biomarkers are available, including neurophysiological measurements of motor neuron damage, imaging techniques (e.g. PET, functional magnetic resonance imaging) and CSF markers of blood–brain barrier dysfunction, axonal degeneration and neuroinflammation.11 Even so, early stage ALS is difficult to diagnose, with more than 20 years and 50 clinical trials since the only approved mildly efficacious drug, riluzole, was approved.12 For many other neurological diseases, post-mortem brain tissue is still the only source of definitive diagnosis, but it is not the best tool to study the mechanisms of disease etiology or course of pathology.
The norm is still a great reliance on studies using non-human genetically engineered model systems that address specific aspects of disease at a cellular or molecular level, but fail to develop other important phenotypes of human disease and that do not fully reflect drug efficacy.3 It is now clear that to succeed in CNS drug development, research findings need to be verified against accurate disease models. In this context, progress in the field of human stem cell research, and in particular implementation of patient-specific cellular models, is shifting translational medicine (Fig.1).13
Figure 1.
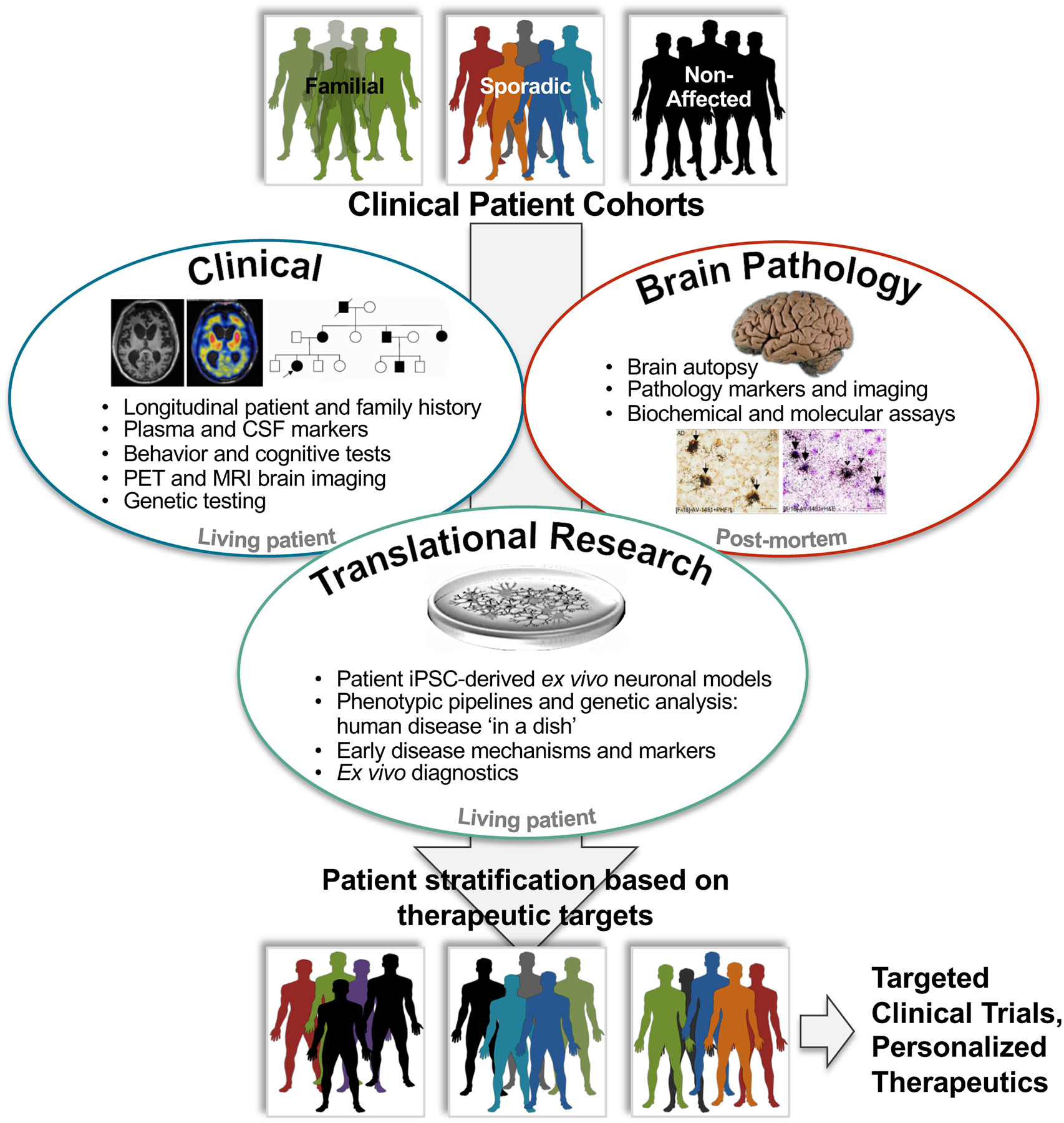
CNS research integrative approaches, between longitudinal clinical phenotyping information (top left, images from Ghetti et al. 2015), post-mortem brain pathology data (top right, images from Marquié et al. 2015) and patient iPSC-derived cellular models and research (bottom middle). Patient iPSC-derived cell models are a tool in the study of early disease molecular and cellular mechanisms, and therefore translational research. Results interpretation in the context of clinical and pathological data allows discerning the most relevant phenotypes, identification of biomarkers for pre-clinical and clinical diagnostics, as well as therapeutics development. Patient stratification based on clinical, pathological and iPSC molecular and cellular data aids in determining drug screening targets in defined subpopulations and may increase overall clinical trials success.
In 1998, Thomson et al. published for the first time a methodology for isolating and culturing human ESCs from blastocysts.14 These pluripotent cells, or embryonic stem cells (ESCs), showed capacity to differentiate into any cell type, however, the need to harvest cells from human embryos raised major ethical concerns. In 2006, Yamanaka and colleagues demonstrated that human mature somatic cells, such as dermal fibroblasts, could be reprogrammed into iPSC through cellular retroviral transduction of four key transcription factors: OCT3/4, SOX2, c-MYC, and KLF4, now commonly referred to as ‘Yamanaka factors’.15–16 In 2012 Dr. Shinya Yamanaka, and Dr. John Gurdon for his earlier work on the concept of reprogramming, were awarded the Nobel Prize in Physiology & Medicine. Subsequent progress has increased the efficiency and robustness of reprograming strategies from fibroblasts, blood, hair and endothelial cells (e.g. urine), as well as implementation of integration-free reprogramming with DNA plasmids or non-integrating Sendai virus,17–19 mRNA transcription factors, micro-RNAs (miRNA) and small molecule cocktails.17, 19–23 Human stem cells are proving to be fundamental tools in cell-replacement therapies, study of human neuro-development, and as genetically-accurate systems to study human disease.13, 24–36
Stem cells have the capacity for self-renewal in culture and are able, in theory, to differentiate into any cell type of the body, provided the appropriate culture conditions.15–16, 37 Given these properties, stem cells are highly relevant for the development of ex vivo models of human disease affecting tissues and cell types that are of a limited availability and cannot be accessed non-invasively, namely the nervous system. As advantages of certain technologies over others become more evident, continued advances focus on refining the robustness and accuracy of iPSC-derived cellular models. The urgent need for better therapies has also spiked interest in the use of patient-specific iPSC-derived cells in the drug discovery process, particularly in the early testing of efficacy and toxicity of new drug candidates, with the goal of increasing clinical trials success (Fig.2).38–39 With traditional approaches, transgenic animal and immortalized cell models are relied upon for drug screening, efficacy, toxicity and mode-of-action studies, and where the ‘human context’ is often only implemented later at the clinical trial phase. In contrast, patient iPSC-derived cell models fundamentally introduce the ‘human context’ early in the discovery pipeline at a pre-clinical phase. Genotypic and phenotypic molecular signatures, together with drug response profiles in a physiological- and disease-relevant context, fundamentally aid in understanding complex disease molecular mechanisms (Fig.2).
Figure 2.
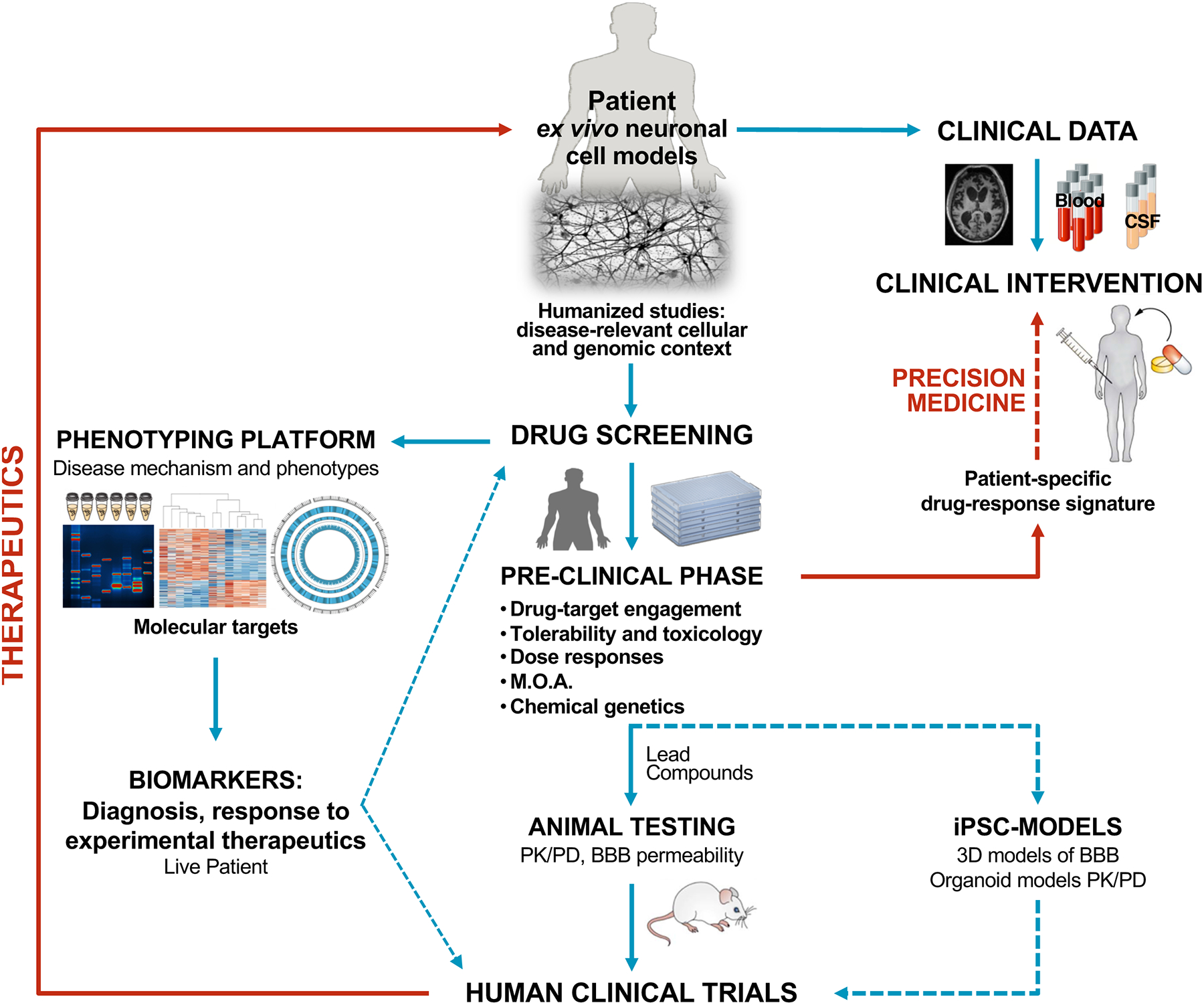
Proposed research pipeline, from human ex vivo models to in human clinical trials, with the goal of implementing human testing as early as possible. Patient-derived disease models focus on endogenous physiologically relevant proteins and molecular pathways, with associated genetic context. Coupled with extensive phenotypic analysis and validation across an increasing number of cell lines, iPSC-derived cells will have an important role in clinical diagnostics and identification of biomarkers that can guide drug screening as well as patient stratification for clinical trials. Most remarkably, iPSC-derived cells show increasing potential for small-molecule screening and secondary testing of novel therapeutics’ efficacy, toxicity and mode of action, in a human context and in advance of animal testing and human clinical trials. Novel human 3D models of BBB and organoids tissues like liver and kidneys may also be implemented in place of animal testing for PK/PD analysis. Predicted drug success vs. failure based on correlations derived from ex vivo cell models and patient-response will undoubtedly contribute to precision medicine efforts.
Here, we will review several approaches to generate patient iPSC-derived cellular models, genotypic and phenotypic methodologies to identify therapeutic targets, high throughput (HTS) and high content (HCS) approaches for drug screening, as well drug efficacy and toxicity testing. A number of key challenges will also be addressed.
Strategies for human ex vivo stem cell differentiation into neurons
To aid in the study of neurological diseases and for drug screening, human iPSCs are converted into the cell types of interest, i.e. the most relevant and affected cells in any particular disease. Generation of functionally specialized neural subtypes relies on manipulation of culture formats in the presence of specific factors that promote the conversion of pluripotent cells into neural progenitors, neurons and glia (Fig.3). Several protocols have been developed to differentiate human stem cells into neurons40–45 and microglia,46–48 but cellular maturation and aging for disease modeling are still challenging. Initially, many typically labor-intensive and time-consuming protocols that lacked scalability and reproducibility, gave rise to sparse numbers of neurons in culture, which was a major challenge for phenotypic studies or drug screening. Then, work such as the one by Li et al.45 showed that small molecule-based (GSK-3β, TGFβ and Notch inhibitors, plus LIF3) neuronal differentiation directly from stem cells, through high efficiency derivation of multipotent neural stem cells, could originate forebrain, midbrain and hindbrain neuronal and glial subtypes. Otherwise, to circumvent the low throughput associated with stem cells direct differentiation, methods relying on expansion of intermediate stages have been developed. Presently, two-dimensional (2D) or three-dimensional (3D) methodologies of neural differentiation show incredible potential for generation of suitable cultures for different goals of disease study and small molecule screens, examples of which are covered below (Fig.3). Notably, it is at the discretion of the investigator to judge the most suitable protocol for a specific disease model, depending on the molecular pathways possibly involved in disease that should not be affected by the protocol implemented.
Figure 3.
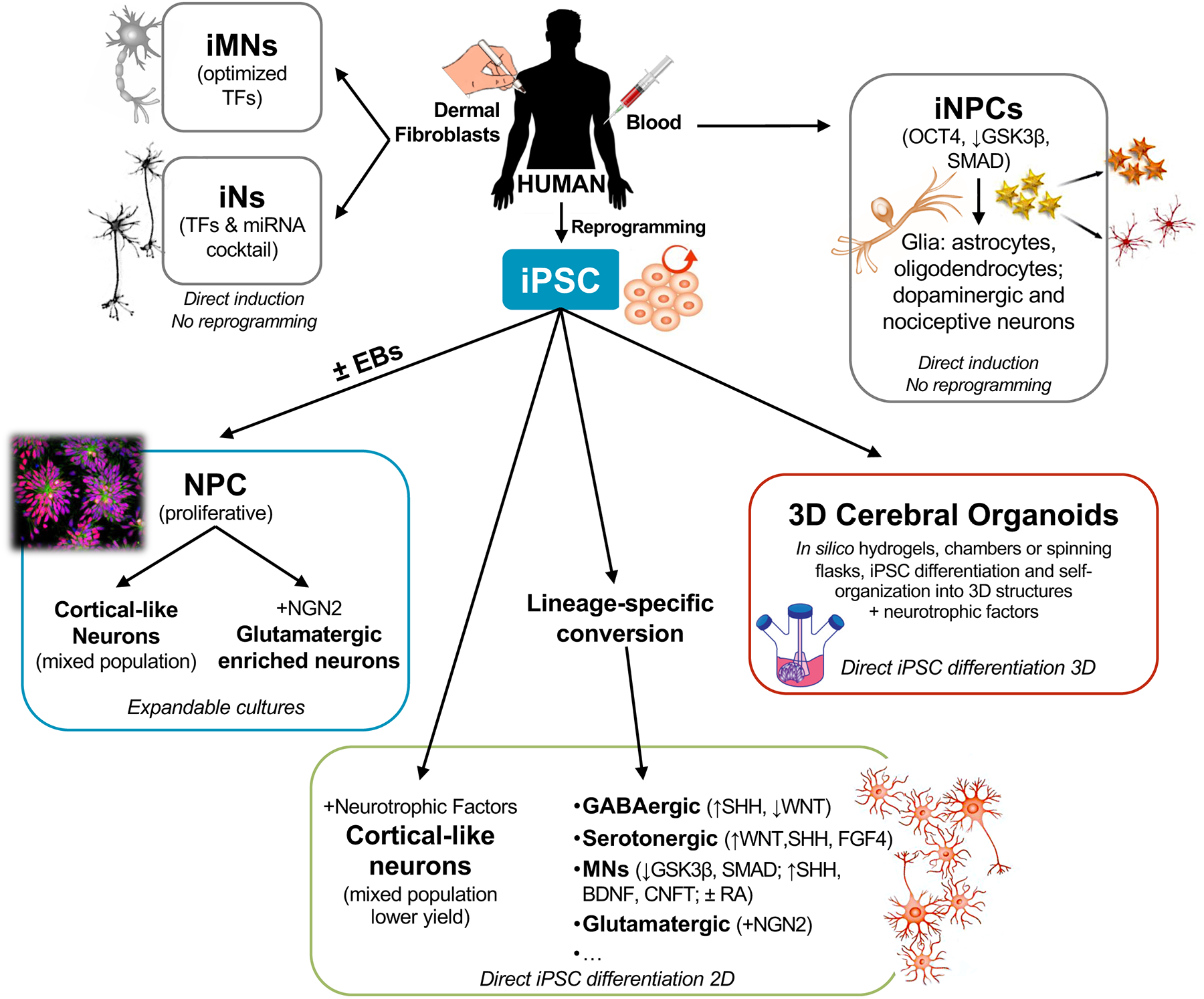
Summarized, non-exhaustive, schematic of the current landscape for human iPSC-derived or directed differentiation of somatic cells into neurons in 2D and 3D culture formats.
Neural progenitor cell-based differentiation
Stem cells can be differentiated through capture of an intermediate state of neural progenitor cells (NPCs) that consist of homogenous, expandable and self-renewable multipotent cells under a defined medium with growth factors (EGF, FGF).49–57 NPCs can then be differentiated into astrocytes, oligodendrocytes, and functional, electrically active neurons. A key breakthrough by Chambers et al. revealed that, early in iPSC differentiation, inhibition of BMP and TGFβ signaling, termed dual-SMAD inhibition, selectively blocked endoderm and mesodermal cell fates and significantly enriched cultures for neural ectoderm lineage progenitors, i.e., NPCs.49 Alternatively, iPSCs can form embryoid body-like aggregates (EBs) and subsequently neural rosette structures that are precursors of NPCs (Fig.3). Differentiation capacity of iPSCs can be enhanced by promoting the formation of EBs under non-adherent culture conditions58 or in microwell plates designed to promote uniform EB size, maximize neural rosette formation and NPC output (StemCell Technologies, Inc.).51, 54 Then, selection and dissociation of neural rosettes into a monolayer of NPCs can be further enriched for cortical precursors by fluorescence-activated cell sorting (FACS) of specific cell-surface markers (CD184-positive, CD133-positive and CD271-negative).51, 54, 56 This is important for producing CNS-enriched NPCs with high homogeneity and extensive proliferative capacity, over peripheral nervous system neural crest cells.50–51, 56, 59 These NPCs have been stably propagated in culture for up to one year,50 or at least 50 passages,51 maintaining differentiation potential and karyotype integrity.50, 54 By withdrawal of growth factors from the culture medium, differentiation can be initiated and maintained for more than 5 months, without selectivity for a specific neuronal subtype.51, 53–55 Alternatively, neural rosettes derived from EBs are dissociated into NPCs, passaged up to 3 – 4 times and immediately differentiated in media supplemented with neurotrophic factors (e.g. BDNF, GDNF).60 Although the latter is a quicker protocol, the neuronal count obtained is lower because of the earlier limitations in NPC propagation. Both methods originate mature neurons with production of strong action potentials. Variations of these protocols include elimination of EB formation, where iPSCs directly originate neural rosettes and NPCs, and subsequently somewhat scalable cultures of mainly glutamatergic neurons in a total of 8 weeks.61 A drawback of these methods is that neuronal maturation requires a period of more than 7 months, and cells need to be re-plated due to loss of plate-coating adherence, relying then on the regenerative capacity of each cell line to continue differentiating.62
Directly induced neurons
One limitation associated with iPSCs is reprogramming itself, which has led to the development of methods of direct conversion of somatic cells, like dermal fibroblasts, into induced neurons (iNs). This is accomplished by overexpression of a set of transcription factors33, 63–66 or miRNAs23, 43, 67 that promote chromatin remodeling (transcriptional programming) and drive direct neural lineage differentiation, bypassing the need for iPSC generation (Fig.3). Perhaps one of the most attractive advantages might be that bypassing reprogramming reduces disruption of epigenetic marks associated with the age of the somatic cells, allowing to create neuronal models at “pathogenic ages”.68 The ability to recapitulate age-related characteristics of human neurons in culture has been demonstrated by conversion of fibroblasts from postnatal and near centenarian donors. Direct comparison of the resulting iNs demonstrated that multiple age-associated marks were preserved, including DNA methylation patterns, transcriptomic and microRNA profiles, oxidative stress, DNA damage (loss of heterochromatin and nuclear organization) and telomere length, and were highly predictive of the age of the donor fibroblasts.69 Transdifferentiation of patient somatic cells (fibroblasts) into induced motor neurons (iMNs) has also been done successfully, through transgenic co-expression of seven to eight transcription factors (BRN2, ASCL, MYT1l, LHX3, ISL1, NGN2, HB9).41, 70 iMNs also display unique age-related cellular characteristics not observed in iPSC-derived MNs. Moreover, co-culture with ALS patient-derived glial cells reveals drastic phenotypes such as cell death, suggesting that this system is suitable for modeling late-onset pathogenesis.41, 70 Alternatively, Lee et al.71 developed a strategy to generate induced NPCs (iNPCs) directly from neonatal and adult peripheral blood using a single-factor OCT4 reprogramming with dual-SMAD and GSK-3β inhibition. Blood-derived iNPCs were successfully differentiated into glia (astrocytes and oligodendrocytes) and both CNS (dopaminergic) and peripheral nociceptive neurons. More recently, Miskinyte et al.72 demonstrated that BRN2, MYT1L and FEZF2 transcription factors can convert fibroblasts into functional excitatory cortical neurons with electrophysiological properties, pyramidal-like cell morphology and expression of cortical projection neuronal markers. Concomitant treatment with small molecules and microRNAs was able to increase initial efficiency by 16%. By combining directed differentiation and transcription factor reprogramming, Nehme et al.73 showed that NGN2 together with SMAD and WNT inhibition generates iNs of mixed differentiation states, ranging from NPC-like to mature excitatory glutamatergic neurons with measurable AMPAR and NMDAR-mediated synaptic transmission.
While undoubtedly promising, technical restrictions of iNs include genomic instability resulting from increased somatic cell expansion requirement (need for larger starting number of cells), deficient maturation and relatively low yields. All of these are limiting factors for routine or large-scale studies, such as drug screening.
iPSC-derived neuronal subtypes and co-culture systems
The necessity to study neuronal-specific mechanisms of pathology has contributed to significant advances in creating robust and scalable protocols for derivation of specific neuronal types, many based on overexpression of different transcription factor combinations (Fig.3).33, 74 Still, more “mixed” types of cultures and co-cultures allow dissecting differential phenotypes within a population of neurons, without losing possible cell non-autonomous mechanisms of disease etiology (e.g. glia contribution to neuronal death in ALS).75–76
Regarding brain region specificity, Sarkar et al.77 established a protocol to generate hippocampal-patterned NPCs (by co-inhibition of WNT, TGFß, SHH and BMP signaling pathways) and differentiate CA3 pyramidal neurons by treatment with recombinant WNT3A, BDNF, ascorbic acid and cAMP. The resulting cells included secretagogin (SCGN)-expressing CA3 neurons, which were employed to study dendate gyrus CA3 mossy fiber electrophysiological activity deficits in schizophrenia models. Meanwhile, Rajamani et al.78 optimized a chemically-based inducing media, without the standard neuronal supplements, to generate hypothalamic-like neurons from human iPSCs, in a three-phase method that encompasses specification of neural ectoderm, patterning toward ventral diencephalon, and maturation of hypothalamic-like neurons. The formed cells were shown to express hypothalamus genetic- and protein-specific markers, validating this cellular system to study neuronal dysfunction in anorexia nervosa, diabetes, obesity and anxiety disorder.
With relevance to several neuropsychiatric diseases, iPSC can be differentiated through medial ganglionic eminence progenitors into GABAergic interneurons, using a chemical-based system,79 or sonic hedgehog (SHH) enhancers in concert with WNT suppressors.80–81 Within approximately 25 days of differentiation, neurons express ventral (ISL1, OLIG2, ASCL1), forebrain (FOXG1), and GABAergic markers (LHX8, LHX6).82–83 Also, iPSCs can be converted into rostral hindbrain neural stem cells and differentiated into serotonin neurons by activating simultaneously WNT (GSK-3β inhibition), SHH and FGF4 signaling pathways with a >60% success rate.84–85 Zhang et al.22 developed a protocol for human ESCs/iPSCs conversion into glutamatergic neuronal cultures with a “nearly 100% yield”, through stable overexpression of a single transcription factor, neurogenin-2 (iNGN2), that converts stem cells or NPCs into excitatory glutamatergic forebrain neurons.22, 51, 86 The resulting cells express neuronal-specific genes and morphology as early as 2 weeks, and are electrophisiologically responsive to glutamate receptor antagonists within 3 weeks.22 This method has already demonstrated an incredible consistency and ease of use in large-scale production of neuronal cultures for small molecule HTS assays (Cheng C. and Haggarty S.J. et al. in preparation).86
To study motor neuron degenerative diseases such as ALS and spinal muscular atrophy (SMA), a number of protocols have been developed to differentiate ESCs/iPSCs into functional corticospinal motor neurons (MNs).87–88 Directing pluripotent stem cells toward MN fate relies on the use of small molecules and recombinant signaling molecules, in three main phases: neuralization through GSK-3β and dual-SMAD inhibition, caudalization with retinoic acid (SHH agonist) and ventralization by SHH activation. Then, MN progenitors rely on neurotrophic factors (GDNF, BDNF, CNTF) for axon projection and differentiation into MNs.88–89 Within 2–4 weeks electrophisiologically active, HB9+/ISL1+ MNs can be detected.49, 87, 90 To further increase efficiency, Hester et al. coupled direct differentiation by overexpression of LHX3, ISL1 and NGN2 (LIN factors), with RA and SHH,91 enabling differentiation of mature MNs with measurable electrophysiological activity at 11 days. Dedicated literature on MNs generation clearly suggests that subtle differences in timing, cell plating and media composition can strongly influence yield, purity and phenotype.87–88, 90, 92
Neuronal-only cultures are limited in their ability to accurately re-capitulate network functionality, neuronal development, pruning and activity. In this regard, glial cells, including astrocytes, oligodendrocytes and microglia, are of great importance.48, 93–94 For example in ALS, the extent to which neuronal death is an intrinsic process exacerbated by pathological interactions with other surrounding cell types, such as microglia and astroglia, has become an area of intense investigation.75 Eggan et al. pioneered a method for large-scale production of MNs from hESCs in a co-culture system with glial cells to study ALS;75–76 while others have focused on astrocytes co-cultures with human iPSC-derived neurons, with promising results for maturation and synaptic function.56
iPSC-derived three-dimensional cerebral organoids
Human neurons differentiated in 2D formats have limited spatial network organization, have restricted inter-cellular interactions, and have reduced cell-extracellular matrix interactions, and in this regard, do not recapitulate brain physiological characteristics. To circumvent these limitations, over the past decade there has been a great effort in deriving 3D neural cultures, termed organoids (Fig.3).24–25, 95–99 Basal hydrogels and culture chambers that attempt to recreate the physics of the brain environment have been used to promote iPSCs differentiation into 3D structures.24–25, 100–101 Based on the premises that iPSCs have self-organizing capacity, Lancaster et al. developed the first protocol for growing human iPSC-matrigel droplets and then transfer these 3D neural structures into spinning bioreactors to strengthen the exchange of nutrients and oxygen across the cell matrix.25, 97 In the 3D microenvironment, cell-to-cell and cell-to-extracellular matrix interactions influence spatial arrangement, as well as cellular functions such as proliferation, differentiation, morphology, gene and protein expression, and cellular responses to external stimuli that may be more relevant when modeling brain disease.102 Organoids have also been shown to allow faster neuronal maturation and higher degree of cellular diversity, with more rapid formation of dendritic spines and spontaneously active neuronal activity, recapitulating key aspects of cortical cellular identity and multi-regional architecture, including an outer radial glia population similar to human early fetal brain.29 However, despite an impressive self-organization of layered cellular structures, organoids generally lack GABA interneurons, and organoid-to-organoid heterogeneity is still high without reproducible anatomical structures that resemble in vivo tissue spatial distribution.103 Also, the cells in the core of the organoids tend to reveal some necrosis and loss of differentiation capacity after about 3 months in culture, due to the lack of nutrients and gas exchange.25, 104
The utility of organoids for disease model phenotyping and drug screening depends upon robust and reproducible standardized protocols for generating homogeneous populations of organoids at large scale. For this reason, investment has been made towards increasing homogeneity and scalability of 3D cultures for long-term cell maintenance and maturation, using miniaturized plate-based spinning bioreactors.97, 104–106 Rigamonti et al.62 described a method for large-scale production of “high purity” cerebral organoids, maintained for an extensive period of time (>40 days) in suspension in spinner flasks, originating mature and electrophisiologically functional cortical and spinal cord MNs at a scale amenable to drug screening. These spin cultures dissociated spheres could then be further differentiated in monolayer cultures for as long as 4 months. Meanwhile, Sloan et al.107 developed a strategy to generate astrocytic cells in 3D, starting from iPSC-derived cerebral cortical spheroids. Through elegant strategies of cell purification and selection up to 20 months in vitro, the resulting glia cells closely resemble primary human fetal astrocytes and, over time, transitioned from a predominantly fetal to an increasingly mature astrocyte state, concomitant with phagocytic capacity and calcium signaling.
Medical research and therapeutic potential of organoids have been areas of great interest, with the offer of new platforms for brain neurological disorders investigation, regenerative medicine and drug testing.97, 105, 108 The expectation is that drug testing in disease-specific organoids may not only better recapitulate patient’s response and tolerability but could, potentially, exclude the use of animal testing where many drugs seem efficacious only to later fail in clinical trials.3, 24–25, 109–110 Still, 3D cerebral organoids lack vasculature, which plays a vital role in neurophysiology and drug delivery.
Stem cell-derived models advancing the study of human CNS disorders
A major obstacle in studying human CNS diseases has been the inaccessibility to diseased tissue or other specimens for research, especially if it requires routine patient sample collection. Even in cases where tissue is available, terminally differentiated cells such as neurons cannot be maintained nor propagated in culture, prohibiting the possibility of experimental repetition and scalability. Conversely, readily accessible tissues, such as lymphocytes, do not necessarily recapitulate the molecular pathways and proteins involved in disease. Traditional approaches have relied on transgenic animals and immortalized cell lines, through overexpression of human transgenes predicted to be causal to disease, in a particular cell-type amenable to phenotypic analysis. These have been instrumental in identification of genetic and protein causal relationships to specific phenotypes and have led experimental therapeutics through pre-clinical efficacy and safety tests. But sole reliance on these model systems has had poor success and led frequently to high-cost failed human clinical trials.1, 5, 30 This is thought to be the result of poor translation of phenotypic findings and small molecule properties in physiological and genomic heterologous model systems. This translational gap is starting to be addressed by human iPSC-derived model systems that allow access to physiologically relevant cell types and protein complexes without the need for overexpression of heterologous genes. Thus, patient iPSC-derived neurons represent unique ex vivo neuronal 2D or 3D networks that allow investigating disease-relevant genetic and molecular pathways in endogenous and physiological cellular microenvironments.24, 30, 97, 111 By taking into account the patient genomic background, one can generate both familial and sporadic forms of disease models, without need to have a priori knowledge of the genetic cause to create a useful model to study disease.27, 112–114 By recapitulating molecular and cellular phenotypic aspects of the disease ex vivo, iPSC-derived neuronal and glial cells have started to show great impact in the understanding of particular aspects of neurological diseases. Furthermore, genetic manipulation techniques, such as mediated by clustered regularly interspaced short palindromic repeats (CRISPR)-associated protein 9 (Cas9), have also been employed to create isogenic iPSC lines, allowing the elucidation of disease and risk alleles effect on molecular and cellular phenotypes.115 A comprehensive description of the multitude of human iPSC-derived CNS disease models can be found in focused literature,24, 33, 35, 52, 116–126 with key examples mentioned bellow.
Neurodegenerative disorders
Neurodegenerative diseases are characterized by age-dependent focal, i.e. with regional and neuronal type specific vulnerability, and progressive loss of neuronal structure and function, characteristically associated with the presence of aberrant protein inclusions in the affected regions of the brain.127–128 Protein aggregation can result from a mutation(s) in a disease-related gene(s), leading to expression and accumulation of aberrant misfolded proteins in the cell or, in the absence of genetic alterations, it is thought to be the result of environmental stressors (e.g. traumatic brain injury) and aging.129–131 The relative percentages of sporadic and familial cases are different amongst diseases such as frontotemporal dementia (FTD), ALS and Parkinson’s disease (PD), whereas, for instance, Huntington’s disease (HD) is inherited in a fully autosomal dominant manner.13, 129, 132–136 Although the principal gene and protein associated with each disease are thought to account for the majority of pathological phenotypes, the underlying genetics not always match the same phenotypes, diagnosis or pathology, due to additional gene loci and rare variants that confer different disease vulnerability and progression.132–133, 135–143 Moreover, a growing body of evidence suggests that preceding the formation of late stage protein inclusions, misfolded monomers and oligomeric species initiate molecular events of pathway disruption and neuronal toxicity, and might be the actual triggers of neuronal death.127, 130, 144 In fact, model systems and emerging clinical data, such as CSF biomarkers and PET imaging that can detect brain alterations ahead of symptomology, have revealed that cellular and biochemical alterations occur at a pre-symptomatic stage, pointing towards the period in the disease when treatment will ultimately prove critical.145–149 Therefore, a better understanding of the early biochemistry and biophysics of protein conformational changes and aberrant interactions with cellular pathways and toxicity, in patient-specific model systems, is indispensable to understand disease and to determine which mechanisms should be exploited for drug discovery. The effort to build large patient-derived cell line cohorts and integrative phenotyping pipelines, will determine relevant targets for therapeutics (Figs.1, 2). Patient or gene mutation-positive iPSCs have now shown to recapitulate disease-relevant phenotypes, as corroborated by post-mortem pathology and CSF markers, providing insight into early disease mechanisms and common processes of neurodegeneration across diseases.33, 54, 114, 150–157
Stem cell-derived models have been widely employed to study AD and FTD. AD is characterized by the presence of Aβ plaques and neurofibrillary tau tangles in areas of the neocortex and hippocampus, where primarily pyramidal, glutamatergic neurons seem to be affected. So, a major goal has been to generate patient-specific glutamatergic neuronal models. 2D and 3D AD models are extensively reviewed by Arber et al.158 Phenotypic observations include Aβ accumulation and toxicity in the ER, endosomes and lysosomes, oxidative stress and swollen early endosomes in the cell body of patient-derived neurons as well as astrocytes, which could be reversed by γ-secretase inhibitory drugs and docosahexaenoic acid, without affecting Aβ levels.113–114, 159 Differentially regulated genes identified in sporadic AD iPSC-derived neurons were also validated in post-mortem brain, testifying to the validity of the cellular model. AD 3D organoids, either expressing familial AD mutations (PSEN1, APP) or sporadic iPSC-derived, revealed Aβ40 aggregates together with phosphorylated, insoluble, silver-positive tau aggregates, and endosome abnormalities, after long-term culture and for the first time in an AD cellular model.108, 160 Treatment with β- and γ-secretase inhibitors reduced Aβ and insoluble tau, whereas GSK-3β inhibitors reduced phosphorylated tau levels, suggesting suitability of 3D models for drug screening.
Familial FTD genes include MAPT and GRN, intronic expansion in C9orf72, and less frequently TARDBP, FUS, CHMP2B and VCP. Neuropathologically, there are characteristic patterns of abnormal deposition of tau, TDP-43, FUS and ubiquitin proteins in neurons and/or glia in the affected regions of the brain.126 Several reports have described neuronal phenotypes in patient-derived cells with tau mutations, including premature maturation;153 increased oxidative stress and activation of the unfolded protein response;151 and upregulation of different forms of tau (cleaved, oligomeric and detergent-insoluble), with somatodendritic tau redistribution and increased vulnerability to stress.54, 161 Cerebral organoids generated from an FTD patient iPSC with a tau P301L mutation exhibited increased levels of p25.162 CRISPR-mediated inhibition of p35 conversion into p25 resulted in lower levels of total tau and phosphorylated tau, together with increase in synaptophysin levels, suggestive of a rescue in synapse formation in 2-month-old organoids. These results propose new mechanisms of tau-mediated neuronal toxicity through p25.162 Meanwhile, the first study of progranulin (PGRN)-deficient FTD using iPSC-derived neurons, revealed a ~50% decrease in levels of both secreted and intracellular PGRN protein, recapitulating the haploinsufficiency disease phenotype, as well as compromised PI3K/AKT and MEK/MAPK signaling pathways, which were rescued by overexpression of wild-type PGRN.163 Considerable research has also focused on C9orf72 expansions (GGGGCC intronic repeats), shown to form RNA aggregates in the nuclei of cortex and motor neurons. In the first C9-FTD iPSC study, patient-derived neurons recapitulated aspects of C9orf72 pathology such as high levels of p62 protein and compromised autophagy.164 Similarly, C9orf72 MNs revealed disruption of the lysosomal pathway, increase in glutamate receptors and decreased viability.165–166 Analysis of electrophysiological properties of either C9orf72 or mutant TARDBP MNs also showed abnormal patterns of synaptic activity.167–168 The majority of TDP-43 iPSC models focus on FTD-ALS and cell-autonomous phenotypes in MNs or glia, showing increased levels and mislocalization of soluble and insoluble TDP-43, decreased survival, neuritic abnormalities, and increased vulnerability of the PI3K pathway.169–170 DiGiogio et al.75 were the first to show that human ESC-derived MNs are selectively sensitive to the toxic effect of glial cells carrying an ALS-causing mutation in SOD1 (superoxide dismutase 1), revealing cell non-autonomous effects of pathology.
Patient iPSC-derived models of HD have also been attempted, but the differentiated neurons do not exhibit neuropathological phenotypes (e.g. mutant huntingtin/HTT aggregates) or compromised survival, unless they are treated with stressors such as proteasome inhibitors or kept in culture for very long periods.171 Conversely, directly induced striatal medium spinal neurons from fibroblasts of HD patients did recapitulate age-associated disease signatures, HTT aggregation and neurodegeneration, supporting the potential for pathology modelling using iNs of HD patients.172
Neurodevelopmental and psychiatric disorders
The current understanding of the pathophysiology of psychiatric diseases remains limited, and because these are highly polygenic in nature and influenced by environmental factors,173–174 the genetic and molecular causes of disease are challenging to uncover.30, 175 Conventional approaches have relied on post-mortem brain analysis,176 but these tissue samples not only have a highly stringent requirement for acquisition and cannot be investigated under flexible experimental conditions, they represented end stage pathology, which is highly confounded by long-term medications and individuals’ lifestyle, and consequently may not represent disease-specific deficits. It is also very difficult to create genetically accurate models of psychiatric disorders in animals, with neuroanatomical differences representing real challenges for extrapolation of phenotypes177–178. Patient iPSC-derived NPCs, neurons and glia have paved new opportunities for research, drug discovery and potentially personalized medicine in this area.179 These models capture the complex genetic architecture of polygenic susceptibility, and allow epigenetic, chemical, proteomic and signaling pathways dysregulation to be measured in a cell-specific manner, helping dissect underlying disease molecular and cellular phenotypes,26, 53, 177, 180–181 which can then be determined to be permissive to disease state or not through clinical cohort-based data (Fig.1).179, 182 Following the selection of patients and control subjects, and the generation of iPSC lines, the next step is to determine the cell type of interest for targeted differentiation. Several neural subtypes have been linked to pathology from postmortem analyses of patients (GABAergic, dopaminergic, serotonergic and glutamatergic), as well as particular neural regions (hippocampus and prefrontal cortex).
Schizophrenia was the first complex psychiatric disorder to be modeled using human iPSCs, leading to the first reports of reduction in neuronal connectivity in patient-derived neurons.183–185 Schizophrenia NPCs also exhibit elevated levels of reactive oxygen species,183 consistent with proteomic analysis of brain tissue that revealed disruption of mitochondrial function and oxidative stress186. These were also the first neuronal models where antipsychotic drugs were tested and some were found to rescue neuronal phenotypes ex vivo.183 Soon after, other reports of patient iPSC-derived models emerged for ASD, bipolar disorder (BD)187–190, attention-deficit hyperactivity disorder (ADHD), fragile X syndrome,53, 191 and Rett syndrome;192–194 focusing either on genetically defined or unknown genetics patients. Overall these studies revealed major neural developmental and aberrant differentiation patterns, alterations in gene and protein expression,27 decreased neuronal connectivity and synaptic dysfunction,195 as well as the recognition of DISC1 as a risk gene for psychiatric disorders.182, 184, 196 These models have provided an unprecedented foundation for phenotypic assays of disease biology and for functional chemical screens.177 Mariani et al.197 used forebrain organoids to study idiopathic ASD, strongly linked to dysregulated neurogenesis, and found an enrichment of inhibitory neurons likely as a consequence of increased FOXG1 expression, abnormal synapses and neurites. With access to larger cohorts of patients iPSC-derived cell models, common disease phenotypes, molecular and gene expression changes are starting to emerge and guide screens of disease-modifying or corrective therapeutics (Fig.1).26–27, 35, 44, 53, 177, 179–181, 183, 189, 193
Down Syndrome (DS or trisomy 21), the most common genetic disorder of intellectual disability, exhibits fewer interneurons in the cerebral cortex and has unknown underlying cause. A recent study tested the hypothesis that fewer cortical interneurons are a result of decreased production, proliferation, and/or migration of medial ganglionic eminence NPCs.198 This study compared forebrain GABAergic interneurons differentiated from DS and control iPSCs, and found critical structural and molecular abnormalities in DS GABAergic interneurons, including smaller size cells and fewer processes, possibly contributing to the smaller brain size and impaired cognition.
Organoids are a versatile system to study brain development and functional networks, serving as 3D models of corticogenesis, further allowing the study of the contribution of environmental factors to neurodevelopment.98 Leveraging these features, organoids have been used to model microcephaly, a neurodevelopment disorder in some cases caused by genetic mutations or by Zika virus exposure.25, 199 Organoids derived from microcephaly patients harboring truncating mutations of CDK5RAP2 were found to be significantly smaller than the ones generated from controls, associated with ‘precocious’ neural differentiation.
Transformation of neural progenitor cells and cancer
Stem cells are a hallmark of cancer biology, but understanding brain cancer pathogenesis has been hindered by limited access to samples, tumor heterogeneity and the lack of reliable model systems.200 As an example, glioblastoma (GBM) is the most aggressive type of malignant brain tumor with no cure and a bleak prognosis.201 Research shows that GBMs are derived from cells with features of neural stem and progenitor cells, which also contribute to treatment resistance and disease relapse.202 In fact, glioma tumor cells originate from transformation of NPCs and it is possible to propagate these cells, in defined, feeder-free, adherent cultures,203 even if isolation of these cells from patients remains restricted to tissue obtained at surgery or post-mortem.201 Both primary and NPC-derived GBM cell lines represent human disease models to study genetics, epigenetics and biology of cell propagation, and to execute drug or genetic screens to identify tumor-specific vulnerabilities.201 For example, while tumor-associated genetic alterations have now been well documented,203 the role of epigenetic alterations is not so well understood. Thus, by combining reprogramming technologies that introduce epigenome changes without affecting the genomic sequence, and CRISPR-based genome editing, researchers can now precisely study genetic and epigenetic effects in GBM-patient iPSC or NPC-derived, and isogenic matched neural stem cells.204
Phenotypic analysis of patient ex vivo neuronal models: disease etiology, biomarkers and therapeutic targets
CNS disorders cellular models need to replicate disease phenotypes to an extent that confidently predicts translation of a given target to clinically relevant features of illness. Incorporation of human ex vivo cellular assays at the earliest stages of drug discovery may improve the likelihood of successfully translating preclinical discoveries into clinical trials success (Fig.2). This is how iPSC-derived models are starting to impact the drug discovery process. As reviewed above, scalable cultures of iPSC- and NPC-derived neurons and cerebral organoids are valuable phenotypic platforms for testing genotype-phenotype correlations in complex genetic disorders, and to support approaches to human chemical neurobiology and drug screening.205–206 So far, expandable NPCs might be the best platform to provide large scale and consistent sampling of human neurons compatible with HTS, proteomic and genomic studies, with NGN2-derived neurons showing advantages for HTS of small molecules libraries (Table 1).13, 26, 51, 55, 177, 207 State-of-the-art phenotyping assays, including functional genomics, quantitative biochemistry and proteomics, and high-content multiplexed screening assays, offer an unprecedented potential for understanding human disease (Fig.4).26, 38, 97, 105, 152, 177, 206, 208 These approaches have also benefited from accessible libraries of small molecules and oligonucleotides that allow probing disease-related genes, proteins and molecular pathways, towards identification of valid biomarkers. When employed consistently across patient neuronal models, this will lead to cumulative evidence of the molecular and cellular alterations most-relevant to disease and, therefore, of the targets with highest potential for a positive therapeutic outcome (Fig.2). These same assays are important to implement downstream in the validation of novel pharmacological agents.
Table 1.
Representative examples of low- and high-throughput drug screens with relevance to CNS disorders, using human ESC/iPSC model systems. Cases are presented in chronological order to emphasize the evolution of the methodologies.
| Model | Screening Strategy | Experimental Drugs, Library | Lead Hits | Reference |
|---|---|---|---|---|
| ESC survival | Human ESC ↑ survival in vitro | Candidate approach | ROCK inhibitor Y-27632 | Watanabe K. et al. (2007) Nature Biotech. |
| ESC proliferation and differentiation | Human ESC self-renewal and differentiation based on OCT4/NANOG markers | 2,880 small molecules (known bioactive and 748 FDA-approved) | 22 activators of proliferation/differentiation (THEA, SNM, GTFX, FBP, RA, SLG, CYM, SRM) | Desbordes S.C. et al. (2008) Cell Stem Cell |
| Human ESC fate | ↑ differentiation and viability | 1,040 compounds | 22 hits (e.g. steroids, pinacidil) | Barbaric I. et al. (2010) Stem Cell Res. |
| ESC-derived neurons potentiation | AMPA glutamatergic neuronal potentiation | 2.4 million compounds | 37 hits, 2 novel molecular series (CE-382349) | McNeish J. et al. (2010) JBC |
| SMA patient fibroblasts and iPSC-derived MNs | Rescue of MNs survival | 3,500 bioactive compounds | 188 hits (RTK-PI3K-AKT-GSK-3 signaling modulators) | Makhortova N.R. et al. (2011) Nat Chem Biology |
| Schizophrenia patient iPSC-derived neurons | ↑ neuronal synaptic markers, electrophysiology | Candidate approach | Loxapine | Brennand K.J. et al. (2011) Nature |
| Psychiatry patient iPSC-derived NPCs | Rescue of WNT signaling (WNT reporter assay) | 1,500 FDA-approved and known bioactive compounds | 45 hits (Riluzole, Trifluridine, Kaempferol, FG7142) | Zhao W.N. et al. (2012) J Biomol Screen |
| Familial dysautonomia patient iPSC-derived neural-crest precursors | Rescue of IKBKAP gene expression | 6,912 compounds | 8 hits SKF-86466 | Lee G. et al. (2012) Nature Biotechnology |
| ALS (TDP-43 or sporadic) patient iPSC-derived MNs | Rescue of MN phenotypes | Candidate approach | Anacardic acid | Egawa N. et al. (2012) Sci Transl Med |
| Human GBM stem cells | ↓Glioma stem cells proliferation | 2,000 compounds | 78 hits Disulfiram (DSF) | Hothi P. et al. (2012) Oncotarget |
| Human GBM-derived neural stem cells | ↓proliferation, induced mitotic arrest | Candidate approach | PLK1 inhibitor JNJ-10198409 (J101) | Danovi D. et al. (2013) PLoS One |
| iPSC-derived neural stem cells viability | Modulators of proliferation and viability | 1,000 compounds | 5 hits (Cdk-2 modulator) | McLaren D. et al. (2013) J Biomol Screen |
| Human iPSC-derived forebrain neurons +Aβ42 aggregates (AD) | Rescue of Aβ42 toxicity (viability) and electrophysiology | GSK proprietary compound library | 19 hits (CDK2 inhibitor) | Xu X. et al. (2013) Stem Cell Res. |
| AD patients (familial and sporadic) iPSC-derived neurons | ↓Aβ oligomers-induced ER and oxidative stress | Candidate approach | BSI (β-secretase inhibitor IV), DHA (docosahexaenoic acid) | Kondo T. et al. (2013) Cell Stem Cell |
| Sporadic ALS patients’ iPSC-derived MNs | ↓TDP-43 aggregation | 1757 bioactive compounds | CDK inhibitors JNK inhibitors Triptolide, Cardiac glycosides | Burkhardt M.F. et al. (2013) Mol and Cellul Neuroscience |
| iPSC-derived dopaminergic neurons; MTT+ and rotenone PD-like models | ↑viability | Candidate approach 44 compounds | 16 hits (indomethacin, nicotine, EGCG, resveratrol, taurine) | Peng J. et al. (2013) J Biomol Screen |
| PD patient iPSC-derived cortical neurons with α-Syn(A53T) mutation | Rescue of ER processing, ↓nitric oxide levels | Hit validation (yeast screen >180,000 small molecules) | NAB2 | Chung C.Y. et al. (2013) Science |
| ALS (familial and sporadic) patient iPSC-derived MNs | Rescue of hyperexcitability | Candidate approach | Retigabine | Wainger B.J. et al. (2014) Cell Rep. |
| Fragile X Syndrome patient iPSC-derived neural cells | ↑FMR1/FMRP expression | 5,000 bioactive compounds | 6 hits | Kumari D. et al. (2015) Stem Cells Transl Med |
| Fragile X Syndrome patient iPSC-derived NPCs | ↑FMR1 expression | 50,000 compounds | 790 hits | Kaufmann M. et al. (2015) J Biomol Screen |
| Patient-derived GBM cell lines, adherent or neurosphere-grown | Growth inhibition | Candidate approach Drug combinations | Bortezomib ABT-263+AZD-8055 | Quartararo C.E. et al. (2015) ACS Med Chem Lett |
| ASD SHANK3 haploinsufficiency iPSC-derived neurons | Rescue of neurite length and synaptic function | 202 compounds | 2 hits Lithium, Valproic acid | Darville H. et al. (2016) EBioMedicine |
| Friedreich Ataxia patients’ iPSC-derived neurons | ↑FXN (frataxin) | Hypothesis test | HDAC inhibitor 109 | Codazzi F. et al. (2016) Hum Mol Genet |
| FTD-PGRN (sporadic or PGRN-S116X) patient iPSC-derived cortical neurons | ↑PGRN expression and secretion | Proof-of-concept | SAHA | Almeida S. et al. (2016) Neurobiol Aging |
| FTD-PGRN fibroblasts and iPSC-derived neurons, from GRN mutation carriers | ↑PGRN expression | Candidate approach Autophagy modulators | Trehalose | Holler C.J. et al. (2016) Mol Neurodegener |
| FTD Tau-A152T patient iPSC-derived neurons | ↓Tau, rescue of neuronal stress vulnerability | Proof-of-concept | Rapamycin | Silva M.C. et al. (2016) Stem Cell Reports |
| ESC-derived astrocytes oxidative stress model (hydrogen peroxide-induced) | Protection against oxidative stress | 4,100 bioactive and approved drugs | 9 hits (Norcantharidin, Tyrphostin A1, Oxyphenbutazone, Enzastaurin) | Thorne N. et al. (2016) Stem Cells Transl Med |
| Gaucher disease (± Parkinsonism) patients’ iPSC-derived macrophages and dopaminergic neurons | ↑glucocerebrosidase levels and activity, ↓glycolipid storage, ↓α-Syn in dopaminergic neurons | Hit validation | NCGC607 (small molecule chaperone) | Aflaki E. et al. (2016) J Neurosci |
| SMA patient iPSC-derived MNs and astrocytes | ↑SMN expression, ↑dendrite and axon development | Candidate Approach (drugs in Clinical trial) | Top hit: TRH analog, 5-oxo-l-prolyl-l-histidyl-l-prolinamide | Ohuchi K. et al. (2016) Stem Cells Transl Med |
| ALS SOD1-L144FVX patient iPSC-derived MNs | Rescue of SODl-mediated neuronal cell death | 1416 FDA-approved drugs or in clinical trial | Bosutinib | Imamura K. et al. (2017) Sci Transl Med |
| ALS SOD1-E100G patient iPSC-derived MNs | ↑MN survival | Candidate approach | FR180204, Pifithrin-α hydrobromide, SB203580, SP600125, XAV 939 | Bhinge A. et al. (2017) Stem Cell Reports |
| AD PSEN1-G384A patient iPSC-derived cortical neurons | ↓Amyloid-β (↓Aβ42/40) | 1,258 FDA-approved drugs (repurposing) | 27 hits Bromocriptine, Cromolyn, Topiramate | Kondo T. et al. (2017) Cell Rep |
| AD/Trisomy 21 iPSC-derived cortical neurons | Modulators of APP processing: ↑short Aβ peptides, ↓Aβ42/38 | 1280 molecules (Prestwick library) | Avermectin: Selamectin | Brownjohn P.W. et al. (2017) Stem Cell Reports |
| Fragile X Syndrome patient iPSC-derived NPCs | ↑FMR1 gene expression | 1262 Compounds | 5-aza-dC 5-aza-C | Li M. et al. (2017) Stem Cells |
| HD patient iPSC-derived medium spiny-like neurons | ↓Cell death | Candidate approach | Bexarotene (PPARgamma activator) | Dickey A.S. et al. (2017) Sci Transl Med |
| HD patient iPSC-derived neurons | ↓Cell death, ↑neurite length, ↑NEUROD1 expression | Candidate approach | Isoxazole-9 | Consortium H.D.I. (2017) Nat Neurosci |
| HD (HTT-66Q, -71Q,-109Q) patient iPSC-derived BMECs | Rescue of HTT-induced defects in angiogenesis | Candidate approach | XAV939 | Lim R.G. et al. (2017) Cell Rep |
| PD patients (idiopathic or DJ-1 c.192G>C) iPSC-derived dopaminergic neurons | Rescue of mitochondrial oxidative stress, ↓oxidized dopamine, ↓α-Syn, ↑lysosomal function | Candidate approach | FK506 Isradipine Mito-TEMPO N-acetylcysteine | Burbulla L.F. et al. (2017) Science |
| PD (LRRK2-G2019S) patient iPSC-derived dopaminergic neurons | ↓Aggregates of Amyloid-β and ↓P-APP | Candidate approach | LRRK2-IN-1 | Chen Z.C. et al. (2017) Sci Signal |
| PD (α-Syn-G209A) patient iPSC-derived dopaminergic, GABAergic and glutamatergic neurons | ↓Protein aggregation, ↑neurite outgrowth | Hypothesis testing α-Syn oligomerization | NPT100-18A NPT100-14A ELN484228 | Kouroupi G. et al. (2017) PNAS |
| PD patient-derived NPCs and neurons | ↓SNCA expression and rescue of oxidative stress | 1,126 Compounds | Clenbuterol | Mittal S. et al. (2017) Science |
| Human iPSC-derived NPCs and neurons | ↑GRN/PGRN expression | Candidate approach | Class I HDAC inhibitors Apicidin, Valproate | She A. et al. (2017) Cell Chem Biol |
| Human iPSC-derived cortical NPCs and organoids infection by the Zika virus | ↓Zika virus infection, ↑growth and differentiation | >1,000 FDA-approved drug candidates | Hippeastrine hydrobromide (HH) | Zhou T. et al. (2017) Cell Stem Cell |
| SMA patient iPSC-derived neurons | ↑SMN2/SMN expression | Candidate approach | Compound 3 HDAC inhibitor | Lai J.I. et al. (2017) Bioorg Med Chem Lett |
| ALS-C9orf72 patient iPSC-derived MNs | ↓Cell death | Hit validation (mouse primary spinal cord cultures screen) | Vardenafil | Osborn T.M et al. (2018) Neurobiol of disease |
| ALS-C9orf72 MNs and cortical neurons | ↓RNA and protein repeat foci | Hit validation (in silico screen) | DB1246 DB1247 DB1273 | Simone R. et al. (2018) EMBO Mol Med |
| ALS (FUS-P525L) iPSCs | ↓Stress granules | 2600 compounds | Rapamycin, Torkinib, Paroxetine, Trimipramine | Marrone L. et al. (2018) Stem Cell Reports |
| AD-APOE4 iPSC-derived neurons | ↓Aβ40 and 42, ↓P-Tau, ↓GABAergic neuron degeneration | Candidate approach | PH002 | Wang C. et al. (2018) Nature Medicine |
| AD (sporadic) patient iPSC-derived neurons | RetromerVPS35, VPS29, VPS26 stabilizer compounds, ↓APP processing, ↓Aβ peptides, ↓P-Tau | Candidate approach | R33, R55 chemical chaperones | Young J.E. et al. (2018) Stem Cell Reports |
| AD (PSEN1-P117L) patient iPSC-derived neurons | ↓β-Amyloid (intra- and extra-cellular) | Candidate approach | Nobiletin | Kimura J. et al. (2018) Biol Pham Bull |
| Human iPSC-derived sensory neurons excitability (pain disorder) | Inhibition of veratridine-evoked calcium flux (excitability) assay | 2700 compounds | 9 hits (Astemizole, Loperamide, Sertraline, Fluoxetine, Amitriptyline, Paroxetine) | Stacey P. et al. (2018) SLAS Discov |
| Human iPSC-derived neurons (neural networks and regeneration) | Modulation of neurite growth | 4421 bioactive small molecules | 108 hits (37 FDA approved) | Sherman S.P., Bang A.G. (2018) Dis Model Mech |
| Patient-derived GBM stem cells 3D-spheroids | ↓Proliferation, ↑cytotoxicity | ~3,300 FDA approved drugs | Molecules anti-proliferation | Quereda V. et al. (2018) SLAS Discov |
| Psychiatry patients’ iPSC-derived NPCs | ↓WNT signaling | 300,000 compounds | Multiple scaffolds | Zhao W.N., Haggarty S.J. et al. (manuscript in preparation) |
Key: AD/Alzheimer’s disease, ALS/amyotrophic lateral sclerosis, APOE4/apolipoprotein E variant E4, APP/amyloid precursor protein, ASD/autism spectrum disorders, BMECs/brain microvascular endothelial cells, DJ-1/nucleic acid deglycase gene or PARK7, ESC/embryonic stem cells, FDA/U S Food and Drug Administration, FMR1/fragile X mental retardation 1 gene, FUS/fused in sarcoma RNA-binding protein, GBM/Glioblastoma, GRN/progranulin gene, iPSC/induced pluripotent stem cells, LRRK2/leucine-rich repeat kinase 2, MNs/motor neurons, MTT/tetrazolium dye, NPCs/neural progenitor cells, PD/Parkinson’s disease, PGRN/progranulin protein, PSEN1/presenilin 1, SMA/spinal muscular atrophy, SOD1/superoxide dismutase 1, αSyn/α-Synuclein, TDP-43/TAR DNA-binding protein 43.
Figure 4.
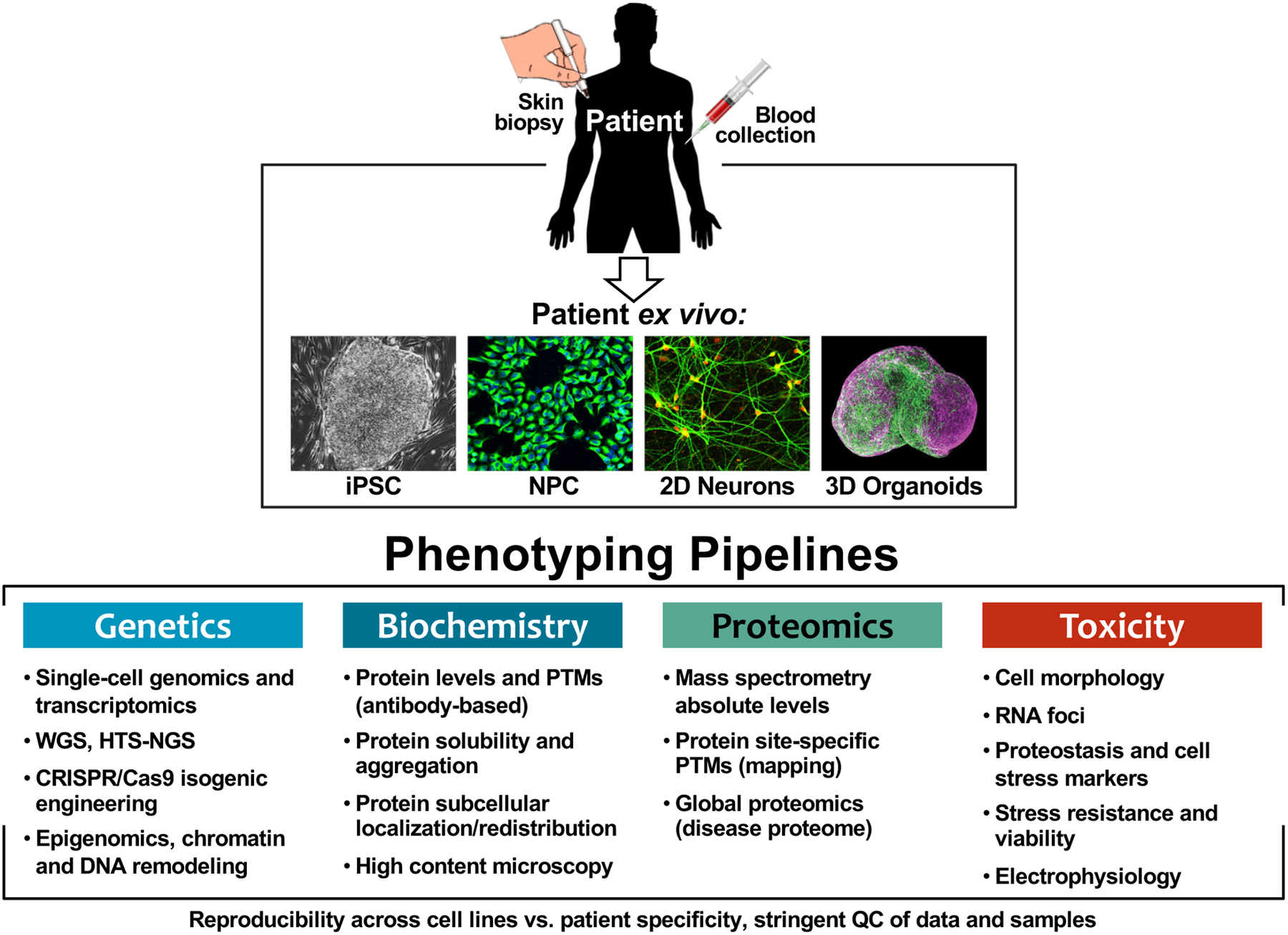
Phenotypic platforms for human iPSC-derived neuronal models. Optimization and quality control (QC) of CNS cell model derivation paired with development and integration of standardized phenotypic pipelines, including genetic, biochemical, proteomic and toxicity analysis of neurons ex vivo, for determining molecular mechanisms of disease. Organoid image adapted from sciencenews.org 2018.
While many quantifiable phenotypes may prove adaptable to screening formats, it is important to define which ex vivo cellular phenotypes are relevant to disease pathophysiology and should, therefore, be the focus of experimental small-molecule testing.26, 120, 177 One promising strategy to help recognize disease-relevant phenotypes and understand disease in a broader context (e.g. common aspects of neuronal death among related neurodegenerative diseases), consists of integration of molecular data from cellular models across different studies of the same or related disorders with genetic and clinical longitudinal patient data (Fig.1).1, 13 In parallel, the creation of large cohorts of human iPSCs (e.g. CIRM Human Pluripotent Stem Cell Repository, NIMH Stem Cell Center, NYSCF Research Institute Stem Cell Repository, StemBANCC, HipSci Consortium, Stem Cells Network DPUK Cohorts) has facilitated the bridge between studies of different disease models and technologies.209–210 As evidence starts to build from patient models and clinical data, a better understanding of the early disease processes could also aid in diagnosis by contributing new molecular biomarkers. We briefly review advances in genomic, biochemical and cellular assays that are employed in the study of disease molecular mechanisms and in testing new molecules emerging from drug screens (Figs.1, 2).
Genomics and Epigenomics
Remarkable developments in high throughput next generation (NGS) sequencing, whole exome sequencing (WES), genome-wide association studies (GWAS) and RNA profiling have allowed for probing the human genome and transcriptome with considerable ease.135, 138–139, 141, 143 This has led to the identification of common genetic variants, rare variants (<1% frequency), and high-risk variants segregating with multifactorial, complex genetic disorders.138–139 Particularly in psychiatric illnesses, all traits are genetically multi-factorial and complex due to multiple risk alleles, gene-gene interaction effects and epigenetic effects. But the wealth of knowledge is growing exponentially due to more routine application of GWAS, high throughput sequencing, and gene expression studies by RNA profiling that can inform on splicing defects or changes in expression of non-coding RNAs.211 Furthermore, the ability to survey the genome and transcriptome at the single-cell level is starting to offer unique advances in precision studies of cell type contribution and vulnerability to disease.212
As elucidation of genetic contribution to disease is critical to understanding disease etiology, accurate conclusions can only be obtained through large clinical cohorts, especially if the goal is to define causal or risk association for de novo mutations or rare variants.112, 133, 140–141, 177, 213 Sequencing programs within patient cohorts (e.g. Precision Medicine Initiative (PMI) led by the FDA/United States Food and Drug Administration)214 start to offer important interpretive guidance in the mapping of pathways and cellular components with relevance to neuronal integrity, homeostasis and function, contributing to the development of gene-panel-based diagnostics. Moreover, gene allelic variants can provide correlative evidence of how modulating a specific gene-target will affect human disease biology.213 In certain cases, an allelic series can be identified that supports the directionally of what a therapy is desired to do (loss-of-function or gain-of-function), guiding efforts to identify a therapeutic modality that can mimic the effect of the genetic variation. Then, being able to directly measure the biochemical and cellular consequence of specific allelic series using patient-derived cell models holds tremendous promise for elucidating translatable markers of target modulation for drug screening.
As multiple disease-associated genes, proteins and entire molecular pathways emerge, it is also becoming increasingly evident that epigenetic regulation: DNA methylation, histone acetylation and micro-RNAs (miRNAs), can be direct modifiers of disease, or that chemical epigenetic modulation can indirectly regulate pathways affecting disease. For instance, in ALS patients and animal models, histones are generally hypo-acetylated in association with neuronal death through apoptosis;215–216 whereas in AD, global DNA hypo-methylation and gene-specific hyper-methylation have been observed.217 Large patient cohorts genomic profiling can equally help in the identification of epigenetic modifications that either contribute or protect against disease. As such, epigenetic therapy is also a growing area of research for correcting gene expression in disease.177
Biochemical, proteomic and cellular phenotypes
Biochemical profiling of patient iPSC-derived neuronal cells has relied on quantitative measurement of gene expression (qRT-PCR), analysis of steady-state levels and post-translational modifications (PTMs) of specific proteins (western blotting, ELISA) and, in particular for neurodegenerative diseases, analysis of high-order protein species, i.e. oligomers and aggregates of decreased solubility (Fig.4). Beyond antibody-based approaches, quantitative methodologies using high-resolution mass spectrometry (MS) are powerful phenotyping tools. First, MS can measure specific protein-variant levels far surpassing accuracy of any other method. Second, MS generates proteome profiles (“bar-coding”) from patient ex vivo neurons that can be compared with post-mortem brain tissue to reveal disease-specific patterns of protein biochemistry, overall contributing to disease categorization and possibly accuracy of diagnostics.54, 60, 218–221 Combining proteomic and pharmacological profiling of patient neuronal models is an innovative strategy to discover chemical probes that unveil network or pathway vulnerabilities in patient-only cells. That is, disease-affected pathways uncovered by proteome profiling can be pharmacologically probed in a temporally conditional manner to confirm altered vulnerability in disease, contributing to the dissection of toxicity mechanisms.54, 114, 151, 153–154, 157, 161, 163 Advances in automated microscopy and high-content imaging now allow single-cell and subcellular analysis of phenotypes with increased throughput, adaptable to iPSC-derived neuronal cultures.222–224 Parallel increased sophistication in computational methods for image analysis with elevated number of parameters (multiplex) enables quantification of a diverse range of cellular features.86, 222, 225 Combined, automated high-content microscopy can reveal a range of quantifiable phenotypes, including changes in developmental and differentiation patterns, cellular anatomical abnormalities, differential expression and subcellular localization of functional markers, as well as (mis)localization of molecules and proteins that are relevant for disease. This is well illustrated by recent work.29, 33, 53–54, 86, 105, 113, 117, 153–154, 157, 160–161, 163, 165, 169 For cell models of psychiatric diseases, with associated polygenicity or altogether unknown genetics, patient-derived cell models allow access to measures of signaling pathways and genotype-phenotype relationships through pathway-selective reporter genes,55 high-content imaging,53 multiplexed gene expression and proteomic profiling assays.177, 208, 226 Complementarily, multi-electrode arrays (MEAs) that measure electrophysiological activity and evoked action potentials in ex vivo neuronal networks, can capture disease-associated alterations and monitor pharmacological effects on synaptic activity.227
One constraint is the inherent variability in both the iPSC derivation process and differentiation protocols, with an increased variability in 3D cultures. This is particularly relevant for diseases where individuals show essentially normal neurodevelopment and cellular function and only present symptoms after birth, old age or exposure to environmental triggers. In this case, iPSC-derived neurons are expected to show weak and variable phenotypes. It is therefore necessary to perform comparative studies across several independently generated cell lines, from both healthy and diseased individuals, in order to identify robust phenotypes amongst technical variability and noise. CRISPR/Cas9-mediated generation of isogenic cell pairs that differ in a single genetic variation, either mutation or correction to wild-type, could allow dissection of the molecular and cellular phenotypes directly related to this gene, establishing a causal relationship. However, if no effect is observed, no conclusion can be ascertained for disease contribution, and also the process of genome editing can itself introduce off-target mutations and clonal variability within a particular iPSC line.161, 228–229
Integration with clinical and pathology information
Arnerić et al.1 reviews all up-to-date efforts (or lack of) on integration of data across disease studies, clinical and cohort-based entities and repositories, and how this impacts drug development across diseases that share common mechanisms. Stem cell-derived cellular models of human disease now introduce the human context into the earliest stages of CNS drug discovery (Fig.2). However, to identify the most disease-relevant phenotype for drug screening, it might be necessary to adopt standardized phenotypic pipelines (biochemical, imaging and physiological), select corroborated phenotypes across multiple models of disease, and increase the scrutiny and quality control of emerging data. Then, robust assay pipelines might just become new diagnostic tools using patient iPSCs for pre-clinical assessment and guidance of clinical trials.148–149 But to succeed, these efforts require data interpretation in the context of clinico-pathological information emerging from biomarker, imaging and post-mortem studies (Fig.1). Patient clinical cohorts that are extensively and longitudinally phenotyped by a variety of psychometric assessments, structural, functional and molecular neuroimaging (PET, MRI), fluid biomarker sampling (CSF, blood), and neuropathological analysis, offer a rich dataset context in which to interpret results from iPSC models. Emerging data from these integrative studies is starting to reveal alterations in living patients, that can also be measured in patient cellular models, reporting on earlier than expected disruption of specific molecular pathways.137, 148–149, 230–231 This now offers guidance for undisputed phenotypic relevance and therapeutic targets for drug screens and clinical trials. Conceptually, testing experimental drugs in patient cell models provides a powerful proof-of-concept for correlating physiologically relevant phenotypes to pre-existing disease biomarkers (e.g. AD148–149), establishing the role for iPSC-based models in drug discovery, driven by human disease biology at each step of the process (Fig.2).
A role for patient-specific neuronal cells in clinical diagnosis?
A key challenge in today’s medicine is not only the development of effective, disease-modifying and preventative treatments for CNS disorders, but also improved diagnostics so treatment could be initiated early, potentially at a time when it can be most effective. While biomarker and genetic testing for certain diseases can be done, many diseases lack precise molecular biomarkers or are polygenic in nature rendering a strict genetic diagnosis not yet feasible. So, there is an increasing interest in the concept of generating ex vivo drug-response signatures to diagnose patients and be able to provide a prognosis (Fig.2). For example, we and others have observed that coupled to early dysregulation of protein homeostasis, patient-derived neurons show increased vulnerability to a panel of proteotoxic, excitotoxic and mitochondrial stressor compounds.54, 151, 153, 169, 232–233 Since a majority of neurodegenerative diseases are sporadic in nature, panels of cell stressors generating drug response signatures, coupled with transcriptional and proteomic profiles,208, 226 could conceivably reveal pathway-based susceptibility grouping of disorders, revealing common and new mechanisms of pathogenesis. When coupled with PET imaging, peripheral blood or CSF analysis, these assays could increase accuracy of diagnostics at very early stages of the disease.
Drug screens focusing on CNS disorders using iPSC-derived cell models
Drug screens have been performed in a variety of human cell lines and have contributed significantly to novel therapeutic discovery. However, CNS drug discovery could be accelerated if more accurate cell models were developed to displace less relevant, exploratory and heterologous ones, and if the discrepancy between simplified in vitro assays and the complexity of in vivo pathologies could be addressed. Generally, drug screens focus on the identification of hits that satisfy specific molecular targets or phenotypic requirements, that are then validated and optimized in similarly over-simplified models, and later tested in animal models, which often fail.3 Due to well-known differences in physiology, metabolism and tolerability between species, there is a significant lack of fidelity between current testing procedures and clinical outcomes. In 2008, the President’s Council of Advisors on Science and Technology (PCAST) articulated the concept of “personalized medicine,” which encourages medical research and treatment based on a patient’s genetic background and specific disease characteristics, in order to increase therapeutic success and benefits.234 In line with this, patient iPSC-derived cell models enable identification of relevant mechanisms and targets in a “human context”, offering a unique and unlimited platform that recapitulates aspects of human disease and where disease-associated phenotypes and physiologically-relevant assays can lead preclinical drug discovery. Being of patient origin, these cell models theoretically allow development of personalized drug testing.32 Another emerging concept of clinical-trial-in-a-dish proposes to utilize large cohorts of patient iPSC-derived models to test efficacy of experimental drugs (Figs.1, 2).235–236 In particular, phenotypic-based drug screens are becoming increasingly popular because, unlike target-based screens, there is no a priori need to understand the molecular mechanism of action, and the effect of compounds on patient-specific cell phenotypes is observed directly.13, 30, 36, 86, 155, 183
When using iPSC-derived models for drug screening, two key aspects are: 1) the disease biology supporting the relevance of the ex vivo cellular phenotype being screened, and 2) the translatability of this phenotype. As already mentioned, while many cellular phenotypes could in principle be used for drug screening, demonstration of the importance of a particular phenotype to the underling disease often requires knowledge integration at multiple levels, an impossibility for many diseases, and ultimately only achievable through in-human clinical trials. Still, in many cases, new therapeutics being developed will likely benefit from being optimized in the context of human biology, rather than based on probes to measure human homologs in heterologous systems. Moreover, 3D cell models thought to be more reflective of in vivo cellular responses, have also started to be implemented in early drug discovery process, mainly for cancer and viral infection studies, with slower progress in CNS due to scalability and variability, as mentioned before.
Two strategies are commonly used for drug screening with iPSC-derived disease models, candidate-focused hypothesis-driven screening or discovery-based library screening (Table 1). Focused screens involve testing a relatively small number of experimental drugs to rescue disease phenotypes in the cellular model, simultaneously uncovering dysregulated pathways or targets. Often, these candidate drugs are selected based on predicted targets that are relevant to disease. This approach provides proof-of-concept in establishing cellular models, either accelerating the identification of disease-modulators or establishing a basis for follow-up unbiased screening using large compound libraries. In contrast, discovery-based HTS of large compound libraries allows for identification of novel molecules that can rescue disease phenotypes, which is especially useful for diseases of undefined molecular mechanisms, with the potential to reveal previously unappreciated disease mechanisms. A compromising strategy consists of HTS of FDA-approved drugs for novel disease applications, where repurposing of well characterized drugs could fast-track the transition from bench to bedside.237 HTS using iPSC-derived models is still rare, and many iPSC-based chemical screens have used libraries containing clinically approved or experimental drugs with annotated targets that can be easily validated in downstream studies, with known safety and toxicity profiles, and with potentially rapid clinical translation (Table 1).238 As already mentioned, expandable NPCs and NGN2-derived neurons offer a good platform for large scale and consistent sampling of human neurons compatible with HTS. To address the limitations of 2D models, new advances in 3D cerebral organoids, microfluidics-based organ-on-chip systems and direct transdifferentiation237 should also soon enable testing and pre-clinical validation of new lead compounds in CNS.96 Below we review representative examples of drug screens for CNS disorders using iPSC-derived models (comprehensive list in Table 1).
Small molecule screens in psychiatric disorder models
Development of effective therapies for psychiatric disorders is one of the greatest challenges of the pharmaceutical industry,239 because the cause of disease is complex, heterogeneous, and still not well understood.26, 173, 177 There are now a number of human psychiatric patient-derived cell models compatible with phenotypic assays and functional chemical screens,177 that in fact have demonstrated translational potential by showing responsiveness to pharmacological agents. Examples include rescue of abnormal calcium signaling with the voltage-dependent L-type calcium channel antagonist nimodipine and rescue of neuronal differentiation patterns by roscovitine in Timothy syndrome neurons, a multi-systemic disorder with autism features;26, 44 rescue of glutamatergic synapses by the insulin-like growth factor 1 (IGF-1) in Rett syndrome neurons;192, 240 and rescue of gene expression and connectivity abnormalities in schizophrenia neurons with the antipsychotic loxapine183 (Table 1). These were candidate-approach studies that established the value of the cell models for small molecule screening, with the potential for follow-up mechanistic studies of the mode-of-action in human cells, at a pre-clinical phase.
One of the first large-scale (>2 million compounds) drug screens in human stem cell-derived neurons, relevant to multiple psychiatric and degenerative disorders, is that of McNeish et al.241 It aimed to identify new AMPA glutamate receptors potentiators, with a Fluorimetric Imaging Plate Reader (FLIPR) calcium influx assay (Table 1). Given the importance of glutamatergic signaling in nervous system plasticity, and the challenge of faithfully accessing the biology of ion channels in heterologous systems, this work reflects the potential for chemical screening and the scalability of human cell assays. Then, with the goal of identifying novel pro-neurogenic compounds, McLaren et al.242 reported on a ~1,000 compound screen for enhanced proliferation and/or viability of human NPCs using a bioluminescence-based assay. The compounds identified were new leads for pro-neurogenic drugs relevant to a range of psychiatric and degenerative disorders, as well as research tools to aid in the expansion of NPC for HTS. Drug screens targeting genetic psychiatric disorders have also been successfully executed. Two examples are the HTS of compounds that restore expression of the silenced FMR1 gene, encoding the Fragile X mental retardation protein (FMRP) that causes Fragile X syndrome. Using a homogenous, time-resolved fluorescence resonance energy transfer (FRET) assay to measure FMRP levels in a high-density, 1536-well plate format, Kumari et al.243 screened 5,000 compounds and identified six structurally diverse molecules capable of enhancing FMRP expression. Meanwhile, Kaufmann et al.207 screened over 50,000 compounds by HCS imaging and found a small number of compounds also capable of reactivating FMRP expression (Table 1). In both cases the level of FMRP upregulation was modest and perhaps not sufficient to rescue neurodevelopmental phenotypes in vivo,53 but the success of these screens motivates further screening for FMRP expression enhancers.
An alternative approach to discovering therapeutic targets for psychiatric disorders has been to use phenotypic assays and to probe entire pathways for phenotypic rescue, with the potential to be relevant for a range of diseases. The WNT signaling pathway is a key regulator of the nervous system development and plasticity, and WNT dysregulation has been associated with many psychiatric disorders, most notably ASD.244–245 Zhao et al. executed a screen for modulators of the WNT/β-catenin signaling pathway in human NPCs, using a 384-well plate format and a stably integrated HTS-compatible, luciferase-based WNT pathway reporter (Table 1).55 From a library of ~1,500 FDA-approved drugs and known bioactive compounds, both known and novel regulators of WNT signaling were identified, including enhancers of lithium action, selective mGLUR1 (metabotropic glutamate receptor 1) antagonists, and the FDA-approved drug riluzole (Rilutek). Further automation of this assay has enabled screening >300,000 compounds for novel activators and inhibitors of WNT signaling (Zhao W.N., Haggarty S.J., manuscript in preparation).
Drug discovery in neurodegeneration patient cell models
While symptomatic treatments are available for some neurodegenerative diseases, there are no effective disease-modifying therapies, and timely diagnosis continues to be challenging.127, 129, 246 Patient or gene-positive iPSCs are being used to derive human cell models to investigate the early molecular and cellular mechanisms of disease and to advance drug screens based upon specific disease-relevant phenotypes. In one of the first studies, Cooper et al.247 examined neural cells derived from PD patients iPSCs, carrying mutations in the PINK1 (PTEN-induced putative kinase 1) or LRRK2 (leucine-rich repeat kinase 2) genes, focusing specifically on mitochondrial function and integrity. This work showed that PD-derived neural cells were particularly vulnerable to chemical stressors and toxins targeting mitochondrial function and the proteasome machinery, also exhibiting higher levels of oxidative stress. This neuronal vulnerability was pharmacologically rescued by coenzyme Q10, rapamycin and the LRRK2 kinase inhibitor GW5074, which not only provided disease mechanistic insight, but also a role for this model system in drug testing. Peng et al.248 took a more neuronal type-specific approach and utilized human iPSC-derived dopaminergic neurons cultured in 96-well plates in the presence of 1-methyl-4-phenylpyridium (MPP+), a neurotoxin widely used to model PD, and executed a low-throughput neurotoxic/neuroprotective automated screen of 44 compounds predicted to target different mechanisms of neuroprotection. Secondary assays included a rotenone-induced cell death assay that confirmed that iPSC-derived dopaminergic neurons can be used for screening and validation of potential neuroprotective agents in PD. Later, in a similar iPSC-derived neuronal PD model system utilizing MPP+, Wimalasena et al.249 carried out an in-silico gene expression-based screen to identify small molecules associated with gene-expression profiles that are anti-correlated with a PD profiles (literature curated). The rationale was that normalizing aberrant gene expression to healthy-control levels is of therapeutic value. This study identified the cyclin-dependent kinase 2/5 (CDK2/5) inhibitor GW8510, protective against MPP+ toxicity in a dose-dependent manner, and protective against small-molecule mitochondrial and endoplasmic reticulum stressors.
In FTD-tau patient iPSC-derived neurons, we have demonstrated the ability of mTORC1 inhibition by rapamycin treatment to cause tau clearance, through autophagy, and to rescue stress vulnerability.54 More recently, Wang et al.86 developed an HCS assay utilizing human iPSC-derived and Ngn2-induced cortical glutamatergic neurons, to identify tau-lowering compounds in LOPAC (Library of Pharmacologically Active Compounds) containing 1,280 bioactive small molecules that affect most signaling pathways and cover major drug target classes. This screen employed an integrated Ngn2-inducible and isogenic human iPSC line (i3N) that can be differentiated mainly into functional glutamatergic neurons amenable to HTS in microplates and imaging-based HCS of neuronal and tau specific markers. Top hits from the screen included adrenergic receptor (AR) agonists (moxonidine, metaproterenol, clonidine, dexmedetomidine, isoproterenol) that reduced tau levels in human neurons. Conversely, AR inhibition led to tau accumulation. For another FTD class, Lee et al.250 focused on rescue of FTD-PGRN patient iPSC-derived neuronal phenotypes with pharmacological agents such as 1-[2-(2-tert-butyl-5-methylphenoxy)-ethyl]-3-methylpiperidine (MPEP), which decreases Sortilin (SORT1) expression and rescued extracellular PGRN secretion. Using a similar model, Almeida et al.163 provided proof-of-concept that GRN mRNA and PGRN protein levels can be upregulated by suberoylanilide hydroxamic acid (SAHA), a histone deacetylase (HDAC) inhibitor. Characterization of the epigenetic regulation of GRN expression was further explored by She et al.,251 with a comprehensive chemogenomic strategy to probe the selectivity and kinetic requirements of HDAC inhibition for upregulation of PGRN expression and secretion. Additionally, Holler et al.252 performed a screen of known autophagy-lysosome modulators in human fibroblasts and iPSC-derived neurons from GRN mutation carriers, and identified multiple novel activators of human GRN expression, including mTOR inhibitors and an mTOR-independent activator of autophagy, trehalose. Secondary assays validated trehalose as the most promising lead compound, increasing endogenous PGRN and neuroprotection in all cell lines tested. Altogether, these screens provide concrete evidence for the potential of FTD iPSC-derived models for novel target and drug discovery (Table 1).
In AD, various anti-amyloid drugs targeting different pathways of Aβ42 production and/or aggregation have been developed and gone through clinical trials. Unfortunately, these candidates have so far failed to produce the expected therapeutic breakthroughs.9–10 In an AD-patient iPSC model carrying an APP mutation, increased levels of APP and Aβ, aberrant β and γ secretase cleavage of APP, and increased levels of total and phospho-tau, were rescued by treatment with specific Aβ antibodies early during the differentiation process.159 In a different model, where AD patient-derived neurons were co-cultured with activated murine microglial cells, treatment with the anti-inflammatory small molecule apigenin promoted a reversal of neuronal morphological deficiencies and hyper-excitability, and offered protection against apoptosis.253 Brownjohn et al.254 took a different approach and designed a phenotypic small-molecule screen to identify secretase-independent, small molecule modulators of APP processing that would shift the production of fragments from Aβ42 to shorter, non-toxic forms, in human cortical neurons from DS patients, a complex genetic model of AD. Hit compounds included a family of macrocyclic lactone anthelminthic compounds, the avermectins, commonly used as anthelmintics, revealing an indirect and secretase-independent mechanism of modulating APP processing.
In the context of the motor neuron disorder ALS (Table 1), Egawa et al.170 generated MNs from familial ALS patients with TDP-43 mutations, which produced aberrant, insoluble, cytosolic aggregates of TDP-43, along with abnormal neurite outgrowth. Anacardic acid, a compound with histone acetyltransferase inhibitory activity, rescued MN phenotypes, providing the first evidence that patient iPSC-derived MNs could be used for drug screening. In turn, Yang et al.255 performed a large-scale screen on control and mutant SOD1 MNs, which revealed that the multi-kinase inhibitor kenpaullone prolonged survival of human MNs with both SOD1 and TDP-43 mutations. A second compound in this study, dexpramipexole, was inactive and interestingly failed clinical trials in ALS. A third compound, olesoxime, that also failed in ALS clinical trials was only able to mildly rescue SOD1 but not TDP-43 neuronal survival. Burkhardt et al.256 performed a HCS of >1,700 compounds targeting TDP-43 aggregation in iPSC-derived MNs from sporadic ALS patients. This screen led to the identification of FDA-approved cardiac glycosides drugs digoxin, lanatoside C and proscillaridin A, two CDK inhibitors (flavopiridol, Cdk1/2 inhibitor III), four JNK inhibitors (Incedis-01, BP-11767, NSN01, NSN02), and the natural product triptolide. All were capable of reducing TDP-43 aggregation through mechanisms as of yet unclear. Also, notably, Wainger et al.257 identified a hyper-excitable phenotype in ALS patient iPSC-derived MNs, which could be reversed by treatment with the anti-epileptic drug retigabine. Here, the use of electrophysiological phenotypes drove the selection of a pharmacological agent for subsequent clinical investigation. As retigabine is an approved antiepileptic drug, this facilitated rapid initiation of a clinical trial in ALS. Still in the context of ALS, associated with the C9orf72 intronic expansions, Simone et al.258 performed a small molecule screen in iPSC-derived motor and cortical neurons to identify small molecules that specifically stabilize GGGGCC repeat G-quadruplex RNA, a secondary-fold state proposed to be associated with neuronal toxicity through repeat RNA-forming foci and toxic dipeptide repeat proteins. Hit compounds significantly reduced RNA foci burden, levels of dipeptide repeat proteins, and improved survival in a fly model of ALS, providing proof-of-principle that targeting GGGGCC repeat G-quadruplexes has therapeutic potential. Shi et al.166 identified alterations in the lysosomal pathway of MNs derived from C9ORF72 patients, prompting a screen of potential therapeutic targets in this pathway. This work identified a PIKfyve inhibitor that decreased cell death of patient-specific MNs when neurotrophic factors were removed. PIKfyve is an enzyme involved in phospholipid metabolism, critical in the process of endosomal maturation and lysosome formation. Consequently, inhibiting PIKfyve rescued lysosome numbers, decreasing glutamate receptors and dipeptide-repeat proteins.
Altogether, these representative examples demonstrate the growing impact of human, scalable cell models in the identification of potential novel and repurposing therapeutics.
Other neurological disorders stem-cell based drug screens
Pioneering work from Lee et al.259 demonstrated the ability of conducting a screen of >6,000 small molecules in patient-derived neural crest precursor cells for the ability to rescue expression of the IKBKAP gene, which is disrupted in the rare autonomic neurological disorder familial dysautonomia. The lead agents identified target cAMP/protein kinase A (PKA) activity and phosphorylation of the transcription factor CREB, that play key roles in gene transcription and neuroplasticity. Meanwhile, Pfizer generated iPSC-derived peripheral sensory neurons from patients with inherited erythromelalgia, caused by gain-of-function mutations in the SCN9A gene encoding the sodium channel NAV1.7.260 Here, the disease clinical severity could be recapitulated by ex vivo neuronal hyper-excitability, and this altered electrophysiological phenotype could be suppressed by NAV1.7 antagonists providing a therapeutic target for a sensory neuron disorder.
For diseases like GBM, similarly to psychiatric diseases, phenotypic-based drug screens have the advantage of not relying on prior knowledge of molecular targets, increasing the probability to identify new and better targets. Using patient-derived GBM cells, Hothi et al.261 performed an HTS of 2,000 compounds to identify molecules that inhibit stem cell proliferation and that could extend patient survival. Although the identified compounds consisted of 47 FDA approved drugs, and 31 already used as experimental therapeutics, follow-up studies introduced disulfiram as a new inhibitor of GBM stem cells survival. DSF is an FDA approved drug for treatment of alcoholism, it is relatively non-toxic, penetrates the blood-brain barrier, and it was significantly potent across multiple patient-derived cell models to reduce cell proliferation and viability. Danovi et al.262 took a candidate approach and tested 160 kinase inhibitors in human GBM-derived neural stem cells, using live-cell imaging and high content image analysis to measure cell proliferation and survival. This screen identified J101 (JNJ-10198409) as a small-molecule that induced mitotic arrest at prometaphase in GBM cells but not in control neural cells, possibly through modulation of the PLK1 kinase. In fact, potent and specific PLK1 inhibitors are already in clinical development (BI2536, BI6727 and GSK461364) corroborating the immediate therapeutic value of new molecules coming through the pipeline. Taking a slight different approach, Quartararo et al.263 first optimized a drug-sensitive culture system of patient-derived GBMs for HTS to identify new experimental drugs. They screened seven patient-derived cell models against “56 chemical agents in 17-point dose-response curves in 384-well format in triplicate”, revealing a range of effects and sensitivities of the seven patient-derived models to the chemotherapeutic agents tested. For instance, while MEK inhibitors caused only patient-specific growth inhibition, bortezomib (FDA-approved proteasome inhibitor) was potently lethal across all patient models. Most interestingly, follow-up studies revealed that combination of the BCL-2 inhibitor ABT-263 and the mTOR inhibitor AZD-8055, had a synergistic effect in a subset of patient-derived models. Given low efficacy of current therapies, drug combinations are very relevant strategies to enhance efficacy and delay onset of drug resistance, with focused research on drug pair interaction screens across GBM patient-derived cell lines.264 This approach also highlighted correlations between drug-pair effects with cell line molecular profiles, offering novel biomarker signatures for anticancer drug-drug interactions in glioblastoma. Recently Quereda et al.265 used patient-derived GBM stem cells cultured as neurospheres, and developed a 1536-well 3D-spheroid, ultra-high-throughput proliferation assay. A pilot screen of ~3300 compounds identified specific molecules with anti-GBM stem cell proliferation.
Drug responders and non-responders, toxicology and drug metabolism
As patient-specific iPSC-derived cellular models started to reveal disease-relevant phenotypes, the concept of “clinical trials in a dish” was immediately considered as a strategy for the identification of subpopulations of patients that may respond to particular therapeutic agents. It was also considered as a potential means to generate patient-specific cellular signatures of response to pharmacological agents and create diagnostic subgroups (Fig.1).26, 205, 266 These goals ultimately rely on the generation of sufficiently large numbers of patient cell models representative of the heterogeneity of each disorder, to segregate responders and non-responders. It also requires the development of molecular and cellular biomarkers with sufficient predictive power and accuracy to translate between ex vivo assays and in vivo treatment (Fig.2). For diseases where efficacy in the clinic is poorly predicted by existing animal models, this strategy may provide an alternative path forward to gain confidence in efficacy. Furthermore, by identifying ex vivo responders using patient-specific iPSC models prior to a Phase II trial, the clinical trial itself could be performed with subjects already predicted to respond to the drug. Theoretically, this would increase the power of studies to detect drug efficacy in smaller patient populations and minimize the negative effects of exposing patients to ineffective treatments. While this has yet to be implemented, conceptually the cost and timeline of generation of such a patient cohort and respective cell lines is significantly lower compared to the costs and setbacks of a failed Phase II or Phase III trial. Additionally, ex vivo patient cellular responses could be used to decide patient treatment. For example, being able to predict which subjects respond to the mood stabilizer lithium on the basis of their own neuronal cell assays, could greatly improve the standard of care in bipolar disorder. In fact, for lithium, this approach has been performed retrospectively with the observation of a correlation between clinical response and the ability to suppress a hyperexcitability phenotype in hippocampal-like neurons only in lithium responders vs. non-responders derived iPSC models190. This approach could also be used, for example, to dissect the relationship between epigenetic status and drug response using directly iNs.267 With time, the systematic generation of ex vivo experimental drug responses from large numbers of patients may ultimately allow the discovery of predictive rules for future therapeutics. Large numbers of patients will be crucial given that not all patients with the same disease will respond to drug therapy in a uniform manner or with the same level of efficacy, due to a high contribution of genomic background to drug response, i.e. heritability of drug response.268–269 But, focusing on the drug response of individual patient cells provides a practical realization of the concepts of personalized and precision medicine,214 impacting how future pre-clinical and clinical trials are performed (Fig.2).
While current regulatory policies prevent the replacement of standard clinical trials with an iPSC trial, it may soon be feasible to establish such a framework if sufficient retrospective predictions of drug response and efficacy are demonstrated for complex disorders205. Such studies would not obviate the need for careful safety and toxicology studies, which themselves are beginning to benefit from patient-specific iPSC-derived models270, for example for cardiotoxicity271, hepatoxicity272, and neurotoxicity271, creating new avenues to demonstrate as early as possible the efficacy and safety of medicines.109–110
The fact that many approved drugs have been subsequently withdrawn from the market based on toxicity, a failure attributable at least in part to conventional drug-safety assays using animal models, has led to an increased focus on human iPSC-derived models’ predictive capacity of adverse drug response. The use of high-throughput assays that detect alterations in cellular processes of relevance to neurodevelopment has been proposed as one approach to screen chemicals for developmental neurotoxicity. Recent work has established HTS based on measures of cell viability273 and apoptosis274, to test compounds across human NPCs, neurons, as well as animal model systems. Results so far reveal that neurotoxins have clear species and cell-type specificity, strongly encouraging the use of human neural cells in neurotoxicity testing. Toxicology studies have also started to rely on stem-cell derived organoids that more closely resemble human tissues in silico.275 Early toxicity and metabolism tests using iPSC-differentiated specific cell types, such as hepatocytes, cardiomyocytes, neurons and intestinal tissue, as well as liver microsomes, serve as a cost-effective alternative to traditional in vivo testing.13, 34, 205, 276–279
Models of the blood–brain barrier (BBB) are also starting to emerge as tools for testing brain penetrance of new CNS drugs. 3D BBB organoid models have been generated by co-culturing human primary brain endothelial cells, pericytes and astrocytes under low-adhesion conditions. The resulting spheroids contain astrocytes in the core with endothelial cells and pericytes in the exterior surface, reproducing features of the BBB, including expression of tight junctions, molecular transporters and drug efflux pumps. These BBB organoids have been shown to be reliable and predictive systems of brain-permeable compounds and could accelerate the decision process at a pre-clinical stage for drugs with poor penetrance.280–282
Drug target validation
Another valuable use for human iPSC-derived models in drug discovery is in the rigorous assessment of target engagement in physiologically and disease relevant cellular systems.283 It is well recognized that in vitro systems frequently utilized are far removed from the native drug target in a complex cellular and tissue context, whereas human iPSC-derived neurons, glia and organoids potentially provide access to the native target and physiological protein complexes, without the need for overexpression in artificial cellular contexts. These considerations are particularly relevant for CNS targets, many of which are cell-surface receptors or ion-channels, often within large protein and lipid complexes, highly dependent on the expression of multiple subunits, and regulated by cellular function and activity.284–285 Drug screening and pharmacological assays are also likely to benefit from CRISPR/Cas9 and related genome-editing techniques for the creation of isogenic cell models to aid in the understanding of genotype-phenotype correlations, and to assist in target identification and validation.286–288 These assays include the tagging of gene promoters for the creation of endogenous multiplexed reporter genes or reporter proteins without the need to express them as transgenes. Such assays enable the investigation of gene products and protein complexes in the presence or absence of a drug, with minimized risk of altering their composition and stoichiometry.289
Complementarity between human stem cell-derived models and animal models
Traditional approaches in drug development use biochemical in vitro, animal or human immortalized cell culture models for initial screening of small-molecule libraries. Then, animal models are employed for in vivo testing of drug delivery to target organs and cells, drug efficacy and toxicity. This has been debated intensively over the past decade and reviewed elsewhere.3, 290–291 The main initial explanation for the high failure rate was toxicology and safety issues, but thanks to the implementation of additional toxicology and drug properties testing earlier in the process, this has become less of a problem. Instead, efficacy of the new therapy in the patient is now the main issue and seems to be the consequence of inappropriate screening assays and misuse of animal models that contribute to the large gap in translational efficacy. Although rodent and human nervous systems share a number of evolutionary conserved properties, there are also critical differences in terms of neurogenesis, neural patterning and expression of specific types of neurons, that are critical for human diseases. Additionally, many aspects of gene epigenetic regulation, particularly relevant in polygenic psychiatric diseases, are unique to humans and cannot be adequately investigated in other systems. Finally, the immortalization process of animal or human cells to derive stable cell lines (e.g. SH-SY5Y) interferes with the normal mechanisms and molecular pathways of cell differentiation. The ‘human context’ has typically only been implemented late in the discovery process, during clinical trials, after lead compounds have been identified in animal models. Approaches using human stem cell-derived disease models represent a paradigm shift that introduces the ‘human context’ early in the drug discovery pipeline (Fig.2). Now, disease relevance and toxicity of new experimental drugs tested in iPSC-derived neurons or organoids are predicted to be more indicative of human tolerability than rodent animals, at a pre-clinical stage. Recent examples of successful FDA approved drugs in disease areas where animal models were deemed inadequate (e.g. Kalydeco drug for cystic fibrosis),292 combined with the uncertainties of modeling polygenic and sporadic disorders in animals,293 provides strong motivation to pursue human stem cell-based drug discovery. With potential for increasing translational pharmacology and accelerate the overall drug development process, human ex vivo systems may be a powerful step to determine efficacy prior to commencing clinical trials. But animal models are still critical for testing drug mode of administration, blood-brain barrier penetration, pharmacokinetics (PK) and pharmacodynamics (PD) in a whole organism. The reality is that due to the complexity of disease mechanisms in whole organisms, only a small percentage of initially promising drugs may qualify for humans. The complementarity between “humanized” screening platforms and animal models PK/PD testing, is becoming clear and is probable to lead to maximum drug discovery success (Fig.2).
Challenges in stem cell-based CNS drug discovery
The poor translation between phenotypic-driven drug screens in cellular and animal models and drug efficacy in human trials, is a challenge to drug discovery. To minimize the costs of failed experimental therapeutics, the dismissal of ineffective or highly toxic small molecules should occur as early as possible, preferably even before animal PK/PD testing. Therefore, it is imperative to establish reliable and scalable cell-based model systems that can more accurately resemble in vivo molecular and cellular events and that are predictive of clinical outcome. That is, cellular models need to replicate disease phenotypes to an extent that confidently predicts translation of a given molecular mechanism to clinically relevant features of illness.206, 294 While human stem cell-derived models present promising avenues in this regard and for pharmacological development, there are still a number of limitations and challenges.
One challenge relates to the choice itself of which patients to derive iPSCs from, to then study fundamental aspects of disease. Given the variability amongst CNS disorders and limited diagnostics, it is always necessary to generate multiple independent cell lines from multiple healthy and diseased individuals in order to detect disease phenotypes of relevance for drug screening. A supporting strategy has been the reliance on patient cohorts with in-depth longitudinal clinical assessment. Longitudinal studies gather clinical data over time for multi-disciplinary research into the causes and progression of disease. Comparison between studies, for any given disease, can help researchers gain a deeper understanding of disease and to make informed decisions regarding patients for cell model generation and drug screens. However, significant methodological variations across cohort studies, including data collection, metrics and analysis, may impede cross-studies comparisons.295–297 Thus integration of clinical data with genetic information or molecular data from patient iPSC-derived cells may ultimately be what enables the segregation or stratification of patients for clinical trials. A related topic is the best practice to choose control samples. Traditionally, control iPSCs are chosen amongst age-matched healthy individuals, both genetically and symptomatically, under the erroneous assumption in the case of neurodegenerative diseases, that these individuals will never suffer from the disease in the old age. Then should instead healthy centenarians be used as controls? As an alternative, healthy relatives that share a high percentage of genomic background are used as controls for individuals carrying a disease-causing mutation. Presently, isogenic CRISPR-corrected cell lines are at the forefront choice as controls for monogenic and potentially oligogenic disorders.
Adding to variability in disease, there is high variability amongst pluripotent cell lines that directly influence the developmental, functional and phenotypic properties of the neurons that are derived from each iPSC clone, even if the same exact protocols are used.298 Moreover, derivation of functional neurons from iPSCs or ESCs in a scalable manner is still dependent on months-worth of tissue culture work, which makes large scale screens somewhat limited. For any new emerging methodology, there is an urgent need to improve conversion and differentiation efficiencies and to generate cultures in larger scale before translatable drug screening can be fully implemented.
iPSC-derived models also present some limitations at the level of genetic and physiological representation of the human tissue affected by disease. From a genetic point of view, iPSC-derived neurons do not recapitulate brain cells genetic mosaicism accurately, as most protocols rely on clonal iPSC amplification and differentiation.299–302 Physiologically, the relative immature nature of any iPSC-derived cells is a strong limitation for CNS studies where the cell types of interest mature over an individual’s lifetime. Differentiated cells can be matured to a degree but are still conceptually different from the brain in regard to development, physiological complexity and age. As neurons age, there is increase in DNA damage, decreased in transcriptional output, increase in oxidative stress and altered metabolism. These aspects are difficult to recapitulate in stem-cell differentiated neurons because iPSC reprogramming resets the age clock. Transcriptome profiles of iPSC-derived neuronal cells have revealed a greater similarity to primary fetal brain cells albeit expression of cortical layer markers.303–304 Some strategies to circumvent this aspect, and promote faster maturation and aging, include the generation of cerebral organoids, iNs, and co-cultures over longer periods to capture features of more mature cells, although often at the expense of diminished throughput and scalability. Less used approaches include overexpression of progerin, a protein that causes Hutchinson-Gilford progeria syndrome and promotes accelerated aging features, and finally telomerase inhibition.68, 305–306 It will be interesting to determine how different strategies can be combined to provide a more complete representation of actual cellular aging. For now, the undeniable “young age” of these cellular and organoid models dictates the type of therapeutics being screened for. For neurodegeneration in particular, depending on the phenotypic progression achievable with differentiation methodology, drug screens can better identify preventive therapeutics (pre-symptomology) than disease-modifying (post-symptoms onset) therapies.
Generation of relatively homogenous neuronal type populations poorly reflects the heterogeneity in the brain and selective neuronal vulnerability to disease. It is important to keep in mind that patient-derived cellular models address mainly cell-autonomous aspects of disease, neglecting potentially relevant aspects of other cell types present in the native environment that can certainly influence the outcome of therapeutic interventions. For example, interactions between neurons and glia, inter-neurons synaptic transmission, and higher order circuitries in the brain are challenging to reproduce in ex vivo cellular models. But even if a disease is most apparent at the network level, it does not necessarily follow that deficits will not be observed in individual cells. Along these lines, mixed neural cell populations that result from standard differentiation protocols that were initially considered a limitation, might instead represent an opportunity to interrogate multiple cell types simultaneously without losing possible cell non-autonomous mechanisms of disease etiology. Complementary validation of phenotypes with patient clinical data and post-mortem pathology, should circumvent some of these concerns (Fig.1).
Despite progress in identifying and being able to promote the developmental of specific cell types, such as neurons, glia and MNs, these protocols are still difficult to adapt to drug screening. The requirement for strict and methodical implementation of protocols to successfully obtain desired healthy cell types, without introducing variables like cell stress and viability loss, continue to affect the consistency of results. Current efforts focus on developing robust methodology for reproducible, large-scale production of disease-relevant specific cell types, compatibility with high-throughput screening modalities, including high-content imaging, multiplexed screening, and electrophysiology with multielectrode arrays.13, 33, 38, 111, 170, 177, 247, 307 But relative high experimental costs keep driving research to small scale screens with a minimum number of case and control lines, which leads to limited representation of genetically heterogeneous patient populations, particularly for complex, polygenic disorders, and possibly inaccurate translation of the compounds identified.
Finally, collective coordination and integration of research across diseases is minimal to non-existent, when possibly data from different diseases with high degree of co-morbidities could be relevant in cross-study analysis.1 This has to do, in part, with research costs and funding that are awarded by distinct patient/disease advocacy groups and non-profit organizations or NIH “allocated funding lines” (e.g., Alzheimer disease, Aging, Schizophrenia, Depression, etc.). As collections of patients’ iPSC lines start to be implemented at different geographic locations, a process that includes access and compilation of corresponding clinical and pathological data, it is expected that new integrative approaches are implemented, within specific diseases as well as across diseases that share important features (e.g. protein aggregation-associated neurodegeneration). However, despite efforts to support data sharing from the academic, industry and funding sectors, there is still a general reluctance, including the sharing of negative results that could be highly informative to the community in general.308
Concluding Remarks
CNS drug development has traditionally been associated with tremendous high costs and low success, with drugs failing especially during phase III, the most expensive phase of clinical trials. This is largely attributed to poor clinical efficacy and/or unacceptable toxicity. For every drug screen pipeline covering thousands of small molecules, hundreds of compounds can reach the pre-clinical phase, with about 10 making it to human trials, and less than 1 being approved, in a process that can take 10 to 15 years.309 This undoubtedly has also had a negative impact in research, but it is important to learn from negative trials results and understand the reasons that led to failure. One of the main aspects still has to do with the limited understanding of the underlying biology of disease, with few validated molecular targets. This is also linked to a lack of disease-specific biomarkers of predictive power, which limits the ability of interrogating new compounds’ pharmacology. From a research point of view, the major challenge over the last decade has been the development of disease models that accurately represent aspects of the disease biology, without recurring to heterologous systems. Human iPSC-derived 2D and 3D systems have caused a paradigm shift for CNS research, offering more biologically relevant disease models. Consequently, cellular, molecular and genetic phenotypes are providing screening assays useful for drug discovery, introducing the ‘human context’ earlier in the pipeline. This is expected to drive more successful drug development programs. Still, in order to improve usefulness of human iPSC modeling for drug discovery, it is important to focus on standardization of protocols for iPSCs generation, differentiation and maturation, with implementation of phenotypic assays with strict quality control parameters. The main driving force for advancing human iPSC-derived models in drug discovery will continue to be the demonstration that these cellular systems can be effectively used to understand the molecular and cellular mechanisms of disease, can be predictive of clinical outcomes, and ultimately can be employed in functional cellular and biochemical assays towards diagnostics and discovery of therapeutic agents for the treatment of human diseases.
Acknowledgements
Members of the Haggarty laboratory are thanked for their continued dedication to advancing patient-specific iPSC protocols and models of psychiatric and neurodegeneration disorders. We apologize for the inability to cite a number of other important papers in the field due to space limitations. Research in the Haggarty laboratory on human stem cell models has been supported in part by the FRAXA Research Foundation, Harvard Stem Cell Institute Seed Grant, Marigold Foundation, Stanley Medical Research Institute, Tau Consortium, Association for Frontotemporal Degeneration, Bluefield Project to Cure FTD, Brace Cove Foundation, Pitt-Hopkins Research Foundation, LouLou Foundation, Defense Medical Research and Development Program, National Institute of Mental Health (R33MH087896; R01MH091115; R01MH095088), National Institute of Aging (RF1AG042978), National Institute of Neurological Disorders & Stroke (R21NS085487, R01NS108115), NHGRI (P50MH106933), National Center for Complementary and Integrative Health (R01AT009144), and Stuart & Suzanne Steele MGH Research Scholars Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing Interests
SJH is a scientific advisory board member of Rodin Therapeutics, Psy Therapeutics, and Frequency Therapeutics. None of these entities were involved in manuscript preparation.
References
- 1.Arneric SP, Kern VD&Stephenson DT 2018. Regulatory-accepted drug development tools are needed to accelerate innovative CNS disease treatments. Biochem Pharmacol. 151: 291–306. [DOI] [PubMed] [Google Scholar]
- 2.Kinch MS 2015. An analysis of FDA-approved drugs for neurological disorders. Drug Discov Today. 20: 1040–1043. [DOI] [PubMed] [Google Scholar]
- 3.Cummings J 2018. Lessons Learned from Alzheimer Disease: Clinical Trials with Negative Outcomes. Clin Transl Sci. 11: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Group, G. B. D. N. D. C. 2017. Global, regional, and national burden of neurological disorders during 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 16: 877–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Association., A. s. 2016. Alzheimer’s Disease Facts and Figures. [DOI] [PubMed]
- 6.Newton CR 2018. Global Burden of Pediatric Neurological Disorders. Semin Pediatr Neurol. 27: 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marder SR, Laughren T&Romano SJ 2017. Why Are Innovative Drugs Failing in Phase III? Am J Psychiatry. 174: 829–831. [DOI] [PubMed] [Google Scholar]
- 8.Reardon S 2014. NIH rethinks psychiatry trials. Nature. 507: 288. [DOI] [PubMed] [Google Scholar]
- 9.Congdon EE&Sigurdsson EM 2018. Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol. 14: 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panza F, Solfrizzi V, Seripa D, et al. 2016. Tau-Centric Targets and Drugs in Clinical Development for the Treatment of Alzheimer’s Disease. Biomed Res Int. 2016: 3245935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vejux A, Namsi A, Nury T, et al. 2018. Biomarkers of Amyotrophic Lateral Sclerosis: Current Status and Interest of Oxysterols and Phytosterols. Front Mol Neurosci. 11: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petrov D, Mansfield C, Moussy A, et al. 2017. ALS Clinical Trials Review: 20 Years of Failure. Are We Any Closer to Registering a New Treatment? Front Aging Neurosci. 9: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silva MC, Cross A, Brandon NJ, Haggarty SJ, Human iPSC Models in Drug Discovery: Opportunities and Challenges. In Comprehensive Medicinal Chemistry III, Samuel Chackalamannil, D. P. R. a. S. E. W, Ed. Elsevier: Oxford, 2017; Vol. Vol. 1, pp 48–73. [Google Scholar]
- 14.Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. 1998. Embryonic stem cell lines derived from human blastocysts. Science. 282: 1145–1147. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi K, Tanabe K, Ohnuki M, et al. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131: 861–872. [DOI] [PubMed] [Google Scholar]
- 16.Yu J, Vodyanik MA, Smuga-Otto K, et al. 2007. Induced pluripotent stem cell lines derived from human somatic cells. Science. 318: 1917–1920. [DOI] [PubMed] [Google Scholar]
- 17.Fusaki N, Ban H, Nishiyama A, et al. 2009. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 85: 348–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okita K, Matsumura Y, Sato Y, et al. 2011. A more efficient method to generate integration-free human iPS cells. Nat Methods. 8: 409–412. [DOI] [PubMed] [Google Scholar]
- 19.Stadtfeld M, Nagaya M, Utikal J, et al. 2008. Induced pluripotent stem cells generated without viral integration. Science. 322: 945–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warren L, Manos PD, Ahfeldt T, et al. 2010. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 7: 618–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu J, Hu K, Smuga-Otto K, et al. 2009. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 324: 797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Pak C, Han Y, et al. 2013. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 78: 785–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Victor MB, Richner M, Hermanstyne TO, et al. 2014. Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron. 84: 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lancaster MA&Knoblich JA 2014. Organogenesis in a dish: modeling development and disease using organoid technologies. Science. 345: 1247125. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster MA, Renner M, Martin CA, et al. 2013. Cerebral organoids model human brain development and microcephaly. Nature. 501: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haggarty SJ, Silva MC, Cross A, et al. 2016. Advancing drug discovery for neuropsychiatric disorders using patient-specific stem cell models. Mol Cell Neurosci. 73: 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brennand KJ&Gage FH 2012. Modeling psychiatric disorders through reprogramming. Dis Model Mech. 5: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brennand KJ, Marchetto MC, Benvenisty N, et al. 2015. Creating Patient-Specific Neural Cells for the In Vitro Study of Brain Disorders. Stem Cell Reports. 5: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Di Lullo E&Kriegstein AR 2017. The use of brain organoids to investigate neural development and disease. Nat Rev Neurosci. 18: 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolmetsch R&Geschwind DH 2011. The human brain in a dish: the promise of iPSC-derived neurons. Cell. 145: 831–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ebert AD, Liang P&Wu JC 2012. Induced pluripotent stem cells as a disease modeling and drug screening platform. J Cardiovasc Pharmacol. 60: 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engle SJ&Puppala D 2013. Integrating human pluripotent stem cells into drug development. Cell Stem Cell. 12: 669–677. [DOI] [PubMed] [Google Scholar]
- 33.Ichida JK&Kiskinis E 2015. Probing disorders of the nervous system using reprogramming approaches. EMBO J. 34: 1456–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ko HC&Gelb BD 2014. Concise review: drug discovery in the age of the induced pluripotent stem cell. Stem Cells Transl Med. 3: 500–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marchetto MC, Winner B&Gage FH 2010. Pluripotent stem cells in neurodegenerative and neurodevelopmental diseases. Hum Mol Genet. 19: R71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sayed N, Liu C&Wu JC 2016. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J Am Coll Cardiol. 67: 2161–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reubinoff BE, Pera MF, Fong CY, et al. 2000. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 18: 399–404. [DOI] [PubMed] [Google Scholar]
- 38.Desbordes SC&Studer L 2013. Adapting human pluripotent stem cells to high-throughput and high-content screening. Nat Protoc. 8: 111–130. [DOI] [PubMed] [Google Scholar]
- 39.Jensen J, Hyllner J&Bjorquist P 2009. Human embryonic stem cell technologies and drug discovery. J Cell Physiol. 219: 513–519. [DOI] [PubMed] [Google Scholar]
- 40.Pang ZP, Yang N, Vierbuchen T, et al. 2011. Induction of human neuronal cells by defined transcription factors. Nature. 476: 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Son EY, Ichida JK, Wainger BJ, et al. 2011. Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell. 9: 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsunemoto RK, Eade KT, Blanchard JW, et al. 2015. Forward engineering neuronal diversity using direct reprogramming. EMBO J. 34: 1445–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoo AS, Sun AX, Li L, et al. 2011. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 476: 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasca SP, Portmann T, Voineagu I, et al. 2011. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 17: 1657–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li W, Sun W, Zhang Y, et al. 2011. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A. 108: 8299–8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen W, Abud EA, Yeung ST, et al. 2016. Increased tauopathy drives microglia-mediated clearance of beta-amyloid. Acta Neuropathol Commun. 4: 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heneka M, Andreasson KI, Bachstetter AD, et al. 2016. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J Neurochem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muffat J, Li Y, Yuan B, et al. 2016. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 22: 1358–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chambers SM, Fasano CA, Papapetrou EP, et al. 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 27: 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nemati S, Hatami M, Kiani S, et al. 2011. Long-term self-renewable feeder-free human induced pluripotent stem cell-derived neural progenitors. Stem Cells Dev. 20: 503–514. [DOI] [PubMed] [Google Scholar]
- 51.Cheng C, Fass DM, Folz-Donahue K, et al. 2017. Highly Expandable Human iPS Cell-Derived Neural Progenitor Cells (NPC) and Neurons for Central Nervous System Disease Modeling and High-Throughput Screening. Curr Protoc Hum Genet. 92: 21 28 21–21 28 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee S&Huang EJ 2015. Modeling ALS and FTD with iPSC-derived neurons. Brain Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sheridan SD, Theriault KM, Reis SA, et al. 2011. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 6: e26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Silva MC, Cheng C, Mair W, Almeida S, Fong H, Biswas MHU, Zhang Z, Huang Y, Temple S, Coppola G, Geschwind DH, Karydas A, Miller BL, Kosik KS, Gao FB, Steen JA, Haggarty SJ 2016. Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability. Stem Cell Reports. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao WN, Cheng C, Theriault KM, et al. 2012. A high-throughput screen for Wnt/beta-catenin signaling pathway modulators in human iPSC-derived neural progenitors. J Biomol Screen. 17: 1252–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muratore CR, Srikanth P, Callahan DG, et al. 2014. Comparison and optimization of hiPSC forebrain cortical differentiation protocols. PLoS One. 9: e105807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koch P, Opitz T, Steinbeck JA, et al. 2009. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci U S A. 106: 3225–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Odorico JS, Kaufman DS&Thomson JA 2001. Multilineage differentiation from human embryonic stem cell lines. Stem Cells. 19: 193–204. [DOI] [PubMed] [Google Scholar]
- 59.Yuan SH, Martin J, Elia J, et al. 2011. Cell-surface marker signatures for the isolation of neural stem cells, glia and neurons derived from human pluripotent stem cells. PLoS One. 6: e17540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sato C, Barthelemy NR, Mawuenyega KG, et al. 2018. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron. 98: 861–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.D’Aiuto L, Zhi Y, Kumar Das D, et al. 2014. Large-scale generation of human iPSC-derived neural stem cells/early neural progenitor cells and their neuronal differentiation. Organogenesis. 10: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rigamonti A, Repetti GG, Sun C, et al. 2016. Large-Scale Production of Mature Neurons from Human Pluripotent Stem Cells in a Three-Dimensional Suspension Culture System. Stem Cell Reports. 6: 993–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vierbuchen T, Ostermeier A, Pang ZP, et al. 2010. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 463: 1035–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ambasudhan R, Talantova M, Coleman R, et al. 2011. Direct reprogramming of adult human fibroblasts to functional neurons under defined conditions. Cell Stem Cell. 9: 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu W, Qiu B, Guan W, et al. 2015. Direct Conversion of Normal and Alzheimer’s Disease Human Fibroblasts into Neuronal Cells by Small Molecules. Cell Stem Cell. 17: 204–212. [DOI] [PubMed] [Google Scholar]
- 66.Wapinski OL, Lee QY, Chen AC, et al. 2017. Rapid Chromatin Switch in the Direct Reprogramming of Fibroblasts to Neurons. Cell Rep. 20: 3236–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abernathy DG, Kim WK, McCoy MJ, et al. 2017. MicroRNAs Induce a Permissive Chromatin Environment that Enables Neuronal Subtype-Specific Reprogramming of Adult Human Fibroblasts. Cell Stem Cell. 21: 332–348 e339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mertens J, Reid D, Lau S, et al. 2018. Aging in a Dish: iPSC-Derived and Directly Induced Neurons for Studying Brain Aging and Age-Related Neurodegenerative Diseases. Annu Rev Genet. 52: 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mertens J, Paquola ACM, Ku M, et al. 2015. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell. 17: 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang Y, Liu ML, Zang T, et al. 2017. Direct Reprogramming Rather than iPSC-Based Reprogramming Maintains Aging Hallmarks in Human Motor Neurons. Front Mol Neurosci. 10: 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee JH, Mitchell RR, McNicol JD, et al. 2015. Single Transcription Factor Conversion of Human Blood Fate to NPCs with CNS and PNS Developmental Capacity. Cell Rep. 11: 1367–1376. [DOI] [PubMed] [Google Scholar]
- 72.Miskinyte G, Devaraju K, Gronning Hansen M, et al. 2017. Direct conversion of human fibroblasts to functional excitatory cortical neurons integrating into human neural networks. Stem Cell Res Ther. 8: 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nehme R, Zuccaro E, Ghosh SD, et al. 2018. Combining NGN2 Programming with Developmental Patterning Generates Human Excitatory Neurons with NMDAR-Mediated Synaptic Transmission. Cell Rep. 23: 2509–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tao Y&Zhang SC 2016. Neural Subtype Specification from Human Pluripotent Stem Cells. Cell Stem Cell. 19: 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Giorgio FP, Boulting GL, Bobrowicz S, et al. 2008. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell. 3: 637–648. [DOI] [PubMed] [Google Scholar]
- 76.Dimos JT, Rodolfa KT, Niakan KK, et al. 2008. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 321: 1218–1221. [DOI] [PubMed] [Google Scholar]
- 77.Sarkar A, Mei A, Paquola ACM, et al. 2018. Efficient Generation of CA3 Neurons from Human Pluripotent Stem Cells Enables Modeling of Hippocampal Connectivity In Vitro. Cell Stem Cell. 22: 684–697 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rajamani U, Gross AR, Hjelm BE, et al. 2018. Super-Obese Patient-Derived iPSC Hypothalamic Neurons Exhibit Obesogenic Signatures and Hormone Responses. Cell Stem Cell. 22: 698–712 e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y, Liu H, Sauvey C, et al. 2013. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat Protoc. 8: 1670–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chen YJ, Vogt D, Wang Y, et al. 2013. Use of “MGE enhancers” for labeling and selection of embryonic stem cell-derived medial ganglionic eminence (MGE) progenitors and neurons. PLoS One. 8: e61956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maroof AM, Keros S, Tyson JA, et al. 2013. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 12: 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Flames N, Pla R, Gelman DM, et al. 2007. Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci. 27: 9682–9695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Manabe T, Tatsumi K, Inoue M, et al. 2005. L3/Lhx8 is involved in the determination of cholinergic or GABAergic cell fate. J Neurochem. 94: 723–730. [DOI] [PubMed] [Google Scholar]
- 84.Lu H, Yu J, Wang J, et al. 2016. Simultaneous quantification of neuroactive dopamine serotonin and kynurenine pathway metabolites in gender-specific youth urine by ultra performance liquid chromatography tandem high resolution mass spectrometry. J Pharm Biomed Anal. 122: 42–51. [DOI] [PubMed] [Google Scholar]
- 85.Ye W, Shimamura K, Rubenstein JL, et al. 1998. FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell. 93: 755–766. [DOI] [PubMed] [Google Scholar]
- 86.Wang C, Ward ME, Chen R, et al. 2017. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem Cell Reports. 9: 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davis-Dusenbery BN, Williams LA, Klim JR, et al. 2014. How to make spinal motor neurons. Development. 141: 491–501. [DOI] [PubMed] [Google Scholar]
- 88.Sances S, Bruijn LI, Chandran S, et al. 2016. Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat Neurosci. 19: 542–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li XJ, Du ZW, Zarnowska ED, et al. 2005. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 23: 215–221. [DOI] [PubMed] [Google Scholar]
- 90.Amoroso MW, Croft GF, Williams DJ, et al. 2013. Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci. 33: 574–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hester ME, Murtha MJ, Song S, et al. 2011. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol Ther. 19: 1905–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maury Y, Come J, Piskorowski RA, et al. 2015. Combinatorial analysis of developmental cues efficiently converts human pluripotent stem cells into multiple neuronal subtypes. Nat Biotechnol. 33: 89–96. [DOI] [PubMed] [Google Scholar]
- 93.Haenseler W, Sansom SN, Buchrieser J, et al. 2017. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Reports. 8: 1727–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhang PW, Haidet-Phillips AM, Pham JT, et al. 2016. Generation of GFAP::GFP astrocyte reporter lines from human adult fibroblast-derived iPS cells using zinc-finger nuclease technology. Glia. 64: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bellin M, Marchetto MC, Gage FH, et al. 2012. Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol. 13: 713–726. [DOI] [PubMed] [Google Scholar]
- 96.Grskovic M, Javaherian A, Strulovici B, et al. 2011. Induced pluripotent stem cells--opportunities for disease modelling and drug discovery. Nat Rev Drug Discov. 10: 915–929. [DOI] [PubMed] [Google Scholar]
- 97.Wang Z, Wang SN, Xu TY, et al. 2017. Organoid technology for brain and therapeutics research. CNS Neurosci Ther. 23: 771–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Amin ND&Pasca SP 2018. Building Models of Brain Disorders with Three-Dimensional Organoids. Neuron. 100: 389–405. [DOI] [PubMed] [Google Scholar]
- 99.Hartley BJ&Brennand KJ 2017. Neural organoids for disease phenotyping, drug screening and developmental biology studies. Neurochem Int. 106: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bershteyn M&Kriegstein AR 2013. Cerebral organoids in a dish: progress and prospects. Cell. 155: 19–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lindborg BA, Brekke JH, Vegoe AL, et al. 2016. Rapid Induction of Cerebral Organoids From Human Induced Pluripotent Stem Cells Using a Chemically Defined Hydrogel and Defined Cell Culture Medium. Stem Cells Transl Med. 5: 970–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Simao D, Silva MM, Terrasso AP, et al. 2018. Recapitulation of Human Neural Microenvironment Signatures in iPSC-Derived NPC 3D Differentiation. Stem Cell Reports. 11: 552–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sun AX, Ng HH&Tan EK 2018. Translational potential of human brain organoids. Ann Clin Transl Neurol. 5: 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Quadrato G&Arlotta P 2017. Present and future of modeling human brain development in 3D organoids. Curr Opin Cell Biol. 49: 47–52. [DOI] [PubMed] [Google Scholar]
- 105.Brawner AT, Xu R, Liu D, et al. 2017. Generating CNS organoids from human induced pluripotent stem cells for modeling neurological disorders. Int J Physiol Pathophysiol Pharmacol. 9: 101–111. [PMC free article] [PubMed] [Google Scholar]
- 106.Pasca AM, Sloan SA, Clarke LE, et al. 2015. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 12: 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sloan SA, Darmanis S, Huber N, et al. 2017. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron. 95: 779–790 e776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Choi SH, Kim YH, Hebisch M, et al. 2014. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 515: 274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shinde V, Sureshkumar P, Sotiriadou I, et al. 2016. Human Embryonic and Induced Pluripotent Stem Cell Based Toxicity Testing Models: Future Applications in New Drug Discovery. Curr Med Chem. 23: 3495–3509. [DOI] [PubMed] [Google Scholar]
- 110.Sirenko O, Hancock MK, Hesley J, et al. 2016. Phenotypic Characterization of Toxic Compound Effects on Liver Spheroids Derived from iPSC Using Confocal Imaging and Three-Dimensional Image Analysis. Assay Drug Dev Technol. 14: 381–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shi Y, Kirwan P, Smith J, et al. 2012. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 15: 477–486, S471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Geschwind DH&Flint J 2015. Genetics and genomics of psychiatric disease. Science. 349: 1489–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Israel MA, Yuan SH, Bardy C, et al. 2012. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 482: 216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kondo T, Asai M, Tsukita K, et al. 2013. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. 12: 487–496. [DOI] [PubMed] [Google Scholar]
- 115.Hockemeyer D&Jaenisch R 2016. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell. 18: 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liu L, Huang JS, Han C, et al. 2015. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol Neurobiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mattis VB&Svendsen CN 2015. Modeling Huntingtons disease with patient-derived neurons. Brain Res. [DOI] [PubMed] [Google Scholar]
- 118.Spitalieri P, Talarico VR, Murdocca M, et al. 2016. Human induced pluripotent stem cells for monogenic disease modelling and therapy. World J Stem Cells. 8: 118–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Torrent R, De Angelis Rigotti F, Dell’Era P, et al. 2015. Using iPS Cells toward the Understanding of Parkinson’s Disease. J Clin Med. 4: 548–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zeltner N&Studer L 2015. Pluripotent stem cell-based disease modeling: current hurdles and future promise. Curr Opin Cell Biol. 37: 102–110. [DOI] [PubMed] [Google Scholar]
- 121.Cobb MM, Ravisankar A, Skibinski G, et al. 2018. iPS cells in the study of PD molecular pathogenesis. Cell Tissue Res. 373: 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Liu L, Huang JS, Han C, et al. 2016. Induced Pluripotent Stem Cells in Huntington’s Disease: Disease Modeling and the Potential for Cell-Based Therapy. Mol Neurobiol. 53: 6698–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ghaffari LT, Starr A, Nelson AT, et al. 2018. Representing Diversity in the Dish: Using Patient-Derived in Vitro Models to Recreate the Heterogeneity of Neurological Disease. Front Neurosci. 12: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hibaoui Y&Feki A 2012. Human pluripotent stem cells: applications and challenges in neurological diseases. Front Physiol. 3: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Liu YN, Lu SY&Yao J 2017. Application of induced pluripotent stem cells to understand neurobiological basis of bipolar disorder and schizophrenia. Psychiatry Clin Neurosci. 71: 579–599. [DOI] [PubMed] [Google Scholar]
- 126.Preza E, Hardy J, Warner T, et al. 2016. Review: Induced pluripotent stem cell models of frontotemporal dementia. Neuropathol Appl Neurobiol. 42: 497–520. [DOI] [PubMed] [Google Scholar]
- 127.Ross CA&Poirier MA 2005. Opinion: What is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol. 6: 891–898. [DOI] [PubMed] [Google Scholar]
- 128.Skovronsky DM, Lee VM&Trojanowski JQ 2006. Neurodegenerative diseases: new concepts of pathogenesis and their therapeutic implications. Annu Rev Pathol. 1: 151–170. [DOI] [PubMed] [Google Scholar]
- 129.Hardy J&Gwinn-Hardy K 1998. Genetic classification of primary neurodegenerative disease. Science. 282: 1075–1079. [DOI] [PubMed] [Google Scholar]
- 130.Powers ET, Morimoto RI, Dillin A, et al. 2009. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 78: 959–991. [DOI] [PubMed] [Google Scholar]
- 131.Uryu K, Chen XH, Martinez D, et al. 2007. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp Neurol. 208: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cruts M, Theuns J&Van Broeckhoven C 2012. Locus-specific mutation databases for neurodegenerative brain diseases. Human mutation. 33: 1340–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cruts M&Van Broeckhoven C, Genetics of frontotemporal dementia and related disorders. 2nd Edition ed.; 2015. [Google Scholar]
- 134.Polymeropoulos MH, Lavedan C, Leroy E, et al. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 276: 2045–2047. [DOI] [PubMed] [Google Scholar]
- 135.Rademakers R, Neumann M&Mackenzie IR 2012. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 8: 423–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ross CA, Aylward EH, Wild EJ, et al. 2014. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 10: 204–216. [DOI] [PubMed] [Google Scholar]
- 137.Ghetti B, Oblak AL, Boeve BF, et al. 2015. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: a chameleon for neuropathology and neuroimaging. Neuropathol Appl Neurobiol. 41: 24–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jin SC, Pastor P, Cooper B, et al. 2012. Pooled-DNA sequencing identifies novel causative variants in PSEN1, GRN and MAPT in a clinical early-onset and familial Alzheimer’s disease Ibero-American cohort. Alzheimer’s research & therapy. 4: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Metzker ML 2010. Sequencing technologies - the next generation. Nat Rev Genet. 11: 31–46. [DOI] [PubMed] [Google Scholar]
- 140.Rohrer JD, Guerreiro R, Vandrovcova J, et al. 2009. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 73: 1451–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tsuji S 2010. Genetics of neurodegenerative diseases: insights from high-throughput resequencing. Hum Mol Genet. 19: R65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hudry E, Dashkoff J, Roe AD, et al. 2013. Gene transfer of human Apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 5: 212ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Vilarino-Guell C, Rajput A, Milnerwood AJ, et al. 2014. DNAJC13 mutations in Parkinson disease. Hum Mol Genet. 23: 1794–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kirkitadze MD, Bitan G&Teplow DB 2002. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 69: 567–577. [DOI] [PubMed] [Google Scholar]
- 145.Cheng HC, Ulane CM&Burke RE 2010. Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol. 67: 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chien DT, Szardenings AK, Bahri S, et al. 2014. Early clinical PET imaging results with the novel PHF-tau radioligand [F18]-T808. J Alzheimers Dis. 38: 171–184. [DOI] [PubMed] [Google Scholar]
- 147.Dani M, Brooks DJ&Edison P 2016. Tau imaging in neurodegenerative diseases. Eur J Nucl Med Mol Imaging. 43: 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jack CR Jr.&Holtzman DM 2013. Biomarker modeling of Alzheimer’s disease. Neuron. 80: 1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jack CR Jr., Knopman DS, Jagust WJ, et al. 2013. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12: 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Abisambra JF, Jinwal UK, Blair LJ, et al. 2013. Tau accumulation activates the unfolded protein response by impairing endoplasmic reticulum-associated degradation. J Neurosci. 33: 9498–9507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Ehrlich M, Hallmann AL, Reinhardt P, et al. 2015. Distinct Neurodegenerative Changes in an Induced Pluripotent Stem Cell Model of Frontotemporal Dementia Linked to Mutant TAU Protein. Stem Cell Reports. 5: 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Heywood WE, Galimberti D, Bliss E, et al. 2015. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol Neurodegener. 10: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Iovino M, Agathou S, Gonzalez-Rueda A, et al. 2015. Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain. 138: 3345–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Usenovic M, Niroomand S, Drolet RE, et al. 2015. Internalized Tau Oligomers Cause Neurodegeneration by Inducing Accumulation of Pathogenic Tau in Human Neurons Derived from Induced Pluripotent Stem Cells. J Neurosci. 35: 14234–14250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wan W, Cao L, Kalionis B, et al. 2015. Applications of Induced Pluripotent Stem Cells in Studying the Neurodegenerative Diseases. Stem Cells Int. 2015: 382530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wen X, Tan W, Westergard T, et al. 2014. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 84: 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wren MC, Zhao J, Liu CC, et al. 2015. Frontotemporal dementia-associated N279K tau mutant disrupts subcellular vesicle trafficking and induces cellular stress in iPSC-derived neural stem cells. Mol Neurodegener. 10: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arber C, Lovejoy C&Wray S 2017. Stem cell models of Alzheimer’s disease: progress and challenges. Alzheimer’s research & therapy. 9: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Muratore CR, Rice HC, Srikanth P, et al. 2014. The familial Alzheimer’s disease APPV717I mutation alters APP processing and Tau expression in iPSC-derived neurons. Hum Mol Genet. 23: 3523–3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Raja WK, Mungenast AE, Lin YT, et al. 2016. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS One. 11: e0161969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Fong H, Wang C, Knoferle J, et al. 2013. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports. 1: 226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Seo J, Kritskiy O, Watson LA, et al. 2017. Inhibition of p25/Cdk5 Attenuates Tauopathy in Mouse and iPSC Models of Frontotemporal Dementia. J Neurosci. 37: 9917–9924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Almeida S, Zhang Z, Coppola G, et al. 2012. Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep. 2: 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Almeida S, Gascon E, Tran H, et al. 2013. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 126: 385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pasca SP 2018. The rise of three-dimensional human brain cultures. Nature. 553: 437–445. [DOI] [PubMed] [Google Scholar]
- 166.Shi Y, Lin S, Staats KA, et al. 2018. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 24: 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Devlin AC, Burr K, Borooah S, et al. 2015. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 6: 5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sareen D, O’Rourke JG, Meera P, et al. 2013. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med. 5: 208ra149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bilican B, Serio A, Barmada SJ, et al. 2012. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 109: 5803–5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Egawa N, Kitaoka S, Tsukita K, et al. 2012. Drug screening for ALS using patient-specific induced pluripotent stem cells. Sci Transl Med. 4: 145ra104. [DOI] [PubMed] [Google Scholar]
- 171.Jeon I, Lee N, Li JY, et al. 2012. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 30: 2054–2062. [DOI] [PubMed] [Google Scholar]
- 172.Hou PS, Chuang CY, Yeh CH, et al. 2017. Direct Conversion of Human Fibroblasts into Neural Progenitors Using Transcription Factors Enriched in Human ESC-Derived Neural Progenitors. Stem Cell Reports. 8: 54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sullivan PF, Daly MJ&O’Donovan M 2012. Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat Rev Genet. 13: 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Neale BM&Sklar P 2015. Genetic analysis of schizophrenia and bipolar disorder reveals polygenicity but also suggests new directions for molecular interrogation. Curr Opin Neurobiol. 30: 131–138. [DOI] [PubMed] [Google Scholar]
- 175.Giegling I, Hosak L, Mossner R, et al. 2017. Genetics of schizophrenia: A consensus paper of the WFSBP Task Force on Genetics. World J Biol Psychiatry. 18: 492–505. [DOI] [PubMed] [Google Scholar]
- 176.Bakhshi K&Chance SA 2015. The neuropathology of schizophrenia: A selective review of past studies and emerging themes in brain structure and cytoarchitecture. Neuroscience. 303: 82–102. [DOI] [PubMed] [Google Scholar]
- 177.Haggarty SJ&Perlis RH 2014. Translation: screening for novel therapeutics with disease-relevant cell types derived from human stem cell models. Biol Psychiatry. 75: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Seok J, Warren HS, Cuenca AG, et al. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 110: 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Soliman MA, Aboharb F, Zeltner N, et al. 2017. Pluripotent stem cells in neuropsychiatric disorders. Mol Psychiatry. 22: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Bhattacharyya A&Zhao X 2016. Human pluripotent stem cell models of Fragile X syndrome. Mol Cell Neurosci. 73: 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Telias M, Kuznitsov-Yanovsky L, Segal M, et al. 2015. Functional Deficiencies in Fragile X Neurons Derived from Human Embryonic Stem Cells. J Neurosci. 35: 15295–15306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Prytkova I&Brennand KJ 2017. Prospects for Modeling Abnormal Neuronal Function in Schizophrenia Using Human Induced Pluripotent Stem Cells. Front Cell Neurosci. 11: 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brennand KJ, Simone A, Jou J, et al. 2011. Modelling schizophrenia using human induced pluripotent stem cells. Nature. 473: 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chiang CH, Su Y, Wen Z, et al. 2011. Integration-free induced pluripotent stem cells derived from schizophrenia patients with a DISC1 mutation. Mol Psychiatry. 16: 358–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Pedrosa E, Sandler V, Shah A, et al. 2011. Development of patient-specific neurons in schizophrenia using induced pluripotent stem cells. J Neurogenet. 25: 88–103. [DOI] [PubMed] [Google Scholar]
- 186.Prabakaran S, Swatton JE, Ryan MM, et al. 2004. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 9: 684–697, 643. [DOI] [PubMed] [Google Scholar]
- 187.Bavamian S, Mellios N, Lalonde J, et al. 2015. Dysregulation of miR-34a links neuronal development to genetic risk factors for bipolar disorder. Mol Psychiatry. 20: 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chen HM, DeLong CJ, Bame M, et al. 2014. Transcripts involved in calcium signaling and telencephalic neuronal fate are altered in induced pluripotent stem cells from bipolar disorder patients. Transl Psychiatry. 4: e375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Madison JM, Zhou F, Nigam A, et al. 2015. Characterization of bipolar disorder patient-specific induced pluripotent stem cells from a family reveals neurodevelopmental and mRNA expression abnormalities. Mol Psychiatry. 20: 703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mertens J, Wang QW, Kim Y, et al. 2015. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature. 527: 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Urbach A, Bar-Nur O, Daley GQ, et al. 2010. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 6: 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Marchetto MC, Carromeu C, Acab A, et al. 2010. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell. 143: 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Farra N, Zhang WB, Pasceri P, et al. 2012. Rett syndrome induced pluripotent stem cell-derived neurons reveal novel neurophysiological alterations. Mol Psychiatry. 17: 1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kim KY, Hysolli E&Park IH 2011. Neuronal maturation defect in induced pluripotent stem cells from patients with Rett syndrome. Proc Natl Acad Sci U S A. 108: 14169–14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wen Z, Nguyen HN, Guo Z, et al. 2014. Synaptic dysregulation in a human iPS cell model of mental disorders. Nature. 515: 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Thomson PA, Malavasi EL, Grunewald E, et al. 2013. DISC1 genetics, biology and psychiatric illness. Front Biol (Beijing). 8: 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Mariani J, Coppola G, Zhang P, et al. 2015. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell. 162: 375–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Huo HQ, Qu ZY, Yuan F, et al. 2018. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Reports. 10: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Qian X, Nguyen HN, Song MM, et al. 2016. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell. 165: 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gingold J, Zhou R, Lemischka IR, et al. 2016. Modeling Cancer with Pluripotent Stem Cells. Trends Cancer. 2: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Sancho-Martinez I, Nivet E, Xia Y, et al. 2016. Establishment of human iPSC-based models for the study and targeting of glioma initiating cells. Nat Commun. 7: 10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Bradshaw A, Wickremsekera A, Tan ST, et al. 2016. Cancer Stem Cell Hierarchy in Glioblastoma Multiforme. Front Surg. 3: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.O’Duibhir E, Carragher NO&Pollard SM 2017. Accelerating glioblastoma drug discovery: Convergence of patient-derived models, genome editing and phenotypic screening. Mol Cell Neurosci. 80: 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Semi K&Yamada Y 2015. Induced pluripotent stem cell technology for dissecting the cancer epigenome. Cancer Sci. 106: 1251–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Inoue H, Nagata N, Kurokawa H, et al. 2014. iPS cells: a game changer for future medicine. EMBO J. 33: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Inoue H&Yamanaka S 2011. The use of induced pluripotent stem cells in drug development. Clin Pharmacol Ther. 89: 655–661. [DOI] [PubMed] [Google Scholar]
- 207.Kaufmann M, Schuffenhauer A, Fruh I, et al. 2015. High-Throughput Screening Using iPSC-Derived Neuronal Progenitors to Identify Compounds Counteracting Epigenetic Gene Silencing in Fragile X Syndrome. J Biomol Screen. 20: 1101–1111. [DOI] [PubMed] [Google Scholar]
- 208.Abelin JG, Patel J, Lu X, et al. 2016. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Mol Cell Proteomics. 15: 1622–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Soares FA, Sheldon M, Rao M, et al. 2014. International coordination of large-scale human induced pluripotent stem cell initiatives: Wellcome Trust and ISSCR workshops white paper. Stem Cell Reports. 3: 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Morrison M, Klein C, Clemann N, et al. 2015. StemBANCC: Governing Access to Material and Data in a Large Stem Cell Research Consortium. Stem Cell Rev. 11: 681–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Gelernter J 2015. Genetics of complex traits in psychiatry. Biol Psychiatry. 77: 36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Macaulay IC, Ponting CP&Voet T 2017. Single-Cell Multiomics: Multiple Measurements from Single Cells. Trends Genet. 33: 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Plenge RM, Scolnick EM&Altshuler D 2013. Validating therapeutic targets through human genetics. Nat Rev Drug Discov. 12: 581–594. [DOI] [PubMed] [Google Scholar]
- 214.Collins FS&Varmus H 2015. A new initiative on precision medicine. N Engl J Med. 372: 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Rouaux C, Loeffler JP&Boutillier AL 2004. Targeting CREB-binding protein (CBP) loss of function as a therapeutic strategy in neurological disorders. Biochem Pharmacol. 68: 1157–1164. [DOI] [PubMed] [Google Scholar]
- 216.Ryu H, Smith K, Camelo SI, et al. 2005. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 93: 1087–1098. [DOI] [PubMed] [Google Scholar]
- 217.Chouliaras L, Mastroeni D, Delvaux E, et al. 2013. Consistent decrease in global DNA methylation and hydroxymethylation in the hippocampus of Alzheimer’s disease patients. Neurobiol Aging. 34: 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.MacLean B, Tomazela DM, Shulman N, et al. 2010. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26: 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Mair W, Muntel J, Tepper K, et al. 2016. FLEXITau: Quantifying Post-translational Modifications of Tau Protein in vitro and in Human Disease. Anal Chem. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sanders DW, Kaufman SK, DeVos SL, et al. 2014. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 82: 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Singh S, Springer M, Steen J, et al. 2009. FLEXIQuant: a novel tool for the absolute quantification of proteins, and the simultaneous identification and quantification of potentially modified peptides. J Proteome Res. 8: 2201–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Boutros M, Heigwer F&Laufer C 2015. Microscopy-Based High-Content Screening. Cell. 163: 1314–1325. [DOI] [PubMed] [Google Scholar]
- 223.Sirenko O, Hesley J, Rusyn I, et al. 2014. High-content high-throughput assays for characterizing the viability and morphology of human iPSC-derived neuronal cultures. Assay Drug Dev Technol. 12: 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Wheeler HE, Wing C, Delaney SM, et al. 2015. Modeling chemotherapeutic neurotoxicity with human induced pluripotent stem cell-derived neuronal cells. PLoS One. 10: e0118020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Finkbeiner S, Frumkin M&Kassner PD 2015. Cell-based screening: extracting meaning from complex data. Neuron. 86: 160–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Lamb J, Crawford ED, Peck D, et al. 2006. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 313: 1929–1935. [DOI] [PubMed] [Google Scholar]
- 227.Heikkila TJ, Yla-Outinen L, Tanskanen JM, et al. 2009. Human embryonic stem cell-derived neuronal cells form spontaneously active neuronal networks in vitro. Exp Neurol. 218: 109–116. [DOI] [PubMed] [Google Scholar]
- 228.Kiskinis E, Sandoe J, Williams LA, et al. 2014. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 14: 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bassett AR 2017. Editing the genome of hiPSC with CRISPR/Cas9: disease models. Mamm Genome. 28: 348–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Chhatwal JP, Schultz AP, Marshall GA, et al. 2016. Temporal T807 binding correlates with CSF tau and phospho-tau in normal elderly. Neurology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Marquie M, Normandin MD, Vanderburg CR, et al. 2015. Validating novel tau positron emission tomography tracer [F-18]-AV-1451 (T807) on postmortem brain tissue. Ann Neurol. 78: 787–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Nguyen HN, Byers B, Cord B, et al. 2011. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 8: 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Stoveken BJ 2013. Tau pathology as a cause and consequence of the UPR. J Neurosci. 33: 14285–14287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Executive Office of the President, O. o. S. a. T. P. Priorities for personalized medicine. Report of the President’s Council of Advisors on Science and Technology. http://www.whitehouse.gov/files/documents/ostp/PCAST/pcast_report_v2.pdf.
- 235.Garbes L, Heesen L, Holker I, et al. 2013. VPA response in SMA is suppressed by the fatty acid translocase CD36. Hum Mol Genet. 22: 398–407. [DOI] [PubMed] [Google Scholar]
- 236.Haston KM&Finkbeiner S 2016. Clinical Trials in a Dish: The Potential of Pluripotent Stem Cells to Develop Therapies for Neurodegenerative Diseases. Annu Rev Pharmacol Toxicol. 56: 489–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hung SSC, Khan S, Lo CY, et al. 2017. Drug discovery using induced pluripotent stem cell models of neurodegenerative and ocular diseases. Pharmacol Ther. 177: 32–43. [DOI] [PubMed] [Google Scholar]
- 238.Elitt MS, Barbar L&Tesar PJ 2018. Drug screening for human genetic diseases using iPSC models. Hum Mol Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kessler RC, Aguilar-Gaxiola S, Alonso J, et al. 2009. The global burden of mental disorders: an update from the WHO World Mental Health (WMH) surveys. Epidemiol Psichiatr Soc. 18: 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Pini G, Scusa MF, Benincasa A, et al. 2014. Repeated insulin-like growth factor 1 treatment in a patient with rett syndrome: a single case study. Front Pediatr. 2: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.McNeish J, Roach M, Hambor J, et al. 2010. High-throughput screening in embryonic stem cell-derived neurons identifies potentiators of alpha-amino-3-hydroxyl-5-methyl-4-isoxazolepropionate-type glutamate receptors. J Biol Chem. 285: 17209–17217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.McLaren D, Gorba T, Marguerie de Rotrou A, et al. 2013. Automated large-scale culture and medium-throughput chemical screen for modulators of proliferation and viability of human induced pluripotent stem cell-derived neuroepithelial-like stem cells. J Biomol Screen. 18: 258–268. [DOI] [PubMed] [Google Scholar]
- 243.Kumari D, Swaroop M, Southall N, et al. 2015. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl Med. 4: 800–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.O’Roak BJ, Vives L, Fu W, et al. 2012. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 338: 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Okerlund ND&Cheyette BN 2011. Synaptic Wnt signaling-a contributor to major psychiatric disorders? J Neurodev Disord. 3: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Price DL, Sisodia SS&Borchelt DR 1998. Genetic neurodegenerative diseases: the human illness and transgenic models. Science. 282: 1079–1083. [DOI] [PubMed] [Google Scholar]
- 247.Cooper O, Seo H, Andrabi S, et al. 2012. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 4: 141ra190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Peng J, Liu Q, Rao MS, et al. 2013. Using human pluripotent stem cell-derived dopaminergic neurons to evaluate candidate Parkinson’s disease therapeutic agents in MPP+ and rotenone models. J Biomol Screen. 18: 522–533. [DOI] [PubMed] [Google Scholar]
- 249.Wimalasena NK, Le VQ, Wimalasena K, et al. 2016. Gene Expression-Based Screen for Parkinson’s Disease Identifies GW8510 as a Neuroprotective Agent. ACS Chem Neurosci. 7: 857–863. [DOI] [PubMed] [Google Scholar]
- 250.Lee WC, Almeida S, Prudencio M, et al. 2014. Targeted manipulation of the sortilin-progranulin axis rescues progranulin haploinsufficiency. Hum Mol Genet. 23: 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.She A, Kurtser I, Reis SA, et al. 2017. Selectivity and Kinetic Requirements of HDAC Inhibitors as Progranulin Enhancers for Treating Frontotemporal Dementia. Cell Chem Biol. 24: 892–906 e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Holler CJ, Taylor G, McEachin ZT, et al. 2016. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: a novel therapeutic lead to treat frontotemporal dementia. Mol Neurodegener. 11: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Balez R, Steiner N, Engel M, et al. 2016. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci Rep. 6: 31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Brownjohn PW, Smith J, Portelius E, et al. 2017. Phenotypic Screening Identifies Modulators of Amyloid Precursor Protein Processing in Human Stem Cell Models of Alzheimer’s Disease. Stem Cell Reports. 8: 870–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Yang YM, Gupta SK, Kim KJ, et al. 2013. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 12: 713–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Burkhardt MF, Martinez FJ, Wright S, et al. 2013. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 56: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Wainger BJ, Kiskinis E, Mellin C, et al. 2014. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 7: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Simone R, Balendra R, Moens TG, et al. 2018. G-quadruplex-binding small molecules ameliorate C9orf72 FTD/ALS pathology in vitro and in vivo. EMBO Mol Med. 10: 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Lee G, Ramirez CN, Kim H, et al. 2012. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 30: 1244–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Cao L, McDonnell A, Nitzsche A, et al. 2016. Pharmacological reversal of a pain phenotype in iPSC-derived sensory neurons and patients with inherited erythromelalgia. Sci Transl Med. 8: 335ra356. [DOI] [PubMed] [Google Scholar]
- 261.Hothi P, Martins TJ, Chen L, et al. 2012. High-throughput chemical screens identify disulfiram as an inhibitor of human glioblastoma stem cells. Oncotarget. 3: 1124–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Danovi D, Folarin A, Gogolok S, et al. 2013. A high-content small molecule screen identifies sensitivity of glioblastoma stem cells to inhibition of polo-like kinase 1. PLoS One. 8: e77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Quartararo CE, Reznik E, deCarvalho AC, et al. 2015. High-Throughput Screening of Patient-Derived Cultures Reveals Potential for Precision Medicine in Glioblastoma. ACS Med Chem Lett. 6: 948–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Schmidt L, Kling T, Monsefi N, et al. 2013. Comparative drug pair screening across multiple glioblastoma cell lines reveals novel drug-drug interactions. Neuro Oncol. 15: 1469–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Quereda V, Hou S, Madoux F, et al. 2018. A Cytotoxic Three-Dimensional-Spheroid, High-Throughput Assay Using Patient-Derived Glioma Stem Cells. SLAS Discov. 2472555218775055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Watmuff B, Berkovitch SS, Huang JH, et al. 2016. Disease signatures for schizophrenia and bipolar disorder using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 73: 96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Jacquemont S, Curie A, des Portes V, et al. 2011. Epigenetic modification of the FMR1 gene in fragile X syndrome is associated with differential response to the mGluR5 antagonist AFQ056. Sci Transl Med. 3: 64ra61. [DOI] [PubMed] [Google Scholar]
- 268.Stern S, Linker S, Vadodaria KC, et al. 2018. Prediction of response to drug therapy in psychiatric disorders. Open Biol. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Tortelli R, Seripa D, Panza F, et al. 2016. Pharmacogenetics in Neurodegenerative Diseases: Implications for Clinical Trials. Front Neurol Neurosci. 39: 124–135. [DOI] [PubMed] [Google Scholar]
- 270.Shinde V, Sureshkumar P, Sotiriadou I, et al. 2016. Human Embryonic and Induced Pluripotent Stem Cell Based Toxicity Testing Models: Future Applications in New Drug Discovery. Curr Med Chem. [DOI] [PubMed] [Google Scholar]
- 271.Deshmukh RS, Kovacs KA&Dinnyes A 2012. Drug discovery models and toxicity testing using embryonic and induced pluripotent stem-cell-derived cardiac and neuronal cells. Stem Cells Int. 2012: 379569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Medine CN, Lucendo-Villarin B, Storck C, et al. 2013. Developing high-fidelity hepatotoxicity models from pluripotent stem cells. Stem Cells Transl Med. 2: 505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Malik N, Efthymiou AG, Mather K, et al. 2014. Compounds with species and cell type specific toxicity identified in a 2000 compound drug screen of neural stem cells and rat mixed cortical neurons. Neurotoxicology. 45: 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Druwe I, Freudenrich TM, Wallace K, et al. 2015. Sensitivity of neuroprogenitor cells to chemical-induced apoptosis using a multiplexed assay suitable for high-throughput screening. Toxicology. 333: 14–24. [DOI] [PubMed] [Google Scholar]
- 275.Leite SB, Wilk-Zasadna I, Zaldivar JM, et al. 2012. Three-dimensional HepaRG model as an attractive tool for toxicity testing. Toxicol Sci. 130: 106–116. [DOI] [PubMed] [Google Scholar]
- 276.Del Alamo JC, Lemons D, Serrano R, et al. 2016. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim Biophys Acta. 1863: 1717–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Du Y, Wang J, Jia J, et al. 2014. Human hepatocytes with drug metabolic function induced from fibroblasts by lineage reprogramming. Cell Stem Cell. 14: 394–403. [DOI] [PubMed] [Google Scholar]
- 278.Ieda M, Fu JD, Delgado-Olguin P, et al. 2010. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 142: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Phillips BW, Hentze H, Rust WL, et al. 2007. Directed differentiation of human embryonic stem cells into the pancreatic endocrine lineage. Stem Cells Dev. 16: 561–578. [DOI] [PubMed] [Google Scholar]
- 280.Bergmann S, Lawler SE, Qu Y, et al. 2018. Blood-brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat Protoc. 13: 2827–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Cho CF, Wolfe JM, Fadzen CM, et al. 2017. Blood-brain-barrier spheroids as an in vitro screening platform for brain-penetrating agents. Nat Commun. 8: 15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Aday S, Cecchelli R, Hallier-Vanuxeem D, et al. 2016. Stem Cell-Based Human Blood-Brain Barrier Models for Drug Discovery and Delivery. Trends Biotechnol. 34: 382–393. [DOI] [PubMed] [Google Scholar]
- 283.Plenge RM 2016. Disciplined approach to drug discovery and early development. Sci Transl Med. 8: 349ps315. [DOI] [PubMed] [Google Scholar]
- 284.Kuenzel K, Friedrich O&Gilbert DF 2016. A Recombinant Human Pluripotent Stem Cell Line Stably Expressing Halide-Sensitive YFP-I152L for GABAAR and GlyR-Targeted High-Throughput Drug Screening and Toxicity Testing. Front Mol Neurosci. 9: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Zhang WB, Ross PJ, Tu Y, et al. 2016. Fyn Kinase regulates GluN2B subunit-dominant NMDA receptors in human induced pluripotent stem cell-derived neurons. Sci Rep. 6: 23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Dominguez AA, Lim WA&Qi LS 2016. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 17: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Doudna JA&Charpentier E 2014. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 346: 1258096. [DOI] [PubMed] [Google Scholar]
- 288.Hsu PD, Lander ES&Zhang F 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 157: 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Pei Y, Sierra G, Sivapatham R, et al. 2015. A platform for rapid generation of single and multiplexed reporters in human iPSC lines. Sci Rep. 5: 9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Cook D, Brown D, Alexander R, et al. 2014. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 13: 419–431. [DOI] [PubMed] [Google Scholar]
- 291.Waring MJ, Arrowsmith J, Leach AR, et al. 2015. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov. 14: 475–486. [DOI] [PubMed] [Google Scholar]
- 292.Hadida S, Van Goor F, Zhou J, et al. 2014. Discovery of N-(2,4-di-tert-butyl-5-hydroxyphenyl)-4-oxo-1,4-dihydroquinoline-3-carboxamide (VX-770, ivacaftor), a potent and orally bioavailable CFTR potentiator. J Med Chem. 57: 9776–9795. [DOI] [PubMed] [Google Scholar]
- 293.Kaiser T&Feng G 2015. Modeling psychiatric disorders for developing effective treatments. Nat Med. 21: 979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Heilker R, Traub S, Reinhardt P, et al. 2014. iPS cell derived neuronal cells for drug discovery. Trends Pharmacol Sci. 35: 510–519. [DOI] [PubMed] [Google Scholar]
- 295.Biundo R, Weis L, Bostantjopoulou S, et al. 2016. MMSE and MoCA in Parkinson’s disease and dementia with Lewy bodies: a multicenter 1-year follow-up study. J Neural Transm (Vienna). 123: 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Moody CJ, Mitchell D, Kiser G, et al. 2017. Maximizing the Potential of Longitudinal Cohorts for Research in Neurodegenerative Diseases: A Community Perspective. Front Neurosci. 11: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Thompson PM, Stein JL, Medland SE, et al. 2014. The ENIGMA Consortium: large-scale collaborative analyses of neuroimaging and genetic data. Brain Imaging Behav. 8: 153–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wu H, Xu J, Pang ZP, et al. 2007. Integrative genomic and functional analyses reveal neuronal subtype differentiation bias in human embryonic stem cell lines. Proc Natl Acad Sci U S A. 104: 13821–13826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.D’Gama AM&Walsh CA 2018. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci. 21: 1504–1514. [DOI] [PubMed] [Google Scholar]
- 300.Lin M, Lachman HM&Zheng D 2016. Transcriptomics analysis of iPSC-derived neurons and modeling of neuropsychiatric disorders. Mol Cell Neurosci. 73: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.McConnell MJ, Lindberg MR, Brennand KJ, et al. 2013. Mosaic copy number variation in human neurons. Science. 342: 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rohrback S, Siddoway B, Liu CS, et al. 2018. Genomic mosaicism in the developing and adult brain. Dev Neurobiol. 78: 1026–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Handel AE, Chintawar S, Lalic T, et al. 2016. Assessing similarity to primary tissue and cortical layer identity in induced pluripotent stem cell-derived cortical neurons through single-cell transcriptomics. Hum Mol Genet. 25: 989–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Stein JL, de la Torre-Ubieta L, Tian Y, et al. 2014. A quantitative framework to evaluate modeling of cortical development by neural stem cells. Neuron. 83: 69–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Miller JD, Ganat YM, Kishinevsky S, et al. 2013. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 13: 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Vera E, Bosco N&Studer L 2016. Generating Late-Onset Human iPSC-Based Disease Models by Inducing Neuronal Age-Related Phenotypes through Telomerase Manipulation. Cell Rep. 17: 1184–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Sherman SP&Bang AG 2018. High-throughput screen for compounds that modulate neurite growth of human induced pluripotent stem cell-derived neurons. Dis Model Mech. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Stefaniak JD, Lam TCH, Sim NE, et al. 2017. Discontinuation and non-publication of neurodegenerative disease trials: a cross-sectional analysis. Eur J Neurol. 24: 1071–1076. [DOI] [PubMed] [Google Scholar]
- 309.DiMasi JA&Grabowski HG 2007. Should the patent system for new medicines be abolished? Clin Pharmacol Ther. 82: 488–490. [DOI] [PubMed] [Google Scholar]