

Abstract
Glutamate decarboxylase (GAD) catalyzes the production of γ-aminobutyric acid (GABA), a major inhibitory neurotransmitter. The mammalian brain contains two forms of GAD, with Mrs of 67,000 and 65,000 (GAD67 and GAD65). Using a new antiserum specific for GAD67 and a monoclonal antibody specific for GAD65, we show that the two forms of GAD differ in their intraneuronal distributions: GAD67 is widely distributed throughout the neuron, whereas GAD65 lies primarily in axon terminals. In brain extracts, almost all GAD67 is in an active holoenzyme form, saturated with its cofactor, pyridoxal phosphate. In contrast, only about half of GAD65 (which is found in synaptic terminals) exists as active holoenzyme. We suggest that the relative levels of apo-GAD65 and holo-GAD65 in synaptic terminals may couple GABA production to neuronal activity.
Keywords: Glutamate decarboxylase, γ-Aminobutyric acid, Intraneuronal distribution
A substantial fraction of the neurons in the vertebrate brain use glutamate decarboxylase (GAD; EC 4.1.1.15) to produce the inhibitory neurotransmitter γ-aminobutyric acid (GABA). Recent evidence shows that GAD is also present in the β-cells of the pancreas and that immunoreactive GAD polypeptides are major autoantigens in insulin-dependent diabetes mellitus (Baekkeskov et al., 1990; D. L. Kaufman et al., submitted). Immunoblotting of brain extracts with conventional antisera to GAD reveals two polypeptides with M,s of approximately 65,000 and 67,000 (GAD65 and GAD67) (Fig. 1; Kaufman et al., 1986; Legay et al., 1987; Chang and Gottlieb, 1988). Other workers have estimated the molecular sizes of these same two forms to be 59,000 and 62,000 or 63,000 (Legay et al., 1987; Chang and Gottlieb, 1988; Martin et al., 1990). Adsorption chromatography also resolves several forms of mammalian GAD, with distinctive kinetic properties (for reviews, see Erlander and Tobin, 1990; Martin, 1990), but it is not known how these forms correspond to those seen on immunoblots. The inability to study the cellular and intracellular distribution of these individual forms of GAD has frustrated attempts to understand their significance in regulating GABA production.
FIG. 1.
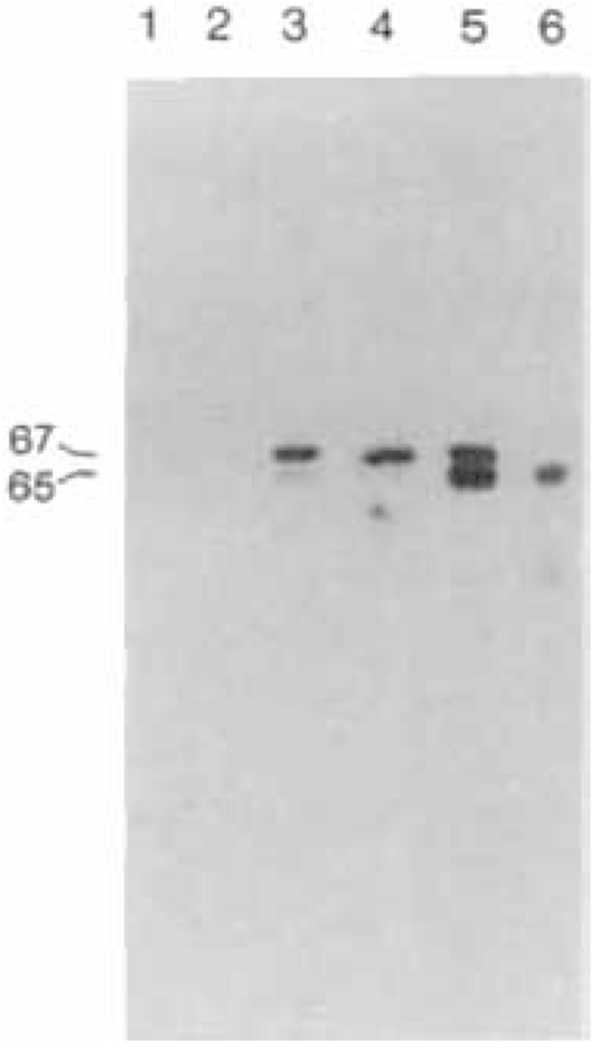
Immunoblots of mouse brain extracts. Lane 1, preimmune serum from rabbit K-1; lane 2, preimmune serum from rabbit K-2; lane 3, immune serum from rabbit K-1; lane 4, immune serum from rabbit K-2; lane 5, sheep antiserum to GAD (Oertel et al., 1981a); lane 6, GAD-6 monoclonal antibody (Chang and Gottlieb, 1988). Similar results are obtained with extracts of rat and human brain.
We previously reported the isolation of a cDNA encoding GAD67 (Kaufman et al., 1986). Using this GAD67 cDNA in a bacterial expression system, we prepared milligram amounts of enzymatically active GAD(|J, which we used to generate an antiserum specific for GAD67. We used this antiserum, together with a monoclonal antibody specific for GAD65 (Chang and Gottlieb, 1988) to show that the two forms of GAD differ in intraneuronal distribution and interaction with pyridoxal phosphate (PLP). We discuss the implications of these findings on the regulation of GABA synthesis.
MATERIALS AND METHODS
To obtain the K-1 and K-2 antisera, we used feline GAD cDNA to direct GAD synthesis in E. coli, using the bacterial expression system of Studier and Moffat (1986). Nucleotides 122-2,265 were inserted into the BamHI site of pET3B (Kobayashi et al., 1987). We purified the resulting GAD polypeptide by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and injected it into rabbits.
For immunoblotting, antisera and the GAD-6 ascite fluid were used at dilutions of 1:2,000, using methods described in Kaufman et al. (1986). For immunocytochemistry, anesthetized rats were perfused with 4% paraformaldehyde in phosphate buffer, pH 7.3. Free-floating 30-μm sections were processed for immunohistochemistry with an unlabeled antibody, using the peroxidase-antiperoxidase (PAP) method (Houser et al., 1988, 1990). Triton was omitted from all steps since its inclusion decreased cell body staining. Adjacent sections were incubated in either of the following two series of reagents: (1) normal goat serum for both series (1:30, 1 h); (2) K-2 antiserum (1:2,000–3,000, 20 h) or GAD-6 monoclonal ascites fluid (1:500–1,000, 20 h); (3) goat anti-rabbit IgG or goat anti-mouse IgG (1:75; 1 h); (4) PAP, rabbit or mouse (1:100; 1 h). The sections were then processed for 15 min in 0.006% 3,3′-diaminobenzidine-HCl and 0.006% H202, diluted in phosphate buffered saline, pH 7.3. After rinsing, the sections were treated for 30 s in 0.05% osmium tetroxide, mounted on slides, dehydrated, and coverslipped. Control sections were incubated in the same series of reagents, except for the substitution of preimmune rabbit serum or the omission of the GAD-6 antibody, and showed no specific staining.
For GAD assays, mouse brains were extracted as described previously (Kaufman et al., 1986), except that the extraction buffer did not contain PLP. Extract (100 μl) was incubated at 0°C for 40 min with K-2 antiserum (2 μl) or with GAD-6 ascites fluid (0.1 μl), followed by appropriate secondary antibodies (20 min), and precipitated with Staph A. Supernatant and precipitate were analyzed by immunoblotting to verify selective removal. Assays of the supernatant were performed in triplicate as described previously (Kaufman et al., 1986), but with 0.5 mM glutamate and 5 min assays. The (+PLP) assays contained 100 μM PLP in the assay buffer.
RESULTS AND DISCUSSION
Bacteria programmed with GAD67 cDNA produce enzymatically active GAD
On immunoblots, the bacterially produced feline GAD is specifically recognized by the GAD antisera of Oertel et al. (1981) and comigrates with GAD67 in extracts of mouse, rat, and human brains (data not shown). Lysates of GAD67-producing bacteria contained about 2,000 times more GAD activity than lysates of host bacteria. We isolated the bacterially produced GAD by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and used it to generate two rabbit antisera, which we call K-1 and K-2.
Immunoblotting with antiserum K-1 (Fig. 1, lane 3) labels both GAD65 and GAD67, as does the widely used antiserum of Oertel et al. (1981a) (Fig. 1, lane 5). The labeling of both forms by K-1 shows that the feline GAD67 cDNA encodes epitopes that are also present in GAD65. The K-1 antiserum, however, exhibited greater labeling of GAD67, whereas the Oertel antiserum showed stronger labeling of GAD65. Immunoblotting with our K-2 antiserum (Fig. 1, lane 4) specifically labels GAD67, and not GAD65. In contrast to K-2, the GAD-6 monoclonal antibody of Chang and Gottlieb (1988) specifically labels only GAD65 (Fig. 1, lane 6). We used the differences in the specificity of K-2 and GAD-6 to investigate the intraneuronal distributions and kinetic properties of GAD65 and GAD67. The specificity of K-2 for GAD67 exceeds that of previously described antisera and may in part depend upon the presence, in the bacterially produced GAD67, of an amino-terminal polypeptide consisting of 12 amino acid residues of the T7 coat protein, which may have made the amino-terminal region especially antigenic.
The overall regional distribution of GAD immunoreactivity in rat brain, as revealed by K-2, is the same as that observed with other GAD antisera (Ribak et al., 1978; Oertel et al., 1981b). At the cellular level, K-2 produced strong, specific cell body labeling of many known GAD-containing neurons, even at low concentrations of the antiserum (Fig. 2A). Previously available GAD antisera (which preferentially label GAD65) generally do not label neuronal somata, unless special fixatives are used or the animals are pretreated with colchicine to block axonal transport of GAD65 (Ribak et al., 1978).
FIG. 2.
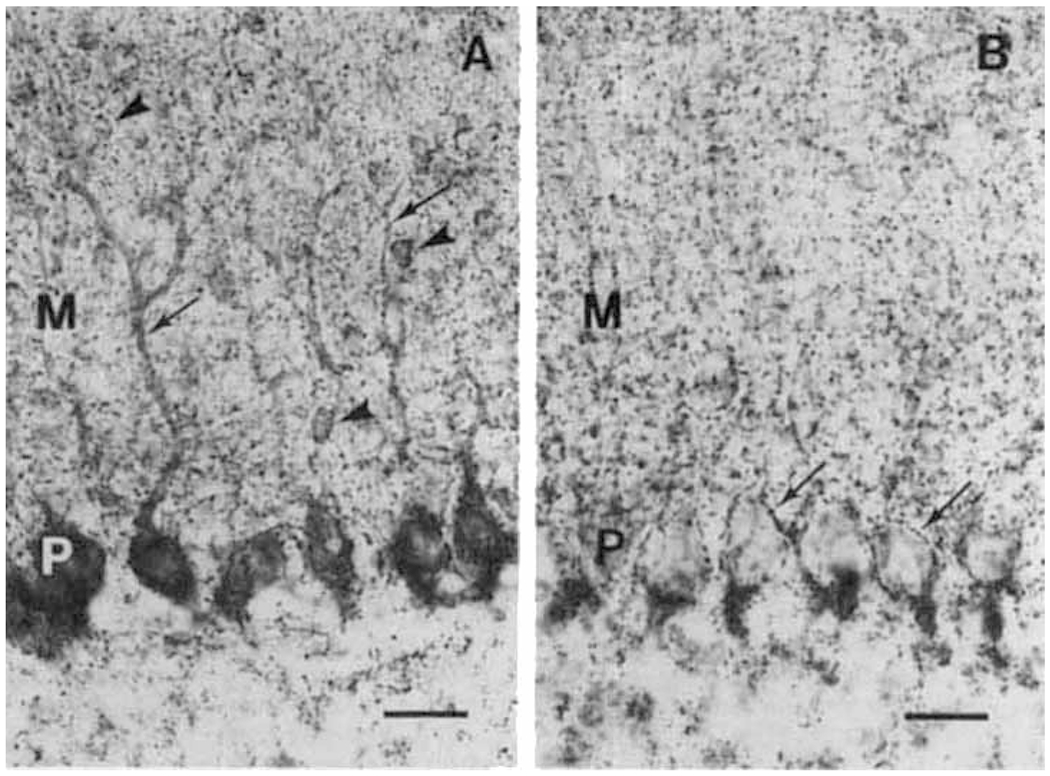
Immunohistochemical localization of GAD65 and GAD67 in rat cerebellar cortex. A. With K-2 antiserum, immunoreactivity is present in the somata of Purkinje cells (P) and extends into the primary and secondary dendrites (arrows). Labeling is also present in the somata of stellate/basket cells (arrowheads) within the molecular layer (M). Labeled puncta (presumptive axon terminals) are evident in all layers. B. With the GAD-6 monoclonal antibody, immunoreactive puncta (arrows) are present around the cell bodies of Purkinje cells (P) and are highly concentrated near the axon initial segments at the base of these neurons. Presumptive axon terminals are also distributed throughout the molecular layer (M). Little immunoreactivity is evident in neuronal cell bodies of all layers. Scale lines = 25 μm.
In the cerebellar cortex, for example, K-2 antiserum clearly labels the somata of all Purkinje neurons, as well as primary and secondary dendrites and punctate structures, which are presumably axon terminals (Fig. 2A). In contrast, GAD-6, which specifically recognizes GAD65, produces little cell body labeling in the cerebellar cortex but recognizes punctate structures (Fig. 2B). (In other brain regions, however, GAD-6 labels selected somata as well as axon terminals.) The cerebellar cortex neuronal somata thus appear to contain primarily GAD67, whereas axon terminals contain both GAD65, and GAD67.
Our data demonstrate that virtually all Purkinje cells contain immunoreactive GAD67. Additional unpublished work with colchicine-treated preparations, using GAD-6, indicates that the vast majority of Purkinje cells also contain GAD65 (C. R. Houser, in preparation). The two forms of GAD must therefore be coexpressed within Purkinje neurons but have different intracellular distributions.
In order to investigate the possible biological significance of the differential distribution of the two forms of GAD, we examined the interactions of each form of GAD with its cofactor, PLP. The association of apo-GAD and PLP to form active holo-GAD is an important regulator of GAD activity, both in vitro and in vivo (Martin, 1987).
We used the K-2 and GAD-6 antibodies to remove either GAD67 or GAD65 from brain extracts by immunoprecipitation. We then determined the remaining enzymatic activity with and without added PLP (Table 1). Assays containing added PLP should saturate apo-GAD with cofactor, to give estimates of total GAD (apo-GAD + holo-GAD) activity; assays without added PLP should measure the levels of holo-GAD present in the extract. Addition of PLP to an extract containing only GAD67 caused only a slight increase in GAD activity, suggesting that GAD67 is already saturated with cofactor. In contrast, the addition of exogenous PLP increased the activity of extracts containing only GAD65 by a factor of 2.2. These data show that GAD65 is only partly saturated with PLP. GAD65 is thus subject to regulation by PLP itself or by effectors that influence the rate of its association and dissociation with PLP (for reviews, see Martin, 1987; Erlander and Tobin, 1990). GAD67, on the other hand, is nearly saturated with PLP and its activity is probably less subject to such regulation.
TABLE 1.
Dependence of GAD65 and GAD67 activity on added PLP
| Sample | −PLP | +PLP | Stimulation by added PLP |
|---|---|---|---|
| Assay buffer only | 0.06 ± 0.006 | 0.06 ± 0.006 | 1.0 |
| Unfractionated brain extract | 4.1 ± 0.18 | 7.0 ± 0.34 | 1.7 |
| Unfractionated brain extract + preimmune serum + Staph A | 3.9 ± 0.56 | 6.4 ± 0.28 | 1.7 |
| Unfractionated brain extract + rabbit anti-mouse IgG + Staph A | 4.1 ± 0.18 | 6.6 ± 0.34 | 1.6 |
| GAD65, after removal of GAD67 with K-2 | 0.74 ± 0.02 | 1.7 ± 0.04 | 2.2a |
| GAD67, after removal of GAD65 with GAD-6 | 1.4 ± 0.04 | 1.6 ± 0.1 | 1.1a |
K-2 and GAD-6 antisera were used to selectively immunoprecipitate either GAD67 or GAD65 from brain extracts as described in Materials and Methods. The enzymatic activity of the remaining form of GAD in solution was examined with and without the addition of exogenous PLP cofactor. Data are expressed as pmol of CO2 evolved/min/μg of brain extract protein ±SEM (n = 4).
Significantly different from unfractionated brain extract (p < 0.01) by analysis of variance.
In the presence of PLP, extracts containing only GAD65 or only GAD67 have about equal GAD enzymatic activity (Table 1). Immunoblots of the material precipitated from brain extracts by K-2 and GAD-6 reveal that, although the pellet contains mainly the targeted form of GAD, some of the nontargeted form also immunoprecipitates (data not shown). Chang and Gottlieb (1988) obtained similar results with GAD-6 and suggested that the two forms of GAD can associate to form heterodimers. Because of the immunoprecipitation of these heterodimers, the sum of the activity of GAD65 and GAD67 left in solution is less than the total activity of unfractionated brain extracts (Table 1). All GAD activity removed from solution by immunoprecipitation, however, was found in the pellet (data not shown).
The two forms of GAD thus differ in their intraneuronal distributions and PLP saturation. GAD67, which is nearly saturated with PLP, is present throughout the neuron, while GAD65, which is about half-saturated with PLP, is concentrated in axon terminals. Together, these data suggest that GAD65 may act as a synaptic GAD reservoir. By stimulating the conversion of apo-GAD65 to holoenzyme, neuronal activity can increase the synaptic synthesis of GABA. Pharmacological agents that induce seizures by competing with or by binding to PLP may act chiefly by interfering with the association of GAD65 and PLP (Nitsch, 1980; Tapia, 1983).
Several groups have previously shown that Pi and ATP strongly influence the association of PLP and GAD, with Pi, increasing and ATP decreasing the rates of association (reviewed by Martin, 1987). Increases in neuronal activity would be expected to deplete ATP and increase Pi, conditions that would enhance holoenzyme formation, while low neuronal activity should decrease holoenzyme formation. Consistent with this prediction, Gold and Roth (1979) have shown that the depolarization of rat striatal neurons increases total GAD activity, whereas Miller and Walters (1979) have shown that depolarization of synaptosomes increases the level of (unfractionated) GAD holoenzyme. Our data suggest that the modulation of holo-GAD65 levels at synapses may be responsible for the observed coupling of neuronal activity and GAD enzymatic activity.
The differential localization of the two forms of GAD raises the question whether they secrete GABA into different intracellular compartments, which hold GABA for either vesicular or carrier-mediated release (Nicholls, 1989; O’Malley and Masland, 1989). It is of interest that Covarrubias and Tapia (1980) reported that GAD activity in brain homogenates could be fractionated on the basis of its binding to liposomes: liposome-bound GAD responded to PLP activation twice as much as soluble GAD. Future experiments using the K-2 and GAD-6 antisera may help elucidate whether the two forms of GAD are in fact differentially associated with membranes and whether they may release GABA into different subcellular compartments.
GABA is involved in several metabolic processes including the “GABA shunt,” which can provide energy for the cell. The differential expression and intraneuronal distribution of multiple enzymatic forms, each with distinct kinetic and regulatory properties may well represent a general mechanism by which cells regulate the production of small molecules, like GABA, that serve several physiological roles. Specifically in the case of GABA production, understanding the molecular basis of such regulation may be important to designing new therapeutic approaches to seizure and movement disorders, many of which are characterized by altered levels of GAD and GABA.
Acknowledgment:
We are grateful to David Gottlieb and Irwin Kopin for providing us with GAD antisera, to William Studier for the T7 expression system, and to David Martin and members of Allan Tobin’s and Glen Evans’ laboratories for helpful discussions. This work was supported by grants from NINDS to A.J.T. and A. V. Delgado-Escueta, and from VA Medical Research Funds to C.R.H.
Abbreviations used:
- GABA
γ-aminobutyric acid
- GAD
glutamate decarboxylase
- PLP
pyridoxal phosphate
REFERENCES
- Baekkeskov S, Aanstoot H-J, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, and De Camilli P (1990) Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature 347, 151–156. [DOI] [PubMed] [Google Scholar]
- Chang Y-C and Gottlieb DI (1988) Characterization of proteins purified with monoclonal antibodies to glutamic acid decarboxylase. J. Neurosci. 8, 2123–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias M and Tapia R (1980) Brain glutamate decarboxylase: properties of its calcium dependent binding to liposomes and kinetics of the bound and free enzyme. J. Neurochem. 34, 1682— 1688. [DOI] [PubMed] [Google Scholar]
- Erlander MG and Tobin AJ (1990) The structural and functional heterogeneity of glutamic acid decarboxylase: a review. Neurochem. Res. (in press). [DOI] [PubMed] [Google Scholar]
- Gold BI and Roth RH (1979) Glutamate decarboxylase activity in striatal slices: characterization of the increase following depolarization. J. Neurochem. 32, 883–888. [DOI] [PubMed] [Google Scholar]
- Houser CR, Olsen RW, Richards JG, and Möhler H (1988) Immunohistochemical localization of benzodiazepine/GABAA receptors in human hippocampal formation. J. Neurosci. 8, 1370–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR, Miyashiro JE, Swartz BE, Walsh GO, Rich JR, and Delgado-Escueta AV (1990) Altered patterns of dynorphin immunoreactivity suggest mossy fiber reorganization in human hippocampal epilepsy. J. Neurosci. 10, 267–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman DL, McGinnis JF, Krieger NR, and Tobin AJ (1986) Brain glutamate decarboxylase cloned in λgt-11: fusion protein produces γ-aminobutyric acid. Science 232, 1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Kaufman DL, and Tobin AJ (1987) Glutamic acid decarboxylase cDNA: nucleotide sequence encoding an enzymatically active fusion protein. J. Neurosci. 7, 2768–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legay F, Henry S, and Tappaz M (1987) Evidence for two distinct forms of native glutamic acid decarboxylase in rat brain soluble extract: an immunoblotting study. J. Neurochem. 48, 1022–1026. [DOI] [PubMed] [Google Scholar]
- Martin DL (1987) Regulatory properties of brain glutamate decarboxylase. Cell. Molec. Neurobiol. 7, 237–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DL, Wu SJ, and Martin SB (1990) Active site labeling of brain glutamate decarboxylase. J. Neurochem. 55, 524–532. [DOI] [PubMed] [Google Scholar]
- Miller LP and Walters JR (1979) Effects of depolarization on cofactor regulation of glutamic acid decarboxylase in substantia nigra synaptosomes. J. Neurochem. 33, 533–539. [DOI] [PubMed] [Google Scholar]
- Nicholls DG (1989) Release of glutamate, aspartate, and γ-aminobutyric acid from isolated nerve terminals. J. Neurochem. 52, 331–341. [DOI] [PubMed] [Google Scholar]
- Nitsch C (1980) Regulation of GABA metabolism in discrete rabbit brain regions under methoxypyridoxine—regional differences in cofactor saturation and the preictal activation of glutamate decarboxylase activity. J. Neurochem. 34, 822–830. [DOI] [PubMed] [Google Scholar]
- O’Malley DM and Masland RH (1989) Co-release of acetylcholine and γ-aminobutyric acid by a retinal neuron. Proc. Natl. Acad. Sci. USA 86, 3414–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel WH, Schmechel DE, Tappaz ML, and Kopin IJ (1981a) Production of a specific antiserum to rat brain glutamic acid decarboxylase by injection of an antigen-antibody complex. Neuroscience 6, 2689–2700. [DOI] [PubMed] [Google Scholar]
- Oertel WH, Schmechel DE, Mugnaini E, Tappaz ML, and Kopin IJ (1981b) Immunocytochemical localization of glutamate decarboxylase in rat cerebellum with a new antiserum. Neuroscience 6, 2715–2735. [DOI] [PubMed] [Google Scholar]
- Ribak CE, Vaughn JE, and Saito K (1978) Immunocytochemical localization of glutamic acid decarboxylase in neuronal somata following colchicine inhibition of axonal transport. Brain Res. 140, 315–332. [DOI] [PubMed] [Google Scholar]
- Studier F and Moffatt B (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 189, 113–130. [DOI] [PubMed] [Google Scholar]
- Tapia R (1983) Regulation of glutamate decarboxylase activity, in Glutamine, Glutamate, and GABA in the Central Nervous System (Hertz L, Kvamme E, McGeer EG, and Schousboe A, eds), pp. 113–128. Alan R. Liss, New York. [Google Scholar]