

Abstract
Glutamate decarboxylase (GAD; E.C. 4.1.1.15) converts glutamate to γ-aminobutyric acid (GABA), the major inhibitory neurotransmitter in the vertebrate central nervous system. This report describes the isolation of a GAD complementary DNA clone by immunological screening of a λgt-11 brain complementary DNA expression library. The fusion protein produced by this clone catalyzes the conversion of glutamate to GABA and carbon dioxide, confirming its identity as GAD. Antibodies to β-galactosidase remove GAD enzymatic activity from solution, showing that this activity is associated with the fusion protein. In immunoblotting experiments all three available antisera to GAD reacted with the fusion polypeptide and with two major polypeptides (molecular size, 60,000 and 66,000 daltons) in brain extracts.
BECAUSE MOTOR DISORDERS, SEIZURES, and anxiety are often associated with altered concentrations of γ-aminobutyric acid (GABA), the gene for brain glutamate decarboxylase (GAD; E.C. 4.1.1.15) has been suggested as the site of genetic lesions in a wide range of diseases (1, 2). Studies of GAD in disease and development have been hampered by the difficulty of obtaining sufficient enzyme for detailed characterization. We therefore undertook to isolate a recombinant DNA clone containing the GAD coding sequence. Although no amino acid sequence data are available for GAD, several groups have reported specific antisera to brain GAD (3–5). We have used one of these antisera to screen a bacterial expression library containing brain messenger RNA (mRNA) sequences.
Although immunological screening of bacterial expression libraries allows the efficient isolation of specific complementary DNA’s (cDNA’s), definitive identification of a cloned cDNA requires additional evidence (6–8). When amino acid sequence data are unavailable, the identity of a clone may be confirmed by characterizing the translation products of hybrid-selected mRNA’s (9, 10). These products may differ from cellular polypeptides, however, because of cross hybridization of nonidentical mRNA’s, posttranslational alterations or proteolysis of cellular polypeptides, reactivity of antibodies with related but nonidentical epitopes, or impurities in the antibody preparation. Here we report the isolation of a cDNA clone (λGAD) encoding brain GAD. After initial immunological identification, we used a functional confirmation of identity—the demonstration of GAD activity in the fusion protein encoded by λGAD.
We constructed a λgt-11 library of more than 2 × 106 recombinant clones with cDNA copied from the polyadenylated RNA of adult cat occipital cortex, a region relatively rich in GABA-containing neurons. The λgt-11 is a bacteriophage expression vector in which cDNA’s are inserted near the 3′ end of the gene for Escherichia coli β-galactosidase (6, 7). Recombinant phage induced with isopropyl-thio-β-d-galactoside (IPTG) produce a fusion protein consisting of an amino terminal β-galactosidase polypeptide linked to a carboxyl terminal polypeptide encoded by the cDNA insert.
Immunological screening was done with a sheep antiserum to GAD, prepared by Oertel et al. (4). Bound antibodies were detected with rabbit antiserum to sheep immunoglobulin G (IgG) and 125I-labeled protein A (11). Initial screening of 400,000 plaques identified 20 immunoreactive candidates, three of which remained immunoreactive in subsequent plaque purifications. DNA isolated from these three clones contained 2.3-kilobase (kb) cDNA inserts, which were identical as determined by restriction mapping.
To characterize the fusion protein, we prepared lysogens of λGAD and λgt-11 in E. coli strain Y1089, induced them with IPTG, and examined the polypeptides of each by electrophoresis and immunoblotting (12, 13). The results (Fig. 1) are consistent with the production of a fusion protein that contains antigenic sites of both β-galactosidase and GAD: (i) antibody to β-galactosidase reacts with the 116-kD β-galactosidase polypeptide in nonrecombinatit λgt-11 infected cells (lane 2) and with a larger polypeptide (>156 kD) in cells infected with λGAD (lane 3); and (ii) antibody to GAD does not react with polypeptides in the λgt-11 control, but does recognize the larger polypeptide in extracts of cells infected with λGAD (lane 5). Synthesis of the GAD antigen depends on induction with IPTG, an inducer of the lac operon (Fig. 2).
Fig. 1.
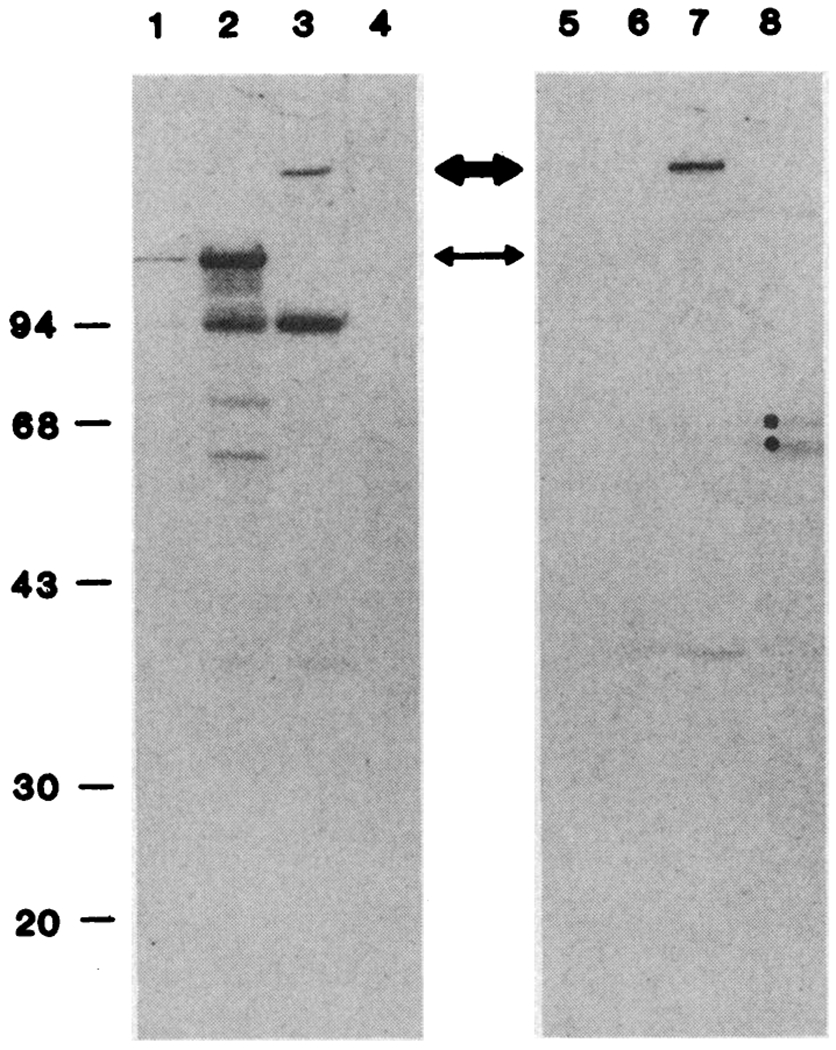
Immunological verification of GAD and β-galactosidase epitopes in the same polypeptide. Protein was extracted from λ-lysogens as described in the legend to Table 1, except that 50 mM tris-HCl, pH 7.2, replaced potassium phosphate. Brain was homogenized in a solution containing 0.2 mM PMSF, 150 mM NaCl, and 50 mM tris-HCl, pH 8.0, and the protein was extracted. Cell debris was removed by centrifugation. The proteins were separated by electrophoresis in a 10% polyacrylamide gel in SDS (23) and electrophoretically transferred to nitrocellulose (13, 24). The unreacted sites were blocked with bovine serum albumin and gelatin, and the nitrocellulose sheet was cut in half. One half was incubated overnight with a 1:1000 dilution of affinity-purified rabbit antibody to β-galactosidase and the other half with a 1:500 dilution of sheep antibody to GAD; the sheets were then incubated for 1 hour with a 1:1000 dilution of rabbit antiserum to sheep IgG. The sheet was extensively washed, and bound antibodies were detected with 125I-labeled protein A and autoradiography. (Lanes 1 to 4) Detection of β-galactosidase epitopes. (Lanes 5 to 8) Detection of GAD epitopes. Each lane contained the following: lanes 1 ana 5, purified β-galactosidase (200 ng); lanes 2 and 6, λgt-11 lysogen extracts (20 μg); lanes 3 and 7, λGAD lysogen extracts (20 μg); and lanes 4 and 8, extracts of cat brain (20 μg). A ninth lane (not shown) contained 125I-labeled molecular weight standards. The thin arrow marks the position of wild-type β-gaiactosidase, and the thick arrow marks die fusion protein produced by λGAD.
Fig. 2.
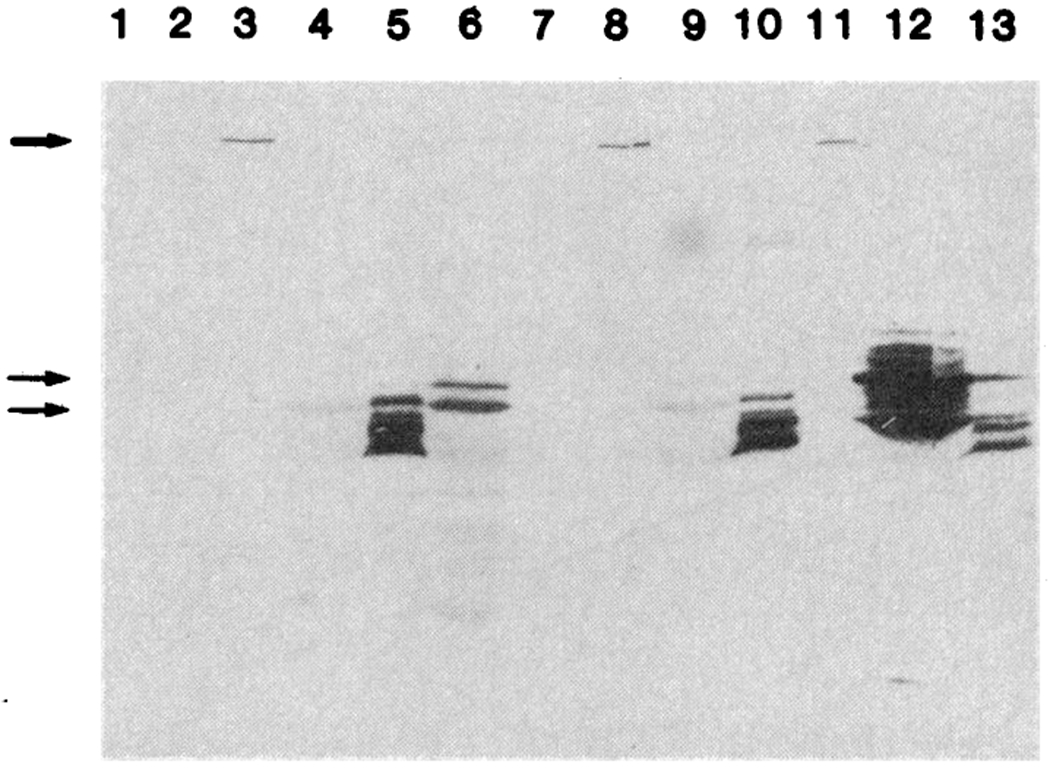
Immunoblotting of brain extracts with three antibodies to GAD. Extracts of lysogens and brain were analyzed by electrophoresis in 10% polyacrylamide gel in SDS followed by immunoblotting as described in the legend to Fig. 1. The nitrocellulose sheet, was cut into three parts, and each was incubated with a 1:500 dilution of a different antiserum to GAD: lanes 1 to 7 with sheep antiserum to rat GAD (4), lanes 8 to 10 with sheep antiserum to pig GAD (25), and lanes 11 to 13 with rabbit antiserum to mouse GAD (3). These antisera specifically recognize the fusion polypeptide (thick arrow) and at least two major polypeptides (60 and 66 kD) in cat brain (thin arrows). Although these two polypeptides are weak in lane 7, they can be seen on longer exposures. Similar results were obtained when electrophoresis was performed in 7.5% polyacrylamide. The following extracts were examined: Lane 1, λgt-11 lysogen (20 μg); lane 2, λGAD lysogen without IPTG induction (20 μg); lanes 3, 8, and 11, λGAD lysogen after IPTG induction (20 μg); lanes 4, 9, and 12, cat brain (20 μg); lanes 5, 10, and 13, partially purified rat GAD (25) (4 μg); lane 6, mouse brain (20 μg); and lane 7, human brain (20 μg).
In cat brain extracts, antibody to β-galactosidase does not react with any polypeptide (lane 4), whereas antibody to GAD reacts with both a 60-kD and a 66-kD polypeptide (lane 8). From the size of the fusion polypeptide we estimate that the GAD polypeptide segment is >400 amino acid residues long, corresponding to more than 60% of the 66-kD GAD polypeptide.
The experiments summarized in Table 1 show that the fusion protein produced in E. coli by λGAD catalyzes the decarboxylation of glutamate. Extracts of bacteria infected with λGAD convert about 20 times as much glutamate to CO2 as extracts of cells infected with nonrecombinant λgt-11. This GAD enzymatic activity can be removed from λGAD cell extracts by treatment with antibody to β-galactosidase. After immuno-precipitation with antibody to β-galactosidase, 14CO2 production in the GAD enzymatic assay falls almost to the control level. Immunoprecipitation of control extracts (from bacteria infected with λgt-11) with antibody to β-galactosidase had no effect on basal production of 14CO2 in the assay. The limited ability of control cells to convert [l4C]glutamate to 14CO2 could result either from endogenous bacterial GAD or from the oxidative deamination of glutamate and subsequent decarboxylations by Krebs cycle enzymes.
Table 1.
GAD enzymatic activity in extracts of induced lysogens. E. coli strain Y1089, lysogenized with λGAD or nonrecombinant λgt-11, was incubated at 32°C until late log phase (A550 = 0.5 of growth), and the temperature was then shifted to 44°C for 20 minutes to induce the lytic cycle (6, 7). The IPTG was added and the bacteria were incubated for an additional 60 minutes at 37°C. Bacteria were harvested, resuspended, and frozen (−70°C) in 1/40 of their original culture volume in a buffer containing 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 mM 2-aminoethylisothiouronium bromide (AET), 0.5 mM pyridoxal phosphate, and 60 mM potassium phosphate, pH 7.1. After being stored and thawed, the cells were lysed by sonication, and debris was removed by centrifugation. Protein concentration ranged from 23 to 35 mg/ml (20). The GAD enzymatic activity was determined as described (21, 22). Production of 14CO2 was linear with respect to both time of incubation and protein concentration. Enhanced GAD activity in extracts of cells infected with λGAD depended on IPTG induction. The cell lysate (200 μl) was incubated with 4 μl of IgG (20 mg/ml, Cappel) to β-galactosidase for 1 hour on ice. Enough Formalin-fixed Staphylococcus A (BRL Immunoprecipitin) was added to precipitate all IgG and the mixture was incubated again for 1 hour on ice. The bound antibody-antigen complexes were removed by centrifugation. The supernatant was recovered, reincubated with antibody and Immunoprecipitin, and centrifuged. The second supernatant was then assayed for GAD activity. Immunoprecipitation with antibody to GAD was accomplished similarly with a 1:25 dilution of sheep antiserum to rat GAD (1440-4 from I. J. Kopin) and subsequent treatment with rabbit antibody to sheep IgG (0.74 mg/ml) and Immunoprecipitin. These results were obtained from fusion protein isolated from a single 10-liter culture of λGAD in E. coli Y1089. Partially purified GAD-fusion protein was obtained by fractionation on Sepharose 6B, DEAE-cellulose, and hydroxyapatite. The fusion protein was monitored on the basis of its immunoreactivity with antibody to β-galactosidase and its GAD enzymatic activity. Purified fusion protein was stored in liquid nitrogen. After 4 months of storage, its specific activity fell by a factor of about 10 between the two experiments shown in Table 1. In the control experiments (without antibody), an equal volume of normal goat serum replaced the antibody preparations; the samples were otherwise processed as above.
| Exact | GAD activity in extracts* | |||
|---|---|---|---|---|
| Without antibody |
With antibody |
|||
| Activity | Specific activity | Activity | Specific activity | |
| Precipitation of activity with antibody to β-galactosidase | ||||
| λgt-11 lysogen | 414 ± 12 | 1.1 | 378 ± 22 | 1.0 |
| λGAD lysogen | 7772 ± 360 | 20.8 | 639 ± 24 | 1.7 |
| Fusion protein | 64463 ± 3418 | 246 | 767 ± 93 | 2.9 |
| Precipitation of activity with antibody to GAD | ||||
| λgt-11 lysogen | 327 ± 5 | 1.0 | 343 ± 17 | 1.1 |
| λGAD lysogen | 4010 ± 84 | 12.7 | 2013 ± 33 | 6.4 |
| Fusion protein | 5794 ± 215 | 23.5 | 2555 ± 50 | 10.4 |
Enzyme activity is reported as counts per minute (and range) in 14CO2 per 60 μl of reaction mixture, and specific activity as counts per minute per microgram of protein.
To purify the fusion protein away from bacterial decarboxylases, we performed gel filtration with Sepharose 6B, followed by chromatography on hydroxyapatite and DEAE-cellulose. After approximately 50-fold purification of the fusion protein, approximately 99% of the GAD activity could be removed by antibody to β-galactosidase. This result confirms that the GAD enzymatic activity and β-galactosidase epitopes are physically linked.
GAD enzymatic activity can also be removed with antibody to GAD (Table 1). Treatment with this antibody, however, was less efficient in removing enzyme activity than precipitation with antibody to β-galactosidase. This antibody removes 85% of the GAD enzymatic activity in brain extracts (4). Its lower efficiency with the fusion protein may result from (i) lack of a complete GAD coding sequence in the cDNA, (ii) the masking of epitopes by the β-galactosidase polypeptide, or (iii) altered folding or posttranslational modification in E. coli.
To confirm that the partially purified fusion protein catalyzes the production of GABA as well as of CO2, we measured GABA production by high-performance liquid chromatography of phenylthiocarbamyl (PTC)–amino acids on a Waters PICO-TAG reversed-phase column (14). PTC-GABA eluted at 5.8 minutes. In one experiment, for example, uniformly labeled [14C]glutamate was converted to equimolar amounts of 14CO2 (10,600 dis/min) and [14C]GABA (45,300 dis/min).
Studies of GAD from mice, rats, and humans suggest subunit sizes ranging from 15 to 67 kD, as judged by sedimentation equilibrium under denaturing conditions and electrophoresis of purified GAD in sodium dodecyl sulfate (SDS) (15, 16). Because of this uncertainty, we tested antisera produced to different preparations of purified GAD to ascertain whether they would all react with our fusion protein and if they would recognize the same brain polypeptides. We have used three such antisera in the immunoblotting experiments shown in Fig. 2. These antisera all reacted specifically with the fusion protein produced by GAD. All three also recognized two major polypeptides of 60 and 66 kD in human, cat, rat, and mouse brain extracts. These three antisera (two of which have been extensively used to map GABA-containing neurons) seem to recognize overlapping sets of epitopes. The smaller polypeptides recognized by these antisera may represent degradation products of a single 66-kD polypeptide.
The antigenicity and enzymatic activity of the fusion protein establishes the identity of the GAD cDNA. In addition, labeled GAD DNA hybridizes to a single electrophoretic component in polyadenylated RNA of cat and human brain, but not of liver or kidney, a result consistent with the expected distribution of GAD in the brain (1, 2, 17). Furthermore, the pattern of in situ hybridization with single-stranded GAD RNA probes with frozen sections of mouse brain is consistent with the immunocytochemically determined distribution of GABA-containing neurons (18).
Of particular note is the enzymatic activity of the fusion protein, which consists of 1006 amino acid residues of β-galactosidase and at least 400 amino acid residues of GAD, representing two-thirds or more of the total length of one of the brain GAD polypeptides. Apparently the attachment of the GAD polypeptide segment to the β-galactosidase polypeptide segment is sufficiently flexible to allow the assembly not only of immunologically detectable domains but also of the active site. Direct assays of enzyme activity or ligand binding (19) may be useful for identifying other members of bacterial expression libraries.
Acknowledgments
The antiserum to GAD used in the screening was provided through the Laboratory of Clinical Science, National Institute of Mental Health, where it was developed under the supervision of I. J. Kopin, with W. Oertel, D. Schmechel, and M. Tappaz. We thank D. Martin and J. Vaughn for providing antibodies; P. Comiso for assistance in the immunoblotting experiments; Y. Kobayashi for advice on the protein purification; J. Collopy for help in manuscript preparation; and N. Buchwald, R. Fisher, A. Fowler, S. Huttner, I. Kopin, E. Roberts, E. Askanas, K. Bugra, R. Koelling, R. Scott, L. Sussman, N. Tillakaratne, D. Wandres, T. Wood, and C. Wuenschell for help and advice. Supported by a grant from, the Dystonia Medical Research Foundation and NINCDS grants NS20356 and NS22256 (A.J.T.), NICHD grant HD05615 (J.F.M.), and USPHS National Research Service Award GM07185 (D.L.K.).
Contributor Information
Daniel L. Kaufman, Department of Biology, University of California, Los Angeles, CA 90024
James F. McGinnis, Mental Retardation Research Center and Departments of Anatomy and Obstetrics and Gynecology, UCLA School of Medicine, Los Angeles, CA 90024
Neil R. Krieger, Department of Anesthesia, Harvard Medical School and Brigham and Women’s Hospital, Boston, MA 02115
Allan J. Tobin, Department of Biology, Molecular Biology Institute, and Brain Research Institute, University of California, Los Angeles, CA 90024.
REFERENCES
- 1.Roberts E, Chase TN, Tower DB, Eds., GABA in Nervous System Function (Raven, New York, 1976). [Google Scholar]
- 2.Hertz L, Kvamme E, McGeer EG, Schousboe A, Eds., Glutamine, Glutamate and GABA in the Central Nervous System (Liss, New York, 1983). [Google Scholar]
- 3.Saito K et al. , Proc. Natl. Acad. Sci. U.S.A 71, 269 (1974).4131274 [Google Scholar]
- 4.Oertel WH, Schmechel DE, Tappaz ML, Kopin IJ, Neuroscience 6, 2689 (1981). [DOI] [PubMed] [Google Scholar]
- 5.Oertel WH, Schmechel DE, Mugnaini E, Tappaz ML, Kopin IJ, ibid., p. 2715. [DOI] [PubMed] [Google Scholar]
- 6.Young RA and Davis RW, Proc. Natl. Acad. Sci. U.S.A 80, 1194(1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.——, Science 222, 778 (1983). [DOI] [PubMed] [Google Scholar]
- 8.Kaufman DL and Tobin AJ, in Molecular and Chemical Characterization of Membrane Receptors, Harrison LC and Venter JC, Eds. (Liss, New York, 1984), pp. 241–259. [Google Scholar]
- 9.Harpold MM, Dobner PR, Evans RM, Bancroft EL, Nucleic Acids Res. 5, 2039 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ricciardi RP, Miller JS, Roberts BE, Proc. Natl. Acad. Sci. U.S.A 76, 4927 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarzbauer JE, Tamkun JW, Lemischka IR, Hynes RO, Cell 35, 421 (1983). [DOI] [PubMed] [Google Scholar]
- 12.Towbin H, Staehelin T, Gordon J, Proc. Natl. Acad. Sci. U.S.A 76, 4350 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burnette NW, Anal. Biochem 112, 195 (1981). [DOI] [PubMed] [Google Scholar]
- 14.Bidlingmeyer BA, Cohen SA, Tarvin TL, J. Chromatogr 336, 93 (1984). [DOI] [PubMed] [Google Scholar]
- 15.Wu J-Y, Matsuda T, Roberts E, J. Biol. Chem 248, 3029 (1973). [PubMed] [Google Scholar]
- 16.Blinderman J-M, Maitre M, Ossola L, Mandel P, Eur. J. Biochem 86, 143 (1978). [DOI] [PubMed] [Google Scholar]
- 17.Wood TL, Frantz GD, Menkes JH, Tobin AJ, J. Neurosci. Res, in press. [DOI] [PubMed] [Google Scholar]
- 18.Wuenschell CW, Fisher RS, Kaufman DL, Tobin AJ, Proc. Natl. Acad. Sci. U.S.A, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.For example, Sikela JM and Hahn WE, Soc. Neurosci. Abstr 11, 353 (1985). [Google Scholar]
- 20.Bradford MM, Anal. Biochem 72, 248 (1976). [DOI] [PubMed] [Google Scholar]
- 21.Krieger NR and Heller JS, J. Neurochem 33, 299 (1979). [DOI] [PubMed] [Google Scholar]
- 22.Tappaz ML, Brownstein MJ, Palkovits M, Brain Res. 108, 371 (1976). [DOI] [PubMed] [Google Scholar]
- 23.Laemmli UK, Nature (London) 227, 680 (1970). [DOI] [PubMed] [Google Scholar]
- 24.McGinnis JF and Leveille PJ, Curr. Eye Res 4, 1127 (1985). [DOI] [PubMed] [Google Scholar]
- 25.Spink DC, Porter TG, Wu SJ, Martin DL, Biochem. J 231, 695 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]