

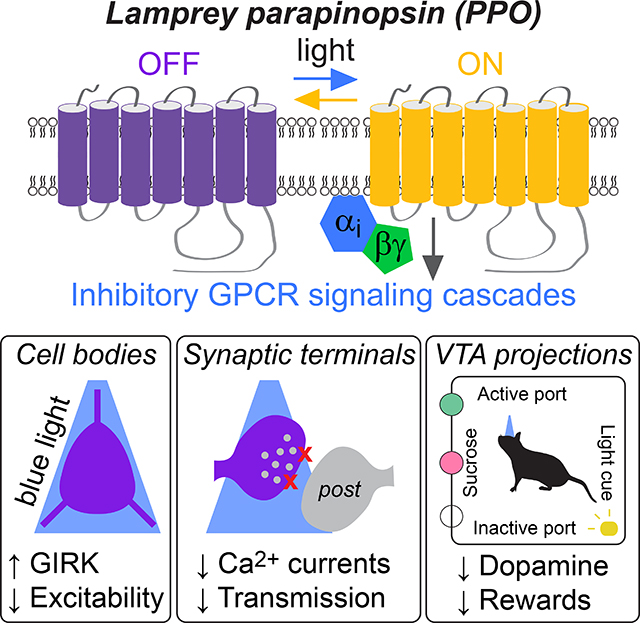
SUMMARY
Optical manipulations of genetically defined cell types have generated significant insights into the dynamics of neural circuits. While optogenetic activation has been relatively straightforward, rapid and reversible synaptic inhibition has proven more elusive. Here we leveraged the natural ability of inhibitory presynaptic GPCRs to suppress synaptic transmission and characterize parapinopsin (PPO) as a GPCR-based opsin for terminal inhibition. PPO is a photoswitchable opsin that couples to Gi/o-signaling cascades and is rapidly activated by pulsed blue light, switched off with amber light, and effective for repeated, prolonged, and reversible inhibition. PPO rapidly and reversibly inhibits glutamate, GABA, and dopamine release at presynaptic terminals. Furthermore, PPO alters reward behaviors in a time-locked and reversible manner in vivo. These results demonstrate that PPO fills a significant gap in the neuroscience toolkit for rapid and reversible synaptic inhibition and has broad utility for spatiotemporal control of inhibitory GPCR signaling cascades.
Graphical Abstract
eTOC
Optical approaches to rapidly and reversibly inhibit neuronal projections have lagged behind those for activation. Copits et al. identify a photoswitchable GPCR-based opsin that couples to inhibitory effectors. This opsin leverages the natural ability of presynaptic GPCRs to inhibit transmitter release, providing an alternative strategy to manipulate distinct synaptic projections.
INTRODUCTION
The development of molecular tools to monitor and control defined cell types has greatly accelerated our understanding of fundamental biological processes. Within the field of neuroscience, these tools have granted access to the circuit dynamics underlying complex behaviors. Approaches to manipulate neuronal activity have largely focused on optogenetic strategies using light-activated ion channels and pumps (Copits et al., 2016; Kim et al., 2017; Rost et al., 2017; Spangler and Bruchas, 2017; Wiegert et al., 2017), or chemogenetic implementations with engineered G protein coupled receptors (GPCRs) (Atasoy and Sternson, 2018; Burnett and Krashes, 2016; Roth, 2016). The primary strength of optogenetic activators has been their spatiotemporal precision in controlling neuronal firing, and more importantly their distinct projections, at millisecond timescales. However, optogenetic inhibition using ion-based opsins at synaptic terminals has proven to be far more problematic. These challenges are largely due to biophysical limitations resulting in direct depolarization or rebound spiking (Mahn et al., 2016, 2018; Raimondo et al., 2012; Wiegert et al., 2017). An alternative strategy for inhibiting synaptic projections with chemogenetic receptors took advantage of the natural ability of Gi-coupled GPCRs to inhibit synaptic transmission (Stachniak et al., 2014). However, while powerful in many applications, these chemogenetic tools lack spatial and temporal precision compared to optical approaches.
Engineered rhodopsin-GPCR (opto-XR) chimeras, in which light sensitivity is granted to different GPCRs by splicing them to the light-sensitive visual protein rhodopsin, bridges opto- and chemogenetic approaches to gain spatiotemporal control of physiological signaling cascades (Airan et al., 2009; Kleinlogel, 2016). However, existing opto-XRs are limited by high photosensitivity and irreversible activation of the rhodopsin chromophore (Ernst et al., 2014). Evolution has generated tremendous diversity in visual and non-visual opsins throughout the animal kingdom (Davies et al., 2010; Koyanagi and Terakita, 2014; Marshall et al., 2015; Terakita et al., 2015). We posited that these opsins may have evolved unique spectral and/or kinetic properties that could make them superior candidates to manipulate neural circuits. The ideal opto-GPCR would be (1) rapidly reversible, (2) exhibit moderate light sensitivity to avoid activation by ambient light, and (3) have spectral properties that overlap with existing optogenetic equipment. Additionally, opsin coupling to GPCR signaling cascades that have been evolutionarily co-opted to suppress neurotransmitter release would directly address a major limitation with currently available tools – inhibiting synaptic projections (Wiegert et al., 2017).
We tested various opsins that might possess these features, and identified lamprey parapinopsin (PPO) as a potential candidate (Eickelbeck et al., 2019; Kawano-Yamashita et al., 2015; Koyanagi and Terakita, 2014; Koyanagi et al., 2004, 2017; O’Neill et al., 2018). PPO is a non-visual G protein coupled opsin expressed in the lamprey pineal gland that can be photoswitched into on and off states when illuminated with ultraviolet vs. green/amber light (Kawano-Yamashita et al., 2015; Koyanagi and Terakita, 2014). This bistable property is thought to confer color discrimination abilities in these ancient vertebrates to sense diurnal or seasonal changes (Dodt and Meissl, 1982; Koyanagi et al., 2004; Morita et al., 1992; Uchida and Morita, 1994). We hypothesized that this feature might be harnessed for more precise manipulations of neuronal circuits.
Here we establish PPO as a photoswitchable GPCR-based opsin for rapid and reversible control of inhibitory G-protein pathways and synaptic terminal inhibition in vivo. PPO represents a unique GPCR-based tool for projection-specific presynaptic inhibition and for dissecting fundamental aspects of cellular biology and GPCR pharmacology.
RESULTS
Spectral characterization of a photoswitchable GPCR-based opsin
Lamprey parapinopsin (PPO) is a GPCR-based opsin that interconverts between on and off states by absorption of ultraviolet (UV) and amber light, respectively (Figure 1A) (Koyanagi et al., 2004). PPO was shown to couple to mammalian Gi/o proteins (Eickelbeck et al., 2019; Kawano-Yamashita et al., 2015), however UV irradiation is a potential limiting factor for use in vivo. We recently used violet light control cell migration with PPO in vitro (O’Neill et al., 2018) and hypothesized that PPO may be sensitive to even longer wavelengths, and thus more suitable in vivo.
Figure 1. Spectral characterization of a photoswitchable GPCR-based opsin.
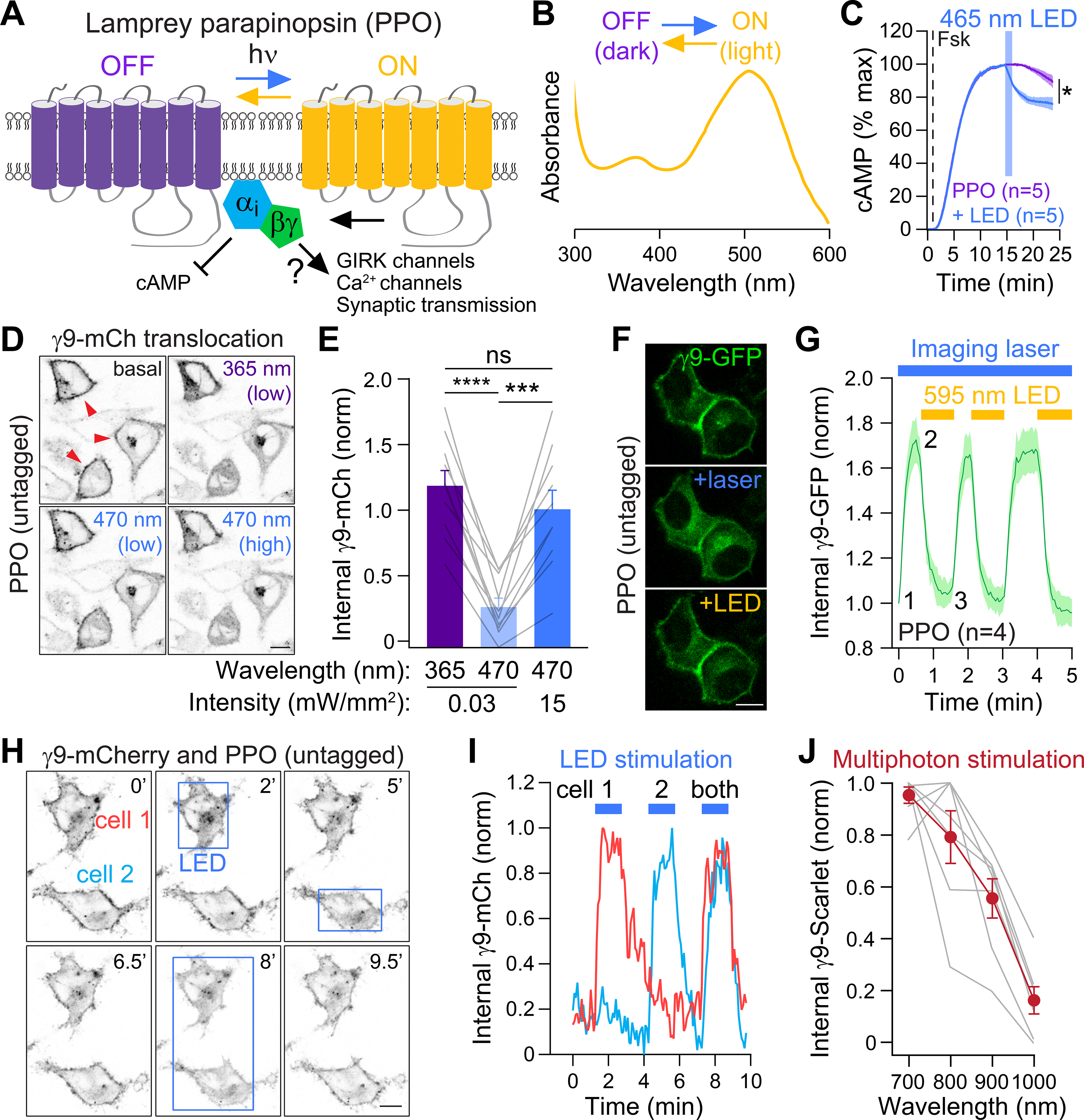
(A) Cartoon of parapinopsin (PPO), a photoswitchable GPCR that is activated by UV light and turned off by amber light.
(B) Absorption spectra of purified PPO protein in the dark off state (purple) and after UV light (amber). Graph is modified from (Koyanagi et al., 2004) (copyright 2004 National Academy of Sciences, USA)
(C) Optical stimulation with blue LED light (465 nm, 15.6 mW/cm2) inhibits forskolin-induced cAMP luminescence in PPO-expressing HEK cells. n=5
(D) Live-cell images of Gβγ translocation assays. In the dark γ9-mCherry is localized to the plasma membrane (PM) (red arrows) but translocates after stimulation of PPO with UV (365 nm) or blue (470 nm) light. Scale=10 μm
(E) Quantification of γ9-mCherry translocation following stimulation with UV vs. blue light. n=10, Paired t-test, ns=not significant, ***p<0.001, ****p<0.0001
(F) Translocation assays of PPO photoswitching. GFP-γ9 was imaged every 3s with a 488 nm laser (~45 μW), which also activated PPO and caused GFP-γ9 translocation from the PM (+laser). This was reversed by widefield illumination with an amber LED (590 nm, ~150 μW). Scale=10 μm.
(G) Quantification of γ9-GFP translocation. n=4 cells
(H) Individual photoactivation of cells expressing γ9-mCherry and untagged PPO. Boxes depict regions of blue LED photoactivation. Scale=10 μm
(I) Traces of γ9-mCherry translocation from panel H.
(J) Multiphoton activation of PPO using γ9-translocation assays. γ9-mScarlet was imaged with a Ti:Sapphire laser at 1080 nm, and multiphoton stimulation was delivered by a second laser at 700–1000 nm. n=7
We observed a tail in the blue wavelengths of the PPO absorption spectra (Figure 1B, leading us to ask if blue light sources commonly used in optogenetic experiments (Copits et al., 2016; Spangler and Bruchas, 2017) can activate PPO. We confirmed that purified PPO absorbs non-UV light (Figure S1A) and that blue LED stimulation inhibited forskolin-induced cAMP (Figures 1C, S1B). To more precisely define the spectral and temporal properties of PPO signaling in vitro, we used imaging assays of G protein translocation to monitor engagement of downstream Gβγ subunits (Karunarathne et al., 2012; O’Neill et al., 2012). We co-expressed PPO with fluorescently tagged γ9 subunits in HeLa cells and quantified γ9 fluorescence changes from the plasma membrane (PM). PPO was well expressed on the PM of both HEK cells (Figure 1D) striatal neurons (Figure S1C). Imaging of mCherry-tagged γ9 with a 595 nm laser did not affect PM localization (red arrows, Figure 1D), consistent with the action spectra of PPO in the off state (Figure 1B). Widefield illumination with low intensity UV light (365 nm, 0.03 mW/mm2) produced rapid intracellular translocation of γ9-mCherry from the PM (Figure 1D–E; Video S1). Photoactivation with blue light of the same intensity caused minimal translocation; however, at 15 mW/mm2 we observed equivalent translocation compared to UV illumination (Figure 1D–E). To examine the photoswitching properties of PPO, we imaged GFP-tagged γ9 subunits with a 488 nm blue laser (45 μW power through the spinning disk, typical for GFP imaging), which activated PPO and decreased the PM localization of GFP-γ9 (Figure 1F–G). Simultaneous illumination with an amber LED (595 nm, 150 μW), rapidly reversed GFP-γ9 back to the PM even while photostimulating PPO. We could repeatedly photoswitch PPO on and off without any apparent desensitization (Figure 1G). Together, these data indicate that UV-sensitive PPO can also be activated by blue light and turned off with amber light.
Multiplexed optical GPCR activation and imaging
The lack of spontaneous γ9 translocation while imaging, and the ability to photoswitch PPO into the off state, suggested that PPO could be used in parallel with red fluorescent proteins or sensors. We imaged γ9-mCherry and then illuminated individual cells with blue light. We observed rapid translocation of γ9-mCherry in stimulated regions without affecting adjacent cells (Figure 1H–I; Video S2). We could also achieve subcellular resolution of G protein activation (Figure S1D–E), indicating that PPO may be a useful tool for probing the subcellular dynamics of GPCR signaling (Eichel and von Zastrow, 2018; Lobingier and von Zastrow, 2019).
We next determined if this GPCR-based opsin can be combined with multiphoton imaging. We imaged γ9-mScarlet at 1080 nm, while photostimulating at different infrared wavelengths with a second tunable laser. We observed maximal G protein activation around 700 nm, which decreased at longer wavelengths tested up to 1000 nm (Figures 1J and S1F). While PPO may have utility in multiphoton optogenetics, care must be taken when combined with imaging particular fluorophores at these wavelengths to avoid unintended activation.
UV and blue light differentially engage PPO signaling
We next tested whether PPO engages downstream effectors like G-protein coupled inwardly rectifying K+ (GIRK) channels, which was recently demonstrated with UV/violet light (Eickelbeck et al., 2019; Rodgers et al., 2021) and other mammalian opsins (Siuda et al., 2015a) (Figure 2A). We co-expressed PPO-Venus with GIRK2A channels in HEK cells and recorded whole-cell currents (Figure 2B). Both blue and UV light (10 mW/mm2) produced equivalent activation of GIRK currents (Figure 2B–C). Surprisingly, even very low intensity blue light could achieve ~60% of the maximal activation seen with UV light (Figure 2C). We observed no evidence of tonic activity (Figure S2A–B) and limited activation by ambient room lighting (Figure S2C), but continued to minimize light exposure. We next tested the kinetics of PPO coupling to GIRK channels. 100 ms flashes of UV or blue light (both 10 mW/mm2) rapidly activated GIRK currents, with UV light activating PPO more rapidly (Figure 2D–E and S2D), but with similar peak amplitudes (Figure S2E). The off rates after UV illumination exhibited the most striking difference however, with a much longer decay and large steady-state currents that were switched off with amber light (Figure 2F–H and S2F). A single UV pulse caused GIRK activation with a Taudecay of 5 minutes and a persistent current for >10 minutes (Figure 2I).
Figure 2. UV and blue light cause differential coupling of PPO to GIRK channels.
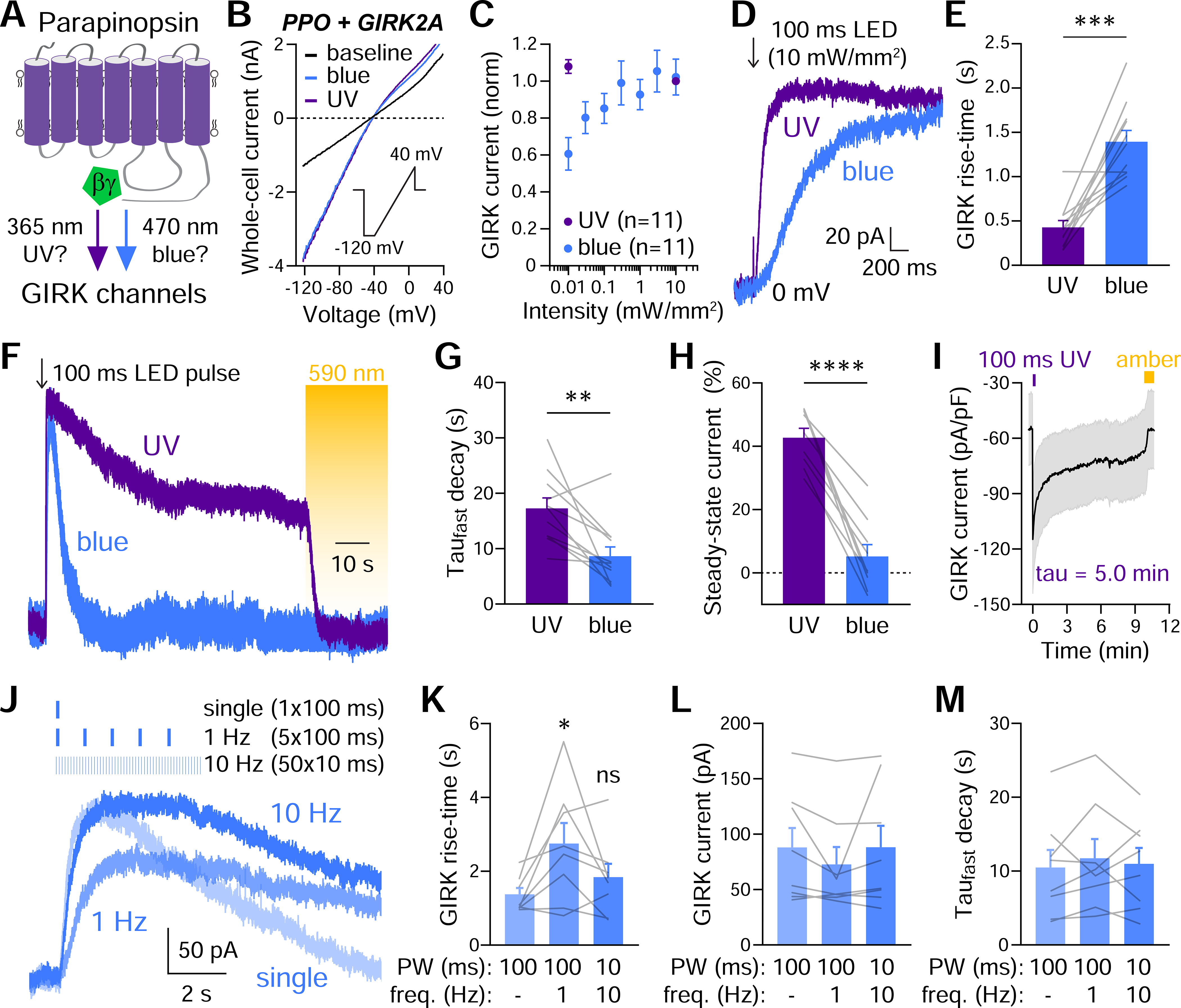
(A) Cartoon depicting UV and/or blue light coupling PPO to GIRK channels.
(B) GIRK current to voltage ramps in HEK cells transfected with PPO-Venus and GIRK2A. Baseline trace (black) and after photoactivation with 365 nm UV (purple) and 470 blue light (both 10 mW/mm2). Inset depicts the voltage protocol.
(C) GIRK dose-response curves with UV and blue light. Data are normalized to 10 mW/mm2 UV light.
(D) Outward GIRK currents recorded at 0 mV. Currents were activated more rapidly by UV (purple) than blue light (both 10 mWmm2)
(E) Summary graph of 10–90% GIRK rise-times with UV and blue light. n=11, paired t-test, ***p<0.001
(F) Prolonged GIRK channel activation by UV but not blue light (both 100 ms, 10 mW/mm2). Constant amber light (590 nm, 1 mW/mm2) was used to deactivate PPO 75 seconds after photoactivation.
(G) Summary graph of the fast decay after photoactivation with UV or blue light. n=11, paired t-test, **p<0.001
(H) Summary graph of the steady-state current sensitive to amber light. n=11, paired t-test, ****p< 0.0001
(I) GIRK currents after a single 100 ms UV pulse. GIRK activation had a decay constant of 5.0 min, remained activated for >10 minutes, and could be switched off with amber light.
(J) GIRK current traces in response to blue light pulses at 1 or 10 Hz. The number of pulses and pulse widths are indicated.
(K) Summary graph of 10–90% GIRK current rise-times with pulsed blue light. n=8, 1-way ANOVA, ns=not statistically significant, *p<0.05
(L) Summary graph of GIRK amplitudes to pulsed blue light. n=8
(M) Summary graph of the fast decay tau to pulsed blue light. n=8
Due to the need for a second wavelength to photoswitch PPO off, we next tested if sustained activation could be achieved using agonist pulses of blue light. Interestingly, repeated 100 ms pulses at 1 Hz slightly slowed activation, likely due to simultaneous activation and deactivation of receptors (Figure 2J–K). In contrast, shorter pulses (10 ms, 10 Hz) activated GIRK channels more rapidly, with no difference in latency, peak amplitude, or desensitization (Figure 2J–M, S2G), suggesting that pulsed blue light could be used for persistent activation of PPO while retaining rapid off kinetics.
PPO couples to neuronal GIRK channels to decrease excitability and suppress reward seeking behavior
We next determined whether PPO can be used for neuronal inhibition, akin to chemogenetic Gi-coupled DREADDs (Roth, 2016) (Figure 3A). To test this, we created a Cre-dependent AAV construct (AAV5:Ef1α:DIO:PPO-Venus) and injected viruses into the ventral tegmental area (VTA) of DAT-Cre mice to target midbrain dopamine (DA) neurons (Figure 3B). We observed strong expression of PPO-Venus in DA neurons of the VTA (Figure 3C–D) and recorded from these neurons to determine if PPO couples to native GIRK channels to decrease excitability. Pulsed blue LED light produced outward currents, consistent with GIRK channel activation at −60 mV (Figure 3E). Photoactivation of PPO strongly inhibited these neurons (Figure 3F–H), and increased rheobase, decreased input resistance, and reduced firing, which were all fully reversed with bath application of the GIRK channel blocker Ba2+ (Figure 3I–L). Inwardly rectifying currents were similarly activated by blue light and blocked by Ba2+ (Figure 3M). These results indicate that PPO can be used for photoactivation of Gi-coupled signaling cascades in neurons with blue light, consistent with a recent report using violet light (Rodgers et al., 2021).
Figure 3. PPO inhibits somatodendritic excitability to suppresses reward behaviors.

(A) Schematic of PPO coupling to GIRK channels to inhibit neuronal activity.
(B) Experimental strategy for expressing Cre-dependent PPO-Venus (AAV5:Ef1α:DIO:PPO-Venus) in DAT-Cre+ dopamine neurons in the ventral tegmental area (VTA).
(C) Horizontal slice depicting the VTA (purple shaded region). Overlayed IR-DIC and fluorescence images of PPO-Venus in acute horizontal slices. Patch pipette is in dashed lines. Scale=100 μm
(D) 40x IR-DIC images (top) and PPO-Venus (bottom) in cell bodies of the VTA. Scale=10 μm
(E) Plot of normalized holding currents in response to blue LED stimulation (10 Hz, 10 ms, 10 mW/mm2) n=6
(F) Voltage traces from PPO-expressing VTA DA neurons in response to step current injections. Neurons were held at −60 mV.
(G) Same neuron in (F) during photostimulation with 10 Hz blue LED light.
(H) Inhibition was reversed by bath application of the GIRK channel blocker Ba2+ (1 mM)
(I-J) Summary graph of the change in rheobase (I) and input resistance (J) in response to 10 Hz LED stimulation and Ba2+. n=6, Paired t-tests of baseline to LED stimulation, p<0.05
(K) Plot of AP frequency to depolarizing current injections before (purple), during 10 Hz LED (blue) and after bath application of Ba2+ (red).
(L) Summary graph of input-output gain (AP# divided by the cumulative current injected). n=6, paired t-test p<0.05
(M) Current elicited by a voltage ramp from −40 to −140 mV at baseline (purple), during 10 Hz LED (blue) and after Ba2+ (red).
(N) Experimental strategy for expressing Cre-dependent PPO-Venus in DAT-Cre+ DA neurons in the VTA. Optical fibers were implanted bilaterally above the VTA to stimulate cell bodies.
(O) Confocal micrograph of PPO-Venus expression in VTA DA neurons. Scale=400 μm.
(P) Experimental timeline for operant task training for sucrose rewards. Mice were trained on fixed ratio (FR)-1 and FR-3 schedules.
(Q) Cartoon of an operant chamber. Mice nose poke into an active port in response to a light cue to receive a sucrose reward.
(R) Experimental design of the operant task where a sucrose reward is given for nose pokes in the active port after the light cue. No reward is given for pokes in the inactive port. FR-1 requires 1 nose poke for reward while FR-3 requires 3 nose pokes.
(S) Summary graphs of operant behaviors. 10 Hz pulsed laser light (473 nm, 10 ms, 5–8 mW) decreased both the number of nose pokes (left) and rewards (right). n=9 mice each, paired t-test **p<0.01
We next assessed if PPO could suppress reward-seeking behaviors mediated by VTA DA neurons in vivo (Morales and Margolis, 2017; Parker et al., 2019). We bilaterally injected Cre-dependent AAVs into DAT-Cre mice (Bäckman et al., 2006) to express PPO in VTA DA neurons, and implanted optical fibers above their somata (Figure 3N). We observed PPO expression throughout VTA cell bodies and along neuronal processes (Figures 3O and S3A). Reward-seeking behavior was tested in mice trained to perform operant tasks for a sucrose reward (Figure 3P–R). Food-restricted mice underwent Pavlovian conditioning to associate a light cue with access to a sucrose sipper (Figure 3Q) and were trained on fixed ratio (FR) schedules to nose poke for sucrose rewards (Figure 3R). We then assessed reward seeking behavior in FR-3 tests. 10 Hz laser light significantly suppressed both the number of nose pokes and rewards (Figure 3S). These data indicate reduced reward-seeking behavior when VTA DA neuron cell bodies were inhibited by PPO, consistent with other inhibitory approaches (Corre et al., 2018; Tye et al., 2013).
PPO rapidly and reversibly inhibits neuronal voltage-gated calcium channels
The spatiotemporal control of Gi signaling suggested that PPO could also have applications as an optogenetic inhibitory tool at synaptic terminals, akin to the well-established role of presynaptic inhibitory GPCRs. To test this, we determined whether PPO inhibits neuronal voltage gated Ca2+ channel currents (Figure 4A). We transduced cultured dorsal root ganglia (DRG) neurons from AvilCre mice (da Silva et al., 2011; Zhou et al., 2010) with AAVs to express Venus-tagged PPO (Figure S4A), and found that PPO-Venus was effectively trafficked along tau-labeled axons where it colocalized with the presynaptic marker synapsin-1 at terminals (Figure 4B).
Figure 4. PPO inhibits neuronal calcium channel currents.
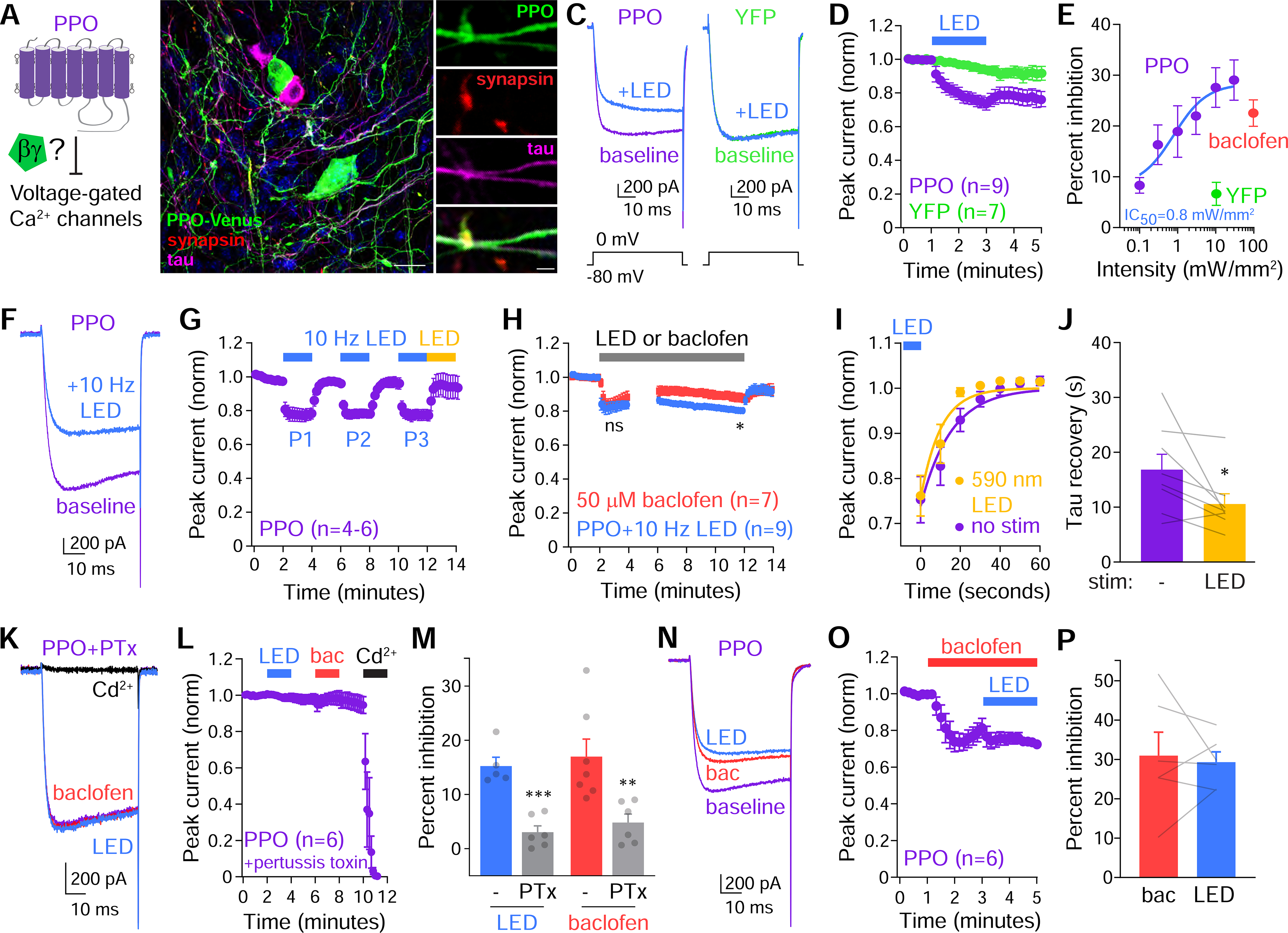
(A) Cartoon of PPO coupling to voltage-gated Ca2+ channels.
(B) Confocal micrograph of cultured DRG neurons from AvilCre mice 7 days after transduction with Cre-dependent AAV5 (CAG:DIO:PPO-Venus). PPO-Venus (green) was present at cell bodies, tau+ axons (magenta), and synapsin-1+ axonal boutons (red). Scale=20 μm (left) and 5 μm (right).
(C) Ca2+ channel current traces elicited by voltage steps from −80 to 0 mV in DRG neurons. LED illumination (470 nm, 10 mW/mm2) inhibited currents in PPO-expressing neurons (purple) but not YFP+ controls (green).
(D) Quantification of normalized VGCC currents in PPO+ (purple) vs. YFP control (green) neurons. Blue bar indicates stimulation with constant blue LED light (10 mW/mm2).
(E) Dose-response curve of VGCC inhibition by blue light stimulation of PPO+ neurons (purple) or YFP+ controls (green). The GABABR agonist baclofen (50 μM, coral) was used to compare efficacy of PPO. n=6–11 for PPO, n=7 for YFP controls, n=20 for baclofen.
(F) Ca2+ channel trace of PPO expressing neuron before (purple) and after (blue) stimulation with pulsed blue light (10 Hz, 10 ms, 10 mW/mm2).
(G) Time course of peak current inhibition to repeated LED pulses (blue bars) and enhanced recovery in amber light (yellow, 590 nm LED). n=4–6 neurons
(H) Plot of normalized VGCC currents in response to prolonged stimulation of PPO with 10 Hz LED light (blue) or 50 μM baclofen (coral). During minutes 4–6, we ran pre-pulse protocols (Figure S4). t-test, *p<0.05, ns=not significant
(I) Normalized recovery time-course of VGCC currents with amber LED (590 nm) or in the dark (no stim, purple). The blue bar depicts LED for inhibition. n=8 cells.
(J) Summary graph of the recovery tau with (amber) and without (purple) subsequent illumination with a 590 nm LED. Paired t-test, *p<0.05
(K) Ca2+ channel trace of PPO expressing neuron treated with 200 ng/ml pertussis toxin (PTx, purple) and responses to 10 Hz blue light (blue), 50 μM baclofen (coral) and 100 μm Cd2+ to block Ca2+ channels (black).
(L) Time course of normalized peak VGCC currents in PPO-expressing neurons treated with PTx (purple) in response to 10 Hz LED stimulation (blue), 50 μm baclofen (coral), and 100 μm Cd2+ (black).
(M) Summary graph of VGCC inhibition in neurons treated with PTx. Blue (LED) and coral (baclofen) bars represent untreated controls. Gray bars represent PTx-treated PPO+ neurons stimulated with blue LED (left) or baclofen (right). n=5–7, t-test **p<0.01, ***p<0.001
(N) Ca2+ channel trace of PPO expressing neuron (purple) stimulated with 50 μM baclofen (coral) followed by 10 Hz LED stimulation (blue).
(O) Time course of normalized peak VGCC currents in PPO-expressing neurons (purple) stimulated by 50 μM baclofen (coral) and 10 Hz LED pulses (blue). n=6
(P) Summary graph of the percent inhibition of VGCC currents by baclofen and subsequent LED stimulation. n=6, paired t-test, not significant
We recorded voltage gated Ca2+ channel (VGCC) currents in DRG neurons, where activation of endogenous Gi-coupled GPCRs inhibits Ca2+ currents through Gβγ-mediated voltage-dependent mechanism (Bean, 1989; Bourinet et al., 1996; Currie, 2010; Dunlap and Fischbach, 1981). Constant illumination of PPO-expressing DRG neurons with blue LED light rapidly suppressed VGCC currents in a light intensity-dependent manner (Figure 4C–E). Maximal inhibition plateaued around 10 mW/mm2 and was equivalent to that observed with the GABAB receptor agonist baclofen, a well-established Gi-coupled inhibitor of neurotransmitter release (Figure 4E). Blue LED light inhibited VGCC currents with an IC50 of 0.8 mW/mm2, and no inhibition was observed in YFP-expressing controls (10 mW/mm2, Figure 4C–E).
In initial experiments VGCC currents did not fully recover after constant blue light at higher intensities (Figures 4D and S3B–C), and we were unable to reverse this with amber LEDs (Figure S3C). LED stimulation did not change voltage-dependent gating (Figure S3D–F), so we reasoned that PPO might be photodamaged by constant illumination. We tested whether agonist pulses of blue light could affect Ca2+ currents, as they did for GIRK channels (Figure 2J–M). We used the same 10 Hz pulsed protocol and achieved similar efficacy of inhibition, but currents recovered rapidly (Figure 4F–G). Ca2+ channel inhibition and reversal were similar at both physiological and room temperature (Figure S3G–H). We could rapidly and repeatedly inhibit currents (Figures 4G and S3I) suggesting that PPO undergoes little functional desensitization. To more rigorously test this, we compared VGCC inhibition during prolonged optical stimulation to activation of endogenous GABABRs with baclofen. We observed similar inhibition with PPO and GABABR after 2 minutes, however the baclofen response desensitized ~50% over 10 minutes, while PPO did not exhibit any observable functional desensitization (Figure 4H and S3J). Consistent with our reversal of Gβγ localization, we were able to speed the recovery of VGCC currents approximately two-fold with 590 nm LED light (Figure 4I–J). The recovery tau of ~10 seconds reflects both photoconversion to the off state and G protein deactivation, with the latter likely to be the limiting step (Vilardaga, 2010).
We next treated DRG neurons with pertussis toxin (PTx) to inactivate Gαi/o subunits and inhibit G protein signaling (Dolphin and Scott, 1987; Katada and Ui, 1982; Scott and Dolphin, 1987). PTx completely blocked VGCC inhibition by both blue LED stimulation of PPO and the activation of endogenous GABABRs with baclofen (Figure 4K–M). We also inquired as to whether PPO couples to Gαo subunits. To answer this, we used imaged γ9 translocation in cells treated with PTx to inactivate Gαi/o proteins, and co-expressed a PTx-resistant Gαo mutant (Hunt et al., 1994; Wise et al., 1997). PTx treatment prevented γ9-mScarlet translocation during light stimulation of PPO and this was partially restored by expression of PTx-resistant Gαo subunits (Figure S4A–B), demonstrating that PPO also can couple to Gαo signaling pathways in certain cellular settings.
Following activation of Gi-coupled GPCRs, Gβγ is the primary downstream modulator of channel function in neurons (Lüscher and Slesinger, 2010; Zamponi and Currie, 2013). Thus we tested whether PPO inhibits Ca2+ channels through a voltage-dependent mechanism (Bean, 1989; Herlitze et al., 1996; Ikeda, 1996). Depolarizing pre-pulse steps fully reversed Ca2+ channel inhibition by PPO and baclofen (Figure S4C–D) consistent with well-established mechanisms of VGCC inhibition by Gi-coupled GPCRs. Our previous development of opto-XRs suggested that rhodopsin-based chimeras converge with endogenous receptor signaling (Siuda et al., 2015b, 2015a). To test this with PPO we recorded VGCC currents and activated GABABRs with baclofen. Inhibition plateaued within ~1 minute and then desensitized slightly (Figure 4N–O). Subsequent illumination of PPO did not increase the peak inhibition observed with baclofen (Figure 4N–P). These results indicate that PPO couples to the same pool of G proteins in neurons as native Gi-coupled GABABRs to inhibit Ca2+ channels.
While we did not observe functional desensitization, illumination of PPO with UV light induced translocation of lamprey β-arrestin (Kawano-Yamashita et al., 2011; Wilden et al., 1986). To determine whether PPO undergoes arrestin-dependent processes following blue light activation in mammalian cells, we performed total internal reflection fluorescence (TIRF) microscopy of GFP-tagged arrestin-3 and mCherry-clathrin to mark endocytic zones. Activation of PPO induced arrestin clustering at these clathrin+ sites (Figure S5A–B), confirming that PPO can engage mammalian arrestin. However, PPO did not accumulate with arrestin (Figure S5C), and we did not observe any internalization of PPO even after 90 minutes of stimulation with pulsed blue light (Figure S5D). We also did not detect any internalization of PPO in DRG neurons, despite strong endocytosis of the GFP-tagged Mu-opioid receptor (MOR-GFP) with DAMGO (Figure S5E–G), which is well-established (Siuda et al., 2015a; Whistler et al., 1999). While PPO clearly engages and mobilizes arrestin, we did not detect receptor internalization or functional desensitization, previously observed with other types of GPCRs (Eichel and von Zastrow, 2018; Eichel et al., 2016, 2018; Nuber et al., 2016; Siuda et al., 2015b). Collectively, these data demonstrate that PPO can be used to rapidly and reversibly inhibit neuronal Ca2+ channels with equivalent efficacy to endogenous GABABRs but minimal canonical desensitization.
Reversible optical inhibition of presynaptic release
Presynaptic Gi/o-coupled GPCRs, like GABABRs, exert powerful control over synaptic transmission by inhibiting vesicle release through multiple mechanisms, including decreased VGCC function and interference with release machinery (Blackmer et al., 2001; Browning et al., 2002; Heinke et al., 2011; Stachniak et al., 2014; Takahashi et al., 1998; Zurawski et al., 2019a). The similar Gi/o coupling between PPO and GABABRs, suggested that PPO might represent a unique optical approach for presynaptic inhibition that takes advantage of these conserved mechanisms (Figure 5A). We first tested this in thalamic projections to the barrel cortex, which was used previously to test synaptic efficacy of inhibitory Cl− and H+ pumps (Mahn et al., 2016). We injected Cre-dependent AAVs in the ventral posterior medial nucleus (VPM) of the thalamus in Vglut2-Cre mice to express PPO in these glutamatergic projections (Figure 5B) (Vong et al., 2011). We observed PPO-Venus in thalamocortical axons innervating the barrel cortex with dense projections to layer IV neurons (Figure 5C). Imaging of these terminals revealed colocalization of PPO-Venus with the presynaptic glutamate transporter vGluT2 which was opposed to PSD-95+ excitatory synapses (Figure 5C). These results indicate that PPO traffics effectively to axon terminals in vivo, making it well-positioned to control presynaptic release.
Figure 5. PPO inhibits presynaptic release at glutamatergic and GABAergic terminals.
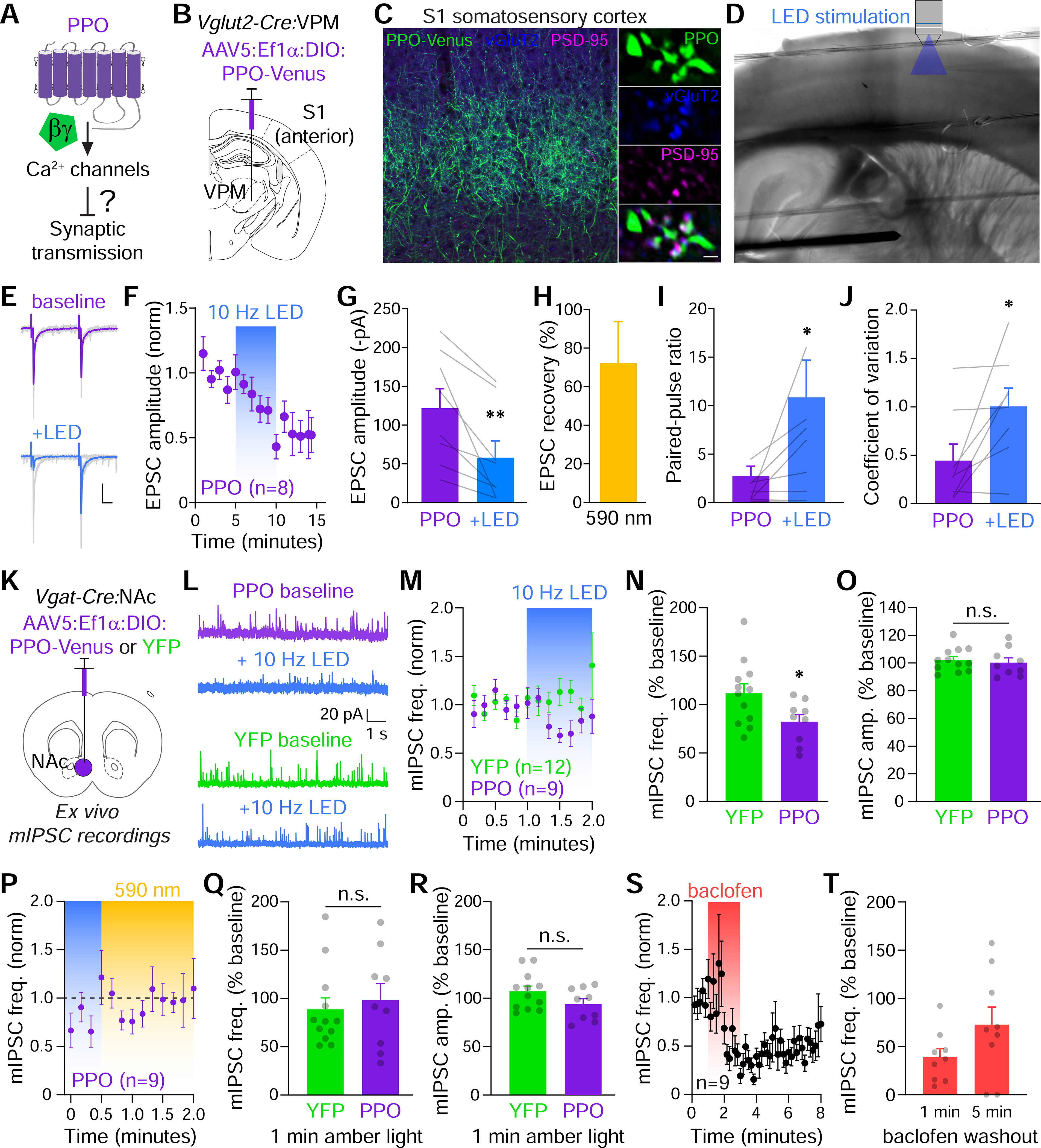
(A) Experimental question asking if PPO can inhibit synaptic transmission at axon terminals.
(B) Cartoon depicting the injection site of Cre-dependent AAV5 (Ef1α:DIO:PPO-Venus) into the ventral posterior medial nucleus (VPM) of thalamus in Vglut2-Cre mice to target excitatory projections to primary somatosensory cortex (S1).
(C) Confocal micrograph of S1 from a Vglut2-Cre mouse expressing PPO-Venus in the VPM. PPO-Venus+ axons (green) densely innervated layer IV of the barrel cortex. Scale=100 μm. PPO (green) colocalized at presynaptic vGluT2+ terminals (blue) opposed to postsynaptic sites marked by PSD-95 (magenta). Scale=1 μm.
(D) IR-DIC images of an acute thalamocortical slice depicting the stimulating electrode in VPM and the patch pipette and objective for optical stimulation of terminals in S1.
(E) Representative EPSCs (gray) and averaged responses during baseline (purple) and 10 Hz LED stimulation (blue). Paired pulses were delivered at 50 ms intervals.
(F) Plot of normalized EPSC amplitudes which were inhibited by pulsed blue LED light (blue bar). Tau of inhibition was 2.6 minutes.
(G) Summary graph of EPSC amplitudes before and after 10 Hz photostimulation. n=8, paired t-test **p<0.01
(H) Summary graph of the reversal of inhibition after 30s of constant amber light, n=6 cells.
(I-J) Summary graph of EPSC paired pulse ratios (I), and coefficients of variation (J) before (purple) and after LED stimulation (blue). n=8, paired t-tests, *p<0.05
(K) Cartoon of the strategy to express Cre-dependent PPO-Venus or YFP in the NAc of Vgat-Cre mice. Acute slices were then used to record mIPSCs.
(L) Representative mIPSC traces recorded at 0 mV from non-fluorescent neurons in PPO or YFP-injected mice.
(M) Plot of normalized mISPC frequencies which were rapidly inhibited by the pulsed LED (blue bar) in PPO (n=9) but not YFP controls (n=12).
(N) Summary graph of the effect of 10 Hz LED on mIPSC frequencies (normalized to baseline) in YFP (n=12) and PPO (n=9) slices. t-test, *p<0.05
(O) Summary graph of normalized rmIPSC amplitudes in YFP (n=12) and PPO (n=9) slices. n.s. not significant
(P) Plot of normalized mIPSC frequency recovery in constant amber light (590 nm, 1 mW/mm2). n=9 slices.
(Q) Summary graph of mIPSC frequency recovery after 1 min. of amber light in YFP (n=12) and PPO (n=9) slices. n.s. not significant
(R) Summary graph of mIPSC amplitudes after 1 min. of amber light in YFP (n=12) and PPO (n=9) slices. n.s. not significant
(S) Plot of normalized mIPSC frequencies in response to 50 μM baclofen for 2 min. (coral bar) in slices from YFP-injected mice (n=9).
(T) Summary graph of mIPSC frequency recovery 1 and 5 min. after washout of baclofen, n=9 slices.
We tested PPO at long-range thalamocortical projections in acute slices, which permits electrical stimulation of cell bodies in the VPM while limiting photostimulation to terminals in the barrel cortex (Agmon and Connors, 1991; Cruikshank et al., 2007) (Figure 5D). We electrically evoked monosynaptic excitatory postsynaptic currents (EPSCs) in VPM, and then photostimulated PPO-expressing terminals (Figure 5E–G). Blue light pulses inhibited EPSC amplitudes which could be reversed by illumination with amber light (Figure 5H). Paired-pulse ratios and EPSC variation were both significantly increased (Figure 5I,J), consistent with a presynaptic inhibitory mechanism (Bagnall et al., 2011; Johnson and Lovinger, 2016; Kreitzer and Malenka, 2007; Nanou and Catterall, 2018; Zucker and Regehr, 2002; Zurawski et al., 2019b).
To further test if PPO acts at presynaptic terminals to inhibit neurotransmitter release, we injected AAVs to express Cre-dependent PPO or YFP into the NAc of Vgat-Cre mice and recorded action potential-independent GABAergic mIPSCs in acute slices (Figure 5K). Photostimulation rapidly inhibited mIPSC frequencies (Figure 5L–N), but not amplitudes (Figure 5O), in PPO injected mice, consistent with a presynaptic mechanism of action (Nicola and Malenka, 1997; Tejeda et al., 2017). We did not observe any effect on mIPSCs in YFP-injected controls (Figure 5L–O). Importantly, mIPSC frequencies recovered rapidly to baseline within 1 minute when slices were illuminated by constant amber light (Figure 5P–R). We also compared PPO to activation of endogenous GABABRs with baclofen at these synapses. In contrast to PPO, GABABRs produced a much slower onset and recovery of mIPSC frequencies (Figure 5S–T). Taken together, these results demonstrate that optical stimulation of PPO reversibly couples to endogenous GPCR signaling cascades to inhibit presynaptic release at terminals.
PPO is functional in vivo, and reversibly inhibits synaptic transmission at terminals.
To determine the efficacy of PPO in silencing terminal activity in vivo, we tested whether PPO could suppress GABAergic projections from the bed nucleus of the stria terminalis (BNST) to the lateral hypothalamus (LH), where inhibition of BNST terminals leads to disinhibition of postsynaptic LH activity (Jennings et al., 2013). We injected AAVs to express PPO in the BNST of Vgat-Cre mice and performed single-unit recordings in the LH (Figures 6A and S6A). PPO inhibition of BNST terminals in the LH resulted in a rapid increase in frequency and average firing rate of the postsynaptic LH neurons which completely recovered in 30/33 units (Figures 6B–C and S6B–C). Three of these units did not recover and remained elevated throughout the recording. These results indicate that in vivo photoactivation of PPO at GABAergic terminals leads to disinhibition, consistent with endogenous presynaptic Gi-coupled GPCRs (St. Laurent et al., 2020; Mahn et al., 2021; Matsui and Williams, 2011).
Figure 6. PPO inhibits synaptic transmission at terminals in vivo.
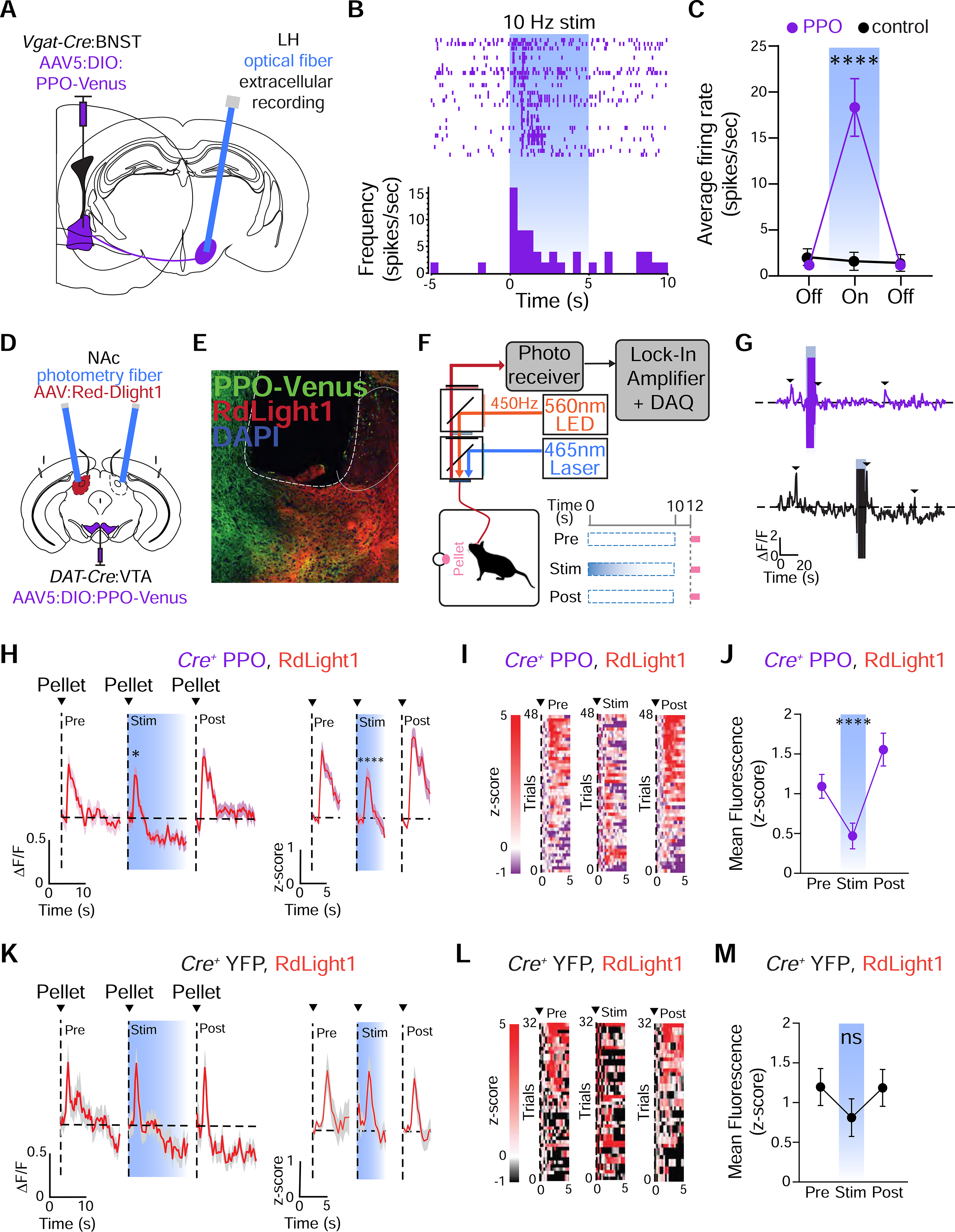
(A) Schematic illustrating single-unit recordings of lateral habenula (LH) neurons modulated by PPO-expressing BNST-LH terminals
(B) Raster plot from recorded LH units (top) and the peri-event histogram from a representative unit (bottom) during 5 sec of 10 Hz photoinhibition of BNST-GABA terminals expressing PPO.
(C) Average firing rate of LH units from PPO (purple) or control (black) groups; Mixed-effects 1-way ANOVA, ****p<0.0001, n=30 units from 6 mice. Firing rate in controls was unchanged; Mixed-effects 1-way ANOVA, p>0.05, n=19 units from 3 mice.
(D) Cartoon depicting injection of Cre-dependent AAV5 (Ef1α:DIO:PPO-Venus) into the VTA of DAT-Cre mice, injection of RdLight1 (AAVDJ:CAG:RdLight1) into the NAc core and fiber implants.
(E) Confocal micrograph depicting PPO-Venus (green), RdLight1 (red) and DAPI (blue) expression in the NAc core. The placement of the optical fiber is outlined. Scale=100 μm.
(F) Schematic of fiber photometry set-up and behavioral design.
(G) Representative ΔF/F traces for PPO (top) and YFP controls (bottom) showing RdLight1 activity during reward delivery (black arrows) and stimulation artifact (blue bar).
(H) RdLight1 (ΔF/F, left or z-score, right) from PPO mice during reward delivery averaged across all trials in pre (left), stim (middle) or post (right) trials. n=6 mice.
(I) Heatmap raster plot of RdLight1 fluorescence (z-score) from PPO mice during reward delivery averaged across all trials in pre (left), stim (middle) or post (right) (n=6 mice). Trials are in ascending order of activity following reward.
(J) Mean fluorescence following reward delivery (1–5s). n=6 mice; Mixed-effects one-way ANOVA, ****p<0.0001. Mult. comparisons: pre vs. stim, **p<0.01 and post vs. stim, ****p<0.0001
(K) RdLight1 (ΔF/F, left or z-score, right) from YFP mice during reward delivery averaged across all trials in pre (left), stim (middle) or post (right). n=5 mice
(L) Heatmap raster plot of RdLight1 fluorescence (z-score) from YFP controls during reward delivery averaged across all trials in pre (left), stim (middle) or post (right) (n=5 mice). Trials are in ascending order of activity following reward.
(M) Mean fluorescence following reward delivery (1–5s). n = 5 mice; Pre vs. Stim vs. Post, Mixed-effects one-way ANOVA, p>0.05, Mult. comparisons all not significant
Next, we asked whether PPO can reversibly inhibit neuromodulatory transmission using in vivo fiber photometry with a fluorescent dopamine biosensor. We virally injected PPO (or YFP, for controls) bilaterally into the VTA of DAT-Cre mice. We also injected RdLight1, a red-shifted fluorescent dopamine biosensor (Patriarchi et al., 2020) unilaterally in the NAc, and bilaterally implanted photometry fibers into the NAc core (to ensure a within-animal control for RdLight1 function) (Figure 6D). We observed expression of PPO in DA terminals projecting from the VTA opposed to RdLight1 on somas and dendrites in the NAc core (Figure 6E). Animals were trained on an uncued reward delivery task, where sucrose pellets were delivered at random intervals (Figure 6F). On the recording day, blue light stimulation was delivered for 10 seconds unilaterally through the photometry fiber to inhibit PPO+ DA terminals, and sucrose pellets were delivered 2 seconds later. Blue light was delivered every third trial, to ensure within-animal and within-session comparisons of RdLight1 activity preceding (pre), during (stim) and following (post) PPO-mediated inhibition of DA terminals (Figure 6F). Blue light delivery caused a stimulation artifact in our recordings but did not impact our ability to record RdLight1 activity during sucrose delivery (Figure 6G). As shown previously (Patriarchi et al., 2020), sucrose delivery in trained animals elicited robust elevations in DA, as measured by RdLight1 activity (Figures 6H–M). Moreover, we did not detect any fluorescence changes in recordings from RdLight1-negative controls (Figures S6E–F). RdLight1 activity was significantly reduced during photostimulation trials in PPO-injected animals (Figures 6H–J), as compared to YFP controls (Figures 6K–M). However, unilateral blue light stimulation did not alter reward seeking behavior during the stimulation trials (Figure S6G). This suggests that there is a necessity for bilateral inhibition for these behaviors, as is typically the case for chemo- and optogenetic methods. Altogether, these results indicate that PPO is compatible with red-shifted biosensors and can efficiently inhibit terminals in vivo.
Terminal photoinhibition with PPO induces state-dependent, time-locked and reversible changes in motivated behaviors
We next assessed the ability of PPO to cause changes in behavior via inhibition of synaptic terminals. We first injected PPO in the VTA of DAT-Cre mice and bilaterally implanted optical fibers in the NAc core (Figure 7A). We observed strong PPO expression in VTA neuron terminals in the NAc (Figure 7B), consistent with the efficient trafficking of PPO we observed in all other neuron types. Animals were tested on an uncued reward task (Figure 6F) and received blue light stimulation bilaterally to inhibit DA terminals via PPO for 10 seconds prior to reward delivery on every third trial, to ensure within-animal and session comparisons. In contrast to the YFP-injected controls, we observed a significant increase in the latency to retrieve pellets on the trials that received blue light stimulation prior to reward delivery (Figure 7C). We then performed similar operant tasks for sucrose rewards in these mice as described previously (Figure 3O–Q). In the FR-3 tasks, photoinhibition of DA projections to the NAc core during the whole session significantly suppressed active total nose poke behavior (Figure S7A) and reduced the number of rewards obtained in PPO-expressing Cre+ mice compared to Cre− controls (Figure 7D). In contrast, photoinhibition of DA terminals in the NAc core via constant green light illumination with halorhodopsin (eNPHR3.0) resulted in no apparent differences in nose pokes or rewards earned (Figure S7B–C). Additionally, we also performed PR tests, where the number of nose pokes required to receive a reward was escalated exponentially to assess the point at which the animal is no longer willing to work for a reward (Figure 7E) (Hodos, 1961; Parker et al., 2019; Richardson and Roberts, 1996). During these PR tests, pulsed blue light was either delivered throughout the session, or was triggered by a nose poke (1 second). PPO photostimulation depressed both nose pokes (Figure S7D–E) and the number of rewards in Cre+ mice expressing PPO but not in Cre− controls, when blue light was delivered either continuously (Figure 7F) or time-locked to the nose poke (Figures 7G–H). These data are consistent with the role of this DA projection in both reward-seeking and motivated behaviors (Corre et al., 2018; Kim et al., 2013; Tsai et al., 2009; Tye et al., 2013).
Figure 7. PPO inhibits dopamine neuron terminals to suppress reward seeking behaviors.
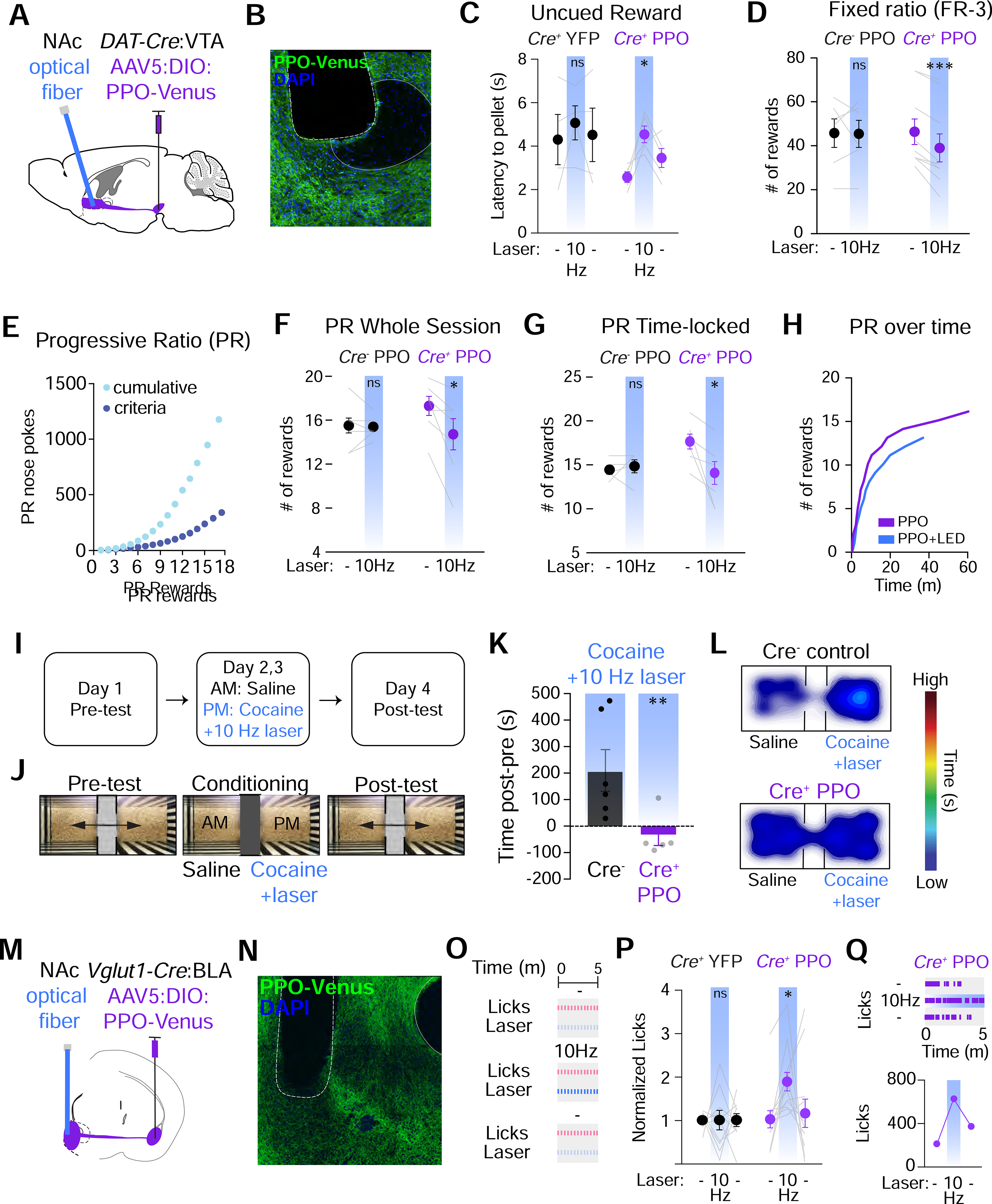
(A) Cartoon of the strategy to inhibit DA neuron projections to the NAc. Cre-dependent AAVs were injected into the VTA of DAT-Cre mice and optical fibers were placed above their terminals in the NAc core.
(B) Confocal micrograph of PPO-Venus in DA neuron terminals of the NAc. The placement of the optical fiber is shown. Scale=100 μm
(C) Summary graphs of uncued reward delivery task. Terminal illumination (blue bars) increased the latency to retrieve pellets in PPO mice (right, n=5), but not YFP controls (left, n=4). Mixed-effects 1-way ANOVA, *p<0.05. Mult. comparisons: (−) vs. 10 Hz, *p<0.05
(D) Summary graphs of FR-3 testing. Terminal illumination (blue bars) decreased the # of rewards that mice received in Cre+ mice (right, n=9), but not Cre− controls (left, n=6). Paired t-test ***p<0.001
(E) Example graph of the escalating nose pokes to receive a reward in progressive ratio (PR) testing.
(F) Summary graph of PR testing. Terminal illumination (blue bars) decreased the # of rewards received in Cre+ mice (right, n=7), but not Cre− mice (left, n=8). Paired t-test *p<0.05.
(G) Summary graph of nose poke-triggered, time-locked PR testing. Terminal illumination (blue bars) decreased the # of rewards received in Cre+ mice (right, n=5), but not Cre−controls (left, n=6). Paired t-test *p<0.05.
(H) Representative trace of rewards earned in a PR session over time +/− blue light stimulation.
(I-J) Experimental timeline and set-up for testing cocaine preference behaviors. During pre-testing mice could freely explore the 2 chambers. During pairing, mice were given saline in 1 chamber and cocaine (10 mg/kg) + 10 Hz blue light in the other. After 2 days of pairing, preference for either chamber was determined.
(K) Summary graph of difference in time spent in the cocaine-paired chamber. Cre− control mice (n=6) exhibit strong preference for the cocaine-paired chamber which was completely blocked by photoinhibition in PPO-expressing mice (n=5). Both groups received optical stimulation during cocaine-pairing. t-test **p<0.01
(L) Heat maps of relative time spent in each chamber.
(M) Cartoon depicting glutamatergic neuron projections to the NAc. Cre-dependent AAVs were injected into the BLA of Vglut1-Cre mice and optical fibers were placed above terminals in the NAc shell.
(N) Confocal micrograph of PPO-Venus expression in BLA neuron terminals of the NAc. The placement of the optical fiber is shown. Scale=100 μm
(O) Schematic depicting sucrose licking task and experimental design.
(P) Normalized sipper licks. n=12 Cre+ and n=10 Cre− mice. For Cre+: Mixed-effects 1-way ANOVA, *p<0.05. Mult. comparisons: (−) vs. 10Hz, *p<0.05 and 10Hz vs. (−), *p<0.05. All other comparisons ns
(Q) Top: Representative lick raster plots for (−) vs. 10Hz vs. (−) conditions. Bottom: Raw licks for a single animal
We next tested whether PPO could inhibit behavioral states mediated by increased dopaminergic tone by examining whether we could alter cocaine-induced conditioned place preference behavior. We used the same approach as above to express PPO and target DA terminals in both DAT-Cre+ mice and Cre− controls (Figure 7A). After pre-testing, mice were paired with saline injections in one chamber followed by cocaine pairing in the other chamber (Figure 7I–J, 10 mg/kg). During cocaine pairing however, both groups also received 10 Hz photoinhibition of VTA DA terminals in the NAc. After 2 days of cocaine pairing and photoinhibition, we assessed the expression of preference behavior. Cre− control mice strongly preferred the cocaine-paired chamber, and this was completely absent in DAT-Cre+ mice expressing PPO (Figure 7K–L). Importantly, locomotor activity (distances traveled) were similar between the two groups (Figure S7F), demonstrating the selectively of PPO-mediated inhibition of DA terminals in the NAc and not collateral DA projections to dorsal striatum.
Finally, we assessed the role of PPO to reversibly alter reward consumption. Glutamatergic projections from the BLA to the NAc shell have been implicated in reward consumption, and inhibition of this circuit results in an increase in sucrose consumption (Reed et al., 2018; Stuber et al., 2011). To test this, we injected PPO into the BLA of Vglut1-Cre mice and bilaterally implanted optical fibers in the NAc shell (Figure 7M–N). We next trained these animals to lick for 10% sucrose solution through a sipper, available for 5-minute intervals across a 30-minute session. On the test day, animals were then able to engage with the sucrose sipper for three 5-minute sessions, and in second session, sipper licks triggered a time-locked 1 second blue light delivery to activate PPO (Figure 7O). In this experiment, optical stimulation time-locked to licks, resulted in a significant increase in the licks (normalized to Cre− controls), as compared to the first session without optical stimulation (Figures 7P–Q). Importantly, animals showed recovery of licking behavior in the subsequent session without laser stimulation suggesting rapid reversal of PPO inhibition (Figures 7P–Q). Collectively, these data indicate that PPO is an effective optical silencing tool for state-dependent, rapid, time-locked and reversible inhibition of synaptic projections that regulate behavioral states in vivo.
DISCUSSION
Here we identify and characterize PPO as a photoswitchable Gi/o-coupled GPCR for inhibiting presynaptic terminals. This opsin fills a major gap in cell biology, pharmacology, and neuroscience fields, providing a unique optical tool for probing GPCR dynamics that can also achieve rapid and reversible synaptic inhibition (Lin et al., 2013; Liu et al., 2019; Mahn et al., 2016; Wiegert et al., 2017). Indeed, the strength of this Gi-coupled opsin approach for synaptic inhibition was independently demonstrated in a parallel study from the Yizhar lab using OPN3, a bistable Gi/o-coupled opsin from mosquitos (Mahn et al., 2021). Together these and related light sensitive GPCRs represent developmental scaffolds for constructing new and improved photoswitchable opto-GPCR chimeras, similar to other channel opsins and biosensors (Boyden et al., 2005; Nagai et al., 2001; Nakai et al., 2001; Patriarchi et al., 2018; Sun et al., 2018; Yizhar et al., 2011). PPO and OPN3 are thus likely to provide critical insights into developing future GPCR-based opsins with faster kinetics, improved reversibility, spectrum shifts for multimodal control of different opsins, and/or engagement of discrete intracellular pathways.
Spectral properties of PPO and technical considerations
PPO was first identified as the UV sensitive pigment in lamprey pineal cells (Koyanagi et al., 2004). Despite the optogenetic potential of a photoswitchable GPCR-based opsin, PPO seemed limited by the requirement for UV or violet irradiation (Eickelbeck et al., 2019; Kawano-Yamashita et al., 2015; Rodgers et al., 2021). We took advantage of the tail in the activation spectrum and used blue light sources, readily available in many biology labs, to stimulate PPO at light intensities comparable to those used in typical optogenetic experiments or GFP imaging. This was an important advance, as we were able to integrate PPO into all of our optogenetic setups without needing to purchase new equipment. Also, while many GPCR-based opsins possess exquisite light sensitivities that that can be problematic due to activation by ambient light, we did not find any evidence of this with PPO. Importantly we were able to use pulsed light for persistent inhibition with PPO, potentially avoiding caveats of tissue heating with the constant illumination required by the widely used inhibitory pumps (Owen et al., 2019; Stujenske et al., 2015). Moreover, we observed that PPO is largely kept in the off state by ambient light, limiting concerns of unintended activation or constitutive activity prior to, or during, experiments, a significant limitation with rhodopsins (Ernst et al., 2014; Siuda et al., 2015b, 2015a). It is important to consider that not all blue light sources are the same, particularly when working with LEDs. In some cases, their broad emission spectra may include UV or green photons, which could switch PPO into unintended states. As with all optical tools, precise turning and calibration of PPO are required before its use (Rodgers et al., 2021).
Stimulation with UV or violet light constitutively activates PPO until subsequent illumination with longer light wavelengths, including blue light (Eickelbeck et al., 2019; Kawano-Yamashita et al., 2015; Rodgers et al., 2021). In contrast to Eickelbeck et al., who observed reversal of UV activation of PPO with blue light, we found that blue light can activate PPO in a variety of systems tested here. Surprisingly, we also observed that photoactivation with UV and blue light differ greatly in their kinetics. UV light triggered a persistent activated state while PPO turned off much more rapidly after blue light illumination. The long-lasting effects of UV light may indeed be advantageous for chronic optogenetic inhibition experiments. We hypothesize that this is due to a unique GPCR conformational states (Hilger et al., 2018), triggered by the overlapping activation/deactivation curves in the blue light range, however this requires further exploration. While PPO off rates can be sped up by amber light, the active state induced by pulsed blue light could reflect intermediate conformations (Ernst et al., 2014; Hilger et al., 2018; Kouyama and Murakami, 2010) which has recently been demonstrated for GPCRs with various chemical ligands (Du et al., 2019; Kato et al., 2019; Wingler et al., 2019). We find this possibility particularly exciting, as it suggests that PPO coupling to G proteins could be dynamically altered with different durations/colors of agonist light pulses, something akin to receptor signaling bias.
In vivo application of GPCR-based inhibitory opsins
PPO couples to mammalian Gαi/o proteins to engage canonical signaling cascades to inhibit adenylyl cyclase (Kawano-Yamashita et al., 2015), activate GIRK channels (Eickelbeck et al., 2019; Rodgers et al., 2021), and inhibit Ca2+ channels (this report). We focused here on the latter mechanism to test whether PPO could fill a major gap in the systems neuroscience toolbox – that is, rapid and reversible inhibition of presynaptic neurotransmitter release (Wiegert et al., 2017). PPO rapidly and reversibly inhibited neuronal voltage-gated Ca2+ channels with equivalent efficacy to the GABABR agonist baclofen (Figure 4), which has well established presynaptic effects. While decreased Ca2+ influx by presynaptic Gi/o-coupled GPCRs can directly inhibit vesicle release, it is possible that PPO may also activate Gβγ subunits to also inhibit SNARE proteins (Blackmer et al., 2001; Zurawski et al., 2019b). In the hippocampus, presynaptic GPCRs can inhibit synaptic transmission through distinct mechanisms with GABABRs primarily inhibiting Ca2+ channels while serotonin 5-HT1B receptors exert SNARE-dependent effects (Zurawski et al., 2019a). We suspect that one or both of these mechanisms is responsible for inhibiting initial DA release and thus preventing of cocaine place-preference behavior, which has been established for other presynaptic Gi-coupled GPCRs (Chefer et al., 2005; Thompson et al., 2000).
Presynaptic manipulations that alter release probability at terminals can act as high-pass filters and become less effective during bursts of activity (Burke et al., 2018). This is not unique to PPO and extends to endogenous presynaptic Gi-coupled GPCRs. How this alters circuit dynamics however is still poorly understood, and the spatiotemporal control of presynaptic release using PPO, together with genetically encoded sensors, can help shed light on this (Sabatini and Tian, 2020). When implementing PPO for in vivo studies, it is essential to directly test PPO at the projections being targeted, as presynaptic GPCRs can modulate synapses in an input specific manner, with varying degrees of efficacy (Bagnall et al., 2011; Burke et al., 2018; Creed et al., 2016; Kreitzer and Malenka, 2007; Marcott et al., 2018; Tejeda et al., 2017). Whether PPO can couple to distinct signaling cascades in different cell types or engage presynaptic plasticity mechanisms certainly warrants future investigation.
Conclusions and future directions
Here we show that PPO is an effective optogenetic tool for rapid and reversible control of neuromodulation via inhibitory GPCR signaling cascades. By coupling to evolutionarily conserved endogenous inhibitory mechanisms, PPO is effective both at inhibiting excitability through somatodendritic GIRK channel activation and presynaptic inhibition of vesicle release. PPO may also represent a new scaffold for engineering photoswitchable GPCR chimeras to mimic their signaling with greater spatiotemporal precision (Airan et al., 2009; Gunaydin et al., 2014; Li et al., 2015; Oh et al., 2010; Siuda et al., 2015b, 2015a; van Wyk et al., 2015). These molecular bioengineering efforts require a more detailed understanding of the structural dynamics of PPO photoswitching, which in turn may also lead to faster or spectrally shifted variants. More broadly, achieving precise optical control over neuromodulatory signaling will provide fundamental insights into the spatiotemporal nature of GPCR signaling as it relates to these clinically important drug targets in vivo.
RESOURCE AVAILABILITY
Lead contact
Further information and requestions for resources and reagents should be directed to the Lead Contact Michael Bruchas (mbruchas@uw.edu).
Materials Availability
All DNA and viral constructs generated here are deposited on Addgene and are also available upon request from the Lead Contact.
Data and code availability
All data and analysis codes are available from the Lead Contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Model organisms
All experiments were conducted in accordance with the National Institutes of Health guidelines and with approval from the Institutional Animal Care and Use Committee at Washington University School of Medicine and the Office of Animal Welfare at University of Washington. 4–6 week old male Avil-Cre mice (Aviltm2(cre)Fawa; Jackson labs #032536) were used for DRG cultures (da Silva et al., 2011; Zhou et al., 2010). Male and female Vglut2-IRES-Cre mice (Slc17a6tm2(Cre)Lowl; Jackson labs #016963) and Vgat-IRES-Cre (Slc32a1tm2(cre)Lowl) were used for slice electrophysiology and in vivo recordings (Vong et al., 2011). Male and female DAT-IRES-Cre mice (Slc6a3tm1.1(Cre)Bkmn; Jackson labs #006660) (Bäckman et al., 2006) or Vglut1-IRES-Cre (Slc17a7tm1.1(cre)Hze; Jackson labs #023527) (Harris et al., 2014) were used for behavioral experiments. For slice electrophysiology, mice were injected with viruses at 5–6 weeks old and used for recordings 4–6 weeks later. For behavioral experiments, 12–16 week old mice were used. All mice were group housed with ad libitum access to food and water before experiments and maintained on a 12hr light:dark cycle. We used mice of both sexes and did not observe any effects of sex on these findings.
METHOD DETAILS
DNA and viral constructs
Parapinopsin in pcDNA3.1 was kindly provided by Akihisa Terakita (Osaka City University, Japan). EGFP-γ9 and mCherry-γ9 were previously generated (Karunarathne et al., 2012, 2013), and the PTx-resistant mutant GαOA(C351I)-CFP was previously described (Azpiazu et al., 2006). Arrestin3-GFP was a gift from Ken Mackie (Indiana University Bloomington). Clathrin-mCherry was obtained from Addgene (#55019). Red dLight was a gift from Lin Tian (Patriarchi et al., 2020).
mScarlet-γ9 was generated by PCR amplification of mScarlet using forward primer 5’-CCC AAG CTT ATG GTG AGC AAG GGC GAG GCA G-3’ (HindIII) and reverse primer 5’-CGC GGT ACC CTT GTA CAG CTC GTC CAT GCC G-3’ (KpnI). mCherry was excised from mCherry-γ9 (in pcDNA3.1) after HindIII and KpnI digest, and mScarlet was ligated in frame with γ9.
For in vitro characterization of purified protein, parapinopsin was subcloned into pFastbac vector (ThermoFisher) with an N-terminal FLAG tag and 10xHis tag. The construct was cloned using cut sites NcoI and XhoI through PCR amplification using forward primer 5’-CCA TCA CGC CAT GGA GAA CTT GAC C-3’ and reverse primer 5’-CCT CTA GAT GCA TGC TCG AGT CTA GCT CG-3’. PPO-Venus was generated by PCR amplification of PPO using forward primer 5’-GCG GAA TTC ATG GAG AAC TTG ACC TCG CTC GAC C-3’ (EcoR1) and reverse primer 5’-ATA GTT TAG CGG CCG CGC TCG GGG AGA CCT GCC CCG-3’ (NotI) and amplification of Venus using forward primer 5’-ATA AGA ATG CGG CCG CCA TGG TGA GCA AGG GCG AGG AGC-3’ (Not1 with extra base after the RE site to bring sequence into frame) and reverse primer 5’-GCT CTA GAT TAC TTG TAC AGC TCG TCC ATG CCG-3’ (XbaI site followed by a stop codon). Purified PCR products were ligated into pcDNA3.1 after vector digest with EcoR1 and XbaI.
Cre-dependent viral vectors were generated by amplifying PPO-Venus from pcDNA3.1. Resulting amplification products were restriction digested, then gel-purified and ligated into a Cre-dependent pAAV vector digested with the same enzymes used for the amplification product. The Cre-dependent viral vector using the CAG promoter to drive expression (kindly provided by Larry Zweifel, University of Washington) was digested with AgeI and AscI. PPO-Venus was amplified using the forward primer 5’-CGG ATC CAC TAG TCC AGA CCG GTG GAA TTC ATG G-3’ and the reverse primer 5’-GGC ACA GTC GAG GCG CGC CAG CGG GTT TAA ACG-3’, then digested with AgeI and AscI. The Cre-dependent viral vector using the EF1α promoter to drive expression (available from Addgene, plasmid # 27056) was digested with NheI and AscI. PPO-Venus was PCR amplified using the T7 promoter as a forward primer and the reverse primer 5’-GGC ACA GTC GAG GCG CGC CAG CGG GTT TAA ACG-3’, then ligated into the vector digested with NheI and AscI. All constructs were verified by Sanger sequencing.
AAVs were produced in the Hope Center Viral Vector Core. AAV5:CAG:DIO:PPO-Venus was used for all in vitro studies at a titer of 2*1013 vg/ml and AAV5:Ef1α:DIO:PPO-Venus for all in vivo studies at 1.3×1013 vg/ml. AAV5:Ef1α:DIO:eYFP was used for all control experiments at a titer of 5×1012 vg/ml.
Antibodies
The following primary antibodies were used: mouse anti-synapsin-1 (Synaptic Systems cat. #106011, RRID:AB993033), guinea pig anti-tau (Synaptic Systems cat. # 314004, RRID:AB_A1547385), guinea pig anti-vGluT2 (Synaptic Systems cat. # 135404, RRID:AB_887884), and mouse anti-PSD-95 (NeuroMAb cat. # 75–028 RRID:AB_2292909). Secondary antibodies were both from Life Technologies: donkey anti-mouse Alexa Fluor 555 (cat. #31570) and goat anti-guinea pig Alexa Fluor 647 (cat. # 21450).
In vitro protein characterization
Production of purified parapinopsin
The construct was grown in DH10Bac (ThermoFisher, 10361012) cells and transfected into Sf9 insect cells using Cellfectin II reagent (ThermoFisher, 10362100). Baculovirus was generated according to the manufacturer’s protocol. From this point onward all experiments were completed in the dark or under dim red light.
Sf9 cells were grown to a density of 2 million/mL at which time they were transduced with the baculovirus at a MOI=1.0. Protein was expressed for 48 hours in the dark. At 24 hours post-transduction, 10 μM 9-cis-retinal was added to the culture. Cells were harvested and spun down at 1000xg for 10 minutes. They were then resuspended in buffer containing 65mM NaCl, 50mM HEPES, 1mM EDTA (pH=7.4), with protease inhibitors partial thromboplastin time (PTT) and leupeptin hemisulfate (LS). The suspension was placed in a nitrogen cavitation chamber at 600 PSI for 30 minutes at 4°C. Upon release, lysed cells were centrifuged at 1000xg for 15 minutes followed by the supernatant being spun at 125,000xg for 35 minutes. The pellet was resuspended in buffer containing 50 mM NaCl, 50 mM HEPES, 5 mM MgCl2 (pH=7.4) with protease inhibitors (PTT/LS) and centrifuged at 125,000xg for 40 minutes. Resuspension of the pellet and centrifugation were repeated. Finally, the membranes were homogenized with a dounce and protein concentration was measured.
Membranes were diluted to 5mg/ml of total protein and solubilized in 200 mM NaCl, 20 mM HEPES, 5mM MgCl2, and 1% DDM (pH=7.4) at 4°C for 1 hour, at which point they were centrifuged at 125,000xg for 40 minutes. After centrifugation, the supernatant was applied to a Talon affinity column. The column was washed with 20 column volumes of wash buffer containing 50 mM NaCl, 20 mM HEPES, 5 mM MgCl2, 5 mM imidazole, and 0.1% DDM (pH=7.4). Protein was eluted with elution buffer containing 150 mM imidazole, 40 mM NaCl, 20 mM HEPES, 1mM MgCl2, 0.1% DDM (pH=7.4).
Spectroscopy
Absorption spectra of enriched protein samples were recorded on a SpectraMax 190 Microplate Reader (Molecular Devices) at 23°C. Cuvette was blanked with elution buffer. The protein sample was initially scanned in a dark state. A 1 kW halogen lamp (Phillips) with a shortpass filter (Edmund Optics) was used for blue light (470 nm) photoactivation of the protein and protein deactivation was achieved using filtered amber light (594nm). Absorbance measurements were plotted using Graphpad Prism.
Cell culture and transfection
Real-time cAMP dynamics
Luminescence assays to monitor cAMP levels were performed as we described previously (Siuda et al., 2015a). Briefly, HEK293 cells (ATCC, cat. # CRL-1573) were co-transfected with PPO-Venus and pGloSensor-22F (Promega E2301) plasmids using JetPrime reagent (Polyplus, cat. # 114–07). Two hours before the experiment, cells were incubated with 2% of the GloSensor reagent (Promega, cat. # E1290). Relative luminescence units were recorded using a SynergyMx microplate reader (Biotek). Adenylyl cyclase was activated with 1 μm forskolin, and cells were stimulated with constant blue LED light (465 nm, 5 mW, Plexon) for 60 seconds before being returned to the plate reader. Data, expressed as relative luminescence units, were normalized to the peak levels and plotted using GraphPad Prism.
Imaging assays of γ9 translocation
The HeLa cell line was obtained from ATCC and cultured in MEM supplemented with 10% dialyzed FBS (Atlanta Biologicals) and 1x pen-strep at 37°C and 5% CO2. Cells were cultured and transiently transfected in 35-mm glass bottom dishes (CellVis), using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Transfections used 2 μl of lipofectamine per dish along with the following DNA amounts:
GFP-γ9 experiments: 1μg PPO (untagged), 0.1μg EGFP-γ9
Two-photon experiments: 1μg PPO-Venus, 0.1μg mScarlet-γ9
Gαo/PTX experiments: 1μg PPO-Venus, 0.1μg mScarlet-γ9, with or without 1 μg αOA(C351I)-CFP
Arrestin experiments: 1μg PPO (untagged), 1μg Arrestin3-GFP, 0.5μg mCh-clathrin, or 1μg PPO-Venus, 1μg Arrestin3-mCh.
Cells were imaged 1–2 days after transfection.
Dorsal root ganglia (DRG) neuron culture
DRG cultures were prepared from 1–2 day old wildtype C57BL/6J or 4–6 week old AvilCre mice as described previously (Siuda et al., 2015a). Mice were anesthetized with isoflurane, spinal columns were removed, and DRG were dissected in HBSS + 10 mM HEPES (HBSS+H). Tissue was enzymatically digested with papain (45U, Worthington, cat. # LS003126) for 20 minutes at 37°C, rinsed in HBSS+H, followed by collagenase (1.5 mg/mL; Sigma, cat. #C6885) for 20 minutes at 37°C. Ganglia were washed and resuspended in culture media consisting of Neurobasal A (Gibco), 5% FBS (Life Technologies), 1x B27 (Gibco), 2 mM glutamax (Life Technologies) and 100 μg/mL penicillin/streptomycin (Life Technologies). Neurons were dissociated by mechanical trituration through glass pipettes, filtered with 40 μm filters, and plated at a density of 10,000 cells/well onto poly-D-lysine and collagen coated 12 mm coverslips. For imaging, neurons were transfected 24 hours after plating with 1 μg plasmid cDNA : 2 μl lipofectamine 2000 per well. For electrophysiology, neurons were infected with 2*1010 vg/ml of Cre-dependent AAVs (AAV5:CAG:DIO:PPO-Venus or CAG:DIO:eYFP) 2 hours after plating. For experiments testing G-protein dependence, neurons were incubated overnight in 200 ng/ml pertussis toxin (List Biological Laboratories, cat. # 179A) before recordings.
Surgical procedures
For slice electrophysiology, 5–6 week old mice were anesthetized with isoflurane and secured on a stereotaxic frame. For Vglut2-IRES-Cre mice, 1 μl of AAV5:Ef1α:DIO:PPO-Venus was injected unilaterally into the right VPm (AP: −1.5 mm, ML: +1.8 mm, DV: −3.6 mm) using a beveled tip Hamilton syringe. For DAT-IRES-Cre mice, 1 μl of AAV5:Ef1α:DIO:PPO-Venus was injected unilaterally into the right VTA (AP: −3.3 mm, ML: +0.4 mm, DV: −4.5 mm. Viruses were injected at 75 nl/min and the needle was left in place for 10 minutes after infusion before being slowly withdrawn and suturing the skin.
For anesthetized, in vivo electrophysiology, 12–16 week old Vgat-IRES-Cre (Cre+) mice or Cre− littermate controls were anesthetized with isoflurane and 0.4 μl (per hemisphere) of AAV5:Ef1α:DIO:PPO-Venus was injected bilaterally at 100 nl/min into the bed nucleus of the stria terminalis (BNST) (AP: +0.16 mm, ML: +/−0.9 mm, DV: +4.8 mm).
For in vivo fiber photometry experiments, 12–16 week old DAT-IRES-Cre (Cre+) mice were anesthetized with isoflurane and 0.4 μl (per hemisphere) of AAV5:Ef1α:DIO:PPO-Venus or AAV5:Ef1α:DIO:YFP was injected bilaterally at 100 nl/min into the ventral tegmental area (VTA) (AP: −3.4 mm, ML: ±0.4 mm, DV: −4.4 mm). 0.4 μl (per hemisphere) of AAVDJ:CAG:RdLight1 was injected unilaterally at 100 nl/min into the the nucleus accumbens core (NAc core) (AP: +1.4 mm, ML: +/−1.5 mm, DV: −4.0 mm, 10° angle) 400 μm optic fibers were placed bilaterally targeting the NAc core (coordinates same as above).
In behavior experiments, 12–16 week old DAT-IRES-Cre (Cre+) mice or Cre−littermate controls were anesthetized with isoflurane and 0.4 μl (per hemisphere) of AAV5:Ef1α:DIO:PPO-Venus was injected bilaterally at 100 nl/min into the ventral tegmental area (VTA) (AP: −3.4 mm, ML: ±0.4 mm, DV: −4.4 mm). 220 μm optic fibers were placed bilaterally targeting either the VTA (coordinates same as above) or the nucleus accumbens core (NAc core) (AP: +1.4 mm, ML: +/−1.5 mm, DV: −4.0 mm, 10° angle). 12–16 week old Vglut1-IRES-Cre (Cre+) mice or Cre− littermate controls were anesthetized with isoflurane and 0.4 μl (per hemisphere) of AAV5:Ef1α:DIO:PPO-Venus was injected bilaterally at 100 nl/min into the basolateral amygdala (BLA) (AP: −1.35 mm, ML: ±3.0 mm, DV: −4.5 mm). 220 μm optic fibers were placed bilaterally targeting either the nucleus accumbens shell (NAc core) (AP: +1.4 mm, ML: +/−1.5 mm, DV: −4.5 mm, 10° angle).
Histology
DRG cultures
One to 7 days after AAV infection, cultured DRG neurons were rinsed in PBS and fixed in 4% PFA/sucrose for 10 minutes on ice. After rinsing in PBS, neurons were blocked and permeabilized in PBS containing 3% donkey serum and 0.3% Triton X-100 for 15 minutes at room temperature. Cultures were incubated in primary antibodies (mouse anti-synapsin-1 and guinea pig anti-tau both at 1:2000 dilution) in PBS with 3% donkey serum overnight at 4°C. After washing, primary antibodies were labeled with donkey anti-mouse AF555 and goat anti-guinea pig AF647 secondary antibodies (both 1:2000 in 3% donkey serum) for 1 hour at room temperature. Coverslips were rinsed five times before being mounted on slides with Prolong Gold Antifade reagent with DAPI (Life Technologies, cat. # 36931). Neurons were imaged on a Leica TCS SPE confocal microscope with a 63x oil immersion objective (NA=1.4).
Thalamocortical projections
Two to 6 weeks after viral injections of Cre-dependent PPO into the VPM, mice were deeply anesthetized with ketamine/xylazine and transcardially perfused with PBS followed by 4% PFA. Brains were removed and cut at a 45° angle to the right of the midline to preserve thalamocortical axon projections in the plane of slicing. They were then post-fixed in PFA overnight, cryoprotected in 30% sucrose and frozen in OCT media. 50 μm thick thalamocortical slices were then cut parallel to the 45° plane and stored in PBS with 0.2% NaN3. Sections were rinsed thoroughly in PBS before being blocked and permeabilized in PBS with 2% donkey serum, 2% goat serum and 0.3% Triton X-100 for 1 hour at room temperature. Primary antibodies (mouse anti-PSD-95 (1:500) and guinea pig anti-vGluT2 (1:500)) were incubated with the sections overnight at 4°C in PBS with 2% each donkey and goat serum. Sections were washed 5 times and incubated with secondary antibodies (donkey anti-mouse AF555 (1:500) and goat anti-guinea pig AF647 (1:500)) for 1 hour at room temperature in PBS with 2% donkey and goat serum. Sections were washed 5 times again and mounted onto slides using Vectashield Hardset Antifade reagent with DAPI (Vector Labs, cat. # H-1400). Thalamocortical slice imaging was performed on an Andor Dragonfly microscope described below.
VTA/BLA neurons projections
Following behavioral experiments, mice were deeply anesthetized with pentobarbital and transcardially perfused with PBS followed by 10% formalin. Heads were placed with optic fibers intact overnight in 10% formalin. Brains were extracted and post-fixed in formalin overnight, followed by cryoprotection in 30% sucrose. 30 μm thick slices containing either the VTA, BLA or NAc core, shell were collected and mounted onto slides using Vectashield Hardset Antifade reagent with DAPI (Vector Labs, cat #H-1400) and imaged using an Olympus FV1000 confocal microscope (Olympus Scientific Solutions).
Imaging assays
Live-cell confocal imaging
Imaging was performed using an Andor Dragonfly 500 spinning disk confocal system built on a Nikon Ti2 inverted fluorescence microscope equipped with Nikon Perfect Focus to actively prevent focal drift. The system is equipped with five solid state lasers (445 nm, 488 nm, 515 nm, 561 nm, and 640 nm), the emission can be collected along one of two paths containing emission filter wheels and either an EMCCD camera (Andor iXon Life) or a sCMOS camera (Andor Xyla). LED light from a multi-LED illumination system (CoolLED pE-4000) was directed through the back port of the microscope by an Andor Mosaic DMD, which enabled spatially patterned illumination at the sample. Blue or UV LED light was coupled into the excitation path with the 515nm and longer wavelength lasers using a dichroic mirror to enable simultaneous imaging and photoactivation. LED pulsing was controlled using the CoolLED touch pad and LED illumination for UV vs. blue activation was a single 150 ms pulse every 5 seconds, or in 10 ms pulses at 10Hz where specified.
HeLa cell imaging used a 60x, 1.4NA oil immersion objective (Nikon MRD01605: CFI Plan Apo Lambda 60x Oil), the EMCCD camera, and single plane confocal imaging. An OkoLabs stage top incubator maintained the samples at 37°C, 5% CO2. HeLa cells were imaged in their culture medium supplemented with 10 μM 9-cis retinal.
Total internal reflection fluorescence (TIRF) imaging
TIRF imaging of PPO-induced arrestin translocation was performed on the Andor Dragonfly system described above, which is equipped for multi-color TIRF imaging. Images were captured using a 60x, 1.49 NA TIRF objective (Nikon: CFI Apochromat TIRF 60XC Oil) and an EMCCD camera (Andor iXon Life). Images were captured at 5 sec intervals for 10 min, and photoactivation with 460 nm LED light (10mW/mm2, 10 ms pulses, 10 Hz) was applied throughout the duration of the image sequence. Images of Arrestin3-GFP and mCh-clathrin were captured using 488 nm and 561 nm laser excitation, respectively. Images of PPO-Venus and Arrestin3-mCh were captured using 515 nm and 561 nm laser excitation, respectively.
Receptor internalization
4–5 days after transfection with either PPO-Venus or MOR-GFP, neurons were transferred to a 37°C heated chamber filled with extracellular solution containing (in mM): 145 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, 10 HEPES, pH to 7.3 with NaOH, 310 mOsm. To avoid prior photoactivation, transfected cells were first identified using low light then coverslips were pretreated with 10 μM 9-cis-retinal for at least 30 minutes in the dark prior to imaging. Live-cell imaging was performed on an Andor Dragonfly spinning disk system described above using the 488 nm laser line for Venus/GFP Z-stack imaging and a sCMOS camera (Andor Zyla) acquired every 5 minutes. Neurons were photoactivated with either a blue LED at 10 Hz described above for TIRF imaging or treated with 1 μM DAMGO, and imaged for 30 minutes.
Thalamocortical slice imaging
Brain slice imaging was performed using the Andor Dragonfly system described above. A 20x, 0.75NA objective (Nikon MRD00205) was used to identify regions of the barrel cortex for higher resolution imaging, which was performed using the 60x objective as above. Z-stack images were acquired using the sCMOS camera and deconvolved using Andor Fusion software. Laser excitation at 488nm, 561nm and 640nm was used to image PPO-Venus, Alexa Fluor 555 and 647 dyes, respectively.
LED-based deactivation during GFP imaging
Imaging was performed using an Andor Revolution spinning disk imaging system build on a Leica DMI6000B inverted microscope with adaptive focus control, with a Yokogawa CSU x1 spinning disk unit, and an Andor iXon EMCCD camera. The system is equipped with four solid state lasers at 445 nm, 488 nm, 514 nm, and 594 nm, combined through an acousto-optic tunable filter to enable rapid switching and control excitation power. An incubation chamber surrounding the entire microscope was maintained at 37°C, 5% CO2. Imaging was performed using a 63x, 1.4NA oil immersion objective (Leica 506187: HCX PL APO 63x). For GFP-γ9 imaging and simultaneous activation of PPO, a single confocal plane was imaged at a rate of one frame every 3 s, with 488 nm excitation through the spinning disk (average power ~45 μW, which is typical for imaging GFP alone) and 300ms exposure time. PPO deactivation was achieved using a 595 nm LED (CoolLED pE-4000). The LED light was directed through the back port of the microscope by an Andor Mosaic DMD operating in “white mask” mode and ~150 μW measured through the objective. The 488nm imaging laser and the 595 nm LED were coupled into the excitation path using a 562nm long pass dichroic mirror (Semrock Brightline FF556-SDi01). GFP emission was collected through the same dichroic mirror, a 488nm notch mirror, and a 525/30nm emission filter.
Multiphoton imaging
Multiphoton imaging was performed on an Olympus Fluoview FVMPE-RS multiphoton imaging system equipped with two wavelength-tunable Ti:Sapphire lasers (Spectra Physics, MaiTai and Insight lasers). HeLa cells transfected with PPO-Venus and mScarlet-γ9 were imaged at room temperature (22°C) in 35mm glass bottom dishes containing culture medium supplemented with 10 μM 9-cis retinal. A water immersion objective (Olympus XLPLN25XWMP2, 25x, 1.05NA) was immersed in the medium in the dish from above for imaging. Imaging and photostimulation were performed by laser scanning at a lateral resolution of 0.4792 μm/pixel. Imaging was performed with a scan speed of 20 μs/pixel, and each line was scanned 3 times and averaged. Two-photon excitation of mScarlet was performed using the Insight and tuned to 1080 nm. Two-photon photoactivation of PPO at each wavelength tested (700, 800, 900, 1000nm), was performed by tuning the wavelength of the MaiTai laser, adjusting the output power to an average of 10 mW measured at the back focal plane of the objective, and scanning the focused laser at a speed of 2 μs/pixel. The two lasers were combined into the same excitation path using a laser coupling dichroic mirror (Olympus LCDM690–1050). Emission was collected through a green-red filter cube (Olympus FV30-FGR) onto GaAsP photomultiplier tubes. In order to reset PPO into the off state after each image acquisition, the sample was illuminated from below through a condenser lens by constant amber LED light (ThorLabs M590L3) for at least 2 min.
Live-cell imaging analysis
Image sequences were first inspected manually for any lateral drift of the sample throughout the time course of the experiment. If necessary, the ImageJ TurboReg plugin was used to correct for lateral drift. Translocation of FP-γ9 from the plasma membrane to intracellular regions was quantified by manually selecting one cytosolic ROI per cell that excluded the plasma membrane (PM) and nucleus. ROIs are manually selected for each cell. Care was taken to select ROIs that clearly exclude any plasma membrane fluorescence and only quantify intracellular fluorescence. The mean fluorescence intensity within the ROI was computed for each frame in a time series, using either ImageJ or Andor iQ software. An ROI selected in a nearby region of the sample lacking fluorescent cells was used for background subtraction. Since the fluorescence intensity varied among the transiently transfected cells, intracellular FP-γ9 intensities were normalized by the value at the first time point. Receptor internalization was quantified by taking the ratio of PM to intracellular fluorescence which was normalized for each cell by blinded investigators. Images were corrected for photobleaching using the bleach correction plug-in for ImageJ.
Electrophysiology
Recombinant G-protein coupled inwardly rectifying K+ (GIRK) channel recordings
GIRK currents were recorded from HEK cells 1 day after transfection with GIRK2A and PPO-Venus at a 3:1 ratio using lipofectamine 2000. Immediately after transfection, cultures were supplemented with 1 μM 9-cis-retinal (Sigma R5754) or 0.01% ethanol (control). Cells were recorded in high K+ external solution containing (in mM): 140 NaCl, 20 KCl, 0.5 mM CaCl2, 2 MgCl2, 10 glucose, 10 HEPES, pH to 7.3 with NaOH, 310–320 mOsm. Whole-cell patch clamp recordings were made using glass pipettes with resistances of 2–3 MΩ when filled with K+ gluconate internal solution containing (in mM): 120 K+ gluconate, 5 NaCl, 0.1 CaCl2, 2 MgCl2, 1.1 EGTA, 10 HEPES, 4 Na2ATP, 0.4 Na2GTP, 15 Na2phosphocreatine, pH=7.28, 290 mOsm. All recordings were performed at 34°C using a heated chamber (ALA Scientific Instruments, HCS) and temperature controller (ALA, HCT-10). Cells were visualized using infrared DIC (IR-DIC 770±25 nm) illumination to prevent photoactivation.
Recordings were performed using Patchmaster software controlling a HEKA EPC10 amplifier. Cells were voltage clamped at −40 mV (near the calculated K+ reversal potential of −46 mV) for measuring ramp currents, or 0 mV to determine activation kinetics. Inwardly rectifying currents were elicited by 200 ms ramp depolarizations from −120 to 40 mV every 2 seconds, filtered at 3 kHz, and digitized at 20 kHz. Whole-cell and pipette capacitance transients were minimized using the compensation circuits of the amplifier, and series resistance was compensated by 70%.
Wide-field photostimulation was delivered through the 40x objective using custom LEDs coupled to the back fluorescence port of the microscope (Olympus, BX-51). 365 nm UV (Thorlabs, M365L3) and 470 nm blue (M470L2), and 590 nm amber (M590L2) light was collimated with aspheric condenser lenses (Thorlabs, cat. #ACL2520U-A). UV and blue light were separated by a 425 nm longpass dichroic mirror (Semrock, T425lpxr) and reflected off a 505 longpass dichroic mirror (Thorlabs, DMLP505R). LEDs were triggered by TTL pulses from the amplifier, Master-9 signal generator (AMP Instruments), or PulsePal to the LED current controller (Thorlabs, DC4104). All light intensities were calibrated using a photodiode (Thorlabs, S121C) and power meter (Thorlabs, PM100D). Ambient light levels were measured from the overhead room lighting using a light meter (HDE, HDE-F86-NEW).
Voltage gated calcium channel recordings
Calcium channel currents were recorded from cultured DRG neurons after 3–6 days in vitro. Neurons were transferred into choline external solution containing (in mM): 135 choline chloride, 10 HEPES, 10 glucose, 2 BaCl2, 4 MgCl2, pH adjusted to 7.3 with CsOH and 300–310 mOsm. 500 nM TTX (Abcam, cat. # ab120055) was included during recordings to block any residual sodium currents. Barium was used as the charge carrier for these recordings to avoid Ca2+ dependent changes in channel gating and physiology, and barium currents are referred to as calcium channel currents in the manuscript. GABABRs were stimulated with bath application of 50 μM R/S-baclofen (Abcam, cat. # ab120149), and 100 μM Cd2+ was routinely added at the end of an experiment to verify isolation of calcium channel currents. DRG neurons were visualized through a 40x objective using IR-DIC illumination to minimize light stimulation on an upright BX-51 microscope, and Venus/YFP-expressing neurons were identified using epifluorescence. Whole-cell patch-clamp recordings were made using fire-polished glass pipettes with resistances of 2–3 MΩ when filled with internal solution containing (in mM): 110 CsCl, 10 TEA chloride, 0.1 CaCl2, 10 EGTA, 10 HEPES, 4 MgATP, 0.4 Na2GTP, 10 Na2phosphocreatine, pH to 7.28 with CsOH, 290 mOsm. The majority of recordings were performed at 25°C. For testing the effects of PPO at physiological temperatures, a heated chamber and temperature controller were used to maintain the bath temperature at 32–34°C.
Whole-cell patch-clamp recordings were made with Patchmaster software controlling a HEKA EPC10 amplifier. Calcium channel currents were elicited by step depolarizations from −80 to 0 mV and monitored every 10 seconds, while leak currents were subtracted with a p/4 protocol of four 20 mV depolarizing test pulses after each sweep. IV curves were generated by step depolarizations from −80 to +60 mV in 10 mV increments. For pre-pulse protocols, neurons were depolarized to +80 mV for 50 ms, followed by a return to −80 mV for 10 ms before 50 ms test pulses to 0 mV. Currents were filtered at 3 kHz and digitized at 20 kHz. Whole-cell and pipette capacitance transients were minimized using the compensation circuits of the amplifier, and series resistance was compensated by 70% in all recordings. Only cells with stable Rs < 15 MΩ that did not change by more than 20% were included. Neurons were discarded from analysis if currents changed by more than 10% during baseline.
Photostimulation was delivered through the 40x microscope objective as described above. Blue LED light (Thorlabs, 470L2) was directed into the microscope by a 505 nm longpass dichroic mirror, while collimated green (Thorlabs, 530L3) and amber (Thorlabs, 590L2) were coupled behind this mirror. LEDs were triggered as described above.
Acute slice preparation
Brain slices for electrophysiology recordings were prepared using a protective cutting and recovery method (Siuda et al., 2015a; Ting et al., 2014). For VTA and NAc recordings, 3–4 weeks after viral injections into the VTA, mice were deeply anesthetized with ketamine and xylazine and transcardially perfused with cold oxygenated choline aCSF containing (in mM): 93 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbic acid, 2 thiourea, 3 sodium pyruvate, 12 N-acetyl-L-cysteine, 0.5 CaCl2, 5 MgCl2, pH=7.3 300–310 mOsm. Brains were rapidly removed, embedded in 2% low-melt agarose (Sigma, A0676) and 200 μm thick horizontal (VTA) or 250 μm thick coronal slices (NAc) were cut using a Compresstome (Precisionary Instruments, cat. # VF210–0Z). Slices were transferred to a recovery chamber containing oxygenated choline aCSF at 32°C for 5 minutes before transfer to a holding chamber filled with room temperature oxygenated holding-aCSF containing (in mM): 92 NaCl, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 2 CaCl2, 2 MgCl2, pH=7.3, 300–310 mOsm. Slices were maintained in the dark at room temperature and allowed to recover >1 hour before recording.
For thalamocortical recordings, 2–6 weeks after viral injections mice were deeply anesthetized with ketamine and xylazine and transcardially perfused with cold oxygenated NMDG-aCSF containing (in mM): 93 N-methyl-D-glucamine, 2.5 KCl, 1.25 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 ascorbic acid, 2 thiourea, 3 sodium pyruvate, 0.5 CaCl2, 5 MgCl2, pH=7.3, 300–310 mOsm. Brains were rapidly removed and blocked with a 45° cut to the right of the midline (Agmon and Connors, 1991; Crandall et al., 2017). The cut surface was then glued to a platform and embedded in 2% low-melt agarose (Sigma, A0676). 350 μm thick slices were cut using a Compresstome (Precisionary Instruments, cat. # VF-200–0Z) and transferred to a recovery chamber containing oxygenated NMDG-aCSF at 32°C for 10–12 minutes. Slices were transferred to a holding chamber as described above and allowed to recover >1 hour before recording.
VTA excitability recordings
Slices were transferred to a heated recording chamber of an upright BX-51 microscope and perfused at a rate of 2 ml/minute with oxygenated aCSF containing (in mM): 124 NaCl, 2.5 KCl, 1.25 NaH2PO4, 24 NaHCO3, 5 HEPES, 12.5 glucose, 2 CaCl2, 1 MgCl2 (pH=7.3 and 300–310 mOSm). Slices were maintained at 32–34°C as described above. Neurons were visualized under IR-DIC illumination and PPO-Venus+ neurons identified using brief low intensity LED light and standard GFP filter cubes. After cells were identified they were illuminated with amber light for 2 minutes to reverse any prior activation of PPO. Whole-cell patch-clamp recordings were made with 3–4 MΩ glass pipettes filled with K+ gluconate internal described above. Recordings were performed using pClamp11 software (Molecular Devices) controlling a Multiclamp 700B amplifier. Neurons were held at −60 mV in both voltage and current clamp configurations in aCSF with synaptic and GABAB receptor antagonists to isolate the effects of PPO including 10 μM NBQX, 50 μM D-APV, 10 μM bicuculline, 100 μM picrotoxin, 50 μM saclofen (all from Abcam). Photostimulation was delivered through the 40x objective as described above. Input resistance, rheobase and input-output curves were quantified from current clamp recordings using 1 second step current injections from −50 to 200 pA in 10 pA increments. Inwardly rectifying currents were measured using 200 ms voltage ramps from −40 to −140 mV. 1 mM Ba2+ was added at the end of all recordings to confirm GIRK channel coupling.
Thalamocortical projection recordings
Oblique thalamocortical slices were perfused and visualized as described above. Whole-cell patch-clamp recordings were made from visually identified neurons in layer IV of the barrel cortex using fire-polished glass pipettes with open tip resistances of 3–5 MΩ when filled with internal solution containing (in mM): 110 cesium gluconate, 8 tetraethylammonium chloride, 3 QX314 bromide, 1.1 EGTA, 0.1 CaCl2, 10 HEPES, 4 MgATP, 0.4 Na2GTP, 10 Na2phosphocreatine, pH to 7.27 with CsOH, 291 mOsm.
Recordings were performed using Patchmaster software controlling a HEKA EPC10. Neurons were voltage-clamped at −70 mV and electrically evoked thalamocortical currents were elicited by a bipolar stimulating electrode (FHC cat. #30202) placed in the VPm. Current pulses (0.2 ms, 50 ms interpulse intervals) was provided by a stimulus isolator (WPI, cat. #A365) and adjusted for each cell to produce ~90% success rates. Excitatory postsynaptic currents were pharmacologically isolated using 10 μM bicuculline (Abcam, cat. # ab120108) and 100 μM picrotoxin (Abcam, cat. # ab120315) and elicited every 10 seconds. LED stimulation (10 Hz, 10 ms pulses, 10 mW/mm2) was delivered through the 40x objective above the recorded barreloid as described above. We confirmed the restricted photostimulation by illuminating PPO-Venus+ fibers within the 40x field of view using constant blue illumination (10 mW/mm2 for ~5 minutes) and inspecting the photobleached area with the 4x objective. Series resistance values were not compensated and were continuously monitored using 5 mV hyperpolarizing test pulses every sweep. Neurons were excluded from analysis if Rs values changed more than 20% during recording. Data were sampled at 20 kHz, filtered at 3 kHz, and analyzed offline. PPO-Venus+ fibers were imaged after recordings using LED epifluorescence through standard GFP filter cubes.
NAc mIPSC recordings
Miniature inhibitory postsynaptic currents (mIPSCs) were recorded in PPO-negative cells of the nucleus accumbens that were innervated by a subset of PPO+ terminals from adjacent inhibitory neurons. Recordings were performed as described above for VTA neurons, using 4–5 MΩ pipettes filled with Cs+ gluconate internal solution. mIPSCs were pharmacologically isolated with 10 μM NBQX, 50 μM D-APV and 500 nM TTX (all from Abcam) and YFP negative neurons were recorded at 0 mV using gapfree protocols with a Multiclamp 700B amplifier. Data were digitized at 10 kHz and filtered at 3 kHz. Cells were discarded if the series resistance was >30 MΩ or changed more than 20%. Light stimulation was delivered through the microscope objective as described above. All recordings and analysis were performed by investigators blinded to the group.
Electrophysiology analysis
Electrophysiology recordings were analyzed using Igor Pro software (Wavemetrics) with the NeuroMatic plug-in (Rothman and Silver, 2018) and custom-written macros. Inwardly rectifying currents from recombinant GIRK channel currents were measured at −120 mV before the voltage ramp. Latencies were quantified from the onset of the LED to 10% of the peak and rise-times are from 10–90% of the peak. Decay constants were determined from single-exponential fits and steady-state currents represent the magnitude of the current change during amber illumination to turn off PPO. Endogenous GIRK currents were elicited with a 200 ms voltage ramp from −20 to −20 mV. Input resistance was determined from the linear regression of the current-voltage plot in response to hyperpolarizing steps. Action potentials were quantified as depolarizations that overshot 0 mV during step current injections.
Calcium channel currents were normalized to membrane capacitance and plotted normalized to the average baseline amplitude. Neurons were discarded if baseline currents drifted by more than 10%. Percent inhibition was quantified as the maximum inhibition observed during optical or pharmacological stimulation relative to baseline amplitudes. EPSC amplitudes were taken as the average amplitude within 0.1 ms of the peak. Latencies and jitter were calculated from the onset of electrical stimulation to 10% of the peak rise-time. Series resistance changes were monitored by calculating the peak amplitudes of the capacitance transients during 5 mV hyperpolarizing test pulses. Miniature IPSCs were detected using template fitting in Clampfit11 software, visually inspected, and confirmed by investigators blinded to the experimental group. Recovery tau values were calculated from single-exponential fits of the normalized peak current amplitudes as they returned to baseline using NeuroMatic. Half maximal activation for IV curves were determined using Boltzmann sigmoidal non-linear regression with Graphpad Prism 8 software. Prism software was also used for all statistical analyses.
Anesthetized electrophysiology experiments and analysis
Single-unit activity was recorded as previously described (McCall et al., 2015; Siuda et al., 2015a) (McCall et al., 2015; Siuda et al., 2015a). Briefly, mice were lightly anesthetized (1% isoflurane), placed in a stereotactic frame and one skull screw was placed on the side of the midline to ground the electrode array. The recording apparatus consisted of a 16-channel (35 μm tungsten wires, 150 μm spacing between wires, 150–33 μm spacing between rows, Innovative Physiology, Durham, NC) electrode array. This array was epoxied to a fiber optic and lowered into the LH (stereotaxic coordinates from bregma: −1.3 mm (AP), −0.9 mm (ML) and −4.85 mm (DV). Extracellular recordings were taken from Vgat-Cre::BNST-LH:PPO mice. Spontaneous neuronal activity and activity that resulted from BNST-LH photoinhibition was recorded from each electrode, bandpass-filtered with activity between 250 and 8000 Hz, and analyzed as spikes. Voltage signals were amplified and digitally converted using OmniPlex and PlexControl (Plexon, Dallas, TX) (RRID:SCR_014803). For Vgat-Cre::BNST-LH:PPO recordings, 2 min of baseline recordings were made followed by 5 seconds of 10 Hz photoinhibition (10 ms pulses, 10 mW) and then another 1 min post- stimulation (off, on, off). Principle component analysis and/or evaluation of t-distribution with expectation maximization was used to sort spikes using Offline Sorter (Plexon)(RRID:SCR_000012) and only cells with distinct clusters away from the noise that remained firing throughout the duration of the recording were included. To determine the latency to fire, we calculated the average time from the onset of first photostimulation to the next spike from each cell.
In vivo behavior and photometry
In vivo fiber photometry and analysis
For fiber photometry studies, mice were initially food deprived to 90% of their body weight and trained in an uncued reward delivery task with a modular test chamber (17.8 × 15.2 × 18.4 cm) (Med Associates Inc.). Fiber photometry recordings were made throughout the entirety of the session as previously described (Gunaydin et al., 2014; Parker et al., 2019). Prior to recording, an optic fiber was attached to the implanted fiber using a ferrule sleeve (Doric, ZR_2.5). A 450-Hz sinusoidal LED light (Thorlabs, LED light: M560F3; LED driver: DC4104) was bandpass filtered (560 ± 20 nm, Doric, FMC4) to excite RdLIght1 and evoke emission. LED intensity for the 560 nm wavelength band was measured at the tip of the optic fiber and adjusted to 50 μW before each day of recording. Fluorescence traveled through the same optic fiber before being bandpass filtered (625 ± 25 nm, Doric, FMC4), transduced by a femtowatt silicon photoreceiver (Newport, 2151) and recorded by a real-time processor (TDT, RZ10X). The 450-Hz signals were extracted in real-time by the TDT program Synapse at a sampling rate of 1017.25 Hz. For the PPO stimulation experiments, a 465 nm laser was used at a 5–7 mW intensity to deliver blue light through the tip of the same optic fiber used to excite RdLight1.
Custom MatLab scripts were developed for analyzing fiber photometry data in context of mouse behavior. The 560 nm excitation signal was extracted and a double exponential curve was fit to the raw trace and subtracted to correct for baseline drift. After baseline correction, the photometry trace was z-scored relative to the mean and standard deviation. All data are normalized to a pre-determined time window (0–1s) to account for issues oowing to the blue light stimulation artifact. The post-processed fiber photometry signal was analyzed in the context of animal behavior during reward delivery. Mean fluorescence was determined during pre-determined time windows following reward delivery (1–4s). Code that supports the analysis will be made available from the corresponding author upon request.
Operant uncued reward delivery behaviors
All behaviors were performed in a sound-attenuated room at least one week after habituation to handling and the holding room. One week prior, DAT-Cre+ (PPO and YFP controls) mice were food restricted down to ~85% of free feeding body weight and habituated to the reward (sucrose pellet) for 2 days prior to training. Mice were trained to to retrieve and consume sucrose pellets for 3–5 days within an operant box (Med-Associates, ENV-307A) under a variable interval 90 paradigm, where they would receive the pellet at a randomized interval of 30, 60 or 90 s separating consecutive trials. A randomized intertrial interval of between 30–90 seconds separated consecutive trials lasting 30 minutes for a total of 24 trials. Every third trial would be preceded by blue light delivery for photoinhibition at 10 Hz (473 nm wavelength, 10 ms pulse width, 5–8 mW of power; matched with optic fiber efficiency), ensuring within-session and within-animal comparisons for behavioral measures (such as latency for pellet retrieval). Pellet retrieval was scored via a custom IR head entry detector.
Operant sucrose licking behaviors
All behaviors were performed in a sound-attenuated room at least one week after habituation to handling and the holding room. One week prior, Vglut1-Cre+ or Cre− mice were water restricted down to ~85% of free feeding body weight and habituated to the reward (10% sucrose solution) for 2 days prior to training. Mice were trained to engage with a sipper, delivering 10% sucrose triggered by licks for 3–5 days within an operant box (Med-Associates, ENV-307A). The sipper is available for 5 minutes, 3 times in a 30 minute session. In the second 5-minute sipper session (but not the first or third sessions), licks would trigger blue light delivery for photoinhibition at 10 Hz (473 nm wavelength, 10 ms pulse width, 5–8 mW of power; matched with optic fiber efficiency), ensuring within-session and within-animal comparisons for behavioral measures (such as latency for pellet retrieval).
Operant FR and PR behaviors
All behaviors were performed in a sound-attenuated room at least one week after habituation to handling and the holding room. Animals were trained on Pavlovian conditioning for 3 days, followed by fixed-ratio 1 (FR-1; 1 nose poke, one sucrose pellet) for 3 days, fixed ratio-3 (FR-3; 3 nose poke, one sucrose pellet) for 3 days and then progressive ratio testing. One week prior to Pavlovian conditioning, DAT-Cre+ or Cre− mice were food restricted down to ~85% of free feeding body weight. Mice were also habituated to the reward (sucrose pellet) for 2 days prior to Pavlovian conditioning. Mice were trained to associate illumination of a house light (CS) with access to a sucrose pellet (US) within an operant box (Med-Associates, ENV-307A) under a variable interval 90 paradigm, where they would receive the pellet at a randomized interval of 30, 60 or 90 s separating consecutive trials. The house light would illuminate concurrently for 20s with reward delivery. A randomized intertrial interval of between 30–90 seconds separated consecutive trials. Pavlovian conditioning sessions lasted for 60 minutes, over which an average of 35 rewards were presented. Following conditioning for 3 days upon which animals consumed all the rewards presented in one session, mice underwent FR-1, where they were trained to perform 1 nose poke for 1 reward. Similar to Pavlovian conditioning, the house light would illuminate concurrently for 20s with reward delivery, following which, the animals would be able to proceed nose-poking. Upon optimum performance in FR-1, mice underwent FR-3 (3 nose-pokes for 1 reward) for 3 days, mice were counterbalanced to receive laser stimulation for photoinhibition at 10 Hz (473 nm wavelength, 10 ms pulse width, 5–8 mW of power; matched with optic fiber efficiency) for the entirety of the FR-3 session. Following photoinhibition during FR-3, mice were retrained on FR-3 without light stimulation and underwent progressive ratio (PR) testing, following the geometric progression, nj = 5e j/5− 5, in which the criteria for rewards increased in an exponential manner (1, 2, 4, 6, 9, 12…) over the training period (Richardson and Roberts, 1996). Mice received laser stimulation for photoinhibition using the aforementioned parameters either for the entirety of the session or where stimulation was triggered by nose pokes for 1 second. Furthermore, during all photoinhibition experiments, mice were counterbalanced to receive laser stimulation during the entire PR session.
Conditioned place preference
DAT-Cre+ or Cre− mice were trained in an unbiased, three-compartment conditioning apparatus as described previously (Al-Hasani et al., 2015; McCall et al., 2015). In brief, we used a modified three-chamber CPP apparatus consisting of two square boxes (27 cm × 27 cm) that served as the conditioning chambers separated by a small center area that served as the passageway (5 cm wide × 8 cm long) between boxes. Boxes had 2.5 cm black-and-white vertical stripes or horizontal stripes and floors were covered with 500 ml of bedding on each side. The floor of the center area was smooth Plexiglas. Mice were habituated to handling, injection restraint and to the testing room for one week prior to testing. On day 1, mice were allowed free access to all 3 compartments of the conditioning apparatus for a 30-minute session. For 2 consecutive days following pre-test, mice received counterbalanced saline or cocaine (10 mg/kg, I.P) injections confined to either the left or right compartment at least 4 hours apart. Mice also received laser stimulation for photoinhibition at 10 Hz (473 nm wavelength, 10 ms pulse width, 5–8 mW of power; matched with optic fiber efficiency) on the cocaine-paired compartment for the entirety of the 30-minute session. Following conditioning, mice underwent a “post-test” day, where they were again allowed free access to all 3 compartments. Time spent in each chamber and total distance traveled for the 30-minute trial was measured using Ethovision 10 (Noldus Information Technologies, Leesburg, VA).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed and plotted using Graphpad Prism. Details of the statistical tests for each experiment are listed in the figure legends. All data were analyzed with post-hoc tests to confirm that they met assumptions of the statistical tests used. Corrections were applied, where appropriate, and indicated in the figure legends. All data are represented as mean ± standard error of the mean and individual data points are shown for all data, where applicable. Statistical significance is denoted *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s.= not statistically significant
Supplementary Material
Video S1. PPO is activated by both UV and blue light (related to Figure 1). Movie of γ9-mCherry translocation in HeLa cells co-expressing untagged parapinopsin (PPO). γ9-mCherry (red) was imaged with a 595 nm laser which did not cause spontaneous translocation. Widefield stimulation with blue (470 nm) and UV (365 nm) light (150 ms pulses every 5 seconds) was delivered when indicated. Note the rapid translocation of γ9-mCherry from the plasma membrane to intracellular compartments during photostimulation and recovery when the light is switched off.
Video S2. PPO can be activated at single-cell resolution (related to Figure 1). Movie of γ9-mCherry (red) translocation in HeLa cells co-expressing untagged parapinopsin (PPO). LED stimulation of individual cells in the boxed regions of interest was delivered through a digital mirror device. Note the rapid translocation in the regions of interest which did not affect translocation in adjacent cells.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| mouse anti-synapsin-1 | Synaptic Systems | Cat. # 106011, RRID: AB_993033 |
| guinea pig anti-tau | Synaptic Systems | Cat. #314004, RRID:AB_1547385 |
| guinea pig anti-vGluT2 | Synaptic Systems | Cat. #135404, RRID:AB_887884 |
| mouse PSD-95 | NeuroMAb | Cat. #75-028, RRID:AB_2292909 |
| donkey anti-mouse AF555 | Life Technologies | Cat. #A-31570, RRID:AB_2536180 |
| goat anti-guinea pig AF647 | Life Technologies | Cat. # A-21450, RRID:AB2735091 |
| Bacterial and Virus Strains | ||
| AAV5:CAG:DIO:PPO-Venus | This study | n/a |
| AAV5:Ef1a:DIO:PPO-Venus | This study | n/a |
| AAV5:Ef1a:DIO:eYFP | Gift from Karl Deisseroth | Addgene cat. # 27056 |
| AAVDJ:CAG:red Dlight | Patriarchi et al., 2020 | n/a |
| AAV5:Ef1a:DIO-eNpHR3.0-eYFP | Gradinaru et al, 2010 | Addgene cat. # 26966 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 9-cis-retinal | Sigma | Cat. # R5754 |
| JetPrime reagent | Polyplus | Cat. # 114-07 |
| Lipofectamine 2000 | Invitrogen | Cat. # 11668027 |
| B27 supplement | Gibco | Cat. # 17504044 |
| Glutamax | Gibco | Cat. # 35050061 |
| Neurobasal A | Gibco | Cat. # 10888022 |
| poly-D-lysine | Sigma | Cat. # P0899 |
| Vectashield Hardset with DAPI | Vector Labs | Cat. # H-1400 |
| pertussis toxin | List Biological Laboratories | Cat. # 179A |
| R/S-baclofen | Abcam | Cat. # Ab120149 |
| DAMGO | Abcam | Cat. # Ab120674 |
| picrotoxin | Abcam | Cat. # Ab120315 |
| bicuculline | Abcam | Cat. # Ab120108 |
| NBQX | Abcam | Cat. # Ab120046 |
| D-APV | Abcam | Cat. # Ab120003 |
| saclofen | Abcam | Cat. # Ab0120327 |
| tetrodotoxin | Abcam | Cat. # Ab120055 |
| cocaine | Sigma | Cat. # C5776 |
| Critical Commercial Assays | ||
| pGloSensor-22F | Promega | Cat. # E2301 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T/17 | ATCC | Cat. # CRL-1573 |
| Human: HeLa | ATCC | Cat. # CCL-2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Advillin-Cre [Aviltm2(Cre)Fawa] | Jackson Labs | Cat. # 32536 |
| Mouse: DAT-IRES-Cre [Slc6a3tm1.1(Cre)Bkmn | Jackson Labs | Cat. # 006660 |
| Mouse: Vgat-IRES-Cre [Slc32a1tm2(cre)Lowl] | Jackson Labs | Cat. # 016962 |
| Mouse: Vglut1-IRES-Cre [Slc17a7tim1.1(cre)Hze | Jackson Labs | Cat. # 23527 |
| Mouse: Vglut2-IRES-Cre [Slc17a6tm2(Cre)Lowl] | Jackson Labs | Cat. # 16963 |
| Mouse: C57BL/6J | Jackson Labs | Cat. # 000664 |
| Rat: Sprague Dawley Crl:CD(SD) | Charles River | Cat. # 001 |
| Recombinant DNA | ||
| pcDNA3:Parapinopsin (PPO) | Koyanagi et al., 2004 PNAS | n/a |
| pcDNA3:PPO-Venus | this paper | Addgene: 139502 |
| pcDNA3:PPO-mScarlet | this paper | Addgene: 139503 |
| pAAV:CAG:DIO:PPO-Venus | this paper | Addgene:139504 |
| pAAV:Ef1a:DIO:PPO-Venus | this paper | Addgene:139505 |
| pcDNA3:mScarlet-gamma9 | this paper | Addgene:140097 |
| pFastBAC:PPO-FLAG-10xHis | this paper | n/a |
| pcDNA3:GFP-gamma9 | Karunarathne et al., 2012 | n/a |
| pcDNA3:mCherry-gamma9 | Karunarathne et al., 2013 | n/a |
| pcDNA3:GaOA(C351I)-CFP | Azpiazu et al., 2006 | n/a |
| Arrestin3-GFP | gift from Ken Mackie (Indiana University) | n/a |
| Arrestin3-mCherry | gift from Mark von Zastrow (UCSF) | n/a |
| mCherry-Clathrin | Rizzo et al., 2009 | Addgene: 55019 |
| pcDNA3:MOR-GFP | gift from Charley Chavkin (University of Washington) | n/a |
| pcDNA3:GIRK1-AU5 | Wydeven et al, 2014 PNAS | n/a |
| pCAG:GIRK2A-myc | Wydeven et al, 2014 PNAS | n/a |
| Software and Algorithms | ||
| HEKA Patchmaster | HEKA Instruments | https://www.heka.com RRID: SCR_000034 |
| pCLAMP11 | Molecular Devices | https://www.moleculardevices.com RRID: SCR_011323 |
| Igor Pro | Wavemetrics | https://www.wavemetrics.com RRID: SCR_00325 |
| Neuromatic | Rothman and Silver, 2018 | http://www.neuromatic.thinkrandom.com RRID: SCR_004186 |
| Matlab | Mathworks | https://www.mathworks.com RRID: SCR 001622 |
| ImageJ with Fiji | NIH | https://imagej.net/Fiji RRID: SCR_002285 |
| Andor iQ | Andor | https://andor.oxinst.com/ RRID: SCR_014461 |
| Synapse | Tucker-Davis Technologies | https://www.tdt.com |
| EthoVision | Noldus | https://www.noldus.com RRID: SCR_014461 |
| Prism | Graphpad | https://www.graphpad.com RRID: SCR_002798 |
| Illustrator | Adobe | https://www.adobe.com |
| Offline Sorter | Plexon | https://plexon.com |
| Other | ||
| 365 nm UV LED | Thorlabs | M365L3 |
| 470 nm blue LED | Thorlabs | M470L2 |
| 590 nm amber LED | Thorlabs | M590L2 and M590L3 |
| 4-channel LED driver | Thorlabs | DC4104 |
| 465 nm blue LED | Plexon | OPT/LED_Blue_TT_FC |
| CoolLED illumination system | CoolLED | pE-4000 |
| 473 nm blue laser | Shanghai Laser and Optics | BL473 |
| 561 nm yellow-green laser | Shanghai Laser and Optics | GL561 |
| Master-9 pulse stimulator | A.M.P. Instruments | n/a |
| PulsePal v2 | Open Ephys | n/a |
| Compresstome | Precisionary Instruments | VF210-0Z |
| HEKA EPC10 | HEKA Instruments | n/a |
| Multiclamp 700B | Molecular Devices | n/a |
| Dragonfly 500 spinning disk confocal | Andor | n/a |
| Revolution spinning disk confocal | Andor | n/a |
| TCS SPE confocal microscope | Leica | n/a |
| FV1000 confocal microscope | Olympus | n/a |
| FVMPE-RS multiphoton system | Olympus | n/a |
| Fiber Photometry System | Tucker-Davis Technologies | LUX RZ10X |
| OmniPlex and PlexControl for single-unit recordings | Plexon | RRID:SCR_014803 |
Highlights.
Parapinopsin (PPO) is a photoswitchable Gi-coupled opsin activated by blue light
At cell bodies, PPO inhibits neuronal activity to suppress reward-seeking behaviors
PPO reversibly inhibits neurotransmitter release at axon terminals
Photoinhibition of projections in vivo rapidly and reversibly alters mouse behavior
Acknowledgements:
This work was supported by a NIH BRAIN Initiative grant R01 MH111520 (MRB and RKS), NIH R21 DA049569 (BAC and PRO), NIH K01 DA042219 (PRO), NIH K01 DK115634 (VKS), T32DA007278 (KSG), P30DA048736 and NIH R35 GM122577 (NG), the Mary Gates Research Fellowship (AE), and Scan Design Research Foundation (SS). We would like to especially thank Akihisa Terakita and Mitsumasa Koyanagi for initial advice in working with parapinopsin, Yu-Qing Cao for advice in recording Ca2+ channel currents, and Ben Suter for suggestions in building LED systems. We also thank Mathias Mahn, Simon Wiegert, and Ofer Yizhar for helpful comments and discussion on the manuscript, and Fletcher Austin and Paul Gaetano for insightful discussions. This work was also supported by the Hope Center Viral Vectors Core at Washington University School of Medicine.
Footnotes
Declaration of interests: R.W.G and M.R.B. are co-founders of Neurolux Inc. a neuroscience technology company, but no related technology was used in this study. All other authors declare no competing interests.
Inclusion and diversity: We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One of more of the authors of this paper received support from a program designed to increase minority representation in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agmon A, and Connors BW (1991). Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41, 365–379. [DOI] [PubMed] [Google Scholar]
- Airan RD, Thompson KR, Fenno LE, Bernstein H, and Deisseroth K (2009). Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029. [DOI] [PubMed] [Google Scholar]
- Al-Hasani R, McCall JG, Shin G, Gomez AM, Schmitz GP, Bernardi JM, Pyo C-O, Park S Il, Marcinkiewcz CM, Crowley NA, et al. (2015). Distinct subpopulations of nucleus accumbens dynorphin neurons drive aversion and reward. Neuron 87, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, and Sternson SM (2018). Chemogenetic tools for causal cellular and neuronal biology. Physiol. Rev 98, 391–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azpiazu I, Akgoz M, Kalyanaraman V, and Gautam N (2006). G protein βγ11 complex translocation is induced by Gi, Gq and Gs coupling receptors and is regulated by the α subunit type. Cell. Signal 18, 1190–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckman CM, Malik N, Zhang Y, Shan L, Grinberg A, Hoffer BJ, Westphal H, and Tomac AC (2006). Characterization of a mouse strain expressing Cre recombinase from the 3’ untranslated region of the dopamine transporter locus. Genesis 44, 383–390. [DOI] [PubMed] [Google Scholar]
- Bagnall MW, Hull C, Bushong EA, Ellisman MH, and Scanziani M (2011). Multiple Clusters of Release Sites Formed by Individual Thalamic Afferents onto Cortical Interneurons Ensure Reliable Transmission. Neuron 71, 180–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (1989). Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340, 153–156. [DOI] [PubMed] [Google Scholar]
- Blackmer T, Larsen EC, Takahashi M, Martin TF, Alford S, and Hamm HE (2001). G protein betagamma subunit-mediated presynaptic inhibition: regulation of exocytotic fusion downstream of Ca2+ entry. Science (80-. ). 292, 293–297. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, and Snutch TP (1996). Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc. Natl. Acad. Sci. U. S. A 93, 1486–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, and Deisseroth K (2005). Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci 8, 1263–1268. [DOI] [PubMed] [Google Scholar]
- Browning KN, Kalyuzhny AE, and Travagli RA (2002). Opioid peptides inhibit excitatory but not inhibitory synaptic transmission in the rat dorsal motor nucleus of the vagus. J. Neurosci 22, 2998–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke KJ, Keeshen CM, and Bender KJ (2018). Two forms of synaptic depression produced by differential neuromodulation of presynaptic calcium channels. Neuron 99, 969–984.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett CJ, and Krashes MJ (2016). Resolving behavioral output via chemogenetic designer receptors exclusively activated by designer drugs. J. Neurosci 36, 9268–9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chefer VI, Czyzyk T, Bolan EA, Moron J, Pintar JE, and Shippenberg TS (2005). Endogenous κ -Opioid Receptor Systems Regulate Mesoaccumbal Dopamine Dynamics and Vulnerability to Cocaine. 25, 5029–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copits BA, Pullen MY, and Gereau RW (2016). Spotlight on pain: optogenetic approaches for interrogating somatosensory circuits. Pain 157, 2424–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre J, van Zessen R, Loureiro M, Patriarchi T, Tian L, Pascoli V, and Lüscher C (2018). Dopamine neurons projecting to medial shell of the nucleus accumbens drive heroin reinforcement. Elife 7, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crandall SR, Patrick SL, Cruikshank SJ, and Connors BW (2017). Infrabarrels are layer 6 circuit modules in the barrel cortex that link long-range inputs and outputs. Cell Rep. 21, 3065–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Ntamati NR, Chandra R, Lobo MK, and Lüscher C (2016). Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron 92, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Lewis TJ, and Connors BW (2007). Synaptic basis for intense thalamocortical activation of feedforward inhibitory cells in neocortex. Nat. Neurosci 10, 462–468. [DOI] [PubMed] [Google Scholar]
- Currie KPM (2010). G protein inhibition of CaV2 calcium channels. Channels 4. [Google Scholar]
- Davies WL, Hankins MW, and Foster RG (2010). Vertebrate ancient opsin and melanopsin: divergent irradiance detectors. Photochem. Photobiol. Sci 9, 1444–1457. [DOI] [PubMed] [Google Scholar]
- Dodt E, and Meissl H (1982). The pineal and parietal organs of lower vertebrates. Experientia 38, 996–1000. [DOI] [PubMed] [Google Scholar]
- Dolphin AC, and Scott RH (1987). Calcium channel currents and their inhibition by (−)-baclofen in rat sensory neurones: modulation by guanine nucleotides. J. Physiol 386, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Duc NM, Rasmussen SGF, Hilger D, Kubiak X, Wang L, Bohon J, Kim HR, Wegrecki M, Asuru A, et al. (2019). Assembly of a GPCR-G protein complex. Cell 177, 1232–1242.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, and Fischbach GD (1981). Neurotransmitters decrease the calcium conductance activated by depolarization of embryonic chick sensory neurones. J. Physiol 317, 519–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, and von Zastrow M (2018). Subcellular organization of GPCR signaling. Trends Pharmacol. Sci 39, 200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, and Von Zastrow M (2016). β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat. Cell Biol 18, 303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichel K, Jullié D, Barsi-Rhyne B, Latorraca NR, Masureel M, Sibarita JB, Dror RO, and Von Zastrow M (2018). Catalytic activation of β-Arrestin by GPCRs. Nature 557, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickelbeck D, Rudack T, Tennigkeit SA, Surdin T, Karapinar R, Schwitalla JC, Mücher B, Shulman M, Scherlo M, Althoff P, et al. (2019). Lamprey parapinopsin (“UVLamP”): a bistable UV-sensitive optogenetic switch for ultrafast control of GPCR pathways. ChemBioChem 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst OP, Lodowski DT, Elstner M, Hegemann P, and Brown LS (2014). Microbial and animal rhodopsins. [DOI] [PMC free article] [PubMed]
- Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, Lammel S, Mirzabekov JJ, Airan RD, Zalocusky KA, et al. (2014). Natural neural projection dynamics underlying social behavior. Cell 157, 1535–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JA, Hirokawa KE, Sorensen SA, Gu H, Mills M, Ng LL, Bohn P, Mortrud M, Ouellette B, Kidney J, et al. (2014). Anatomical characterization of Cre driver mice for neural circuit mapping and manipulation. Front. Neural Circuits 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinke B, Gingl E, and Sandkühler J (2011). Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J. Neurosci 31, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, and Catterall WA (1996). Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature 380, 258–262. [DOI] [PubMed] [Google Scholar]
- Hilger D, Masureel M, and Kobilka BK (2018). Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol 25, 4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodos W (1961). Progressive ratio as a measure of reward strength. Science 134, 943–944. [DOI] [PubMed] [Google Scholar]
- Hunt TW, Carroll RC, and Peralta EG (1994). Heterotrimeric G proteins containing G alpha i3 regulate multiple effector enzymes in the same cell. Activation of phospholipases C and A2 and inhibition of adenylyl cyclase. J. Biol. Chem 269, 29565–29570. [PubMed] [Google Scholar]
- Ikeda SR (1996). Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 380, 255–258. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Rizzi G, Stamatakis AM, Ung RL, and Stuber GD (2013). The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science (80-. ) 341, 1517–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, and Lovinger DM (2016). Presynaptic G protein-coupled receptors: Gatekeepers of addiction? Front. Cell. Neurosci 10, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunarathne WKA, O’Neill PR, Martinez-Espinosa PL, Kalyanaraman V, and Gautam N (2012). All G protein βγ complexes are capable of translocation on receptor activation. Biochem. Biophys. Res. Commun 421, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunarathne WKA, Giri L, Kalyanaraman V, and Gautam N (2013). Optically triggering spatiotemporally confined GPCR activity in a cell and programming neurite initiation and extension. Proc. Natl. Acad. Sci. U. S. A 110, 1565–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada T, and Ui M (1982). Direct modification of the membrane adenylate cyclase system by islet-activating protein due to ADP-ribosylation of a membrane protein (rat C6 glioma cel/3-adrenergic receptor/NAD/guanine nucleotide regulatory protein). Biochemistry 79, 3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato HE, Zhang Y, Hu H, Suomivuori CM, Kadji FMN, Aoki J, Krishna Kumar K, Fonseca R, Hilger D, Huang W, et al. (2019). Conformational transitions of a neurotensin receptor 1–Gi1 complex. Nature 572, 80–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano-Yamashita E, Koyanagi M, Shichida Y, Oishi T, Tamotsu S, and Terakita A (2011). Beta-arrestin functionally regulates the non-bleaching pigment parapinopsin in lamprey pineal. PLoS One 6, 4–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano-Yamashita E, Koyanagi M, Wada S, Tsukamoto H, Nagata T, and Terakita A (2015). Activation of transducin by bistable pigment parapinopsin in the pineal organ of lower vertebrates. PLoS One 10, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CK, Adhikari A, and Deisseroth K (2017). Integration of optogenetics with complementary methodologies in systems neuroscience. Nat. Rev. Neurosci 18, 222–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, McCall JG, Jung YH, Huang X, Siuda ER, Li Y, Song J, Song YM, Pao HA, Kim R-H, et al. (2013). Injectable, cellular-scale optoelectronics with applications for wireless optogenetics. Science 340, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinlogel S (2016). Optogenetic user’s guide to Opto-GPCRs. Front. Biosci. (Landmark Ed 21, 794–805. [DOI] [PubMed] [Google Scholar]
- Kouyama T, and Murakami M (2010). Structural divergence and functional versatility of the rhodopsin superfamily. Photochem. Photobiol. Sci 9, 1458–1465. [DOI] [PubMed] [Google Scholar]
- Koyanagi M, and Terakita A (2014). Diversity of animal opsin-based pigments and their optogenetic potential. Biochim. Biophys. Acta - Bioenerg 1837, 710–716. [DOI] [PubMed] [Google Scholar]
- Koyanagi M, Kawano E, Kinugawa Y, Oishi T, Shichida Y, Tamotsu S, and Terakita A (2004). Bistable UV pigment in the lamprey pineal. Proc. Natl. Acad. Sci 101, 6687–6691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M, Kawano-Yamashita E, Wada S, and Terakita A (2017). Vertebrate bistable pigment parapinopsin: Implications for emergence of visual signaling and neofunctionalization of non-visual pigment. Front. Ecol. Evol 5, 1–7. [Google Scholar]
- Kreitzer AC, and Malenka RC (2007). Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature 445, 643–647. [DOI] [PubMed] [Google Scholar]
- St. Laurent R, Martinez Damonte V, Tsuda AC, and Kauer JA (2020). Periaqueductal Gray and Rostromedial Tegmental Inhibitory Afferents to VTA Have Distinct Synaptic Plasticity and Opiate Sensitivity. Neuron 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Rial D, Canas PM, Yoo J, Li W, Zhou X, Wang Y, van Westen GJP, Payen M, Augusto E, et al. (2015). Optogenetic activation of intracellular adenosine A2A receptor signaling in the hippocampus is sufficient to trigger CREB phosphorylation and impair memory. Mol. Psychiatry 1–11. [DOI] [PubMed] [Google Scholar]
- Lin JY, Knutsen PM, Muller A, Kleinfeld D, and Tsien RY (2013). ReaChR: a red-shifted variant of channelrhodopsin enables deep transcranial optogenetic excitation. Nat. Neurosci 16, 1499–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sinnen BL, Boxer EE, Schneider MW, Grybko MJ, Buchta WC, Gibson ES, Wysoczynski CL, Ford CP, Gottschalk A, et al. (2019). A Photoactivatable Botulinum Neurotoxin for Inducible Control of Neurotransmission. Neuron 101, 863–875.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobingier BT, and von Zastrow M (2019). When trafficking and signaling mix: How subcellular location shapes G protein-coupled receptor activation of heterotrimeric G proteins. Traffic 20, 130–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, and Slesinger P a (2010). Emerging roles for G protein-gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci 11, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn M, Prigge M, Ron S, Levy R, and Yizhar O (2016). Biophysical constraints of optogenetic inhibition at presynaptic terminals. Nat. Neurosci 19, 554–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn M, Gibor L, Patil P, Cohen-Kashi Malina K, Oring S, Printz Y, Levy R, Lampl I, and Yizhar O (2018). High-efficiency optogenetic silencing with soma-targeted anion-conducting channelrhodopsins. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn M, Saraf-Sinik I, Patil P, Pulin M, Bruentgens F, Bitton E, Palgi S, Gat A, Dine J, Wietek J, et al. (2021). Optogenetic silencing of neurotransmitter release with a naturally occurring invertebrate rhodopsin. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcott PF, Gong S, Donthamsetti P, Grinnell SG, Nelson MN, Newman AH, Birnbaumer L, Martemyanov KA, Javitch JA, and Ford CP (2018). Regional Heterogeneity of D2-Receptor Signaling in the Dorsal Striatum and Nucleus Accumbens. Neuron 98, 575–587.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall J, Carleton KL, and Cronin T (2015). Colour vision in marine organisms. Curr. Opin. Neurobiol 34, 86–94. [DOI] [PubMed] [Google Scholar]
- Matsui a., and Williams JT (2011). Opioid-aensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J. Neurosci 31, 17729–17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall JG, Al-Hasani R, Siuda ER, Hong DY, Norris AJ, Ford CP, and Bruchas MR (2015). CRH engagement of the locus coeruleus noradrenergic system mediates stress-induced anxiety. Neuron 87, 605–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales M, and Margolis EB (2017). Ventral tegmental area: Cellular heterogeneity, connectivity and behaviour. Nat. Rev. Neurosci 18, 73–85. [DOI] [PubMed] [Google Scholar]
- Morita Y, Tabata M, Uchida K, and Samejima M (1992). Pineal-dependent locomotor activity of lamprey, Lampetra japonica, measured in relation to LD cycle and circadian rhythmicity. J. Comp. Physiol. A 171, 555–562. [Google Scholar]
- Nagai T, Sawano A, Park ES, and Miyawaki A (2001). Circularly permuted green fluorescent proteins engineered to sense Ca 2 + 98, 3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai J, Ohkura M, and Imoto K (2001). A high signal-to-noise Ca 2 + probe composed of a single green fluorescent protein. 3, 137–141. [DOI] [PubMed] [Google Scholar]
- Nanou E, and Catterall WA (2018). Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 98, 466–481. [DOI] [PubMed] [Google Scholar]
- Nicola SM, and Malenka RC (1997). Dopamine Depresses Excitatory and Inhibitory Synaptic Transmission by Distinct Mechanisms in the Nucleus Accumbens. 17, 5697–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, and Hoffmann C (2016). B-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature 531, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill PR, Karunarathne WKA, Kalyanaraman V, Silvius JR, and Gautama N (2012). G-protein signaling leverages subunit-dependent membrane affinity to differentially control βγ translocation to intracellular membranes. Proc. Natl. Acad. Sci. U. S. A 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill PR, Castillo-Badillo JA, Meshik X, Kalyanaraman V, Melgarejo K, and Gautam N (2018). Membrane Flow Drives an Adhesion-Independent Amoeboid Cell Migration Mode. Dev. Cell 46, 9–22.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Maejima T, Liu C, Deneris E, and Herlitze S (2010). Substitution of 5-HT1A receptor signaling by a light-activated G protein-coupled receptor. J. Biol. Chem 285, 30825–30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen SF, Liu MH, and Kreitzer AC (2019). Thermal constraints on in vivo optogenetic manipulations. Nat. Neurosci 22, 1061–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KE, Pedersen CE, Gomez AM, Spangler SM, Walicki MC, Feng SY, Stewart SL, Otis JM, Al-Hasani R, McCall JG, et al. (2019). A Paranigral VTA Nociceptin Circuit that Constrains Motivation for Reward. Cell 178, 653–-671.e19.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, Folk RW, Broussard GJ, Liang R, Jang MJ, et al. (2018). Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science (80-. ) 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Mohebi A, Sun J, Marley A, Liang R, Dong C, Puhger K, Mizuno GO, Davis CM, Wiltgen B, et al. (2020). An expanded palette of dopamine sensors for multiplex imaging in vivo. Nat. Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo JV, Kay L, Ellender TJ, and Akerman CJ (2012). Optogenetic silencing strategies differ in their effects on inhibitory synaptic transmission. Nat. Neurosci 15, 1102–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SJ, Lafferty CK, Mendoza JA, Yang AK, Davidson TJ, Grosenick L, Deisseroth K, and Britt JP (2018). Coordinated Reductions in Excitatory Input to the Nucleus Accumbens Underlie Food Consumption. Neuron 99, 1260–1273.e4. [DOI] [PubMed] [Google Scholar]
- Richardson NR, and Roberts DC (1996). Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J. Neurosci. Methods 66, 1–11. [DOI] [PubMed] [Google Scholar]
- Rodgers J, Bano-Otalora B, Belle MDC, Paul S, Hughes R, Wright P, McDowell R, Milosavljevic N, Orlowska-Feuer P, Martial FP, et al. (2021). Using a bistable animal opsin for switchable and scalable optogenetic inhibition of neurons. EMBO Rep. e51866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost BR, Schneider-Warme F, Schmitz D, and Hegemann P (2017). Optogenetic Tools for Subcellular Applications in Neuroscience. Neuron 96, 572–603. [DOI] [PubMed] [Google Scholar]
- Roth BL (2016). DREADDs for Neuroscientists. Neuron 89, 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman JS, and Silver RA (2018). NeuroMatic: An Integrated Open-Source Software Toolkit for Acquisition, Analysis and Simulation of Electrophysiological Data. Front. Neuroinform 12, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, and Tian L (2020). Imaging Neurotransmitter and Neuromodulator Dynamics In Vivo with Genetically Encoded Indicators. Neuron 108, 17–32. [DOI] [PubMed] [Google Scholar]
- Scott RH, and Dolphin AC (1987). Activation of a G protein promotes agonist responses to calcium channel ligands. Nature 330, 760–762. [DOI] [PubMed] [Google Scholar]
- da Silva S, Hasegawa H, Scott A, Zhou X, Wagner AK, Han B-X, and Wang F (2011). Proper formation of whisker barrelettes requires periphery-derived Smad4-dependent TGF-beta signaling. Proc. Natl. Acad. Sci. U. S. A 108, 3395–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda ER, Copits BA, Schmidt MJ, Baird MA, Al-Hasani R, Planer WJ, Funderburk SC, McCall JG, Gereau IV RW, and Bruchas MR (2015a). Spatiotemporal control of opioid signaling and behavior. Neuron 86, 923–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda ER, McCall JG, Al-Hasani R, Shin G, Il Park S, Schmidt MJ, Anderson SL, Planer WJ, Rogers JA, and Bruchas MR (2015b). Optodynamic simulation of β-adrenergic receptor signalling. Nat. Commun 6, 8480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangler SM, and Bruchas MR (2017). Optogenetic approaches for dissecting neuromodulation and GPCR signaling in neural circuits. Curr. Opin. Pharmacol 32, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachniak TJ, Ghosh A, and Sternson SM (2014). Chemogenetic Synaptic Silencing of Neural Circuits Localizes a Hypothalamus→Midbrain Pathway for Feeding Behavior. Neuron 82, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuber GD, Sparta DR, Stamatakis AM, van Leeuwen WA, Hardjoprajitno JE, Cho S, Tye KM, Kempadoo KA, Zhang F, Deisseroth K, et al. (2011). Excitatory transmission from the amygdala to nucleus accumbens facilitates reward seeking. Nature 475, 377–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stujenske JM, Spellman T, and Gordon JA (2015). Modeling the Spatiotemporal Dynamics of Light and Heat Propagation for InVivo Optogenetics. Cell Rep. 12, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zeng J, Jing M, Zhou J, Feng J, Owen SF, Luo Y, Li F, Wang H, Yamaguchi T, et al. (2018). A Genetically Encoded Fluorescent Sensor Enables Rapid and Specific Detection of Dopamine in Flies, Fish, and Mice. Cell 174, 481–496.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, and Tsujimoto T (1998). G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABA(B) receptor. J. Neurosci 18, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejeda HA, Wu J, Kornspun AR, Pignatelli M, Kashtelyan V, Krashes MJ, Lowell BB, Carlezon WA, and Bonci A (2017). Pathway- and Cell-Specific Kappa-Opioid Receptor Modulation of Excitation-Inhibition Balance Differentially Gates D1 and D2 Accumbens Neuron Activity. Neuron 93, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terakita A, Nagata T, Sugihara T, and Koyanagi M (2015). Optogenetic Potentials of Diverse Animal Opsins. In Optogenetics: Light-Sensing Proteins and Their Applications, Yawo H, Kandori H, and Koizumi A, eds. (Tokyo: Springer Japan; ), pp. 77–88. [Google Scholar]
- Thompson AC, Zapata A, Jr JBJ, Vaughan RA, Sharpe LG, and Shippenberg TS (2000). κ -Opioid Receptor Activation Modifies Dopamine Uptake in the Nucleus Accumbens and Opposes the Effects of Cocaine. 20, 9333–9340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting JT, Daigle TL, Chen Q, and Feng G (2014). Patch-Clamp Methods and Protocols (New York, NY: Springer New York; ). [Google Scholar]
- Tsai H-C, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, and Deisseroth K (2009). Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science (80-. ) 324, 1080–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, Kim SY, Adhikari A, Thompson KR, Andalman AS, et al. (2013). Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature 493, 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K, and Morita Y (1994). Spectral sensitivity and mechanism of interaction between inhibitory and excitatory responses of photosensory pineal neurons. Pflügers Arch. Eur. J. Physiol 427, 373–377. [DOI] [PubMed] [Google Scholar]
- Vilardaga J-P (2010). Theme and variations on kinetics of GPCR activation/deactivation. J. Recept. Signal Transduct. Res 30, 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S, and Lowell BB (2011). Leptin Action on GABAergic Neurons Prevents Obesity and Reduces Inhibitory Tone to POMC Neurons. Neuron 71, 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang H, Chu P, Jan LY, and von Zastrow M (1999). Functional Dissociation of Opioid Receptor Signaling and Endocytosis: Implications for the Biology of Opiate Tolerance and Addiction of receptors causes profound effects on drug-induced. Neuron 23, 737–746. [DOI] [PubMed] [Google Scholar]
- Wiegert JS, Mahn M, Prigge M, Printz Y, and Yizhar O (2017). Silencing Neurons: Tools, Applications, and Experimental Constraints. Neuron 95, 504–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U, Wüst E, Weyand I, and Kühn H (1986). Rapid affinity purification of retinal arrestin (48 kDa protein) via its light-dependent binding to phosphorylated rhodopsin. FEBS Lett. 207, 292–295. [DOI] [PubMed] [Google Scholar]
- Wingler LM, Elgeti M, Hilger D, Latorraca NR, Lerch MT, Staus DP, Dror RO, Kobilka BK, Hubbell WL, and Lefkowitz RJ (2019). Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell 176, 468–478.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Watson-Koken MA, Rees S, Lee M, and Milligan G (1997). Interactions of the α2A-adrenoceptor with multiple Gi-family G-proteins: Studies with pertussis toxin-resistant G-protein mutants. Biochem. J 321, 721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wyk M, Pielecka-Fortuna J, Löwel S, and Kleinlogel S (2015). Restoring the ON switch in blind retinas: opto-mGluR6, a next-generation, cell-tailored optogenetic tool. PLoS Biol. 13, e1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, and Deisseroth K (2011). Optogenetics in neural systems. Neuron 71, 9–34. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, and Currie KPM (2013). Regulation of Ca(V)2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 1828, 1629–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang L, Hasegawa H, Amin P, Han B-X, Kaneko S, He Y, and Wang F (2010). Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc. Natl. Acad. Sci. U. S. A 107, 9424–9429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, and Regehr WG (2002). Short-Term Synaptic Plasticity. Annu. Rev. Physiol 64, 355–405. [DOI] [PubMed] [Google Scholar]
- Zurawski Z, Gray ADT, Brady LJ, Page B, Church E, Harris NA, Dohn MR, Yim YY, Hyde K, Mortlock DP, et al. (2019a). Disabling the Gβγ-SNARE interaction disrupts GPCR-mediated presynaptic inhibition, leading to physiological and behavioral phenotypes. Sci. Signal 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurawski Z, Yim YY, Alford S, and Hamm HE (2019b). The expanding roles and mechanisms of G protein–mediated presynaptic inhibition. J. Biol. Chem 294, 1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. PPO is activated by both UV and blue light (related to Figure 1). Movie of γ9-mCherry translocation in HeLa cells co-expressing untagged parapinopsin (PPO). γ9-mCherry (red) was imaged with a 595 nm laser which did not cause spontaneous translocation. Widefield stimulation with blue (470 nm) and UV (365 nm) light (150 ms pulses every 5 seconds) was delivered when indicated. Note the rapid translocation of γ9-mCherry from the plasma membrane to intracellular compartments during photostimulation and recovery when the light is switched off.
Video S2. PPO can be activated at single-cell resolution (related to Figure 1). Movie of γ9-mCherry (red) translocation in HeLa cells co-expressing untagged parapinopsin (PPO). LED stimulation of individual cells in the boxed regions of interest was delivered through a digital mirror device. Note the rapid translocation in the regions of interest which did not affect translocation in adjacent cells.
Data Availability Statement
All data and analysis codes are available from the Lead Contact.