

Abstract
A lot of high quality data on the biological activity of chemical compounds are required throughout the whole drug discovery process: from development of computational models of the structure–activity relationship to experimental testing of lead compounds and their validation in clinics. Currently, a large amount of such data is available from databases, scientific publications, and patents. Biological data are characterized by incompleteness, uncertainty, and low reproducibility. Despite the existence of free and commercially available databases of biological activities of compounds, they usually lack unambiguous information about peculiarities of biological assays. On the other hand, scientific papers are the primary source of new data disclosed to the scientific community for the first time. In this study, we have developed and validated a data-mining approach for extraction of text fragments containing description of bioassays. We have used this approach to evaluate compounds and their biological activity reported in scientific publications. We have found that categorization of papers into relevant and irrelevant may be performed based on the machine-learning analysis of the abstracts. Text fragments extracted from the full texts of publications allow their further partitioning into several classes according to the peculiarities of bioassays. We demonstrate the applicability of our approach to the comparison of the endpoint values of biological activity and cytotoxicity of reference compounds.
Graphical Abstract
INTRODUCTION
Drug discovery is a multidisciplinary process involving medicinal chemistry, pharmacology, toxicology, and so forth. Experimental evaluation of the biological activity of a chemical compound is crucial for the development of new drugs. High quality pharmacological and toxicological data are needed through the whole drug discovery and development pipeline: from searching for hits with the presumed needed biological activity by the application of computational models to their final validation in clinical trials.1-3
Data about the biological activity of compounds are available from three main sources: (i) databases of bioactive compounds,4-6 (ii) scientific publications, and (iii) patents.3 Many attempts have been made to analyze the contents and comparability of certain endpoints of the biologically active compounds found in databases.1,7-10 Most databases include a lot of information about the biological activities of chemical substances measured in different bioassays. Although a “definition of reporting guidelines for bioactive entities” has been proposed by Orchard et al. in 2011,11 this has not been applied until now as a standard format for the representation of bioassay details in either databases or scientific publications. The wide variety of representations of such data in different sources significantly restricts the possibilities of comparing the features of bioassay descriptions.1,8,9 Therefore, there is a need for an efficient procedure(s) to enable one identifying the data on biological activity in scientific texts and extract useful information from these data. A comprehensive review recently published by Krallinger et al. describes the approaches to text mining and data retrieval from scientific publications, patents, and electronic resources available via the internet.3 This review is mainly focused on approaches for extraction of chemical structures from scientific texts.
Many studies are dedicated to the integration of chemical and biological data. Different approaches have been proposed to establish drug–target–disease relationships,12 identify protein–protein interactions,13,14 interpret associations between proteins and genes,15 annotate proteins16 as well as protein expression and disease mechanisms,17 perform gene ontology analysis,15,18 search for associations between drugs and diseases;19 and extract data on the melting points of chemical compounds.20
Text mining has also been used to analyze the fragments of texts (FoTs) containing bioassay description in the ChEMBL database. Several applications of such approaches to the generation of new knowledge have been described.21,22 Extraction of high-quality experimental data associated with the chemical–protein interaction is essential for the development of predictive (quantitative) structure–activity relationships [(Q)SAR] and chemogenomics models. Thus, good quality of the extracted experimental data about biological activity provides the basis for building (Q)SAR models with reasonable accuracy and predictivity,23 which is particularly important for the analysis of large chemical libraries.24,25
Moreover, the comprehensiveness of biomedical data that could be extracted from the texts is now questioned, which is confirmed by several studies.3,7,8 Although some approaches aimed at classifying bioassay protocols26,27 have been proposed, there is, to our knowledge, no study directed at the application of analysis of publications and automatic comparison of bioassay descriptions extracted from the scientific literature.
The purpose of our study is to develop and validate a data-mining workflow that allows (i) automatic selection of those scientific publications that contain a description of bioassays (“relevant publications”) and filtering out the papers without such data (“irrelevant publications”) and (ii) automatic categorization of relevant publications into particular bioassay classes. We chose HIV-1 reverse transcriptase (RT) inhibitors for this case study due to the availability of a large amount of experimental data representing different RT inhibiting bioassays.8 We present a detailed description of the workflow developed and a few examples of its application to interpreting data about the biological activity of compounds.
METHODS
Design of the Study.
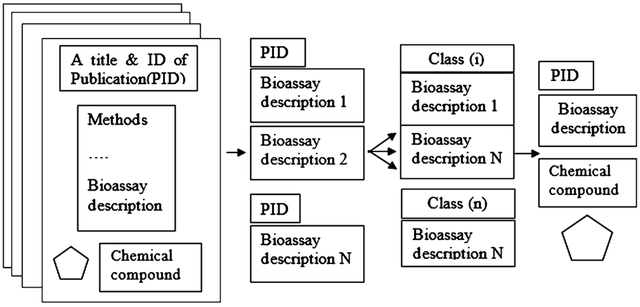
Our purpose was to categorize scientific publications into several classes according to the details of bioassays. To achieve this goal, one should extract each FoT with a description of the bioassay(s) from the full text of a publication and then categorize the fragments into several classes based on the characteristics of bioassays (Figure 1a). Such categorization can represent a link between the compounds obtained from a particular publication and the characteristics of bioassays used to measure their biological activity.
Figure 1.
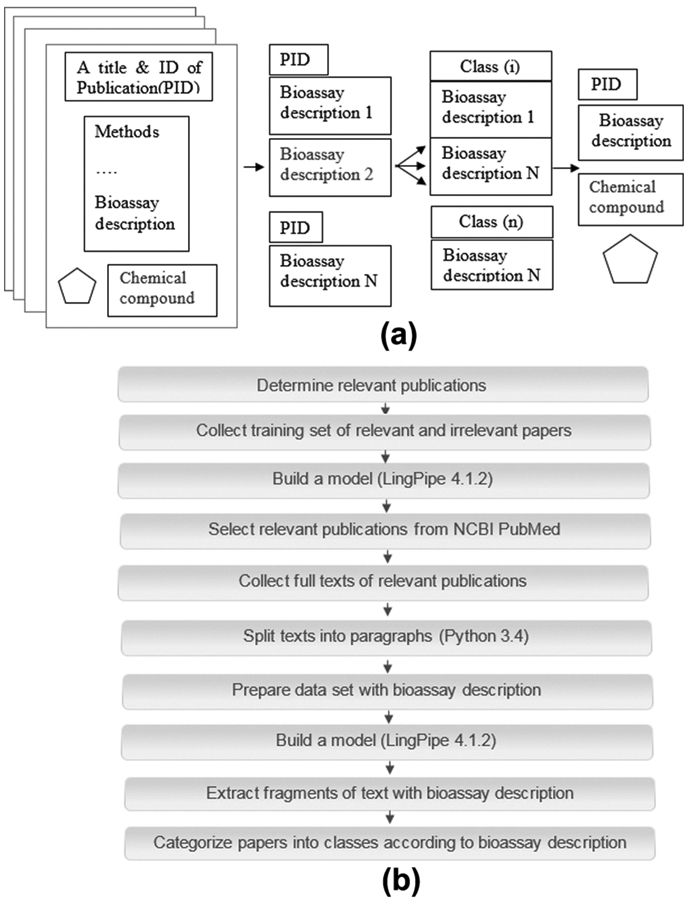
(a) Principal idea of the method; (b) general scheme of text analysis to obtain classification of bioassays.
Several steps should be taken to extract the FoT with assay descriptions reported in the publications. As a first step, one should automatically select relevant publications from bibliographic databases. As the second step, it is necessary to extract bioassay descriptions from the publications chosen. Finally, one needs to categorize the selected relevant publications into specific bioassay classes. Figure 1b represents the whole scheme of our workflow.
Data Set Collected from PubMed Related to the Inhibition of HIV-1 RT.
We have analyzed two ways to facilitate the automatic process of selection: based on (1) analysis of the abstracts retrieved from NCBI PubMed and (2)analysis of the full texts of publications.
We used the following query to find the publications in PubMed related to the experimental testing of HIV-1 RT inhibitors: “HIV-1” AND (“reverse transcriptase” OR “RT”) AND “inhibitors (date of access: February 23, 2019).” This query enabled one to retrieve over 5500 records in NCBI PubMed and comprised what we called the PubMed data set. We note that utilization of extended variants of this query, including synonyms such as “blockers,” did not lead to any substantial increase in the number of the publications selected. The query mentioned above returned both relevant and irrelevant papers associated with HIV-1 RT inhibition.
Manual Classification into Relevant and Irrelevant Publications.
We introduce several concepts related to the selection of relevant and irrelevant publications. We considered the publications relevant if they satisfied the following criteria: (1) contained the results of the study of compounds against RT and (2) had a detailed description of the biological assay. In particular, this detailed description should include information about the cell line used or peculiarities of the cell-free assay (type of primer, containing oligonucleotides) if that was used in the experiments. Additional characteristics could also include the details of the biochemical method of detecting binding, time of incubation, and any peculiarities of the DNA vectors used for delivery of viral material into the cells. This was a mandatory prerequisite to consider the publication relevant.
All publications that did not satisfy the criteria mentioned above were considered irrelevant even though they were related to HIV-1. We chose for consideration only publications directed to HIV-1 studies by application of a PubMed query related to HIV-1 inhibitors. Some of the publications may not be related to HIV occasionally, but most of them are undoubtedly related to it. The only difference between both types of the articles is that in the relevant publications, there are data on the bioassay protocols for testing chemical compounds but in irrelevant ones, there are no such data. We did not consider publications relevant, if they did not contain a detailed description of the method, for instance “using method described in [ref].” Review articles were considered irrelevant as well.
For our computational experiments, we selected only those scientific articles that contained the description of compounds active against the wild type HIV-1 since such information is the most representative. In addition, we focused on single-target (RT) inhibitors, excluding studies of dual RT/integrase or RT/protease inhibitors.
Data Sets Used for Automatic Classification.
Manual Selection of the Data Sets of Relevant and Irrelevant Publications.
In our study, we used two data sets to estimate accuracy of classification in two relevant and irrelevant papers. First, we manually collected relevant and irrelevant papers, using sources of bibliographic information available on the internet. This compiled data set (Data set 1) consisted of 64 (31 relevant and 31 irrelevant, respectively) publications in total. The PubMed identifiers (PMIDs) of the collected publication are given in Supporting Information (Table S1).
To additionally assess the quality of classification, we further selected 100 publications from the PubMed data set and applied our classifier for categorizing them into relevant and irrelevant ones. Those 100 publications containing 50 relevant and 50 irrelevant publications comprised Data set 2. The PMIDs of the collected publications are provided in Supporting Information.
Automatic Selection of Relevant and Irrelevant Publications.
We extracted from PubMed the abstract in XML format (XMLs) for each of the publications of Data set 1 and Data set 2.
We used freely available LingPipe 4.1.0 toolkit28 to build a classifier, which allows for the automatic selection of relevant publications (see Figure 1b). LingPipe uses the principles of dynamic modeling of the natural language processing. An input text is considered in LingPipe in a character-based model. LingPipe uses a set of descriptors, which represent the results of text tokenization followed by an analysis of indexes calculated based on a particular word obtained as a result of tokenization. The program provides a set of tools for text processing based on algorithms of computational linguistics. LingPipe is a Java-based software and allows for customization. LingPipe classification uses several different algorithms, which are based on modified Naïve Bayes approach, and the final selected results are those characterized by the highest accuracy of classification related to a specific algorithm. We chose LingPipe for the usage because it is based on a character-based model, so we expected the high accuracy of text classification.
The classifier was built using LingPipe by the application of its classification procedure to the training set. To assess the quality of classification, standard values of sensitivity (Sens), specificity (Spec), and balanced accuracy (BA) were calculated.
Papers selected using a classifier built on the basis of Data set 1 comprised Relevant data set.
Retrieval of Text Fragments with a Description of Biological Assays.
To retrieve text fragments with a description of biological assays, we split the full texts of publications into several FoTs and automatically selected FoTs identified as containing the description of the biological assay. To test this procedure, Relevant data set containing full texts of publications was divided into the training and test sets in a ratio of about 1:1 comprising Relevant training set and Relevant test set.
The texts of the publications of the training set were manually divided into parts that contained and did not contain a bioassay description. The parts of the texts of the training set and the full texts of the test sets were automatically split into FoTs using Python script where each FoT was represented by one paragraph because this procedure enables one to divide the texts into fragments quickly and efficiently.3 We excluded from consideration automatically generated paragraphs that contained only one short phrase (less than seven words).
We then used LingPipe package to build the classifier based on the FoTs from the training set and used this classifier to categorize the FoTs of the test set. Then, we used the model developed to classify the FoTs obtained from two categories: (i) with and (ii) without a description of the bioassay.
Categorizing the Bioassays.
The FoTs manually selected from the publications of the training set were divided into several classes according to the characteristics of each bioassay. A first-level rough classification included only two groups of bioassays: (1) cell based and (2) enzyme (RT) based. A more detailed classification included the information about HIV strains, biological material, and/or the method of detection of antiretroviral activity. Each class of fragments in the (1) cell-based and (2) enzyme-based groups was subdivided into groups resulting in five different subclasses: (I) cell-based/MT4 (cell line)/HIV-IIIB (HIV-1 strain)/colorimetric assay based on MTT reduction; (II) cell-based/various HIV strains/other cell types (HEK 293 cells, CD4+ cells, C8166 cells, etc.)/other detection method (including fluorescence, p24 antigen detection/colorimetric assay based on XTT, MAGI assay, and syncytium formation assay); (III) RT-based/polymerase activity measurement/poly(rA)-oligo(dT)/dUTP/dTTP(template-primers)//fluorescence/radioactivity; (IV) RT-based/polymerase activity measurement/poly(rC)-oligo(dG)/dGTP(template-primers)/fluorescence/radioactivity; (V) RT-based/other primer-template pairs (including mixed primers and commercial kits)/other detection method.
Some important details related to our method of FoTs creation are given below. During our workflow each FoT is assigned to the PMID. Two distinct FoTs are classified as belonging to two different assay classes, and then one PMID will be linked with two different assay classes too. When we prepared the training sets of fragments containing bioassay description, we selected all available fragments with bioassays from the text of the manuscript. Some publications include more than one bioassay. Then, we classified them using our workflow. Since we generated FoTs for the publications of the test set, one publication is associated with many FoTs, so if one of FoTs was classified as belonging to method A and another one as belonging to method B, then one publication may be linked to several different bioassays.
The FoTs divided according to these classes of bioassays were used as the training set. LingPipe was applied to categorize a particular FoT with the bioassay description as belonging to a certain class.
As a result, we aimed at linking the publication of the test set with a particular class of bioassay. Such categorization should make it possible to compare the biological activity of the compounds reported in these publications, taking into account the characteristics of the bioassays used to evaluate the biological activity.
Analysis of Variability of the Endpoint Values Reported in Publications.
Many studies are dedicated to the analysis of reproducibility and certainty of data about biological activity, retrieved from the databases of chemical compounds.7,9,10 One of the sources of uncertainty is the errors of annotation when data about a compound are loaded into the database. Since publications are often the primary source of information about the biological activity of chemical compounds, we evaluated the consistency of the biological data coming from scientific articles. To investigate the consistency of the data about the biological activity of the same compound, we selected publications with “the most similar experimental conditions” based on two distinct criteria: (i) the set of research papers should share at least one author’s name and (ii) research papers should belong to the same class of bioassays according to our automatic classification. To perform this analysis by criteria (i), we calculated the overlap between the coauthors’ names in various published articles using the XMLs downloaded. The number of coauthors shared by relevant publications and the resulting set of publications obtained are given in Supporting Information.
An examination of the variation of endpoint values can be performed by comparing the average (Av) and standard deviation (SD). We analyzed Av and SD of endpoint values (pIC50, pEC50, and pCC50) of compounds from research papers with “the most similar experimental conditions” if at least 10 measurements of endpoint values were available.
RESULTS AND DISCUSSION
Categorizing the Publications into Relevant and Irrelevant.
The accuracy of classification was estimated as follows: (i) by using fivefold cross-validation applied for Data set 1 and (2) by using Data set 1 as a training set and Data set 2 as a test set.
The values of accuracy obtained in these computational experiments are reported in Figure 2.
Figure 2.
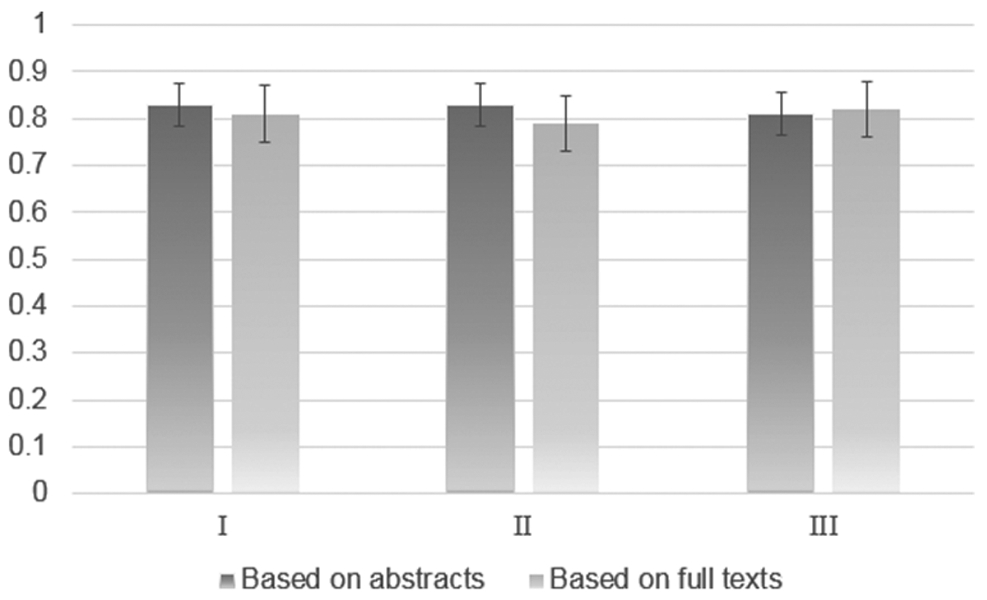
The BA of automatic categorizing publications into relevant and irrelevant for the following datasets: (I) Data set 1, fivefold cross validation; (II) data set 1 (training) and Data set 2 (test); (III) random sampling with replacement (Data set 1 merged with Data set 2). The details about computational experiments are given in the text.
The results were: Sens: 0.79; Spec: 0.87; BA: 0.83 for full texts of publications and Sens: 0.77; Spec: 0.87; BA: 0.82 for the abstracts (see Figure 2 and Table S2 of the Supporting Information). The parameters of classification for Data set 2 as a test set were Sens: 0.66; Spec: 0.92; BA: 0.79 for full texts and Sens: 0.82; Spec: 0.84; BA: 0.83 for abstracts.
Unlike for fivefold cross-validation procedure, the accuracy of classification for the model based on full texts of publications was lower compared to those obtained for abstracts-based model. To find an answer whether classification based on full texts is preferable or not, we also applied random sampling with a replacement procedure.29 We repeated the procedure of random sampling with replacement for Data set 1 merged with Data set 2 1000 times.
By applying such a validation procedure, we expected to estimate differences between abstracts-based models and models based on full texts. We think this is particularly important because in most cases relevant and irrelevant publications of the training set have very similar content except for the parts with bioassay description.
Based on the results of computational experiments using fivefold cross validation, external test set, and random sampling with replacement, accuracy of classification was reasonable in both cases of abstracts and full texts (Figure 2). In general, although the abstracts did not contain any details of a bioassay, one may nevertheless use them for a broad classification of publications into relevant and irrelevant categories. Suitability of abstracts to the extraction of useful data from their texts has been discussed earlier.14,17 In our study, we demonstrated that automatic analysis of abstract contents enables one to categorize the full texts of publications into relevant and irrelevant groups, where the papers analyzed are categorized as relevant if they are related to the reporting of a compound’s biological activity confirmed in the experiment.
Automatic Collection of Relevant Publications.
Then we used the created classifier to automatically select relevant publications from the bibliographic databases. As a result, we collected 420 abstracts from the PubMed data set. A list of the PMIDs of selected publications based on both full-text classifier and abstracts classifier is available in Supporting Information.
Comparison of the Developed Classifier with Baseline Models.
We compared the simple search for the key terms of bioassays in the full texts of papers with our classifier built for distinguishing relevant publications from irrelevant ones. For relevant publications, we defined the following key terms: “HIV, RT,” “inhibitors,” “bioassay,” and “experiment.” For irrelevant publications they were “HIV,” “RT,” and “inhibitors.” The parameters of classification accuracy using a simple search for key terms throughout Data set 2 were as follows: Sens: 0.62, Spec: 0.70, BA: 0.66. These values are lower compared to our method of classification (for our models based on abstracts: Sens: 0.82; Spec: 0.84; BA: 0.83). Therefore, our models provide better accuracy of classification of scientific papers into relevant and irrelevant ones.
Furthermore, we evaluated the possibility of distinguishing relevant publications from irrelevant ones based on the keywords and Medical Subject Heading (MeSH) terms available in NCBI PubMed for each record, associated with a publication.
Keywords and MeSH terms for relevant and irrelevant publications from the abstracts of Data set 1 were extracted. The PubMed data set was also classified into relevant and irrelevant abstracts based on the model created. For a variety of relevant and irrelevant publications selected in this way, we have also extracted keywords and MeSH terms.
We calculated the occurrences of keywords and MeSH terms in both types of scientific articles chosen. The values of occurrences were calculated as the number of terms found in the set of relevant or in the set of irrelevant publications to their number. They are provided in Supporting Information (Table S3). Our second purpose was to check whether the keywords or MeSH terms included those related to the description of a bioassay. That could be done by analyzing the most common keywords and MeSH terms.
First, it is important to note that the keywords were found in less than 30% of the considered publications. The values of occurrence of the most common keywords and MeSH terms are very close to each other for the set of relevant and irrelevant publications, both (i) in the case of manual selection of relevant articles according to the criteria defined and (ii) in the case of automatically dividing them into relevant and irrelevant ones (the difference does not exceed 10% for every term). Therefore, keywords and MeSH terms cannot be used for simple categorization of publications into relevant and irrelevant ones, and our classifier has an advantage over information search using a set of user-defined words in the texts of publications. A search for relevant publications using a query based on common keywords of MeSH terms seemed to be topically significant also lacks precision. Neither MeSH terms nor keywords included the words related to the bioassay details.
As an additional baseline model, we built a random forest classifier with 1000 trees based on the most frequent words of an abstract and the most frequent MeSH terms converted to a set of binary descriptors. We considered a word or MeSH term to be frequently occurred if it occurred in over 10% of abstracts (a list of descriptors is available in Supporting Information). The results of classification were as follows: Sens 0.79, Spec 0.81, BA 0.80, and they do not exceed the results of classification obtained by using our previously developed classifier (for our models based on abstracts: Sens: 0.82; Spec: 0.84; BA: 0.83). This confirms our suggestion about difficulties of utilizing MeSH terms for categorizing publications related to the studies on inhibition of an enzyme into two distinct categories—with and without the description of a bioassay.
Bioassay Categorization.
We downloaded 294 full texts of papers from web servers of the publishers using an automatic search with Python 3.4 script; the remaining 126 texts of publications were not available.
The articles, which came in PDF (Acrobat Reader) format, were converted for further processing into plain text format using the TET PDFlib converter.30 During conversion process, we found out that 39 publications could not be converted properly, because they were damaged. We downloaded them manually and applied TET PDFlib repeatedly. Thus, in total, we obtained 287 out of 294 texts of publications because 7 of them were not successfully converted. The Relevant data set thus comprised 287 selected publications, and the estimated positive predictive value for this set is 0.90.
We divided randomly the Relevant data set into the training and test sets in a ratio of about 1:1, resulting in the training set comprising 147 full texts (Relevant training set) and a test set containing 144 texts (Relevant test set).
In total, this training set comprised 191 FoTs with bioassay description and 16 627 FoTs without any bioassay description. The test set publications were automatically divided into the sets of FoTs yielding 16 623 FoTs in total. Then, we used the classifier developed to categorize the FoTs into fragments associated and not associated with the bioassays description. This yielded an automatic selection of 248 FoTs from the test set as fragments associated with the description of bioassays. On average, 2–3 FoTs containing the description of bioassays were selected per publication. Those FoTs were used as the test set for assessing the quality of categorization according to specific characteristics of bioassays.
We categorized 131 publications of the Relevant test set according to the classes containing very specific description of the bioassays using LingPipe as described above. After automatic selection of the FoTs with the description of a bioassay, we prepared the training set with the FoTs related to a particular bioassay class and built the models. The models obtained were applied to the automatic selection of the FoTs related to the specific bioassays. Accuracy of prediction was calculated automatically using LingPipe module. The BA of classification is reported in Figure 3, and results of classification are given in Table S4 of the Supporting Information.
Figure 3.
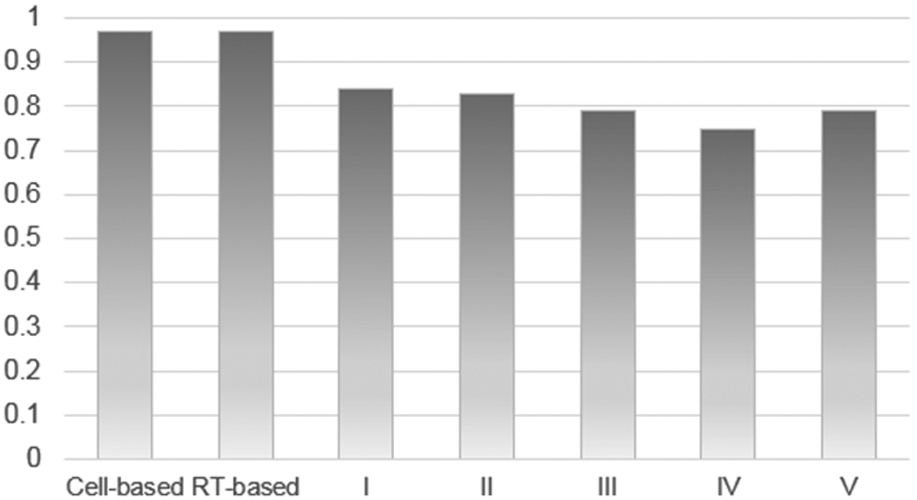
The BA of automatic categorizing FoTs into classes according to bioassay characteristics. Classes of bioassays I–V are described in the text.
The BA of classification for groups III (RT-based/poly(rA)-oligo(dT)/dUTP/dTTP/fluorescence/radioactivity), IV (RT-based/poly(rC)-oligo(dG)/dGTP/fluorescence/radioactivity); V (RT-based/rare descriptors/rare detection method) was lower than 0.80, probably due to a mixture of bioassays in the FoTs of the training set. The comparatively low sensitivity for groups III and IV can be explained by the highly imbalanced training set. In addition, as poly(rA) and oligo(dT) are very common primers used in the RT polymerase reaction, they are often mixed with other primers: this situation can lead to their co-occurrence in the FoT and to low accuracy of recognition. For the three classes with the highest accuracy of classification, I, II, and III (with BA ranging from 0.73 to 0.85), we propose that the method developed provides a basis for the selection of bioassay descriptions from publication texts according to their membership in a particular class. An example of classification of five randomly selected publication texts according to the classes of bioassay description is given in Figure 4a,b.
Figure 4.
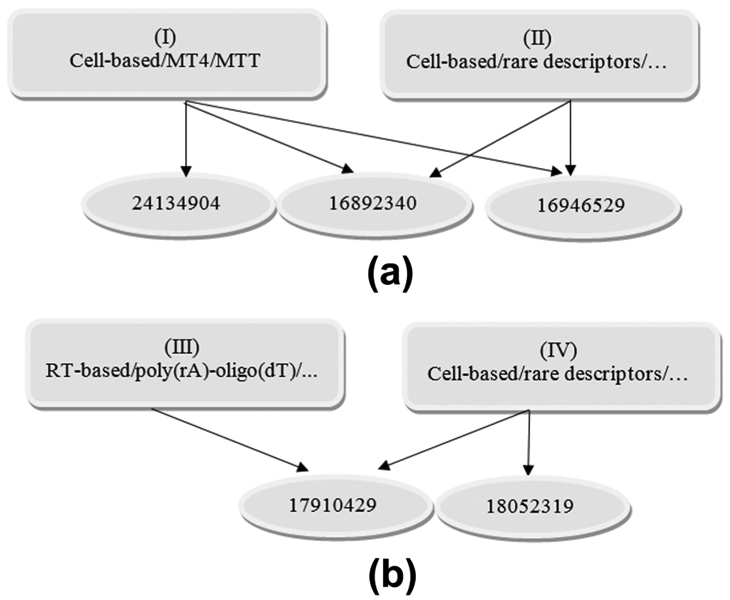
Example of classification of five randomly selected publication texts (represented by PMID) according to classes of bioassay description: for (a) cell-based methods; (b) RT enzyme-based methods.
Summarizing our results, it appears possible to classify publications according to the classes of bioassays they contained, since we successfully identified three classes of bioassays: I, II, and III.
We should add here a few statements regarding the necessity of extracting the FoTs with bioassay description. In approximately 35% of our set a publication describes at least two distinct bioassays: cell-based and RT-based. Therefore, a simple approach aiming at classifying publication without extraction of FoTs can lead to lower values of classification. To check our hypothesis, we built a classification model for categorizing publications into several classes of bioassays (RT-based, cell-based, and I–V) based on full texts of publications and their abstracts without preliminary extraction of FoTs with bioassay description. Indeed, BA of classification is lower almost for all classes of bioassays (mean BA is 0.51 ± 0.03). Results are provided in Supporting Information (Table S5).
To assess the performance of our classifier better, we compared the classification of the bioassays into distinct classes with a search for a few key terms related to the bioassays. Table 1 contains the results for the bioassays group. On the basis of the results given in Table 1, we observe that searching for a few key terms in the FoTs does not lead to the same results as achieved by our models. As one of the reasons for these suggestions, we hypothesize that a keyword-based query does not take into account the context itself, and some FoTs can be selected just by virtue of it describing the state-of-the-art approaches or citing earlier results.
Table 1.
The Comparison of Automatic Classification of the Bioassays into Categories with a Search for Key Terms in the FoTs
| category | combinations of the terms for searching |
Sens | Spec | BA |
|---|---|---|---|---|
| I | MT4 AND MTT | 0.05 | 0.89 | 0.50 |
| I | MT4 OR MTT | 0.72 | 0.5 | 0.62 |
| II | cell AND (fluorescence OR antigen) | 0.01 | 0.84 | 0.42 |
| III, IV, V | enzyme | 0.11 | 0.61 | 0.36 |
| III, IV, V | enzyme AND polymerase AND assay | 0.19 | 0.62 | 0.41 |
On the basis of the results of this comparison with the baseline models used for filtering relevant publications and classifying bioassay description, we can suggest that the workflow we developed provides better recognition of either relevant/irrelevant publications or bioassay categories.
Analysis of Variation of Endpoint Values Reported in Publications.
Several studies have pointed out the high variability of endpoint values for compounds tested in multiple assays.1,8-10,31-34 Such variability implies high inconsistency and low reproducibility of experimentally determined biological activities, probably occurring due to variability of experimental conditions. To simulate similar experimental conditions, we applied the two approaches described in Methods section: (1) research papers sharing names of coauthors and (2) belonging to the same category of bioassays.
According to common practice, any study of biological activities of compounds should also include data on the activity of reference compounds.35 We compared the EC50, CC50, and IC50 values (where available) of the reference compounds within the set of publications of the test set sharing the same authors’ names (Table 2a,b).
Table 2.
Statistics on Endpoints of Reference Compounds Coming from: (a) the Whole Set of Relevant Publications; (b) Sets of Publications That Have Common Co-Authors (i.e. Overlap in Authors’ Names)
| (a) | ||||
|---|---|---|---|---|
| the whole set of publications | ||||
| reference drug | pEC50 Av (SD)a | pIC50 Av (SD) | pCC50 Av (SD) | Nb |
| nevirapine | 0.89 (0.9) | −0.001 (0.79) | −1.53 (0.55) | 32 |
| zidovudine | 2.07 (0.80) | N/D | −1.29 (0.66) | 36 |
| efavirenz | 2.47 (0.55) | N/D | −0.81 (0.04) | 21 |
| etravirine | 2.63 (0.21) | N/D | −0.94 (0.33) | 15 |
| (b) | |||
|---|---|---|---|
| the set of publications with common co-authors | |||
| reference drugc | pEC50a Av (SD) | pCC50 Av (SD) | Nb |
| nevirapine (1) | 0.67 (0.129) | −1.17 (0.01) | 7 |
| nevirapine (1) | 0.80 (0.35) | −1.76 (0.001) | 7 |
| nevirapine (2) | 0.67 (0.129) | −1.17 (0.01) | 7 |
| zidovudine (1) | 2.2 (0.13) | −1.55 (0.46) | 9 |
Average (Av) and SD value of pEC50, pIC50 and pCC50 values.
Number of endpoint values in the set use for the calculation of Av and SD.
Number of common co-authors given in parenthesis: nevirapine (1) retrieved from publications, which shared one coauthor; nevirapine (2)—from publications sharing two co-authors.
Automatic queries in the plain text of publications were applied to identify reference compounds contained therein and their endpoint value data. We found the following known antiretroviral drugs as the reference compounds in the selected research papers that matched the queries: delavirdine, didanosine, efavirenz, etravirine, nevirapine, zidovudine. Zidovudine and nevirapine were the most frequently used reference drugs in the main set: these two drugs occurred in 36 and 32 papers, respectively (Table 2a).
Sets of Publications According to the Overlap between Authors’ Names.
Each set obtained using the identification of authors common to those publications was further used to extract the data on biological activity for the reference compounds and to determine their variations. Table 2b contains Av and SD, calculated for the endpoint values, found for more than 10 test set publications of sharing the name of coauthors (N is the number of publication comprising each subset sharing at least one coauthor).
Upon comparing the data in Table 2a,b one may conclude that, in general, the variability of endpoint values across papers with shared coauthors is lower compared to the whole set of reference compounds. Therefore, an increase of the general consistency is observed when a compound is analyzed under similar experimental conditions. However, too low SD values, even lower than expected experimental error (SD(pKi) = 1 (for details see ref 13)), might be caused by repeatedly published values, which can be found in the literature, even when references to earlier studies were included (and would have sufficed). Such cases make it difficult to analyze the real activity of compounds and should be taken into account in the process of data mining for drug discovery.
Analysis of Variability of the Endpoint Values Related to Similar Bioassays.
Each publication was assigned to the bioassay belonging to one of the groups I–V mentioned above (see Methods section).
Then, we calculated the Av and SD for the endpoint values of reference compounds collected from the published articles classified according to the description of bioassays (see Table 3).
Table 3.
Statistics on Endpoints of Reference Compounds Coming from the Sets of Publications Compiling According to the Belonging to a Certain Class of Bioassaya,b,c
| (a) | |||
|---|---|---|---|
| assay typed | pEC50 Av (SD) | pCC50 Av (SD) | |
| zidovudine | I | 2.14 (0.08) | −1.82 (0.30) |
| II | 2.03 (0.80) | −1.18 (0.67) | |
| nevirapine | I | 0.79 (0.32) | −1.96 (0.09) |
| II | 0.99 (0.38) | −2.26 (0.35) | |
| efavirenz | I | 2.68 (0.67) | −0.88 (0.08) |
| II | 2.12 (0.25) | −0.94 (0.15) | |
| etravirine | I | 2.79 (0.21) | −0.60 (0.14) |
| II | 2.50 (0.25) | −0.94 (0.18) | |
| (b) | ||
|---|---|---|
| assay typed | pIC50 Av (SD) | |
| nevirapine | III | 0.05 (0.33) |
| IV | 0.64 (0.24) | |
The values are given in the table: (a) for pECS0 and pCC50 and (b) for pIC50.
Three values are given for each endpoint: average value and SD in brackets.
N is the number of values.
Classes of bioassay classification are given in roman numbers I cell-based/MT4/MTT; (II) cell-based/rare descriptors/rare detection method; (III) RT-based/poly(rA)-oligo(dT)/dUTP/dTTP/fluorescence/radioactivity (IV) RT-based/poly(rC)-oligo(dG)/dGTP/fluorescence/radioactivity.
Tables 2 and 3 illustrate that the variability of endpoint values observed for the most abundant reference compounds is lower when they are obtained from similar bioassays (see also refs9,10). The variability of endpoint values is lower compared to the whole set of reference compounds. For particular sets of published papers, we observed very small values of SD, which might be the result of repeatedly published values of certain endpoints of reference compounds in several different publications. Such cases make it difficult to analyze the real activity of compounds and should be taken into account in the process of data mining for drug discovery.
By looking at Table 3, it is clear that the variability of pEC50 in the same class of bioassays is lower than that of pCC50 and pIC50 values but is nevertheless comparatively high even within the class. In general, the variability of pEC50 values was higher for nevirapine and zidovudine than that for efavirenz and etravirine. We assume that such a variability factor related to reference compounds can influence reproducibility of biological activity data.
Variability of Text Fragments with a Description of Methods in Publications Related to HIV-1 RT Inhibitors.
We can draw a first conclusion based on the analysis of text fragments with various methods described in them looking at the classes of publications. About 35% of publications contained more than one bioassay protocol description from different clusters and from different classes (RT enzyme based vs cell based) simultaneously. These research papers might be interesting for further analysis because they can contain bioactivity data from different bioassays and provide material for studying the concordance and reproducibility of such data. To analyze such cases and reveal the relationships between the variability of endpoint values and experimental setup, we manually checked the publications that included the description of two different bioassays.
In publication36 Huang et al. compared the results of biological testing on the several HIV strains: SF33 and 1617-1, in the cell lines: TZM-bl, PBMCs, and MT4. Unlike the CC50 values, the IC50 values of the studied compounds did not differ significantly. Also, in the publications by Boyapalle and coauthors, HIV-SF33 was tested in several bioassays with different cell types.37,38 The EC50 results were not significantly different, but the CC50 values were about 1.5–2 times higher than for PBMCs cells. These findings may be tentatively explained by the fact that cytotoxicity itself is a complex biological phenomenon of interaction of a compound with many different molecular pathways, which can be significantly different in different cell lines. Such complex systems might be more sensitive to experimental conditions. This hypothesis corresponds to the results of a recently published study where endpoint values of cytotoxicity were compared.10 The authors concluded that specific cell lines and compounds (including zidovudine) were “more sensitive to experimental setup.” We propose that these findings and concerns should be considered as a reason for careful testing of the downstream effects of an HIV inhibitor on cytotoxicity-related pathways and signaling networks.
Application of the Workflow Developed for the Classification of the Bioassays Related to Other Protein Targets.
We performed computational experiments to check whether our approach can be applied for the classification of a few protein targets besides HIV-1 RT. We selected a few targets yielding a large amount of data related to the biological activities and the bioassays based on the analysis of literature and of the ChEMBL and PubChem databases. Those targets are carbonic anhydrase II (human), cyclooxygenase-2 (human), cytochrome p450 3A4 (human), MAP kinase ERK2 (human), O-acetylserine sulfhydrylase (Mycobacterium tuberculosis), HIV-1 protease (human immunodeficiency virus), and vascular endothelial growth factor receptor 2 (human). We used NCBI PubMed queries (“Protein Name” AND inhibitor) in the same way as for HIV-1 RT.
The results of classification of the scientific papers into relevant and irrelevant based on the training set collected for each protein using the fivefold cross-validation are given in Table 4. Table 5 contains the results of the classification of the relevant publications selected automatically into categories according to the bioassays based on the random division into the training and test set (1:1). For two of the targets chosen, we could not extract enough data to compile reasonable training and test sets of publications. For the remaining five proteins, we managed to collect the data sets and build the models (Tables 4 and 5).
Table 4.
Classification of the Publications Related to the Inhibitors of a Few Selected Targets into Relevant and Irrelevant Onesa
| target | Nr | Ni | abstracts |
full texts |
||||
|---|---|---|---|---|---|---|---|---|
| Sens | Spec | BA | Sens | Spec | BA | |||
| carbonic anhydrase II | 30 | 30 | 0.97 | 0.70 | 0.81 | 0.99 | 0.63 | 0.85 |
| cyclooxygenase-2 | 30 | 30 | 0.71 | 0.77 | 0.74 | 0.76 | 0.81 | 0.78 |
| cytochrome p450 3A4 | 35 | 41 | 0.71 | 0.80 | 0.76 | 0.74 | 0.80 | 0.77 |
| HIV-1 protease | 24 | 30 | 0.95 | 0.84 | 0.90 | 0.97 | 0.85 | 0.91 |
| vascular endothelial growth factor receptor 2 | 30 | 29 | 0.95 | 0.63 | 0.79 | 0.90 | 0.65 | 0.77 |
Nr is the number of relevant publications in the training set; Ni is the number of irrelevant publications in the training set.
Table 5.
Classification of the Publications into Several Groups According to the Bioassays Related to the Selected Targets
| category | a | b | c | d | Sens | Spec | BA |
|---|---|---|---|---|---|---|---|
| target | carbonic anhydrase II (human) | ||||||
| cell-based/pH shift/colorimetric assay | 24 | 94 | 20 | 78 | 0.79 | 0.96 | 0.88 |
| cell-based/human/spectro-photometrical assay under 400 nm | 68 | 50 | 31 | 67 | 0.81 | 0.69 | 0.75 |
| cell-based/miscellaneous/spectro-photometrical assay 400 nm and above | 14 | 104 | 20 | 78 | 0.54 | 0.78 | 0.66 |
| cell-based/other methods | 12 | 106 | 27 | 71 | 0.42 | 0.95 | 0.69 |
| target | cyclooxygenase 2 (human) | ||||||
| cell-based/human/radio immuno-assay | 14 | 102 | 11 | 108 | 0.61 | 0.94 | 0.78 |
| cell-based/human/enzyme immuno-assay | 11 | 105 | 12 | 107 | 0.89 | 0.74 | 0.82 |
| cell-based/other detection methods | 89 | 27 | 94 | 25 | 0.88 | 0.63. | 0.76 |
| target | cytochrome p450 3A4 (Cyp 3A4 human) | ||||||
| Cyp 3A4-based conversion/testosterone/HPLC | 21 | 68 | 24 | 54 | 0.69 | 0.97 | 0.83 |
| Cyp 3A4-based conversion/unknown substrate/LC–MS | 51 | 38 | 34 | 44 | 0.74 | 0.65 | 0.70 |
| Cyp 3A4 activity/luminescence | 13 | 76 | 18 | 50 | 0.79 | 0.96 | 0.88 |
| Cyp 3A4 expression | 4 | 85 | 2 | 76 | N/De | N/D | N/D |
| target | vascular endothelial growth factor 2 (human) | ||||||
| fluorescence/kinase-activated fluorescent peptides/human umbilical cells | 34 | 79 | 38 | 113 | 0.65 | 0.84 | 0.75 |
| cell-based/ELISA | 11 | 102 | 10 | 141 | 0.78 | 0.84 | 0.81 |
| other methods | 21 | 92 | 24 | 127 | 0.46 | 0.68 | 0.57 |
is the number of positive samples (fragments belonging to the particular category) in the training set.
is the number of negative samples (fragments not belonging to the particular category) in the training set.
is the number of positive samples in the test set.
is the number of negative samples in the test set.
N/D: not enough data to build a model.
Based on the results listed in Tables 4 and 5, one may conclude that it is possible to use the proposed workflow for the extraction of the data about various bioassays and different targets from scientific literature if information for them is available in the scientific publications.
Challenges of Machine-Based Bioassay Classification.
For about 10% of the text fragments assigned to the same bioassay class, we noticed that two or more categories were found in a single publication. We analyzed such FoTs corresponding to the particular class of bioassay description.
The FoTs with description sometimes are quite similar, especially if they pertain to measurements of the inhibitory activity on HIV-1 replication and of the cytotoxicity to mock-infected cells, which have different endpoints: EC50 and CC50. For example, publication36 contains a description of an MTT-based test of HIV inhibitory activity in MT4 cells. The experiments on cytotoxicity are described in the same paragraph as a continuation of the efforts to study the HIV-1 inhibitory activity and were categorized as belonging to this class. Strictly speaking, however, the EC50 is a result of the MTT-test endpoint, whereas CC50 is a result of the cytotoxicity test.37 However, in both tests, the same biological materials and the same method of analyzing the growth of virus-infected cells were used. In this example, one can see that a mixture of endpoints in the same description can lead to failure of machine-based algorithms to identify the endpoint of interest.
Some publications were categorized as irrelevant because they did not contain any specific bioassay description. Typically, such research papers include references to earlier published papers, thus providing an extended description of a particular bioassay.
The HIV strain used to evaluate the biological activity was provided in the bioassay description in some papers, whereas it was not specified in others. This leads to the necessity of dividing the proposed classes into several subclasses. For instance, manual inspection of papers assigned by classification to “cell-based/MT4/MTT” revealed that we could assign them to two subclasses: cell-based/HIV-1-IIIB/MT4/MTT and cell-based/unspecified strain/MT4/MTT. Unfortunately, the number of samples in both groups is small; thus, the training/test set lacks sufficient data to build accurate and predictive models.
Low consistency between the activity values is due to poor reproducibility of biological data and might be either a property of the biological object (as in our case, HIV), or occur due to the characteristics of the bioassay.39 There are several studies where the authors compared qualitative values from databases of biologically active compounds and evaluated the consistency between endpoints for the compounds in the overlapping sets.9,10 These authors also pointed out that the inconsistency of the data between bioassays for overlapping compounds is often very high and may additionally vary depending on the biological target and individual compound. The texts of scientific publications are typically not well structured, do not contain sufficient details for their processing by machine-based algorithms and, sometimes, even manual inspection, and do not provide the reader with unambiguous information about certain endpoints and experimental details. As Fourches et al. observed earlier, the data about biological activity may be incomplete, inaccurate, incompatible, and/or irreproducible.40 Our findings are consistent with this conclusion. Today, the description of bioassays, representation of compounds, and different endpoints in publications comprise massive amount of data that lack a unified format of representation. Therefore, an exhaustive systematic analysis of such data, coming from various sources (the so-called big data in chemistry41), may be helpful to address this problem in further investigations.
More information on bioassays should be included in scientific publications so that all the pertinent specific details of every experiment lead to accurate and complete data in databases of biologically active compounds. Such data, for instance, may include: (1) endpoint; (2) extended description of experimental conditions including material, biochemical method and the process parameters such as temperature, time of exposition, pH, and any other parameter; (3) specific description of the HIV strain for every experiment. At the current level of bioassay description, full text mining of publications permits one to build only a rough classification of bioassays. Nevertheless, this may help with the clarification of reasons for low reproducibility (see the examples of reference drugs). We believe that if more details about biological assays were included in research papers, better automatic classification of bioassays might be possible hopefully with the consequence that the reasons for low reproducibility might be determined with a higher level of confidence.
Efforts to create tools that can help with annotating bioassays with better, that is, more comprehensive and standardized terminologies for assay methodology and bioactivity result set descriptions, including the use of a number of controlled vocabularies such as the BioAssay Ontology, Drug Target Ontology, and Cell Line Ontology are underway but in their early stage.42-44 Such approaches can, at least in principle, be applied to both existing contents of bioassay databases and, by original authors, to newly written manuscripts (and/or their accompanying supplementary materials) of publications that contain bioassay results.
Since new molecular and biochemical methods for analyzing biological activities emerge permanently, training sets of research articles should be subdivided by the years of publication; however, this requires a high number of papers, and thus this may be an unrealistic goal. Finally, there is no open access to the full texts of many publications, which poses a significant limitation of the method proposed here.
CONCLUSIONS
We have developed and validated an approach of bioassay classification based on the analysis of the full texts of publications. To perform classification, we identified full texts of publications containing the results of biological assays for HIV RT inhibitors. A simple text-splitting into paragraphs, where one paragraph is treated as one bioassay description, allowed us to perform a supervised classification of bioassay descriptions into several main groups. The average BA of bioassay classification into this rather simple categorization of bioassay descriptions was 0.84. A comparison of the biological activity for the reference compounds from the selected publications yielded more satisfactory results for the compounds assayed using similar methods, especially if done by the same authors. The variations of endpoint values within the same class of biological assays showed different patterns for each reference drug, which allows one to suppose that a factor associated with the compound itself can significantly influence the reproducibility of biological data. On the other hand, very small variability of bioactivity values can be a result of the repeatedly published values of certain endpoints of reference compounds in multiple papers. Therefore, new approaches should not be directed only at the chemical and biological integration using “big published data” but also aimed at the filtering out repeated data from publications. In addition, we have pinpointed some of the difficulties of the bioassay classification using text analysis and the limitations of current text structure that may contribute to the high discrepancy of biological activity values in databases of biologically active substances.
Supplementary Material
ACKNOWLEDGMENTS
This work is supported by the grant of Russian Foundation for Basic Research no. 17-54-30015 NIH_a.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jcim.9b00164.
List of PMIDs related to HIV-1 RT inhibitors selected based on the built model in the MS EXCEL file; the list of PMIDs sharing coauthors, which we used for analysis (MS EXCEL file); PMIDs with manual annotation of bioassays reported in corresponding papers; and PMIDs used for training set and detailed results of classification (ZIP)
The authors declare no competing financial interest.
REFERENCES
- (1).Kramer C; Dahl G; Tyrchan C; Ulander J A Comprehensive Company Database Analysis of Biological Assay Variability. Drug Discovery Today 2016, 21, 1213–1221. [DOI] [PubMed] [Google Scholar]
- (2).Jorgensen WL Computer-Aided Discovery of Anti-HIV Agents. Bioorg. Med. Chem 2016, 24, 4768–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Krallinger M; Rabal O; Lourenço A; Oyarzabal J; Valencia A Information Retrieval and Text Mining Technologies for Chemistry. Chem. Rev 2017, 117, 7673–7761. [DOI] [PubMed] [Google Scholar]
- (4).NCBI Resource Coordinators. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gaulton A; Hersey A; Nowotka M; Bento AP; Chambers J; Mendez D; Mutowo P; Atkinson F; Bellis LJ; Cibrián-Uhalte E; Davies M; Dedman N; Karlsson A; Magariños MP; Overington JP; Papadatos G; Smit I; Leach AR The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wishart DS; Feunang YD; Guo AC; Lo EJ; Marcu A; Grant JR; Sajed T; Johnson D; Li C; Sayeeda Z; Assempour N; Iynkkaran I; Liu Y; Maciejewski A; Gale N; Wilson A; Chin L; Cummings R; Le D; Pon A; Knox C; Wilson M DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kramer C; Lewis R QSARs, Data and Error in the Modern Age of Drug Discovery. Curr. Top. Med. Chem 2012, 12, 1896–1902. [DOI] [PubMed] [Google Scholar]
- (8).Tarasova OA; Urusova AF; Filimonov DA; Nicklaus MC; Zakharov AV; Poroikov VV QSAR Modeling Using Large-Scale Databases: Case Study for HIV-1 Reverse Transcriptase Inhibitors. J. Chem. Inf. Model 2015, 55, 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kalliokoski T; Kramer C; Vulpetti A; Gedeck P Comparability of Mixed IC50 Data—A Statistical Analysis. PLoS One 2013, 8, No. e61007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Cortés-Ciriano I; Bender A How Consistent Are Publicly Reported Cytotoxicity Data? Large-Scale Statistical Analysis of the Concordance of Public Independent Cytotoxicity Measurements. ChemMedChem 2016, 11, 57–71. [DOI] [PubMed] [Google Scholar]
- (11).Orchard S; Al-Lazikani B; Bryant S; Clark D; Calder E; Dix I; Engkvist O; Forster M; Gaulton A; Gilson M; Glen R; Grigorov M; Hammond-Kosack K; Harland L; Hopkins A; Larminie C; Lynch N; Mann RK; Murray-Rust P; Lo Piparo E; Southan C; Steinbeck C; Wishart D; Hermjakob H; Overington J; Thornton J Minimum Information about a Bioactive Entity (MIABE). Nat. Rev. Drug Discovery 2011, 10, 661–669. [DOI] [PubMed] [Google Scholar]
- (12).Capuzzi SJ; Thornton TE; Liu K; Baker N; Lam WI; O’Banion CP; Muratov EN; Pozefsky D; Tropsha A Chemotext: A Publicly Available Web Server for Mining Drug-Target-Disease Relationships in PubMed. J. Chem. Inf. Model 2018, 58, 212–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Li Y; Liu Z; Han L; Li C; Wang R Mining the Characteristic Interaction Patterns on Protein-Protein Binding Interfaces. J. Chem. Inf. Model 2013, 53, 2437–2447. [DOI] [PubMed] [Google Scholar]
- (14).Lee G; Romo Bucheli DE; Madabhushi A Adaptive Dimensionality Reduction with Semi-Supervision (AdDReSS): Classifying Multi-Attribute Biomedical Data. PLoS One 2016, 11, No. e0159088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Finkel J; Dingare S; Manning CD; Nissim M; Alex B; Grover C Exploring the boundaries: gene and protein identification in biomedical text. BMC Bioinf. 2005, 6, S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).McDonald R; Pereira F Identifying Gene and Protein Mentions in Text Using Conditional Random Fields. BMC Bioinf. 2005, 6, S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Evans JA; Rzhetsky A Advancing Science through Mining Libraries, Ontologies, and Communities. J. Biol. Chem 2011, 286, 23659–23666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Li C; Jimeno-Yepes A; Arregui M; Kirsch H; Rebholz-Schuhmann D PCorral–Interactive Mining of Protein Interactions from MEDLINE. Database 2013, 2013, bat030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Doğan RI; Leaman R; Lu Z NCBI Disease Corpus: A Resource for Disease Name Recognition and Concept Normalization. J. Biomed. Inf 2014, 47, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tetko IV; Lowe DM; Williams AJ The Development of Models to Predict Melting and Pyrolysis Point Data Associated with Several Hundred Thousand Compounds Mined from PATENTS. J. Cheminf 2016, 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Oprea TI; Nielsen SK; Ursu O; Yang JJ; Taboureau O; Mathias SL; Kouskoumvekaki I; Sklar LA; Bologa CG Associating Drugs, Targets and Clinical Outcomes into an Integrated Network Affords a New Platform for Computer-Aided Drug Repurposing. Mol. Inf 2011, 30, 100–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tari LB; Patel JH Systematic Drug Repurposing through Text Mining. Methods Mol. Biol 2014, 1159, 253–267. [DOI] [PubMed] [Google Scholar]
- (23).Fourches D; Muratov E; Tropsha A Trust, but Verify II: A Practical Guide to Chemogenomics Data Curation. J. Chem. Inf. Model 2016, 56, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tetko IV; Engkvist O; Koch U; Reymond J-L; Chen H BIGCHEM: Challenges and Opportunities for Big Data Analysis in Chemistry. Mol. Inf 2016, 35, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kontijevskis A Mapping of Drug-like Chemical Universe with Reduced Complexity Molecular Frameworks. J. Chem. Inf. Model 2017, 57, 680–699. [DOI] [PubMed] [Google Scholar]
- (26).Visser U; Abeyruwan S; Vempati U; Smith RP; Lemmon V; Schürer SC BioAssay Ontology (BAO): A Semantic Description of Bioassays and High-Throughput Screening Results. BMC Bioinf. 2011, 12, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Howe EA; de Souza A; Lahr DL; Chatwin S; Montgomery P; Alexander BR; Nguyen D-T; Cruz Y; Stonich DA; Walzer G; Rose JT; Picard SC; Liu Z; Rose JN; Xiang X; Asiedu J; Durkin D; Levine J; Yang JJ; Schürer SC; Braisted JC; Southall N; Southern MR; Chung TDY; Brudz S; Tanega C; Schreiber SL; Bittker JA; Guha R; Clemons PA BioAssay Research Database (BARD): Chemical Biology and Probe-Development Enabled by Structured Metadata and Result Types. Nucleic Acids Res. 2015, 43, D1163–D1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Carpenter B LingPipe for 99.99% Recall of Gene Mentions. Proceedings of the 2nd BioCreative Workshop: Valencia, Spain, 2007. [Google Scholar]
- (29).Vorberg S; Tetko IV Modeling the Biodegradability of Chemical Compounds Using the Online CHEmical Modeling Environment (OCHEM). Mol. Inf 2014, 33, 73–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).TET PDFLib. (https://www.pdflib.com/products/tet/, accessed May 30, 2019).
- (31).Zakharov A; Tarasova O; Poroikov V; Nicklaus MC Mix- and-match (Q) SAR modelability. Abstracts of 250th American Chemical Society Meeting: Boston, MA, USA, Aug 16–20, 2015. [Google Scholar]
- (32).Tarasova O; Poroikov V HIV Resistance Prediction to Reverse Transcriptase Inhibitors: Focus on Open Data. Molecules 2018, 23, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Qin B; Jiang X; Lu H; Tian X; Barbault F; Huang L; Qian K; Chen C-H; Huang R; Jiang S; Lee K-H; Xie L Diarylaniline Derivatives as a Distinct Class of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem 2010, 53, 4906–4916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Tarasova O; Mayorov IS; Filimonov D; Poroikov V; Mayzus I; Rzhetsky A Computational analysis of publications’ texts for bioassay protocol classification. Abstracts of 256th American Chemical Society Meeting: Boston, MA, USA, Aug 16–20, 2015. [Google Scholar]
- (35).Judson R; Kavlock R; Martin M; Reif D; Houck K; Knudsen T; Richard A; Tice RR; Whelan M; Xia M; Huang R; Austin C; Daston G; Hartung T; Fowle JR 3rd; Wooge W; Tong W; Dix D Perspectives on Validation of High-Throughput Assays Supporting 21st Century Toxicity Testing. ALTEX 2013, 30, 51–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Huang Y; Wang X; Yu X; Yuan L; Guo Y; Xu W; Liu T; Liu J; Shao Y; Ma L Inhibitory Activity of 9-Phenylcyclohepta-[d]Pyrimidinedione Derivatives against Different Strains of HIV-1 as Non-Nucleoside Reverse Transcriptase Inhibitors. Virol. J 2011, 8, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Boyapalle S; Xu W; Raulji P; Mohapatra S; Mohapatra SS A Multiple SiRNA-Based Anti-HIV/SHIV Microbicide Shows Protection in Both In Vitro and In Vivo Models. PLoS One 2015, 10, No. e0135288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wu H-Q; Yao J; He Q-Q; Chen W-X; Chen F-E; Pannecouque C; De Clercq E; Daelemans D Synthesis and Biological Evaluation of DAPY-DPEs Hybrids as Non-Nucleoside Inhibitors of HIV-1 Reverse Transcriptase. Bioorg. Med. Chem 2015, 23, 624–631. [DOI] [PubMed] [Google Scholar]
- (39).Lipinski CA Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. [DOI] [PubMed] [Google Scholar]
- (40).Fourches D; Muratov E; Tropsha A Curation of Chemogenomics Data. Nat. Chem. Biol 2015, 11, 535. [DOI] [PubMed] [Google Scholar]
- (41).Tetko IV; Engkvist O; Chen H Does “Big Data” exist in medicinal chemistry, and if so, how can it be harnessed? Future Med. Chem 2016, 8, 1801–1806. [DOI] [PubMed] [Google Scholar]
- (42).BioAssay Express. (https://www.bioassayexpress.com/), date of access: May 30, 2019.
- (43).Clark AM; Bunin BA; Litterman NK; Schürer SC; Visser U Fast and Accurate Semantic Annotation of Bioassays Exploiting a Hybrid of Machine Learning and User Confirmation. PeerJ 2014, 2, No. e524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Clark AM; Litterman NK; Kranz JE; Gund P; Gregory K; Bunin BA BioAssay Templates for the Semantic Web. PeerJ Comput. Sci 2016, 2, No. e61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.