

Abstract
This international multidisciplinary document intends to provide clinicians with evidence-based practical patient-centered recommendations for evaluating patients and decedents with (aborted) sudden cardiac arrest and their families. The document includes a framework for the investigation of the family allowing steps to be taken, should an inherited condition be found, to minimize further events in affected relatives. Integral to the process is counseling of the patients and families, not only because of the emotionally charged subject, but because finding (or not finding) the cause of the arrest may influence management of family members. The formation of multidisciplinary teams is essential to provide a complete service to the patients and their families, and the varied expertise of the writing committee was formulated to reflect this need. The document sections were divided up and drafted by the writing committee members according to their expertise. The recommendations represent the consensus opinion of the entire writing committee, graded by Class of Recommendation and Level of Evidence. The recommendations were opened for public comment and reviewed by the relevant scientific and clinical document committees of the Asia Pacific Heart Rhythm Society (APHRS) and the Heart Rhythm Society (HRS); the document underwent external review and endorsement by the partner and collaborating societies. While the recommendations are for optimal care, it is recognized that not all resources will be available to all clinicians. Nevertheless, this document articulates the evaluation that the clinician should aspire to provide for patients with sudden cardiac arrest, decedents with sudden unexplained death, and their families.
Keywords: Brugada syndrome, Cardiac arrest, Cardiac genetics, Catecholaminergic polymorphic ventricular tachycardia, Defibrillator, Expert consensus statement, Genetic counseling, Guidelines, Long QT syndrome, Postmortem, Resuscitation, Sudden arrhythmic death syndrome, Sudden cardiac death, Sudden unexplained death, Ventricular arrhythmia
Section 1. Introduction
1.1. Purpose
This expert consensus statement represents an international multidisciplinary effort led by the Asia Pacific Heart Rhythm Society (APHRS), in partnership with the Heart Rhythm Society (HRS) and in collaboration with the Association for European Cardiovascular Pathology (AECVP), the European Heart Rhythm Association (EHRA), the European Society of Human Genetics (ESHG), the Latin American Heart Rhythm Society (LAHRS), the National Society of Genetic Counselors (NSGC) (USA), the Pediatric and Congenital Electrophysiology Society (PACES), and the European Reference Network for Rare and Low Prevalence Complex Diseases of the Heart: ERN GUARD-Heart. The intent is to provide clinicians with practical patient-centered recommendations for evaluating patients with sudden cardiac arrest (SCA), decedents with sudden cardiac death (SCD), and their families, based on all available evidence. Although the recommendations are for optimal care, the writing committee recognizes that not all resources will be available to all clinicians. Nevertheless, this document articulates the evaluation that the clinician should aspire to provide.
1.2. Organization of the Writing Committee
The writing committee consisted of internationally recognized experts from 14 countries in the fields of cardiac electrophysiology, cardiology, pediatric cardiology, genetic counseling, community genetics and public health genomics, and cardiac pathology, representing APHRS, HRS, AECVP, EHRA, ESHG, LAHRS, NSGC, PACES, and ERN GUARD-Heart and selected according to each society’s procedures. In addition, a patient representative was chosen to provide a consumer viewpoint. Each partner society nominated a chair, who did not have relevant relationships with industry and other entities (RWIs). In accordance with the APHRS policies, disclosure of any RWIs was required from the writing committee members (Appendix 1) and from the peer reviewers (Appendix 2); of the 28 committee members, 23 (82%) had no relevant RWIs. Recommendations were drafted by the writing committee members who did not have relevant RWIs.
1.3. Methodology and Evidence Review
After development of a preliminary outline, committee members were given writing assignments and a schedule of conference calls. Writing committee members conducted a comprehensive evidence search using MEDLINE/PubMed, Embase, and the Cochrane Library and summarized the evidence in standardized tables (Appendix 3), with attention to the study type, size, inclusion criteria, and key findings. The writing committee reviewed evidence and established consensus to generate recommendations, which are presented in a modular knowledge chunk format, with each chunk including a table of recommendations, a brief synopsis, recommendation-specific supportive text, flow diagrams or tables as appropriate, and references. Recommendations were formulated according to the American College of Cardiology (ACC)/American Heart Association (AHA) Class of Recommendation (COR) and Level of Evidence (LOE) system1 (Table 1) and were subject to a period of public comment. The COR indicates the strength of a recommendation based on assessment of the estimated benefits and risks; LOE rates the quality of evidence that supports the recommendation based on type, quantity, and consistency of data from clinical trials and other sources. Case reports were not used to support recommendations. The threshold for consensus was considered as 80% or higher agreement. The 74 recommendations were balloted by the 28 writing committee members and approved by an average of 94%. A quorum of two-thirds of the writing committee was met for all votes.
Table 1.
ACC/AHA recommendation system: Applying Class of Recommendation and Level of Evidence to clinical strategies, interventions, treatments, and diagnostic testing in patient care*
| CLASS (STRENGTH) OF RECOMMENDATION | |
| CLASS 1 (STRONG) | Benefit >>> Risk |
| Suggested phrases for writing recommendations: | |
| • Is recommended | |
| • Is indicated/useful/effective/beneficial | |
| • Should be performed/administered/other | |
| • Comparative-Effectiveness Phrases†: | |
| – Treatment/strategy A is recommended/indicated in preference to treatment B | |
| – Treatment A should be chosen over treatment B | |
| CLASS 2a (MODERATE) | Benefit >> Risk |
| Suggested phrases for writing recommendations: | |
| • Is reasonable | |
| • Can be useful/effective/beneficial | |
| • Comparative-Effectiveness Phrases†: | |
| – Treatment/strategy A is probably recommended/indicated in preference to treatment B | |
| – It is reasonable to choose treatment A over treatment B | |
| CLASS 2b (WEAK) | Benefit ≥ Risk |
| Suggested phrases for writing recommendations: | |
| • May/might be reasonable | |
| • May/might be considered | |
| • Usefulness/effectiveness is unknown/undear/uncertain or not well-established | |
| CLASS 3: No Benefit (MODERATE) (Generally, LOE A or B use only) | Benefit = Risk |
| Suggested phrases for writing recommendations: | |
| • Is not recommended | |
| • Is not indicated/useful/effective/beneficial | |
| • Should not be performed/administered/other | |
| Class 3: Harm (STRONG) | Risk > Benefit |
| Suggested phrases for writing recommendations: | |
| • Potentially harmful | |
| • Causes harm | |
| • Associated with excess morbidity/mortality | |
| • Should not be performed/administered/other | |
| LEVEL (QUALITY) OF EVIDENCE‡ | |
| LEVEL A | |
| • High-quality evidence‡ from more than 1 RCT | |
| • Meta-analyses of high-quality RCTs | |
| • One or more RCTs corroborated by high-quality registry studies | |
| LEVEL B-R | (Randomized) |
| • Moderate-quality evidence‡ from 1 or more RCTs | |
| • Meta-analyses of moderate-quality RCTs | |
| LEVEL B-NR | (Nonrandomized) |
| • Moderate-quality evidence‡ from 1 or more well-designed, well-executed nonrandomized studies, observational studies, or registry studies | |
| • Meta-analyses of such studies | |
| LEVEL C-LD | (Limited Data) |
| • Randomized or nonrandomized observational or registry studies with limitations of design or execution | |
| • Meta-analyses of such studies | |
| • Physiological or mechanistic studies in human subjects | |
| LEVEL C-EO | (Expert Opinion) |
| • Consensus of expert opinion based on clinical experience | |
COR and LOE are determined independently (any COR may be paired with any LOE).
A recommendation with LOE C does not imply that the recommendation is weak. Many important clinical questions addressed in guidelines do not lend themselves to clinical trials. Although RCTs are unavailable, there may be a very clear clinical consensus that a particular test or therapy is useful or effective.
The outcome or result of the intervention should be specified (an improved clinical outcome or increased diagnostic accuracy or incremental prognostic information).
For comparative-effectiveness recommendations (COR 1 and 2a; LOE A and B only), studies that support the use of comparator verbs should involve direct comparisons of the treatments or strategies being evaluated.
The method of assessing quality is evolving, including the application of standardized, widely-used, and preferably validated evidence grading tools; and for systematic reviews, the incorporation of an Evidence Review Committee.
COR indicates Class of Recommendation; EO, expert opinion; LD, limited data; LOE, Level of Evidence; NR, nonrandomized; R, randomized; and RCT, randomized controlled trial.
1.4. Document Review and Approval
After review by the entire writing committee, the recommendations were opened for public comment; the draft document was reviewed by the International Scientific Document Writing Committee of the APHRS and the Scientific and Clinical Documents Committee of the HRS and was revised prior to external review. The document underwent external peer review by reviewers appointed by the APHRS and HRS and each of the collaborating societies. After subsequent revisions and endorsement by the participating societies, the document was ready for publication.
1.5. Scope of the Document
This document provides a framework for the investigation of 1) patients with SCA, 2) decedents with sudden unexplained death (SUD), and 3) families of both SCA survivors and SUD victims, as many conditions responsible for the cardiac arrest or unexplained death may be familial. Identifying a cause is important for preventing further events in the family, should an inherited condition be found. Integral to the process is the counseling of the patients and families, not only because of the emotionally charged subject, but because finding (or not finding) the cause of the arrest may influence the futures of the family members. The disciplines of cardiology, pediatrics, radiology, pathology, counseling, psychology, and genetics are all involved in this process. Therefore, the formation of multidisciplinary teams is essential to provide a complete service to the patients and their families.
While this document endeavors to provide clinicians with practical recommendations for evaluating patients with SCA, decedents with SUD, and their families, the best approach will vary with the situation and will be influenced by, for example, the subject’s age and results of initial testing. Although some of the recommendations do specify an age cutoff, it is recognized that this age is somewhat arbitrary and may not be always appropriate for the disease being investigated for or the demographics of the patient’s country. Nevertheless, where an age is specified in a recommendation, it has passed the consensus voting of the writing group. Referral to a center with a multidisciplinary team experienced in such evaluations is recommended because it can facilitate navigation of these complexities. A multidisciplinary team can also help organize interval follow-up evaluations for SCA survivors and their family members. Repeated interval follow-up can reveal important new clinical data and allows for integration of new knowledge into the continued evaluation and care of these patients. The writing committee members recognize that not all investigative modalities recommended will be available in all circumstances; however, this document is an attempt to outline an approach to which the clinician should aspire.
1.6. Relevant Clinical Practice Documents
Table 2 lists pertinent guidelines and consensus statements that the writing committee considered for this document. The included documents contain relevant information for the diagnosis of patients with SCA and SCD.
Table 2.
Relevant clinical practice documents
| Title | Publication year |
|---|---|
| European Recommendations Integrating Genetic Testing into Multidisciplinary Management of Sudden Cardiac Death2 | 2019 |
| 2019 HRS/EHRA/APHRS/LAHRS Expert Consensus Statement on Catheter Ablation of Ventricular Arrhythmias3 | 2019 |
| 2019 HRS Expert Consensus Statement on Evaluation, Risk Stratification, and Management of Arrhythmogenic Cardiomyopathy4 | 2019 |
| 2018 ESC Guidelines for the Diagnosis and Management of Syncope5 | 2018 |
| 2017 AHA/ACC/HRS Guideline for Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death6 | 2017 |
| Pre-participation Cardiovascular Evaluation for Athletic Participants to Prevent Sudden Death: Position Paper from the EHRA and the EACPR, Branches of the ESC7 | 2016 |
| 2015 ESC Guidelines for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death8 | 2015 |
| EHRA/HRS/APHRS Expert Consensus on Ventricular Arrhythmias9 | 2014 |
| HRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromes10 | 2013 |
| HRS/EHRA Expert Consensus Statement on the State of Genetic Testing for the Channelopathies and Cardiomyopathies11 | 2011 |
1.7. Definitions
The terms used in the consensus statement are defined in Table 3.
Table 3.
Definitions
| Term | Definition |
|---|---|
| Sudden cardiac arrest (SCA) | Sudden cessation of cardiac activity with hemodynamic collapse, typically due to sustained ventricular arrhythmia |
| Sudden cardiac death (SCD) | Death that occurs within 1 hour of onset of symptoms in witnessed cases, and within 24 hours of last being seen alive when it is unwitnessed |
| Sudden unexplained death (syndrome) (SUD[S]) | Unexplained sudden death occurring in an individual older than 1 year |
| Sudden unexplained death in infancy (SUDI) | Unexplained sudden death occurring in an individual younger than 1 year with negative pathological and toxicological assessment Note: Synonymous with “sudden unexplained infant death” (SUID) |
| Sudden arrhythmic death (syndrome) (SAD[S]) | Unexplained sudden death occurring in an individual older than 1 year with negative pathological and toxicological assessment Note: Synonymous with “autopsy-negative sudden unexplained death” |
| Sudden unexplained death in epilepsy (SUDEP) | Sudden and unexpected, nontraumatic and nondrowning death of a person with epilepsy, without a toxicological or anatomical cause of death detected during the postmortem examination |
Section 2. Epidemiology
2.1. Epidemiology: Sudden Death
“Sudden unexplained death” refers to an unexpected and sudden death in an individual older than 1 year. Sudden death occurring unexpectedly within the first year of life is termed “sudden unexplained death in infancy” (SUDI). Multiple definitions have been in use over the past decades, although most recent studies implement a definition that differs between witnessed and unwitnessed events; in witnessed cases, death has to occur within 1 hour of change in cardiovascular status, whereas unwitnessed cases have to be seen alive and functioning normally within 24 hours of being found dead.12,13
SCD constitutes the majority of SUD.14–16 Reported overall SCD incidence rates vary across studies and countries, in part due to large difference in SCD definitions and methods for estimation of SCD rates. Previous studies report overall SCD rates ranging from 15 to 159 SCD per 100,000 persons per annum, corresponding to 6–20% of all deaths.17–24 However, both incidence and causes of SCD vary markedly with age. Lowest SCD incidence is observed in children and adolescents.15,18,25–28 SCD incidence is low in children and the young under 35 years and increases dramatically up until the age of approximately 60–80 years.15,19,20,29,30
In young persons aged 1–35 years, most SCDs are caused by potentially inherited heart diseases, including primary arrhythmogenic disorders (eg, congenital long QT syndrome and catecholaminergic polymorphic ventricular tachycardia [CPVT]), hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy, and dilated cardiomyopathy;15,18,25–29,31,32 however, coronary artery disease, anomalous coronary arteries, aortic dissection, congenital heart disease, and myocarditis are also potential causes, potentially with a non-negligible genetic component (Figure 1). From the age of 35 years, coronary artery disease becomes the most common cause of SCD, although potentially inherited heart diseases remain a common cause of SCD at least until the age of 50 years.27,29,33 Individuals with SUD who subsequently have negative pathological and toxicological assessment may be assumed to have sudden arrhythmic death (syndrome), or SAD(S), a term synonymous with “autopsy-negative SUD.”
Figure 1.
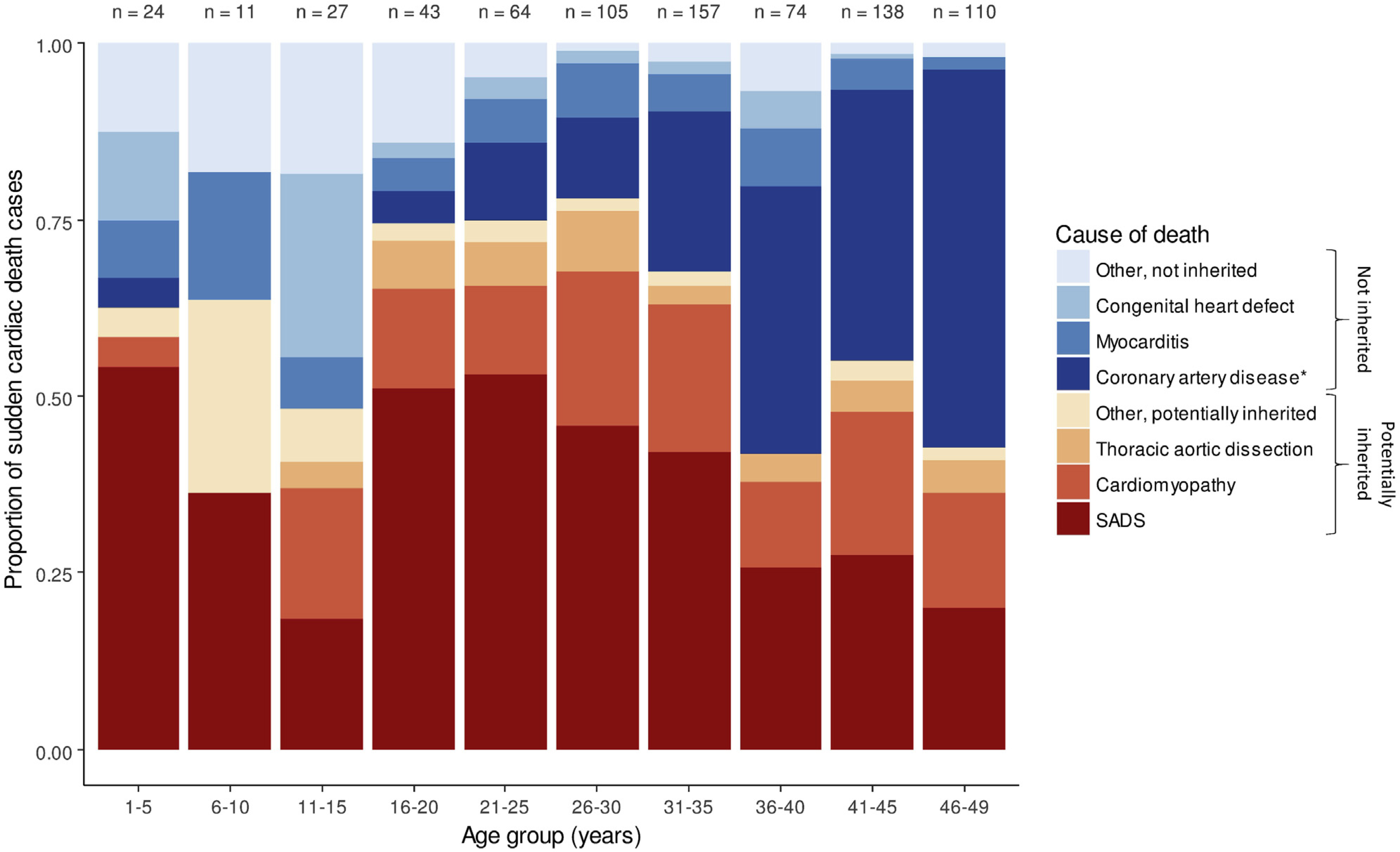
Distribution of causes of death among autopsied cases of sudden cardiac death (n = 753) according to age in persons aged 1–49 years in Denmark (J.T.-H., unpublished data). SADS = sudden arrhythmic death syndrome. *Coronary artery disease, especially in young persons, may be due to inherited disease (eg, familial hypercholesterolemia).
At any age, males have higher SCD rates compared with females, even after adjustment for risk factors of coronary heart disease.34 Ethnic background seems to have large effect.35,3,6
Recommendations for improving outcomes from sudden death
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Investigation of SUD at a young age should be made a public health priority due to the combined prevalence of inherited cardiac diseases of at least 1:500, the years of potential life lost, and the significant impact on the family and community; therefore, public funding should be allocated for relevant investigations. | 15,25,37 |
| 1 | C-EO | 2. Identification of inherited cardiac conditions that predispose to SCD should be made a public health priority, as diagnosis may prevent future cardiac events in affected family members. | |
| 1 | B-NR | 3. The burden of SUD and varied outcomes in relation to sex, different ethnic populations, and socioeconomic backgrounds should be investigated worldwide. | 35,36,38,39 |
Synopsis
SUD is a tragedy and, in the case of an underlying genetic predisposition, may be preventable. The main cause of SUD is SCD. SCD in the young often occurs in people who were thought to be well, may occur without warning symptoms, and is often the first presentation of an underlying genetic heart disease. Across all ages, estimates differ from 5% to 20% of all deaths, and ethnicity-specific data on SCD incidences worldwide are sparse. Cause of death changes according to age (Figure 1). Exact estimates of the burden of SCD are crucial in order to adjudicate public health spending.
Recommendation-Specific Supportive Text
1. and 2. Inherited cardiac disorders are the main cause of SCD in the young. Sudden death is SCD in 60–90% depending on age, of which the majority is potentially from inherited cardiac disease.2,15,40 Exact estimates of the burden of SCD are crucial in order to adjudicate public health spending.25
3. Estimates of SCD among different ethnic backgrounds are sparse worldwide.35,36,38,39
2.2. Epidemiology: Sudden Cardiac Arrest Survivors
2.2.1. Background
Out-of-hospital cardiac arrest (OHCA) is a leading cause of mortality globally41–43 and is defined as the loss of functional cardiac mechanical activity in association with an absence of systemic circulation, occurring outside of a hospital setting. The exact burden of OHCA remains unknown, since a considerable number of cases are not attended by emergency medical services (EMS) and regional variations are prevalent in both reporting systems and survival.17,44,45 Approximately 275,000 people in Europe have cardiac arrest treated by EMS per year, with only 29,000 (10.5%) surviving hospital discharge.46 In England, 28,729 EMS-treated OHCA cases were reported in 2014 (53 cases per 100,000 of the resident population), with only 7.9% surviving to hospital discharge.47 In the United States, reports from 35 communities suggested an incidence of 55 per 100,000 person-years48 or approximately 155,000 individuals having an EMS-treated all-rhythm OHCA per year.48 Globally, the weighted incidence estimates per 100,000 person-years of EMS-treated OHCA are 34.4 in Europe, 53.1 in North America, 59.4 in Asia, and 49.7 in Australia. For reported survival estimates, the percentage survival to discharge was 7.6% in Europe, 6.8% in North America, 3.0% in Asia, and 9.7% in Australia.42
Significant geographical variation in the incidence of OHCA associated with poor outcomes has remained unchanged in the past 3 decades.17,41,42,44 However, implementation of coordinated efforts targeted at improving the local chain of survival in some cities has improved regional survival to 20–40%.49,50 This survival benefit can be partially attributed to varying definitions of OHCA,42 but it is primarily due to a coordinated effort to optimize the effectiveness of the local chain of survival.51 Identifying and improving weak links in the local chain of survival, paired with targeted approaches to improve the effectiveness, has resulted in positive outcomes achieved in several geographic regions.49,52–54
2.2.2. Causes of Out-of-Hospital Cardiac Arrest
OHCA causes are classified into cardiac and noncardiac causes.47,55,56 Approximately 80% of individuals presenting with OHCA reached by EMS, and in whom resuscitation is considered possible, have a cardiac cause.56
OHCA can affect seemingly fit and healthy athletes, young adults, or children. The incidence of SCD in athletes can range from 1 in 23,000 to 1 in 200,000 athletes per year, depending on a number of factors including populations studied.57,58 In a retrospective analysis of the Rescu Epistry database of consecutive OHCA attended by EMS in a specific area of Ontario, Canada, the incidence of SCD during participation in competitive sports was reported to be 0.76 cases per 100,000 athlete-years.59 The main causes of SCD were stratified by age. In those younger than 35 years, structural heart and primary arrhythmic causes were most common. In those aged between 35 and 45 years, coronary artery disease was the most frequent underlying pathology.59 In a prospective study of children and young adults aged 1–35 years, 490 cases of SCD were identified from centers in Australia and New Zealand.25 The cause of death was unexplained in 40% of these cases at autopsy, in whom a structurally normal heart was reported.25 In this study, the annual incidence of SCD was calculated to be 1.3 cases per 100,000 people. When stratified according to age group, the highest incidence (3.2 cases per 100,000 people per year) was observed in those aged 31–35 years. Coronary artery disease was the most common cause ascribed. Younger age and SCD occurring at night were independently associated with unexplained SCD, probably due to congenital channelopathies. Less common causes were inherited cardiomyopathies (eg, dilated, hypertrophic, and arrhythmogenic right ventricular), myocarditis, and aortic dissection.
The Cardiac Arrest Registry to Enhance Survival (CARES), established by the Centers for Disease Control and Prevention (CDC),60 evaluated OHCA events of presumed cardiac etiology that involve persons who received resuscitative effort. OHCA is defined in CARES as a cardiac arrest that occurred in the prehospital setting, had a presumed cardiac etiology, and involved a person who received resuscitative efforts, including cardiopulmonary resuscitation (CPR) or defibrillation. The registry includes 40,274 OHCA records, of which 31,689 OHCA events were presumed to be of cardiac etiology (eg, myocardial infarction or arrhythmia) that received resuscitation efforts in the prehospital setting (mean age 64.0 years [SD 18.2]; 61.1% male). The survival rate to hospital admission was 26.3%, and the overall survival rate from cardiac arrest to hospital discharge was 9.6% (Figure 2). Approximately 36.7% of OHCA events were witnessed by a bystander. Only 33.3% of all patients received bystander CPR, and only 3.7% were treated by bystanders with an automated external defibrillator (AED) before the arrival of EMS providers.
Figure 2.
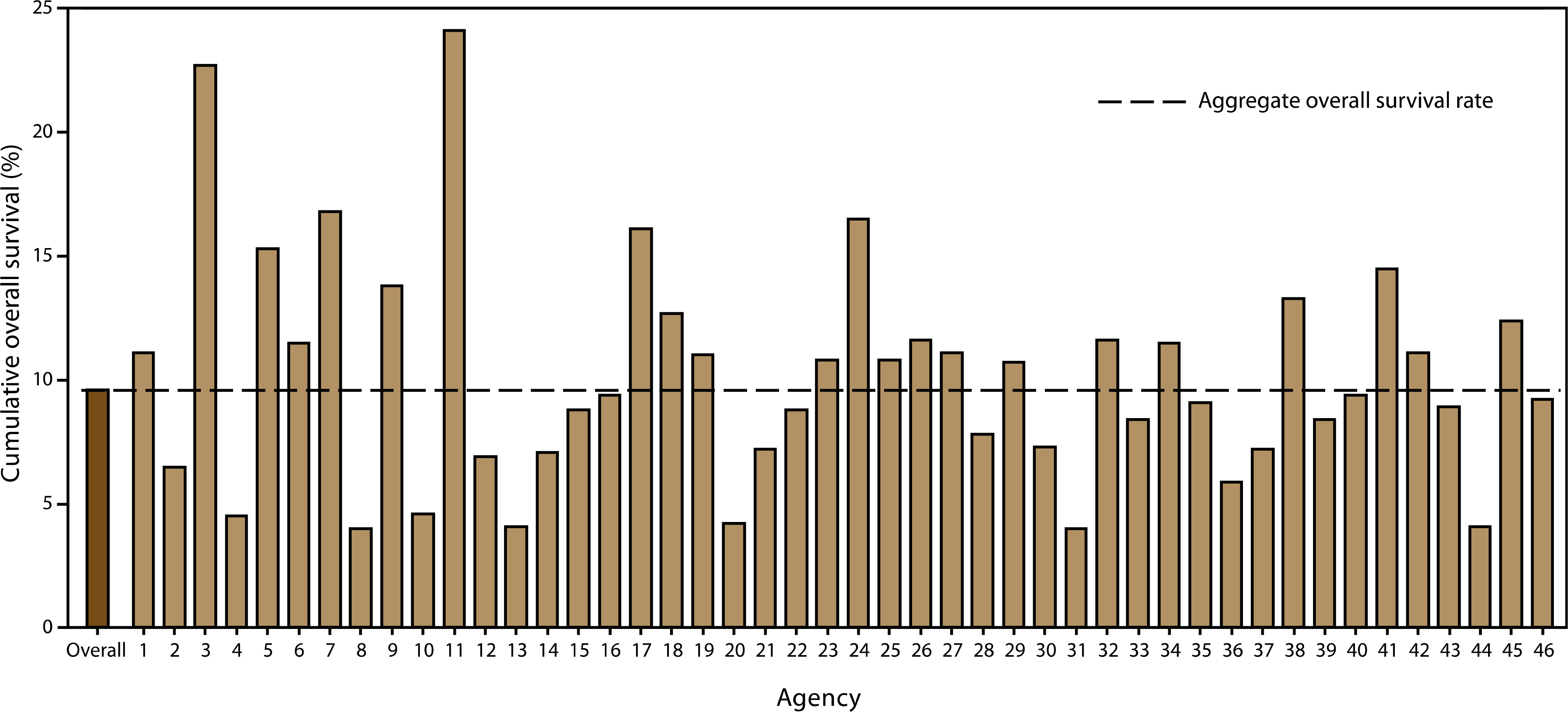
Cumulative overall survival rates, by participating emergency medical services agency—Cardiac Arrest Registry to Enhance Survival (CARES), United States, October 1, 2005–December 31, 2010. Agencies sorted by total number of out-of-hospital cardiac arrest events in CARES (from low to high; range: 18–5,434).60
The group most likely to survive an OHCA is persons who are witnessed to collapse by a bystander and found in a shockable rhythm (ie, arrhythmias leading to ventricular fibrillation or pulseless ventricular tachycardia).60 Among this group, survival to discharge was 30.1% (Figure 3). A subgroup analysis, performed among persons who experienced OHCA events unwitnessed by EMS, revealed that whites were significantly more likely to receive CPR than blacks, Hispanics, or members of other racial/ethnic populations (p < 0.001). Overall survival to hospital discharge of patients whose events were not witnessed by EMS personnel was 8.5%. Of these, patients who received bystander CPR had a significantly higher rate of overall survival (11.2%) than those who did not (7.0%) (p < 0.001).
Figure 3.
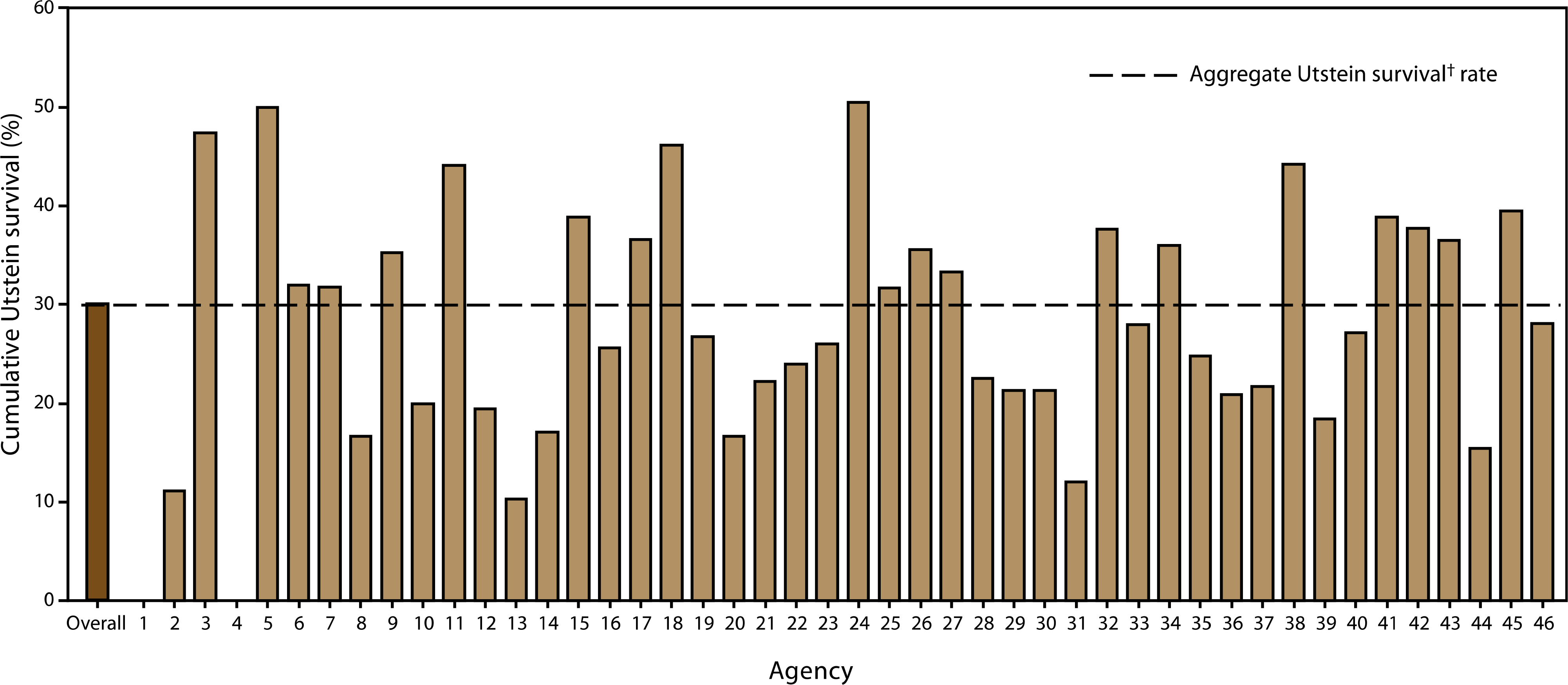
Cumulative Utstein survival rates (patients alive when arriving to hospital) by participating emergency medical services agency—Cardiac Arrest Registry to Enhance Survival (CARES), United States, October 1, 2005–December 31, 2010. Agencies sorted by total number of out-of-hospital cardiac arrest events in CARES (from low to high).60 †Utstein survival refers to survival to hospital discharge of persons whose cardiac arrest events were witnessed by a bystander and had an initial rhythm of ventricular fibrillation or pulseless ventricular tachycardia (range: 0–598).
Figure 4 shows bystander CPR and lay AED use by percentage of black residents in the area. Directing attention toward improving education, availability of AEDs, and treatment of cardiac arrest in predominantly black neighborhoods may save lives.39
Figure 4.
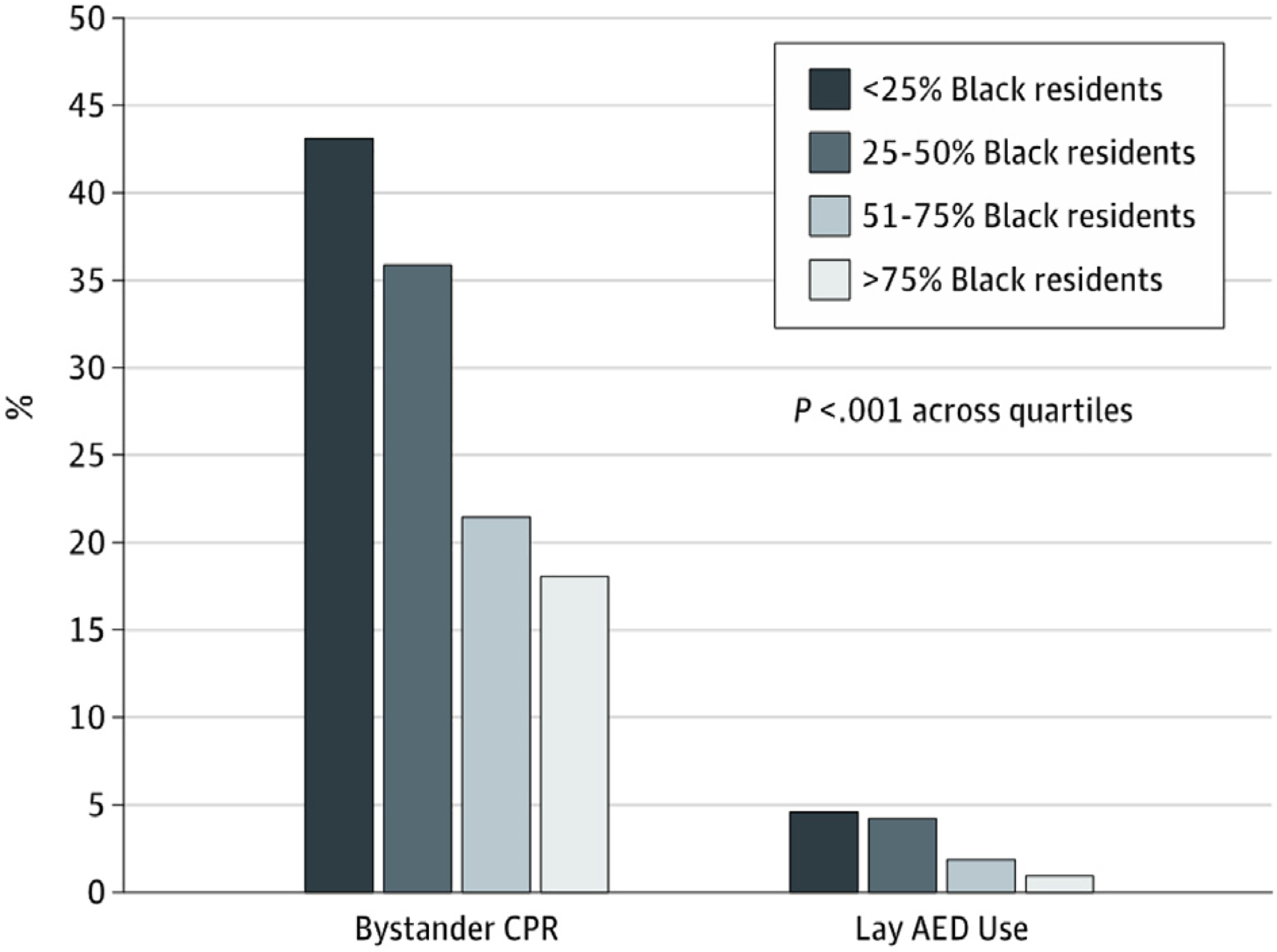
Bystander treatments of patients with out-of-hospital cardiac arrest before emergency medical services arrival among neighborhoods by percentage of black residents. Reprinted with permission from the American Medical Association.39 AED = automated external defibrillator; CPR = cardiopulmonary resuscitation.
Bystander AED use in OHCA in pediatric populations is variable and uncommon, with important variations based on neighborhood characteristics leading to marked disparities in survival and outcomes. Griffis et al.38 reported that AED use (likely due to availability) was more common in neighborhoods with a median household income of >$50,000 per year (12.3%; p = 0.016), <10% unemployment (12.1%; p = 0.002), and >80% high school education (11.8%; p = 0.002). Greater survival to hospital discharge and neurologically favorable survival were among arrests with bystander AED use, varying by neighborhood characteristics.
2.2.3. Public Health Implications
The majority of persons who experience an OHCA event, irrespective of etiology, do not receive bystander CPR or other timely interventions that are known to improve the likelihood of survival to hospital discharge (eg, defibrillation).54 Because nearly half of cardiac arrest events are witnessed, efforts to increase survival rates should focus on timely and effective delivery of interventions by bystanders and EMS personnel (Figure 5).
Figure 5.
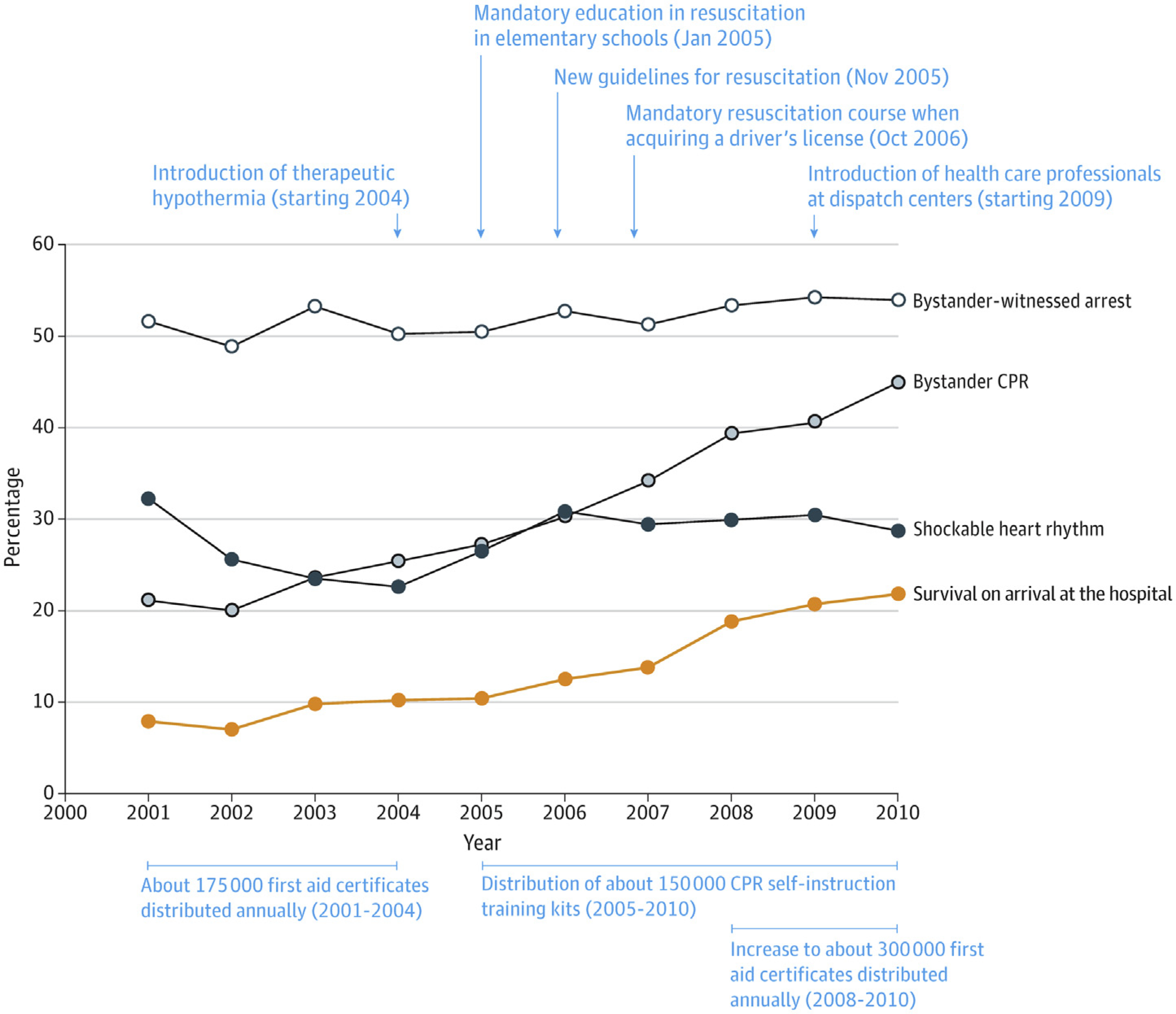
Bystander-witnessed arrest, bystander cardiopulmonary resuscitation (CPR), shockable heart rhythm as first recorded rhythm, and survival on arrival at the hospital, Denmark, 2001–2010. Reprinted with permission from the American Medical Association.54
Education of public officials and community members regarding the importance of increasing rates of bystander CPR and promoting the use of early defibrillation by lay and professional rescuers is critical to increasing survival rates. Reporting at local and national levels can enable local and national public health and EMS agencies to coordinate their efforts to target improving emergency response for OHCA events, regardless of etiology, which can lead to improvement in OHCA survival rates (Figure 6).
Figure 6.
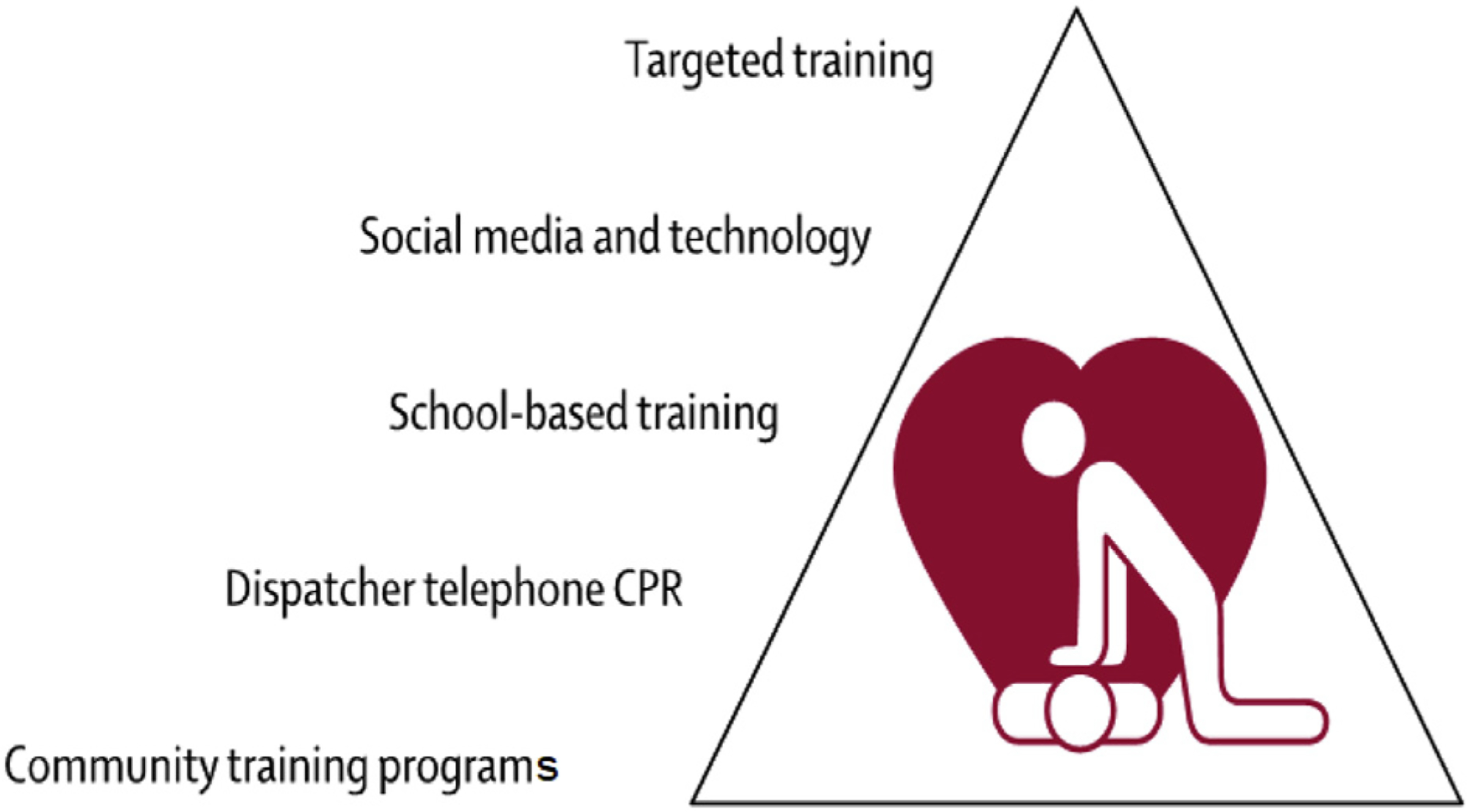
Examples of complimentary bystander cardiopulmonary resuscitation (CPR) programs. Reprinted with permission from Elsevier.64
Recommendations for improving outcomes in SCA survivors
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Targeted CPR training should be widely implemented with particular emphasis on low-income communities, ethnic minorities, and middle- to low-income countries. | 38,60 |
| 1 | B-NR | 2. The burden of out-of-hospital SCA and varied outcomes in different ethnic populations and socioeconomic backgrounds should be investigated worldwide. | 35,38,39 |
| 1 | B-NR | 3. Appropriately maintained AEDs should be readily available at schools, stadiums, public transport stations, casinos, etc, as well as venues where no other access to AEDs is available (eg, trains, ships, planes), with appropriate training of users. | 61,62 |
Synopsis
OHCA remains a significant cause of mortality globally. Despite implementation of cardiac arrest protocols including CPR training and AEDs, only 33% of witnessed OHCA cases receive bystander CPR and less than 4% are defibrillated onsite. OHCA hospital discharge survival remains dismal at around 10% and has remained stagnant for the past 3 decades. Significant geographic variation in OHCA incidence and the role of social disparities merit further research. Public health campaigns promoting CPR training in at-risk communities and greater availability of AEDs are needed.
Recommendation-Specific Supportive Text
Coordinated efforts targeted at improving the local chain of survival have improved regional survival.49,50 Targeted approaches to improve the effectiveness of CPR have resulted in positive outcomes.52–54 The group most likely to survive an OHCA is persons who are witnessed to collapse by a bystander and found in a shockable rhythm, so widespread CPR training is recommended. Subgroup analysis has revealed that whites were significantly more likely to receive CPR than other racial/ethnic populations.60 AED use was more common in neighborhoods with high median household income, <10% unemployment, and >80% high school education.38 Therefore, maximum benefit will be gained from targeting CPR training to groups of high socioeconomic need and ethnic minorities.
The burden of OHCA and the response of bystanders appears to vary according to ethnicity and socioeconomic status.35,38,39,60 Further investigation of these findings may result in targeted approaches to maximize outcome from investment when aimed at these communities.
Availability of AEDs has been shown to improve survival.61–63 Therefore, as the majority of cardiac arrests are witnessed, AEDs at schools, stadiums, stations, etc, may be expected to increase survival. Venues where delivery of AEDs by emergency services is unlikely (eg, trains, ships, planes) are of particular importance. Appropriately maintained equipment and appropriate training of potential AED users are an essential component of this strategy.
Section 3. Multidisciplinary Team
3.1. Introduction
The investigation of SCD and resuscitated SCA requires input from a variety of different disciplines. The coordination and the communication between them mandate the formation of a multidisciplinary team. Numerous consensus statements agree on the importance of a dedicated combined cardiac genetic service in this setting.2,11,65–67
3.2. Key Features of an Effective Multidisciplinary Team
Certain key features can be identified in well-functioning multidisciplinary teams across specialties. Nancarrow et al.68 propose 10 key attributes including positive leadership and management, communication strategies and structures, appropriate resources, appropriate skill mix, and a supportive team climate with a focus on education of each other. There should be open communication and shared decision-making.
The detection of inherited heart conditions by pathologists and by hospital clinicians requires heightened awareness of their existence and a simple referral pathway to a multidisciplinary service with cardiac genetic expertise. Clinical experience shows that the appointment of a coordinator, as well as an enthusiastic team leader, is essential to facilitate this process, and regular meetings increase relevance and improve attendance69 (Figures 7 and 8).
Figure 7.
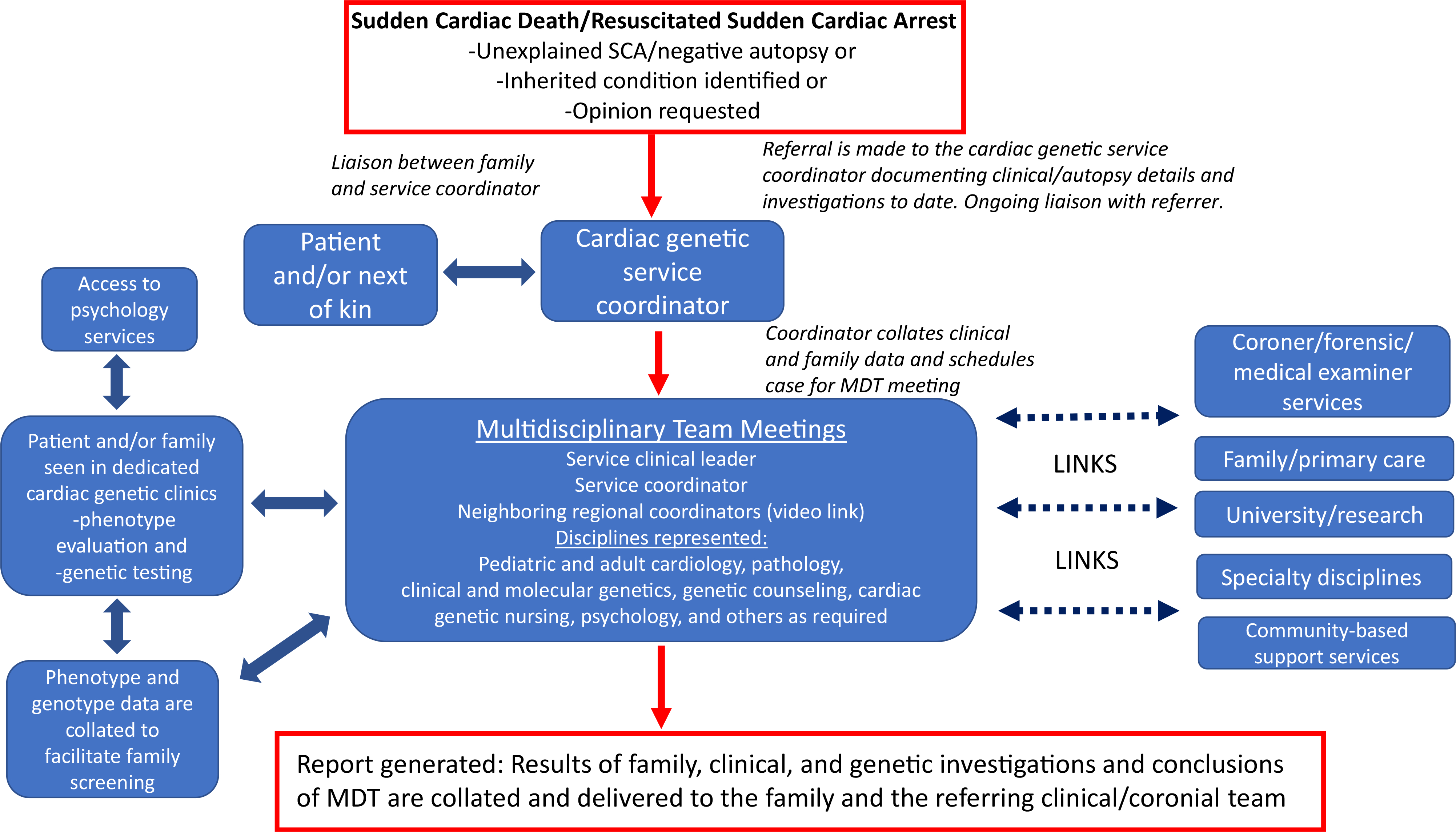
Referral flow for cardiac genetic investigation of sudden cardiac death (SCD) or resuscitated sudden cardiac arrest (SCA). MDT = multidisciplinary team.
Figure 8.
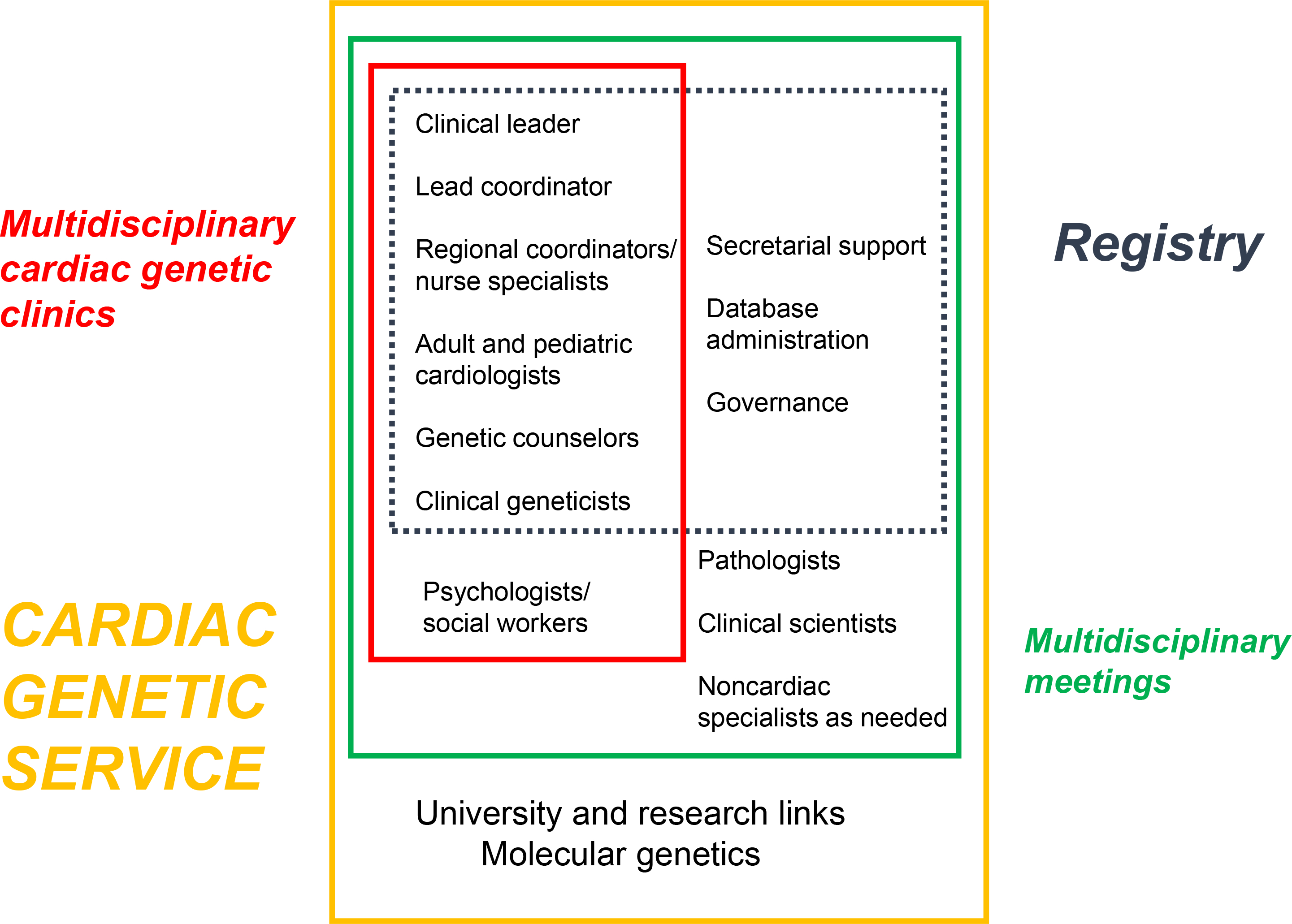
Participants in a cardiac genetic service. “Pathologists” includes forensic pathologists. Modified with permission from Elsevier.69
3.3. Defining Which Disciplines Should Be Represented
The investigation of SUD is led by (forensic) pathology and the investigation of resuscitated SCA by pediatric or adult cardiology, with cardiac heart rhythm specialists and genetic cardiologists often being central. Clinical and molecular genetic specialists and genetic counselors are needed because of the significant role of molecular genetics in achieving a diagnosis and cascade screening, the consideration of multisystem genetic syndromes, and the high prevalence of genetic variants of uncertain significance.70,71 The high levels of psychological morbidity among SCA survivors and family members of both SCA survivors and decedents mandates access to psychology expertise,72–74 and input into the multidisciplinary team helps keep this in focus.75 The presence of a specialist cardiac genetic nurse in the cardiology inpatient setting increases detection of inherited cardiac conditions following SCA.76 Other clinical specialists can be helpful and be drafted in for certain cases—neurologists, pediatricians, metabolic specialists, and intensivists, for example.
3.4. Coordination Across Disciplines and Other Boundaries
The fact that the many disciplines may not be co-located highlights the importance of a coordinator to a multidisciplinary service. Co-location is not critical for effective collaboration, and non-co-location should not be an excuse for failed collaboration. A regional or institutional coordinator could be a nurse specialist, genetic counselor, or other allied professional and is vital to facilitate team meetings and communication between specialists and between centers, with primary care and across regions or between states and countries where necessary to facilitate family screening69,77 (Figure 7).
3.5. Links to Other Services
Links to other services as proposed in a recent scientific statement66 and practiced by some centers already69 include connections to molecular genetic expertise, researchers, primary health providers, between regions, and to a cardiac genetic clinical registry to facilitate family screening and follow-up across traditional boundaries (Figure 8).
Clinical and genetic registries are generally voluntary and consent-based and have a research element. We do not consider that they are compulsory. However, in this setting they do have particular relevance because many cases remain unresolved after the initial investigation and families may find comfort in knowing that efforts to find a diagnosis continue. The multidisciplinary team also provides a mechanism to revisit family members if new findings appear in the wider family or if the pathogenicity of a genetic variant is redefined.
Recommendations for the role of a multidisciplinary team for investigation of SUD and SCA
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. The investigation of SUD and SCD due to a potentially heritable condition should be overseen by a multidisciplinary team with, as a minimum, appropriate expertise in pediatric and/or adult cardiology, genetics, genetic counseling, and pathology. | 78–82 |
| 1 | B-NR | 2. The investigation of a sudden cardiac arrest survivor where a heritable condition is possible should be overseen by a multidisciplinary team with, as a minimum, appropriate expertise in pediatric and/or adult cardiology, genetics, and genetic counseling. | 79,82–85 |
Synopsis
The cardiac and genetic investigation of SUD and resuscitated SCA should be overseen by a multidisciplinary team with appropriate expertise in this area. Recommendations include adequate resourcing, a dedicated coordinator, strong leadership, and a mutually supportive team that meets regularly.
Recommendation-Specific Supportive Text
For regions where coordinated cardiac genetic services exist that include the investigation of SUD, detection of inherited heart conditions is higher than in regions where they are not.78 Families prefer specialized clinics that combine co-located cardiac and genetic expertise and genetic counseling.79 Many such dedicated clinics internationally have led to the detection of inherited heart conditions following SCD and resuscitated SCA.80–82 It is therefore logical that multidisciplinary teams should have links to such clinics. Continued productive dialogue among pathology, coronial, police, and cardiac genetic services is recommended to improve the quality and relevance of forensic pathologists’ reports.86
Genetic testing in this context leads to a significant proportion of both pathogenic and unclassified variants, and precise evaluation of clinical phenotype is imperative for the correct assignation of such variants, so that a service that combines specialist cardiology and genetic expertise is essential.83,84 Specialized clinics that combine co-located cardiac and genetic expertise and genetic counseling are preferred,79 and such combined clinics have a high detection of inherited cardiac conditions following resuscitated SCA.82,85
Section 4. Counseling Families, the Bereaved and the Nearly Bereaved
Genetic counseling is a process that aims to assist patients and their families to understand and adapt to the medical, psychosocial, and familial impact of inherited diseases.87,88 Genetic counseling goes beyond the discussion of genetic testing and is important for all patients with a genetic condition, at all stages of management.89 Although genetic counseling may be performed by any number of health professionals, genetic counselors are specifically trained in this role and have grown to a large allied health workforce worldwide.90,91 In some institutions, this role may be performed by a clinical/medical geneticist, genetic nurse, or other appropriately trained specialist.
In the setting of SCD or resuscitated SCA where a genetic cause is suspected, the inclusion of genetic counselors in the multidisciplinary team is widely advocated. The role of the cardiac genetic counselor includes taking a detailed family history, investigating and confirming details such as postmortem reports, providing education and awareness, assisting in coordinating family clinical screening, and providing psychosocial support.74,92–95 Throughout the process of genetic testing, genetic counselors provide important pre- and post-test genetic counseling, assist with interpretation of the results, help communicate this information to relatives, and assist with cascade genetic testing75,93,96,97 (Table 4).
Table 4.
Key goals of genetic counseling following sudden cardiac death/resuscitated sudden cardiac arrest
| Goal | Description |
|---|---|
| Genetic counseling about inheritance risks | Provide information tailored specifically to the family about their inheritance risks. |
| Provide education and awareness | Educate about inheritance risks, the need for clinical surveillance, and options for genetic testing to allow the family to make subsequent important medical decisions. Conveying information is not straightforward, given varying health literacy and competing health concerns;however, genetic counseling can support effective communication.108 Genetic counseling can also include connection of families with advocacy organizations and relevant research studies. |
| Pre- and post-test genetic counseling | Explain the process and discuss the options of genetic testing, all possible outcomes of testing, implications for patients and/or their family members, and worries and fears about testing;ensure consideration of all possible results and implications.109 Care should be taken in conveying test results of uncertain significance,101,110 specifically ensuring adequate understanding and confidence to communicate key risk information to family members. |
| Pre- and post-test genetic counseling for cascade testing of asymptomatic relatives | There are ethical, legal, and social implications when considering cascade genetic testing of asymptomatic at-risk relatives. Careful pre-test genetic counseling should explore the individual’s feelings toward their risk, how they might feel if they are gene positive or gene negative, and implications for their own health and clinical management based on their genetic result. Discussion about the potential for reclassification of the genetic result is also important.100,111 |
| Provide input regarding classification of genetic variants | Knowledge of variant and gene curation processes will enable review of any genetic test findings at all stages of family management.112 Clinicians involved in family management (including genetic counselors) are more likely to provide conservative variant classifications compared to clinical laboratories,113 and processes to guarantee regular review of variants will ensure appropriate reclassifications are made.114,115 |
| Obtain detailed three-generation family history and confirm details | Record family history information in a pedigree and interpret the information and the risk posed to family members. Taking a detailed family history can allow development of rapport, elucidate family relationships and social circumstances, and inform clinical care.116 |
| Assist with coordination of family clinical screening | Ensure adequate understanding of the clinical screening recommendations for family members and provide assistance with communicating this to relatives as needed. Provide support in organizing cardiology appointments with appropriate tests.92 |
| Provide psychosocial support and identify when referral to clinical psychologist is required | Although genetic counseling is unlikely to resolve any significant psychopathologies, the process of providing information and a big picture perspective allowing a patient to normalize their experience and emotional response can have a positive impact, including patient empowerment.103,106,107 |
Where there are significant emotional difficulties (see Section 5), the process of effectively conveying genetic information can be challenging.98 For families who have experienced a young SCD where a genetic cause is suspected, learning the potential inheritance risk to family members and need for clinical screening can add an additional stressor at a time of intense grief. Furthermore, with the increasing availability of postmortem genetic testing (see Sections 6.4 and 6.5), the need for complex genetic discussions with families is more commonplace.99 Genetic counseling prior to and after genetic testing is important, particularly where genetic test results are not straightforward such as identification of variants of uncertain significance or in the event of a variant reclassification.100,101
There is wide acknowledgment that genetic counseling as a process should go beyond just provision of information.10,2,103 The psychosocial aspects of genetic counseling include psychological support, empathic listening, crisis intervention skills, knowledge of family dynamics, coping models, processes of grief, and adjustment to disease diagnoses, all of which align with the core competencies of genetic counseling accreditation.10,4,105 Attending to the psychosocial needs, in addition to provision of education and information, has been demonstrated to positively impact patient outcomes, largely based around knowledge and recall, but healthy adjustment, empowerment, behavioral change, and satisfaction with decision-making also reduce anxiety and worry.103,104,106,10,7
Recommendations for counseling families affected by SUD and SCA
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Genetic counseling is strongly recommended for all families where there has been an SUD or resuscitated SCA and a heritable cause is suspected, and should include antemortem and postmortem data collection and evaluation, so that risks, benefits, results, and the clinical significance of genetic testing can be discussed. | 80–82,84,85,103,106,107,109 |
| 1 | C-EO | 2. It is recommended that genetic testing in families where an SUD or resuscitated SCA due to a heritable cause is suspected is performed only with appropriate genetic counseling. |
Synopsis
Genetic counseling of patients and their families with genetic conditions is recommended, including those with SCD or resuscitated SCA where a genetic cause is suspected. Key aspects of the process include discussion of inheritance risks, education and awareness, pre- and post-test genetic counseling, interpretation of genetic results, taking a family history, coordination of clinical screening, and psychosocial support. Genetic counseling is focused on both information provision and psychosocial support and together has been shown to improve knowledge and recall; promote healthy adjustment, empowerment, and behavioral change; increase satisfaction with decision-making; and reduce anxiety and worry. While genetic counseling is a process often performed by a variety of health professionals, ideally a specifically trained genetic counselor or genetic nurse with appropriate skills in information provision and psychosocial support would perform this role.
Recommendation-Specific Supportive Text
Genetic counseling includes both information provision and psychosocial support. It is ideally performed by health professionals with specific training and experience; this includes genetic counselors, genetic nurses, or other qualified health professionals. 80–82,84,85,103,106,107,109
In the context of genetic testing, pre- and post-test genetic counseling must be performed.75,95,96,99 In cases where there is uncertainty in the findings, such as a variant of uncertain significance or a variant reclassification, this is of particular importance.100,101
Section 5. Psychological Care
SCD where a genetic cause is suspected has a profound psychological impact on the surviving members of the family. Grief is a normal emotional response to the loss of a loved one. Individuals will grieve differently, and while there is no single trajectory, many will experience disbelief, yearning, anger, sadness, and acceptance.117 After a death, an individual will not return to normal, but rather create a revised meaningful life without the deceased. In a small proportion of bereaved individuals, the initial grief response does not resolve and may result in prolonged grief, or persistent complex bereavement disorder according to the Diagnostic and Statistical Manual of Mental Disorders, 5th Edition (DSM-5).118,119 This occurs in approximately 7% of the general bereaved population,120 and in 21% of first-degree relatives following SCD in the young.121 Posttraumatic stress symptoms can also be experienced by family members. Posttraumatic stress occurs in response to a specific trigger, typically one that threatens one’s own or a loved one’s well-being. It is characterized by avoidance with hyperarousal and intrusive thoughts, including persistent and extreme fear and panic similar to that experienced by family members at the time of the event.122 Posttraumatic stress has been shown in 44% of first-degree relatives following a young SCD.121 Individuals with prolonged grief and/or posttraumatic stress symptoms can benefit from intervention with a clinical psychologist or other appropriately trained clinicians, and there is extensive evidence to support the efficacy of psychological treatments for these conditions in other settings.120 Further, there is greater risk of other psychiatric comorbidities,118 suicide,123 and development of chronic medical conditions.124
Factors associated with poor psychological outcomes have been investigated. One study showed that mothers of the deceased were more likely to report anxiety and depression symptoms.72 In total, 53% of the mothers surveyed reported probable anxiety disorder on average 4 years after the death. In a larger study, after adjusting for factors including relationship to the decedent, those family members who witnessed the death or discovered the decedent’s body had a 3-fold risk of posttraumatic stress symptoms (OR 3.3, 95% CI 1.2–8.7, p = 0.02) and a 4-fold risk of prolonged grief (OR 4.0, 95% CI 1.3–12.5, p = 0.02).121 Given that half report symptoms indicating psychological difficulties, all first-degree relatives should be offered psychological evaluation and treatment. Although the evidence for psychological support is derived from studies investigating SCD where a genetic cause is suspected, it may logically apply to those individuals who have survived SCA and their families (Figure 9).
Figure 9.
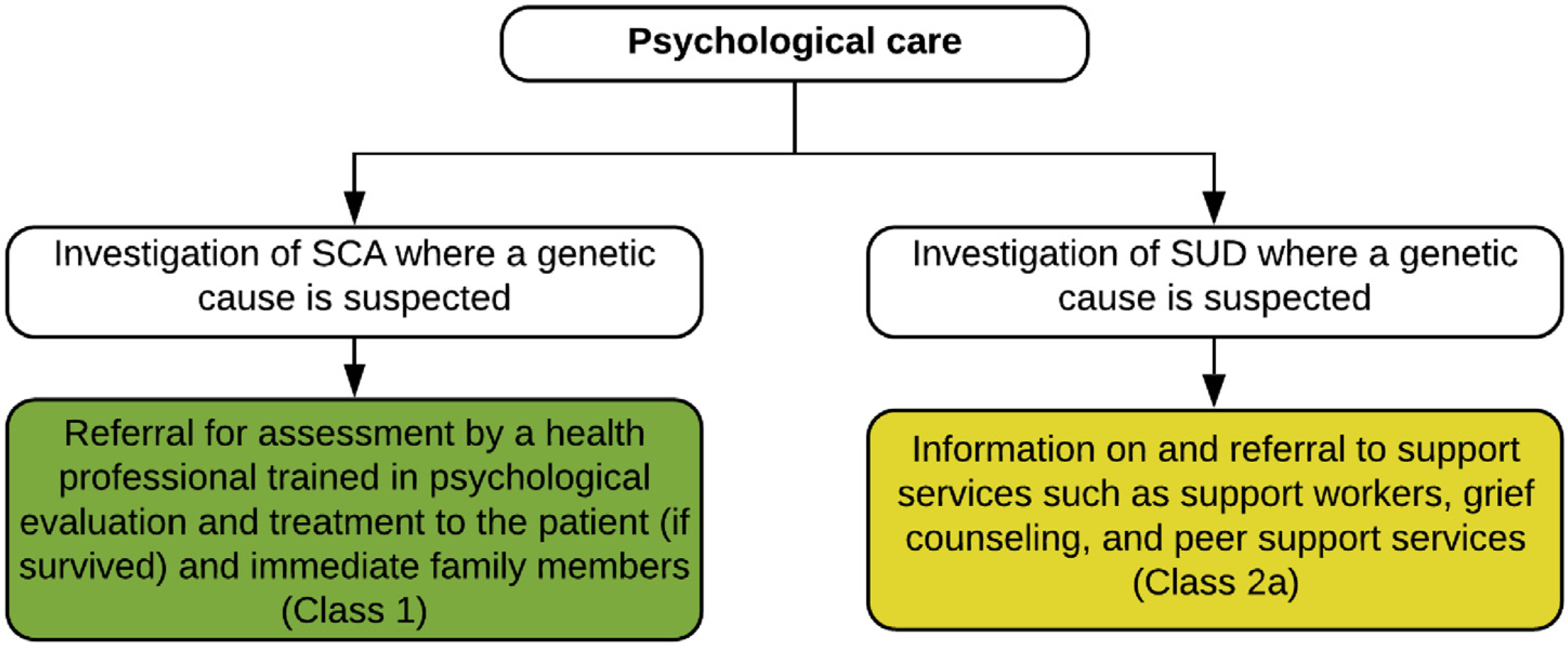
Psychological care following a sudden cardiac arrest (SCA) or a sudden unexplained death (SUD) where a genetic cause is suspected. Colors correspond to the Class of Recommendation in Table 1.
There may be initial reluctance to seek psychological support given community stigma around mental health. Indeed, a recent study investigating families who had experienced a young SCD found that only 12% had sought psychological support, with most of those being self-referrals.125 In discussing options for ongoing psychological support with patients and families, normalizing their response to a significant psychological stressor and describing common symptoms of prolonged grief and posttraumatic stress may reduce any perceived sense of stigma and increase interest in seeking support.
A recent needs analysis of parents who had experienced the SCD of their child (including adult children) found that while medical information and support were the most important need, psychological information and support were the most unmet need.126 Nearly three-quarters reported wanting access to professional counseling or psychological services. Further, many indicated access to genetic testing or understanding the genetic cause to be an important need, highlighting the importance of maintaining realistic expectations regarding the diagnostic yield of postmortem genetic testing with families.101 At present, this is likely not greater than 15%, and there is a high likelihood of uncertain genetic findings especially with increasing gene panel sizes.25,70 A Swedish study of parents whose children died suddenly between 15 and 35 years of age likewise showed a critical lack of information and support in the acute grief stage.127 This included a need for better communication of the postmortem examination process (how long it would take, when they would get results), time with a health professional to discuss the death, and information about the cause of death. There was a lack of psychological support in the immediate aftermath, with many family members seeking their own care, including grief counselors and support groups. The need for support in the early aftermath has been shown to be important in other studies examining suddenly bereaved parents.128,129
Community or peer-based bereavement support groups can also enhance social support.120,130 Peer support programs come in many different forms but always involve people with similar backgrounds providing emotional, social, or practical support to each other.131 Peer supporters draw on their shared experiences to provide empathic understanding, information, and advice to those they are helping. A key aim is to promote hope, recovery from illness or trauma, improved life skills, psychological well-being, and social integration.132 A recent systematic review of peer support services for bereaved survivors of the sudden death of a loved one in multiple settings found evidence of reductions in grief and increased well-being and personal growth among participants, and improved personal growth and positive meaning in life among peer providers.133 There is a current gap in care in addressing psychological support needs of families after the SCD of a young relative.
Recommendations for psychological care
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. In the investigation of SCA where a genetic cause is suspected, it is recommended that referral be offered for assessment by a health professional trained in psychological evaluation and treatment to the patient (if survived) and immediate family members. | 72,121,126,134 |
| 2a | C-LD | 2. In the investigation of SUD where a genetic cause is suspected, provision of information and referral to support services such as support workers, grief counseling, and peer support services can be useful. | 127,131–133 |
Synopsis
The psychological impact to the family following an SCD where a genetic cause is suspected can be significant. Although many family members will navigate their way through this traumatic experience, up to 44% may require additional psychological support from an appropriately trained health professional such as a clinical psychologist. Addressing community stigma around mental health needs to be considered and discussed with families. In addition, support services such as social workers, grief counselors, psychosocial teams, and peer support groups may be useful to many families. Whereas the evidence for psychological support is derived from studies investigating SCD where a genetic cause is suspected, it may logically apply in those families where there has been an SCA.
Recommendation-Specific Supportive Text
1. and 2. A clinical psychologist or appropriately trained health professional includes those equipped to assess and treat trauma; for example, those experienced in delivering cognitive behavioral therapies. While the evidence to date supports a need for psychological support in family members following a young SCD where a genetic cause is suspected, this may likewise be important for relatives of a patient who suffers an SCA.72,121,134 There is a need to train personnel in psychological care for SUD and SCA, as this is an area where the need is not currently met.
Section 6. Investigation of Sudden Death
6.1. Investigation of Sudden Death: History—Personal and Family
Despite being “low-tech” and inexpensive, the history, as a tool for clinical phenotyping, is the essential and fundamental basis of approaching a patient with SCA because it can guide appropriate use and interpretation of other diagnostic modalities. The history should be focused toward both the decedent proband and also the wider family for evidence of other potentially affected members prior to investigations. Surviving family members should be investigated by a multidisciplinary team within a specialist program for cardiovascular genetic disorders with the all appropriate medical, genetic, and psychological personnel and ability for comprehensive investigations2,10 (Figure 10).
Figure 10.
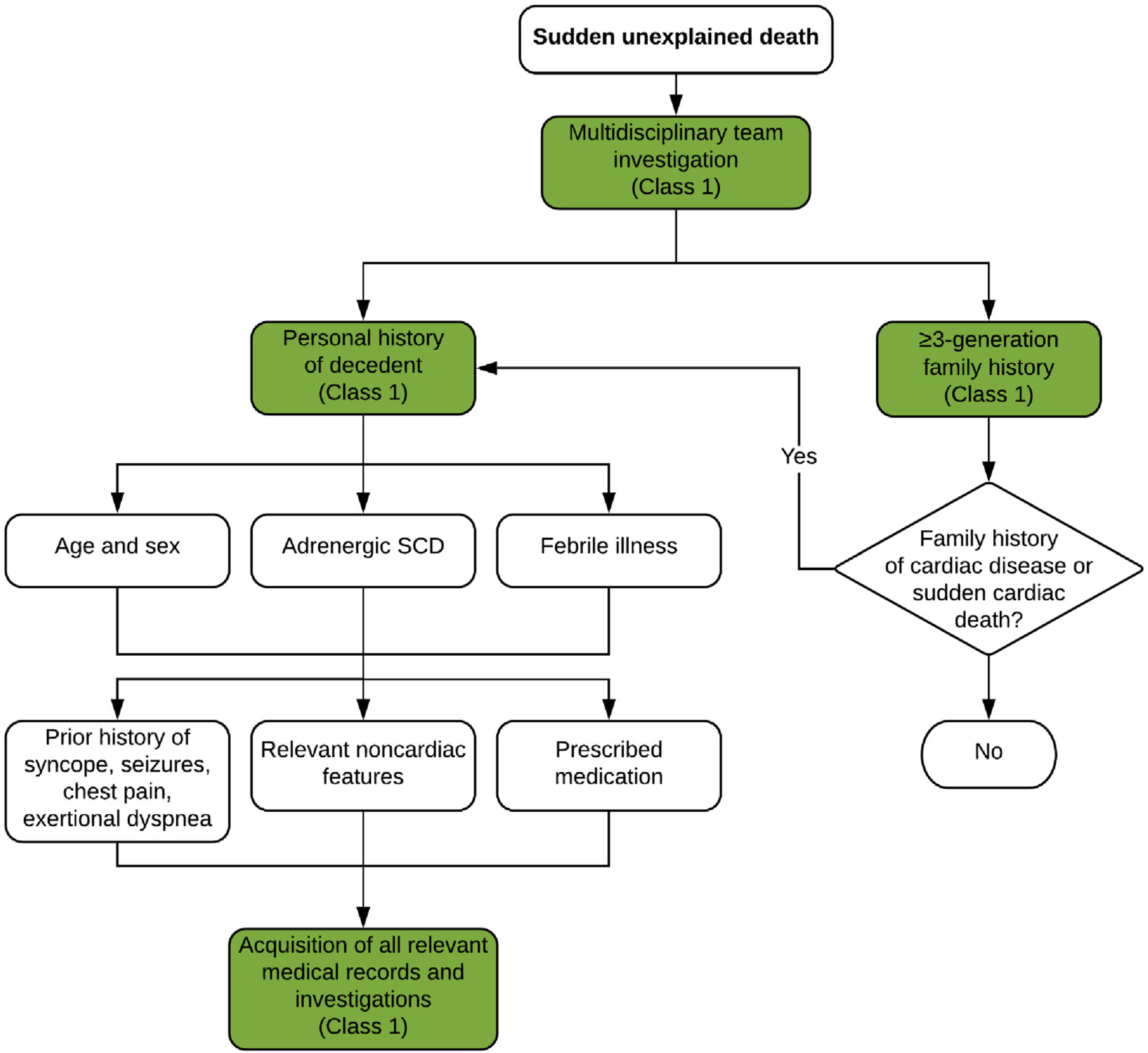
Investigation of sudden unexplained death: personal and family history. Colors correspond to the Class of Recommendation in Table 1. SCD = sudden cardiac death.
The proband age may help define potential etiologies; CPVT and long QT syndrome are typically diseases of the young, whereas coronary artery disease and cardiomyopathies become more common with age (Figure 1). Although most deaths occur at rest or during sleep,25 death during exertion may point to specific etiologies such as CPVT, long QT syndrome type 1, or arrhythmogenic cardiomyopathy. In addition to a detailed prior medical and medication history (including potential drugs of abuse), the decedent’s health in the 24–48 hours preceding death including the presence of any viral prodrome or fever, as well as any prescribed medication, may be relevant. Myocarditis secondary to viral infection may be associated with viral and gastrointestinal symptoms, and both Brugada syndrome and long QT syndrome may be exacerbated by specific pharmacological agents through further inhibition of ion channel function.135 Fever is a well-recognized trigger of ECG changes and arrhythmia in Brugada syndrome136 and in some long QT syndrome subtypes,137 and in young children may be misdiagnosed as febrile seizures.138
Between 18% and 45% of sudden death cases may have experienced prior relevant symptoms, typically palpitations, chest pain, pre-syncope, or syncope, and may have undergone relevant investigations.29,70,139 All medical records relevant to the sudden death etiology should be sought.
Relevant information from the family history should be collected by a health professional with specific experience in cardiovascular genetic disease (preferably a genetic counselor) and by an appropriately trained cardiologist. Symptoms and diagnoses in other family members as well as prior cardiovascular investigations should be sought. Noncardiac findings may be highly pertinent including unexplained epilepsy unresponsive to conventional therapy; skeletal muscle weakness; curled hair and subtle palmoplantar hyperkeratosis/keratoderma (arrhythmogenic cardiomyopathy)140; attention deficit disorder and intellectual disability (CPVT)141; and history of pneumothoraces, vascular disease, and gastrointestinal and uterine rupture (vascular Ehlers-Danlos syndrome).142 Any other deaths or major cardiac events in the family should be recorded including those related to drowning in good swimmers, unexplained motor vehicle accidents, and sudden infant death or late fetal demise. If the SUD was observed, it is useful to collect witness accounts about the events occurring immediately prior to the collapse and during any resuscitation attempts.
Recommendations for investigation of sudden death: personal and family history
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. In the investigation of SUD, an effort should be taken to obtain detailed personal and three-generation family history (as a minimum) with the assistance of a multidisciplinary team, including witness accounts. | 25,29,70,77,81,135,139,143,144 |
| 1 | B-NR | 2. In the investigation of SUD, prior medical records and relevant investigations from the decedent proband and family members should be retrieved. | 25,29,70,77,81,135,139,143–145 |
Synopsis
The personal medical and three-generation family history provides the initial information on which subsequent investigations will be based. Specific features within the wider family may suggest diagnoses and help direct subsequent investigation. The history should be recorded by cardiologists, specialist nurses, and geneticists or genetic counselors experienced in cardiovascular genetic diseases, ideally within the confines of a multidisciplinary program that can address the medical, genetic, and psychological needs of the family (see Section 3).
Recommendation-Specific Supportive Text
The personal and three-generation family history may provide critical information relevant to the etiology of SCD and provide a starting point for further investigations in both the decedent proband and surviving family members.77 Multiple studies show a significant proportion of children and adults experience relevant cardiac symptoms prior to sudden death,10,25,29,70,135,138,139 and some may have sought medical attention and undergone investigations. Available ECGs and cardiac imaging, together with autopsy findings, may allow a diagnosis to be made (or excluded) in the proband who, until family investigations have been performed, is the one definitively affected member of the family. Noncardiac features and symptoms may also provide important diagnostic information.140–142
Further investigation is necessary where sudden death occurs in specific circumstances such as when a cardiac event may have triggered an apparently environmental death. Examples include road traffic accidents with no apparent cause and drowning in competent swimmers.
The presence and associated investigations for other noncardiac conditions should also be evaluated, specifically epilepsy. Failure to identify a neurological etiology or abnormality would suggest seizures may have had a cardiac etiology. Overlap syndromes exist between true neurological epilepsy and long QT syndrome type 2.146,147
6.2. Investigation of Sudden Death: Examination of Premorbid Investigations
Individuals who have succumbed to SUD may have had pertinent investigations prior to their death that aid in the diagnosis of the cause of their SUD. Twelve-lead electrocardiogram (ECG) is the most useful pre-SUD investigation. Although long or short QT interval, spontaneous type 1 Brugada pattern, and early repolarization pattern are associated with fatal arrhythmias due to congenital long QT syndrome, short QT syndrome, Brugada syndrome, or early repolarization syndrome,148–152 many patients with SUD without structural heart disease have a normal or near-normal ECG,145,153 particularly women.154 Additional ECG findings suggestive of arrhythmic syncope include bifascicular block; intraventricular conduction abnormalities (QRS duration >0.12 s); Mobitz I second-degree atrioventricular block and first-degree atrioventricular block with markedly prolonged PR interval; sinus bradycardia (<40 bpm) or slow atrial fibrillation (<40 bpm); nonsustained ventricular tachycardia; pre-excited QRS complexes; negative T waves in right precordial leads or epsilon waves; and left ventricular hypertrophy,5 any of which may indicate potential diagnoses of inherited arrhythmia syndromes such as progressive cardiac conduction defect, familial pre-excitation, arrhythmogenic cardiomyopathy, or hypertrophic cardiomyopathy.4,151 In the general population, premature ventricular complexes (PVCs) are mostly benign; however, some frequent or complex PVCs significantly increase the risk of SCD.155–157 If an ECG is recorded by the AED or EMS just before SCD, features such as J-wave or ST segment elevation (especially if augmented after a long pause) may help in the diagnosis of coronary spasm, early repolarization syndrome, or Brugada syndrome.158,159 Interpretation of ECGs obtained immediately after resuscitation/defibrillation should be performed with great caution (see Section 7.4).
Syncope is a sentinel clinical symptom before SUD and may prompt investigations subsequently useful in making a retrospective diagnosis of the cause of SUD. In particular, the trigger for the syncopal event bears useful information. Ambulatory ECG monitoring during life may provide clues to the cause of SUD and should be sought.
If transthoracic echocardiography, cardiac computed tomography (CT), or cardiac magnetic resonance imaging (CMR) are performed during the patient’s life, detailed review may indicate features of dilated cardiomyopathy, hypertrophic cardiomyopathy, or arrhythmogenic cardiomyopathy.160 If blood or other tissue sample has been taken before SUD, this may be a source of DNA for genetic testing, should there not be a postmortem collection of tissue.8,11,67,15,1 Neurological findings such as developmental delay or seizures thought to be suspicious for epilepsy during life may contribute to a diagnosis of a cardiac channelopathy, such as CPVT or long QT syndrome.141,161,16,2 If a patient with SUD has a cardiovascular implantable electronic device (CIED) implanted, postmortem interrogation of the CIED is useful to determine the cause and timing of SCD.163
Recommendations for investigation of sudden death: examination of premorbid investigations
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. All relevant cardiac investigations, including 12-lead ECGs, echocardiography, CT, CMR, genetic analyses, and ambulatory monitoring recorded before SUD, should be reviewed and analyzed. | 145,148–150,153–158,164 |
| 1 | B-NR | 2. Any blood or DNA sample (eg, blood in EDTA, blood on filter paper card) taken before SUD should be stored for future genetic analysis. | 84,165–167 |
| 1 | B-NR | 3. Neurological events such as seizures suspicious for epilepsy before SUD should be reviewed and studied for a potential cardiac etiology. | 70,146,147,161,162,168,169 |
| 2b | C-LD | 4. ECG information from the AED or ECG monitor recorded around the time of SCD may be useful for review and analysis. | 158,170 |
| 1 | B-NR | 5. Any implanted cardiac electronic device in an individual with SCD should be reviewed and analyzed. | 163,171,172 |
Synopsis
During the investigation of SUD, pertinent investigations performed prior to death can aid in establishing the cause. Although ECG features such as QT interval, type 1 Brugada pattern, and early repolarization may be critical for diagnosis, many ECGs taken during life will be normal. Ambulatory ECG monitoring and cardiac imaging should be sought to provide clues to the diagnosis of SCD. Symptoms attributed to a neurological cause may be re-evaluated, in collaboration with neurologists. Any potential DNA sample before SUD should be stored if tissue is not gained at autopsy. ECG information from the AED, emergency services, or CIEDs may also be useful to determine the cause of SCD.
Recommendation-Specific Supportive Text
Investigations during life may provide clues to the cause of SUD and should be sought to aid in diagnosis.173 These include resting, exercise and ambulatory ECG tracings, and cardiac imaging studies (echocardiography, CT, and CMR).145,148–150,153–158,164
Samples for potential DNA testing taken during life may subsequently prove invaluable should they be the only source of DNA. Although it is a Class 1 recommendation that patients with SUD have an autopsy and material for DNA testing collected (see Section 6.3), it is recognized that sometimes this is not done, making blood or tissue samples taken during life the only remaining source for molecular autopsy.84,165–167 Using such samples requires appropriate consent from family, unless ordered by the coroner. It should be recognized that success varies depending on the storage method, but attempts to gather useful DNA may be worthwhile even from suboptimal sources. Future extraction methods may improve the yield so continued storage is advisable.
Symptoms such as seizures thought to be suspicious for epilepsy during life may in fact be attributable to a cardiac channelopathy when further investigation is done. Other neurological findings such as developmental delay hold significance for diagnoses such as CPVT. Thus, meticulous recording of neurological events during life may lead to a diagnosis in SUD.70,146,147,161,162,168,169
Recordings from emergency services continuous ECG monitoring or from interrogation of AEDs, when available, may provide clues to the etiology of SUD. However, it should be acknowledged that a finding of ventricular fibrillation is often due to this rhythm being a final common rhythm in arrhythmic death, regardless of the initial rhythm causing hemodynamic collapse. Nevertheless, at points such as reinitiation of arrhythmia and glimpses of normal rhythm in between arrhythmia may suggest a specific diagnosis.158,170
The memory function of CIEDs may reveal the initiation pattern of cardiac arrhythmia and aid in the diagnosis of SCD. Therefore, if an SUD victim has a CIED implanted in life, interrogation of this device can provide useful clues in the diagnosis of SCD.163,171,172
6.3. Investigation of Sudden Death: The Postmortem Examination and Imaging
The critical components to the investigation of SUD include examining the circumstances of the death and the autopsy (Figure 11). Identification of SUD relies on the reporting of EMS, police, hospitals, and witnesses. Investigation of a death is determined by the jurisdiction in which the death occurs.14,174,175 Unexpected or unexplained deaths, when the individual was in apparent good health, should be carried out by a trained pathologist who has a thorough knowledge of cardiac pathology.176 Autopsies vary not only by country but also by individual jurisdictions within countries. The autopsy should be comprehensive, examining all organs and conducted in a systematic and objective method with a focus on standardized reporting.2,67,177,178 Cases should be referred to a cardiac pathologist when a cardiac cause is suspected.2,176
Figure 11.
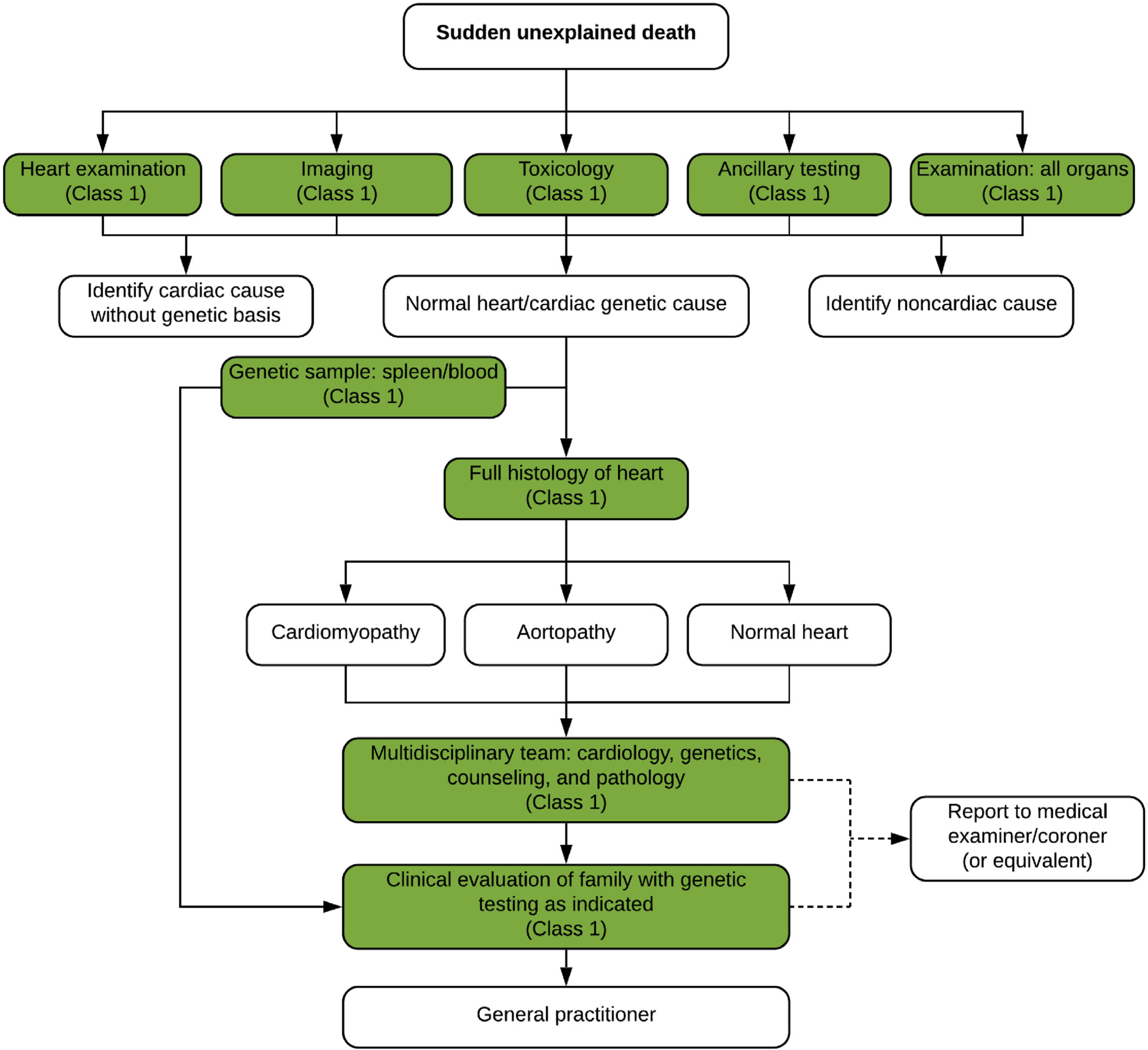
Investigation of sudden unexplained death: the postmortem examination and imaging. Colors correspond to the Class of Recommendation in Table 1.
Imaging includes X-rays and photography. Photography is useful in providing documentation of syndromic features and highlighting individual organ pathology. Postmortem CT and magnetic resonance imaging (MRI) have been shown to be useful179 but are not universally available. Noncardiac causes should be looked for including infection, thromboembolism, tumors, intracerebral lesions, respiratory disease, and abdominal causes such as ruptured abdominal aneurysm. Body mass index should be recorded along with waist circumference.
Ancillary testing should be performed including microbiology/cultures for infectious disease, metabolic screening (particularly in younger children), toxicology, vitreous testing for biochemistry, genetic testing (see Sections 6.4 and 6.5), and other testing as indicated by the autopsy findings. Taking a sample for toxicology is recommended in all sudden unexpected deaths.180
Samples for genetic testing should be saved at the time of autopsy from every sudden death case.2,181 Ideally, two of the following three should be saved: a small piece of fresh frozen heart, a small piece of fresh frozen spleen/liver/thymus, and EDTA blood. If RNAlater (ThermoFisher Scientific, Waltham, MA, USA) or similar reagent to preserve DNA at room temperature is available, fresh tissue can be transported in this to the referral genetic center without need for freezing.
The heart should be examined thoroughly67 and at least 7–10 samples taken for histology. Cardiovascular disease is the leading cause of sudden death in the young and is divided into two major groups: morphologically positive (eg, congenital heart disease, coronary artery disease, and cardiomyopathy) and morphologically normal hearts. Combined with negative toxicology, those with morphologically normal hearts have been labeled as having “autopsy-negative sudden unexplained death” or “sudden arrhythmic death (syndrome) or SAD(S).”182 Samples should always be taken, even from a macroscopically normal heart, as histology may reveal inflammation and cardiomyopathies. Always consider sudden unexpected death in epilepsy (SUDEP) and sudden death in alcohol misuse (SUDAM)183 where clinical history and circumstances are important. Pathologists and clinicians should not overinterpret findings in the heart at autopsy such as nonsignificant coronary artery disease, etc.184
Recommendations for investigation of sudden death: the postmortem examination and imaging
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. An autopsy is strongly recommended in individuals with an SUD. | 14,25,37,175–177 |
| 1 | B-NR | 2. Autopsies for SUD should be comprehensive, including photography, imaging, toxicology, gross examination of all organs, and detailed examination of the brain, heart, and thorax, with histology being essential. | 14,175–177,180 |
| 1 | B-NR | 3. EDTA blood and/or one type of fresh tissue (heart, liver, spleen, skeletal muscle) should be saved at autopsy for SUD and banked at −20°C or −80°C for potential genetic analysis; two sources are ideal, if possible. | 25,70,166,185 |
| 2b | C-LD | 4. Storing frozen myocardial tissue may be considered at autopsy for SUD, as it may aid in assessing the significance of future genetic findings. | 186,187 |
| 1 | C-EO | 5. Findings of an autopsy for SUD should be communicated to the family in a timely fashion in accordance with local legal requirements. | |
| 1 | B-NR | 6. Cases with likely cardiac causes for SUD should be referred to a pathologist with expertise in cardiac disease, as the finding of an abnormal or normal heart is important for family screening. | 176,177,184 |
| 1 | C-LD | 7. When an autopsy for SUD reveals a possible genetic cause, or the heart is normal, then referral for clinical and genetic investigation of the family is recommended. | 80,81,143,188,189 |
Synopsis
A comprehensive autopsy is an essential part of the investigation of SUD and should include collection and storage of tissue suitable for genetic analysis. When the autopsy suggests a possible genetic cause, or no cause and the heart is normal, referral to a multidisciplinary team for further investigation is indicated (see Section 3).
Recommendation-Specific Supportive Text
SUD should have an autopsy done by a trained pathologist.174–176 Studies have shown that autopsies performed by pathologists who have a thorough knowledge of cardiac pathology have a superior diagnostic yield.174–176 In cases where autopsy is not possible (eg, for religious reasons), a full body MRI or CT scan is recommended.179
Samples should be taken for infection and toxicology. Histological sampling of all important organs especially the heart (from multiple sites) is essential even when macroscopically normal.174,175
Blood or tissue suitable for DNA extraction and postmortem genetic testing should be obtained at all autopsies.176 Following the initial investigation, DNA should be extracted and banked if genetic disease is suspected or if the cause remains unknown.2,176 Ideally, lack of cost coverage should not be a reason not to comply with these recommendations.
Frozen myocardial tissue may be useful for subsequent RNA analysis or expression studies of aberrant proteins.2,67
Cause of death should be discussed in a multidisciplinary meeting (see Section 3) and provided by a pathologist to the medical examiner/coroner. The findings and follow-up recommendations should be communicated to the family.178,190,191
Finding of an abnormal or normal heart is important for family screening and directs much of the subsequent investigation (see Sections 6.4 and 6.5).
Autopsy phenotype should be established at a multidisciplinary meeting of pathologist, medical examiner, cardiologist, and clinical geneticist.
6.4. Investigation of Sudden Death: Genetic Evaluation Where the Phenotype Is Known
Genetic evaluation may be appropriate following SCD in two scenarios: most commonly, where the deceased individual is the proband with no prior medical history, or alternatively the deceased is part of a family where diagnosis is established but he/she has not yet undergone genetic evaluation. Initially, pathological examination should be performed by an experienced pathologist to ensure that all cardiac and extracardiac features relevant to the potential diagnosis are recognized (see Section 6.3).
In cases where the deceased is the proband and a postmortem diagnosis is established, identification of a pathological variant may facilitate genetic testing in the wider family evaluation. Genetic testing of DNA from the deceased proband may be performed directly after autopsy or deferred until first-degree family members have been clinically evaluated (Figure 12). As part of familial evaluation, a three-generation pedigree (at a minimum) performed by a practitioner knowledgeable in the genetics of cardiovascular disease (eg, a genetic counselor or specialist nurse) is mandatory and should cover all potentially relevant cardiac and extracardiac features within the family (see Section 6.1). Genetic testing of deceased individuals may not be covered by health insurance in certain countries; in this instance, using a clinically affected family member as the testing proband with confirmatory testing in the deceased may be a more feasible strategy. Clinical and genetic testing in the proband and multiple family members will define segregation of the identified genetic variant(s), adding to the validity of the genetic findings. The yield of genetic testing in cases where a diagnosis of cardiomyopathy is established postmortem is significantly greater than where structural changes are uncertain.192
Figure 12.
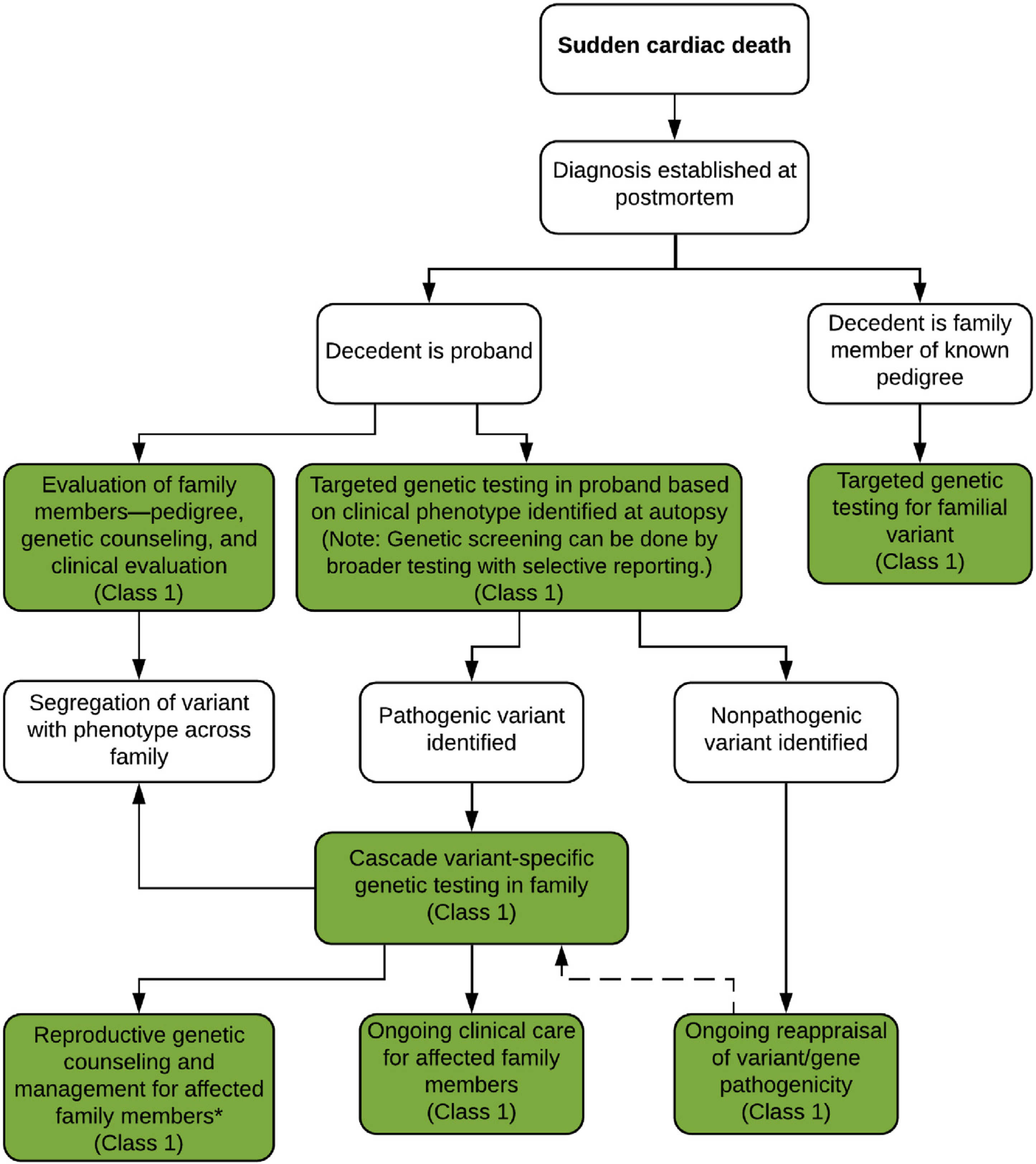
Clinical and genetic evaluation after sudden death where a phenotype is known. Colors correspond to the Class of Recommendation in Table 1. *See Section 8.2.
In cases where no other family members are clinically affected and the deceased proband is an apparently isolated case, genetic testing can be used as evaluation of the single definitely affected individual within the family. Families should be counseled about the expected benefits and potential outcomes of genetic investigations prior to testing. If identified variants are considered likely pathogenic or pathogenic, cascade testing across the family can be considered to identify at-risk individuals with no current clinical features.
In cases where the deceased is part of a family with a prior diagnosis of cardiovascular disease and known pathological variant, confirmatory genetic testing may be performed.
Recommendations for investigation of sudden death: genetic evaluation where the phenotype is known
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. For SCD where the phenotype is suspected to be heritable, genetic testing is recommended to attempt to elucidate the genetic basis and to facilitate the identification of first-degree family members at risk for developing the same disease (cascade testing). | 25,80–82,84,85,143 |
| 1 | C-LD | 2. Genetic testing in the deceased proband with SCD and known phenotype should include only genes with robust evidence of gene–disease association. | 192 |
| 1 | B-NR | 3. In first-degree relatives of a proband with SCD from a suspected heritable cause, phenotype-guided clinical screening is recommended and, where a genetic diagnosis is available, cascade genetic testing should be offered. | 25,80–82,84,85,143,189,193 |
| 1 | B-NR | 4. In families affected by SCD who have undergone genetic testing, periodic re-evaluation of the genetic test results is recommended. | 115,194–201 |
| 1 | C-LD | 5. A genetic diagnosis made in a relative of a proband with SCD should be considered together with the clinical findings. | 70,143,189 |
Synopsis
A postmortem diagnosis following SCD significantly facilitates further clinical evaluation of family members and may provide an explanation for the family as to the underlying etiology. Genetic testing targeted toward the clinical diagnosis and phenotype is an important component of the overall evaluation of both the proband and family and provides additional support to the clinical diagnosis. Further investigations can be performed as clinically indicated. In cases where no other individuals within the family are clinically affected, identification of a definitively pathogenic variant in the proband facilitates cascade genetic testing in family members.
Recommendation-Specific Supportive Text
Genetic testing in the decedent proband is recommended to support the clinical diagnosis and facilitate cascade genetic testing within the family.10,11 The yield of genetic testing is significantly higher when associated with a specific postmortem diagnosis.84,192
If a cardiac phenotype has been identified in the deceased proband, genetic testing should be targeted toward that specific phenotype to maximize the chances of a clinically actionable result and minimize the risk of ambiguous secondary findings. Targeted panels for cardiomyopathy, channelopathies, familial thoracic aortic aneurysm, and familial hyperlipidemia are the preferred option. However, given the limited availability of proband DNA and potential financial implications, broader genetic testing including whole exome and genome with selective reporting based on the phenotype may be considered. Robust evidence for gene–disease association is currently probably only available in the Clinical Genome Resource (ClinGen) expert panels for long QT syndrome, Brugada syndrome, hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy.195,196,202 Wider panels and whole-exome sequencing/whole-genome sequencing share the potential disadvantage of a high burden of variants of uncertain/unknown significance, but in experienced hands (ie, expert centers), this will not lead to unintended follow-up.203
Clinical evaluation for family members of a proband with an evident or likely phenotype can be targeted based on that phenotype, although due to varied expressivity and overlap syndromes should be sufficiently broad to provide a comprehensive cardiovascular evaluation. If a pathogenic or likely pathogenic variant has been identified in the deceased proband in a gene consistent with the clinical diagnosis, genetic counseling and testing should be offered to family members.25,80–82,84,85,143,189,193 Cascade testing for likely pathogenic variants should be done at the discretion of an experienced provider after reviewing the data. In cases with limited supporting data from the family, a more conservative approach may be appropriate.
Serial re-evaluation of variants should be performed based on new phenotype data in the family pedigree, or new data regarding both specific variants and whole genes, the pathogenicity of which may have been up- or downgraded based on contemporaneous evidence.115,194–196,204 Responsibility for re-evaluation is unclear, but the re-evaluation is best carried out in a center of expertise.
Genetic variants identified in deceased probands and subsequently in family members should be correlated with clinical findings to determine segregation patterns within the family.
6.5. Investigation of Sudden Death: Genetic Evaluation Where the Phenotype Is Unknown
SCD may occur in an individual without any prior medical history or medical data. As indicated above, a three-generation pedigree including examination of all potentially relevant cardiac and extracardiac features within the family should be performed by a cardiologist experienced in genetic heart disease or clinical geneticist experienced in cardiology in an effort to optimize the identification of subtle clinical features before defining the cause of death as unknown. Specific triggers (eg, competitive athlete, emotional or physical stress, drug use, swimming, acoustic triggers, seizure) leading to the SCD event may help focus clinical and genetic investigation. Collection of blood and/or suitable tissue for molecular autopsy/postmortem genetic testing is recommended in all victims of SUD.2,10 Local protocols are recommended to ensure proper handling and extended storage of biosamples, as well as processes and consent allowing for contact of families for genetic testing in the future.
Family members of sudden death victims where the phenotype remains unknown should be instructed to request re-evaluation of the index event in case of future developments in the family. Clinical signs or symptoms in first-degree family members leading to a suspected phenotype of a sudden death victim may occur over many years and sometimes skip generations, so an unknown phenotype might prompt more extensive questioning of older family members.
In an SCD case where the phenotype remains unknown after expert evaluation, re-evaluation of family members to assess for new information that may impact diagnosis should be performed periodically, although the yield is low.205 We suggest every 3 to 5 years, but shorter intervals should be considered if there is more than one SCD event in the family. Periodic re-evaluation should be stopped for individuals after age 45 years, unless the decedent died in this age range or new findings emerge.
Three scenarios may trigger arrhythmia syndrome–focused genetic evaluation of SCD even if the phenotype remains unknown: 1) documented arrhythmic death suggestive of an arrhythmia syndrome; 2) specific triggers associated with familial arrhythmia syndromes; 3) young age (<40 years) (Figure 13).
Figure 13.
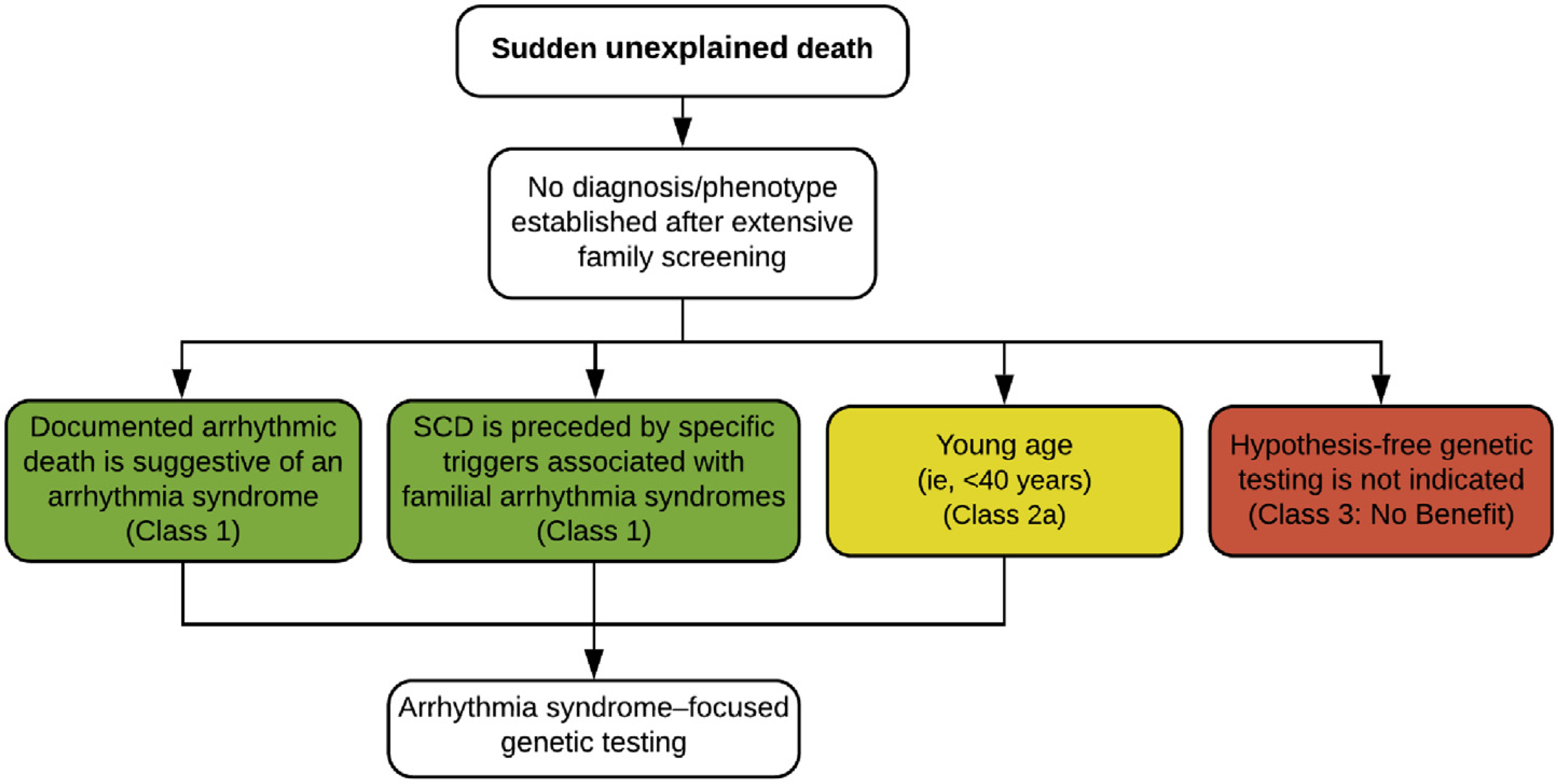
Investigation of sudden death: genetic evaluation where the phenotype is unknown. Colors correspond to the Class of Recommendation in Table 1. SCD = sudden cardiac death.
Families should be counseled about the expected benefits and potential outcomes of genetic investigations prior to testing. When genotyping is performed, the identification of a genetic variant as causal remains challenging and requires reassessing the correct classification or potential reclassification periodically. With rapidly advancing genotyping technologies and the availability of large gene panels, the identification of genetic variants of uncertain/unknown significance becomes more frequent. Medical uncertainty in general elicits a variety of responses from patients. It is important to consider patients’ responses to the ambiguous nature of genetic testing. Medical professionals ordering genetic testing should be prepared for the possibility of their patients’ misinterpretation of such results. Pre-test counseling should include a discussion of the possibility of a variant of uncertain significance and what it would mean for the patient’s care and its potential psychosocial impacts. When a variant of uncertain significance is found, post-test counseling should include additional education and a discussion of the variant’s implications and medical management recommendations based on the results. If identified variants are considered likely pathogenic or pathogenic, cascade testing across the family should be offered to identify at-risk individuals with no current clinical features. Cascade testing should not be performed with variants of uncertain significance; however, careful investigation within a multidisciplinary team (including genetic counseling) may allow eventual reclassification of the variant so that it may then be used for cascade testing (see Sections 3 and 4).
Recommendations for investigation of sudden death: genetic evaluation where the phenotype is unknown
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. In an SUD case at a young age where the phenotype remains unknown after expert evaluation, re-evaluation of first-degree relatives to assess for new information that may achieve diagnosis should be performed every 3 to 5 years (shorter intervals should be considered if there is more than one SCD event in the family) until at least age 45 years. | 25,70,80,85,205,206 |
| 1 | B-NR | 2. In an SUD case where the phenotype is unknown, arrhythmia syndrome–focused genetic testing is recommended if 1) documented arrhythmic death is suggestive of an arrhythmia syndrome, and 2) SCD is preceded by specific triggers associated with familial arrhythmia syndromes. | 25,70,80,84,85,185,205–208 |
| 2a | B-NR | 3. In an SUD case occurring in a patient younger than 40 years where the phenotype is unknown, arrhythmia syndrome–focused genetic testing can be useful. | 25,70,80,84,85,205,206,208,209 |
| 3: No Benefit | B-NR | 4. In an SUD case where the phenotype is unknown, hypothesis-free genetic testing using exome or genome sequencing is not indicated in routine patient care, as this may lead to misinterpretation of genetic variants (specifically variants of uncertain significance). | 210,211 |
Synopsis
Collection and storage of blood and/or suitable tissue for postmortem genetic testing is recommended in all victims of SUD. In a large number of cases, the phenotype underlying SCD remains unknown despite comprehensive evaluation of the victim and their family. In SCD cases where the phenotype is determined as unknown after expert evaluation, re-evaluation should be performed periodically to assess for new information that may impact diagnosis. While hypothesis-free genetic testing is not indicated in cases of SCD where the phenotype remains unknown, arrhythmia syndrome–focused genetic evaluation of SCD is advised if 1) an arrhythmic death is documented suggestive of an inherited arrhythmia syndrome, 2) specific triggers associated with familial arrhythmia preceded the SCD, and/or 3) SCD occurred at young age.
Recommendation-Specific Supportive Text
Genetic diseases may express with reduced penetrance. Hence, a negative clinical screening does not exclude the (silent) presence of a genetic disorder. It is, therefore, reasonable to suggest repeated screening with a time interval between 3 to 5 years until at least age 45 years. Studies demonstrating the yield of clinical screening after the sudden death of a close relative usually report only the result of the first screening.25,70,80,85,205,206 Responsibility for the repeated (infrequent) screening lies in the hands of the individual, but the local team should make sure that he/she is appropriately informed.
In an SCD case where the phenotype is unknown, arrhythmia syndrome–focused genetic testing of the proband should be considered if 1) documented arrhythmic death (such as torsades de pointes arrhythmias leading to ventricular fibrillation) is suggestive of an arrhythmia syndrome, and/or 2) SCD is preceded by specific triggers (eg, competitive athlete, emotional or physical stress, swimming, drug use, acoustic triggers, seizure) associated with familial arrhythmia syndromes.10,25,70,80,85,205,206,212 Collection and storage of blood and/or suitable tissue for postmortem genetic testing is recommended in all victims of SUD irrespective of an identified phenotype at the time of death. Long-term storage of biosamples of SCD victims is recommended in expert centers to allow for genetic testing if indicated at present or in the future.10,25,70,80,85,205,206,212
In an SCD case where the phenotype is unknown, arrhythmia syndrome–focused genetic testing of the proband can be considered if SCD occurred at young age. Testing for cardiomyopathy genes (such as LMNA) has been studied and can increase the diagnostic rate, although it should be recognized that the yield is lower.10,25,70,80,85,205,206,212
Hypothesis-free genetic testing is not indicated in cases of SCD where the phenotype remains unknown. Genetic testing using any range from large unfocused gene panels to whole-exome or whole-genome sequencing in the absence of a clinical phenotype or diagnosis may be considered in the context of a scientific effort but is not recommended for routine patient care and counseling.213,214 The aim of discouraging hypothesis-free testing in clinical settings is to reduce the misinterpretation of genetic variants and their causality, specifically, variants of uncertain significance. A specific problem in this field is the nonuniformity in calling variants across different laboratories.215
Figure 14 summarizes the recommendations from Section 6.
Figure 14.
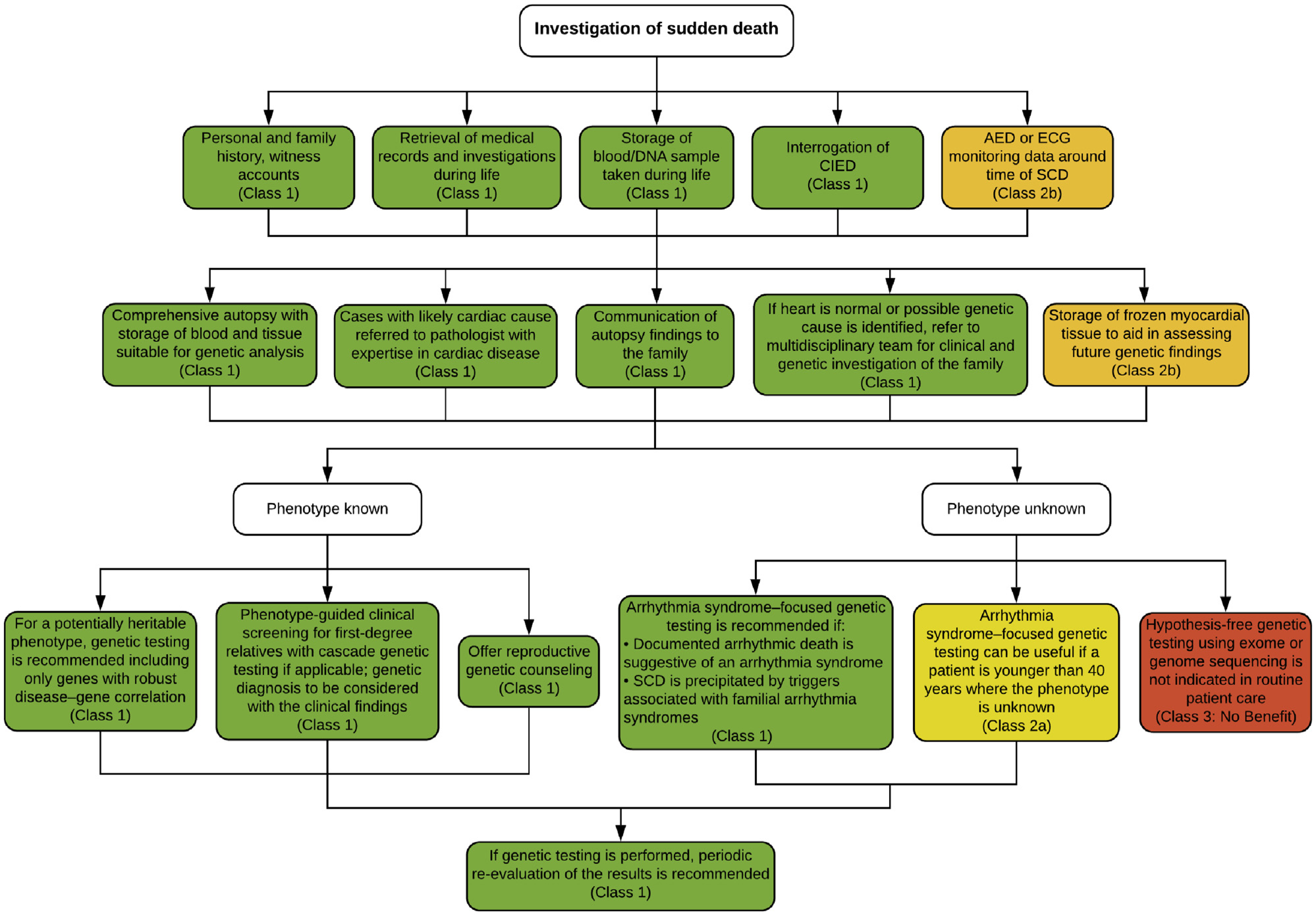
Investigation of sudden death. Colors correspond to the Class of Recommendation in Table 1. AED = automated external defibrillator; CIED = cardiovascular implantable electronic device; ECG = electrocardiogram; SCD = sudden cardiac death.
Section 7. Investigation of Sudden Cardiac Arrest Survivors
7.1. Investigation of Sudden Cardiac Arrest Survivors: History—Personal and Family
When an individual has been resuscitated after SCA, the clinician must try to define the likely underlying cause, similar to that discussed in Section 6.1 focused on SCD. This should include information on age, sex, past medical history, recent symptoms, activity or emotional status at the time of SCA (eg, sleeping, exercising, or emotion), time of onset (eg, morning or night), and environment (eg, public or private location), exposure to medicinal or recreational drugs (particularly those that block potassium or sodium channels) or alcohol, and a detailed family history of three generations at least.
To focus the history taking, one must consider the differential diagnosis of SCA, which is similar to that discussed in Section 6.1. If the SCA was observed, a description of the event by the observer can add useful information. All records of the primary event, including initial rhythm recordings and details of the resuscitation, should be collected in addition to any prior ECGs and imaging studies. A family history of heart disease, syncope, or sudden death may point to a genetic cause. A study by Waddell-Smith et al.77 showed that while inpatient cardiology teams identified a familial condition in only about 8% of cases, nurses trained in taking a family history detected a familial condition in 32% of cases. Thus, although most patients with SCA will be in a nonspecialty environment at first, ideally specially trained members of the cardiology and genetic counselor team with experience in genetic heart disorders should be utilized to elicit relevant details of the family history. Practical educational assistance can be found at https://www.primarycaregenetics.org.
Recommendations for investigation of SCA survivors: personal and family history
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. In the investigation of an SCA survivor, detailed personal and three-generation family history should be taken with the assistance of a multidisciplinary team, including witness accounts. | 77 |
| 1 | B-NR | 2. All possible details surrounding an SCA event should be sought, including patient’s recollection, witness accounts, and medical records. | 216–220 |
Synopsis
Observational studies have demonstrated that the cause of SCA can be determined in a substantial proportion of patients. Historical features, especially age, coronary risk factors, symptoms, activity at the time of SCA, exposure to drugs, and family history frequently provide important clues to the diagnosis and point the way to further investigation.
Recommendation-Specific Supportive Text
In a study of 37 patients with history of SCD, cardiomyopathy, or ventricular tachycardia, a family history obtained by specially trained personnel was far more likely to elicit a history of inherited cardiac disease than one obtained by an inpatient cardiac team.77
A detailed history is a crucial component of diagnosing the cause of SCA. Studies utilizing a comprehensive, systematic approach including history, physical examination, ECGs (eg, 12-lead ECG, treadmill, 24-hour Holter, signal-averaged ECG, if needed), cardiac imaging (eg, coronary angiography, echocardiogram, CMR, CT), provocative testing, electrophysiological study, cardiac biopsy, and genetic testing have shown that a diagnosis can be established in a substantial proportion of survivors of SCA.216–219
7.2. Investigation of Sudden Cardiac Arrest Survivors: Examination
The next step in cardiac evaluation of SCA survivors is physical examination. The main purpose is to identify signs of syndromic and nonsyndromic diseases that can be associated with SCA. For example, obesity and/or the presence of xanthomata may indicate an increased likelihood of premature coronary atherosclerosis.221,222 Syndromic features that may be relevant to genetic disorders include woolly hair and palmoplantar keratoderma (arrhythmogenic right ventricular cardiomyopathy),140 joint contractures (Emery-Dreifuss muscular dystrophy),223 muscle weakness and atrophy (lamin A/C and desmin cardiomyopathies, Triadin knockout syndrome),224 micrognathia, syndactyly, clinodactyly (Andersen-Tawil and Timothy syndromes),224 chest and limb deformities, and tall stature (Marfan syndrome).225,226
Fever, hypothermia, dehydration, and signs of drug abuse may be detected on physical examination, although these signs may be confounded by neurological impairment after cardiac arrest. These factors can trigger life-threatening arrhythmia in genetic heart disease. For example, fever has been associated with malignant arrhythmias in 6% of patients with cardiac arrest and Brugada syndrome.136
Cardiac murmurs can raise the suspicion of left ventricular outflow tract obstruction, mitral mid-systolic click (valve prolapse) with or without regurgitation, and Ebstein anomaly. Signs of pulmonary edema and hepatosplenomegaly can be detected in patients with severe systolic myocardial dysfunction. Importantly, examination findings may be affected by the SCA event and evolve during a hospital stay, requiring repeated physical examination to determine whether findings are related to the cause of SCA or the effect of SCA.
Recommendation for investigation of SCA survivors: examination
Synopsis
There are no data describing the usefulness of physical examination in resuscitated SCD. However, it is a first basic step in the diagnostic process that will focus subsequent complementary investigations.
Recommendation-Specific Supportive Text
7.3. Investigation of Sudden Cardiac Arrest Survivors: Baseline Investigations
In most emergency settings, a patient resuscitated from cardiac arrest in whom myocardial infarction is suspected will have undergone a coronary reperfusion strategy to treat acute occlusion. It is critical to obtain blood tests (cardiac enzymes, inflammatory markers, glucose, serum electrolytes, and white blood cell count) and pertinent toxicological analysis at presentation. The latter may include testing for drugs of abuse such as ethanol, opiates, and stimulants, as well as levels of prescribed medication that may prolong QT interval/QRS duration or cause respiratory depression.228 While assisting in the acute management of a resuscitated patient, these results will also help to differentiate acute myocardial injury as a cause of cardiac arrest (eg, ischemia without clear evidence of coronary occlusion or myocarditis) and pick up other reversible causes such as drug overdose, electrolyte imbalance, or endocrine and metabolic disorders. Retention and storage of suitable blood samples on patient arrival in the emergency department will allow subsequent diagnostic evaluation including DNA extraction and analysis in a patient who dies prior to diagnosis or for later clinical and family review. In some cases, this may be the only opportunity to obtain genetic material for analysis.
In an OHCA, the use of AEDs is ever more widespread and increases survival.229 Sensitivity for the diagnosis of cardiac rhythm at the time of arrest is about 99%.230 Therefore, routine inspection of data from AED recordings may improve the quality of diagnosis (see Section 6.2). The underlying rhythm of cardiac arrest may provide information on the arrhythmogenic mechanism, assist in diagnosis, and eventually indicate any misdiagnosis of rhythm.231 Any ECG tracings from emergency services, as well as recordings from interrogation of CIEDs or wearables can also contribute to diagnosis.163
The 12-lead ECG in sinus rhythm or during arrhythmia recurrence is fundamental to the diagnostic investigation and should be repeated daily during recovery.216 It may support diagnoses of primary electrical disorders, pre-excitation, and heart muscle diseases. However, abnormalities of cardiac conduction and repolarization may result from myocardial injury during the cardiac arrest and patients undergoing post-arrest hypothermia protocols may have transient ECG changes including QT prolongation and J point elevation that should be interpreted with caution. Information about electrolyte levels, drug prescription, and body temperature should be added to the ECG to prevent misinterpretation of such ECG abnormalities. A high precordial lead ECG is an inexpensive tool to increase detection of Brugada syndrome pattern.184,232–235
In addition to standard ECG, a signal-averaged ECG may demonstrate late potentials. Two or more abnormalities in the absence of a prolonged QRS duration (≥110 ms) on the standard ECG is a minor diagnostic criterion for arrhythmogenic cardiomyopathy and suggests ventricular depolarization abnormality.4
Continuous heart rhythm monitoring is recommended during hospitalization due to the transient nature of some arrhythmias. Recording the onset (including pause-dependent or tachycardia-associated initiation) and late and short-coupled ventricular ectopics as triggers for torsade de pointes, polymorphic ventricular tachycardia, or ventricular fibrillation will elucidate cardiac arrest mechanism and likely diagnosis.236 Evidence of dynamic ST elevation associated with chest pain may also indicate likelihood of coronary vasospasm.216
Echocardiography is the screening tool of choice for structural heart disease, although early myocardial dysfunction may be present after cardiac arrest and, if present early, the test should be repeated later during the patient’s convalescence. CMR allows detection of inflammatory diseases, such as myocarditis and sarcoidosis, through recognition of subepicardial edema. Identification of an inflammatory etiology is important, as it may be self-limiting or treatable. If sarcoidosis is suspected, then positron emission tomography–CT scanning may be indicated.237 The presence of subendocardial edema would suggest ischemic injury.238 Late gadolinium enhancement indicates chronic fibrosis, permitting detection of a cardiomyopathic etiology, and can also contribute to the diagnosis of mitral valve prolapse associated with a risk of SCD.239,240 Coronary imaging (at any age) will be important to exclude coronary artery disease not investigated at presentation as an emergency and ensure that an anomalous coronary circulation or coronary dissection is not missed.241 This may be by cardiac catheterization or by CT coronary angiography.242 Coronary angiography will only be required in select pediatric and young survivors.
Patients in whom the cause of their SCA remains undiagnosed may require periodic re-evaluation of the above investigations, as features may develop later that point to a cause of their SCA (similar to Section 6.5, Recommendation 1).
Recommendations for investigation of SCA survivors: baseline investigations
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Blood samples for electrolytes, toxicology, and EDTA blood stored for future genetic testing are recommended for all SCA survivors on admission to hospital. | 228 |
| 1 | B-NR | 2. Retrieval of recordings from CIEDs and wearable monitors is recommended for all SCA survivors. | 243–245 |
| 2b | C-LD | 3. Retrieval of recordings from AEDs and ambulance services may be useful for all SCA survivors. | 158,170 |
| 1 | B-NR | 4. Recording of 12-lead ECGs during sinus rhythm and, if possible, during arrhythmia, is recommended for all SCA survivors. | 85,150,153,216,246 |
| 1 | C-LD | 5. A high precordial lead ECG is recommended in all undiagnosed SCA survivors to increase detection of a type 1 Brugada ECG pattern. | 189,232–235 |
| 1 | C-LD | 6. Continuous ECG monitoring is recommended for all SCA survivors during the initial hospital stay. | 85,216,246 |
| 2b | C-LD | 7. A signal-averaged ECG may be useful in SCA survivors to aid in the diagnosis of arrhythmogenic cardiomyopathy. | 247 |
| 1 | B-NR | 8. Echocardiography is recommended for evaluation of cardiac structure and function in all SCA survivors. | 85,216 |
| 1 | B-NR | 9. CMR with late gadolinium enhancement is recommended for evaluation of acute or chronic myocardial disease in SCA survivors without a clear underlying cause. | 238,248 |
| 2a | B-NR | 10. CMR can be useful for evaluation of acute or chronic myocardial disease in SCA survivors, when the etiology is primary electrical or there is evidence for acute cardiac ischemia. | 238,248 |
| 1 | B-NR | 11. Coronary imaging is recommended in all adult SCA survivors, to exclude coronary artery disease, dissection, or anomalies not considered fully at first presentation, and in select younger cases. | 241 |
Synopsis
Systematic clinical testing is paramount in SCA survivors. This includes blood testing, toxicology, ECG, signal-averaged ECG, high precordial lead ECG, continuous ECG monitoring, echocardiography, and coronary imaging. If the diagnosis remains elusive and cardiac arrest is deemed unexplained, then CMR is important to identify subtle forms of cardiomyopathy or acquired structural disease.
Recommendation-Specific Supportive Text
The usefulness of blood testing and toxicology is by consensus, and a diagnostic role is unquestionable.2,10,228 Viral studies may be useful, but no systematic evidence is available as yet.249 A potential role for biomarkers specific for one of the arrhythmia syndromes is anticipated.250,251
Results of a forensic study indicate the value of postmortem CIED interrogation to define the cause and timing of death more accurately and to detect potential CIED-related safety issues. CIED interrogation in unexplained deaths clarified the manner of death in 60.8% of the cases including cardiac and nonarrhythmic death and device concerns.163
Sensitivity for the diagnosis of cardiac rhythm via AED at the time of arrest is about 99%.230 However, the AED seldom catches the initial rhythm of cardiac arrest and therefore may not contribute to the etiology of SCA.
Primary electrical disorders and specific cardiomyopathies may be detected by conventional ECG.2,10,216,252 ECG findings in the immediate aftermath of a cardiac arrest, other than ST-segment elevation indicative of an acute coronary syndrome, may, however, have poor diagnostic accuracy.253,254 These could be caused by abnormal repolarization following electrical cardioversion,255 metabolic and electrolyte abnormalities, or even subarachnoid hemorrhage.256 Therapeutic hypothermia may lead to misleading ECG changes such as prolongation in PR, QRS and QT intervals, and J point elevation.257,258 Interpretation of ECGs obtained immediately after resuscitation/defibrillation should be performed with great caution.
Although there are no data describing directly the value of high precordial lead ECGs in SCD survivors, there is ample evidence of an increased yield of the type 1 Brugada ECG pattern.189
Cardiac monitoring during short-term follow-up demonstrates an arrhythmogenic mechanism of SCD in some registries.216,236,246
Signal-averaged ECG is part of the Task Force Criteria for arrhythmogenic right ventricular cardiomyopathy.4 Signal-averaged ECG has been proposed as useful in other conditions (ie, Brugada syndrome), although systematic evaluation has not been performed.
Echocardiography is a valuable screening tool for detection of arrhythmogenic cardiomyopathy and other structural abnormalities useful in elucidating the cause of SCA.85 Patients with functional abnormalities on initial echocardiogram should have this test repeated after recovery, to allow for the effects of the SCA itself and drugs used around the time of the arrest to wear off.
9. and 10. The utility of CMR has been evaluated in a series of studies involving survivors of unexplained arrest and has repeatedly been shown to provide significant incremental diagnostic value. A study of 137 individuals with unexplained aborted cardiac arrest found that CMR provided a diagnosis or identified an arrhythmic substrate in 76% of individuals, including an infarct pattern suggestive of occult myocardial infarction in 44%. Notably, the presence of late gadolinium enhancement, reflective of myocardial fibrosis, was associated with a 6.7 hazard ratio (p < 0.001) of recurrent arrhythmic events on multivariate analysis.248 The presence of subendocardial edema would suggest ischemic injury even when initial coronary imaging excludes significant obstruction. Coronary vasospasm and dissection might be misdiagnosed, and coronary re-evaluation may then be reconsidered.238 The frequency of occult infarcts is not insignificant,248 although the risk of SCD in patients with myocardial infarction and nonobstructive coronary arteries is low.259 For Recommendation 10, primary electrical disease is not referring to an established diagnosis of long QT syndrome or CPVT, where MRI is unlikely to be of use.
Coronary artery disease is the leading cause of SCD in adults and might be treatable. Furthermore, coronary dissection and anomalies may also be relevant in this age group as well as in younger patients.
7.4. Investigation of Sudden Cardiac Arrest Survivors: Provocative Testing
Once a cardiac arrest survivor has undergone initial thorough baseline evaluation, most overt acquired or genetic etiologies will have been diagnosed. However, concealed disorders may be uncovered by provocative maneuvers such as lying to standing ECGs, exercise ECG testing, epinephrine challenge, sodium channel blocker challenge, or ergonovine and acetylcholine testing. Some may even be employed in a resuscitated cardiac arrest survivor who is unlikely to survive due to neurological injury.
Exercise testing may uncover ventricular arrhythmia relevant to the cause of cardiac arrest. For example, evidence of monomorphic ventricular tachycardia arising from the right ventricle is part of Task Force Criteria for diagnosis of arrhythmogenic right ventricular cardiomyopathy (arrhythmogenic cardiomyopathy).260–262 Exercise may also uncover concealed epsilon waves or even a type 1 Brugada ECG pattern.263,264 The generation of bidirectional ventricular ectopy or tachycardia and/or polymorphic ventricular tachycardia in the absence of ischemia, structural disease, or digoxin toxicity is typical of CPVT265 and has been evaluated in the Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER) protocol.216 Abnormal dynamics of repolarization in response to challenges may also inform the likelihood of underlying long QT syndrome. To this end, maximum QT prolongation (QT stretch) and T-wave morphology changes during lying to standing and then return to baseline heart rate (QT stunning) may be useful markers, although they are less specific in children.266–268 These discriminate well in genotyped families and may add to diagnostic utility, but evaluation in unexplained cardiac arrest survivors has not been undertaken.269 The recovery phase of exercise may also reveal readily measurable QT prolongation and T-wave abnormalities and has been validated in families with long QT syndrome for prediction of genotype270,271 and in cardiac arrest survivors216 such that a QTc >480 ms at 4 min of recovery forms part of the long QT syndrome risk score.272
Epinephrine challenge with ECG monitoring has been advocated as an alternative to exercise testing for the diagnosis of long QT syndrome and CPVT, particularly where the patient is unable to exercise. In long QT syndrome, QT prolongation and secondary T-wave changes have been able to discriminate LQT1 and LQT2 patients from unaffected family members even though they have normal baseline QTc intervals.273,274 The test has been assessed in unexplained cardiac arrest survivors and has suggested a low specificity for long QT syndrome.275 The finding would be unlikely to provide a secure diagnosis in isolation and was proposed as useful in association with exercise testing and genetic testing. Indeed, in normal subjects, pharmacological sympathetic stimulation does produce significant prolongation of QTc.276 Epinephrine testing has also shown some diagnostic utility for CPVT in cardiac arrest survivors by inducing ventricular ectopic activity, bidirectional couplets, and ventricular tachycardia.275 However, there is uncertainty as to the ideal cutoff for epinephrine-induced arrhythmia, and the diagnostic sensitivity compared to exercise testing in CPVT families is low.277 Isoproterenol challenge for the diagnosis of arrhythmogenic right ventricular cardiomyopathy has also been advocated but has not been tested by other groups or in the unexplained cardiac arrest survivor without overt phenotype.278
Sodium channel blocker challenge (ajmaline, procainamide, flecainide, and pilsicainide) has been used extensively for investigating the possibility of Brugada syndrome in cardiac arrest survivors, although these are mainly reported in series of patients with a strong suspicion of Brugada syndrome279–281 rather than in unexplained cardiac arrest survivors.282 The use of leads V1 and V2 in the second and third intercostal space or high right precordial ECG leads during provocation increases the diagnostic yield.233,283 In the CASPER registry, there was a yield from procainamide challenge,284 although this may underestimate the true burden, as different sodium channel blockers have different potencies for inducing the type 1 Brugada ECG pattern. For example, ajmaline is associated with an odds ratio of 8 for inducing the type 1 Brugada ECG pattern compared with procainamide285 and a 4% yield in a small group of “healthy” controls.286 Furthermore, while recent consensus guidelines would give a definite diagnosis to a cardiac arrest survivor with a type 2 or 3 pattern converting to a type 1, the implication of a drug-induced type 1 pattern without a baseline type 2 or 3 is not addressed.152 Yet cardiac arrest survivors from CASPER without a type 2 or 3 pattern had positive procainamide challenges.284 Other tests such as the full stomach test have been proposed but have not been taken up in general.287
Different approaches for provocation of an underlying repolarization abnormality have been employed including drug challenge with quinidine and sotalol and mental stress tests.288–291 These may offer utility in the future but have not been tested in the cardiac arrest survivor.
Coronary vasospasm, while a recognized cause of cardiac arrest, may not be picked up clinically at presentation.292 Ergonovine or acetylcholine challenge has been proposed as a Class 1 indication by recent guidelines293 and was employed selectively in the CASPER experience.216 Hyperventilation has also been used as a diagnostic test.294 Recent experience from the Paris Sudden Death Expertise Center investigators suggests that pharmacological challenge is useful for diagnosis and could be better employed.295 Nonetheless, there are few centers with extensive experience in the use of the test, and there is a possibility of false-positive findings in the cardiac arrest survivor population.
Adenosine challenge has been used to unmask pre-excitation that may otherwise be missed.296,29,7 In the absence of other causes, it will indicate the need for electrophysiological study to evaluate the risk of the accessory pathway (rapidity of antegrade conduction) followed by ablation therapy. However, electrophysiological study is not routinely included in the workup of unexplained cardiac arrest.8 Indeed, previous consensus guidelines proposed a Class 3 indication when assessing a suspected primary electrical disorder.8 It does not add additional diagnostic or prognostic value unless there is evidence to indicate otherwise. For example, pre-excited atrial fibrillation, bundle branch re-entrant ventricular tachycardia, and rapid supraventricular tachycardias that degenerate into ventricular fibrillation have all previously been described as culprits requiring an invasive approach to diagnosis and curative ablation therapy.298,29,9 Electroanatomic voltage mapping of the right ventricle is a discretionary tool that may be considered to detect evidence of subclinical arrhythmogenic right ventricular cardiomyopathy.300 More recently, extensive endocardial and epicardial mapping of unexplained cardiac arrest cases has been employed to identify cases with either Purkinje triggers and/or subtle depolarization abnormalities that may be suitable for ablation therapy.301
Recommendations for investigation of SCA survivors: provocative testing
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Exercise testing is recommended in all undiagnosed SCA survivors to induce arrhythmias that may support the diagnoses of arrhythmogenic cardiomyopathy and CPVT and to evaluate dynamic depolarization or repolarization features that may support the diagnoses of Brugada syndrome, arrhythmogenic cardiomyopathy, and long QT syndrome. | 216,220,263–265,270,271,302,303 |
| 2a | B-NR | 2. Lying to standing ECGs can be useful in SCA survivors for the diagnosis of long QT syndrome, but must be interpreted with caution in children. | 266–270 |
| 2b | B-NR | 3. Epinephrine challenge may be considered for the diagnosis of long QT syndrome and CPVT, in those unable to exercise. | 273–275,277,304 |
| 1 | B-NR | 4. Sodium channel blocker challenge with standard and high precordial ECG leads is recommended for the diagnosis of Brugada syndrome in undiagnosed SCA survivors with suggestive clinical characteristics, including a type 2 or 3 Brugada ECG pattern. | 233,273–275,277,286 |
| 2a | B-NR | 5. Sodium channel blocker challenge with standard and high precordial ECG leads can be useful for the diagnosis of Brugada syndrome in SCA survivors where no other disorder has been identified. | 233,279–282 |
| 2b | B-NR | 6. Ergonovine, acetylcholine, or hyperventilation testing when performed in experienced centers may be considered for the diagnosis of coronary vasospasm as the cause of SCA in a survivor where no other disorder has been identified. | 216,294,295 |
| 2b | C-LD | 7. Adenosine challenge may be useful for the unmasking of ventricular pre-excitation and therefore the diagnosis of rapidly conducted atrial arrhythmia as the likely cause of SCA in a survivor where no other disorder has been identified. | 297 |
| 2a | C-LD | 8. An electrophysiological study can be considered if bundle branch re-entrant ventricular tachycardia, pre-excited atrial fibrillation, or supraventricular tachycardia are suspected in an SCA survivor. | 298,299 |
| 2b | C-LD | 9. Electroanatomic right ventricular voltage mapping may be considered for detection of subclinical arrhythmogenic cardiomyopathy in an SCA survivor where no other disorder has been identified. | 300 |
| 2b | C-LD | 10. An electrophysiological study may be considered in an SCA survivor where no other disorder has been identified to evaluate potential underlying substrate. | 301 |
Synopsis
Concealed Brugada syndrome, long QT syndrome, CPVT, arrhythmogenic cardiomyopathy, pre-excitation, and coronary vasospasm may be uncovered by provocative maneuvers in the cardiac arrest survivor whose cause of cardiac arrest remains unknown after baseline clinical, ECG, and imaging investigations. Exercise ECG testing and sodium channel blocker challenge appear to offer most potential utility, whereas lying to standing ECGs; epinephrine, isoproterenol, and adenosine challenge; and hyperventilation, ergonovine, and acetylcholine testing may be considered in specific patients. Electrophysiological study and electroanatomic mapping may be useful to provide patient-specific insights into the mechanism of cardiac arrest and offer therapeutic options but should be avoided in the routine investigation of channelopathy. However, data in general are limited to case reports or case series, some with validation cohorts, and there are no randomized studies.
Recommendation-Specific Supportive Text
Exercise testing is a versatile, straightforward, and readily available test that may yield diagnoses due to the arrhythmic challenge or effects on depolarization and repolarization. As such, it should be a standard part of the investigative armamentarium.216,220,263–265,270–272,302,303
Lying to standing ECGs may offer some insights into the likelihood of long QT syndrome, but in isolation a positive result may not fulfill diagnostic criteria and therefore may only complement the evaluation.266–270
Similarly, epinephrine challenge has low specificity for long QT syndrome and may be insensitive for CPVT. In isolation, a positive result may not fulfill diagnostic criteria and therefore may only complement the evaluation.273–275,277,304 There are risks to this test, both in inducing ventricular arrhythmia and in false diagnosis.
Sodium channel blocker testing has a clear role to play in evaluating the cardiac arrest survivor, although the implication of a positive result in patients with or without a high prior likelihood of Brugada syndrome is unclear, eg, male vs. female, cardiac arrest during sleep vs. exercise, type 2 or 3 ECG pattern vs. no Brugada pattern at baseline. This reflects the lack of a gold standard for the diagnosis of Brugada syndrome, and therefore the utility of sodium channel blocker challenge in patients without a clear prior likelihood of Brugada syndrome is less certain and merits a lower utility in the evaluation cascade.233,273–275,277,286 Furthermore, sodium channel blocking agents differ in their sensitivity and specificity for inducing ECG changes and availability varies worldwide (see Section 7.4 text).285
The utility of ergonovine or acetylcholine challenge or hyperventilation testing in all cardiac arrest survivors is unclear, as there are no studies of systematic testing in all cardiac arrest survivors. In particular, the specificity of the test is unknown, especially in the cardiac arrest survivor population. It can, however, lead to diagnoses when employed in a protocol and therefore should be considered as part of the armamentarium until more evidence is available.216,293–295
Adenosine challenge has not been tested systematically in cardiac arrest survivors, but there are limited data suggesting that it will uncover concealed pre-excitation that may cause cardiac arrest in the setting of rapidly conducted pre-excited atrial tachyarrhythmias.296,297
There are limited case series describing the use of electrophysiological study in diagnosis and treatment of pre-excited atrial arrhythmias and bundle branch re-entrant ventricular tachycardia.298,299
Studies of endocardial mapping report a higher sensitivity for detection of arrhythmogenic cardiomyopathy, but this has not been explored in SCA survivors.300 Sensitivity and specificity are unknown, particularly in patients where no other tests are abnormal.
Extensive endocardial and epicardial mapping of unexplained cardiac arrest cases has been employed to identify cases with either Purkinje triggers and/or subtle depolarization abnormalities that may be suitable for ablation therapy.301
7.5. Investigation of Sudden Cardiac Arrest Survivors: Genetic Evaluation
Although most SCA survivors will have an indication for an implantable defibrillator for secondary prevention of a cardiac arrest,6 genetic evaluation may influence final diagnosis, treatment recommendations, and family screening (Figure 15). In some cases, genetic evaluation may enable therapy specific to the disease mechanism. Recent technological advances in genetic evaluations, establishment of reference databases of genetic variants, systematic annotation of causal genes,305 and standardization of variant interpretation306 have enabled efficient and comprehensive genetic assessment. However, the pace of genetic discovery and variant interpretation is evolving rapidly, creating a complex landscape surrounding genetic evaluation of SCA survivors.
Figure 15.
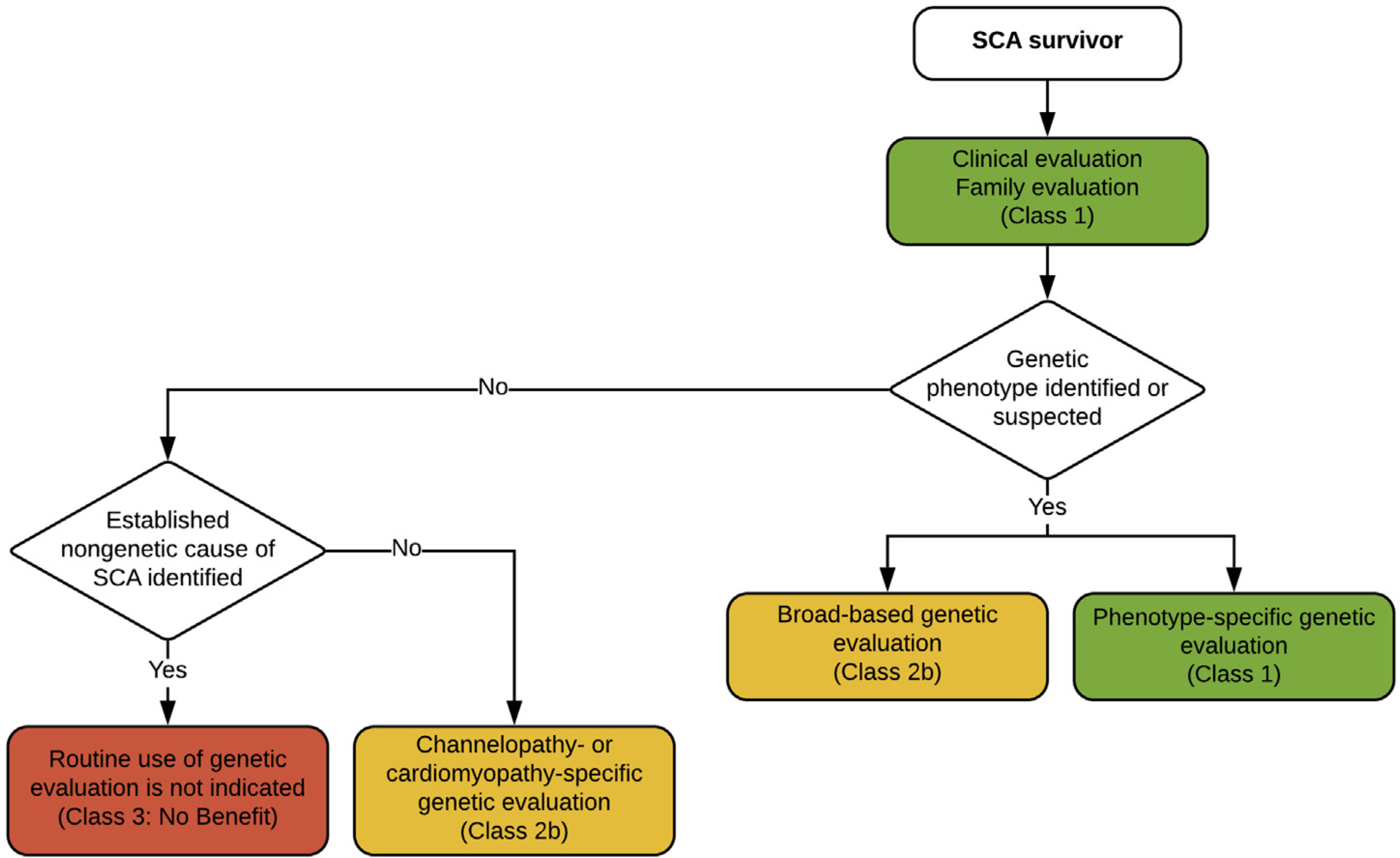
Investigation of sudden cardiac arrest (SCA) survivors: genetic evaluation. Colors correspond to the Class of Recommendation in Table 1.
The decision to pursue genetic evaluation is an individualized one in which the patient, with proper (clinical and genetic) counseling, must weigh the benefits, limitations, and personal and familial implications. The genetic basis of SCA and most individual conditions that predispose to SCA remain incompletely understood. As such, caution is advised when interpreting negative genetic test results or when examining genes with low likelihood of causing SCA. Prior clinical practice guidelines have specifically addressed the diagnostic considerations regarding specific genetic cardiovascular conditions.4,10,307 The ClinGen represents a systematic effort to enumerate causal genes and variation related to human disease, including SCA-related conditions.305 Previous reports have been developed for Brugada syndrome,195 long QT syndrome,196 and hypertrophic cardiomyopathy.202
The yield of genetic testing varies substantially by condition.188 Variant interpretation may differ by laboratory215 despite recent efforts to standardize variant interpretation.306 Given the rapid changes in available technology to evaluate the genome, complexities in variant interpretation, and nuances in the ethical and legal framework surrounding genetic testing in some settings,66,115,215,308–310 it is recommended that SCA survivors undergoing genetic assessment have evaluations performed at centers with multidisciplinary experience in counseling, variant interpretation, and management of genetic heart disease (see Section 3). For SCA survivors who have undergone genetic testing, an offer of periodic re-evaluation of the genetic test results is advocated (similar to Section 6.4, Recommendation 4).
Recommendations for genetic evaluation of SCA survivors
| COR | LOE | Recommendations | References |
|---|---|---|---|
| 1 | B-NR | 1. Genetic evaluation of SCA survivors is recommended for those with a diagnosed or suspected genetic cardiac disease phenotype when the results are likely to influence diagnosis, management, or family screening. | 84,311–322 |
| 1 | B-NR | 2. When genetic evaluation is performed in an SCA survivor with a suspected or diagnosed genetic cardiac disease phenotype, it is recommended that evaluations include only genes where there is robust gene–disease association. | 84,308 |
| 2b | B-NR | 3. When genetic evaluation is performed in an SCA survivor with a suspected or diagnosed genetic cardiac disease phenotype, assessment of genes or genomic regions that are not known to be causally related may be considered in select circumstances. | 84 |
| 2b | B-NR | 4. Genetic evaluation of SCA survivors without a distinct genetic cardiac disease phenotype may be considered in select circumstances. | 82,84,209,246,323–330 |
| 3: No Benefit | C-EO | 5. Genetic testing in SCA survivors with a well-established nongenetic cause of SCA is not recommended. |
Synopsis
SCA can be caused by diverse etiologies, some of which may be predominantly or partially influenced by genetic predisposition. Whereas a thorough clinical evaluation leads to a diagnosis of the cause of SCA for most individuals, the cause of SCA may remain uncertain in others.216 In some cases, genetic evaluation of SCA survivors can confirm a molecular etiology that predisposed to the SCA event, support the diagnosis of a specific phenotype, influence management, and facilitate screening in family members at risk via cascade genetic testing.
Recommendation-Specific Supportive Text
Genetic testing, after appropriate genetic counseling and informed consent, may facilitate identification of a molecular cause of SCA by identifying pathogenic variants in genes associated with specific phenotypes and fulfilling formal disease-based diagnostic criteria.4,10 Examples of scenarios in which discovery of a genetic cause of SCA may influence management recommendations include administration of beta blockade for patients with long QT syndrome,311 sodium channel inhibition in long QT syndrome type 3,312–314 flecainide administration for patients with CPVT,315,331 or exercise restriction recommendations in patients with arrhythmogenic cardiomyopathy.316,317 Genetic evaluation may also influence family screening by facilitating cascade genetic testing and clinical surveillance in relatives at greatest risk for disease.
2. and 3. Genetic tests have variable yield and may result in discovery of variants of uncertain clinical significance, which can be frequent and challenging to interpret. Using genetic tests that comprise well-established genes related to a suspected or diagnosed genetic phenotype is most likely to result in discovery of disease-causing variants in an individual or family, while minimizing the probability of discovering a variant of uncertain clinical significance. In contrast, genetic tests with more comprehensive genomic coverage may lead to moderately increased diagnostic yield but at the expense of increased rates of discovery of variants of uncertain clinical significance.84,308 Nevertheless, given a rapidly evolving understanding of the molecular causes of specific phenotypes and increased yield of broader genetic assessment,82,84,246,323,324 tests that include broader coverage may be considered in select circumstances, such as when a heritable phenotype is being mapped within a family or assessment for de novo variation is sought through sequencing of multiple family members. The latter examples would only be pertinent when it becomes apparent that a familial trait is likely or there has been exome sequencing in a trio of confirmed parents and the index case.
Genetic evaluation appears to be highest for individuals with a phenotype consistent with a genetic cause84,323,324 and is lower among SCA survivors without a clearly identifiable genetic phenotype.82,84,246,323–326 Nevertheless, individuals with an idiopathic cause of SCA may eventually develop a diagnosis of a genetic etiology during long-term follow-up.327 Genetic testing can identify variants during the concealed phase of a genetic disease such as arrhythmogenic cardiomyopathy,328 or in individuals with conditions otherwise regarded as nongenetic such as in drug-induced long QT syndrome.329 As such, a low but non-negligible yield for genetic testing appears to be present among individuals with idiopathic SCA.
In individuals with a well-established nongenetic cause of SCA, the routine use of genetic evaluation is not recommended owing to the potential for discovery of variants of uncertain significance and misdiagnosis.
Figure 16.
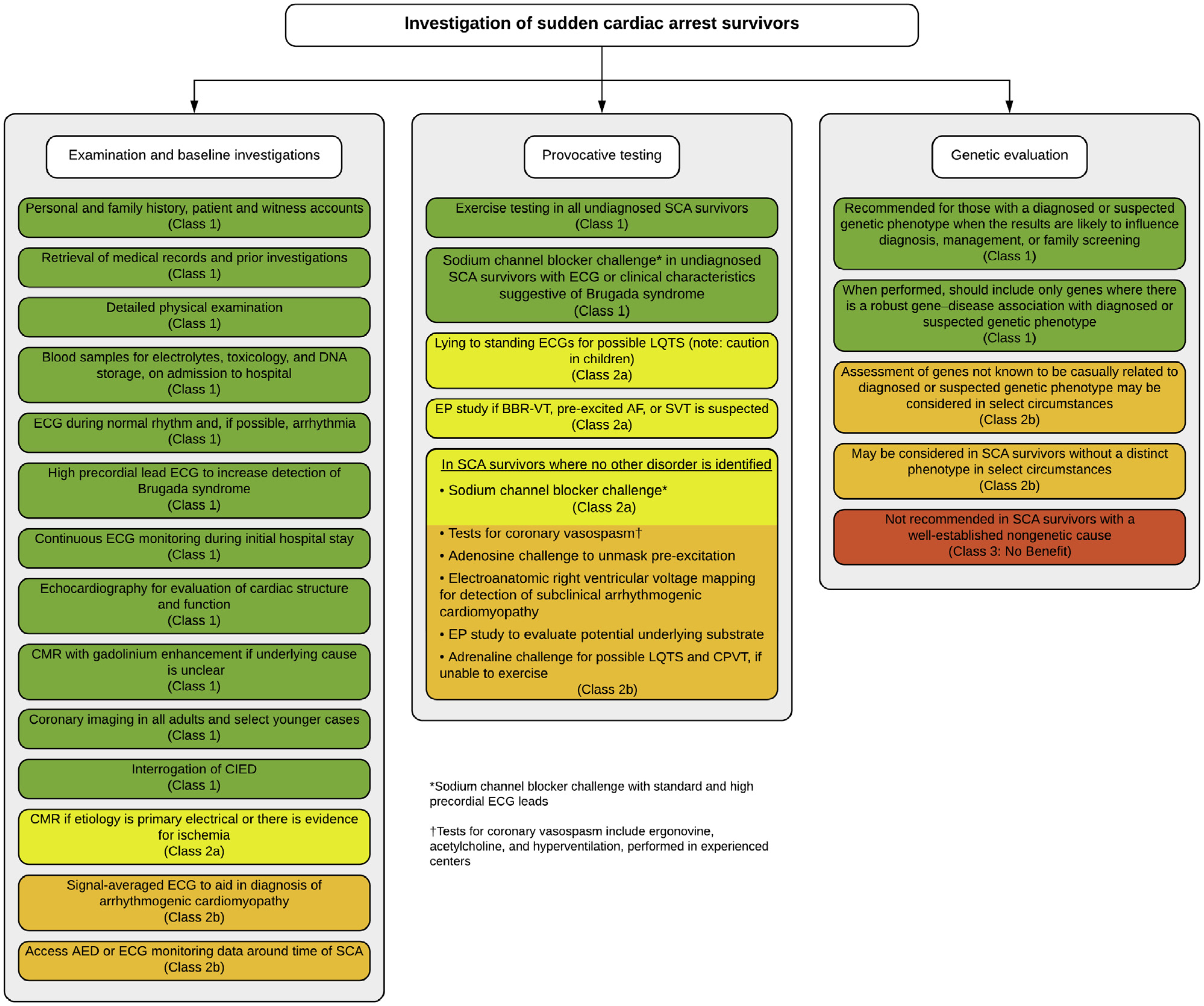
Investigation of sudden cardiac arrest survivors. Colors correspond to the Class of Recommendation in Table 1. AED = automated external defibrillator; AF = atrial fibrillation; BBR-VT = bundle branch re-entry ventricular tachycardia; CIED = cardiovascular implantable electronic device; CMR = cardiac magnetic resonance imaging; CPVT = catecholaminergic polymorphic ventricular tachycardia; ECG = electrocardiogram; EP = electrophysiological; LQTS = long QT syndrome; SCA = sudden cardiac arrest; SVT = supraventricular tachycardia.
Section 8. Investigation of the Family
8.1. Background
The sudden death of a young, apparently healthy individual raises many questions for family members. Apart from mourning and the question “could we have done something to avoid this,” a very relevant question is whether family members could be affected as well. Roles for health care providers include providing psychological support for the family, identifying a cause for the sudden death, and understanding the implications for family members (see Sections 4 and 5). For this chapter, it is especially relevant that families need support to organize clinical and genetic testing for family members and sometimes postmortem genetic testing of the deceased.2,101,332
Currently, many cases and even familial forms of unexplained cardiac disease remain insufficiently investigated.333 When clinical symptoms indicate that a cardiomyopathy or arrhythmia may have contributed to the death, further steps are needed to specify the diagnosis. If blood or tissue of the deceased is available, DNA testing can be done for a range of arrhythmia syndromes and cardiomyopathies, nowadays often using gene panels. Without a specific diagnosis in the deceased, clinical investigation of the first-degree relatives (parents, siblings, and children) can identify a person with similar symptoms or signs, although sometimes mild. This relative of the deceased can be the proband for DNA testing and thus provide the key to a diagnosis for the family.
Efforts are needed to increase the proportion of postmortem examinations (either forensic or medical autopsy) to clarify whether or not an underlying cause can be suspected or proven. The postmortem result should be communicated to the family as per local protocols. Without a postmortem diagnosis, efforts are needed to evaluate eventual clinical symptoms of the parents and other first-degree family members.80,81,143,144,334 This is outlined in Section 6. If DNA of the deceased person can be used for testing, or a relative who has similar symptoms can be tested, a monogenic form of cardiomyopathy or arrhythmia may be recognized that may also be present in family members. Typically, first-degree relatives are at 50% risk of carrying the same pathogenic mutation, since many of these conditions follow an autosomal dominant pattern of inheritance.
Depending on whether or not autopsy has been performed, whether DNA is available, whether a relative is already diagnosed with a cardiogenetic condition, and whether symptoms were noticed during life, there are different situations possible: 1) sudden death, with clinical observations or DNA testing suggesting a specific diagnosis, and 2) sudden death, with no cause identified. The first situation will be discussed in Section 8.2, the second in Section 8.3 (Figure 17).
Figure 17.
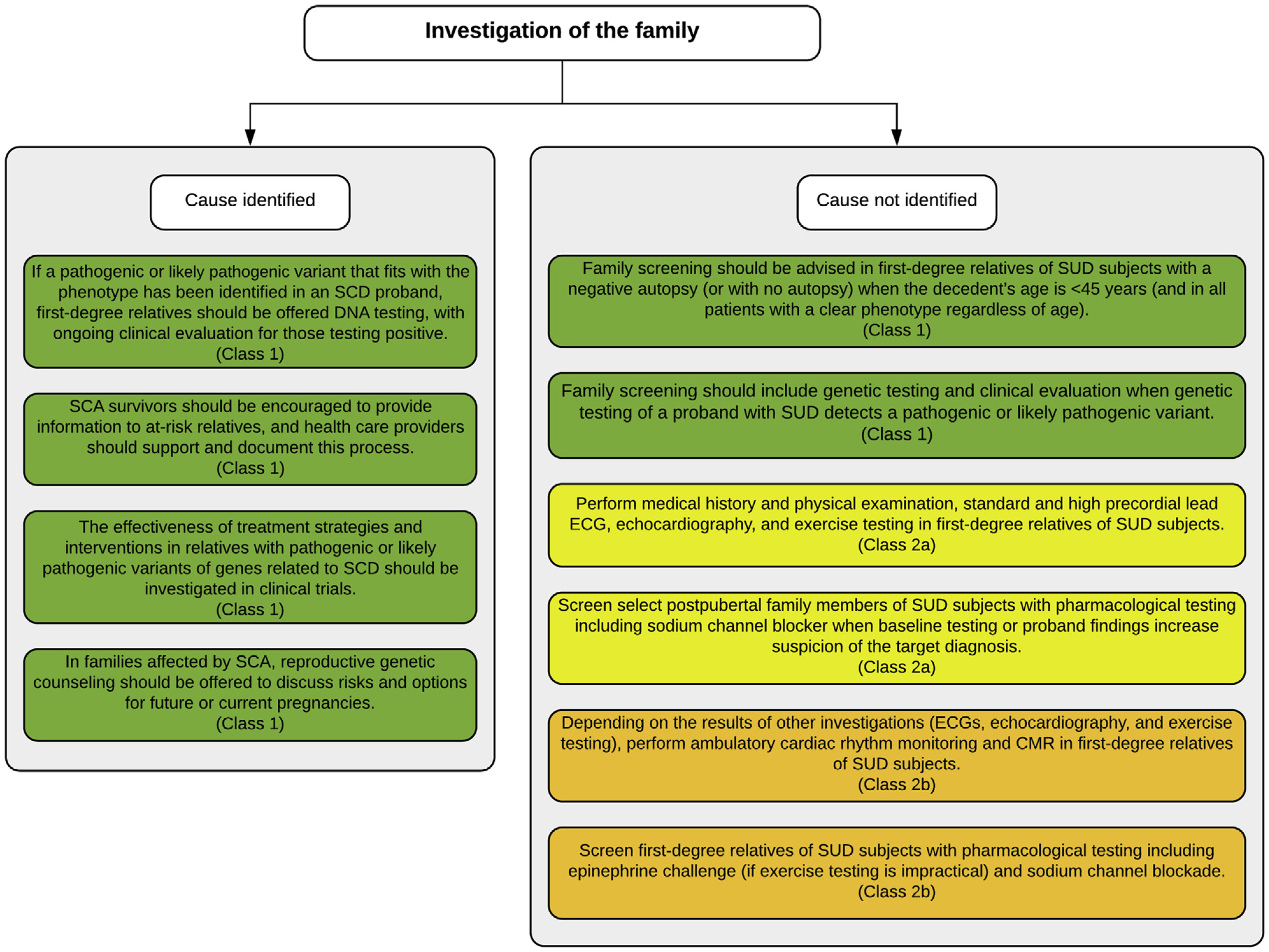
Investigation of the family affected by sudden cardiac arrest and/or sudden unexplained death when cause is identified or not identified. Colors correspond to the Class of Recommendation in Table 1. CMR = cardiac magnetic resonance imaging; ECG = electrocardiogram; SCA = sudden cardiac arrest; SCD = sudden cardiac death; SUD = sudden unexplained death.
8.2. Investigation of the Family: Cause Identified—Cascade Testing, Clinical and Genetic Investigations
In cascade testing, after the postmortem diagnosis in an index patient by a DNA test, an invitation can be sent to parents, brothers and sisters, and children.335 If any of the relatives is also diagnosed with the condition, a next circle of first-degree relatives is invited. If one of the first-degree relatives is not available (either deceased or not wanting to participate), second-degree relatives are invited, for instance, children of a deceased sibling. This systematic approach is very effective in autosomal dominant conditions, since first-degree relatives are at 50% a priori risk to carry the same pathogenic mutation and second-degree relatives at 25% risk. For minors, the age at which treatment starts determines the age before which DNA testing is advised. This may differ between countries and conditions. DNA testing of minors is not advised if the result would have no consequences in childhood.
Presymptomatic DNA testing makes it possible to organize preventive measures, such as regular cardiological follow-up, use of medication (eg, beta blockers), lifestyle advice (eg, avoid intensive sports), implantable cardioverter-defibrillators, or reproductive planning.80,193,336–342 Presymptomatic DNA testing also puts an end to uncertainty and fear for those family members who test negative.339 For them, follow-up investigations are no longer indicated.
In studies describing cascade screening, often the participation of relatives is limited.343–348 This implies that many persons carrying pathogenic variants remain undiagnosed and are at continued risk of sudden death. Increasingly, geneticists and other stakeholders plea for an active approach to cascade testing for conditions where interventions are available.349–351 Stakeholders agree on the importance of early diagnosis and informing the family.2,340,352 Barriers to cascade screening include out-of-pocket expenses for the patient, limited resources for informing relatives, and privacy regulations.339,351,353,354 To benefit from predictive, personalized, and preventive medicine, the roles and responsibilities of stakeholders in genetic testing as a preventive strategy need to be carefully aligned.
If clinical signs and symptoms suggest an inherited condition but no DNA test has been performed or no pathogenic variant has been identified, then history, examination, and clinical investigations of first-degree relatives are required to identify those at risk for SCD.
Recommendations for investigation of the family: cause identified—cascade testing, clinical and genetic investigations
| COR | LOE | Recommendations | References |
| 1 | C-LD | 1. If a pathogenic or likely pathogenic variant that fits with the phenotype has been identified in an SCD proband, first-degree relatives should be offered DNA testing, with ongoing clinical evaluation for those testing positive. | 70,84,346,355,356 |
| 1 | C-LD | 2. SCA survivors should be encouraged to provide information to at-risk relatives, and health care providers should support and document this process. | 350,357 |
| 1 | C-LD | 3. The effectiveness of treatment strategies and interventions in relatives with pathogenic or likely pathogenic variants of genes related to SCD should be investigated in clinical trials. | 333,358 |
| 1 | B-NR | 4. In families affected by SCA, reproductive genetic counseling should be offered to discuss risks and options for future or current pregnancies. | 336–338 |
Synopsis
A postmortem diagnosis in a victim of SCD implies the possibility of avoiding sudden death in relatives. This requires an active approach to inform parents, siblings, and children of the index case and offer clinical evaluation and potential genetic testing. A multidisciplinary service should support all elements of recognizing an inherited cardiac disorder in a victim (pathologist), identifying pathogenic mutations (geneticist), clinical evaluation and surveillance of carriers of the mutation (cardiologist), and supporting the patients and relatives (psychologist).
Recommendation-Specific Supportive Text
Since undiagnosed cardiogenetic conditions can be life-threatening and since interventions are available to reduce the risk of sudden death, first-degree relatives who are at 50% risk of carrying the pathogenic variant need to be informed about the possibilities of clinical investigations and genetic testing.70,84,346,355,356 Second-degree relatives whose intervening first-degree relative refuses genetic testing (or is not available) should be offered testing due to a 25% risk.
Patients often can inform their relatives at risk, but medical professionals increasingly feel ethically responsible to support this process.349,350,357 Supplying a letter for the patient to share with relatives is advised. Occasionally, directly contacting relatives may be possible in some jurisdictions. Studies have shown that there is substantial room for improvement of the uptake of family screening. Barriers include reluctance to consent to postmortem investigations, lack of information, and lack of funding for the services. While the services needed should involve many disciplines (pathologist, cardiologist, geneticist), the systematic approaching of family members fits the specialty of clinical genetics well. To achieve a high uptake, a systematic approach is needed.
The identification of at-risk individuals leads to the question of whether all require active treatment. Prospective clinical studies are needed to answer this question. To balance pros and cons for survival and for quality of life on treatment, long-term follow-up of persons following the suggested surveillance and interventions is needed. Precision prevention advice should build on this evidence.
In families with genetically affected individuals (with or without clinical findings), detailed prenatal counseling and guidance regarding inheritance patterns, variant penetrance, and risk should be offered, and other options including preimplantation genetic diagnosis should be explored.336–338
8.3. Investigation of the Family: Cause Not Identified—Clinical and Genetic Investigations
Sudden death in the young is always a tragedy for those lost and the remaining family. When the cause of sudden death is not identified, either because there was no postmortem examination or because the autopsy was negative, significant anxiety in the family focuses on two major questions: why did the subject die suddenly and what risks apply to the remaining family members? An autopsy is typically requested, and in some jurisdictions mandated, but may not be completed because of cultural, family, or logistical limitations. When a postmortem examination establishes a cause of death, diagnosis transitions from SUD to death attributed to autopsy-related findings (see Section 8.2). When no underlying anatomic or toxicologic cause of death is identified with forensic autopsy, the description of the death goes from SUD to sudden arrhythmic death (syndrome)/SAD(S), since death is attributed to a presumed arrhythmia, or autopsy-negative SCD. In the case of nonspecific findings, follow-up of families should be similar to that with a negative autopsy.184,359
When sudden death is classified as SAD or autopsy-negative SCD (ie, SUD), the differential diagnosis includes a breadth of inherited conditions that are predominantly ion channelopathies, with latent cardiomyopathy a consideration based on subtle autopsy findings. Causes include long QT syndrome, CPVT, short QT syndrome, Brugada syndrome, and arrhythmogenic cardiomyopathy. Careful review by a trained cardiac pathologist is recommended to ensure that assigned causes or absent causes are accurate (see Section 6.3).2
Recent studies have shown that a genetic evaluation of the deceased subject’s DNA associated with a clinical evaluation of first-degree relatives of the deceased subject retrospectively identified the cause of death in 20–40% of cases.70,80,81,143,144,189,3,3,4 In this situation, the identification of the cause of sudden death provides an explanation to the family and facilitates further cascade screening. In a second step, it will enable prevention measures in the family to limit the risk of a second death.
Recommendations for investigation of the family: cause not identified—clinical and genetic investigations
| COR | LOE | Recommendations | References |
| 1 | B-NR | 1. Family screening should be advised in first-degree relatives of SUD subjects with a negative autopsy (or with no autopsy) when the decedent’s age is <45 years (and in all patients with a clear phenotype regardless of age). | 80,81,143,144,334 |
| 1 | B-NR | 2. Family screening should include genetic testing and clinical evaluation when genetic testing of a proband with SUD detects a pathogenic or likely pathogenic variant. | 70,80,81,84,143,144,189,334 |
| 2a | B-NR | 3. It is reasonable to take a medical history and perform physical examination, standard and high precordial lead ECG, echocardiography, and exercise testing in first-degree relatives of SUD subjects. | 80,81,143,144,334 |
| 2b | C-LD | 4. Depending on the results of other investigations (ECGs, echocardiography, and exercise testing), it may be reasonable to perform ambulatory cardiac rhythm monitoring and CMR in first-degree relatives of SUD subjects. | 70,81,143,189,360 |
| 2a | B-NR | 5. It is reasonable to screen select postpubertal family members of SUD subjects with pharmacological testing including sodium channel blocker when baseline testing or proband findings increase suspicion of the target diagnosis. | 143,189,205,275,288,334 |
| 2b | B-NR | 6. It may be reasonable to screen first-degree relatives of SUD subjects with pharmacological testing including epinephrine challenge (if exercise testing is impractical) and sodium channel blockade. | 189,205,275,288,334 |
Synopsis
Screening of first-degree relatives of the SCD victim is informed by findings from the forensic investigation. Though the yield of genetic testing is relatively low, results should be applied to all first-degree relatives in conjunction with clinical assessment. In the absence of genetic results, screening tests should include a medical history, standard and high precordial lead ECG, 24-hour ambulatory monitoring, echocardiography, and exercise test, with select use of pharmacological provocation and advanced imaging.
Recommendation-Specific Supportive Text
Although autopsy is recommended, family screening after sudden death in young patients is effective even when an autopsy is not conducted. Broad screening of first-degree relatives with systematic testing is warranted.80,81,143,144,334 Combining molecular autopsy with clinical evaluation in surviving families increases diagnostic yield.70 The value of surveillance testing after negative evaluation is uncertain, though commonly undertaken until age 45 years (range: 40–50 years), with decreasing frequency with age.85 The context of SCD, the family history, and existing findings should inform the potential merits of ongoing surveillance, including frequency and duration. The age at which and from which onward surveillance is warranted depends on the (suspected) underlying condition.
When decedent genetic testing detects a pathogenic or likely pathogenic variant, the result enables identification of all family members at risk of SCD.80,81,143,144,334 It is important to combine genetic and clinical evaluation, especially when the pathogenicity of the detected variant is uncertain, to evaluate the correlation between the genetic finding and clinical diagnosis for each family.83 If DNA is not available from the decedent and no clinical phenotype is present in the family, genetic testing of family members should be strongly discouraged.
Several studies have demonstrated that family screening should include at least a medical history, standard and high precordial lead ECG (to improve the detection of the Brugada syndrome), echocardiography, exercise test,80,81,143,144,334 and Holter monitoring on a case-by-case basis.
CMR and 24-hour ambulatory monitoring of family members can inform diagnosis.4,5
5. and 6. A careful assessment of the circumstances of sudden death may point to a specific diagnosis. Sudden death in a young or middle-aged male occurring during a febrile illness or sleep suggests the diagnosis of Brugada syndrome, whereas sudden death occurring in a subject during physical activity suggests the diagnosis of long QT syndrome or CPVT. As Brugada syndrome may be masked or intermittent in some patients, sodium channel blocker challenge may unmask the type 1 pattern and increase the effectiveness of family screening. It should be recognized that there is a potential high rate of false positives, as data on the specificity and sensitivity of the test are not available, and a positive ECG may be induced in 4–5% of normal subjects.286,361 In the pediatric age group, a negative test may convert to a positive test after puberty.362 Long QT syndrome may also be unmasked by standing, exercise test, epinephrine test, or mental stress test.266,269,270,275,288 In first-degree relatives of young SUD victims with no manifest abnormalities during the initial examination, the risk of developing manifest inherited cardiac disease or cardiac events during follow-up is low.205
Section 9. Future Directions
Many of the recommendations in this document seem intuitive, obvious, and straightforward; however, much of what is being recommended within this document is seldom routinely performed even in well-resourced countries. Many decedents of SUD never receive an autopsy, and the evaluation of first-degree relatives of an SUD victim or an SCA survivor ranges widely from no evaluation to ordering a multitude of tests that are then repeated regularly and indefinitely. The extensive variability in practice indicates that developing common sense processes and multidisciplinary teams remains a considerable challenge in many areas and guidance is required. Developing these processes and teams involves leaders challenging many medico-political barriers that obstruct and delay best medical practice. We hope that this document will empower those who wish to achieve such changes for the better, including efforts for continuous improvement of practice. So, assuming the recommendations in this document are embraced and implemented, what are the next steps and future directions in this field?
Firstly, unlike cancer statistics and even SCD in the elderly due to coronary artery disease, the precise prevalence, epidemiology, and etiologies of either SUD or SCA in the young remain obscure in most countries. Only if these conditions become a notifiable event will the true scale and scope ever be captured.
Secondly, communities, states/provinces, and countries must advocate for and expect that a true comprehensive autopsy, including postmortem genetic testing (ie, molecular autopsy), occurs whenever an SUD occurs in a young person. The current dismal rate of autopsy must be reversed. Only when this becomes the standard of care will the true epidemiology/etiology of SCD be determined.
Thirdly, the basic occurrence and subsequent extent of an evaluation of the living, whether she/he is an SCA survivor or the first-degree relative of an SUD victim or an SCA survivor, must become standard of care. After initial cardiological evaluation (examination, ECGs, stress test, and echocardiogram) has been completed, the true contribution of SCA-predisposing genetic heart disease will be exposed. Furthermore, the composition and contribution of advanced investigations ranging from sodium channel drug provocation studies, to MRI, to genetic testing requires further study, as does the recommended interval for a repeat cardiological evaluation of the first-degree relatives when their first evaluation is either normal or inconclusive.
Finally, when such investigations are commenced, future studies to minimize the collateral damage from uncertain clinical findings and genetic “variants of uncertain significance” will be needed. While it is recognized that the correct necropsy diagnosis of the SUD decedent and the correct diagnosis of the SCA survivor may give patients and their families some answers and resolution to the event, the premature and erroneous diagnosis due to excessive confidence in or overinterpretation of clinical or genetic findings of uncertain significance can cause remarkable harm. If we are to “first do no harm,” the model of multidisciplinary teams with the expertise to correctly evaluate all the investigations and potential (mis)diagnoses should be made accessible to all.
Supplementary Material
Top 10 Take-Home Messages.
Sudden cardiac death (SCD) is an important public health issue and warrants further study to better quantify its occurrence, its impact on society, and the opportunities for improving outcomes through public education and provision of automated external defibrillators and cardiopulmonary resuscitation (CPR) training.
For survivors of sudden cardiac arrest (SCA), victims of sudden unexplained death (SUD), and their relatives, a multidisciplinary team is central to thorough investigation, so as to maximize the opportunity to make a diagnosis. Where there has been an SCD or resuscitated SCA and a genetic cause is suspected, genetic testing and counseling is essential for families, to ensure that risks, benefits, results, and the clinical significance of genetic testing can be discussed.
The psychological care of families affected by SUD and survivors of SCA (and their families) should run in parallel with the investigation process. Assessment by professionals trained in psychological care should be offered, as well as grief counseling and peer support, where appropriate.
For the investigation of SUD, a detailed personal and family history is essential, with attention to sentinel symptoms during life such as syncope or seizures, witness accounts, premorbid investigations, and inspection of any cardiac rhythm monitoring around the time of death.
A comprehensive autopsy is an essential part of the investigation of SUD and should include collection and storage of tissue suitable for genetic analysis. When the autopsy suggests a possible genetic cause, or no cause and the heart is normal, referral to a multidisciplinary team for further investigation is indicated.
For victims of SCD or survivors of cardiac arrest where the phenotype is known, genetic testing of the proband focused on likely candidate genes, along with clinical evaluation of family members, aids in identifying family members with, or at risk of developing, the same condition.
For victims of SCD or survivors of cardiac arrest where the phenotype is not known, arrhythmia syndrome–focused genetic testing may help arrive at a secure diagnosis, whereas wider testing without careful consideration of the implications of indeterminate results by experienced clinicians may only serve to add uncertainty and lead to misinterpretation of results.
For the investigation of SCA survivors, essential inquiry includes detailed personal and family history, witness accounts, physical examination, multiple electrocardiograms (ECGs), and cardiac imaging. Ambulatory monitoring and/or provocative testing (exercise, pharmacological, and invasive electrophysiological) may provide additional useful information. A sample suitable for future DNA testing should be taken early in the patient’s course and stored.
Genetic investigation of SCA survivors is best undertaken at a center with multidisciplinary care infrastructure and should focus on likely candidate genes known to be causally related to the suspected phenotype. In some cases, genetic evaluation without a suspected phenotype may be undertaken with appropriate genetic counseling, although genetic evaluation of patients with a known nongenetic cause of cardiac arrest is discouraged.
The investigation of the families of victims of SUD and survivors of SCA should include clinical and, if known, genetic cascade testing. If the cause of SUD (or rarely, SCA) is unknown, then clinical investigation of first-degree relatives may include physical examination, ECGs, cardiac imaging, ambulatory monitoring, and provocative testing (exercise, pharmacological, and rarely invasive electrophysiological) with multidisciplinary team supervision. Follow-up and periodic re-evaluation are important and are directed by initial findings.
Acknowledgments
Developed in partnership with and endorsed by the Asia Pacific Heart Rhythm Society (APHRS) and the Heart Rhythm Society (HRS). Developed in collaboration with and endorsed by the Association for European Cardiovascular Pathology (AECVP), the European Society of Human Genetics (ESHG), the Latin American Heart Rhythm Society (LAHRS), the National Society of Genetic Counselors (NSGC), and the Pediatric and Congenital Electrophysiology Society (PACES). Developed in collaboration with the European Heart Rhythm Association (EHRA).
ABBREVIATIONS
- AED
automated external defibrillator
- CIED
cardiovascular implantable electronic device
- CMR
cardiac magnetic resonance imaging
- COR
Class of Recommendation
- CPR
cardiopulmonary resuscitation
- CPVT
catecholaminergic polymorphic ventricular tachycardia
- CT
computed tomography
- ECG
electrocardiogram
- EMS
emergency medical services
- LOE
Level of Evidence
- MRI
magnetic resonance imaging
- OHCA
out-of-hospital cardiac arrest
- PVC
premature ventricular complex
- RWI
relationship with industry and other entities
- SAD(S)
sudden arrhythmic death (syndrome)
- SCA
sudden cardiac arrest
- SCD
sudden cardiac death
- SUD
sudden unexplained death
Appendix 1. Author Disclosure Table
| Writing group member | Employment | Honoraria/Speaking/Consulting | Speakers’ bureau | Research* | Fellowship support* | Ownership/Partnership/Principal/Majority stockholder | Stock or stock options | Intellectual property/Royalties | Other |
|---|---|---|---|---|---|---|---|---|---|
| Martin K. Stiles, MBChB, PhD, FHRS (APHRS Chair) | Waikato Clinical School, Faculty of Medicine and Health Science, The University of Auckland, Hamilton, New Zealand | None | None | None | None | None | None | None | None |
| Arthur A. M. Wilde, MD, PhD (HRS Chair) | Amsterdam University Medical Center, University of Amsterdam, Heart Center, Department of Clinical and Experimental Cardiology, Amsterdam, the Netherlands | None | None | None | None | None | None | None | None |
| Dominic J. Abrams, MBBS, MD, MBA | Boston Children’s Hospital, Boston, Massachusetts, USA | None | None | None | None | None | None | None | None |
| Michael J. Ackerman, MD, PhD | Mayo Clinic, Rochester, Minnesota, USA | 0: Abbott; 0: Audentes Therapeutics; 0: Boston Scientific; 0: Gilead Sciences; 0: MyoKardia; 1: InVitae; 1: Medtronic | None | 5: NIH | None | None | None | 0: AliveCor; 0: Blue Ox Healthcare; 0: StemoniX | None |
| Christine M. Albert, MD, MPH, FHRS | Cedars-Sinai Medical Center, Los Angeles, California, USA | 1: Roche Diagnostics | None | 5: Abbott; 5: NIH; 5: Roche Diagnostics | None | None | None | None | None |
| Elijah R. Behr, MA, MBBS, MD | Cardiovascular Clinical Academic Group, Molecular and Clinical Sciences Institute, St George’s, University of London, and St George’s University Hospitals NHS Foundation Trust, London, United Kingdom | None | None | None | None | None | None | None | None |
| Sumeet S. Chugh, MD, FHRS | Cedars-Sinai Medical Center, Los Angeles, California, USA | None | None | 5: NIH | 4: Abbott; 4: BIOTRONIK; 4: Boston Scientific; 4: Medtronic | None | None | None | President, Cardiac Electrophysiology Society (2020–2022) |
| Martina C. Cornel, MD, PhD | Amsterdam University Medical Center, Vrije Universiteit Amsterdam, Clinical Genetics, Amsterdam Public Health Research Institute, Amsterdam, the Netherlands | None | None | None | None | None | None | None | 0: European Society of Human Genetics (Vice Chair of Public and Professional Policy Committee); 0: VSOP (Dutch Genetic Alliance) (Board Membership) |
| Karen Gardner, MPH, PhD | University of New South Wales, Canberra, Australia | None | None | None | None | None | None | None | None |
| Jodie Ingles, GradDipGenCouns, PhD, MPH, FHRS | Agnes Ginges Centre for Molecular Cardiology at Centenary Institute, The University of Sydney, Sydney, Australia | None | None | 3: MyoKardia | None | None | None | None | None |
| Cynthia A. James, ScM, PhD, CGC | Johns Hopkins University, Baltimore, Maryland, USA | None | None | 1: NSGC; 2: Boston Scientific | None | None | None | None | 0: NSGC (Chair of the Practice Guidelines Committee) |
| Jyh-Ming Jimmy Juang, MD, PhD | Cardiovascular Center and Division of Cardiology, Department of Internal Medicine, National Taiwan University Hospital and National Taiwan University College of Medicine, Taipei, Taiwan | None | None | None | None | None | None | None | None |
| Stefan Kääb, MD, PhD | Department of Medicine I, University Hospital, LMU Munich, Munich, Germany | 1: AstraZeneca; 1: Bayer | None | 1: BIOTRONIK; 1: Boston Scientific; 1: Medtronic | None | None | None | None | None |
| Elizabeth S. Kaufman, MD, FHRS | MetroHealth Campus, Case Western Reserve University, Cleveland, Ohio, USA | None | None | None | None | None | None | None | None |
| Andrew D. Krahn, MD, FHRS | The University of British Columbia, Vancouver, British Columbia, Canada | 1: Medtronic | None | None | None | None | None | None | None |
| Steven A. Lubitz, MD, MPH | Massachusetts General Hospital, Boston, Massachusetts, USA | 1: Bayer; 1: Bristol-Myers Squibb | None | 5: AHA; 5: Bayer; 5: Boehringer Ingelheim; 5: Bristol-Myers Squibb; 5: Fitbit; 5: IBM; 5: NIH | None | None | None | None | None |
| Heather MacLeod, MS, CGC | Data Coordinating Center for the Sudden Death in the Young Case Registry, Okemos, Michigan, USA | None | None | None | None | None | None | None | None |
| Carlos A. Morillo, MD, FHRS | Libin Cardiovascular Institute, Calgary, Alberta, Canada | 1: Abbott; 1: Medtronic; 1: Servier | None | None | None | None | None | None | None |
| Koonlawee Nademanee, MD, FHRS, CCDS | Chulalongkorn University, Faculty of Medicine, and Pacific Rim Electrophysiology Research Institute at Bumrungrad Hospital, Bangkok, Thailand | 1: Biosense Webster | None | 4: Medtronic | None | None | None | 3: Biosense Webster | None |
| Vincent Probst, MD, PhD | CHU de Nantes, Nantes, France | 1: BIOTRONIK; 1: Boston Scientific; 1: Medtronic | None | 5: Boston Scientific | None | None | None | None | None |
| Elizabeth V. Saarel, MD, FHRS, CEPS-P | Cleveland Clinic Lerner College of Cardiology at Case Western Reserve University, Cleveland, Ohio, and St Luke’s Medical Center, Boise, Idaho, USA | None | None | None | None | None | None | None | None |
| Luciana Sacilotto, MD, PhD | Heart Institute, University of São Paulo Medical School, São Paulo, Brazil | None | None | None | None | None | None | None | None |
| Christopher Semsarian, MBBS, MPH, PhD, FHRS | Agnes Ginges Centre for Molecular Cardiology at Centenary Institute, The University of Sydney, Sydney, Australia | None | None | None | None | None | None | None | None |
| Mary N. Sheppard, MB, BCH, BAO, MD, FRCPath | Cardiovascular Clinical Academic Group, Molecular and Clinical Sciences Institute, St George’s, University of London, and St George’s University Hospitals NHS Foundation Trust, London, United Kingdom | None | None | None | None | None | None | None | AECVP (Past President) |
| Wataru Shimizu, MD, PhD | Department of Cardiovascular Medicine, Nippon Medical School, Tokyo, Japan | None | None | None | None | None | None | None | None |
| Jonathan R. Skinner, MBChB, MD, FHRS | Cardiac Inherited Disease Group, Starship Hospital, Auckland, New Zealand | None | None | None | None | None | None | None | None |
| Jacob Tfelt-Hansen, MD, DMSc | Department of Forensic Medicine, Faculty of Medical Sciences, Rigshospitalet, Copenhagen, Denmark | None | None | None | None | None | None | None | None |
| Dao Wu Wang, MD, PhD | The First Affiliated Hospital of Nanjing Medical University, Nanjing, China | None | None | None | None | None | None | None | None |
Number value: 0 = $0; 1 = ≤ $10,000; 2 = > $10,000 to ≤ $25,000; 3 = > $25,000 to ≤ $50,000; 4 = > $50,000 to ≤ $100,000; 5 = > $100,000.
Research and fellowship support are classed as programmatic support. Sources of programmatic support are disclosed but are not regarded as a relevant relationship with industry for writing group members or reviewers. AHA = American Heart Association; AECVP = Association for European Cardiovascular Pathology; NIH = National Institutes of Health; NSGC = National Society of Genetic Counselors.
Appendix 2. Reviewer Disclosure Table
| Peer reviewer | Employment | Honoraria/Speaking/Consulting | Speakers’ bureau | Research* | Fellowship support* | Ownership/Partnership/Principal/Majority stockholder | Stock or stock options | Intellectual property/Royalties | Other |
|---|---|---|---|---|---|---|---|---|---|
| Takeshi Aiba, MD, PhD | National Cerebral and Cardiovascular Center, Osaka, Japan | None | None | None | None | None | None | None | None |
| Allison L. Cirino, MS, CGC | Brigham and Women’s Hospital, Boston, Massachusetts, USA | None | None | None | None | None | None | None | None |
| Florence Fellmann, MD, PhD | The Collaboratory, University of Lausanne, Lausanne, Switzerland | None | None | None | None | None | None | None | None |
| Michael H. Gollob, MD | Toronto General Hospital, University of Toronto, Toronto, Canada | None | None | None | None | None | None | None | None |
| Yung-Kuo Lin, MD, PhD | Taipei Municipal Wanfang Hospital, Taipei Medical University, Taipei, Taiwan | None | None | None | None | None | None | None | None |
| Joaquín S. Lucena, MD, PhD | Institute of Legal Medicine and Forensic Sciences, Seville, Spain | None | None | None | None | None | None | None | Association for European Cardiovascular Pathology President (2018–2021) |
| Ciorsti MacIntyre, MD | QEII Health Science Centre, Halifax, Canada | 1: Abbott; 1: Medtronic | None | 0: Abbott | None | None | None | None | None |
| Jens Cosedis Nielsen, MD, DMSc, PhD, FESC, FEHRA | Aarhus University Hospital, Aarhus, Denmark | None | None | 5: Novo Nordisk Foundation | None | None | None | None | None |
| Juan David Ramirez, MD | Clínica Cardio VID, Medellín, Colombia | None | None | None | None | None | None | None | 0: Medtronic |
| Gregory Webster, MD, MPH, CCDS | Ann & Robert H. Lurie Children’s Hospital of Chicago, Chicago, Illinois, USA | None | None | 4: AHA; 5: NIH | None | None | None | None | None |
Number value: 0 = $0; 1 = ≤ $10,000; 2 = > $10,000 to ≤ $25,000; 3 = > $25,000 to ≤ $50,000; 4 = > $50,000 to ≤ $100,000; 5 = > $100,000.
Research and fellowship support are classed as programmatic support. Sources of programmatic support are disclosed but are not regarded as a relevant relationship with industry for writing group members or reviewers. AHA = American Heart Association; NIH = National Institutes of Health.
Footnotes
Appendix
Supplementary Data
Supplementary data (Appendix 3) associated with this article can be found in the online version at https://doi.org/10.1016/j.hrthm.2020.10.010.
Document Reviewers: Takeshi Aiba, MD, PhD; Allison L. Cirino, MS, CGC; Florence Fellmann, MD, PhD; Michael H. Gollob, MD; Yung-Kuo Lin, MD, PhD; Joaquín S. Lucena, MD, PhD; Ciorsti MacIntyre, MD; Jens Cosedis Nielsen, MD, DMSc, PhD, FESC, FEHRA; Juan David Ramirez, MD; Gregory We1bster, MD, MPH, CCDS
References
- 1.Halperin JL, Levine GN, Al-Khatib SM, et al. Further evolution of the ACC/AHA clinical practice guideline recommendation classification system: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol 2016;67:1572–1574. [DOI] [PubMed] [Google Scholar]
- 2.Fellmann F, van El CG, Charron P, et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur J Hum Genet 2019;27:1763–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cronin EM, Bogun FM, Maury P, et al. 2019 HRS/EHRA/APHRS/LAHRS expert consensus statement on catheter ablation of ventricular arrhythmias. Heart Rhythm 2020;17:e2–e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm 2019;16:e301–e372. [DOI] [PubMed] [Google Scholar]
- 5.Brignole M, Moya A, de Lange FJ, et al. 2018 ESC guidelines for the diagnosis and management of syncope. Eur Heart J 2018;39:1883–1948. [DOI] [PubMed] [Google Scholar]
- 6.Al-Khatib SM, Stevenson WG, Ackerman MJ, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Heart Rhythm 2018;15:e73–e189. [DOI] [PubMed] [Google Scholar]
- 7.Mont L, Pelliccia A, Sharma S, et al. Pre-participation cardiovascular evaluation for athletic participants to prevent sudden death: position paper from the EHRA and the EACPR, branches of the ESC. Europace 2017;19:139–163. [DOI] [PubMed] [Google Scholar]
- 8.Priori SG, Blomstrom-Lundqvist C, Mazzanti A, et al. 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J 2015;36:2793–2867. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen CT, Kay GN, Kalman J, et al. EHRA/HRS/APHRS expert consensus on ventricular arrhythmias. Heart Rhythm 2014;11:e166–e196. [DOI] [PubMed] [Google Scholar]
- 10.Priori SG, Wilde AA, Horie M, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;10:1932–1963. [DOI] [PubMed] [Google Scholar]
- 11.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies. Heart Rhythm 2011;8:1308–1339. [DOI] [PubMed] [Google Scholar]
- 12.Zipes DP, Camm AJ, Borggrefe M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (writing committee to develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). Circulation 2006;114:e385–e484. [DOI] [PubMed] [Google Scholar]
- 13.Fishman GI, Chugh SS, Dimarco JP, et al. Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010;122:2335–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Risgaard B, Lynge TH, Wissenberg M, et al. Risk factors and causes of sudden noncardiac death: a nationwide cohort study in Denmark. Heart Rhythm 2015; 12:968–974. [DOI] [PubMed] [Google Scholar]
- 15.Winkel BG, Holst AG, Theilade J, et al. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur Heart J 2011;32:983–990. [DOI] [PubMed] [Google Scholar]
- 16.Sanchez O, Campuzano O, Fernandez-Falgueras A, et al. Natural and undetermined sudden death: value of post-mortem genetic investigation. PLoS One 2016;11:e0167358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nichol G, Thomas E, Callaway CW, et al. Regional variation in out-of-hospital cardiac arrest incidence and outcome. JAMA 2008;300:1423–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chugh SS, Jui J, Gunson K, et al. Current burden of sudden cardiac death: multiple source surveillance versus retrospective death certificate-based review in a large U.S. community. J Am Coll Cardiol 2004;44:1268–1275. [DOI] [PubMed] [Google Scholar]
- 19.de Vreede-Swagemakers JJ, Gorgels AP, Dubois-Arbouw WI, et al. Out-of-hospital cardiac arrest in the 1990’s: a population-based study in the Maastricht area on incidence, characteristics and survival. J Am Coll Cardiol 1997; 30:1500–1505. [DOI] [PubMed] [Google Scholar]
- 20.Stecker EC, Reinier K, Marijon E, et al. Public health burden of sudden cardiac death in the United States. Circ Arrhythm Electrophysiol 2014;7:212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wong CX, Brown A, Lau DH, et al. Epidemiology of sudden cardiac death: global and regional perspectives. Heart Lung Circ 2019;28:6–14. [DOI] [PubMed] [Google Scholar]
- 22.Kong MH, Fonarow GC, Peterson ED, et al. Systematic review of the incidence of sudden cardiac death in the United States. J Am Coll Cardiol 2011;57:794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura T, Iwami T, Kawamura T, et al. Nationwide improvements in survival from out-of-hospital cardiac arrest in Japan. Circulation 2012;126:2834–2843. [DOI] [PubMed] [Google Scholar]
- 24.Gillum RF. Sudden coronary death in the United States: 1980–1985. Circulation 1989;79:756–765. [DOI] [PubMed] [Google Scholar]
- 25.Bagnall RD, Weintraub RG, Ingles J, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med 2016;374:2441–2452. [DOI] [PubMed] [Google Scholar]
- 26.Wisten A, Krantz P, Stattin EL. Sudden cardiac death among the young in Sweden from 2000 to 2010: an autopsy-based study. Europace 2017;19:1327–1334. [DOI] [PubMed] [Google Scholar]
- 27.Risgaard B, Winkel BG, Jabbari R, et al. Burden of sudden cardiac death in persons aged 1 to 49 years: nationwide study in Denmark. Circ Arrhythm Electrophysiol 2014;7:205–211. [DOI] [PubMed] [Google Scholar]
- 28.Byrne R, Constant O, Smyth Y, et al. Multiple source surveillance incidence and aetiology of out-of-hospital sudden cardiac death in a rural population in the West of Ireland. Eur Heart J 2008;29:1418–1423. [DOI] [PubMed] [Google Scholar]
- 29.Winkel BG, Risgaard B, Sadjadieh G, Bundgaard H, Haunso S, Tfelt-Hansen J. Sudden cardiac death in children (1–18 years): symptoms and causes of death in a nationwide setting. Eur Heart J 2014;35:868–875. [DOI] [PubMed] [Google Scholar]
- 30.Burns KM, Cottengim C, Dykstra H, et al. Epidemiology of sudden death in a population-based study of infants and children. J Pediatr: X 2020;2:100023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eckart RE, Shry EA, Burke AP, et al. Sudden death in young adults: an autopsy-based series of a population undergoing active surveillance. J Am Coll Cardiol 2011;58:1254–1261. [DOI] [PubMed] [Google Scholar]
- 32.Thiene G, Corrado D, Basso C. The Northeast Italy, Veneto Region experience. In: Thiene G, Corrado D, Basso C, eds. Sudden Cardiac Death in the Young and Athletes. Springer-Verlag Milan; 2016. p. 171–181. [Google Scholar]
- 33.Chappex N, Schlaepfer J, Fellmann F, Bhuiyan ZA, Wilhelm M, Michaud K. Sudden cardiac death among general population and sport related population in forensic experience. J Forensic Leg Med 2015;35:62–68. [DOI] [PubMed] [Google Scholar]
- 34.Deo R, Albert CM. Epidemiology and genetics of sudden cardiac death. Circulation 2012;125:620–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Becker LB, Han BH, Meyer PM, et al. Racial differences in the incidence of cardiac arrest and subsequent survival: the CPR Chicago Project. N Engl J Med 1993;329:600–606. [DOI] [PubMed] [Google Scholar]
- 36.Zhao D, Post WS, Blasco-Colmenares E, et al. Racial differences in sudden cardiac death. Circulation 2019;139:1688–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Behr ER, Casey A, Sheppard M, et al. Sudden arrhythmic death syndrome: a national survey of sudden unexplained cardiac death. Heart 2007;93:601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Griffis H, Wu L, Naim MY, et al. Characteristics and outcomes of AED use in pediatric cardiac arrest in public settings: the influence of neighborhood characteristics. Resuscitation 2020;146:126–131. [DOI] [PubMed] [Google Scholar]
- 39.Starks MA, Schmicker RH, Peterson ED, et al. Association of neighborhood demographics with out-of-hospital cardiac arrest treatment and outcomes: where you live may matter. JAMA Cardiol 2017;2:1110–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basso C, Calabrese F, Corrado D, Thiene G. Postmortem diagnosis in sudden cardiac death victims: macroscopic, microscopic and molecular findings. Cardiovasc Res 2001;50:290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sasson C, Rogers MA, Dahl J, Kellermann AL. Predictors of survival from out-of-hospital cardiac arrest: a systematic review and meta-analysis. Circ Cardiovasc Qual Outcomes 2010;3:63–81. [DOI] [PubMed] [Google Scholar]
- 42.Berdowski J, Berg RA, Tijssen JG, Koster RW. Global incidences of out-of-hospital cardiac arrest and survival rates: systematic review of 67 prospective studies. Resuscitation 2010;81:1479–1487. [DOI] [PubMed] [Google Scholar]
- 43.Otsuki S, Aiba T, Tahara Y, et al. Intra-day change in occurrence of out-of-hospital ventricular fibrillation in Japan: the JCS-ReSS study. Int J Cardiol 2020; 318:54–60. [DOI] [PubMed] [Google Scholar]
- 44.Zive D, Koprowicz K, Schmidt T, et al. Variation in out-of-hospital cardiac arrest resuscitation and transport practices in the Resuscitation Outcomes Consortium: ROC Epistry-Cardiac Arrest. Resuscitation 2011;82:277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girotra S, van Diepen S, Nallamothu BK, et al. Regional variation in out-of-hospital cardiac arrest survival in the United States. Circulation 2016; 133:2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atwood C, Eisenberg MS, Herlitz J, Rea TD. Incidence of EMS-treated out-of-hospital cardiac arrest in Europe. Resuscitation 2005;67:75–80. [DOI] [PubMed] [Google Scholar]
- 47.Hawkes C, Booth S, Ji C, et al. Epidemiology and outcomes from out-of-hospital cardiac arrests in England. Resuscitation 2017;110:133–140. [DOI] [PubMed] [Google Scholar]
- 48.Rea TD, Eisenberg MS, Sinibaldi G, White RD. Incidence of EMS-treated out-of-hospital cardiac arrest in the United States. Resuscitation 2004;63:17–24. [DOI] [PubMed] [Google Scholar]
- 49.Becker L, Gold LS, Eisenberg M, White L, Hearne T, Rea T. Ventricular fibrillation in King County, Washington: a 30-year perspective. Resuscitation 2008; 79:22–27. [DOI] [PubMed] [Google Scholar]
- 50.Bunch TJ, White RD, Gersh BJ, et al. Long-term outcomes of out-of-hospital cardiac arrest after successful early defibrillation. N Engl J Med 2003; 348:2626–2633. [DOI] [PubMed] [Google Scholar]
- 51.Cummins RO, Ornato JP, Thies WH, Pepe PE. Improving survival from sudden cardiac arrest: the “chain of survival” concept. A statement for health professionals from the Advanced Cardiac Life Support Subcommittee and the Emergency Cardiac Care Committee, American Heart Association. Circulation 1991; 83:1832–1847. [DOI] [PubMed] [Google Scholar]
- 52.Iwami T, Nichol G, Hiraide A, et al. Continuous improvements in “chain of survival” increased survival after out-of-hospital cardiac arrests: a large-scale population-based study. Circulation 2009;119:728–734. [DOI] [PubMed] [Google Scholar]
- 53.Lund-Kordahl I, Olasveengen TM, Lorem T, Samdal M, Wik L, Sunde K. Improving outcome after out-of-hospital cardiac arrest by strengthening weak links of the local Chain of Survival; quality of advanced life support and post-resuscitation care. Resuscitation 2010;81:422–426. [DOI] [PubMed] [Google Scholar]
- 54.Wissenberg M, Lippert FK, Folke F, et al. Association of national initiatives to improve cardiac arrest management with rates of bystander intervention and patient survival after out-of-hospital cardiac arrest. JAMA 2013; 310:1377–1384. [DOI] [PubMed] [Google Scholar]
- 55.Moriwaki Y, Tahara Y, Kosuge T, Suzuki N. Etiology of out-of-hospital cardiac arrest diagnosed via detailed examinations including perimortem computed tomography. J Emerg Trauma Shock 2013;6:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Engdahl J, Holmberg M, Karlson BW, Luepker R, Herlitz J. The epidemiology of out-of-hospital ‘sudden’ cardiac arrest. Resuscitation 2002;52:235–245. [DOI] [PubMed] [Google Scholar]
- 57.D’Silva A, Sharma S. Management of young competitive athletes with cardiovascular conditions. Heart 2017;103:463–473. [DOI] [PubMed] [Google Scholar]
- 58.Asatryan B, Vital C, Kellerhals C, et al. Sports-related sudden cardiac deaths in the young population of Switzerland. PLoS One 2017;12:e0174434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Landry CH, Allan KS, Connelly KA, Cunningham K, Morrison LJ, Dorian P. Sudden cardiac arrest during participation in competitive sports. N Engl J Med 2017;377:1943–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McNally B, Robb R, Mehta M, et al. Out-of-hospital cardiac arrest surveillance: Cardiac Arrest Registry to Enhance Survival (CARES), United States, October 1, 2005-December 31, 2010. MMWR Surveill Summ 2011;60:1–19. [PubMed] [Google Scholar]
- 61.Hallstrom AP, Ornato JP, Weisfeldt M, et al. Public-access defibrillation and survival after out-of-hospital cardiac arrest. N Engl J Med 2004;351:637–646. [DOI] [PubMed] [Google Scholar]
- 62.Capucci A, Aschieri D, Piepoli MF, Bardy GH, Iconomu E, Arvedi M. Tripling survival from sudden cardiac arrest via early defibrillation without traditional education in cardiopulmonary resuscitation. Circulation 2002;106:1065–1070. [DOI] [PubMed] [Google Scholar]
- 63.Blom MT, Beesems SG, Homma PC, et al. Improved survival after out-of-hospital cardiac arrest and use of automated external defibrillators. Circulation 2014; 130:1868–1875. [DOI] [PubMed] [Google Scholar]
- 64.Myat A, Song KJ, Rea T. Out-of-hospital cardiac arrest: current concepts. Lancet 2018;391:970–979. [DOI] [PubMed] [Google Scholar]
- 65.Skinner JR, Duflou JA, Semsarian C. Reducing sudden death in young people in Australia and New Zealand: the TRAGADY initiative. Med J Aust 2008; 189:539–540. [DOI] [PubMed] [Google Scholar]
- 66.Ahmad F, McNally EM, Ackerman MJ, et al. Establishment of specialized clinical cardiovascular genetics programs: recognizing the need and meeting standards: a scientific statement from the American Heart Association. Circ Genom Precis Med 2019;12:e000054. [DOI] [PubMed] [Google Scholar]
- 67.Basso C, Aguilera B, Banner J, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the Association for European Cardiovascular Pathology. Virchows Arch 2017;471:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nancarrow SA, Booth A, Ariss S, Smith T, Enderby P, Roots A. Ten principles of good interdisciplinary team work. Hum Resour Health 2013;11:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Earle NJ, Crawford J, Hayes I, et al. Development of a cardiac inherited disease service and clinical registry: a 15-year perspective. Am Heart J 2019; 209:126–130. [DOI] [PubMed] [Google Scholar]
- 70.Lahrouchi N, Raju H, Lodder EM, et al. Utility of post-mortem genetic testing in cases of sudden arrhythmic death syndrome. J Am Coll Cardiol 2017; 69:2134–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Marcondes L, Crawford J, Earle N, et al. Long QT molecular autopsy in sudden unexplained death in the young (1–40 years old): lessons learnt from an eight year experience in New Zealand. PLoS One 2018;13:e0196078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yeates L, Hunt L, Saleh M, Semsarian C, Ingles J. Poor psychological wellbeing particularly in mothers following sudden cardiac death in the young. Eur J Cardiovasc Nurs 2013;12:484–491. [DOI] [PubMed] [Google Scholar]
- 73.van der Werf C, Onderwater AT, van Langen IM, Smets EM. Experiences, considerations and emotions relating to cardiogenetic evaluation in relatives of young sudden cardiac death victims. Eur J Hum Genet 2014;22:192–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Caleshu C, Kasparian NA, Edwards KS, et al. Interdisciplinary psychosocial care for families with inherited cardiovascular diseases. Trends Cardiovasc Med 2016; 26:647–653. [DOI] [PubMed] [Google Scholar]
- 75.Rhodes A, Rosman L, Cahill J, et al. Minding the genes: a multidisciplinary approach towards genetic assessment of cardiovascular disease. J Genet Couns 2017;26:224–231. [DOI] [PubMed] [Google Scholar]
- 76.Waddell-Smith K, Donoghue T, Graham M, et al. The inpatient cardiology visit: missing the opportunity to detect inherited heart conditions. Heart Lung Circ 2014;23(Suppl 2):e18–e19. [Google Scholar]
- 77.Waddell-Smith KE, Donoghue T, Oates S, et al. Inpatient detection of cardiac-inherited disease: the impact of improving family history taking. Open Heart 2016; 3:e000329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Earle N, Crawford J, Gibson K, et al. Detection of sudden death syndromes in New Zealand. N Z Med J 2016;129:67–74. [PubMed] [Google Scholar]
- 79.Ingles J, Lind JM, Phongsavan P, Semsarian C. Psychosocial impact of specialized cardiac genetic clinics for hypertrophic cardiomyopathy. Genet Med 2008; 10:117–120. [DOI] [PubMed] [Google Scholar]
- 80.Tan HL, Hofman N, van Langen IM, van der Wal AC, Wilde AA. Sudden unexplained death: heritability and diagnostic yield of cardiological and genetic examination in surviving relatives. Circulation 2005;112:207–213. [DOI] [PubMed] [Google Scholar]
- 81.Behr E, Wood DA, Wright M, et al. Cardiological assessment of first-degree relatives in sudden arrhythmic death syndrome. Lancet 2003;362:1457–1459. [DOI] [PubMed] [Google Scholar]
- 82.Kumar S, Peters S, Thompson T, et al. Familial cardiological and targeted genetic evaluation: low yield in sudden unexplained death and high yield in unexplained cardiac arrest syndromes. Heart Rhythm 2013;10:1653–1660. [DOI] [PubMed] [Google Scholar]
- 83.Giudicessi JR, Lieve KVV, Rohatgi RK, et al. Assessment and validation of a phenotype-enhanced variant classification framework to promote or demote RYR2 missense variants of uncertain significance. Circ Genom Precis Med 2019;12:e002510. [DOI] [PubMed] [Google Scholar]
- 84.Mellor G, Laksman ZWM, Tadros R, et al. Genetic testing in the evaluation of unexplained cardiac arrest. Circ Cardiovasc Genet 2017;10:e001686. [DOI] [PubMed] [Google Scholar]
- 85.van der Werf C, Hofman N, Tan HL, et al. Diagnostic yield in sudden unexplained death and aborted cardiac arrest in the young: the experience of a tertiary referral center in the Netherlands. Heart Rhythm 2010;7:1383–1389. [DOI] [PubMed] [Google Scholar]
- 86.Wilms HR, Midgley DJ, Morrow P, Stables S, Crawford J, Skinner JR. Evaluation of autopsy and police reports in the investigation of sudden unexplained death in the young. Forensic Sci Med Pathol 2012;8:380–389. [DOI] [PubMed] [Google Scholar]
- 87.Biesecker B. Goals of genetic counselling. Clinical Genetics 2001;60:323–330. [DOI] [PubMed] [Google Scholar]
- 88.Resta R, Biesecker BB, Bennett RL, et al. A new definition of Genetic Counseling: National Society of Genetic Counselors’ Task Force report. J Genet Couns 2006;15:77–83. [DOI] [PubMed] [Google Scholar]
- 89.Charron P, Arad M, Arbustini E, et al. Genetic counselling and testing in cardiomyopathies: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2010;31:2715–2726. [DOI] [PubMed] [Google Scholar]
- 90.Abacan M, Alsubaie L, Barlow-Stewart K, et al. The global state of the genetic counseling profession. Eur J Hum Genet 2019;27:183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laurino MY, Leppig KA, Abad PJ, et al. A report on ten Asia Pacific countries on current status and future directions of the genetic counseling profession: the establishment of the Professional Society of Genetic Counselors in Asia. J Genet Couns 2018;27:21–32. [DOI] [PubMed] [Google Scholar]
- 92.Ingles J, Yeates L, Semsarian C. The emerging role of the cardiac genetic counselor. Heart Rhythm 2011;8:1958–1962. [DOI] [PubMed] [Google Scholar]
- 93.Hershberger RE, Givertz M, Ho CY, et al. Genetic evaluation of cardiomyopathy: a Heart Failure Society of America Practice Guideline. J Card Fail 2018; 24:281–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Skrzynia C, Demo EM, Baxter SM. Genetic counseling and testing for hypertrophic cardiomyopathy: an adult perspective. J Cardiovasc Transl Res 2009; 2:493–499. [DOI] [PubMed] [Google Scholar]
- 95.Ingles J. Psychological issues in managing families with inherited cardiovascular diseases. Cold Spring Harb Perspect Med 2019;September;23:a036558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Aatre RD, Day SM. Psychological issues in genetic testing for inherited cardiovascular diseases. Circ Cardiovasc Genet 2011;4:81–90. [DOI] [PubMed] [Google Scholar]
- 97.Sturm AC. Genetic testing in the contemporary diagnosis of cardiomyopathy. Curr Heart Fail Rep 2013;10:63–72. [DOI] [PubMed] [Google Scholar]
- 98.Burns C, McGaughran J, Davis A, Semsarian C, Ingles J. Factors influencing uptake of familial long QT syndrome genetic testing. Am J Med Genet A 2016; 170A:418–425. [DOI] [PubMed] [Google Scholar]
- 99.Ingles J, James C. Psychosocial care and cardiac genetic counseling following sudden cardiac death in the young. Prog Pediatr Cardiol 2017;45:31–36. [Google Scholar]
- 100.Wong EK, Bartels K, Hathaway J, et al. Perceptions of genetic variant reclassification in patients with inherited cardiac disease. Eur J Hum Genet 2019; 27:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bates K, Sweeting J, Yeates L, McDonald K, Semsarian C, Ingles J. Psychological adaptation to molecular autopsy findings following sudden cardiac death in the young. Genet Med 2019;21:1452–1456. [DOI] [PubMed] [Google Scholar]
- 102.Meiser B, Irle J, Lobb E, Barlow-Stewart K. Assessment of the content and process of genetic counseling: a critical review of empirical studies. J Genet Couns 2008;17:434–451. [DOI] [PubMed] [Google Scholar]
- 103.Edwards A, Gray J, Clarke A, et al. Interventions to improve risk communication in clinical genetics: systematic review. Patient Educ Couns 2008;71:4–25. [DOI] [PubMed] [Google Scholar]
- 104.Austin J, Semaka A, Hadjipavlou G. Conceptualizing genetic counseling as psychotherapy in the era of genomic medicine. J Genet Couns 2014;23:903–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Accreditation Council for Genetic Counseling. Practice based competencies for genetic counselors. Available from https://www.gceducation.org/wp-content/uploads/2019/02/ACGC-Core-Competencies-Brochure_15_Web.pdf. Accessed May 5, 2020. [DOI] [PubMed]
- 106.Michie S, Marteau TM, Bobrow M. Genetic counselling: the psychological impact of meeting patients’ expectations. J Med Genet 1997;34:237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ison HE, Ware SM, Schwantes-An TH, Freeze S, Elmore L, Spoonamore KG. The impact of cardiovascular genetic counseling on patient empowerment. J Genet Couns 2019;28:570–577. [DOI] [PubMed] [Google Scholar]
- 108.Burns C, James C, Ingles J. Communication of genetic information to families with inherited rhythm disorders. Heart Rhythm 2018;15:780–786. [DOI] [PubMed] [Google Scholar]
- 109.Liu G, MacLeod H, Webster G, McNally EM, O’Neill SM, Dellefave-Castillo L. Genetic counselors’ approach to postmortem genetic testing after sudden death: an exploratory study. Acad Forensic Pathol 2018;8:738–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Burns C, Yeates L, Spinks C, Semsarian C, Ingles J. Attitudes, knowledge and consequences of uncertain genetic findings in hypertrophic cardiomyopathy. Eur J Hum Genet 2017;25:809–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ingles J, Semsarian C. Conveying a probabilistic genetic test result to families with an inherited heart disease. Heart Rhythm 2014;11:1073–1078. [DOI] [PubMed] [Google Scholar]
- 112.Reuter C, Grove ME, Orland K, Spoonamore K, Caleshu C. Clinical cardiovascular genetic counselors take a leading role in team-based variant classification. J Genet Couns 2018;27:751–760. [DOI] [PubMed] [Google Scholar]
- 113.Bland A, Harrington EA, Dunn K, et al. Clinically impactful differences in variant interpretation between clinicians and testing laboratories: a single-center experience. Genet Med 2018;20:369–373. [DOI] [PubMed] [Google Scholar]
- 114.Furqan A, Arscott P, Girolami F, et al. Care in specialized centers and data sharing increase agreement in hypertrophic cardiomyopathy genetic test interpretation. Circ Cardiovasc Genet 2017;10:e001700. [DOI] [PubMed] [Google Scholar]
- 115.Das KJ, Ingles J, Bagnall RD, Semsarian C. Determining pathogenicity of genetic variants in hypertrophic cardiomyopathy: importance of periodic reassessment. Genet Med 2014;16:286–293. [DOI] [PubMed] [Google Scholar]
- 116.Dunn KE, Caleshu C, Cirino AL, Ho CY, Ashley EA. A clinical approach to inherited hypertrophy: the use of family history in diagnosis, risk assessment, and management. Circ Cardiovasc Genet 2013;6:118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shear MK. Getting straight about grief. Depress Anxiety 2012;29:461–464. [DOI] [PubMed] [Google Scholar]
- 118.Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry 2007;48:395–399. [DOI] [PubMed] [Google Scholar]
- 119.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), Fifth ed. Washington, DC: American Psychiatric Association Publishing; 2013. [Google Scholar]
- 120.Simon NM. Treating complicated grief. JAMA 2013;310:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ingles J, Spinks C, Yeates L, McGeechan K, Kasparian N, Semsarian C. Post-traumatic stress and prolonged grief after the sudden cardiac death of a young relative. JAMA Intern Med 2016;176:402–405. [DOI] [PubMed] [Google Scholar]
- 122.Shalev A, Liberzon I, Marmar C. Post-traumatic stress disorder. N Engl J Med 2017;376:2459–2469. [DOI] [PubMed] [Google Scholar]
- 123.Latham AE, Prigerson HG. Suicidality and bereavement: complicated grief as psychiatric disorder presenting greatest risk for suicidality. Suicide Life Threat Behav 2004;34:350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Prigerson HG, Bierhals AJ, Kasl SV, et al. Traumatic grief as a risk factor for mental and physical morbidity. Am J Psychiatry 1997;154:616–623. [DOI] [PubMed] [Google Scholar]
- 125.Karam N, Jabre P, Narayanan K, et al. Psychological support and medical screening of first-degree relatives of sudden cardiac arrest victims. JACC Clin Electrophysiol 2020;6:586–587. [DOI] [PubMed] [Google Scholar]
- 126.McDonald K, Sharpe L, Yeates L, Semsarian C, Ingles J. A needs analysis of parents following sudden cardiac death in the young. Open Heart 2020; 7:e001120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wisten A, Zingmark K. Supportive needs of parents confronted with sudden cardiac death: a qualitative study. Resuscitation 2007;74:68–74. [DOI] [PubMed] [Google Scholar]
- 128.Dent A, Condon L, Blair P, Fleming P. A study of bereavement care after a sudden and unexpected death. Arch Dis Child 1996;74:522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Merlevede E, Spooren D, Henderick H, et al. Perceptions, needs and mourning reactions of bereaved relatives confronted with a sudden unexpected death. Resuscitation 2004;61:341–348. [DOI] [PubMed] [Google Scholar]
- 130.Steffen EM, Timotijevic L, Coyle A. A qualitative analysis of psychosocial needs and support impacts in families affected by young sudden cardiac death: the role of community and peer support [published online ahead of print May 5, 2020]. Eur J Cardiovasc Nurs 2020; 10.1177/1474515120922347.1474515120922347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Solomon P. Peer support/peer provided services underlying processes, benefits, and critical ingredients. Psychiatr Rehabil J 2004;27:392–401. [DOI] [PubMed] [Google Scholar]
- 132.Landers GM, Zhou M. An analysis of relationships among peer support, psychiatric hospitalization, and crisis stabilization. Community Ment Health J 2011; 47:106–112. [DOI] [PubMed] [Google Scholar]
- 133.Bartone PT, Bartone JV, Violanti JM, Gileno ZM. Peer support services for bereaved survivors: a systematic review. Omega (Westport) 2019; 80:137–166. [DOI] [PubMed] [Google Scholar]
- 134.Farnsworth MM, Fosyth D, Haglund C, Ackerman MJ. When I go in to wake them ... I wonder: parental perceptions about congenital long QT syndrome. J Am Acad Nurse Pract 2006;18:284–290. [DOI] [PubMed] [Google Scholar]
- 135.Risgaard B, Winkel BG, Jabbari R, et al. Sudden cardiac death: pharmacotherapy and proarrhythmic drugs: a nationwide cohort study in Denmark. JACC Clin Electrophysiol 2017;3:473–481. [DOI] [PubMed] [Google Scholar]
- 136.Michowitz Y, Milman A, Sarquella-Brugada G, et al. Fever-related arrhythmic events in the multicenter Survey on Arrhythmic Events in Brugada Syndrome. Heart Rhythm 2018;15:1394–1401. [DOI] [PubMed] [Google Scholar]
- 137.Amin AS, Herfst LJ, Delisle BP, et al. Fever-induced QTc prolongation and ventricular arrhythmias in individuals with type 2 congenital long QT syndrome. J Clin Invest 2008;118:2552–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Probst V, Denjoy I, Meregalli PG, et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007;115:2042–2048. [DOI] [PubMed] [Google Scholar]
- 139.Glinge C, Jabbari R, Risgaard B, et al. Symptoms before sudden arrhythmic death syndrome: a nationwide study among the young in Denmark. J Cardiovasc Electrophysiol 2015;26:761–767. [DOI] [PubMed] [Google Scholar]
- 140.Maruthappu T, Posafalvi A, Castelletti S, et al. Loss-of-function desmoplakin I and II mutations underlie dominant arrhythmogenic cardiomyopathy with a hair and skin phenotype. Br J Dermatol 2019;180:1114–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lieve KVV, Verhagen JMA, Wei J, et al. Linking the heart and the brain: neurodevelopmental disorders in patients with catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2019;16:220–228. [DOI] [PubMed] [Google Scholar]
- 142.Byers PH, Belmont J, Black J, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet 2017;175:40–47. [DOI] [PubMed] [Google Scholar]
- 143.Behr ER, Dalageorgou C, Christiansen M, et al. Sudden arrhythmic death syndrome: familial evaluation identifies inheritable heart disease in the majority of families. Eur Heart J 2008;29:1670–1680. [DOI] [PubMed] [Google Scholar]
- 144.Steinberg C, Padfield GJ, Champagne J, et al. Cardiac abnormalities in first-degree relatives of unexplained cardiac arrest victims: a report from the Cardiac Arrest Survivors with Preserved Ejection Fraction Registry. Circ Arrhythm Electrophysiol 2016;9:e004274. [DOI] [PubMed] [Google Scholar]
- 145.Raju H, Papadakis M, Govindan M, et al. Low prevalence of risk markers in cases of sudden death due to Brugada syndrome relevance to risk stratification in Brugada syndrome. J Am Coll Cardiol 2011;57:2340–2345. [DOI] [PubMed] [Google Scholar]
- 146.Johnson JN, Hofman N, Haglund CM, Cascino GD, Wilde AA, Ackerman MJ. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology 2009;72:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chahal CAA, Salloum MN, Alahdab F, et al. Systematic review of the genetics of sudden unexpected death in epilepsy: potential overlap with sudden cardiac death and arrhythmia-related genes. J Am Heart Assoc 2020; 9:e012264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Algra A, Tijssen JG, Roelandt JR, Pool J, Lubsen J. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation 1991;83:1888–1894. [DOI] [PubMed] [Google Scholar]
- 149.Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: a marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998;97:457–460. [DOI] [PubMed] [Google Scholar]
- 150.Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med 2008;358:2016–2023. [DOI] [PubMed] [Google Scholar]
- 151.Priori SG, Wilde AA, Horie M, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Heart Rhythm 2013;10:e85–e108. [DOI] [PubMed] [Google Scholar]
- 152.Antzelevitch C, Yan GX, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm 2016;13:e295–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Conte G, Belhassen B, Lambiase P, et al. Out-of-hospital cardiac arrest due to idiopathic ventricular fibrillation in patients with normal electrocardiograms: results from a multicentre long-term registry. Europace 2019;21:1670–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Haukilahti MAE, Holmstrom L, Vahatalo J, et al. Sudden cardiac death in women. Circulation 2019;139:1012–1021. [DOI] [PubMed] [Google Scholar]
- 155.Abdalla IS, Prineas RJ, Neaton JD, Jacobs DR Jr, Crow RS. Relation between ventricular premature complexes and sudden cardiac death in apparently healthy men. Am J Cardiol 1987;60:1036–1042. [DOI] [PubMed] [Google Scholar]
- 156.Ataklte F, Erqou S, Laukkanen J, Kaptoge S. Meta-analysis of ventricular premature complexes and their relation to cardiac mortality in general populations. Am J Cardiol 2013;112:1263–1270. [DOI] [PubMed] [Google Scholar]
- 157.Noda T, Shimizu W, Taguchi A, et al. Malignant entity of idiopathic ventricular fibrillation and polymorphic ventricular tachycardia initiated by premature extrasystoles originating from the right ventricular outflow tract. J Am Coll Cardiol 2005;46:1288–1294. [DOI] [PubMed] [Google Scholar]
- 158.Aizawa Y, Sato A, Watanabe H, et al. Dynamicity of the J-wave in idiopathic ventricular fibrillation with a special reference to pause-dependent augmentation of the J-wave. J Am Coll Cardiol 2012;59:1948–1953. [DOI] [PubMed] [Google Scholar]
- 159.Kawata H, Noda T, Yamada Y, et al. Effect of sodium-channel blockade on early repolarization in inferior/lateral leads in patients with idiopathic ventricular fibrillation and Brugada syndrome. Heart Rhythm 2012;9:77–83. [DOI] [PubMed] [Google Scholar]
- 160.Gandjbakhch E, Redheuil A, Pousset F, Charron P, Frank R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia: JACC state-of-the-art review. J Am Coll Cardiol 2018;72:784–804. [DOI] [PubMed] [Google Scholar]
- 161.Manolis TA, Manolis AA, Melita H, Manolis AS. Sudden unexpected death in epilepsy: the neuro-cardio-respiratory connection. Seizure 2019;64:65–73. [DOI] [PubMed] [Google Scholar]
- 162.MacCormick JM, McAlister H, Crawford J, et al. Misdiagnosis of long QT syndrome as epilepsy at first presentation. Ann Emerg Med 2009;54:26–32. [DOI] [PubMed] [Google Scholar]
- 163.Lacour P, Buschmann C, Storm C, et al. Cardiac implantable electronic device interrogation at forensic autopsy: an underestimated resource? Circulation 2018; 137:2730–2740. [DOI] [PubMed] [Google Scholar]
- 164.Bayes de Luna A, Coumel P, Leclercq JF. Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J 1989;117:151–159. [DOI] [PubMed] [Google Scholar]
- 165.Gladding PA, Evans CA, Crawford J, et al. Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart Rhythm 2010;7:481–486. [DOI] [PubMed] [Google Scholar]
- 166.Carturan E, Tester DJ, Brost BC, Basso C, Thiene G, Ackerman MJ. Postmortem genetic testing for conventional autopsy-negative sudden unexplained death: an evaluation of different DNA extraction protocols and the feasibility of mutational analysis from archival paraffin-embedded heart tissue. Am J Clin Pathol 2008;129:391–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Searles Nielsen S, Mueller BA, De Roos AJ, Checkoway H. Newborn screening archives as a specimen source for epidemiologic studies: feasibility and potential for bias. Ann Epidemiol 2008;18:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Skinner JR, Chong B, Fawkner M, Webster DR, Hegde M. Use of the newborn screening card to define cause of death in a 12-year-old diagnosed with epilepsy. J Paediatr Child Health 2004;40:651–653. [DOI] [PubMed] [Google Scholar]
- 169.Johnson JN, Tester DJ, Bass NE, Ackerman MJ. Cardiac channel molecular autopsy for sudden unexpected death in epilepsy. J Child Neurol 2010; 25:916–921. [DOI] [PubMed] [Google Scholar]
- 170.Tsuda T, Geary EM, Temple J. Significance of automated external defibrillator in identifying lethal ventricular arrhythmias. Eur J Pediatr 2019;178:1333–1342. [DOI] [PubMed] [Google Scholar]
- 171.Riesinger L, Fichtner S, Schuhmann CG, et al. Postmortem interrogation of cardiac implantable electrical devices may clarify time and cause of death. Int J Legal Med 2019;133:883–888. [DOI] [PubMed] [Google Scholar]
- 172.Sinha SK, Crain B, Flickinger K, et al. Clinical inferences of cardiovascular implantable electronic device analysis at autopsy. J Am Coll Cardiol 2016; 68:1255–1264. [DOI] [PubMed] [Google Scholar]
- 173.Hansen BL, Jacobsen EM, Kjerrumgaard A, et al. Diagnostic yield in victims of sudden cardiac death and their relatives. Europace 2020;22:964–971. [DOI] [PubMed] [Google Scholar]
- 174.Peterson GF, Clark SC, National Association of Medical Examiners. Forensic autopsy performance standards. Am J Forensic Med Pathol 2006;27:200–225. [DOI] [PubMed] [Google Scholar]
- 175.Gulino SP, Burns K, Gunther WM, MacLeod H. Improving forensic pathologic investigation of sudden death in the young: tools, guidance, and methods of cardiovascular dissection from the sudden death in the young case registry. Academic Forensic Pathology 2018;8:347–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.de Noronha SV, Behr ER, Papadakis M, et al. The importance of specialist cardiac histopathological examination in the investigation of young sudden cardiac deaths. Europace 2014;16:899–907. [DOI] [PubMed] [Google Scholar]
- 177.Finocchiaro G, Papadakis M, Robertus JL, et al. Etiology of sudden death in sports: insights from a United Kingdom regional registry. J Am Coll Cardiol 2016;67:2108–2115. [DOI] [PubMed] [Google Scholar]
- 178.Brinkmann B. Harmonization of medico-legal autopsy rules: Committee of Ministers. Council of Europe. Int J Legal Med 1999;113:1–14. [DOI] [PubMed] [Google Scholar]
- 179.Blokker BM, Weustink AC, Wagensveld IM, et al. Conventional autopsy versus minimally invasive autopsy with postmortem MRI, CT, and CT-guided biopsy: comparison of diagnostic performance. Radiology 2018;289:658–667. [DOI] [PubMed] [Google Scholar]
- 180.Bjune T, Risgaard B, Kruckow L, et al. Post-mortem toxicology in young sudden cardiac death victims: a nationwide cohort study. Europace 2018; 20:614–621. [DOI] [PubMed] [Google Scholar]
- 181.Middleton O, Atherton D, Bundock E, et al. National Association of Medical Examiners position paper: recommendations for the investigation and certification of deaths in people with epilepsy. Epilepsia 2018;59:530–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mellor G, Raju H, de Noronha SV, et al. Clinical characteristics and circumstances of death in the sudden arrhythmic death syndrome. Circ Arrhythm Electrophysiol 2014;7:1078–1083. [DOI] [PubMed] [Google Scholar]
- 183.Sorkin T, Sheppard MN. Sudden unexplained death in alcohol misuse (SUDAM) patients have different characteristics to those who died from sudden arrhythmic death syndrome (SADS). Forensic Sci Med Pathol 2017;13:278–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Papadakis M, Raju H, Behr ER, et al. Sudden cardiac death with autopsy findings of uncertain significance: potential for erroneous interpretation. Circ Arrhythm Electrophysiol 2013;6:588–596. [DOI] [PubMed] [Google Scholar]
- 185.Tester DJ, Medeiros-Domingo A, Will ML, Haglund CM, Ackerman MJ. Cardiac channel molecular autopsy: insights from 173 consecutive cases of autopsynegative sudden unexplained death referred for postmortem genetic testing. Mayo Clin Proc 2012;87:524–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Gollob MH, Jones DL, Krahn AD, et al. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Engl J Med 2006;354:2677–2688. [DOI] [PubMed] [Google Scholar]
- 187.Leong IUS, Dryland PA, Prosser DO, et al. Splice site variants in the KCNQ1 and SCN5A genes: transcript analysis as a tool in supporting pathogenicity. J Clin Med Res 2017;9:709–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Hofman N, Tan HL, Alders M, et al. Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years’ experience. Circulation 2013; 128:1513–1521. [DOI] [PubMed] [Google Scholar]
- 189.Papadakis M, Papatheodorou E, Mellor G, et al. The diagnostic yield of Brugada syndrome after sudden death with normal autopsy. J Am Coll Cardiol 2018; 71:1204–1214. [DOI] [PubMed] [Google Scholar]
- 190.Elger BS, Michaud K, Fellmann F, Mangin P. Sudden death: ethical and legal problems of post-mortem forensic genetic testing for hereditary cardiac diseases. Clin Genet 2010;77:287–292. [DOI] [PubMed] [Google Scholar]
- 191.Michaud K, Fellmann F, Abriel H, Beckmann JS, Mangin P, Elger BS. Molecular autopsy in sudden cardiac death and its implication for families: discussion of the practical, legal and ethical aspects of the multidisciplinary collaboration. Swiss Med Wkly 2009;139:712–718. [DOI] [PubMed] [Google Scholar]
- 192.Lahrouchi N, Raju H, Lodder EM, et al. The yield of postmortem genetic testing in sudden death cases with structural findings at autopsy. Eur J Hum Genet 2020; 28:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hofman N, Tan HL, Alders M, van Langen IM, Wilde AA. Active cascade screening in primary inherited arrhythmia syndromes: does it lead to prophylactic treatment? J Am Coll Cardiol 2010;55:2570–2576. [DOI] [PubMed] [Google Scholar]
- 194.Manrai AK, Funke BH, Rehm HL, et al. Genetic misdiagnoses and the potential for health disparities. N Engl J Med 2016;375:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Hosseini SM, Kim R, Udupa S, et al. Reappraisal of reported genes for sudden arrhythmic death. Circulation 2018;138:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Adler A, Novelli V, Amin AS, et al. An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 2020;141:418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Ackerman MJ, Tester DJ, Jones GS, Will ML, Burrow CR, Curran ME. Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin Proc 2003;78:1479–1487. [DOI] [PubMed] [Google Scholar]
- 198.Ackerman MJ, Splawski I, Makielski JC, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 2004;1:600–607. [DOI] [PubMed] [Google Scholar]
- 199.Kapa S, Tester DJ, Salisbury BA, et al. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation 2009; 120:1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Giudicessi JR, Roden DM, Wilde AAM, Ackerman MJ. Classification and reporting of potentially proarrhythmic common genetic variation in long QT syndrome genetic testing. Circulation 2018;137:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Giudicessi JR, Wilde AAM, Ackerman MJ. The genetic architecture of long QT syndrome: a critical reappraisal. Trends Cardiovasc Med 2018; 28:453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ingles J, Goldstein J, Thaxton C, et al. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med 2019;12:e002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, Ackerman MJ. The promise and peril of precision medicine: phenotyping still matters most. Mayo Clin Proc 2016;91:1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Carrieri D, Howard HC, Benjamin C, et al. Recontacting patients in clinical genetics services: recommendations of the European Society of Human Genetics. Eur J Hum Genet 2019;27:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.van der Werf C, Stiekema L, Tan HL, et al. Low rate of cardiac events in first-degree relatives of diagnosis-negative young sudden unexplained death syndrome victims during follow-up. Heart Rhythm 2014;11:1728–1732. [DOI] [PubMed] [Google Scholar]
- 206.van der Werf C, Hendrix A, Birnie E, et al. Improving usual care after sudden death in the young with focus on inherited cardiac diseases (the CAREFUL study): a community-based intervention study. Europace 2016;18:592–601. [DOI] [PubMed] [Google Scholar]
- 207.Tester DJ, Spoon DB, Valdivia HH, Makielski JC, Ackerman MJ. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: a molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin Proc 2004;79:1380–1384. [DOI] [PubMed] [Google Scholar]
- 208.Anderson JH, Tester DJ, Will ML, Ackerman MJ. Whole-exome molecular autopsy after exertion-related sudden unexplained death in the young. Circ Cardiovasc Genet 2016;9:259–265. [DOI] [PubMed] [Google Scholar]
- 209.Giudicessi JR, Ackerman MJ. Role of genetic heart disease in sentinel sudden cardiac arrest survivors across the age spectrum. Int J Cardiol 2018; 270:214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Shanks GW, Tester DJ, Ackerman JP, et al. Importance of variant interpretation in whole-exome molecular autopsy: population-based case series. Circulation 2018;137:2705–2715. [DOI] [PubMed] [Google Scholar]
- 211.Narula N, Tester DJ, Paulmichl A, Maleszewski JJ, Ackerman MJ. Post-mortem whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol 2015; 36:768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Beckmann BM, Wilde AA, Kaab S. Dual inheritance of sudden death from cardiovascular causes. N Engl J Med 2008;358:2077–2078. [DOI] [PubMed] [Google Scholar]
- 213.Ackerman MJ. Genetic purgatory and the cardiac channelopathies: exposing the variants of uncertain/unknown significance issue. Heart Rhythm 2015; 12:2325–2331. [DOI] [PubMed] [Google Scholar]
- 214.Clift K, Macklin S, Halverson C, McCormick JB, Abu Dabrh AM, Hines S. Patients’ views on variants of uncertain significance across indications. J Community Genet 2020;11:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Van Driest SL, Wells QS, Stallings S, et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 2016;315:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Krahn AD, Healey JS, Chauhan V, et al. Systematic assessment of patients with unexplained cardiac arrest: Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER). Circulation 2009;120:278–285. [DOI] [PubMed] [Google Scholar]
- 217.Marstrand P, Corell P, Henriksen FL, Pehrson S, Bundgaard H, Theilade J. Clinical evaluation of unselected cardiac arrest survivors in a tertiary center over a 1-year period (the LAZARUZ study). J Electrocardiol 2016;49:707–713. [DOI] [PubMed] [Google Scholar]
- 218.Waldmann V, Bougouin W, Karam N, et al. Characteristics and clinical assessment of unexplained sudden cardiac arrest in the real-world setting: focus on idiopathic ventricular fibrillation. Eur Heart J 2018;39:1981–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Allan KS, Morrison LJ, Pinter A, Tu JV, Dorian P, Rescu I. Unexpected high prevalence of cardiovascular disease risk factors and psychiatric disease among young people with sudden cardiac arrest. J Am Heart Assoc 2019;8:e010330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Herman AR, Cheung C, Gerull B, et al. Outcome of apparently unexplained cardiac arrest: results from investigation and follow-up of the prospective Cardiac Arrest Survivors with Preserved Ejection Fraction Registry. Circ Arrhythm Electrophysiol 2016;9:e003619. [DOI] [PubMed] [Google Scholar]
- 221.Finocchiaro G, Papadakis M, Dhutia H, et al. Obesity and sudden cardiac death in the young: clinical and pathological insights from a large national registry. Eur J Prev Cardiol 2018;25:395–401. [DOI] [PubMed] [Google Scholar]
- 222.Mangili LC, Miname MH, Silva PRS, et al. Achilles tendon xanthomas are associated with the presence and burden of subclinical coronary atherosclerosis in heterozygous familial hypercholesterolemia: a pilot study. Atherosclerosis 2017;263:393–397. [DOI] [PubMed] [Google Scholar]
- 223.Wang S, Peng D. Cardiac involvement in Emery-Dreifuss muscular dystrophy and related management strategies. Int Heart J 2019;60:12–18. [DOI] [PubMed] [Google Scholar]
- 224.Clemens DJ, Tester DJ, Giudicessi JR, et al. International Triadin Knockout Syndrome Registry. Circ Genom Precis Med 2019;12:e002419. [DOI] [PubMed] [Google Scholar]
- 225.Walsh MA, Turner C, Timothy KW, et al. A multicentre study of patients with Timothy syndrome. Europace 2018;20:377–385. [DOI] [PubMed] [Google Scholar]
- 226.Bitterman AD, Sponseller PD. Marfan syndrome: a clinical update. J Am Acad Orthop Surg 2017;25:603–609. [DOI] [PubMed] [Google Scholar]
- 227.Abrams DJ. How to develop a clinic for sudden cardiac arrest survivors and families of non-survivors. Cardiol Young 2017;27:S3–S9. [DOI] [PubMed] [Google Scholar]
- 228.Tseng ZH, Olgin JE, Vittinghoff E, et al. Prospective Countywide Surveillance and Autopsy Characterization of Sudden Cardiac Death: POST SCD Study. Circulation 2018;137:2689–2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Colquhoun MC, Chamberlain DA, Newcombe RG, et al. A national scheme for public access defibrillation in England and Wales: early results. Resuscitation 2008;78:275–280. [DOI] [PubMed] [Google Scholar]
- 230.Zijlstra JA, Bekkers LE, Hulleman M, Beesems SG, Koster RW. Automated external defibrillator and operator performance in out-of-hospital cardiac arrest. Resuscitation 2017;118:140–146. [DOI] [PubMed] [Google Scholar]
- 231.Yamamoto T, Takayama M, Sato N, et al. Inappropriate analyses of automated external defibrillators used during in-hospital ventricular fibrillation. Circ J 2008;72:679–681. [DOI] [PubMed] [Google Scholar]
- 232.Savastano S, Rordorf R, Vicentini A, et al. A comprehensive electrocardiographic, molecular, and echocardiographic study of Brugada syndrome: validation of the 2013 diagnostic criteria. Heart Rhythm 2014;11:1176–1183. [DOI] [PubMed] [Google Scholar]
- 233.Govindan M, Batchvarov VN, Raju H, et al. Utility of high and standard right precordial leads during ajmaline testing for the diagnosis of Brugada syndrome. Heart 2010;96:1904–1908. [DOI] [PubMed] [Google Scholar]
- 234.Curcio A, Mazzanti A, Bloise R, et al. Clinical presentation and outcome of Brugada syndrome diagnosed with the new 2013 criteria. J Cardiovasc Electrophysiol 2016;27:937–943. [DOI] [PubMed] [Google Scholar]
- 235.Miyamoto K, Yokokawa M, Tanaka K, et al. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol 2007;99:53–57. [DOI] [PubMed] [Google Scholar]
- 236.Haïssaguerre M, Shah DC, Jaïs P, et al. Role of Purkinje conducting system in triggering of idiopathic ventricular fibrillation. Lancet 2002;359:677–678. [DOI] [PubMed] [Google Scholar]
- 237.Divakaran S, Stewart GC, Lakdawala NK, et al. Diagnostic accuracy of advanced imaging in cardiac sarcoidosis. Circ Cardiovasc Imaging 2019; 12:e008975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.White JA, Fine NM, Gula L, et al. Utility of cardiovascular magnetic resonance in identifying substrate for malignant ventricular arrhythmias. Circ Cardiovasc Imaging 2012;5:12–20. [DOI] [PubMed] [Google Scholar]
- 239.Basso C, Perazzolo Marra M, Rizzo S, et al. Arrhythmic mitral valve prolapse and sudden cardiac death. Circulation 2015;132:556–566. [DOI] [PubMed] [Google Scholar]
- 240.Basso C, Iliceto S, Thiene G, Perazzolo Marra M. Mitral valve prolapse, ventricular arrhythmias, and sudden death. Circulation 2019;140:952–964. [DOI] [PubMed] [Google Scholar]
- 241.Eckart RE, Scoville SL, Campbell CL, et al. Sudden death in young adults: a 25-year review of autopsies in military recruits. Ann Intern Med 2004;141:829–834. [DOI] [PubMed] [Google Scholar]
- 242.Neumar RW, Shuster M, Callaway CW, et al. Part 1: Executive Summary: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation 2015;132:S315–S367. [DOI] [PubMed] [Google Scholar]
- 243.Avari Silva JN, Bromberg BI, Emge FK, Bowman TM, Van Hare GF. Implantable loop recorder monitoring for refining management of children with inherited arrhythmia syndromes. J Am Heart Assoc 2016;5:e003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Placidi S, Drago F, Milioni M, et al. Miniaturized implantable loop recorder in small patients: an effective approach to the evaluation of subjects at risk of sudden death. Pacing Clin Electrophysiol 2016;39:669–674. [DOI] [PubMed] [Google Scholar]
- 245.Sweeney MO, Ruetz LL, Belk P, Mullen TJ, Johnson JW, Sheldon T. Bradycardia pacing-induced short-long-short sequences at the onset of ventricular tachyarrhythmias: a possible mechanism of proarrhythmia? J Am Coll Cardiol 2007;50:614–622. [DOI] [PubMed] [Google Scholar]
- 246.Jiménez-Jáimez J, Peinado R, Grima EZ, et al. Diagnostic approach to unexplained cardiac arrest (from the FIVI-Gen Study). Am J Cardiol 2015; 116:894–899. [DOI] [PubMed] [Google Scholar]
- 247.Kamath GS, Zareba W, Delaney J, et al. Value of the signal-averaged electrocardiogram in arrhythmogenic right ventricular cardiomyopathy/dysplasia. Heart Rhythm 2011;8:256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Rodrigues P, Joshi A, Williams H, et al. Diagnosis and prognosis in sudden cardiac arrest survivors without coronary artery disease: utility of a clinical approach using cardiac magnetic resonance imaging. Circ Cardiovasc Imaging 2017;10:e006709. [DOI] [PubMed] [Google Scholar]
- 249.Ali-Ahmed F, Dalgaard F, Al-Khatib SM. Sudden cardiac death in patients with myocarditis: evaluation, risk stratification, and management. Am Heart J 2020; 220:29–40. [DOI] [PubMed] [Google Scholar]
- 250.Chatterjee D, Pieroni M, Fatah M, et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur Heart J 2020;41:2878–2890. [DOI] [PubMed] [Google Scholar]
- 251.Wilde AAM, Lodder EM. A highly specific biomarker for Brugada syndrome. Also too good to be true? Eur Heart J 2020;41:2891–2893. [DOI] [PubMed] [Google Scholar]
- 252.Tschabrunn CM, Haqqani HM, Santangeli P, Zado ES, Marchlinski FE. 12-Lead electrocardiogram to localize region of abnormal electroanatomic substrate in arrhythmogenic right ventricular cardiomyopathy. JACC Clin Electrophysiol 2017;3:654–665. [DOI] [PubMed] [Google Scholar]
- 253.M€uller D, Schnitzer L, Brandt J, Arntz HR. The accuracy of an out-of-hospital 12-lead ECG for the detection of ST-elevation myocardial infarction immediately after resuscitation. Ann Emerg Med 2008;52:658–664. [DOI] [PubMed] [Google Scholar]
- 254.Stær-Jensen H, Nakstad ER, Fossum E, et al. Post-resuscitation ECG for selection of patients for immediate coronary angiography in out-of-hospital cardiac arrest. Circ Cardiovasc Interv 2015;8:e002784. [DOI] [PubMed] [Google Scholar]
- 255.Eysmann SB, Marchlinski FE, Buxton AE, Josephson ME. Electrocardiographic changes after cardioversion of ventricular arrhythmias. Circulation 1986; 73:73–81. [DOI] [PubMed] [Google Scholar]
- 256.Kim YJ, Min SY, Lee DH, et al. The role of post-resuscitation electrocardiogram in patients with ST-segment changes in the immediate post-cardiac arrest period. JACC Cardiovasc Interv 2017;10:451–459. [DOI] [PubMed] [Google Scholar]
- 257.Rolfast CL, Lust EJ, de Cock CC. Electrocardiographic changes in therapeutic hypothermia. Crit Care 2012;16:R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Salinas P, Lopez-de-Sa E, Pena-Conde L, et al. Electrocardiographic changes during induced therapeutic hypothermia in comatose survivors after cardiac arrest. World J Cardiol 2015;7:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bière L, Niro M, Pouliquen H, et al. Risk of ventricular arrhythmia in patients with myocardial infarction and non-obstructive coronary arteries and normal ejection fraction. World J Cardiol 2017;9:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Marcus FI, McKenna WJ, Sherrill D, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation 2010;121:1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Hamid MS, Norman M, Quraishi A, et al. Prospective evaluation of relatives for familial arrhythmogenic right ventricular cardiomyopathy/dysplasia reveals a need to broaden diagnostic criteria. J Am Coll Cardiol 2002;40:1445–1450. [DOI] [PubMed] [Google Scholar]
- 262.Nava A, Bauce B, Basso C, et al. Clinical profile and long-term follow-up of 37 families with arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2000;36:2226–2233. [DOI] [PubMed] [Google Scholar]
- 263.Perrin MJ, Angaran P, Laksman Z, et al. Exercise testing in asymptomatic gene carriers exposes a latent electrical substrate of arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol 2013;62:1772–1779. [DOI] [PubMed] [Google Scholar]
- 264.Amin AS, de Groot EA, Ruijter JM, Wilde AA, Tan HL. Exercise-induced ECG changes in Brugada syndrome. Circ Arrhythm Electrophysiol 2009;2:531–539. [DOI] [PubMed] [Google Scholar]
- 265.Priori SG, Napolitano C, Memmi M, et al. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation 2002;106:69–74. [DOI] [PubMed] [Google Scholar]
- 266.Adler A, van der Werf C, Postema PG, et al. The phenomenon of “QT stunning”: the abnormal QT prolongation provoked by standing persists even as the heart rate returns to normal in patients with long QT syndrome. Heart Rhythm 2012;9:901–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Dionne A, Fournier A, Dahdah N, Abrams D, Khairy P, Abadir S. Dynamic QT interval changes from supine to standing in healthy children. Can J Cardiol 2018; 34:66–72. [DOI] [PubMed] [Google Scholar]
- 268.Filippini L, Postema PG, Zoubin K, et al. The brisk-standing-test for long QT syndrome in prepubertal school children: defining normal. Europace 2018; 20:f108–f112. [DOI] [PubMed] [Google Scholar]
- 269.Viskin S, Postema PG, Bhuiyan ZA, et al. The response of the QT interval to the brief tachycardia provoked by standing: a bedside test for diagnosing long QT syndrome. J Am Coll Cardiol 2010;55:1955–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sy RW, van der Werf C, Chattha IS, et al. Derivation and validation of a simple exercise-based algorithm for prediction of genetic testing in relatives of LQTS probands. Circulation 2011;124:2187–2194. [DOI] [PubMed] [Google Scholar]
- 271.Wong JA, Gula LJ, Klein GJ, Yee R, Skanes AC, Krahn AD. Utility of treadmill testing in identification and genotype prediction in long-QT syndrome. Circ Arrhythm Electrophysiol 2010;3:120–125. [DOI] [PubMed] [Google Scholar]
- 272.Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation 2011;124:2181–2184. [DOI] [PubMed] [Google Scholar]
- 273.Shimizu W, Noda T, Takaki H, et al. Diagnostic value of epinephrine test for genotyping LQT1, LQT2, and LQT3 forms of congenital long QT syndrome. Heart Rhythm 2004;1:276–283. [DOI] [PubMed] [Google Scholar]
- 274.Ackerman MJ, Khositseth A, Tester DJ, Hejlik JB, Shen WK, Porter CB. Epinephrine-induced QT interval prolongation: a gene-specific paradoxical response in congenital long QT syndrome. Mayo Clin Proc 2002;77:413–421. [DOI] [PubMed] [Google Scholar]
- 275.Krahn AD, Healey JS, Chauhan VS, et al. Epinephrine infusion in the evaluation of unexplained cardiac arrest and familial sudden death: from the Cardiac Arrest Survivors with Preserved Ejection Fraction Registry. Circ Arrhythm Electrophysiol 2012;5:933–940. [DOI] [PubMed] [Google Scholar]
- 276.Magnano AR, Talathoti N, Hallur R, Bloomfield DM, Garan H. Sympathomimetic infusion and cardiac repolarization: the normative effects of epinephrine and isoproterenol in healthy subjects. J Cardiovasc Electrophysiol 2006; 17:983–989. [DOI] [PubMed] [Google Scholar]
- 277.Marjamaa A, Hiippala A, Arrhenius B, et al. Intravenous epinephrine infusion test in diagnosis of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol 2012;23:194–199. [DOI] [PubMed] [Google Scholar]
- 278.Denis A, Sacher F, Derval N, et al. Diagnostic value of isoproterenol testing in arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol 2014;7:590–597. [DOI] [PubMed] [Google Scholar]
- 279.Meregalli PG, Ruijter JM, Hofman N, Bezzina CR, Wilde AA, Tan HL. Diagnostic value of flecainide testing in unmasking SCN5A-related Brugada syndrome. J Cardiovasc Electrophysiol 2006;17:857–864. [DOI] [PubMed] [Google Scholar]
- 280.Hong K, Brugada J, Oliva A, et al. Value of electrocardiographic parameters and ajmaline test in the diagnosis of Brugada syndrome caused by SCN5A mutations. Circulation 2004;110:3023–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Brugada R, Brugada J, Antzelevitch C, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation 2000; 101:510–515. [DOI] [PubMed] [Google Scholar]
- 282.Krahn AD, Gollob M, Yee R, et al. Diagnosis of unexplained cardiac arrest: role of adrenaline and procainamide infusion. Circulation 2005; 112:2228–2234. [DOI] [PubMed] [Google Scholar]
- 283.Sangwatanaroj S, Prechawat S, Sunsaneewitayakul B, Sitthisook S, Tosukhowong P, Tungsanga K. New electrocardiographic leads and the procainamide test for the detection of the Brugada sign in sudden unexplained death syndrome survivors and their relatives. Eur Heart J 2001;22:2290–2296. [DOI] [PubMed] [Google Scholar]
- 284.Somani R, Krahn AD, Healey JS, et al. Procainamide infusion in the evaluation of unexplained cardiac arrest: from the Cardiac Arrest Survivors with Preserved Ejection Fraction Registry (CASPER). Heart Rhythm 2014;11:1047–1054. [DOI] [PubMed] [Google Scholar]
- 285.Cheung CC, Mellor G, Deyell MW, et al. Comparison of ajmaline and procainamide provocation tests in the diagnosis of Brugada syndrome. JACC Clin Electrophysiol 2019;5:504–512. [DOI] [PubMed] [Google Scholar]
- 286.Hasdemir C, Payzin S, Kocabas U, et al. High prevalence of concealed Brugada syndrome in patients with atrioventricular nodal reentrant tachycardia. Heart Rhythm 2015;12:1584–1594. [DOI] [PubMed] [Google Scholar]
- 287.Ikeda T, Abe A, Yusu S, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol 2006;17:602–607. [DOI] [PubMed] [Google Scholar]
- 288.Huchet F, Kyndt F, Barc J, et al. Familial catecholamine-induced QT prolongation in unexplained sudden cardiac death. J Am Coll Cardiol 2017; 69:1642–1643. [DOI] [PubMed] [Google Scholar]
- 289.Kannankeril PJ, Roden DM, Norris KJ, Whalen SP, George AL Jr, Murray KT. Genetic susceptibility to acquired long QT syndrome: pharmacologic challenge in first-degree relatives. Heart Rhythm 2005;2:134–140. [DOI] [PubMed] [Google Scholar]
- 290.Etienne P, Huchet F, Gaborit N, et al. Mental stress test: a rapid, simple, and efficient test to unmask long QT syndrome. Europace 2018;20:2014–2020. [DOI] [PubMed] [Google Scholar]
- 291.Kaab S, Hinterseer M, Nabauer M, Steinbeck G. Sotalol testing unmasks altered repolarization in patients with suspected acquired long-QT-syndrome: a case-control pilot study using i.v. sotalol. Eur Heart J 2003;24:649–657. [DOI] [PubMed] [Google Scholar]
- 292.Kamakura T, Wada M, Ishibashi K, et al. Significance of coronary artery spasm diagnosis in patients with early repolarization syndrome. J Am Heart Assoc 2018;7:e007942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Beltrame JF, Crea F, Kaski JC, et al. International standardization of diagnostic criteria for vasospastic angina. Eur Heart J 2017;38:2565–2568. [DOI] [PubMed] [Google Scholar]
- 294.Nakao K, Ohgushi M, Yoshimura M, et al. Hyperventilation as a specific test for diagnosis of coronary artery spasm. Am J Cardiol 1997;80:545–549. [DOI] [PubMed] [Google Scholar]
- 295.Waldmann V, Bougouin W, Karam N, et al. Coronary vasospasm-related sudden cardiac arrest in the community. J Am Coll Cardiol 2018;72:814–815. [DOI] [PubMed] [Google Scholar]
- 296.Garratt CJ, Antoniou A, Griffith MJ, Ward DE, Camm AJ. Use of intravenous adenosine in sinus rhythm as a diagnostic test for latent preexcitation. Am J Cardiol 1990;65:868–873. [DOI] [PubMed] [Google Scholar]
- 297.Foo FS, Stiles MK, Heaven D. Unmasking latent preexcitation of a right-sided accessory pathway with intravenous adenosine after unexplained sudden cardiac arrest. J Arrhythm 2020;36:939–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Wang YS, Scheinman MM, Chien WW, Cohen TJ, Lesh MD, Griffin JC. Patients with supraventricular tachycardia presenting with aborted sudden death: incidence, mechanism and long-term follow-up. J Am Coll Cardiol 1991; 18:1711–1719. [DOI] [PubMed] [Google Scholar]
- 299.Roberts JD, Gollob MH, Young C, et al. Bundle branch re-entrant ventricular tachycardia: novel genetic mechanisms in a life-threatening arrhythmia. JACC Clin Electrophysiol 2017;3:276–288. [DOI] [PubMed] [Google Scholar]
- 300.Santangeli P, Hamilton-Craig C, Dello Russo A, et al. Imaging of scar in patients with ventricular arrhythmias of right ventricular origin: cardiac magnetic resonance versus electroanatomic mapping. J Cardiovasc Electrophysiol 2011; 22:1359–1366. [DOI] [PubMed] [Google Scholar]
- 301.Haissaguerre M, Hocini M, Cheniti G, et al. Localized structural alterations underlying a subset of unexplained sudden cardiac death. Circ Arrhythm Electrophysiol 2018;11:e006120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Horner JM, Horner MM, Ackerman MJ. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm 2011;8:1698–1704. [DOI] [PubMed] [Google Scholar]
- 303.Giudicessi JR, Ackerman MJ. Exercise testing oversights underlie missed and delayed diagnosis of catecholaminergic polymorphic ventricular tachycardia in young sudden cardiac arrest survivors. Heart Rhythm 2019;16:1232–1239. [DOI] [PubMed] [Google Scholar]
- 304.Vyas H, Hejlik J, Ackerman MJ. Epinephrine QT stress testing in the evaluation of congenital long-QT syndrome: diagnostic accuracy of the paradoxical QT response. Circulation 2006;113:1385–1392. [DOI] [PubMed] [Google Scholar]
- 305.Rehm HL, Berg JS, Brooks LD, et al. ClinGen: the clinical genome resource. N Engl J Med 2015;372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011;124:2761–2796. [DOI] [PubMed] [Google Scholar]
- 308.Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J. Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ Cardiovasc Genet 2017;10:e001666. [DOI] [PubMed] [Google Scholar]
- 309.Amendola LM, Dorschner MO, Robertson PD, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res 2015;25:305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Mohammed S, Lim Z, Dean PH, et al. Genetic insurance discrimination in sudden arrhythmia death syndromes: empirical evidence from a cross-sectional survey in North America. Circ Cardiovasc Genet 2017;10:e001442. [DOI] [PubMed] [Google Scholar]
- 311.Moss AJ, Zareba W, Hall WJ, et al. Effectiveness and limitations of beta-blocker therapy in congenital long-QT syndrome. Circulation 2000;101:616–623. [DOI] [PubMed] [Google Scholar]
- 312.Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate: implications for gene-specific therapy. Circulation 1995;92:3381–3386. [DOI] [PubMed] [Google Scholar]
- 313.Moss AJ, Windle JR, Hall WJ, et al. Safety and efficacy of flecainide in subjects with Long QT-3 syndrome (DeltaKPQ mutation): a randomized, double-blind, placebo-controlled clinical trial. Ann Noninvasive Electrocardiol 2005; 10:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Moss AJ, Zareba W, Schwarz KQ, Rosero S, McNitt S, Robinson JL. Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol 2008;19:1289–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kannankeril PJ, Moore JP, Cerrone M, et al. Efficacy of flecainide in the treatment of catecholaminergic polymorphic ventricular tachycardia: a randomized clinical trial. JAMA Cardiol 2017;2:759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol 2013; 62:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Ruwald AC, Marcus F, Estes NA 3rd, et al. Association of competitive and recreational sport participation with cardiac events in patients with arrhythmogenic right ventricular cardiomyopathy: results from the North American multidisciplinary study of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J 2015;36:1735–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.van Rijsingen IA, van der Zwaag PA, Groeneweg JA, et al. Outcome in phospholamban R14del carriers: results of a large multicentre cohort study. Circ Cardiovasc Genet 2014;7:455–465. [DOI] [PubMed] [Google Scholar]
- 319.van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in Lamin A/C mutation carriers: a European cohort study. J Am Coll Cardiol 2012;59:493–500. [DOI] [PubMed] [Google Scholar]
- 320.De Ferrari GM, Dusi V, Spazzolini C, et al. Clinical management of catecholaminergic polymorphic ventricular tachycardia: the role of left cardiac sympathetic denervation. Circulation 2015;131:2185–2193. [DOI] [PubMed] [Google Scholar]
- 321.Coleman MA, Bos JM, Johnson JN, et al. Videoscopic left cardiac sympathetic denervation for patients with recurrent ventricular fibrillation/malignant ventricular arrhythmia syndromes besides congenital long-QT syndrome. Circ Arrhythm Electrophysiol 2012;5:782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Bos JM, Crotti L, Rohatgi RK, et al. Mexiletine shortens the QT interval in patients with potassium channel-mediated type 2 long QT syndrome. Circ Arrhythm Electrophysiol 2019;12:e007280. [DOI] [PubMed] [Google Scholar]
- 323.Asatryan B, Schaller A, Seiler J, et al. Usefulness of genetic testing in sudden cardiac arrest survivors with or without previous clinical evidence of heart disease. Am J Cardiol 2019;123:2031–2038. [DOI] [PubMed] [Google Scholar]
- 324.Ashar FN, Mitchell RN, Albert CM, et al. A comprehensive evaluation of the genetic architecture of sudden cardiac arrest. Eur Heart J 2018;39:3961–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Milano A, Blom MT, Lodder EM, et al. Sudden cardiac arrest and rare genetic variants in the community. Circ Cardiovasc Genet 2016;9:147–153. [DOI] [PubMed] [Google Scholar]
- 326.Visser M, Dooijes D, van der Smagt JJ, et al. Next-generation sequencing of a large gene panel in patients initially diagnosed with idiopathic ventricular fibrillation. Heart Rhythm 2017;14:1035–1040. [DOI] [PubMed] [Google Scholar]
- 327.Visser M, van der Heijden JF, van der Smagt JJ, et al. Long-term outcome of patients initially diagnosed with idiopathic ventricular fibrillation: a descriptive study. Circ Arrhythm Electrophysiol 2016;9:e004258. [DOI] [PubMed] [Google Scholar]
- 328.Ingles J, Bagnall RD, Yeates L, et al. Concealed arrhythmogenic right ventricular cardiomyopathy in sudden unexplained cardiac death events. Circ Genom Precis Med 2018;11:e002355. [DOI] [PubMed] [Google Scholar]
- 329.Yang P, Kanki H, Drolet B, et al. Allelic variants in long-QT disease genes in patients with drug-associated torsades de pointes. Circulation 2002; 105:1943–1948. [DOI] [PubMed] [Google Scholar]
- 330.Tester DJ, Ackerman JP, Giudicessi JR, et al. Plakophilin-2 truncation variants in patients clinically diagnosed with catecholaminergic polymorphic ventricular tachycardia and decedents with exercise-associated autopsy negative sudden unexplained death in the young. JACC Clin Electrophysiol 2019;5:120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.van der Werf C, Kannankeril PJ, Sacher F, et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J Am Coll Cardiol 2011;57:2244–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Semsarian C, Ingles J, Wilde AA. Sudden cardiac death in the young: the molecular autopsy and a practical approach to surviving relatives. Eur Heart J 2015; 36:1290–1296. [DOI] [PubMed] [Google Scholar]
- 333.Asatryan B, Medeiros-Domingo A. Molecular and genetic insights into progressive cardiac conduction disease. Europace 2019;21:1145–1158. [DOI] [PubMed] [Google Scholar]
- 334.Quenin P, Kyndt F, Mabo P, et al. Clinical yield of familial screening after sudden death in young subjects: the French experience. Circ Arrhythm Electrophysiol 2017;10:e005236. [DOI] [PubMed] [Google Scholar]
- 335.Cornel MC, van El CG. Barriers and facilitating factors for implementation of genetic services: a public health perspective. Front Public Health 2017;5:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Charron P, Heron D, Gargiulo M, et al. Prenatal molecular diagnosis in hypertrophic cardiomyopathy: report of the first case. Prenat Diagn 2004;24:701–703. [DOI] [PubMed] [Google Scholar]
- 337.Harper JC, Wilton L, Traeger-Synodinos J, et al. The ESHRE PGD Consortium: 10 years of data collection. Hum Reprod Update 2012;18:234–247. [DOI] [PubMed] [Google Scholar]
- 338.Kuliev A, Pomerantseva E, Polling D, Verlinsky O, Rechitsky S. PGD for inherited cardiac diseases. Reprod Biomed Online 2012;24:443–453. [DOI] [PubMed] [Google Scholar]
- 339.van Velzen HG, Schinkel AFL, Baart SJ, et al. Outcomes of contemporary family screening in hypertrophic cardiomyopathy. Circ Genom Precis Med 2018; 11:e001896. [DOI] [PubMed] [Google Scholar]
- 340.Medeiros Domingo A, Bolliger S, Grani C, et al. Recommendations for genetic testing and counselling after sudden cardiac death: practical aspects for Swiss practice. Swiss Med Wkly 2018;148:w14638. [DOI] [PubMed] [Google Scholar]
- 341.Rootwelt-Norberg C, Lie OH, Dejgaard LA, et al. Life-threatening arrhythmic presentation in patients with arrhythmogenic cardiomyopathy before and after entering the genomic era: a two-decade experience from a large volume center. Int J Cardiol 2019;279:79–83. [DOI] [PubMed] [Google Scholar]
- 342.Lafreniere-Roula M, Bolkier Y, Zahavich L, et al. Family screening for hypertrophic cardiomyopathy: is it time to change practice guidelines? Eur Heart J 2019;40:3672–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Christian S, Atallah J, Clegg R, et al. Uptake of predictive genetic testing and cardiac evaluation for children at risk for an inherited arrhythmia or cardiomyopathy. J Genet Couns 2018;27:124–130. [DOI] [PubMed] [Google Scholar]
- 344.Christian S, Somerville M, Huculak C, Atallah J. Practice variation among an international group of genetic counselors on when to offer predictive genetic testing to children at risk of an inherited arrhythmia or cardiomyopathy. J Genet Couns 2019;28:70–79. [DOI] [PubMed] [Google Scholar]
- 345.Harris S, Cirino AL, Carr CW, et al. The uptake of family screening in hypertrophic cardiomyopathy and an online video intervention to facilitate family communication. Mol Genet Genomic Med 2019;7:e940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Knight LM, Miller E, Kovach J, et al. Genetic testing and cascade screening in pediatric long QT syndrome and hypertrophic cardiomyopathy. Heart Rhythm 2020;17:106–112. [DOI] [PubMed] [Google Scholar]
- 347.Shah LL, Daack-Hirsch S, Ersig AL, Paik A, Ahmad F, Williams J. Family relationships associated with communication and testing for inherited cardiac conditions. West J Nurs Res 2019;41:1576–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Adler A, Sadek MM, Chan AY, et al. Patient outcomes from a specialized inherited arrhythmia clinic. Circ Arrhythm Electrophysiol 2016;9:e003440. [DOI] [PubMed] [Google Scholar]
- 349.Boers SN, van Delden JJ, Knoers NV, Bredenoord AL. Postmortem disclosure of genetic information to family members: active or passive? Trends Mol Med 2015;21:148–153. [DOI] [PubMed] [Google Scholar]
- 350.Fox D, Spencer E, Torkamani A. Returning results to family members: professional duties in genomics research in the United States. J Leg Med 2018; 38:201–219. [DOI] [PubMed] [Google Scholar]
- 351.van El CG, Baccolini V, Piko P, Cornel MC. Stakeholder views on active cascade screening for familial hypercholesterolemia. Healthcare (Basel) 2018; 6:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Kauferstein S, Herz N, Scheiper S, et al. Relevance of molecular testing in patients with a family history of sudden death. Forensic Sci Int 2017;276:18–23. [DOI] [PubMed] [Google Scholar]
- 353.Roston TM, Dewar L, Franciosi S, et al. The accessibility and utilization of genetic testing for inherited heart rhythm disorders: a Canadian cross-sectional survey study. J Community Genet 2018;9:257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Shah LL, Daack-Hirsch S. Family communication about genetic risk of hereditary cardiomyopathies and arrhythmias: an integrative review. J Genet Couns 2018;27:1022–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Catchpool M, Ramchand J, Martyn M, et al. A cost-effectiveness model of genetic testing and periodical clinical screening for the evaluation of families with dilated cardiomyopathy. Genet Med 2019;21:2815–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Mak CM, Mok NS, Shum HC, et al. Sudden arrhythmia death syndrome in young victims: a five-year retrospective review and two-year prospective molecular autopsy study by next-generation sequencing and clinical evaluation of their first-degree relatives. Hong Kong Med J 2019;25:21–29. [DOI] [PubMed] [Google Scholar]
- 357.van den Heuvel LM, Huisinga MJ, Hoedemaekers YM, et al. Informing relatives at risk of inherited cardiac conditions: experiences and attitudes of healthcare professionals and counselees. Eur J Hum Genet 2019;27:1341–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Broekhuizen K, van Poppel MN, Koppes LL, Kindt I, Brug J, van Mechelen W. No significant improvement of cardiovascular disease risk indicators by a lifestyle intervention in people with familial hypercholesterolemia compared to usual care: results of a randomised controlled trial. BMC Res Notes 2012;5:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Raju H, Parsons S, Thompson TN, et al. Insights into sudden cardiac death: exploring the potential relevance of non-diagnostic autopsy findings. Eur Heart J 2019;40:831–838. [DOI] [PubMed] [Google Scholar]
- 360.te Riele AS, James CA, Rastegar N, et al. Yield of serial evaluation in at-risk family members of patients with ARVD/C. J Am Coll Cardiol 2014;64:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Hasdemir C, Juang JJ, Kose S, et al. Coexistence of atrioventricular accessory pathways and drug-induced type 1 Brugada pattern. Pacing Clin Electrophysiol 2018;41:1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Conte G, de Asmundis C, Ciconte G, et al. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA 2014;312:2039–2041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.