

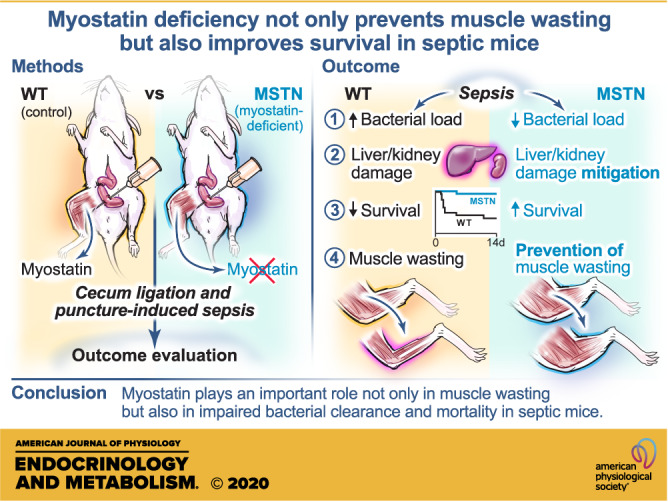
Keywords: bacterial clearance, mortality, muscle wasting, myostatin, sepsis
Abstract
Sepsis remains a leading cause of mortality in critically ill patients. Muscle wasting is a major complication of sepsis and negatively affects clinical outcomes. Despite intense investigation for many years, the molecular mechanisms underlying sepsis-related muscle wasting are not fully understood. In addition, a potential role of muscle wasting in disease development of sepsis has not been studied. Myostatin is a myokine that downregulates skeletal muscle mass. We studied the effects of myostatin deficiency on muscle wasting and other clinically relevant outcomes, including mortality and bacterial clearance, in mice. Myostatin deficiency prevented muscle atrophy along with inhibition of increases in muscle-specific RING finger protein 1 (MuRF-1) and atrogin-1 expression and phosphorylation of signal transducer and activator of transcription protein 3 (STAT3; major players of muscle wasting) in septic mice. Moreover, myostatin deficiency improved survival and bacterial clearance of septic mice. Sepsis-induced liver dysfunction, acute kidney injury, and neutrophil infiltration into the liver and kidney were consistently mitigated by myostatin deficiency, as indicated by plasma concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT), and neutrophil gelatinase-associated lipocalin (NGAL) and myeloperoxidase activity in the organs. Myostatin deficiency also inhibited sepsis-induced increases in plasma high-mobility group protein B1 (HMGB1) and macrophage inhibitory cytokine (MIC)-1/growth differentiation factor (GDF)-15 concentrations. These results indicate that myostatin plays an important role not only in muscle wasting but also in other clinically relevant outcomes in septic mice. Furthermore, our data raise the possibility that muscle wasting may not be simply a complication, but myostatin-mediated muscle cachexia and related changes in muscle may actually drive the development of sepsis as well.
NEW & NOTEWORTHY Muscle wasting is a major complication of sepsis, but its role in the disease development is not known. Myostatin deficiency improved bacterial clearance and survival and mitigated damage in the liver and kidney in septic mice, which paralleled prevention of muscle wasting. These results raise the possibility that muscle wasting may not simply be a complication of sepsis, but myostatin-mediated cachexic changes may have a role in impaired bacterial clearance and mortality in septic mice.
INTRODUCTION
Sepsis, defined as life-threatening organ dysfunction caused by a dysregulated host response to infection (1), is a major cause of morbidity and mortality of critically ill patients. Although in-hospital mortality rate of patients with sepsis decreased during these decades (2, 3), it still remains a leading cause of mortality in critically ill patients. New strategies, therefore, are urgently needed to further reduce the mortality and improve other clinical outcomes, including multiorgan dysfunction and muscle weakness, of patients with sepsis.
Muscle wasting is a major complication of sepsis that negatively affects clinical outcomes. Muscle wasting leads to difficulties in weaning off from mechanical ventilation in patients with sepsis, which, in turn, increases the risk of pneumonia, contributing to prolonged stays in the intensive care unit (ICU) and even death (4). Muscle wasting also affects the long-term clinical outcomes of sepsis. It causes decreased activities of daily living (ADL), prolongs rehabilitation, and worsens the quality of life of sepsis survivors (5). Previous studies have shown that low muscle mass (6), muscle weakness (7), and progressive muscle loss (8) are predictors for mortality in patients with sepsis along with cirrhosis or shock and in mechanically ventilated critically ill patients. These previous studies imply an association between muscle wasting and the prognosis for at least some of critically ill patients. However, it is not known whether these correlations reflect a cause-effect relationship, or whether muscle atrophy is merely a complication that does not have a pathogenic role in the disease development but just mirrors the severity of the disease.
The molecular mechanisms by which critical illness, including sepsis, induces muscle wasting are not fully understood. Myostatin, which is also known as growth differentiation factor (GDF)-8, is primarily produced and secreted by skeletal muscle and mainly acts locally in an autocrine and/or a paracrine manner rather than as an endocrine mode of action (9). Myostatin has been shown to decrease skeletal muscle mass by activating the activin type II receptor pathway (10). Genetic deficiency of myostatin by knockout or loss-of-function mutation of the MSTN (the gene encoding myostatin), therefore, results in increased skeletal muscle mass in mice (11–14), cattle (15–17), sheep (18), dogs (19), and humans (20, 21). Conversely, systemic administration of myostatin is sufficient to induce muscle atrophy in mice (22). Previous studies have shown that genetic or pharmacological inhibition of myostatin prevents or ameliorates muscle atrophy induced by cancer (23, 24), disuse (25), aging (26, 27), and glucocorticoids (1). However, the role of myostatin in muscle wasting has not yet been studied in critical illness, including sepsis.
Skeletal muscle mass adapts depending on physical activity, nutrition, diseases, and aging. Evolutionarily, when accessibility to food is limited, inhibition of muscle hypertrophy by myostatin is an adaptive response for animals to efficiently use limited amounts of available nutrients in the body. Thus, myostatin contributes to muscle homeostasis as a negative regulator. In addition, in case of prolonged starvation, increased muscle proteolysis, which myostatin promotes (28–30), serves as a major source of amino acids that the liver utilizes for gluconeogenesis to help survive under harsh conditions.
Myostatin increases skeletal muscle proteolysis at least in part by increasing muscle-specific RING finger protein 1 (MuRF-1) and atrogin-1 expression (29, 30). Myostatin also inhibits protein synthesis in skeletal muscle (30). However, inhibition of myostatin prevented glucocorticoid-induced muscle wasting by reversing increased proteolysis in mice, whereas suppressed protein synthesis was not altered (31). It is conceivable, therefore, that unlike proteolysis, the role of myostatin in protein synthesis may vary dependent on pathophysiological conditions.
Bacterial clearance and multiorgan dysfunction are major determinants of clinical trajectories in patients with sepsis. Here, we studied the effects of myostatin deficiency on survival, bacterial clearance, and organ damage, as well as muscle wasting, in septic mice. To assess organ damage, we measured indicators of neutrophil infiltration into the liver and kidney, liver dysfunction, acute kidney injury, and circulating level of high-mobility group protein B1 (HMGB1), a damage-associated molecular pattern.
MATERIALS AND METHODS
Animals
Heterozygous myostatin-deficient mice on C57BL/6 background [Mstn lean (Ln)/J mice, Stock No. 009345) were purchased from The Jackson Laboratory (Bar Harbor, ME) and used to generate homozygous myostatin-deficient mice that harbor homozygous MstnLN mutation. Myostatin-deficient mice were backcrossed onto wild-type C57BL/6 mice seven generations. The myostatin deficiency in these mice is the result of a frame shift mutation in the MSTN gene, which results in a premature stop codon and loss of function (11, 14). Both male homozygous myostatin-deficient mice and wild-type (WT) C57BL/6 mice (The Jackson Laboratory) were used for this study. As described previously (11, 14), myostatin-deficient mice have greater body weight (BW) due to the presence of increased lean body mass. We, therefore, used both age- and BW-matched WT mice as controls. The study protocol was approved by the Institutional Animal Care Committee of Massachusetts General Hospital (Protocol No.: 2007N000136). The animal care facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. The mice were housed in a pathogen-free animal facility with 12-h light/dark cycles at 22°C and given ad libitum access to food and water.
Cecum Ligation and Puncture
To induce sepsis, cecum ligation and puncture (CLP) was performed as described previously (32) with minor modifications. In brief, mice were anesthetized by inhalation of 4% isoflurane and maintained under 2% isoflurane inhalation. Laparotomy was performed by making an ∼1.0-cm midline incision in the abdomen to expose the cecum, which was ligated with a 4-0 absorbable suture at 1.5 cm from the tip. The ligated cecum was perforated by one through-to-through puncture with 18-gauge needle, and the feces were gently extruded. Then, the cecum was returned to the peritoneal cavity, and the laparotomy site was closed with a 4-0 absorbable suture. In sham-operated mice, the cecum was located and mobilized as described above but was neither ligated nor punctured. The mice were resuscitated with prewarmed 1 mL of 0.9% saline by subcutaneous injection immediately after CLP. To alleviate pain and distress, we administered buprenorphine (0.1 mg/kg, sc) to the mice at 30 min before CLP, at 6–8-h interval for the first 48 h after CLP, and at 10–14-h interval thereafter up to 72 h after CLP. Myostatin-deficient mice and age-matched WT mice received CLP at 8 wk of age, and the age of BW-matched WT mice was 13 wk at the time of CLP. The body weights of myostatin-deficient mice and BW-matched WT mice were 28.6 ± 0.3 g (means ± SE) and 28.1 ± 0.3 g, respectively, and that of age-matched WT mice was 25.1 ± 0.3 g. Body weight was measured before and at 6 and 24 h and 2, 3, 5, 7, 10, and 14 days after CLP. In all, 19 myostatin-deficient mice, eight age-matched WT mice, and 23 BW-matched WT mice received CLP for the survival study.
Evaluation of Bacterial Clearance
Bacterial loads were determined in the peritoneal lavage and blood at 16 h after CLP as previously described (32). After the laparotomy under anesthesia with isoflurane, 3 mL of sterile PBS was added to the peritoneal cavity and peritoneal lavage was collected. Blood samples were obtained by cardiac puncture. The peritoneal lavage and blood samples were placed on ice and serially diluted with sterile PBS. Then, 100 μL of each diluted sample was placed on trypticase soy agar plates with 5% sheep blood (BD Biosciences, San Diego, CA, BD 221261) and incubated at 37°C for 24 h. The numbers of bacterial colonies were counted and expressed as colony-forming units per milliliter. Seven septic myostatin-deficient and eight septic BW-matched WT mice were used for the assessment of bacterial clearance.
Measurement of Muscle Mass
To evaluate muscle atrophy, mass of gastrocnemius, soleus, and tibialis anterior muscles was measured in all the mice that survived for 14 days after CLP (18 myostatin-deficient mice and 8 BW-matched WT mice) or in naïve mice (n = 7 per group). After the animals were euthanized by CO2 inhalation, gastrocnemius, soleus, and tibialis anterior muscles were harvested without perfusion or fixation and weighed. To minimize possible influences of pathological conditions in dying mice, including serious malperfusion related to heart failure and/or hypotension, we evaluated the effect of myostatin deficiency on skeletal muscle mass in mice that survived for 14 days after CLP.
Measurement of Muscle Fiber Cross-Sectional Area
At 14 days after CLP, to evaluate the muscle fiber cross-sectional area, gastrocnemius, soleus, and tibialis anterior muscles, which were harvested without perfusion or fixation, were embedded in optical cutting temperature (OCT) compound and frozen in liquid nitrogen, and cross-sectioned (4 μm thick) followed by hematoxylin-eosin staining. For quantification, photomicrographs were taken using a Nikon ECLIPSE E800 camera. The muscle cross-sectional areas were measured at least 100 myofibers per mouse at ×100 magnification. Muscle fiber cross-sectional areas were evaluated in six myostatin-deficient and four BW-matched WT mice that survived for 14 days after CLP, and in six naïve myostatin-deficient and four naïve BW-matched WT mice.
Blood and Skeletal Muscle Sample Collection for Biochemical Analyses
At 16 h after CLP or sham operation, tissue and blood samples were collected under anesthesia with isoflurane without perfusion or fixation. No mice died within 16 h after CLP regardless of the genotype. Blood samples were collected by cardiac puncture for evaluation of bacterial loads and measurement of biochemical parameters. The mice were euthanized by exsanguination under anesthesia. Then, gastrocnemius muscle, liver, and kidney were collected and snap frozen in liquid nitrogen immediately after excision. Plasma samples were obtained by centrifugation of heparinized blood at 2,000 g for 20 min at 4°C. Three sham-operated myostatin-deficient and BW-matched WT mice (n = 3 per group), and six septic myostatin-deficient and BW-matched WT mice (n = 6 per group) were used for the biochemical analyses.
Immunoblot Analysis
Immunoblot analysis was performed using gastrocnemius muscle homogenates from mice at 16 h after CLP or sham operation as previously described (33, 34). Anti-muscle RING finger-1 (MuRF-1; R&D, Minneapolis, MN, AF5366), anti-atrogin-1 (Abcam, Cambridge, MA, ab168372), anti-GAPDH (Trevigen, Gaithersburg, MD, 7275-PC-100), anti-signal transducer and activator of transcription 3 (STAT3; Cell Signaling, Danvers, MA, 9132), and anti-phosphorylated STAT3 (Cell Signaling, 9145) antibodies were used as primary antibodies at the dilutions of 1:2,000, 1:25,000, 1:10,000, 1:1,000, and 1:5,000, respectively. The specificity of the primary antibodies was tested by the manufacturers. After the membranes were incubated with primary antibodies for overnight, they were incubated with secondary antibodies for 1 h at room temperature. As secondary antibodies, anti-goat IgG antibody conjugated to horseradish peroxidase (Santa Cruz Biotechnology, Dallas, TX, sc-2354) was used for MuRF-1, and anti-rabbit IgG antibody conjugated to horseradish peroxidase (Cell Signaling, 7077) was used for the rest of the proteins examined.
Measurement of MPO Activity
Myeloperoxidase (MPO) activity was measured at 16 h after CLP or sham operation using the MPO activity assay kit (Abcam, ab111749) according to the manufacturer’s instructions. Snap-frozen liver and kidney samples were homogenized in six volumes of MPO buffer, which was provided in the MPO activity assay kit. After centrifugation at 15,000 g for 15 min at 4°C, the supernatants were used for MPO assay. MPO activity was determined by measuring fluorescence value at 485/535 nm.
Measurement of Plasma Concentrations of HMGB1, AST, ALT, NGAL, and MIC-1/GDF-15
Plasma concentrations of HMGB1, aspartate aminotransferase (AST), alanine aminotransferase (ALT), neutrophil gelatinase-associated lipocalin (NGAL), and macrophage inhibitory cytokine (MIC)-1/growth differentiation factor (GDF)-15 were measured at 16 h post-CLP or sham operation using commercial ELISA kits (HMGB1: Shino-Test Corporation, Tokyo, Japan, ST51011; AST: BioVision, Milpitas, CA, K753-100; ALT: BioVision, K752-100; and NGAL: R&D, MLCN20; MIC-1/GDF15: R&D, MGD150) according to the manufacturers’ instructions.
Statistical Analysis
Log rank test was used to compare survival between the groups in the Kaplan–Meier survival curves. An unpaired two-sided Student’s t test was used to investigate differences between two groups. To study the effects of myostatin deficiency in septic and nonseptic mice, the data were compared using two-way ANOVA followed by Neuman–Keuls test for multiple comparison. Survival time after CLP was compared between myostatin-deficient mice and age- and BW-matched WT mice by one-way ANOVA followed by Neuman–Keuls test for multiple comparison. Body weight over 14-day period post-CLP was compared using a mixed-model ANOVA with the genotype as a between-subjects variable while time was set as a within-subjects variable, which was followed by Fisher’s least significant difference (LSD) test for multiple comparison. A value of P < 0.05 was considered statistically significant. All the data were analyzed by using GraphPad Prism 8.0 and are expressed as means ± SE.
RESULTS
Myostatin Deficiency Improved Survival of Septic Mice
Survival was monitored for 2 wk after the induction of sepsis by cecum ligation and puncture (CLP). Myostatin deficiency caused a significant reduction in CLP-induced mortality compared with age- and BW-matched WT mice (P < 0.0001; Fig. 1A). Consistently, myostatin deficiency significantly increased survival time during the 2-wk observation period after CLP [survival time: myostatin-deficient mice: 325 ± 11 h (means ± SE); age-matched WT mice: 40 ± 3; BW-matched WT mice: 161 ± 29, P < 0.0001, myostatin-deficient vs. age- and BW-matched WT]. Since the age-matched WT mice died within 2 days post-CLP, myostatin-deficient and BW-matched WT mice were compared in the following experiments.
Figure 1.
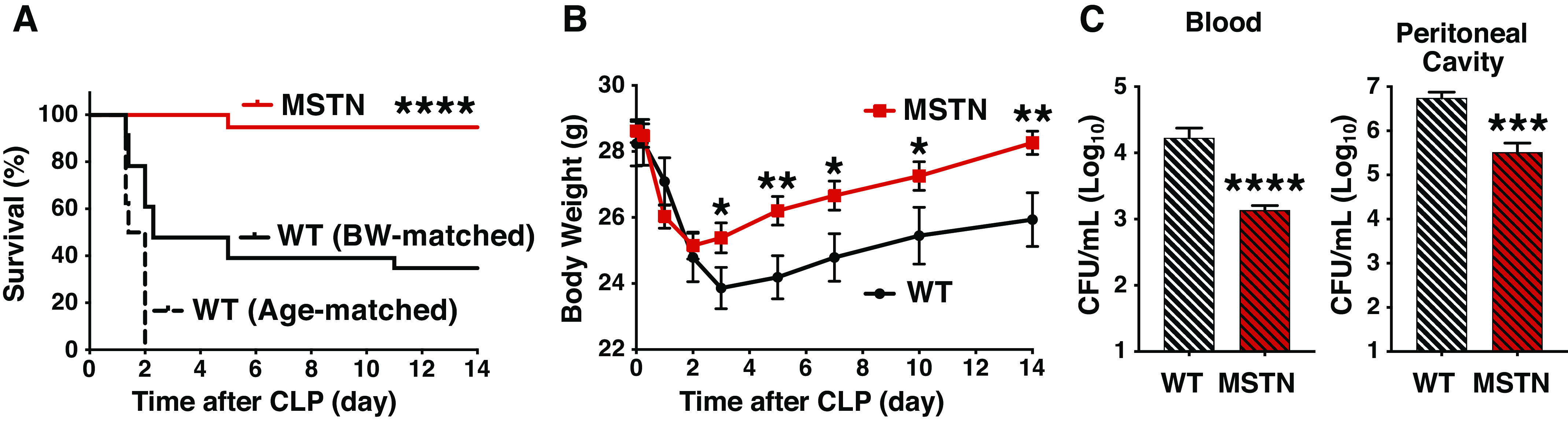
Myostatin deficiency improved survival (A), ameliorated body weight loss (B), and mitigated bacterial loads (C) after cecum ligation and puncture (CLP) in mice. A: survival was improved in myostatin-deficient (MSTN) mice (n = 19) compared with age- and body weight (BW)-matched wild-type (WT) mice (n = 8 and 23 per group, respectively). ****P < 0.0001. B: body weight loss was ameliorated in myostatin-deficient (MSTN) mice (n = 18) compared with body weight-matched wild-type (WT) mice (n = 8). *P < 0.05, **P < 0.01 vs. WT mice. C: bacterial loads in the circulation and peritoneal cavity were significantly lower in myostatin-deficient (MSTN) mice (n = 7) than body weight-matched wild-type (WT) mice (n = 8). ***P < 0.001, ****P < 0.0001 vs. WT mice.
We compared BW of myostatin-deficient and BW-matched WT mice that survived for 2 wk after CLP. Although there was no difference in BW between the two groups before CLP, BW was significantly greater in myostatin-deficient mice than WT mice at 3, 5, 7, 10, and 14 days after CLP (Fig. 1B).
Myostatin Deficiency Improved Bacterial Clearance in Septic Mice
To assess bacterial clearance, we evaluated bacterial loads in the peritoneal cavity, the site of infection, and in the circulation at 16 h after CLP. At 16 h post-CLP, none of the mice died regardless of the genotype. Bacterial loads in the blood and the peritoneal cavity were significantly greater in BW-matched WT mice than in myostatin-deficient mice (Fig. 1C).
Myostatin Deficiency Conferred Resistance to Sepsis-Induced Muscle Wasting
Consistent with previous studies (11, 14), myostatin-deficient mice had greater mass in gastrocnemius, soleus, and tibialis anterior muscles before CLP compared with BW-matched WT mice (Fig. 2A). The mass of both gastrocnemius and soleus muscle was significantly decreased by 27% at 14 days after CLP in WT mice compared with those without CLP (Fig. 2A). Tibialis anterior muscle mass decreased by 18% after CLP in WT mice, but no statistical significance was found. In myostatin-deficient mice, however, gastrocnemius and tibialis anterior mass was not decreased after CLP, and although soleus mass decreased by 9% after CLP in myostatin-deficient mice, the difference was not statistically significant. The CLP-induced percent decreases in mass of gastrocnemius, soleus, and tibialis anterior muscles were significantly greater in WT mice compared with myostatin-deficient mice (Fig. 2B).
Figure 2.

Myostatin deficiency conferred resistance to sepsis-induced muscle mass loss in mice. A: the muscle mass of naïve mice and in septic mice was greater in myostatin-deficient (MSTN) mice than in body weight-matched wild-type (WT) mice. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT mice without CLP, ††††P < 0.0001 vs. WT mice with CLP. B: cecum ligation and puncture (CLP)-induced percent changes in muscle mass were compared between myostatin-deficient (MSTN) and body weight-matched wild-type (WT) mice. **P < 0.01, ****P < 0.0001 vs. WT mice, WT mice without CLP: n = 7, WT mice with CLP: n = 8, MSTN mice without CLP: n = 7, MSTN mice with CLP: n = 18.
The muscle fiber cross-sectional area was greater in gastrocnemius, soleus, and tibialis anterior muscles of myostatin-deficient mice compared with WT mice both before and at 14 days after CLP (Fig. 3 and Supplemental Fig. S1A; see https://doi.org/10.6084/m9.figshare.13129568; and Supplemental Fig. S2A; see https://doi.org/10.6084/m9.figshare.13129577). Myostatin deficiency ameliorated the CLP-induced percent decreases in muscle fiber cross-sectional area of gastrocnemius, soleus, and tibialis anterior muscles compared with WT mice (Fig. 3B and Supplemental Fig. S1B; see https://doi.org/10.6084/m9.figshare.13129568; and Supplemental Fig. S2B; see https://doi.org/10.6084/m9.figshare.13129577).
Figure 3.

Myostatin deficiency inhibited sepsis-induced decrease in gastrocnemius muscle fiber cross-sectional area in mice. A: the cross-sectional area of gastrocnemius muscle fibers was greater in myostatin-deficient (MSTN) mice than in body weight-matched wild-type (WT) mice prior to and at 14 days after CLP. Scale bar: 50 μm. *P < 0.05, **P < 0.01 vs. WT without CLP, †††P < 0.001 vs. WT mice with CLP, §P < 0.05 vs. MSTN without CLP. B: the cecum ligation and puncture (CLP)-induced percent decreases in cross-sectional area of gastrocnemius muscle fibers were less in myostatin-deficient (MSTN) mice than in body weight-matched wild-type (WT) mice. *P < 0.05 vs. WT mice, WT without CLP: n = 4, WT mice with CLP: n = 4, MSTN without CLP: n = 6, MSTN mice with CLP: n = 6.
Previous studies have shown that increased proteolysis is a major contributor to sepsis-induced muscle wasting (35, 36). Induction of skeletal muscle-specific ubiquitin ligases, muscle-specific RING finger protein 1 (MuRF-1) (37) and atrogin-1 (aka muscle atrophy F-box [MAFbx]) (38), collectively known as atrogenes, play a crucial role in increased protein breakdown and subsequent muscle atrophy. We, therefore, evaluated expression of MuRF-1 and atrogin-1. Consistent with previous studies (39–41), protein expression of MuRF-1 and atrogin-1 was increased after CLP in gastrocnemius muscle of WT mice (Fig. 4A). Myostatin deficiency attenuated CLP-induced increased expression of MuRF-1 and atrogin-1 at 16 h after CLP.
Figure 4.

Effects of myostatin deficiency on sepsis-induced expression of MuRF-1 and atrogin-1 and phosphorylation of STAT3 in mouse skeletal muscle. Myostatin deficiency (MSTN) inhibited cecum ligation and puncture (CLP)-induced increases in expression of MuRF-1 and atrogin-1 and phosphorylation of STAT3 in gastrocnemius muscle compared with body weight-matched wild-type (WT) mice. **P < 0.01, ***P < 0.001 vs. WT mice without CLP, †P < 0.05, ††P < 0.01 vs. WT mice with CLP. WT mice without CLP: n = 3, WT mice with CLP: n = 6, MSTN mice without CLP: n = 3, MSTN mice with CLP: n = 6. MuRF-1, muscle-specific RING finger protein 1; STAT3, signal transducer and activator of transcription protein 3.
Activation of the Janus kinase (JAK)-signal transducer and activator of transcription protein 3 (STAT3) pathway has been shown to play an important role in muscle wasting and induction of atrogenes (42–45). To assess the activation status of the JAK-STAT3 pathway, therefore, we evaluated phosphorylation of STAT3 at tyrosine 705, a JAK phosphorylation site. CLP increased phosphorylated STAT3 in gastrocnemius muscle, which was significantly inhibited by myostatin deficiency (Fig. 4B). The total protein expression of STAT3 was not altered by CLP or the genotype.
Myostatin Deficiency Ameliorated Liver Dysfunction and Acute Kidney Injury
CLP increased plasma levels of AST and ALT, biomarkers of liver dysfunction, as well as NGAL, an indicator of acute kidney injury. Myostatin deficiency ameliorated CLP-induced increases in plasma concentrations of AST, ALT, and NGAL (Fig. 5).
Figure 5.

Myostatin deficiency (MSTN) mitigated sepsis-induced increases in plasma levels of AST, ALT and NGAL compared with body weight-matched wild-type (WT) mice. *P < 0.05, **P < 0.01 vs. WT mice without CLP, †P < 0.05 vs. WT mice with CLP. WT mice without CLP: n = 3, WT mice with CLP: n = 6, MSTN mice without CLP: n = 3, MSTN mice with CLP: n = 6. AST, aspartate aminotransferase; ALT, alanine aminotransferase; CLP, cecum ligation and puncture; NGAL, neutrophil gelatinase-associated lipocalin.
Sepsis-Induced Increases in Biomarkers of Inflammatory Response Were Mitigated by Myostatin Deficiency
To evaluate neutrophil organ infiltration, we measured MPO activity in the liver and kidney. CLP increased MPO activity in BW-matched WT mice, which was significantly attenuated in myostatin-deficient mice (Fig. 6A).
Figure 6.

Myostatin deficiency (MSTN) inhibited sepsis-induced increases in MPO activity in the liver and kidney (A) and plasma concentrations of HMGB1 (B) and MIC-1/GDF-15 (C) compared with body weight-matched wild-type (WT) mice. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. WT mice without CLP, †P < 0.05, ††P < 0.01, †††P < 0.001 vs. WT mice with CLP, §§§§P < 0.0001 vs. MSTN mice without CLP. WT mice without CLP: n = 3, WT mice with CLP: n = 6, MSTN mice without CLP: n = 3, MSTN mice with CLP: n = 6. CLP, cecum ligation and puncture; GDF, growth differentiation factor; HMGB1, high-mobility group protein B1; MIC, macrophage inhibitory cytokine; MPO, myeloperoxidase.
Similarly, CLP-induced increase in HMGB1, a damage-associated molecular pattern, was inhibited by myostatin deficiency compared with WT mice (Fig. 6B).
Myostatin Deficiency Blocked Sepsis-Induced Increase in Plasma MIC-1/GDF-15 Concentration
Macrophage inhibitory cytokine-1 (MIC-1)/growth differentiation factor-15 (GDF-15) has inhibitory effects on immune cell function (46, 47), and increased circulating MIC-1/GDF-15 level is associated with higher mortality risk in patients with sepsis (48). In addition, previous studies have shown that MIC-1/GDF-15 is secreted by skeletal muscle under some stressful conditions (49–51). We, therefore, evaluated the effects of CLP on MIC-1/GDF-15 concentration. Plasma concentration of MIC-1/GDF-15 was increased 33-fold after CLP in BW-matched WT mice. In contrast, CLP failed to significantly increase MIC-1/GDF-15 concentration in myostatin-deficient mice (Fig. 6C).
DISCUSSION
Our data showed that myostatin deficiency improved survival and bacterial clearance along with inhibition of muscle wasting in septic mice. The prosurvival effect of myostatin deficiency paralleled the mitigation of BW loss and the inhibition of increases in biomarkers of liver dysfunction and acute kidney injury and neutrophil infiltration into the liver and kidney after CLP. In addition, myostatin deficiency prevented sepsis-induced increased plasma concentrations of HMGB1 and MIC-1/GDF-15, as well. These results indicate that myostatin deficiency mitigates the severity of sepsis after CLP in mice.
Our results indicate that myostatin plays an important role in sepsis-induced muscle wasting in mice. The protective effects of myostatin deficiency on muscle mass in the mice that survived for 14 days after CLP is thought to have clinical relevance because poor long-term clinical outcomes of sepsis survivors, including muscle weakness-associated decreased ADL, is a serious public health problem.
Increased protein breakdown is a major contributor to muscle wasting associated with critical illness, including sepsis (35, 36, 52), in which MuRF-1 and atrogin-1 are involved (37–39). In contrast to sepsis-induced increased proteolysis, previous studies have shown controversial results about the effects of sepsis on protein synthesis, with decreased (53, 54), unchanged (55, 56), or increased protein synthesis (57) being reported. Myostatin deficiency inhibited the CLP-induced increase in MuRF-1 and atrogin-1 protein expression (Fig. 4). In addition, previous studies have shown that the JAK-STAT3 pathway activation plays an important role in muscle wasting of various etiologies, including cancer cachexia, denervation, and chronic kidney disease (42–44). However, it was not known whether sepsis increases the activity of the JAK-STAT3 pathway in skeletal muscle. In this study, CLP markedly activated the JAK-STAT3 pathway in skeletal muscle, as judged by phosphorylation of STAT3, which was ameliorated by myostatin deficiency (Fig. 4). These results suggest that inhibition of the increased expression of MuRF-1 and atrogin-1 and the activation of the JAK-STAT3 pathway may play a role in mitigating sepsis-induced muscle atrophy in myostatin-deficient mice.
MIC-1/GDF-15 induces muscle wasting (58, 59) and inhibits macrophage activation (46). Previous studies have shown that circulating levels of MIC-1/GDF-15 were increased in patients with sepsis (47, 48) and rodent models of sepsis and critical illness (60). Moreover, high MIC-1/GDF-15 levels were associated with a higher risk of mortality in patients with sepsis (48) and with natural killer (NK) cell dysfunction and nosocomial infection in patients with major trauma (47). In our study, plasma MIC-1/GDF-15 concentration was consistently, markedly increased after CLP in WT mice. In myostatin-deficient mice, however, CLP failed to significantly increase MIC-1/GDF-15 level (Fig. 6C). Of note, previous studies indicate that MIC-1/GDF-15 functions as a stress-inducible myokine (49–51, 61). Together, one can speculate that increased MIC-1/GDF-15 secretion may exacerbate muscle wasting and immune dysfunction in septic mice, and that the lack of sepsis-induced increase in MIC-1/GDF-15 may contribute to the beneficial effects of myostatin deficiency. On the other hand, a recent study demonstrated a protective effect of MIC-1/GDF-15 in both septic mice and lipopolysaccharide-challenged mice (60). Further studies are, therefore, required to clarify the effect of blockade of CLP-induced MIC-1/GDF-15 by myostatin deficiency in septic mice.
Previous studies have shown that prevention of muscle wasting by myostatin deficiency and inhibition of the activin type II receptor pathway is associated with improved survival in cancer-bearing mice (24, 62). These studies support the notion that cachexia, a complex metabolic syndrome associated with underlying illness and characterized by loss of skeletal muscle mass with or without loss of fat mass (63), is a significant contributor to mortality in patients with cancer (64). Similar to cancer patients, it is possible that cachexic changes may contribute to short- and long-term morbidity and mortality in patients with sepsis (65). However, this possibility has not been studied in animal models of sepsis or in patients with sepsis. Moreover, the role of muscle wasting or myostatin in infection, bacterial clearance, or organ damage/dysfunction has not been studied in any disease or received significant scientific attention. Our study showed that myostatin deficiency decreased bacterial loads and mortality in parallel with prevention of muscle wasting in septic mice. These results suggest that inhibition of muscle cachexic changes by myostatin deficiency may improve bacterial clearance and survival in septic mice.
Together with previous studies that reported muscle wasting/weakness as an independent predictor of the mortality of critically ill patients (6, 7, 66), the current study raise the possibility that muscle wasting may not simply be a complication of sepsis, but muscle cachexia and related changes in muscle (which include those in secretion of myokines) may actually drive the disease development of sepsis. It should be noted, however, that our data cannot exclude the possibility that the effects of myostatin in cell types other than muscle may also contribute to the protective effects of myostatin deficiency. Overall, our study identified myostatin as a novel molecular target that could potentially be harnessed to improve bacterial clearance and survival and mitigate multiorgan dysfunction in patients with sepsis.
GRANTS
This work was supported by research grants to M. Kaneki from the National Institutes of Health (NIH; R01GM115552, R01GM117298) and Shriners Hospitals for Children (71000, 85800).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M. Kaneki conceived and designed research; M. Kobayashi, S.K., S.S., S.Y., and M. Kaneki performed experiments; M. Kobayashi, S.K., S.S., S.Y., and M. Kaneki analyzed data; M. Kobayashi, S.S., and M. Kaneki interpreted results of experiments; M. Kobayashi, S.K., and M. Kaneki prepared figures; M. Kobayashi and M. Kaneki drafted manuscript; M. Kaneki edited and revised manuscript; M. Kobayashi, S.K., S.S., S.Y., and M. Kaneki approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Mr. Cassio Lynn of Amino Creative, LCC for the graphical abstract.
REFERENCES
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche JD, Coopersmith CM, Hotchkiss RS, Levy MM, Marshall JC, Martin GS, Opal SM, Rubenfeld GD, van der Poll T, Vincent JL, Angus DC. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315: 801–810, 2016. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet 392: 75–87, 2018. doi: 10.1016/S0140-6736(18)30696-2. [DOI] [PubMed] [Google Scholar]
- 3.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, Klompas M; for the CDC Prevention Epicenter Program. Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. Jama 318: 1241–1249, 2017. doi: 10.1001/jama.2017.13836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Callahan LA, Supinski GS. Sepsis-induced myopathy. Crit Care Med 37: S354–367, 2009. doi: 10.1097/CCM.0b013e3181b6e439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batt J, dos Santos CC, Cameron JI, Herridge MS. Intensive care unit-acquired weakness: clinical phenotypes and molecular mechanisms. Am J Respir Crit Care Med 187: 238–246, 2013. doi: 10.1164/rccm.201205-0954SO. [DOI] [PubMed] [Google Scholar]
- 6.Weijs PJ, Looijaard WG, Dekker IM, Stapel SN, Girbes AR, Straaten H, Beishuizen A. Low skeletal muscle area is a risk factor for mortality in mechanically ventilated critically ill patients. Crit Care 18: R12, 2014. doi: 10.1186/cc13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brunello AG, Haenggi M, Wigger O, Porta F, Takala J, Jakob SM. Usefulness of a clinical diagnosis of ICU-acquired paresis to predict outcome in patients with SIRS and acute respiratory failure. Intensive Care Med 36: 66–74, 2010. doi: 10.1007/s00134-009-1645-7. [DOI] [PubMed] [Google Scholar]
- 8.Seo DW, Kim KW, Sohn CH, Ryoo SM, Kim YJ, Shin A, Kim WY. Progressive loss of muscle mass could be an adverse prognostic factor of 28-day mortality in septic shock patients. Sci Rep 9: 16471, 2019. doi: 10.1038/s41598-019-52819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee YS, Huynh TV, Lee SJ. Paracrine and endocrine modes of myostatin action. J Appl Physiol (1985) 120: 592–598, 2016. doi: 10.1152/japplphysiol.00874.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SJ, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA 98: 9306–9311, 2001. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coleman SK, Rebalka IA, D’Souza DM, Deodhare N, Desjardins EM, Hawke TJ. Myostatin inhibition therapy for insulin-deficient type 1 diabetes. Sci Rep 6: 32495, 2016. doi: 10.1038/srep32495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387: 83–90, 1997. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 13.Szabo G, Dallmann G, Muller G, Patthy L, Soller M, Varga L. A deletion in the myostatin gene causes the compact (Cmpt) hypermuscular mutation in mice. Mamm Genome 9: 671–672, 1998. doi: 10.1007/s003359900843. [DOI] [PubMed] [Google Scholar]
- 14.Wilkes JJ, Lloyd DJ, Gekakis N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity-induced insulin resistance. Diabetes 58: 1133–1143, 2009. doi: 10.2337/db08-0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F, Massabanda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet 17: 71–74, 1997. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- 16.Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res 7: 910–916, 1997. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- 17.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA 94: 12457–12461, 1997. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet 38: 813–818, 2006. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- 19.Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 3: e79, 2007. doi: 10.1371/journal.pgen.0030079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 350: 2682–2688, 2004. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- 21.Williams MS. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 351: 1030–1031, 2004. [Author reply 1030–1031]. doi: 10.1056/NEJM200409023511018. [DOI] [PubMed] [Google Scholar]
- 22.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM, Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science 296: 1486–1488, 2002. doi: 10.1126/science.1069525. [DOI] [PubMed] [Google Scholar]
- 23.Benny Klimek ME, Aydogdu T, Link MJ, Pons M, Koniaris LG, Zimmers TA. Acute inhibition of myostatin-family proteins preserves skeletal muscle in mouse models of cancer cachexia. Biochem Biophys Res Commun 391: 1548–1554, 2010. doi: 10.1016/j.bbrc.2009.12.123. [DOI] [PubMed] [Google Scholar]
- 24.Gallot YS, Durieux AC, Castells J, Desgeorges MM, Vernus B, Plantureux L, Remond D, Jahnke VE, Lefai E, Dardevet D, Nemoz G, Schaeffer L, Bonnieu A, Freyssenet DG. Myostatin gene inactivation prevents skeletal muscle wasting in cancer. Cancer Res 74: 7344–7356, 2014. doi: 10.1158/0008-5472.CAN-14-0057. [DOI] [PubMed] [Google Scholar]
- 25.Murphy KT, Cobani V, Ryall JG, Ibebunjo C, Lynch GS. Acute antibody-directed myostatin inhibition attenuates disuse muscle atrophy and weakness in mice. J Appl Physiol (1985) 110: 1065–1072, 2011. doi: 10.1152/japplphysiol.01183.2010. [DOI] [PubMed] [Google Scholar]
- 26.Arounleut P, Bialek P, Liang LF, Upadhyay S, Fulzele S, Johnson M, Elsalanty M, Isales CM, Hamrick MW. A myostatin inhibitor (propeptide-Fc) increases muscle mass and muscle fiber size in aged mice but does not increase bone density or bone strength. Exp Gerontol 48: 898–904, 2013. doi: 10.1016/j.exger.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendias CL, Bakhurin KI, Gumucio JP, Shallal-Ayzin MV, Davis CS, Faulkner JA. Haploinsufficiency of myostatin protects against aging-related declines in muscle function and enhances the longevity of mice. Aging Cell 14: 704–706, 2015. doi: 10.1111/acel.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilson H, Schakman O, Combaret L, Lause P, Grobet L, Attaix D, Ketelslegers JM, Thissen JP. Myostatin gene deletion prevents glucocorticoid-induced muscle atrophy. Endocrinology 148: 452–460, 2007. doi: 10.1210/en.2006-0539. [DOI] [PubMed] [Google Scholar]
- 29.Lokireddy S, Mouly V, Butler-Browne G, Gluckman PD, Sharma M, Kambadur R, McFarlane C. Myostatin promotes the wasting of human myoblast cultures through promoting ubiquitin-proteasome pathway-mediated loss of sarcomeric proteins. Am J Physiol Cell Physiol 301: C1316–C1324, 2011. [Erratum in Am J Physiol Cell Physiol 307: C1152, 2014]. doi: 10.1152/ajpcell.00114.2011. [DOI] [PubMed] [Google Scholar]
- 30.McFarlane C, Plummer E, Thomas M, Hennebry A, Ashby M, Ling N, Smith H, Sharma M, Kambadur R. Myostatin induces cachexia by activating the ubiquitin proteolytic system through an NF-kappaB-independent, FoxO1-dependent mechanism. J Cell Physiol 209: 501–514, 2006. doi: 10.1002/jcp.20757. [DOI] [PubMed] [Google Scholar]
- 31.Wang R, Jiao H, Zhao J, Wang X, Lin H. Glucocorticoids enhance muscle proteolysis through a myostatin-dependent pathway at the early stage. PLoS One 11: e0156225, 2016. doi: 10.1371/journal.pone.0156225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang W, Yamada M, Tamura Y, Chang K, Mao J, Zou L, Feng Y, Kida K, Scherrer-Crosbie M, Chao W, Ichinose F, Yu YM, Fischman AJ, Tompkins RG, Yao S, Kaneki M. Farnesyltransferase inhibitor FTI-277 reduces mortality of septic mice along with improved bacterial clearance. J Pharmacol Exp Ther 339: 832–841, 2011. doi: 10.1124/jpet.111.183558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakazawa H, Chang K, Shinozaki S, Yasukawa T, Ishimaru K, Yasuhara S, Yu YM, Martyn JA, Tompkins RG, Shimokado K, Kaneki M. iNOS as a driver of inflammation and apoptosis in mouse skeletal muscle after burn injury: possible involvement of Sirt1 S-nitrosylation-mediated acetylation of p65 NF-kappaB and p53. PLoS One 12: e0170391, 2017. doi: 10.1371/journal.pone.0170391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakazawa H, Ikeda K, Shinozaki S, Kobayashi M, Ikegami Y, Fu M, Nakamura T, Yasuhara S, Yu YM, Martyn JAJ, Tompkins RG, Shimokado K, Yorozu T, Ito H, Inoue S, Kaneki M. Burn-induced muscle metabolic derangements and mitochondrial dysfunction are associated with activation of HIF-1alpha and mTORC1: Role of protein farnesylation. Sci Rep 7: 6618, 2017. doi: 10.1038/s41598-017-07011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hasselgren PO, James JH, Benson DW, Hall-Angeras M, Angeras U, Hiyama DT, Li S, Fischer JE. Total and myofibrillar protein breakdown in different types of rat skeletal muscle: effects of sepsis and regulation by insulin. Metabolism 38: 634–640, 1989. doi: 10.1016/0026-0495(89)90100-5. [DOI] [PubMed] [Google Scholar]
- 36.Vary TC. Regulation of skeletal muscle protein turnover during sepsis. Curr Opin Clin Nutr Metab Care 1: 217–224, 1998. doi: 10.1097/00075197-199803000-00013. [DOI] [PubMed] [Google Scholar]
- 37.Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA, Poueymirou WT, Panaro FJ, Na E, Dharmarajan K, Pan ZQ, Valenzuela DM, DeChiara TM, Stitt TN, Yancopoulos GD, Glass DJ. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294: 1704–1708, 2001. doi: 10.1126/science.1065874. [DOI] [PubMed] [Google Scholar]
- 38.Gomes MD, Lecker SH, Jagoe RT, Navon A, Goldberg AL. Atrogin-1, a muscle-specific F-box protein highly expressed during muscle atrophy. Proc Natl Acad Sci USA 98: 14440–14445, 2001. doi: 10.1073/pnas.251541198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frost RA, Nystrom GJ, Jefferson LS, Lang CH. Hormone, cytokine, and nutritional regulation of sepsis-induced increases in atrogin-1 and MuRF1 in skeletal muscle. Am J Physiol Endocrinol Metab 292: E501–E512, 2007. doi: 10.1152/ajpendo.00359.2006. [DOI] [PubMed] [Google Scholar]
- 40.Steiner JL, Crowell KT, Kimball SR, Lang CH. Disruption of REDD1 gene ameliorates sepsis-induced decrease in mTORC1 signaling but has divergent effects on proteolytic signaling in skeletal muscle. Am J Physiol Endocrinol Metab 309: E981–E994, 2015. doi: 10.1152/ajpendo.00264.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wray CJ, Mammen JM, Hershko DD, Hasselgren PO. Sepsis upregulates the gene expression of multiple ubiquitin ligases in skeletal muscle. Int J Biochem Cell Biol 35: 698–705, 2003. doi: 10.1016/S1357-2725(02)00341-2. [DOI] [PubMed] [Google Scholar]
- 42.Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, Zimmers TA. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 303: E410–E421, 2012. doi: 10.1152/ajpendo.00039.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, Zimmers TA. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 6: e22538, 2011. doi: 10.1371/journal.pone.0022538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madaro L, Passafaro M, Sala D, Etxaniz U, Lugarini F, Proietti D, Alfonsi MV, Nicoletti C, Gatto S, De Bardi M, Rojas-Garcia R, Giordani L, Marinelli S, Pagliarini V, Sette C, Sacco A, Puri PL. Denervation-activated STAT3-IL-6 signalling in fibro-adipogenic progenitors promotes myofibres atrophy and fibrosis. Nat Cell Biol 20: 917–927, 2018. doi: 10.1038/s41556-018-0151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva KA, Dong J, Dong Y, Dong Y, Schor N, Tweardy DJ, Zhang L, Mitch WE. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J Biol Chem 290: 11177–11187, 2015. doi: 10.1074/jbc.M115.641514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bootcov MR, Bauskin AR, Valenzuela SM, Moore AG, Bansal M, He XY, Zhang HP, Donnellan M, Mahler S, Pryor K, Walsh BJ, Nicholson RC, Fairlie WD, Por SB, Robbins JM, Breit SN. MIC-1, a novel macrophage inhibitory cytokine, is a divergent member of the TGF-beta superfamily. Proc Natl Acad Sci USA 94: 11514–11519, 1997. doi: 10.1073/pnas.94.21.11514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kleinertz H, Hepner-Schefczyk M, Ehnert S, Claus M, Halbgebauer R, Boller L, Huber-Lang M, Cinelli P, Kirschning C, Flohé S, Sander A, Waydhas C, Vonderhagen S, Jäger M, Dudda M, Watzl C, Flohé SB. . Circulating growth/differentiation factor 15 is associated with human CD56(bright) natural killer cell dysfunction and nosocomial infection in severe systemic inflammation. EBioMedicine 43: 380–391, 2019. doi: 10.1016/j.ebiom.2019.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Buendgens L, Yagmur E, Bruensing J, Herbers U, Baeck C, Trautwein C, Koch A, Tacke F. Growth differentiation factor-15 is a predictor of mortality in critically ill patients with sepsis. Dis Markers 2017: 5271203, 2017. doi: 10.1155/2017/5271203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung HK, Ryu D, Kim KS, Chang JY, Kim YK, Yi HS, Kang SG, Choi MJ, Lee SE, Jung SB, Ryu MJ, Kim SJ, Kweon GR, Kim H, Hwang JH, Lee CH, Lee SJ, Wall CE, Downes M, Evans RM, Auwerx J, Shong M. Growth differentiation factor 15 is a myomitokine governing systemic energy homeostasis. J Cell Biol 216: 149–165, 2017. doi: 10.1083/jcb.201607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ito T, Nakanishi Y, Yamaji N, Murakami S, Schaffer SW. Induction of growth differentiation factor 15 in skeletal muscle of old taurine transporter knockout mouse. Biol Pharm Bull 41: 435–439, 2018. doi: 10.1248/bpb.b17-00969. [DOI] [PubMed] [Google Scholar]
- 51.Ost M, Igual Gil C, Coleman V, Keipert S, Efstathiou S, Vidic V, Weyers M, Klaus S. Muscle-derived GDF15 drives diurnal anorexia and systemic metabolic remodeling during mitochondrial stress. EMBO Rep 21: e48804, 2020. doi: 10.15252/embr.201948804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puthucheary ZA, Rawal J, McPhail M, Connolly B, Ratnayake G, Chan P, Hopkinson NS, Phadke R, Dew T, Sidhu PS, Velloso C, Seymour J, Agley CC, Selby A, Limb M, Edwards LM, Smith K, Rowlerson A, Rennie MJ, Moxham J, Harridge SD, Hart N, Montgomery HE. Acute skeletal muscle wasting in critical illness. Jama 310: 1591–1600, 2013. doi: 10.1001/jama.2013.278481. [DOI] [PubMed] [Google Scholar]
- 53.Hobler SC, Williams AB, Fischer JE, Hasselgren PO. IGF-I stimulates protein synthesis but does not inhibit protein breakdown in muscle from septic rats. Am J Physiol 274: R571–R576, 1998. doi: 10.1152/ajpregu.1998.274.2.R571. [DOI] [PubMed] [Google Scholar]
- 54.Lang CH, Frost RA, Vary TC. Regulation of muscle protein synthesis during sepsis and inflammation. Am J Physiol Endocrinol Metab 293: E453–E459, 2007. doi: 10.1152/ajpendo.00204.2007. [DOI] [PubMed] [Google Scholar]
- 55.Hasselgren PO, Talamini M, James JH, Fischer JE. Protein metabolism in different types of skeletal muscle during early and late sepsis in rats. Arch Surg 121: 918–923, 1986. doi: 10.1001/archsurg.1986.01400080064011. [DOI] [PubMed] [Google Scholar]
- 56.Klaude M, Mori M, Tjäder I, Gustafsson T, Wernerman J, Rooyackers O. Protein metabolism and gene expression in skeletal muscle of critically ill patients with sepsis. Clin Sci (Lond) 122: 133–142, 2012. doi: 10.1042/CS20110233. [DOI] [PubMed] [Google Scholar]
- 57.Clowes GH, Jr, George BC, Villee CA, Jr, Saravis CA. Muscle proteolysis induced by a circulating peptide in patients with sepsis or trauma. N Engl J Med 308: 545–552, 1983. doi: 10.1056/NEJM198303103081001. [DOI] [PubMed] [Google Scholar]
- 58.Garfield BE, Crosby A, Shao D, Yang P, Read C, Sawiak S, Moore S, Parfitt L, Harries C, Rice M, Paul R, Ormiston ML, Morrell NW, Polkey MI, Wort SJ, Kemp PR. Growth/differentiation factor 15 causes TGFbeta-activated kinase 1-dependent muscle atrophy in pulmonary arterial hypertension. Thorax 74: 164–176, 2019. doi: 10.1136/thoraxjnl-2017-211440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Patel MS, Lee J, Baz M, Wells CE, Bloch S, Lewis A, Donaldson AV, Garfield BE, Hopkinson NS, Natanek A, Man WD, Wells DJ, Baker EH, Polkey MI, Kemp PR. Growth differentiation factor-15 is associated with muscle mass in chronic obstructive pulmonary disease and promotes muscle wasting in vivo. J Cachexia Sarcopenia Muscle 7: 436–448, 2016. doi: 10.1002/jcsm.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luan HH, Wang A, Hilliard BK, Carvalho F, Rosen CE, Ahasic AM, Herzog EL, Kang I, Pisani MA, Yu S, Zhang C, Ring AM, Young LH, Medzhitov R. GDF15 is an inflammation-induced central mediator of tissue tolerance. Cell 178: 1231–1244.e11, 2019. doi: 10.1016/j.cell.2019.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Montero R, Yubero D, Villarroya J, Henares D, Jou C, Rodríguez MA, Ramos F, Nascimento A, Ortez CI, Campistol J, Perez-Dueñas B, O'Callaghan M, Pineda M, Garcia-Cazorla A, Oferil JC, Montoya J, Ruiz-Pesini E, Emperador S, Meznaric M, Campderros L, Kalko SG, Villarroya F, Artuch R, Jimenez-Mallebrera C. GDF-15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PLoS One 11: e014870, 2016. doi: 10.1371/journal.pone.0148709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142: 531–543, 2010. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 63.Blum D, Stene GB, Solheim TS, Fayers P, Hjermstad MJ, Baracos VE, Fearon K, Strasser F, Kaasa S; Euro-Impact. Validation of the consensus-definition for cancer cachexia and evaluation of a classification model–a study based on data from an international multicentre project (EPCRC-CSA). Ann Oncol 25: 1635–1642, 2014. doi: 10.1093/annonc/mdu086. [DOI] [PubMed] [Google Scholar]
- 64.Han HQ, Zhou X, Mitch WE, Goldberg AL. Myostatin/activin pathway antagonism: molecular basis and therapeutic potential. Int J Biochem Cell Biol 45: 2333–2347, 2013. doi: 10.1016/j.biocel.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 65.Kaneki M. Metabolic inflammatory complex in sepsis: septic cachexia as a novel potential therapeutic target. Shock 48: 600–609, 2017. doi: 10.1097/SHK.0000000000000906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lucidi C, Lattanzi B, Di Gregorio V, Incicco S, D'Ambrosio D, Venditti M, Riggio O, Merli MA. Low muscle mass increases mortality in compensated cirrhotic patients with sepsis. Liver Int 38: 851–857, 2018. doi: 10.1111/liv.13691. [DOI] [PubMed] [Google Scholar]