

Keywords: adipokine, adipsin, atherosclerosis, complement system, obesity
Abstract
Obesity is a potent risk factor for atherosclerotic morbidity and mortality. Cytokines secreted from adipose tissue, namely, adipokines, have been suggested to be actively involved in atherosclerosis. One of the most abundant adipokines, adipsin, is downregulated in obesity. It catalyzes the rate-limiting step of alternative complement activation, which is one of the three complement pathways potentially involved in inflammation in atherosclerosis. Interestingly, adipsin has been identified as a novel biomarker in human coronary artery disease. However, its role in the development of atherosclerosis remains unexplored. We crossed adipsin−/− mice onto an Ldlr−/− background [double-knockout (DKO) mice] and induced atherogenesis by high-fat and high-cholesterol feeding. Metabolic profiles were systemically characterized, and atherosclerotic plaques were measured at both aortic root and arch regions. Western blotting was conducted to assess adipsin level and complement activity. The DKO mice exhibited similar sizes of atherosclerotic lesions as Ldlr−/− control mice at both the aortic root and arch regions. Accordingly, they displayed comparable metabolic parameters, including body weight, insulin sensitivity, and lipid profiles, along with compensated complement activity. Adipsin deficiency does not impact the development of atherosclerosis in Ldlr−/− mice despite its crucial function in alternative complement activation. Therefore, it is unlikely to play an important role in mediating the risk of atherosclerotic complications in obesity.
NEW & NOTEWORTHY Adipsin deficiency does not impact the development of atherosclerosis in Ldlr−/− mice despite its crucial function in alternative complement activation. Therefore, it is unlikely to play an important role in mediating the risk of atherosclerotic complications in obesity.
INTRODUCTION
Cardiovascular diseases (CVDs) are the leading cause of death worldwide and accounted for 17.9 million deaths in 2016 (1). Obesity, along with its primary comorbidity, insulin resistance, is a major risk factor for CVDs (2). In the United States, approximately two-thirds of the adult population is considered overweight or obese (3), which creates an urgent need for developing a better understanding of the connection between obesity and CVDs. Indeed, obesity-associated insulin resistance and type 2 diabetes serve as potent predictors of cardiovascular morbidities and mortalities, the most prominent of which is atherosclerosis (4). Atherosclerosis is a chronic inflammatory disease of large- and medium-sized arteries that is marked by plaques in arterial walls (5). It stands out as the principal cause of myocardial infarctions and stroke. Atherogenesis is initiated by the accumulation of apolipoprotein B-containing lipoproteins in the subendothelial space, which triggers the innate immune response (6). The innate immune response activates the complement cascade to opsonize pathogens and assist in resolving tissue damage (7). There are three main pathways in the complement system that are categorized by their modes of activation: the classical, alternative, and lectin pathways (8). These pathways converge at the formation of C3-convertases, which catalyze the proteolytic cleavage of C3 into C3a and C3b (8). Increasing evidence points toward a potential role of the complement system in the development of atherosclerosis. For example, C3-deficient mice have increased levels of total aortic atherosclerosis (9). However, the contribution of each pathway to atherosclerosis remains unclear.
One emerging connection between obesity and atherosclerosis is the endocrine function of adipose tissue. Beyond its canonical lipid-storage function, adipose tissue actively secretes a variety of adipokines to regulate lipid metabolism, inflammation, host defenses, apoptosis, and more (10); therefore, it is considered an important endocrine organ. Adipose tissue produces proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and leptin to induce vascular injury and immune cell activation. In contrast, other adipokines like adiponectin have anti-inflammatory properties and protect against CVDs (10), and decreased serum levels of adiponectin are associated with more severe atherosclerosis (11). Among the most abundant adipokines is adipsin, which was also the first adipokine to be identified (12, 13). Adipsin is often referred to as complement factor D (CFD), as it catalyzes the rate-limiting step of the alternative pathway in the complement system (14). It is predominantly produced by white adipocytes and is thus considered to be a representative white adipose tissue-specific adipokine (15). Adipsin levels significantly decline in several animal models of obesity and diabetes (16, 17). Notably, high serum adipsin level was recently found to correlate with all-cause death and rehospitalization in coronary artery disease patients, and it is therefore considered a novel prognostic biomarker (18). Despite these interesting connections, the field still lacks direct evidence of adipsin in the development of atherosclerosis.
Given the pivotal functions of adipsin in the alternative complement pathway as well as its close relationship to obesity and diabetes, we sought to determine whether adipsin might play a role in the development of atherosclerosis, and thereby identify a potential mechanism underlying the increased risk of atherosclerosis in obesity.
MATERIALS AND METHODS
Animal Studies
Adipsin−/− mice on the C57BL/6 background (19) were bred with Ldlr–/– mice on the C57BL/6 background (JAX002207) to generate adipsin–/–:Ldlr–/– double-knockout (DKO) mice. The genotypes were verified by genotyping before and after euthanasia and by Western blotting. The animals were housed at 23°C ± 1°C on a 12-h light/dark cycle with access to food and water ad libitum. Three-month-old male mice were fed a Western-type diet (WTD; Envigo, TD. 88137) that contains 42% fat, 42.7% carbohydrates, 15.2% protein, and 0.2% cholesterol to induce atherosclerosis. For intraperitoneal glucose tolerance tests (GTT), glucose (2.0 g/kg body wt) was intraperitoneally injected into the mice after 16-h overnight fasting. Insulin tolerance tests (ITT) were performed after 4-h fasting with an intraperitoneal injection of insulin (0.75 U insulin/kg body wt) (20). Blood glucose levels were determined using a Breeze2 glucometer (Bayer). EchoMRI was used to determine body composition. After 14 wk of WTD feeding, the mice were fasted overnight for 16 h and euthanized with CO2 followed by cervical dislocation to collect tissues and plasma. Mouse Insulin ELISA (Mercodia), Infinity triglyceride reagent (Thermo Scientific), NEFA-HR (Fujifilm Wako), total cholesterol (Fujifilm Wako), and HDL-cholesterol E (Fujifilm Wako) were used to measure plasma insulin, triglyceride, nonesterified fatty acids (NEFAs), cholesterol, and HDL-c levels, respectively. All animal protocols used in this study were reviewed and approved by the Columbia University Animal Care and Utilization Committee.
Mouse Aortic Arch and Root Atherosclerotic Lesion Analyses
Three-month-old male mice were subjected to WTD for 14 wk to induce atherosclerosis and were analyzed as described previously (20). Briefly, after the blood draws through left ventricular puncture and perfusion with PBS, aortas were dissected, aortic arches were photographed, and aortic lesion en face staining by Oil Red O was performed. For aortic root lesional analysis, aortic roots were fixed in paraformaldehyde, embedded in paraffin, and serially sectioned (6 μm each). Then, Harris’ hematoxylin and eosin (H&E) staining was performed on six paraffin sections 60 µm apart from each other to quantify the aortic lesional area and necrotic core area. Fibrous cap thickness was measured at the lesional midpoint in both shoulder regions and quantified as the ratio of fibrous cap to lesional area (21). Image J software (National Institutes of Health) was used for the lesional quantification.
Western Blotting
Equal volumes of diluted plasma samples with loading buffer were used to run SDS-PAGE. Western blotting was performed and detected with ECL (Thermo Scientific). Coomassie Blue staining was used to verify equal protein loading. Adipsin antibody (1:1,000, AF5430, R&D) and C3 antibody (1:1,000, ab48581, Abcam) were used to detect adipsin and cleaved C3 protein, respectively. Western blot quantification was performed using Image J software (National Institutes of Health).
Statistical Analysis
The Mann–Whitney U test was used for the analyses of the aortic lesional area and necrotic core quantifications. Student’s t tests were used for two-group statistical analyses. P < 0.05 was considered to be a significant difference. GraphPad Prism v. 7 (GraphPad Software, San Diego, CA) was used for statistical analyses. Data are presented as means (SD) (standard deviation).
RESULTS
Ablation of Adipsin in Ldlr–/– Mice Does Not Modulate Their Metabolic Profiles
To study the potential role of adipsin in atherogenesis, we crossed adipsin−/− mice with Ldlr−/− mice to generate the adipsin−/−:Ldlr−/− double-knockout mice (DKO). Three-month-old male DKO mice were fed a WTD to induce atherogenesis, using age-matched male Ldlr−/− mice as the controls. Knocking out adipsin had no effect on body weight gain over the course of WTD feeding (Fig. 1, A and B), and at the end of the feeding, DKO mice had the same body weight, fat, and lean body composition as the control mice (Fig. 1C). Likewise, there was no difference in the sizes of inguinal and epididymal white adipose tissue (Fig. 1D). Knocking out adipsin impairs glucose tolerance in high-fat diet-induced obesity due to deficiency in insulin production (19). Surprisingly, the DKO mice showed similar glucose tolerance and insulin sensitivity, as assessed by the glucose tolerance test (GTT) and insulin tolerance test (ITT), respectively (Fig. 1, E–H). Their unaltered glucose tolerance was supported by comparable insulin levels (Fig. 1I). These data collectively indicate that adipsin has little metabolic effects in the atherogenic Ldlr−/− mice.
Figure 1.
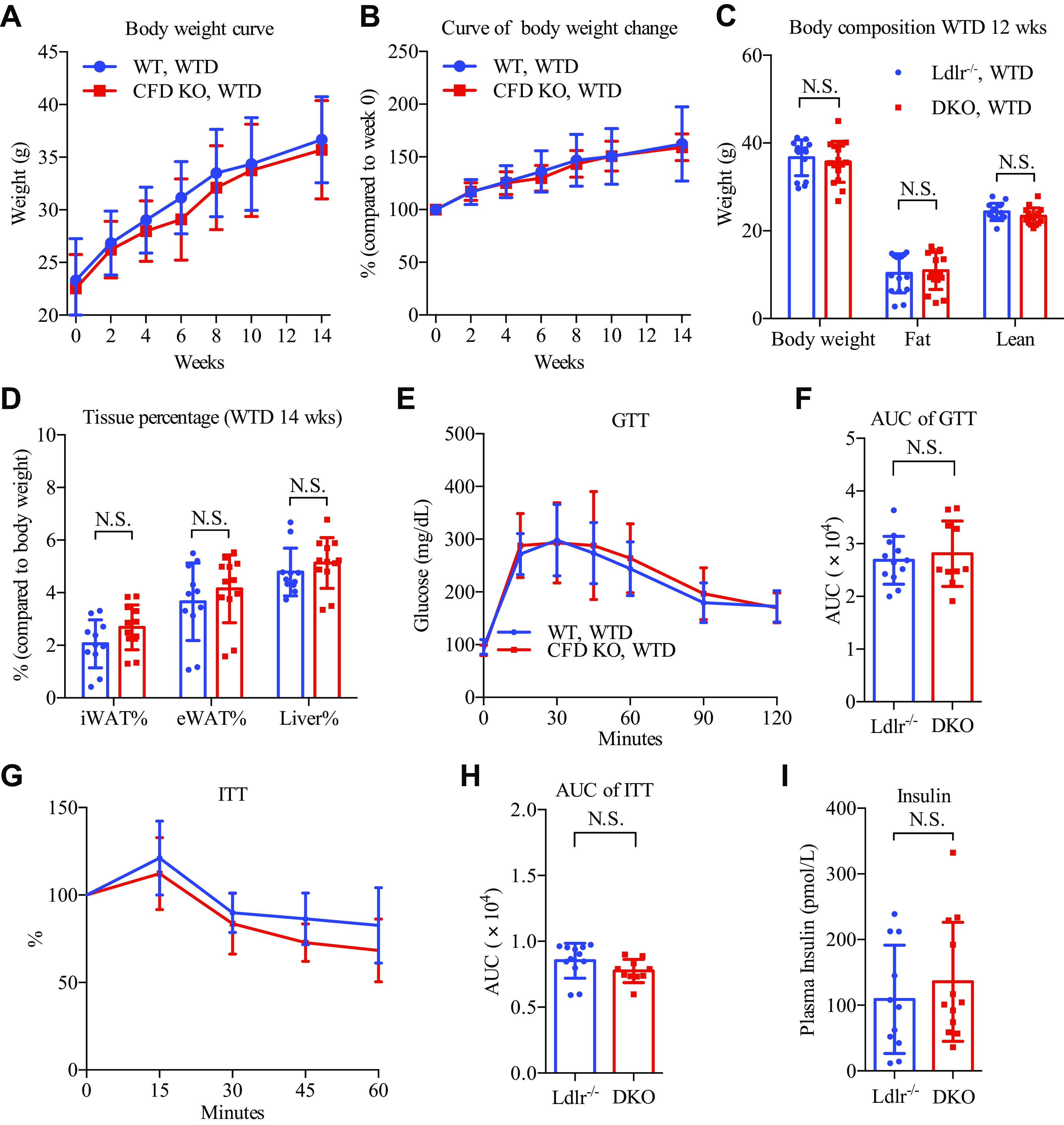
Ablation of adipsin in Ldlr−/− mice does not modulate their metabolic profiles. Three-month-old male adipsin−/−:Ldlr−/− double knockout (DKO) and Ldlr−/− mice were fed a WTD for metabolic characterizations (n = 15 for Ldlr−/−, n = 16 for DKO). Plasma was collected after overnight fasting (16 h). A: body weight curve on WTD feeding. B: curve of body weight change on WTD feeding. C: body composition after 12 wk of WTD feeding. D: tissue sizes (tissue mass normalized to body weight) at euthanasia. E–H: GTT (E) and area under curve (AUC) (F); ITT (G) and AUC (H) after 13 wk of WTD feeding. I: plasma insulin levels. Not significant (N.S.) by two-tailed Student’s t test. Data are presented as means ± SD. CFD, complement factor D; GTT, glucose tolerance tests; ITT, insulin tolerance tests; KO, knockout; WT, wild type; WTD, Western-type diet.
The Development of Aortic Lesions Is Independent of Adipsin
Next, we assessed the development of atherosclerotic lesions in male DKO and control Ldlr−/− mice. The two groups showed comparable aortic root lesional areas, necrotic core areas, and fibrous cap thickness, which indicated similar degrees of atherosclerotic progression and plaque necrosis (Fig. 2, A–D). Adipsin is sensitive to the deacetylation of PPARγ (22, 23), a master regulator of adipocyte biology and an important therapeutic target of insulin resistance. PPARγ deacetylation has been shown to reduce atherosclerotic lesions in the aortic arch but not at the root (20). We, therefore, measured plaque area in the entire aorta by en face staining. No differences were observed in the aortic arches or the descending regions (Fig. 2, E–H). These data indicate that adipsin does not have a significant role in the development of atherosclerosis in Ldlr−/− mice.
Figure 2.

The development of aortic lesions is independent of adipsin in Ldlr−/− mice. Three-month-old male DKO and Ldlr−/− mice were fed the WTD for 14 wk to measure atherosclerosis (n = 24, 24). A: representative H&E-stained aortic root sections, with plaque outlined in black. B and C: quantification of total lesional area (B) and necrotic core area (C) of aortic root sections. D: aortic root fibrous cap thickness (n = 16 for control, n = 23 for DKO). E: representative images of aortic arches with yellow arrows indicating plaques, and F: en face aortic plaque staining by Oil Red O. The quantified arch lesional areas were squared. G and H: quantification of total aortic lesional areas (G) and aortic arch lesional areas (H). The Mann–Whitney U test was used to analyze aortic lesional area and necrotic core quantifications, and the rest were analyzed by two-tailed Student’s t tests. Not significant (N.S.). Data are presented as means ± SD. DKO, double knockout; H&E, hematoxylin-eosin; WTD, Western-type diet.
Ablation of Adipsin Has No Effect on Plasma Lipid and Cholesterol Levels in Ldlr−/− Mice
Dyslipidemia and hypercholesterolemia are the primary driving factors of atherogenesis. To understand the blunted effect of adipsin ablation on atherosclerosis, we measured lipid and cholesterol levels in the plasma. There was no significant difference between the DKO mice and Ldlr−/− controls in their plasma lipid and cholesterol levels, including triglycerides (TGs), NEFAs, total cholesterol, and HDL-c (Fig. 3, A–D). Therefore, adipsin deficiency has minimal effect on lipid and cholesterol metabolism in atherogenic Ldlr−/− mice. These minimal metabolic changes indeed explain why their atherosclerotic lesions were comparable in size with those of the control mice.
Figure 3.

Adipsin deficiency has compensatory activation of C3 without effect on lipid profile. A–D: Three-month-old male DKO and Ldlr−/− mice were fed the WTD for 14 wk. Plasma was collected after overnight (16 h) fasting (n = 12, 12). Plasma triglycerides (TG) (A), nonesterified fatty acids (NEFAs) (B), total cholesterol (C), and HDL cholesterol levels (D). E: Western blotting of adipsin and C3a in plasma from control and DKO mice, using Coomassie Blue staining as the loading control (L.C.). F: quantification of C3a protein level in plasma (n = 6, 6). Not significant (N.S.). ***P < 0.001 by two-tailed Student’s t test. Data are presented as means ± SD. DKO, double knockout; WTD, Western-type diet.
The Compensation of Complement Activity in DKO Mice
Previous studies have demonstrated that complete inactivation of the complement system worsens atherosclerosis (9). However, our finding that adipsin deficiency has minimal effect on atherosclerosis seemed to contradict this. We, therefore, questioned whether complement activity was impaired in DKO mice. We found that the processing of C3, the central player in the complement system, into C3a in DKO mice was not reduced despite the complete absence of adipsin—in fact, there was a 45% increase in C3a levels (Fig. 3, E–F). Therefore, the inactivation of the alternative pathway caused by adipsin deficiency was buffered by the compensatory activation of C3, which likely explains the minimal effect of adipsin deficiency on atherosclerosis.
DISCUSSION
Here we report that an important adipokine, adipsin, is not a determining factor in the pathogenesis of atherosclerosis in Ldlr−/− mice, despite its key role in the alternative complement pathway. Adipsin is unlikely to serve as an important risk factor of atherosclerosis in obesity even though its levels decline under obese conditions (16, 17). Recently, adipsin has been shown to be critical for maintaining pancreatic β cell function—patients with type 2 diabetes with β cell failure are deficient in this adipokine (19, 24). Likewise, adipsin-knockout mice (adipsin−/−) exhibit impaired glucose homeostasis when subjected to prolonged diet-induced obesity (DIO) as a result of impaired β cell function (13, 19). In contrast to its effects in the DIO model, adipsin knockout in the atherogenic Ldlr−/− model showed minimal effects on glucose homeostasis and insulin sensitivity. It also did not alter levels of dyslipidemia or hypercholesterolemia. It is worth noting that the impaired insulin secretion in DIO adipsin−/− mice was not apparent until 16 wk on HFD feeding (19). In contrast, our atherogenic Ldlr−/− mice developed a much milder obesity phenotype, with a final body weight of ∼35 g due to the lower fat content in WTD (42% vs. 60% in HFD) and a shorter feeding period. This could potentially explain the lack of differences observed in metabolic function in our adipsin-deficient mouse model. Overall, our study also builds upon our previous findings that PPARγ deacetylation protects Ldlr−/− mice from atherosclerosis in the aortic arch (20) and is highly repressive of adipsin expression (22, 23). Our current results suggest that the antiatherogenic effect of PPARγ deacetylation occurs through an adipsin-independent mechanism.
Adipsin is among the many cytokines produced by adipocytes, some of which have been shown to be closely associated with atherosclerosis, such as IL-6, IL-8, MCP-1, and adiponectin (10, 25). The effects of adipsin on atherosclerosis, if any, might be compensated for by changes in other adipokines. For example, this occurs for another representative adipokine—adiponectin. Although adiponectin confers insulin-sensitizing and antiatherogenic properties, neither gain- nor loss-of-function of adiponectin affects atherosclerosis development (26). The connection between adipose tissue and atherosclerosis has also been reinforced by recent discoveries in perivascular adipose tissue. Perivascular fat tightly adheres to blood vessels, including the aorta and coronary arteries, and engages in paracrine cross talk with these vessels through the secretion of adipokines (27). It has been suggested that certain vascular dysfunctions such as inflammation, angiogenesis, and coagulation involve cytokines released by perivascular adipocytes (25). Although there may be endocrine regulation of atherosclerosis by adipose tissue, our study indicates that adipsin is unlikely to play a major role in this context, regardless of its function in alternative complement activation. Its potential effects appear to be blunted by the compensatory activation of the complement system. All three complement pathways, including the classical, alternative, and lectin pathways, converge at the formation of C3 convertases, which cleave C3 into C3a (8). C3a has been shown to facilitate platelet activation, which may contribute to arterial thrombus formation (28) and acute myocardial infarction (29), thereby explaining the association between elevated adipsin levels and increased risk of coronary artery disease. However, this would suggest that adipsin ablation would affect plaque stability, though we observed no difference in the lesion cap thickness (data not shown) in our atherogenic model. Further investigation is warranted to understand the potential role of adipsin in advanced plaque structure and formation.
Our findings collectively indicate that adipsin itself may not be critical toward the regulation of vascular health by adipose tissue in Ldlr−/− mice. However, we are aware that the potential effect of adipsin on lipid profiles might be overridden by LDLR deficiency. In these mice, regular lipoprotein uptake is abolished, resulting in abnormally high cholesterol levels and dyslipidemia. The possible role of adipsin in lipoprotein uptake is masked in our present model, and thus, other atherogenic models should also be considered to circumvent this limitation.
GRANTS
This work was supported by National Institutes of Health Grants R01DK112943 (to L. Qiang) and P01HL087123 (to L. Qiang).
DISCLAIMERS
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
L.Q. conceived and designed research; L.L., M.C., and L.Y. performed experiments; L.L., M.C., and L.Q. analyzed data; L.L. and L.Q. interpreted results of experiments; L.L. and M.C. prepared figures; L.L., M.C., and L.Q. drafted manuscript; L.L., M.C., L.Y., W.W., and L.Q. edited and revised manuscript; L.Q. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank G. Kuriakose and S.E. Abramowicz at the Atherosclerosis Phenotyping Core for analyzing atherosclerotic plaque. We also thank Dr. James Lo (Weill Cornell Medicine) for kindly providing the adipsin−/− mice and engaging in helpful discussions with us.
REFERENCES
- 1.World Health Organization. Cardiovascular Disease (CVDs). Geneva, Switzerland: World Health Organization, 2020. [Google Scholar]
- 2.Scherer PE, Hill JA. Obesity, diabetes, and cardiovascular diseases: a compendium. Circ Res 118: 1703–1705, 2016. doi: 10.1161/CIRCRESAHA.116.308999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism 92: 6–10, 2019. doi: 10.1016/j.metabol.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, Tokgozoglu L, Lewis EF. Atherosclerosis. Nat Rev Dis Primers 5: 56, 2019. doi: 10.1038/s41572-019-0106-z. [DOI] [PubMed] [Google Scholar]
- 5.Frostegard J. Immunity, atherosclerosis and cardiovascular disease. BMC Med 11: 117, 2013. doi: 10.1186/1741-7015-11-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity 47: 621–634, 2017. doi: 10.1016/j.immuni.2017.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willows J, Brown M, Sheerin NS. The role of complement in kidney disease. Clin Med 20: 156–160, 2020. doi: 10.7861/clinmed.2019-0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Speidl WS, Kastl SP, Huber K, Wojta J. Complement in atherosclerosis: friend or foe? J Thromb Haemost 9: 428–440, 2011. doi: 10.1111/j.1538-7836.2010.04172.x. [DOI] [PubMed] [Google Scholar]
- 9.Buono C, Come CE, Witztum JL, Maguire GF, Connelly PW, Carroll M, Lichtman AH. Influence of C3 deficiency on atherosclerosis. Circulation 105: 3025–3031, 2002. doi: 10.1161/01.CIR.0000019584.04929.83. [DOI] [PubMed] [Google Scholar]
- 10.Liang W, Ye DD. The potential of adipokines as biomarkers and therapeutic agents for vascular complications in type 2 diabetes mellitus. Cytokine Growth Factor Rev 48: 32–39, 2019. doi: 10.1016/j.cytogfr.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 11.Choi HM, Doss HM, Kim KS. Multifaceted physiological roles of adiponectin in inflammation and diseases. Int J Mol Sci 21: 1219, 2020. doi: 10.3390/ijms21041219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cook KS, Min HY, Johnson D, Chaplinsky RJ, Flier JS, Hunt CR, Spiegelman BM. Adipsin: a circulating serine protease homolog secreted by adipose tissue and sciatic nerve. Science 237: 402–405, 1987. doi: 10.1126/science.3299705. [DOI] [PubMed] [Google Scholar]
- 13.Rosen BS, Cook KS, Yaglom J, Groves DL, Volanakis JE, Damm D, White T, Spiegelman BM. Adipsin and complement factor D activity: an immune-related defect in obesity. Science 244: 1483–1487, 1989. doi: 10.1126/science.2734615. [DOI] [PubMed] [Google Scholar]
- 14.Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol 176: 1305–1310, 2006. doi: 10.4049/jimmunol.176.3.1305. [DOI] [PubMed] [Google Scholar]
- 15.Chan M, Lim YC, Yang J, Namwanje M, Liu L, Qiang L. Identification of a natural beige adipose depot in mice. J Biol Chem 294: 6751–6761, 2019. doi: 10.1074/jbc.RA118.006838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flier JS, Cook KS, Usher P, Spiegelman BM. Severely impaired adipsin expression in genetic and acquired obesity. Science 237: 405–408, 1987. doi: 10.1126/science.3299706. [DOI] [PubMed] [Google Scholar]
- 17.Platt KA, Min HY, Ross SR, Spiegelman BM. Obesity-linked regulation of the adipsin gene promoter in transgenic mice. Proc Natl Acad Sci USA 86: 7490–7494, 1989. doi: 10.1073/pnas.86.19.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ohtsuki T, Satoh K, Shimizu T, Ikeda S, Kikuchi N, Satoh T, Kurosawa R, Nogi M, Sunamura S, Yaoita N, Omura J, Aoki T, Tatebe S, Sugimura K, Takahashi J, Miyata S, Shimokawa H. Identification of adipsin as a novel prognostic biomarker in patients with coronary artery disease. J Am Heart Assoc 8: e013716, 2019. doi: 10.1161/JAHA.119.013716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, Kelly ME, Chatterjee Bhowmick D, Murano I, Cohen P, Banks AS, Khandekar MJ, Dietrich A, Flier JS, Cinti S, Bluher M, Danial NN, Berggren PO, Spiegelman BM. Adipsin is an adipokine that improves beta cell function in diabetes. Cell 158: 41–53, 2014. doi: 10.1016/j.cell.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu L, Fan L, Chan M, Kraakman MJ, Yang J, Fan Y, Aaron N, Wan Q, Carrillo-Sepulveda MA, Tall AR, Tabas I, Accili D, Qiang L. PPARgamma deacetylation confers the anti-atherogenic effect and improves endothelial function in diabetes treatment. Diabetes 69: 1793–1803, 2020. doi: 10.2337/db20-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doran AC, Ozcan L, Cai B, Zheng Z, Fredman G, Rymond CC, Dorweiler B, Sluimer JC, Hsieh J, Kuriakose G, Tall AR, Tabas I. CAMKIIgamma suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest 127: 4075–4089, 2017. doi: 10.1172/JCI94735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kraakman MJ, Liu Q, Postigo-Fernandez J, Ji R, Kon N, Larrea D, Namwanje M, Fan L, Chan M, Area-Gomez E, Fu W, Creusot RJ, Qiang L. PPARgamma deacetylation dissociates thiazolidinedione’s metabolic benefits from its adverse effects. J Clin Invest 128: 2600–2612, 2018. doi: 10.1172/JCI98709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, Accili D. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of PPARgamma. Cell 150: 620–632, 2012. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, Putzel G, Rosselot C, Pabon MA, Camporez JP, Bhambhani V, Hwang SJ, Yao C, Perry RJ, Mukherjee S, Larson MG, Levy D, Dow LE, Shulman GI, Dephoure N, Garcia-Ocana A, Hao M, Spiegelman BM, Ho JE, Lo JC. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat Med 25: 1739–1747, 2019. doi: 10.1038/s41591-019-0610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liberale L, Bonaventura A, Vecchie A, Casula M, Dallegri F, Montecucco F, Carbone F. The role of adipocytokines in coronary atherosclerosis. Curr Atheroscler Rep 19: 10, 2017. doi: 10.1007/s11883-017-0644-3. doi:. [DOI] [PubMed] [Google Scholar]
- 26.Nawrocki AR, Hofmann SM, Teupser D, Basford JE, Durand JL, Jelicks LA, Woo CW, Kuriakose G, Factor SM, Tanowitz HB, Hui DY, Tabas I, Scherer PE. Lack of association between adiponectin levels and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 30: 1159–1165, 2010. doi: 10.1161/ATVBAHA.109.195826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang L, Garcia-Barrio MT, Chen YE. Perivascular adipose tissue regulates vascular function by targeting vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 40: 1094–1109, 2020. doi: 10.1161/ATVBAHA.120.312464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhoch S, Kraft P, Munzer P, Mauler M, Rheinlaender J, Madlung J, Edlich F, Schaffer TE, Meuth SG, Duerschmied D, Geisler T, Borst O, Gawaz M, Kleinschnitz C, Lambris JD, Langer HF. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation 138: 1720–1735, 2018. doi: 10.1161/CIRCULATIONAHA.118.034600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frossard M, Fuchs I, Leitner JM, Hsieh K, Vlcek M, Losert H, Domanovits H, Schreiber W, Laggner AN, Jilma B. Platelet function predicts myocardial damage in patients with acute myocardial infarction. Circulation 110: 1392–1397, 2004. doi: 10.1161/01.CIR.0000141575.92958.9C. [DOI] [PubMed] [Google Scholar]