

Abstract
Severe acute respiratory syndrome coronavirus (SARS-CoV-2), a novel coronavirus, has brought an unprecedented pandemic to the world and affected over 64 million people. The virus infects human using its spike glycoprotein mediated by a crucial area, receptor-binding domain (RBD), to bind to the human ACE2 (hACE2) receptor. Mutations on RBD have been observed in different countries and classified into nine types: A435S, D364Y, G476S, N354D/D364Y, R408I, V341I, V367F, V483A and W436R. Employing molecular dynamics (MD) simulation, we investigated dynamics and structures of the complexes of the prototype and mutant types of SARS-CoV-2 spike RBDs and hACE2. We then probed binding free energies of the prototype and mutant types of RBD with hACE2 protein by using an end-point molecular mechanics Poisson Boltzmann surface area (MM-PBSA) method. According to the result of MM-PBSA binding free energy calculations, we found that V367F and N354D/D364Y mutant types showed enhanced binding affinities with hACE2 compared to the prototype. Our computational protocols were validated by the successful prediction of relative binding free energies between prototype and three mutants: N354D/D364Y, V367F and W436R. Thus, this study provides a reliable computational protocol to fast assess the existing and emerging RBD mutations. More importantly, the binding hotspots identified by using the molecular mechanics generalized Born surface area (MM-GBSA) free energy decomposition approach can guide the rational design of small molecule drugs or vaccines free of drug resistance, to interfere with or eradicate spike-hACE2 binding.
Keywords: SARS-CoV-2, COVID-19, Spike RBD/hACE2, MD simulation, Free energy calculation and decomposition, Hotspot residues, Protein–protein interaction
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a highly pathogenic novel coronavirus, has started a worldwide pandemic since December 2019 [1]. The disease caused by the virus known as coronavirus disease 2019 (COVID-19), with a wide range of symptoms, including dry cough, fever, headache, dyspnea and pneumonia [2]. As of 4 December 2020, the virus has affected more than 216 countries, infected more than 64 million people, and claimed lives for 1.5 million, with the estimated mortality of about 2–5% [3, 4]. The total number of infected people is highly underestimated since part of the mild symptom patients are self-cured without cases recorded.
SARS-CoV-2 belongs to betacoronavirus genus, with family members including SARS-associated coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV). Since the 21st century, these three viruses have trespassed the species barrier and caused an unprecedented epidemic situation [5–7]. Among the three coronaviruses, SARS-CoV-2 has the most severe global impact [8]. The genetic material inside SARS-CoV-2 is single-positive-strand RNA, with 1/3 of the gene codes for structural proteins (SPs) and the rest 2/3 of the gene codes for nonstructural proteins (nSPs) [9]. The major structural proteins expressed by SARS-CoV-2 can be divided into four classes: spike protein (S); envelope protein (E); nucleocapsid protein (N); membrane protein (M) [10]. Same as other betacoronavirus family members [11, 12], SARS-CoV-2 uses spike protein as the cell entry [13, 14]. The spike protein is the protrusion on the virus surface, giving the virus a crown appearance [15]. The spike protein comprises two functional subunits, S1 and S2. The S1 subunit includes the receptor-binding domain (RBD) with a length of about 200 residues targeting a specific protein receptor of host cells called human angiotensin-converting enzyme (hACE2) (see below) [16, 17]. Within RBD area, a short sequence called receptor-binding motif (RBM) makes direct contact with the receptor. The S2 subunit is the fusion machinery that is responsible for membrane fusion [18]. In spike protein, RBD is thought to be essential in viral tropism and infectivity [19–21]. SARS-CoV-2 spike protein provides infusion and penetration of the virus that gives the virus unsmooth surface. Just like SARS-CoV, SARS-CoV-2 infects host cells utilizing hACE2 expressed by HeLa cells as the receptor, [22] and the spike-hACE2 binding triggers a cascade of immune reactions. Normal hACE2 level in the lung is beneficial for the host to combat inflammatory disease, and also crucial for other physiological activities [23, 24]. During the virus infection, hACE2 is one indispensable component and is also considered to be one promising therapeutic target.
Binding mechanism study of spike protein RBD and hACE2 may provide a key to tackle this worldwide health threat. Complementary to wet lab study, computational methods can provide more details about binding pattern and the dynamics of protein–protein interaction. The binding mechanism elucidated by virtual in silico studies can provide hints on designing effective drugs and vaccines to overcome the emergent global challenge. More importantly, mutant models can be easily built through computational mutagenesis and the mutation effect can be easily predicted using the same computational protocol adopted to study the prototype. The hotspot residues that are essential for both the prototype and mutants can be identified by analyzing the binding profiles of their spike RBD-hACE2. Drug molecules or vaccines targeting those hotspot residues are likely to be free of drug resistance. In real world, it is hard to analyze the mutant that has higher infectivity as we will not exactly know which mutant has infected the patient. Moreover, the mutant infectivity analysis is only based on a small fraction of the samples. Therefore, we focused on in silico approaches to study the binding mechanisms of spike protein RBD/hACE2, aiming to provide insights into rational drug and vaccine design.
Spike RBD is the only protein domain initializes the viral infection process of SARS-CoV-2. Its sequence consisting of about 200 amino acid residues is highly conserved [25]. Thus, harmful mutations on this protein domain may cause drug resistance. Under this context, it’s crucial to monitor the mutation dynamics of the virus, and the mutants should be well studied to fully capture their effect on the RBD functions. Strains of SARS-CoV-2 have been collected from multiple countries, and mutants have been detected and classified into nine types according to mutation positions on RBDs, including V341I, F342L, N354D/D364Y, V367F, R408I, A435S, W436R, G476S and V483A. In this study, models of mutant RBDs were generated using the prototype SARS-CoV-2 RBD/hACE2 complex for the computational simulations. To thoroughly study the only dual amino acids mutant, N354D/D364Y, single mutation models of N354D and D364Y were also built for the subsequent studies, including molecular dynamics (MD) simulations of the protein complexes, end-point molecular mechanics-Poisson Boltzmann surface area-weighted solvent accessible surface area (MM-PBSA-WSAS) binding free energy calculations, and end-point molecular mechanics generalized Born surface area (MM-GBSA) binding free energy decomposition analysis (see the Method section). The binding affinity calculations can reveal which mutations are able to strengthen the protein–protein binding and cause drug resistance. Free energy decomposition study further reveals how binding profile is altered by point mutations on RBD. MD simulation technique has been diversely employed in COVID-19 research. Gollapalli et al. [26] reported a structure-based drug design (SBDD) case study using docking and MD simulation to find potential therapeutic targets for wild type of SARS-CoV-2 RBD; Silva de Souza et al. [27] employed MD simulation to help understand the ionic effects on wild RBD/hACE2 complex formation/stability and reported two regions on wild RBD, which can interact with hACE2 differently; de Andrade et al. [28] reported the binding affinities of wild SARS-CoV spike protein and wild SARS-CoV-2 spike protein with hACE2 and provided detailed analysis to help gain a clearer view on the binding process between two viruses. All of these studies focused on the wild type of SARS-Cov-2 spike protein. MD studies on mutant types are scarce. Ou et al. [29] published a preprint stuy on Biorxiv about short MD simulations (10 ns) on mutant RBDs/hACE2 complexes with GROMACS program and OPLS/AA force field. Our study provides a view of mutant RBDs binding with hACE2 through performing multiple MD runs and energy decomposition analysis to help better understand the binding mechanism of SARS-CoV-2 RBD with hACE2. The hotspot residues from both spike RBD and hACE2 were identified by analyzing the free energy decomposition data. The common hotspots occurring to both the wild type and harmful mutants should be major targets in binding pocket definition for structure-based drug design and vaccine development.
Results
We first performed MD simulations to assess the structural stabilities of the wild type and mutant protein complexes, and the collected MD snapshots were then applied to calculate the binding affinities of spike RBD/hACE2 interactions, and to conduct binding free energy decomposition.
Structural stability of the prototype and mutants of SARS-CoV-2 RBD/hACE2 during MD simulations
Five independent MD runs were performed for the prototype spike RBD/hACE2 complex. For each of the mutant types, which include V341I, F342L, N354D, N354D/D364Y, D364Y, V367F, R408I, A435S, W436R, G476S and V483A, two independent MD runs were carried out. Each MD run lasts 100 ns.
The root-mean-square deviations (RMSDs) of the RBD and hACE2 in each system along with the simulation time were calculated from the MD trajectories and shown in Figures S1 and S2, respectively. The RMSD plots showed that both RBD and hACE2 proteins from each system reached equilibria in the equilibrium phase (20 ns). The stable RMSD values were around 1–2.5 Å for most systems. The low RMSD values indicate the complex stability of the MD systems is satisfactory during the simulation time. Although fluctuation can be observed from some different MD runs, the fluctuations are all within 1.5 Å. The fluctuations were understandable because this is MD simulation for two proteins rather than a small ligand with a protein.
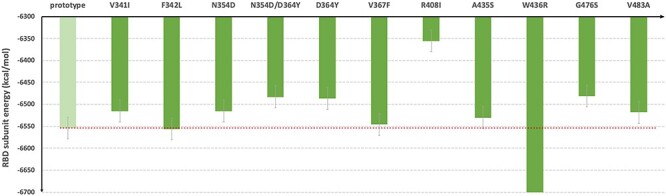
Representative MD structures, which have the smallest RMSDs compared to the average MD structures, were generated for all the MD systems. Interestingly, the representative MD structures are very similar to the crystal structure, especially for the RBD/hACE2 binding interface (Figure 1). For each mutant shown, the key interactions in prototype and in the mutant types are shown in Figure S3 and S4, respectively. As shown in the Figure 1B and Figure S3A, all the mutation sites stay far away from the protein–protein interface except for GLY476 (giving G476S mutant), ARG408 (giving R408I mutant) and VAL484 (giving V483A mutant). Though these three residues were close to the interface, no polar interaction of these three residues with the ACE2 residue was observed. Residues ASN354, ASP364, VAL367, ARG408, ALA435 and TRP436 had strong interactions with nearby residues, whilst residues VAL341, PHE342, GLY476 and VAL483 interacted with surrounded residues weakly. The interaction with nearby residues may be altered due to mutation. For example, ASN354 in the prototype can form multiple hydrogen bonds with ALA348 and SER399; after ASN354 changed to ASP354, the interaction with ALA348 disappeared, which lead to a binding affinity drop. For V483A case, no polar contact with nearby residues has been observed before or after the mutation. And no significant binding affinity alteration showed up for this mutant. By comparing the interaction with nearby residues before and after mutations, we found that the greater interaction change of the mutated residues with its nearby residues, the greater change of the RBD-ACE2 binding affinity. To be specific, V341I, A435S, W436R, G476S and V483I, part of or all of interaction formed before mutation remained after the mutation, and only slight binding affinity change was observed for these types. For cases of F342L, N354D, N354D/D364Y, D364Y, V367F and R408I, old interaction disappeared or new strong interaction formed, thus drastic binding affinity changes were observed (Figure 2). Overall, the residues that had strong interactions with the nearby residues were more likely to alter the binding free energy. This may explain why some of the mutants showed the altered binding affinity, but the other ones had comparable affinity as the prototype. By comparing the changes of conformation and the interaction pattern before and after the mutation, we can estimate the strength of the perturbation induced by a mutation at the mutation site.
Figure 1 .
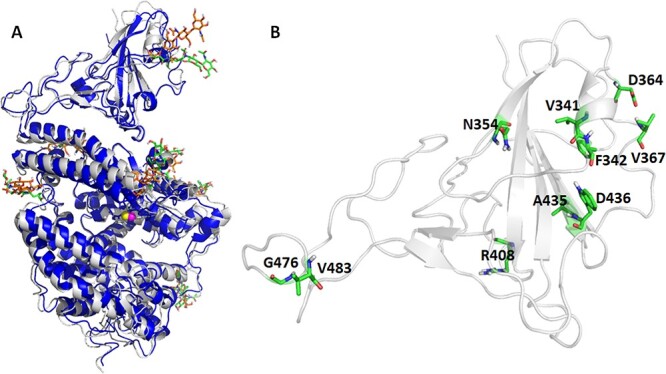
Overlay of crystal and MD representative structure for the wild type RBD/hACE2 (Panel A) and the 10 RBD mutation sites (Panel B). Glycosylated residues are shown in greenish sticks for the X-ray and brownish sticks for representative MD structures. The residues undergo mutations are shown as greenish sticks.
Figure 2 .
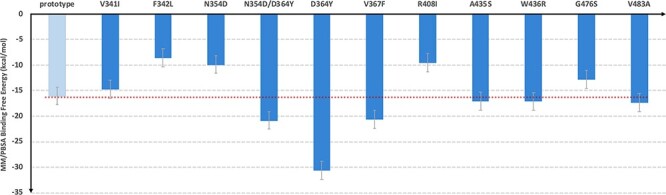
The comparison of binding free energy of each mutant and the prototype SARS-CoV-2 RBDs with hACE2.
Binding energies of RBDs/hACE2
The binding affinity between prototype SARS-CoV-2 spike and human ACE2, or a mutant SARS-CoV-2 spike protein and human ACE2 are represented by the binding affinity (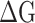 ) between the RBDs and the hACE2. To obtain the
) between the RBDs and the hACE2. To obtain the 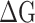 values, we conducted MM-PBSA calculations for the MD snapshots sampled in the product phase of the RBDs-hACE2 simulations. MM-PBSA is a popular end-point free energy calculation method. [30] We estimated the entropic contribution using a method called WSAS [31], as such, we call this free energy method MM-PBSA-WSAS. The results of MM-PBSA-WSAS calculations were summarized in the Table 1, and the comparison between mutants and the prototype was shown in Figure 2. Compared to −16.04 kcal/mol binding free energy of the prototype SARS-CoV-2 RBD system, three mutant types, N354D/D364Y, D364Y and V367F, showed significant lower
values, we conducted MM-PBSA calculations for the MD snapshots sampled in the product phase of the RBDs-hACE2 simulations. MM-PBSA is a popular end-point free energy calculation method. [30] We estimated the entropic contribution using a method called WSAS [31], as such, we call this free energy method MM-PBSA-WSAS. The results of MM-PBSA-WSAS calculations were summarized in the Table 1, and the comparison between mutants and the prototype was shown in Figure 2. Compared to −16.04 kcal/mol binding free energy of the prototype SARS-CoV-2 RBD system, three mutant types, N354D/D364Y, D364Y and V367F, showed significant lower 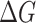 values indicating the significant higher binding affinities with hACE2. Two of these three mutants, N354D/D364Y and V367F, have drawn extensive attention as they showed up in multiple countries, indicating its enhanced binding affinity in the real world [28]. Four mutant types showed comparable binding affinity with the wild type, which are V341I, A435S, W436R and V483A. Four mutant types showed lower binding affinity than the prototype system, which are F342L, N354D, R408I and G476S. From other publication [32], R408I was once reported to have a lower binding affinity, which is consistent with our study. In the case of N354D/D364Y, D364Y mutation significantly enhanced the binding affinity, while N354D lowered the binding affinity, indicating the mutation on Asp364 contributed more to the increment of the mutant N354D/D364Y binding affinity. It is pointed out that the single D364Y mutant has not been observed in any country. The predicted binding free energies were compared with experimental data from multiple reports applying different experimental techniques. Figure 3 shows how well the binding free energies by MM-PBSA-WSAS agree with the experimental values reported by AcroBiosystems [33]. The original experimental values were listed in Table S1 in the Supporting Information (SI). The KD and EC50 values were converted into
values indicating the significant higher binding affinities with hACE2. Two of these three mutants, N354D/D364Y and V367F, have drawn extensive attention as they showed up in multiple countries, indicating its enhanced binding affinity in the real world [28]. Four mutant types showed comparable binding affinity with the wild type, which are V341I, A435S, W436R and V483A. Four mutant types showed lower binding affinity than the prototype system, which are F342L, N354D, R408I and G476S. From other publication [32], R408I was once reported to have a lower binding affinity, which is consistent with our study. In the case of N354D/D364Y, D364Y mutation significantly enhanced the binding affinity, while N354D lowered the binding affinity, indicating the mutation on Asp364 contributed more to the increment of the mutant N354D/D364Y binding affinity. It is pointed out that the single D364Y mutant has not been observed in any country. The predicted binding free energies were compared with experimental data from multiple reports applying different experimental techniques. Figure 3 shows how well the binding free energies by MM-PBSA-WSAS agree with the experimental values reported by AcroBiosystems [33]. The original experimental values were listed in Table S1 in the Supporting Information (SI). The KD and EC50 values were converted into 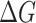 values using equation
values using equation 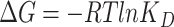 or
or 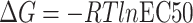 . The simulation results suggested that all the three mutants (N354D/D364Y, V367F and W436R) can enhance the binding between spike RBD and hACE2, which agrees with the experiment. The predicted binding affinities in our study were about 0.5-fold higher than the experimental values, with binding affinity of W436R was slightly higher than that of the prototype, and other two types, V367F and N354D/D364Y, exhibiting more higher binding affinities (Table 1 and Table S1). In summary, our MM-PBSA-WSAS method correctly captured the trend of the binding affinity increment of the aforementioned three mutants than the prototypes measured by experiment, albeit it overestimated the absolute values of the binding free energies. Compared to a limited number of experimental binding affinity data, our in silico method can distinguish the difference of mutation effects for a broad set of mutations, and offers a clearer view on the direction of future studies.
. The simulation results suggested that all the three mutants (N354D/D364Y, V367F and W436R) can enhance the binding between spike RBD and hACE2, which agrees with the experiment. The predicted binding affinities in our study were about 0.5-fold higher than the experimental values, with binding affinity of W436R was slightly higher than that of the prototype, and other two types, V367F and N354D/D364Y, exhibiting more higher binding affinities (Table 1 and Table S1). In summary, our MM-PBSA-WSAS method correctly captured the trend of the binding affinity increment of the aforementioned three mutants than the prototypes measured by experiment, albeit it overestimated the absolute values of the binding free energies. Compared to a limited number of experimental binding affinity data, our in silico method can distinguish the difference of mutation effects for a broad set of mutations, and offers a clearer view on the direction of future studies.
Table 1.
Results of calculated MM-PBSA energy terms and binding free energies of prototype and mutant systems
| Spike |
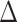 E
vdw
E
vdw
|
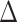 E
eel
E
eel
|
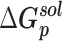
|
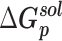
|
-T S S
|
 G
MM-PBSA
G
MM-PBSA
|
|---|---|---|---|---|---|---|
| Prototype | −91.54 ± 0.22 | −615.23 ± 0.90 | 667.81 ± 1.10 | −10.10 ± 0.01 | 33.02 ± 0.04 | −16.04 ± 0.05 |
| V341I | −91.85 ± 0.85 | −584,95 ± 5.67 | 640.29 ± 5.38 | −9.91 ± 0.06 | 31.64 ± 0.26 | −14.77 ± 0.83 |
| F342L | −93.96 ± 0.56 | −564.05 ± 6.68 | 627.49 ± 5.90 | −10.43 ± 0.04 | 32.37 ± 0.22 | −8.59 ± 0.85 |
| N354D | −93.20 ± 0.63 | −415.49 ± 3.40 | 476.86 ± 4.00 | −10.36 ± 0.06 | 32.29 ± 0.14 | −9.90 ± 1.17 |
| N354D/D364Y | −93.19 ± 0.81 | −584.45 ± 5.67 | 634.35 ± 4.27 | −9.94 ± 0.08 | 32.39 ± 0.07 | −20.84 ± 1.41 |
| D364Y | −96.07 ± 0.24 | −754.67 ± 1.89 | 797.13 ± 2.15 | −10.61 ± 0.06 | 36.26 ± 0.03 | −30.61 ± 0.47 |
| V367F | −97.29 ± 0.43 | −604.85 ± 3.1 | 657.86 ± 2.4 | −10.45 ± 0.03 | 34.08 ± 0.21 | −20.66 ± 1.3 |
| R408I | −88.52 ± 0.93 | −410.55 ± 2.13 | 468.19 ± 2.77 | −10.12 ± 0.19 | 31.48 ± 0.26 | −9.53 ± 0.55 |
| A435S | −101.03 ± 0.65 | −554.74 ± 3.41 | 614.91 ± 3.82 | −11.03 ± 0.06 | 34.83 ± 0.16 | −17.07 ± 0.35 |
| W436R | −93.92 ± 0.71 | −831.27 ± 5.51 | 884.57 ± 4.39 | −10.41 ± 0.04 | 33.92 ± 0.17 | −17.12 ± 1.51 |
| G476S | −94.04 ± 0.67 | −585.17 ± 3.49 | 643.48 ± 4.13 | −10.55 ± 0.04 | 33.50 ± 0.18 | −12.79 ± 1.22 |
| V483A | −91.31 ± 0.58 | −622.57 ± 6.54 | 673.45 ± 6.48 | −10.69 ± 0.04 | 33.82 ± 0.15 | −17.30 ± 1.57 |
Figure 3 .
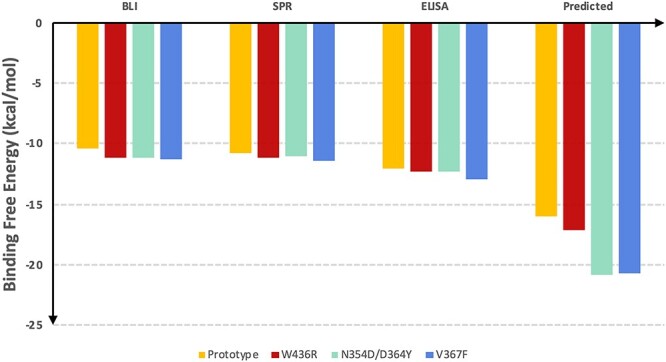
The comparison of the predicted binding free energies and experimental results. BLI is Bio-Layer Interferometry method; SPR is Surface plasmon resonance method; enzyme-linked immunosorbent assay (ELISA) is enzyme-linked immunosorbent assay. 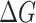 = RTlnKD is used to convert KD to binding free energy for BLI and SPR methods.
= RTlnKD is used to convert KD to binding free energy for BLI and SPR methods. 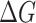 = RTlnEC50 is used to convert EC50 to binding free energy for ELISA method.
= RTlnEC50 is used to convert EC50 to binding free energy for ELISA method.
RBDs free energies
Unlike the first principal method, molecular mechanics energies are not comparable for different molecules. In Rosetta force field, a score12 term was introduced to account for different amino acid type in protein design. This term reflects the energy of an amino acid in unfolded state [34, 35]. This score12 has a narrow range (<2 kcal/mol) for different amino acid types. Considering the larger variation of the MM-PBSA-WSAS complexation energies between different mutants, the reference energy correction applied in the Rosetta force field was not made in this work. The free energy of a RBD mainly reflects the stability of the complex, as the hACE2 is well-folded and remaining the same in all of the systems. The MM-PBSA-WSAS free energies of the prototype and 11 mutant RBDs were listed in Table 2 and shown in Figure 4. As both the RBD and hACE2 are well folded proteins, we only discussed the RBD free energies to better compare the mutation effect on RBD. The hACE2 free energies are listed in Table S2 to show the consistency of hACE2 energies. Overall, most of the RBDs had comparable free energies with that of the prototype RBD except for R408I and W436R. R408I exhibited a significantly higher RBD free energy than the prototype, while W436R RBD had a significantly lower free energy than the prototype. This observation is understandable as the former mutant has a charged amino acid (ARG408) being replaced with a neutral one (ILE408), and the latter mutant has a neutral amino acid (TRP436) being replaced with a charged one (ARG436). For the R408I case, in the prototype ARG408 forming a salt-bridge with the ASP405, moreover, it can also interact with GLN414 by forming a side chain-side chai hydrogen-bond (Figure S3G). From ARG to ILE, the basic amino turned into one hydrophobic amino acid, and the mutation disrupted the interactions with the polar amino acids, explaining why a single mutation has such a significant effect on the whole complex free energy. This change in one protein might not have an effect on the binding affinity, but it can affect the stability of the protein complex. The unfavorable RBD energy and less binding affinity make R408I less risk compared to other mutants. On the other hand, as for W436R, the TRP436 formed two hydrogen bonds with ARG509 in the wild type (Figure S3I); after it was mutated into ARG, four hydrogen bonds formed between ARG436 and ARG509/SER373 (Figure S4H). For this reason, the W436R mutant achieves the lowest RBD free energy than the other RBDs, suggesting its high RBD stability. In addition, the mutant’s binding affinity was slightly higher than the prototype according to the MM-PBSA-WSAS calculation. Therefore, this mutant is relatively risky.
Table 2.
The free energies of prototype RBD and mutant type RBDs (kcal/mol)
| Prototype | V341I | F342L | N354D | N354D/D364Y | D364Y | V367F | R4081 | A435S | W436R | G476S | V483A |
|---|---|---|---|---|---|---|---|---|---|---|---|
| −6554 ± 2 | −6515 ± 7 | −6556 ± 5 | −6515 ± 4 | −6483 ± 3 | −6487 ± 5 | −6546 ± 5 | −6356 ± 4 | −6530 ± 2 | −6739 ± 7 | −6481 ± 5 | −6518 ± 11 |
Figure 4 .
The comparison of RBD free energies of each RBD/hACE2 system.
Binding pattern of prototype SARS-CoV-2 RBD/hACE2
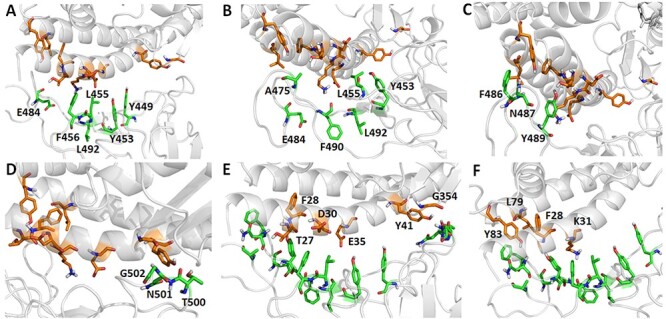
Through the MM-PBSA-WSAS calculation, the binding free energy for the prototype SARS-CoV-2 spike protein with human ACE2 protein is −16.04 kcal/mol, which is close to −12.16 kcal/mol reported [22]. As MM-GBSA-WSAS method tends to overestimate the absolute values of binding free energies, it’s reasonable that the predicted binding free energies are more negative than experimental values. We conducted binding free energy decomposition for the prototype and mutants of RBD/hACE2 using the MM-GBSA method since its computational cost is much lower than MM-PBSA-WSAS. Hotspot residues were identified according to its interaction energy with the protein binder, ΔΔGinter. The ΔΔGinter of a RBD residue is its interaction energy with hACE2, while ΔΔGinter of a hACE2 residue is its interaction energy with the RBD domain of the SARS-CoV-2 spike protein. The hotspot residues for the wild type were listed in Table S3 and illustrated in Figure 5. We used a color scheme to indicate the interaction strength measured by ΔΔGinter, and it showed that most strong interactions occurred to the binding interface residues that are colored in red and blue. The hotspot residues that strongly interact with their protein binder were labeled and shown in Figure 5B-5F. Termination of these polar/nonpolar contacts may directly interfere with the binding between two proteins. The residues positions of the selected hotspots were compared with RBM sequence and shown in the Figure 6. RBM sequence was from 438–506 and most of the significant hotspot residues were within RBM.
Figure 5 .
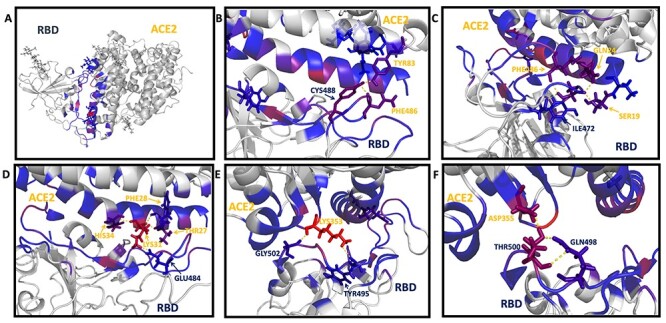
The binding hotspot of prototype SARS-CoV-2 RBD with the human ACE2. The binding interface was colored in red and blue to show the interaction strength between the prototype RBD/hACE2 complex. The red color indicates strong interaction; the blue color indicates mild interaction. The grey color indicates no interaction or the negligible interaction. The binding interactions between the interfaces were labeled with residue names. The residues from RBD are labeled in dark blue, and the residues from ACE2 are labeled in yellow. Panel B shows the interaction between Cys488 (RBD), Tyr83 (ACE2) and Phe486. Panel C shows the interaction of Ile472 (RBD) with Gln24 (ACE2), Ser19 (ACE2) and Phe486 (ACE2). Panel D shows the interaction of Glu484 (RBD) with Thr27 (ACE2), Phe28 (ACE2), Lys31 (ACE2) and His34 (ACE2). Panel E shows the interaction of Lys353 (ACE2) with Tyr495 (RBD) and Gly502 (RBD). Panel F shows the interaction of Asp355 (ACE2) with Gln498 (RBD) and Thr500 (RBD).
Figure 6 .

RBD sequence starting from Cys336 to Lys535. The residues selected in the hotspot were highlighted; the residues starting from Thr438 to Gln506 in RBM were colored in red to show the overlap between the binding area and the RBM motif.
Key binding residues in prototype and mutant RBD/hACE2
The key residues for RBDs/hACE2 binding were identified by analysis of the MM-GBSA binding free energy decomposition results. The residues with a decomposed binding energy ΔΔGinter < −0.1 kcal/mol were selected as key residues and listed in Tables S4 and S5. We generated heatmaps to show the overall contributions of each key residue to the protein–protein binding for both the prototype and mutants (Figures 7 and 8). In a heatmap, the selected key residues were listed vertically and their interaction energies in different systems were mapped into colors using a colormap shown below. Two heatmaps, for RBD and hACE2, were generated. In the RBD heatmaps, ΔΔGinter is the interaction energy between an RBD residue with the whole hACE2 protein (Figure 7), while in the hACE2 heatmap, ΔΔGinter is the interaction energy between an hACE2 residue with the whole RBD domain (Figure 8). For most mutants, the residues at the mutant positions did not take a crucial role in RBD-ACE2 binding and were not recognized as the key residues. However, GLY476, VAL483 and ARG408 were selected and shown in the heatmap since they interacted with hACE2. GLY476 made a significant contribution (−0.8 to −3.8 kcal/mol, Table S5) in protein binding compared to other mutation residues, as it is a part of the protein–protein binding interface (Figure 1). Unlike GLY476, the ΔΔGinter of ARG408 is neglectable (~ −0.2 kcal/mol) and the ΔΔGinter of VAL483 is only significant for a few mutants including A435S and G476S. We further selected some residues that are key residues in both the prototype and mutants. Drugs that target those key residues may block RBD-hACE2 binding for not only the prototype, but also the mutants, and are likely free of the drug resistance problem. Among all RBD key residues, sevens were colored in dark color, indicating their important role in RBD-hACE2 binding: PHE486, ASN487, TYR489, GLN493, THR500, ASN501 and TYR505. Similarly, seven hACE2 residues were colored in dark color indicating their essential role in RBD-hACE2 binding: GLN24, THR27, ASP30, LYS31, HIS34, LYS353 and ASP355. Those key residues are shown in Figure 9. These residues should be considered as hotspots in rational drug and vaccine development.
Figure 7 .
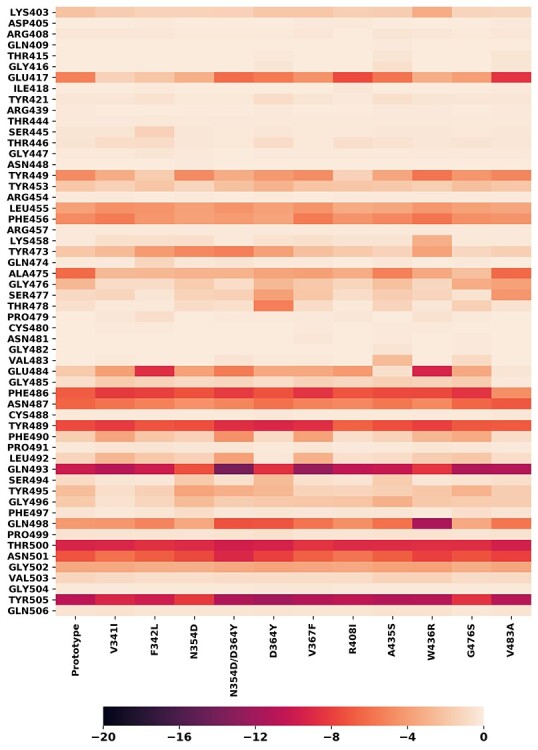
The heatmaps of residues selected from RBDs. The residue was selected if the energy contribution in RBD/hACE2 binding exceeded −0.1 kcal/mol. The y axis label presents the selected residue names; the x axis presents mutant system name. The bar on the bottom represent the relation between energy contribution and the color: darker color on the heatmap indicates bigger contribution of the residue in the binding process.
Figure 8 .

The heatmaps of residues selected from hACE2. The residue was selected if the energy contribution in RBD/hACE2 binding exceeded −0.1 kcal/mol. The y axis label presents the selected residue names; the x axis presents mutant system names. The bar on the bottom represents the relation between energy contribution and the color: darker color on the heatmap indicates bigger contribution in the binding process.
Figure 9 .
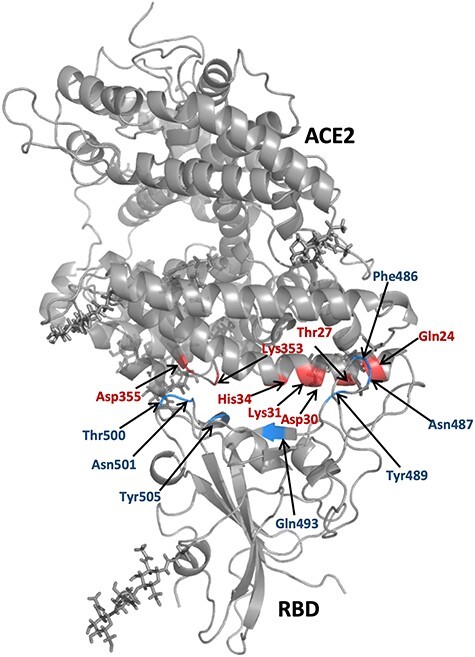
The residues selected from ACE2 and RBD, which contributed the most in RBD and hACE2 binding. The residues located in ACE2 protein were colored in red, and the residues located in RBD protein were colored in blue. These colored residues are defined as essential binding residues.
Discussion
During the COVID-19 pandemic, social economy and technology development were drastically staggered due to the extensively applied quarantine measure. Even more severe, the numbers of infection and death related to the virus are still growing. Facing this global health threat, it is essential to investigate viral infectivity and binding mechanism using all the resources we have. With the virus travel, mutants emerged, and some of the mutants are believed to have an elevated binding affinity with the hACE2 target. The emerging SARS-CoV-2 mutants may jeopardize the effort researchers put on the vaccine, since all the vaccine targets at the prototype and known mutants. This paper provides computational insights into infectivity change of SARS-CoV-2 RBDs caused by mutations, the binding interface of SARS-CoV-2 RBD and hACE2, and the key/essential residues for RBD/hACE2 binding.
Validated using in-vitro experimental values, our simulation and free energy method showed a great promise. We successfully predicted three mutants, i.e. V367F, W436R and N354D/D364Y, had more potent binding affinities than the prototype, and the findings agreed with the KD and EC50 values measured by different methods by ACRO biosystem (https://www.acrobiosystems.com). The trend of the measured binding affinities was: prototype < W436R < N354D/D364Y < V367F. Our prediction result showed that N354D/D364Y and V367F were two mutants with binding affinity significantly more potent than the prototype (−20.84 kcal/mol and − 20.66 kcal/mol versus −16.04 kcal/mol), while W436R was comparable with the prototype (−17.12 kcal/mol versus −16.04 kcal/mol). Our predicted values are more negative than the measured ones, which is satisfactory given the measurement is for protein–protein binding and the MM-PBSA-WSAS technique still needs further improvement on its accuracy.
According to our computational results, two mutant types showed significantly higher binding affinity than the prototype, V367F and N354D/D364Y. V367F mutant was reported by multiple countries and found to be more potent than the prototype, agreeing with our prediction. To understand the binding mechanism of N354D/D364Y, the only double-mutation system, we also performed simulations and conducted free energy calculations separately for N354D and D364Y. Interestingly, we found that the binding free energy of D364Y is much more potent than the prototype, while that of N354D is much less potent (Table 1). We hypothesized that there is compensation mechanism governing the double mutations: too strong interaction between RBD and hACE2 in D364Y might actually prevent the sequential process of SARS-CoV2 entering host cells, and the N354D mutation attenuate the strong interaction between RBD and hACE2 to facilitate the viruses to enter the host cells. As to the other mutants, most of them showed a comparable binding affinity with the prototype, and some specific types, such as F342L, R408I and G476S, showed lower binding affinity than the prototype. As of August 2020, some countries have shown trace of second outbreak caused by mutant SARS-CoV-2, we should be alert that the virus has evolved and selected to be more infective during the travel.
The RBD stability can be altered due to a single amino acid change. For mutant R408I and W436R, the RBD free energy was changed dramatically involved by a charged amino acid being mutated to a neutral one or vice versa. When we evaluate the risk of a mutation, both the changes on the binding affinity and protein stability should be taken into consideration.
The hotspot residues that make significant contributions to protein–protein binding for both prototype and mutants were identified by MM-GBSA binding free energy decomposition analysis. Seven residues in key residues in RBD and seven residues in hACE2 were selected as the key residues (the essential set of key residues) that should be targeted in design inhibitors and vaccines to block the RBD binding to hACE2 (Figure 9). We conducted cluster analysis on the essential set of key residues and the other hotspots (the expanded set of key residues) that occur in at least one mutant and prototype (Tables S2–S4), with an aim to identify possible binding sites where small-molecule drugs bind to RBD or hACE2. We first identified potential binding pockets of RBD, hACE2 and the complex using the SiteID module of the Sybyl software (https://www.certara.com/) and six clusters were identified. Using the center of a cluster, we expanded its residue coverage and identified the key residues within the cluster. As shown in Figure 10, there are four possible binding sites for the RBD, among which Clusters 1 and 2 can accommodate relatively large-sized organic molecules and Clusters 3 and 4 are more suitable to screen regular-sized organic molecules. Most key residues in the essential and expanded sets were covered by the four clusters. As to hACE2, we identified two possible binding sites, which can accommodate both large (Figure 10E) and small (Figure 10F) molecules.
Figure 10 .
Top clusters of binding sites for Spike RBD domain and hACE2. A: Cluster 1 for RBD, B: Cluster 2 for RBD, C: Cluster 3 for RBD, D: Cluster 4 for RBD, E: Cluster 1 for hACE2 and F: Cluster 2 for hACE2. All key residues of RBD are shown as greenish sticks, while those of hACE2 as brownish sticks.
With the reliable computational approaches established in this work, we can easily identify new RBD mutations that may enhance RBD/hACE2 binding. Different from a wet lab study, the aim of computational simulation and free energy calculations focuses on identification of risky mutants and understanding of the underlying molecular mechanisms, which may guide us to develop inhibitors of the protein–protein interaction through allostery.
Conclusion
This paper applied a series of computational methods to explore the binding pattern of SARS-CoV-2 RBD with the human ACE2. Molecular mechanics models of SARS-CoV-2 RBD/hACE2 prototype and mutants were built after a set of force field parameterizations of modified resides. Through MD Simulations and MM-PBSA calculations, the binding free energies of the prototype and mutant type RBDs binding to hACE2 were predicted. Two mutants, V367F and N354D/D364Y, were predicted to have significantly higher binding affinities, and the prediction was validated by experiment and the virus epidemiology. Moreover, the RBD and hACE2 free energies were applied by us to predict structural stability altered by mutagenesis. Two mutants, R408I and W436R were found to decrease and enhance the structural stability, respectively. Last, we conducted MM-GBSA free energy decomposition analysis using the snapshots collected during MD simulations. A set of hotspot residues was identified for both the prototype and mutants. After cluster analysis, a set of potential binding sites where the key residues from both the essential and expanded sets are located, were identified. Those binding sites may be applied to develop inhibitors of the RBD/hACE2 binding through virtual screenings. The key residues identified by us can also provide guidance on vaccine development for the spike protein.
Methods
Molecular simulation system setup
The complex of prototype SARS-CoV-2 RBD and hACE2 structure (PDBID 6 M17) [36] was obtained from Protein Data Bank. [37] The models of mutant type SARS-CoV-2 spike glycoprotein were built based on the prototype RBD/hACE2 complex by mapping the common atoms of the original and mutation residues and manually rotating side chains to maximize the favorable interactions with the surrounding residues. Three types of glycosylated residues, called AS1, AS2 and AS3, which are modified ASN residue with one N-acetylglucosamine (NAG), two NAG and three NAG residues, respectively, were introduced. A modified GLU and HIS residues that are covalently bonded to Zn2+ were also prepared using programs in the Antechamber package [38]. The atomic partial charges of those nonstandard residues were derived by the RESP [39] program to fit the HF/6-31G* electrostatic potentials generated using the Gaussian 16 software package [40]. FF14SB [41] were used for modeling proteins except for NAG, which was described by GAFF [42]. In total, there are 11RBD/hACE2 systems studied and each system contains a copy of RBD/hACE2 protein, 120 Cl− and a certain number of Na+, which neutralize the whole MD system and about 40 000 TIP3P [43] water molecules. The simulation systems were rectangles with sizes of roughly 110 × 110 × 110 Å after equilibrium.
Molecular dynamics simulations
Molecular mechanics (MM) minimization and the sequential molecular dynamics (MD) simulations were performed using the AMBER18 package. [44] First, five 10 000-step restrained minimizations were performed with the restraining forces on the main chain atoms gradually decreased from 20 to 10, 5, 1 and 0 kcal/mol. The followed MD simulations have four phases, including the relaxation phase, the heating up phase, the equilibrium phase and the sampling phase. In the relaxation phase, five 200-ps MD simulations using the same restraining forces to the main chain atoms as in minimization stage. Then the MD system was heated up progressively from 50 to 250 K at steps of 50 K in a series of 1-nanosecond MD simulations. In the next equilibrium phase, the system was equilibrated at 298 K, 1 bar for 10 ns. Last, a 100-ns MD simulation was performed at 298 K, 1 bar to produce isothermal–isobaric ensemble. We repeated the last phase four more times for the prototype and one more time for each mutant. The repeated MD runs produced independent MD trajectories by using different random number seeds for temperature regulation using Langevin dynamics [45] with a collision frequency of 5 ps−1. The integration of the equations of motion was conducted at a time step of 1 fs for the relaxation phase and 2 fs for the other three phases. The particle mesh Ewald (PME) method [46] was used to calculate the full electrostatic energy of a unit cell in a macroscopic lattice of repeating images. In total, 10 000 snapshots were collected from the sampling phase for post-analysis using the Cpptraj module implement in the AMBER software package.
MM-PBSA-WSAS free energy calculation and MM-GBSA energy decomposition
Molecular mechanics/Poisson Boltzmann surface area (MM-PBSA) is an end-point method [47–53] for free energy calculations with the solvation free energy being calculated using the PBSA method and the conformational entropy being estimated using the WSAS method. For a molecule in a solvent, the free energy is calculated using the following equations.
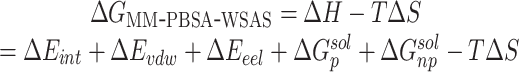 |
 stands for the internal energy contribution, which is cancelled out when applying the ‘Single Trajectory’ sampling protocol as we did in this study [54];
stands for the internal energy contribution, which is cancelled out when applying the ‘Single Trajectory’ sampling protocol as we did in this study [54]; 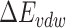 and
and 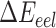 are the van der Waals and gas phase electrostatic energies, respectively;
are the van der Waals and gas phase electrostatic energies, respectively; 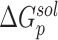 and
and 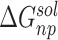 stand for the polar and nonpolar components of the solvation free energy, respectively. T is the absolute temperature;
stand for the polar and nonpolar components of the solvation free energy, respectively. T is the absolute temperature; 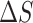 is the change of the conformational entropy.
is the change of the conformational entropy. 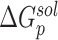 is calculated by solving the Poisson–Boltzmann equations using the Delphi program [50].
is calculated by solving the Poisson–Boltzmann equations using the Delphi program [50]. 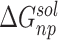 is estimated using solvent accessible surface area with the surface tension coefficient of 0.00542 kcal/(mol·Å2) and a constant of 0.92 kcal/mol. [55] For each MD trajectory, MM-PBSA-WSAS calculations were performed for 200 evenly selected snapshots. MM-GBSA energy decomposition [56], on the other hand, was performed for all the 10 000 snapshots. For MM-GBSA analysis, the polar component of solvation free energy is calculated using the Generalized Born model developed by Hawkins et al. [56]. The internal and external dielectric constants were set to 1 and 80, respectively, for both PBSA and GBSA calculations. The free energy decomposition analysis was performed using an internal program. An RBD residue becomes a hotspot when its interaction energy with hACE2 is smaller than a cutoff, −0.1 kcal/mol, while a hACE2 residue is a hotspot when its interaction energy with RBD is smaller than the cutoff.
is estimated using solvent accessible surface area with the surface tension coefficient of 0.00542 kcal/(mol·Å2) and a constant of 0.92 kcal/mol. [55] For each MD trajectory, MM-PBSA-WSAS calculations were performed for 200 evenly selected snapshots. MM-GBSA energy decomposition [56], on the other hand, was performed for all the 10 000 snapshots. For MM-GBSA analysis, the polar component of solvation free energy is calculated using the Generalized Born model developed by Hawkins et al. [56]. The internal and external dielectric constants were set to 1 and 80, respectively, for both PBSA and GBSA calculations. The free energy decomposition analysis was performed using an internal program. An RBD residue becomes a hotspot when its interaction energy with hACE2 is smaller than a cutoff, −0.1 kcal/mol, while a hACE2 residue is a hotspot when its interaction energy with RBD is smaller than the cutoff.
Key Points
We provided an efficient computational protocol to predict binding affinities of SARS-CoV-2 spike glycoprotein RBD to hACE2 receptor, and our computational results agree with the experimental data well.
Three mutant type RBDs with enhanced binding affinities were identified.
We provided the RBD free energies of different RBD/hACE2 systems to predict the relative structural stability of the mutants.
We identified binding hotspots through MM-GBSA free energy decomposition analysis. Cluster analysis on the hotspot residues were conducted to predict potential binding sites for binding inhibitor screening.
The binding heatmaps were generated to facilitate the identification of key residues for both the prototype and mutant type RBDs and hACE2. Small molecule drugs or vaccines that target those essential residues are likely to be free of drug resistance.
Supplementary Material
Acknowledgments
The authors also thank the computing resources provided by the Center for Research Computing (CRC) at University of Pittsburgh.
Yuzhao Zhang is a MS student in the School of Pharmacy, University of Pittsburgh. Her research interest focuses on computational chemistry and medicinal chemistry.
Xibing He is a Research Scientist in the School of Pharmacy, University of Pittsburgh. His research interests include force-field development, free-energy calculations and computer-aided drug design.
Jingchen Zhai is a MS student in the School of Pharmacy, University of Pittsburgh. Her research interests include molecular dynamic simulation, free energy calculations, large-scale virtual screening and PBPK modeling.
Beihong Ji is a PhD student in the School of Pharmacy, University of Pittsburgh. Her research interest is the development of tools for drug discovery and target identification in drug abuse.
Viet Hoang Man is a Research Scientist in the School of Pharmacy, University of Pittsburgh. His research focuses on amyloid aggregation, protein-ligand/protein interactions and force field development
Junmei Wang is an associate professor in Department of Pharmaceutical Sciences and Computational Chemical Genomic Screening Center, School of Pharmacy, University of Pittsburg. His research interest includes computer modeling and simulation of protein–ligand interactions and other biological events, and pharmacometrics and computational systems pharmacology.
Contributor Information
Yuzhao Zhang, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Xibing He, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Jingchen Zhai, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Beihong Ji, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Viet Hoang Man, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Junmei Wang, Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, PA 15261, USA.
Funding
The National Science Foundation (1955260); the National Institutes of Health (R01GM079383 and P30DA035778).
Conflict of Interests
The authors declare no competing financial interest.
Reference
- 1. Li Q, Guan X, Wu P, et al. Early transmission dynamics in Wuhan, China, of novel coronavirus-infected pneumonia. N Engl J Med 2020;382:1199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang C, Wang Y, Li X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang W, Tang J, Wei F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J Med Virol 2020;92:441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu YC, Chen CS, Chan YJ. The outbreak of COVID-19: an overview. J Chin Med Assoc 2020;83:217–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ksiazek TG, Erdman D, Goldsmith CS, et al. A novel coronavirus associated with severe acute respiratory syndrome. N Engl J Med 2003;348:1953–66. [DOI] [PubMed] [Google Scholar]
- 6. Zaki AM, Boheemen S, Bestebroer TM, et al. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med 2012;367:1814–20. [DOI] [PubMed] [Google Scholar]
- 7. Mohd HA, Al-Tawfiq JA, Memish ZA. Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J 2016;13:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tang B, Bragazzi NL, Li Q, et al. An updated estimation of the risk of transmission of the novel coronavirus (2019-nCov). Infect Dis Model 2020;5:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak – an update on the status. Mil Med Res 2020;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol 2020;92:418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang N, Shi X, Jiang L, et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res 2013;23:986–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Walls AC, Park Y-J, Tortorici MA, et al. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020;181:281–292.e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271–280.e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tortorici MA, Veesler D. Structural insights into coronavirus entry. Adv Virus Res 2019;105:93–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu N, Zhang D, Wang W, et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020;382:727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou P, Yang X-L, Wang X-G, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020;579:270–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020;395:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wan Y, Shang J, Graham R, et al. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol 2020;94:e00127–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen Y, Guo Y, Pan Y, et al. Structure analysis of the receptor binding of 2019-nCoV. Biochem Biophys Res Commun 2020;525:135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol 2020;5:562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang Q, Zhang Y, Wu L, et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020;181:894–904.e899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002;417:822–8. [DOI] [PubMed] [Google Scholar]
- 24. Yagil Y, Yagil C. Hypothesis. Hypertension 2003;41:871–3. [DOI] [PubMed] [Google Scholar]
- 25. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020;117:11727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gollapalli P, Sharath BS, Rimac H, et al. Pathway enrichment analysis of virus-host interactome and prioritization of novel compounds targeting the spike glycoprotein receptor binding domain-human angiotensin-converting enzyme 2 interface to combat SARS-CoV-2. J Biomol Struct Dyn 2020;38:1–14. doi: 10.1080/07391102.2020.1841681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Silva de Souza A, Rivera JD, Almeida VM, et al. Molecular dynamics reveals complex compensatory effects of ionic strength on the severe acute respiratory syndrome coronavirus 2 spike/human angiotensin-converting enzyme 2 interaction. The journal of physical chemistry letters 2020;11:10446–53. [DOI] [PubMed] [Google Scholar]
- 28. Andrade J, Gonçalves PFB, Netz PA. Why does the novel coronavirus spike protein interact so strongly with the human ACE2? A thermodynamic answer. Chembiochem 2021;22:865–75. [DOI] [PubMed] [Google Scholar]
- 29. Ou J, Zhou Z, Dai R, et al. Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. bioRxiv 2020; 2020.2003.2015.991844. [Google Scholar]
- 30. Wang E, Weng G, Sun H, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein-protein interactions. Phys Chem Chem Phys 2019;21:18958–69. [DOI] [PubMed] [Google Scholar]
- 31. Wang J, Hou T. Develop and test a solvent accessible surface area-based model in conformational entropy calculations. J Chem Inf Model 2012;52:1199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jia Y, Shen G, Zhang Y, et al. Analysis of the mutation dynamics of SARS-CoV-2 reveals the spread history and emergence of RBD mutant with lower ACE2 binding affinity. bioRxiv 2020; 2020.2004.2009.034942. [Google Scholar]
- 33. ACROBIosystems . Infectivity of SARS-CoV-2 with Spike Protein Mutations. Newark, US: Expanding the Limits of Computational Chemistry. https://www.news-medical.net/whitepaper/20200618/Infectivity-of-SARS-CoV-2-with-Spike-Protein-Mutations.aspx. (Last accessed date: Aug 24th, 2020). [Google Scholar]
- 34. Leaver-Fay A, O'Meara MJ, Tyka M, et al. Scientific benchmarks for guiding macromolecular energy function improvement. Methods Enzymol 2013;523:109–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Park H, Bradley P, GreisenP, Jr, et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J Chem Theory Comput 2016;12:6201–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full- length human ACE2. Science 2020;367:1444–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Berman HM, Westbrook J, Feng Z, et al. The protein data Bank. Nucleic Acids Res 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J, Wang W, Kollman PA, et al. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 2006;25:247–60. [DOI] [PubMed] [Google Scholar]
- 39. Bayly CI, Cieplak P, Cornell W, et al. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 1993;97:10269–80. [Google Scholar]
- 40. Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 16 Rev. C.01. Wallingford, CT: Expanding the Limits of Computational Chemistry, 2016. [Google Scholar]
- 41. Maier JA, Martinez C, Kasavajhala K, et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput 2015;11:3696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang J, Wolf RM, Caldwell JW, et al. Development and testing of a general amber force field. J Comput Chem 2004;25:1157–74. [DOI] [PubMed] [Google Scholar]
- 43. Jorgensen WL, Chandrasekhar J, Madura JD, et al. Comparison of simple potential functions for simulating liquid water. J Chem Phys 1983;79:926–35. [Google Scholar]
- 44. Case DA, Ben-Shalom IY, Brozell SR, et al. AMBER 2018. San Francisco: University of California, 2018. [Google Scholar]
- 45. Larini L, Mannella R, Leporini D. Langevin stabilization of molecular-dynamics simulations of polymers by means of quasisymplectic algorithms. J Chem Phys 2007;126:104101. [DOI] [PubMed] [Google Scholar]
- 46. Darden T, Perera L, Li L, et al. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure 1999;7:R55–60. [DOI] [PubMed] [Google Scholar]
- 47. Gohlke H, Case DA. Converging free energy estimates: MM-PB(GB)SA studies on the protein-protein complex Ras-Raf. J Comput Chem 2004;25:238–50. [DOI] [PubMed] [Google Scholar]
- 48. Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000;33:889–97. [DOI] [PubMed] [Google Scholar]
- 49. Hou T, Wang J, Li Y, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rocchia W, Alexov E, Honig B. Extending the applicability of the nonlinear Poisson−Boltzmann equation: multiple dielectric constants and multivalent ions. J Phys Chem B 2001;105:6507–14. [Google Scholar]
- 51. Kuhn B, Gerber P, Schulz-Gasch T, et al. Validation and use of the MM-PBSA approach for drug discovery. J Med Chem 2005;48:4040–8. [DOI] [PubMed] [Google Scholar]
- 52. Swanson JMJ, Henchman RH, McCammon JA. Revisiting free energy calculations: a theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys J 2004;86:67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang E, Sun H, Wang J, et al. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem Rev 2019;119:9478–508. [DOI] [PubMed] [Google Scholar]
- 54. Junmei W, Tingjun H, Xiaojie X. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr Comput Aided Drug Des 2006;2:287–306. [Google Scholar]
- 55. Knight JL, BrooksCL, 3rd.. Surveying implicit solvent models for estimating small molecule absolute hydration free energies. J Comput Chem 2011;32:2909–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem 1996;100:19824–39. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.