

Abstract
Background/Aims:
Signaling and metabolic perturbations contribute to dysregulated skeletal muscle protein homeostasis and secondary sarcopenia in response to a number of cellular stressors including ethanol exposure. Using an innovative multiomics-based curating of unbiased data, we identified molecular and metabolic therapeutic targets and experimentally validated restoration of protein homeostasis in an ethanol-fed mouse model of liver disease.
Methods:
Studies were performed in ethanol-treated differentiated C2C12 myotubes and physiological relevance established in an ethanol-fed mouse model of alcohol-related liver disease (mALD) or pair-fed control C57BL/6 mice. Transcriptome and proteome from ethanol treated-myotubes and gastrocnemius muscle from mALD and pair-fed mice were analyzed to identify target pathways and molecules. Readouts including signaling responses and autophagy markers by immunoblots, mitochondrial oxidative function and free radical generation, and metabolic studies by gas chromatography-mass spectrometry and sarcopenic phenotype by imaging.
Results:
Multiomics analyses showed that ethanol impaired skeletal muscle mTORC1 signaling, mitochondrial oxidative pathways, including intermediary metabolite regulatory genes, interleukin-6, and amino acid degradation pathways are β-hydroxymethyl-butyrate targets. Ethanol decreased mTORC1 signaling, increased autophagy flux, impaired mitochondrial oxidative function with decreased tricarboxylic acid cycle intermediary metabolites, ATP synthesis, protein synthesis and myotube diameter that were reversed by HMB. Consistently, skeletal muscle from mALD had decreased mTORC1 signaling, reduced fractional and total muscle protein synthesis rates, increased autophagy markers, lower intermediary metabolite concentrations, and lower muscle mass and fiber diameter that were reversed by β-hydroxymethyl-butyrate treatment.
Conclusion:
An innovative multiomics approach followed by experimental validation showed that β-hydroxymethyl-butyrate restores muscle protein homeostasis in liver disease.
Keywords: Autophagy, Mitochondria, Pathway-analyses, Protein synthesis, Proteomics, Transcriptomics
Introduction
Current approaches to drug development/repurposing use ADMET modeling and screening strategies using compounds from large existing libraries with retrospective identification of potential therapeutic targets and applications [1]. Instead, we developed an innovative approach using untargeted data analyses to identify specific molecule(s) a priori followed by experimental validation in established preclinical models and applied these to reverse ethanol-induced dysregulated protein homeostasis. Skeletal muscle is the largest protein store in mammals and ethanol is an established cellular stressor that perturbs mRNA translation and protein synthesis [2]. Ethanol impairs mTORC1 signaling that causes decreased protein synthesis and increased autophagy-proteolysis and consequent dysregulated protein homeostasis (proteostasis) and sarcopenia [2–6]. Skeletal muscle mitochondrial oxidative dysfunction, with increased free radical generation and reduction in ATP content, also contributes to decreased muscle protein synthesis and increased autophagy in alcohol related liver disease (ALD) [7, 8]. Our recent findings show synergistic adverse effects of hyperammonemia with ethanol on skeletal muscle signaling, organelle and protein regulatory functions [9]. Therefore, strategies using non-nitrogenous amino acid derived anabolic molecules (branched chain keto acids, β-hydroxy-β-methylbutyrate-HMB) [10, 11], have the potential to restore protein homeostasis in a number of disorders including ALD.
Since ethanol and ammonia effect alterations in multiple pathways that mediate muscle loss [3, 12], we used an integrated multiomics approach to determine potential interventions that target these abnormalities following which we manually curated known non-nitrogenous anabolic molecules. Of the number of physiological anabolic molecules, HMB is a physiological metabolite of leucine that is generated in small quantities during L-leucine catabolism [10, 13]. The initial step of L-leucine catabolism is transamination by mitochondrial branched chain amino transferase to generate ketoisocaproic acid (KIC), that is subsequently metabolized in the liver, primarily by mitochondrial a-ketoacid dehydrogenase (BCKDH) to form isovaleryl CoA. Subsequent catalytic reactions generate acetyl CoA and acetoacetate [14–16]. A small quantity (~5%) of KIC forms HMB in the cytosol, catalyzed by the enzyme 4-Hydroxyphenyl pyruvate dioxygenase (4HPPD or KIC dioxygenase) [16]. Protein modifications, altered activity and reduced fluxes across these pathways in response to ethanol have been reported [17, 18], but the preferred pathway for KIC in the liver during metabolic stress needs further evaluation to determine HMB kinetics in patients with liver disease like ALD. Unlike hepatocytes, skeletal muscle has a very limited ability to convert L-leucine into HMB, but responds both in vitro and in vivo to HMB supplementation with reduced proteolysis and increased protein synthesis [10, 13]. During cellular stress, including ethanol exposure, a number of adaptive and maladaptive mechanisms are initiated with alteration in substrate metabolism including increased muscle oxidation of branched chain amino acids (BCAA) to meet cellular energy demand [17–19]. Therefore, the generation of endogenous HMB may be lower than that during physiological states, providing the rationale for the use of HMB as an anabolic molecule during states of cellular stress.
Despite the published data on anabolic effects of HMB on skeletal muscle in multiple conditions [13], there are very limited data on HMB in sarcopenia in liver disease [10]. Administration of HMB to rats with carbon tetrachloride-induced cirrhosis or partial hepatectomy did not restore muscle mass, and the molecular and functional responses were not reported [20, 21]. Our multiomics approach, followed by manual curation, showed HMB as a potential anabolic molecule to reverse muscle perturbations in ALD. We therefore tested if HMB would reverse the molecular, metabolic and functional abnormalities in skeletal muscle in preclinical models of ALD. We observed that HMB supplementation in ethanol-treated myotubes and mice fed ethanol restored signaling responses, mitochondrial oxidative function, and reversed dysregulated proteostasis and the sarcopenic phenotype. These observations provide the mechanistic basis for using HMB to reverse stress-induced cellular responses and secondary sarcopenia.
Materials and Methods
All chemicals were obtained from Sigma Aldrich (St. Louis, MO) unless specified. All antibodies were obtained from Cell Signaling Technologies (Danvers, MA) unless stated (Supplementary Table 1 - for all supplementary material see www.cellphysiolbiochem.com).
In vitro culture studies
Cellular studies were performed in differentiated C2C12 murine myotubes as previously reported [7]. In brief, C2C12 myoblasts (ATCC; CRL 1772) were grown in Dulbecco’s modified Eagle’s medium (DMEM), containing 10% fetal bovine serum (proliferation medium) to ~80% confluence and differentiated in DMEM with 2% horse serum for 48 h. Myotubes were then exposed to 100 mM of ethanol for 6h based on our previous studies [6, 7]. Importantly, we have previously shown that this concentration and duration of ethanol exposure elicits the metabolic and functional perturbations that are observed with ethanol ingestion in vivo [7]. Cell lysates and protein extraction were carried out using protocols standard in our laboratory [6, 7]. Calcium HMB (Bulk supplements; Henderson, NV) at 50mM final concentration in the medium was used. Cellular experiments were performed in at least 3 biological replicates. We chose the calcium salt instead of the free acid due to the suggested better systemic bioavailability over the free form [22, 23].
Animal studies
Animal procedures in mice were approved by the Cleveland Clinic Institutional Animal Care and Use Committee and have been described previously [7]. Female C57BL/6 mice (Jackson Laboratory) at 8–10 wk age were allowed free access to a Lieber DeCarli liquid diet (Dyets Inc, 710260) containing ethanol (mice model of ALD, or mALD; n=6 in each group) or isocalorically substituted maltodextrins (pair-fed- PF; n=4 in each group). Animals were housed in the biological resource unit with a 12 h light/dark cycle. After adjusting to the control liquid diet for 2d, the mALD group was given a liquid diet with 1% ethanol (5.5% total calories) for 2 d, 6% ethanol (32% total calories) for 2 d. Calcium HMB was administered by gavage once daily between 9:30–10 AM in the dose of 230 mg/kg/day for the entire duration of the study. Mice were then euthanized between 9–11 AM to avoid the effect of circadian changes. Gastrocnemius muscle was harvested, weighed and a part of the muscle was used for cryosections and mitochondrial studies as previously reported [7, 24]. The remaining tissue was flash-frozen in liquid nitrogen and stored at −80°C for subsequent assays.
Multiomics analyses
Unbiased approaches including transcriptome and translatome data were evaluated to compare the effects of ethanol as previously described [7] with a focus on metabolic and translational regulation. Briefly, myotubes treated with 100 mM ethanol for 6h (n= 3 biological replicates) and gastrocnemius muscle from mALD or PF mice (n= 4 mice in each group) were used. Significance for proteomics and transcriptomics expression levels for cells and tissue was limited to p<0.05. Significance for pathway enrichment analysis was limited to -log(p-value)= ≥ 1.3.
Total RNA was extracted, quality evaluated, RNA libraries generated, sequenced and bioinformatics analyses were performed as reported by us [7]. Reference genome for mouse, GRCm38 released by the Genome Reference Consortium in 2012 based on the C57BL/6J Mus musculus was used. Sequence data have been uploaded to GitHub (github.com/atomadam2). Pathway analyses for the tricarboxylic acid (TCA) cycle were performed using Qiagen’s Ingenuity Pathway analysis (IPA) system® for core analyses. g:Profiler subfunction, gGOSt [25], was used for functional profiling of RNA sequencing and proteomics. Functional enrichment analyses were performed by over-representational analyses on input genes from the Ensembl database. This analysis includes Gene Ontology [26], pathways from KEGG reactome [27] and regulatory motif matches from TRANSFAC [28]. Regulation of amino acid metabolism including catabolism of all and synthesis of non-essential amino acids were determined using the RNA sequencing data. For metabolic pathway regulatory analyses, differentially expressed genes regulating amino acid metabolism were analyzed using IPA® and data were expressed as fold change of each component of the catabolism of the all 20 amino acids and 10 essential proteinogenic amino acids.
Liver microarray analyses
We also used the transcriptome analysis in the liver from another model of mALD and PF mice (n=4 for PF and n=6 for mALD) using the commercial ThermoFisher Clariom S assay (ThermoFisher Scientific, Waltham, MA) to determine the expression of enzymes regulating metabolic pathways. Mice were pair fed for 2 days followed by 1% ethanol for 2 days, 2% ethanol for 2 days, 4% ethanol for 1 week, 5% ethanol for 1 week, and then 6% ethanol for 1 week. Significance levels for these assays were also the same as for the muscle samples.
In brief, genomic DNA free RNA was placed in bar coded plates, annotations performed and differentially expressed genes regulating amino acid metabolism were analyzed using Ingenuity Pathway Analyses and data were expressed as fold change of each component of the catabolism of all 20 amino acids and 10 essential amino acids.
Immunoblot analysis
Immunoblots were performed using protocols standard in our laboratory [7]. In brief, tissue or cellular protein were extracted, their concentrations measured, separated by gel electrophoresis, electro transferred onto PVDF membranes (Bio-Rad, Hercules, CA, USA) and incubated with primary antibody at 1:1000 concentration, washed in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated with appropriate secondary antibodies. Immunoreactivity was detected with a chemiluminescent horse radish peroxidase substrate (Millipore, Billerica, MA, USA) and densitometry quantification done using Image J.
Protein synthesis
In vitro.
Rates of protein synthesis in myotubes were quantified by incorporation of puromycin using methods previously reported [7]. In brief, myotubes were treated with 100 mM ethanol with/without HMB and 2 μg/ml puromycin was added to the medium 30 min before harvesting the cells. Cells were then washed in ice-cold phosphate-buffered saline (PBS), lysed using RIPA buffer (25 mM Tris•HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) with protease and phosphatase inhibitors (Sigma Aldrich, St Louis, MO), protein extracted and immunoblotted with antipuromycin antibody. All bands in each lane were quantified as a measure of global protein synthesis.
In vivo.
Protein synthesis in gastrocnemius muscle was quantified by the flooding dose method as previously described with minor modifications [29, 30]. In brief, on the study day, [D5]-phenylalanine (150μM/100g) was injected intraperitoneally 30 min before euthanasia. After euthanasia, a part of the frozen gastrocnemius muscle was used to quantify the fractional and total muscle synthesis rates using the free and protein-bound [D5]-phenylalanine (tracer to trace ratio) in muscle samples. Frozen muscle samples (25–40 mg) were precisely weighed, homogenized in ice-cold perchloric acid and centrifuged at 10,000 g for 15 min at 4°C and the supernatant collected and kept on ice. This process was repeated two more times and all supernatants combined as the cytosolic fraction (free phenylalanine) to provide a measure of the precursor pool. The pellet was washed in MilliQ water, 100% ethanol and centrifuged at 10,000 g for 15 min at 4°C. The pellet with the bound muscle proteins was dried overnight and hydrolyzed in 2 ml of 6N hydrochloric acid (HCl) for 24 h at 100°C to hydrolyze the protein bound D5-phenylalanine. After washing the hydrolysates over a cation exchange column (Dowex AG 50W-8X, 200–400 mesh, H+ form, Sigma), amino acids were eluted with 4N ammonium hydroxide, derivatized with N-tert-Butyldimethylsilyl-N-methyltrifluoroacetamide with 1% tert-Butyl-dimethylchlorosilane (MTBSTFA with 1% TBDMS; Restek, Bellefonte, PA) and enrichment of D5-phenylalanine quantified by gas-chromatography mass spectrometry (GC-MS) coupled with electron impact ionization (Agilent Technologies; Santa Clara, CA). The fractional synthesis rate (FSR) was calculated as the rate of [2H5]-phenylalanine tracer incorporated into muscle protein using the formula: FSR (%)=SBx 100/SA x t, where, SB is the protein bound-phenylalanine, SA is the free phenylalanine, and t is the incorporation time in hours. When muscle mass is decreased, FSR may not be an accurate reflection of the rate of protein synthesis [29]. Hence, the entire muscle (total) synthesis rate was calculated using the formula: FSR*muscle mass/(mass of tissue used for analysis) and expressed as protein synthesis in gastrocnemius muscle (mg/h).
Ex vivo responses.
The SUnSET is a validated method for non-radioactive quantification of protein synthesis using parental puromycin in-vivo [31]. However, puromycin could affect regulatory molecule expression due to mRNA translation inhibition. We, therefore, used an ex vivo method described earlier [31] and compared the results with the incorporation of D5 phenylalanine after a flooding dose as described above. About 5 mg of fresh gastrocnemius muscle was incubated in 10 ml of DMEM with 1mM puromycin with oxygen bubbling into the medium continuously. After a precisely documented time of 30 min incubation, muscle tissue was washed in cold PBS, frozen in liquid nitrogen and stored at −80° C for subsequent protein extraction and immunoblotting for quantifying puromycin incorporation.
Autophagy flux.
Autophagy flux was determined in vitro in differentiated myotubes treated with ethanol or medium with/without 10mM chloroquine a lysosomal fusion inhibitor, as previously reported by us [6]. In brief, following ethanol with/without HMB and chloroquine, immunoblots for Beclin-1 and LC3 were performed.
Mitochondrial respiration using high resolution respirometry
Mitochondrial oxygen consumption in situ in myotubes and tissue was quantified using high resolution respirometry at 37°C on a high sensitivity fluororespirometer (Oroboros, Innsbruck, Austria) using protocols described by us previously in ethanol treated intact and permeabilized myotubes and muscle tissue from mice with ALD[7].
Intact cell respiration
Differentiated C2C12 myotubes were treated with 100 mM ethanol with or without 50μM HMB for 6h, trypsinized and intact cell respiration was measured in cells suspended in differentiation medium as previously described [7]. ATP-linked respiration, proton leak, maximum respiratory capacity were measured in response to the protonophore, carbonyl cyanide-4-(tri fluoromethoxy) phenylhydrazone (FCCP), and the response to complex I inhibitor, rotenone, and Antimycin A, an inhibitor of cytochrome c reductase, were quantified. To determine the specific defects in the ETC, responses to substrates and inhibitors of the different electron transport chain (ETC) complexes in permeabilized myotubes were determined.
Mitochondrial OXPHOS in permeabilized myotubes
Standard substrate, uncoupler, inhibitor, titration (SUIT) protocols were used to quantify mitochondrial function in digitonin permeabilized myotubes maintained in mitochondrial respiration medium, MiR05 (0.5 mMEGTA, 3 mM MgCl2.6H2O, 60 mM potassium lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, 1g/L bovine serum albumin, pH 7.1). The sequence, dose and details of these experiments have been described in detail previously [7]. In brief, malate, pyruvate and glutamate were provided as complex I substrates, followed by succinate as a complex II substrate, to quantify oxidative phosphorylation which reflects the capacity of oxidation (electron transport down the gradient along the ETC). ATP synthesis and oxidative phosphorylation were subsequently quantified in response to ADP administration. Oxidation and phosphorylation were then uncoupled using a protonophore, FCCP, to measure the maximum oxidation capacity, or, maximum respiration. The rotenone-sensitive rate (uncoupled complex I rate of oxygen consumption) and rotenone-insensitive rate (uncoupled complex II rate of oxygen consumption) were measured. Antimycin A-insensitive oxygen consumption is equivalent to the non-mitochondrial residual oxygen consumption. Uncoupled complex IV oxidation was calculated as the difference between the azide-insensitive and the tetramethyl phenylene diamine (TMPD) with ascorbate oxygen consumption rates.
DatLab2 Oroboros (Innsbruck, Austria) was used to measure oxygen consumption rates by recording oxygen concentration and flow rates at 2 sec intervals and corrected for residual oxygen consumption as previously described [7]. Studies were performed in at least 4 biological replicates after the oxygen sensors were calibrated and corrections for instrumental background were made. Oxygen consumption was expressed in pmol.sec-1.10−6 cells in myotubes or expressed per milligram of muscle weight to allow for comparisons across experiments.
Mitochondrial supercomplex assembly
Since mitochondrial electron transport chain complexes are lipophilic and believed to exist as a supercomplex on the inner mitochondrial membrane, we evaluated if supercomplexes were disrupted on blue native gel electrophoresis using methods reported by others [32]. Mitochondria from differentiated C2C12 myotubes were isolated by a modification of the protocol described previously. Myotubes were washed in ice cold PBS, treated with 0.025% trypsin and collected in DMEM with 10% fetal calf serum. Cells were then centrifuged at 800g for 2 min at 4°C and resuspended in 1 ml. lysis buffer (250mM sucrose, 20mM HEPES-NaOH (pH 7.9), 10mM KCl, 1.5mM MgCl2, 1mM EDTA, 1mM EGTA, protease inhibitors (1X);) for 20 min. Cell suspension was then homogenized with repeated aspiration and injection 20x using a 26G syringe and centrifuged at 800g for 10 min at 4°C to remove the nuclear pellet. The supernatant was collected and centrifuged at 10,000g at 4°C for 20 min and mitochondrial pellet was collected. The mitochondrial pellet was then dissolved in lysis buffer and protein content was quantified. Mitochondrial membrane was then permeabilized using digitonin (4g digitonin/g protein), incubated at 4°C for 20 minutes, and centrifuged at 10,000g for 10min. The supernatant was collected and mixed with native gel sample buffer (5% Coomassie blue G, 500mM a-amino n-caproic acid, 100mM bis-tris pH7.0). Blue native gel electrophoresis (BNE) was performed using cathode buffer (50mM Tricine, 15mM Bis-Tris and 0.2% of Coomassie blue G) and anode buffer 50mM bis-tris (pH7.0) at 90V for 20 minutes and later 150V until dye front was drained out and the gel was stained with Coomassie blue and then distained to identify mitochondrial complexes using protocols reported by others [32].
Free radical generation and oxidative modifications
Using assays previously described by us, we quantified mitochondrial free radical generation, lipid peroxidation and protein carbonylation [7]. Mitochondrial free radical generation was quantified with a mitochondrial targeted fluorescent dye, MitoSOX and flow cytometry. Lipid peroxidation was quantified by thiobarbituric acid reactive substances (TBARS) measured using a fluorometric assay. Protein carbonylation, an oxidative stress induced post translational modification, was detected by derivatization of the carbonyl group to 2,4 dinitrophenyl (DNP) by immunoblot assay using a primary antibody specific to the DNP.
Quantification of metabolites
Metabolic intermediates were quantified in ethanol-treated and untreated myotubes and gastrocnemius muscle from mALD and PF mice using GC-MS as previously described [7]. In brief, lysates were extracted from cells or muscle tissue using RIPA buffer and protein samples of a known concentration were spiked with labeled internal standards for intermediary metabolites and [D8]-HMB and [1–13C1]-KIC) for HMB and KIC respectively followed by extraction of metabolites with cold ethyl acetate. The extracts were then dried under a nitrogen stream, derivatized using TBDMS and run on an Agilent GC-MS under electron ionization and metabolites quantified using Chemstation (Agilent) as previously reported by us [7]. Concentrations of HMB and KIC in culture medium, cell lysate and muscle tissue were quantified using the area under the curve of samples and their corresponding area of spiked internal standards: 175(M0) and 183(M+8) for HMB and 316(M0) and 317(M+1) for KIC. Intermediary metabolite concentrations were expressed as nmoles/mg protein while those of HMB and KIC in medium were expressed as nmoles/mL of medium and pmol/mg protein in cell and tissue lysates.
Quantitative measurements of amino acids from ethanol-treated myotubes and gastrocnemius muscle in ALD
Free amino acids were quantified in differentiated myotubes treated with/without 100mM ethanol for 6h and 24h and gastrocnemius muscle from mALD and PF mice treated with and without HMB as described previously [33]. Lysates were extracted using RIPA buffer and ~500μg of protein samples were spiked with 4.1 mmol each of the following standards: [D4]-alanine, [D2]-glycine, [D8]-valine, [D10]-leucine, [U-13C6]-isoleucine, [D3]-proline, [U-13C5]-glutamate, [D3]-methionine, [D3]-serine, [D2]-threonine, [D5]-phenylalanine, and [D3]- aspartate. Samples were homogenized in 6% formic acid. After centrifugation, the supernatants were diluted with an equal volume of deionized, HPLC grade water and loaded on a cation exchange column (Dowex AG 50W-8X, 200–400 mesh, H+ form, Sigma-Aldrich, St. Louis, MO). The column was washed with water (5ml) and amino acids were eluted with 4N ammonium hydroxide. The eluate was dried and the residue was derivatized with 80μl of MTBSTFA with 1% TBDMS (Restek, Bellefonte, PA) at 80°C for 4h. Samples were centrifuged at 10,000 ×g for 10 min and the sample (65μl) was transferred into a GC vial for analysis. The N-silyl derivatives of amino acids were analyzed using GC-MS. Under electron ionization, ions were monitored in scan mode from 50 to 500 m/z and spectra were recorded at an isolation width of 1 m/z and normalized collision energy of 35%. Amino acid concentrations were quantified using the area under the curve (AUC) of the sample and the corresponding AUC of spiked internal standard amino acids as follows: 260(M0) and 264(M+4) for alanine, 246(M0) and 248(M+2) for glycine, 288(M0) and 296(M+8) for valine, 274(M0) and 284(M+10) for leucine, 302(M0) and 308(M+6) for isoleucine, 286(M0) and 289 (M+3) for proline, 342 (M0) and 347 (M+5) for glutamate, 320(M0) and 323(M+3) for methionine, 390(M0) and 393 (M+3) for serine, 404(M0) and 406(M+2) for threonine, 336(M0) and 341 (M+5) for phenylalanine, and 418(M0) and 421(M+3) for aspartate.
ATP content measurement
Total cellular ATP content in the mouse gastrocnemius muscle and myotubes were quantified usinga bioluminescence assay with a commercial kit (Molecular Probes, Eugene, OR, USA) as previously described [7].
Sarcopenic phenotype
Myotube diameter was quantified as a measure of myotube size in response to interventions as previously reported [7] from at least 120 myotubes from 3 independent plates. Untreated myotubes were used as controls and the mean diameter was obtained that was used to normalize the diameters for each myotube. This would generate a range of values for each group to allow for variance to be calculated. Since we used low passage number myotubes, and used identical methods for all cell culture experiments, we noted a low variance within each group as has been reported by us previously [7]. Puromycin incorporation was used as a readout for protein synthesis as described earlier [7]. In brief, myotubes or muscle tissue were incubated with 1 μM puromycin for 30 min followed by immunoblotting with antipuromycin antibody (1:10,000 dilution). Densitometry of all the bands in each lane across the entire molecular weight range of puromycin-incorporated proteins (20–250 Kd) was quantified and normalized to total protein loading by Ponceau stain. We have previously established differentiation with our protocol and proliferation was not observed as reported earlier using cytosine arabinoside treatment [7]. Autophagy flux was measured by methods previously described [6]. Lipidation of LC3 and expression of Beclin1 were determined by immunoblots in myotubes treated with and without 100 mM ethanol for 6h. All experiments were done in at least 3 biological replicates.
The overall workflow is shown in Supplementary Fig. 1.
Statistical Analyses
Data were expressed as mean±standard deviation unless specified, and means were compared using analysis of variance with Bonferroni post hoc analyses for quantitative data that were normally distributed and the Kruskal-Wallis test for skewed data. Differences were considered statistically significant at a p value <0.05. For myotube diameters, frequency histograms were also generated.
Results
Unbiased, multiomics data to identify potential interventions targeting skeletal muscle molecular perturbations in ALD
RNA sequencing and proteomics data from ethanol-treated myotubes and gastrocnemius muscle from mALD and PF mice were analyzed to identify specific therapeutic targets. Analyses of differentially expressed genes regulating muscle protein homeostasis in response to ethanol treatment in myotubes at different time points showed distinct patterns on transcriptomics and proteomics (Fig. 1A; Supplementary Table 2). Integrated pathway analyses of signaling components targeting mRNA translation including tRNA charging, translation initiation, insulin-like growth factor 1 (IGF-1) signaling, mitochondrial TCA cycle regulatory enzymes, free radical scavenging pathways, interleukin-6 (IL-6) and ubiquitination pathways were altered in ethanol-treated myotube transcriptomics and proteomics (Fig. 1B; Fig. 2A, 2B; Fig. 3A; Supplementary Tables 3–6). Transcriptomic analyses of ethanol treated myotubes were evaluated using g:Profiler incorporating Gene Ontology, Human Phenotype Ontology, and transcription factor motifs (TRANSFAC) and showed that the genes which were significantly upregulated with ethanol were mechanistically linked to phenotypes including muscle atrophy, weakness, and abnormal muscle fiber morphology (Fig. 3B; Supplementary Table 7). Open Targets [34] was used to determine potential molecular targets in sarcopenia that were compared to our unbiased data from RNA sequencing and proteomics and identified specific molecular perturbations that contribute to skeletal muscle loss (Fig. 3C; Supplementary Table 8). Perturbations in specific muscle protein homeostasis pathways including translation (ribosomal proteins, mTORC1 signaling), mitochondrial oxidative phosphorylation (OXPHOS) and free radical generation and scavenging were identified in mALD (Fig. 3D; Supplementary Table 9) and have been reported to be responsive to HMB supplementation in cellular, preclinical and human studies [13, 35–37]. Endogenous HMB is generated by a multistep process with the initial transamination of L-leucine to KIC catalyzed by BCAT (Supplementary Fig. 2), primarily in the skeletal muscle [10, 16]. Transcriptomic and proteomic expression of BCAT (mitochondrial and cytosolic), the first committed step in branched-chain amino acids (BCAA) showed differential responses to ethanol exposure in a time and context dependent manner (Fig. 4A; Supplementary Table 10). Of the BCAA, L-leucine catabolism generates KIC (HMB precursor) in the skeletal muscle and a small proportion of the KIC generated is then converted to HMB in hepatocytes [10, 13, 16]. Other amino acid metabolism pathways in ethanol-treated myotubes or muscle from mice with ALD also showed significant alterations in regulatory enzymes for catabolism of amino acids and synthesis of non-essential amino acids (Fig. 4B, 4C; Supplementary Tables 11, 12). A manual curation to identify biological molecules that can reverse ethanol-induced perturbations of different pathways was performed. The combination of changes in BCAT expression and impaired HMB targets on transcriptomics and proteomics suggest that a potent, multistep regulatory molecule such as HMB has the potential to restore skeletal muscle protein homeostasis and mitochondrial dysfunction in response to ethanol (Supplementary Fig. 3).
Fig. 1.

Multiomics analyses identified HMB responsive targets in ethanol-treated myotubes. (A). Differentially expressed genes on transcriptomics and proteomics in myotubes treated with 100 mM ethanol for different times compared with responses in untreated myotubes. (B). Integrated network of transcriptome from ethanol-treated myotubes of HMB targets- mTORC1 signaling with nodes including mitochondrial dysfunction, p70S6k signaling, AMPK signaling, and senescence pathways connected via shared molecules.
Fig. 2.

Regulatory networks in ethanol treated myotubes identify HMB targets. (A). Integrated network of the proteome from ethanol-treated myotubes of HMB targets- mTORC1 signaling with nodes including mitochondrial dysfunction, p70S6k signaling, AMPK signaling, and senescence pathways connected via shared molecules. (B). Integrated network of IGF-1 signaling pathway in ethanol treated myotubes. N= 3 biological replicates for untreated and ethanol-treated myotubes. Significance for proteomics and transcriptomics expression levels for cells was limited to p<0.05. Significance for pathway enrichment analysis was limited to -log(p-value)= > 1.3.
Fig. 3.

Ethanol induced transcriptome and proteome perturbations in ethanol-treated myotubes and mice with alcohol-related liver disease. (A). Heatmap of the transcriptome and proteome of the ubiquitination pathway in ethanol treated myotubes and muscle from mALD. (B). g:Profiler results for RNAseq data from differentially upregulated genes in 6h EtOH treated C2C12 myotubes compared to untreated myotubes. (C). Venn diagram of differentially expressed unique and shared HMB target genes on the transcriptome and proteome in ethanol-treated myotubes at different timepoints aligned with genes involved in sarcopenia on OpenTarget. (D). Differentially expressed genes regulating muscle protein homeostasis pathways in the transcriptome of mALD.
Fig. 4.

Ethanol induced alterations in amino acid metabolism enzyme expression. (A). Log fold change expression levels of BCAT1 and BCAT2 in the transcriptome and proteome in C2C12 myotubes and mALD versus untreated controls and PF mice. (B). Heatmap of differentially expressed genes regulating non-essential amino acid biosynthesis pathways. (C). Heatmap of differentially expressed genes regulating proteinogenic (essential and non-essential) amino acid degradation pathways. EtOH/E: ethanol; HMB: β-hydroxy-β-methyl butyrate; UnT: untreated controls; PF: pair-fed mice; mALD: mouse model of ALD; Prot, P.: proteomics; RNA, R.: RNAseq. N = 3 biological replicates for untreated and ethanol-treated myotubes; N = 4 mice in mALD and PF groups. Significance for proteomics and transcriptomics expression levels for cells and tissue (muscle and liver) was limited to p<0.05. Significance for pathway enrichment analysis was limited to -log(p-value)= ≥ 1.3.
We analyzed our microarray data in the liver from mALD and PF mice to determine if there was a relation between the transcriptomic responses for amino acid metabolism between the muscle and liver, specifically for the BCAA that undergo initial transamination in the muscle followed by sequential catabolism in the liver. We noted that in response to ethanol, there was decreased expression of hepatic BCKDH, the key enzyme for the oxidation of KIC to isovaleryl-CoA, which is then ultimately metabolized to acetyl CoA and acetoacetate [15, 38]. The alternative mechanism of KIC metabolism in the liver is via KIC dioxygenase (Brenda alias 4-hydroxypheynylpyruvate dioxygenase; 4HPPD) whose expression was not different between mALD and PF mouse livers. Microarray data in the liver from mALD and PF mice showed changes in expression of essential amino acid degradation pathways and synthesis of non-essential amino acids (Fig. 4B, 4C; Supplementary Table 13, 14). Since HMB is a metabolic product of L-leucine, we specifically evaluated the expression of L-leucine degradation pathways in the liver which showed no significant alterations.
Our data showed no clear direction in alterations in transcriptomics of amino acid metabolism in either the muscle or liver. Since skeletal muscle metabolites regulate signaling and functional responses, we measured the intermediary metabolite and amino acid responses with HMB supplementation in myotubes and muscle from mALD and PF mice. In addition to the metabolic alterations, ethanol inhibits signaling responses to mTORC1, mitochondrial oxidative function, and ATP generation, all of which regulate protein homeostasis and were validated experimentally.
Ethanol inhibits mTORC1 signaling that is restored by HMB treatment
Consistent with prior reports [2], ethanol inhibited phosphorylation of mTOR, the critical component of mTORC1 and downstream targets P70S6 kinase and ribosomal S6 protein in the skeletal muscle (Fig. 5A). HMB alone did not have a significant effect on signaling responses in untreated myotubes, but reversed these signaling abnormalities in ethanol-treated myotubes. Consistently, in vivo, mALD had lower mTOR phosphorylation and decreased mTORC1 signaling as noted by reduced phosphorylation of P70S6 kinase and its target, ribosomal S6 protein (Fig. 5B). mRNA translation is a bioenergetically demanding cellular process and ethanol impairs mitochondrial oxidative function and ATP synthesis [7]. Others have reported increased mitochondrial biogenesis and function with HMB [36, 39], and so we tested muscle mitochondrial responses and tissue metabolic intermediate concentrations with HMB treatment in ALD.
Fig. 5.

HMB reversed impaired mTORC1 signaling in alcoholic liver disease. Representative immunoblots and densitometry of phosphorylated mTOR (Ser2448) and signaling response to mTORC1 activation (phosphorylation of P70S6 kinase and ribosomal S6 protein) shown. (A). Differentiated C2C12 myotubes that were untreated or treated with 100 mM ethanol, with or without 50 μM HMB. (B) Studies in gastrocnemius muscle from PF or mALD mice with or without HMB. All data expressed as mean±SD from at least three biological replicates for experiments in C2C12 myotubes; n=6 mALD and n=4 PF mice in each group. * p<0.05; ** p<0.01, *** p<0.001. EtOH: ethanol; HMB β-hydroxy-β-methyl butyrate; UnT: untreated controls; PF: pair-fed mice; mALD: mouse model of ALD.
HMB restores ethanol-induced reduction in skeletal muscle mitochondrial respiration
Ethanol-impaired mitochondrial oxygen consumption in intact, differentiated myotubes was reversed by HMB treatment (Fig. 6A, 6B). Consistently, impaired oxygen consumption in response to different substrates in permeabilized myotubes were also reversed by HMB treatment (Fig. 6C, 6D). Specifically, ethanol reduced oxygen consumption in response to substrates for complex I, II and IV in myotubes and these abnormalities were reversed by HMB treatment. These perturbations were accompanied by disruption of the mitochondrial supercomplex assembly (Fig. 6E). Consistently, cellular ATP content was decreased in response to ethanol in myotubes (Fig. 6F) and in the gastrocnemius muscle of mALD than PF mice (Fig. 6G). HMB treatment partially restored ATP content in both models. Interestingly, ATP content in myotubes or skeletal muscle was not increased in PF mice treated with HMB. Thus, changes in muscle ATP levels occurred only in response to ethanol-induced mitochondrial oxidative dysfunction.
Fig. 6.

Ethanol impairs and HMB restores mitochondrial respiration in differentiated C2C12 myotubes and ATP content in muscle from mice model of ALD. (A). Representative tracings of high-resolution respirometry to quantify intact cell respiration of differentiated C2C12 myotubes. After initial stabilization, ATP synthetase inhibitor, oligomycin (O) was added, and oxygen consumption quantified to determine the oligomycin-sensitive and -insensitive respiration. Uncoupler of oxidative phosphorylation, FCCP (U) at 0.5 μM increments was then added to quantify maximum respiratory capacity. This was followed by rotenone (R) 375 nM final concentration, to inhibit complex I of the ETC, and then 2.5 μM antimycin A (Aa), which inhibits complex III, was added to determine non-mitochondrial respiration. (B) Intact, non-permeabilized C2C12 myotubes in basal DMEM medium were either untreated or treated with 100 mM ethanol for 6h with and without 50μM HMB for 6h. Basal cell respiration, proton leak, ATP-linked respiration, maximum respiratory capacity (Max. Resp.) from the response to FCCP and reserve respiratory capacity (Reserve Resp.) were measured. (C) Representative tracings of high-resolution respirometry to quantify respiration of permeabilized differentiated C2C12 myotubes. After initial stabilization, 2 mM malate (M) and 2.5 mM pyruvate (P) were added. This was followed by 4.1 mM digitonin (Dig) to permeabilize the cell membrane without losing the integrity of cells or mitochondria for permitting entry of mitochondrial substrates inside the cells; 2.5 mM ADP (D); 10 mM glutamate (G); 10 mM succinate (S); 2 mM increments of FCCP (U) for measuring maximum respiration; 375 nM rotenone (R); 125 nM antimycin A (Aa); 2 mM ascorbate and 2 mM TMPD (tetramethyl p-phenylene diamine) (AT) to test complex IV activity; 50 mM sodium azide (Az) to inhibit complex IV activity. (D) Oxygen consumption was measured in intact non-permeabilized C2C12 myotubes treated with 100 mM ethanol with and without HMB in mitochondrial respiration buffer followed by digitonin permeabilization and ETC complex specific substrates and inhibitors sequentially in the concentrations as stated above. Intact cell respiration, oxidative phosphorylation (OXPHOS) in response to M, P, D, G and S, and Max. R and RR capacity (response to U) were quantified. Rotenone-sensitive and -insensitive respiration and complex II and IV function were measured. (E) Blue native gel electrophoresis of isolated mitochondria to evaluate mitochondrial supercomplex assembly. (F)Total ATP content in C2C12 myotubes either untreated or treated with 100mM ethanol for 6hours with and without 50 μM HMB. (G) ATP content in gastrocnemius muscle from mALD mice with and without HMB compared to that from PF mice with and without HMB. * p<0.05; ** p<0.01, *** p<0.001. EtOH: ethanol; HMB β-hydroxy-β-methyl butyrate; UnT: untreated controls; PF: pair-fed mice; mALD: mouse model of ALD.
Ethanol exposure increased mitochondrial free radical generation in myotubes (Fig. 7A). Treatment with HMB partially reversed ethanol-induced free radicals, but did not have an effect in untreated myotubes. Consistent with ethanol-induced generation of free radicals, oxidative modifications of lipids (TBARS) and proteins (carbonylated protein) were increased in myotubes and skeletal muscle from mALD that were partially reversed by HMB (Fig. 7B, 7C). Interestingly, despite these abnormalities, no changes in mitochondrial mass were noted either in ethanol-treated myotubes or mALD compared to controls (Fig. 7D, 7E).
Fig. 7.

Ethanol-induced increased in free radical generation is reversed by HMB. Studies in differentiated C2C12 myotubes treated with/without 100mM ethanol with/without 50 mM HMB for 6h. (A). Representative flow cytometry of myotubes gated for MitoSox fluorescence and percentage of MitoSox fluorescent cells. (B). Representative immunoblots of carbonylated proteins and densitometry, and TBARS in myotubes treated with/without 100mM ethanol with/without 50 mM HMB for 6h. (C) Representative immunoblots and densitometry of carbonylated proteins, and tissue concentration of TBARS in gastrocnemius muscle from mALD and PF mice treated with/without HMB. (D) Representative immunoblots and densitometry of voltage dependent anion channel (VDAC) and citrate synthase (CS) expression in myotubes as measures of mitochondrial mass. (E). Representative immunoblots of VDAC and CS in gastrocnemius muscle from PF mice and mALD and PF mice treated with and without HMB. All data expressed as mean±SD from at least three biological replicates for experiments in C2C12 myotubes and n=4 PF and n=6 mice. *p<0.05; **p<0.01; ***p<0.001. EtOH: ethanol, TBARS: thiobarbituric acid reactive substances; UnT: untreated controls; PF: pair-fed; mALD: mouse model of ALD.
Metabolic intermediates of the TCA cycle were lower with ethanol treatment in myotubes and muscle tissue from mALD and PF mice and some of these were reversed by HMB treatment (Fig. 8A, 8B). Interestingly, HMB treatment increased the concentrations of some TCA cycle intermediates in untreated myotubes and pair-fed mice. Consistent with our transcriptomics data on amino acid metabolism, cellular and tissue amino acid concentrations were also not different except for higher valine in ethanol-treated myotubes (Tables 1, 2). HMB was measurable and the concentrations were not different in the medium of ethanol-treated and -untreated myotubes (Fig. 8C). However, HMB could not be detected in the cell lysate or muscle tissue from mice treated with HMB. Concentrations of KIC, a leucine metabolite were increased in response to HMB in untreated myotubes and in the culture medium (Fig. 8D). Ethanol did not alter KIC concentrations in myotubes and the HMB-induced increase in KIC in myotubes was not observed in ethanol treated myotubes. Unlike that in myotubes, HMB did not alter KIC in skeletal muscle from PF mice but KIC was increased in the skeletal muscle from mALD vs. PF mice (Fig. 8E). Representative chromatograms are shown in Fig. 8F, 8G. As expected in ALD, mALD had increased plasma triglycerides, aspartate and alanine transaminases, but these were not altered by HMB treatment (Table 3).
Fig. 8.

Intermediary metabolites decreased in ethanol treated myotubes and skeletal muscle in mice with ALD reversed by HMB. (A). Pyruvate and TCA cycle intermediates were quantified by mass spectrometry in C2C12 myotubes that were either untreated or treated with 100 mM EtOH with/without 50 μM HMB for 6hours (B) Pyruvate and TCA cycle intermediates were quantified by mass spectrometry in gastrocnemius muscle from PF mice and mALD treated with and without HMB. (C) Concentration of HMB in medium from myotubes treated with/without HMB. (D) Concentration of KIC in medium and myotube lysates treated with and without HMB. (E) Concentration of KIC in gastrocnemius muscle from mALD and PF mice treated with/without HMB. (F) Representative chromatograms and mass spectra of HMB in medium. (G) Representative chromatograms and mass spectra of KIC in gastrocnemius muscle from mALD. All data expressed as mean±SD from at least 6 biological replicates for experiments in myotubes and n=4 for PF and n=6 for mALD in each group. * p<0.05; ** p<0.01, *** p<0.001. EtOH: ethanol; HMB β-hydroxy-β-methyl butyrate; UnT: KIC a-keto isocaproic acid; untreated controls; PF: pair-fed; mALD: mouse model of ALD; TCA:Tricarboxylic acid.
Table 1.
Amino acid levels in C2C12 myotubes treated with or without ethanol and HMB.
| Amino acids | 6h treatments Concentration (nmol/μg of protein) | 24h treatments Concentration (nmol/μg of protein) | ||||||
|---|---|---|---|---|---|---|---|---|
| CTL | EtOH | HMB | HMB+EtOH | CTL | EtOH | HMB | HMB+EtOH | |
| Alanine | 2.20 ± 0.10 | 2.20 ± 0.09 | 2.23 ± 0.11 | 2.16 ± 0.11 | 2.27 ± 0.15 | 2.27 ± 0.07 | 2.28 ± 0.21 | 2.03 ± 0.10 |
| Glycine | 19.38 ± 0.65 | 19.47 ± 0.62 | 19.79 ± 0.44 | 19.40 ± 0.42 | 19.30 ± 0.35 | 18.85 ± 0.45 | 19.70 ±1.44 | 18.6 ± 0.58 |
| Valine | 4.11 ± 0.10a | 1.25 ± 0.10b | 1.32 ± 0.13c | 1.26 ± 0.15d | 1.29 ± 0.01 | 1.21 ± 0.05 | 1.45 ± 0.12 | 1.07 ± 0.05 |
| Leucine | 1.37 ± 0.15 | 1.31 ± 0.11 | 1.35 ± 0.08 | 1.41 ± 0.05 | 1.39 ± 0.15 | 1.21 ± 0.02 | 1.38 ± 0.08 | 1.31 ± 0.06 |
| Isoleucine | 1.41 ± 0.12 | 1.48 ± 0.03 | 1.47 ± 0.08 | 1.53 ± 0.17 | 1.52 ± 0.20 | 1.33 ± 0.03 | 1.96 ± 0.50 | 1.32 ± 0.07 |
| Proline | 6.08 ± 0.52 | 6.72 ± 0.44 | 5.73 ± 0.75 | 6.15 ± 0.66 | 5.95 ± 0.44 | 5.45 ± 0.26 | 7.98 ± 1.49 | 5.08 ± 0.35 |
| Aspartate | 1.11 ± 0.10 | 1.31 ± 0.13 | 1.04 ± 0.08 | 1.27 ± 0.08 | 1.28 ± 0.32 | 1.53 ± 0.18 | 0.98 ± 0 | 0.93 ± 0.09 |
| Serine | 0.96 ± 0.08 | 0.82 ± 0.08 | 0.85 ± 0.03 | 0.81 ± 0.05 | 0.83 ± 0.10 | 0.79 ± 0.05 | 0.86 ± 0.15 | 0.70 ± 0.03 |
| Glutamate | 28.15 ± 2.55 | 29.76 ± 0.65 | 20.34 ± 1.08 | 29.58 ± 1.54 | 28.13 ± 1.54 | 27.43 ± 1.52 | 30.60 ± 0.51 | 26.15 ± 0.95 |
| Methionine | 0.53 ± 0.10 | 0.41 ± 0.04 | 0.55 ± 0.17 | 0.36 ± 0.05 | 0.40 ± 0.03 | 0.39 ± 0.02 | 0.46 ± 0.01 | 0.31 ± 0.04 |
| Threonine | 3.88 ± 0.33 | 4.09 ± 0.21 | 4.29 ± 0.39 | 4.33 ± 0.34 | 4.13 ± 0.28 | 3.97 ± 0.20 | 6.07 ± 2.28 | 3.82 ± 0.48 |
| Phenylalanine | 1.03 ± 0.17 | 0.83 ± 0.05 | 0.85 ± 0.09 | 1.09 ± 0.38 | 1.00 ± 0.23 | 0.75 ± 0.03 | 0.91 ± 0.04 | 1.58 ± 0.20 |
Values shown are mean±SD (nmol/μg of protein) n=3/group; CTL: Control; EtOH: ethanol; HMB: β-Hydroxy β-methyl butyrate
Table 2.
Amino acid levels in gastrocnemius muscle in mice.
| Amino acids | Coneentration (pmol/μg of protein) | |||
|---|---|---|---|---|
| PF | mALD | PF+HMB | mALD+HMB | |
| Alanine | 39.25 ± 7.07 | 36.21 ± 3.52 | 37.16 ±5.16 | 29.72 ± 3.89 |
| Glycine | 54.49 ± 7.10 | 46.64 ± 6.60 | 48.43 ±4.55 | 34.04 ± 2.61 |
| Valine | 3.53 ± 0.81 | 4.40 ± 0.93 | 3.60 ± 0.71 | 3.92 ± 0.81 |
| Leucine | 3.32 ± 0.56 | 3.80 ±0.78 | 3.14 ±0.51 | 3.14 ± 0.58 |
| Isoleucine | 2.18 ± 0.50 | 2.42 ± 0.38 | 1.99 ± 0.26 | 2.04 ± 0.31 |
| Proline | 6.16 ± 1.62 | 8.10 ± 1.97 | 5.92 ± 1 | 6.56 ±0.98 |
| Aspartate | 19.58 ±4.58 | 12.85 ± 1.77 | 13.51 ± 1.91 | 9.46 ±2.83 |
| Serine | 1278.3 ± 240.41 | 1016.3 ± 230.39 | 1236.6 ±248.37 | 841.87 ±93.53 |
| Glutamate | 14.62 ± 2.87 | 9.08 ±2.10 | 16.73 ± 1.77 | 12.32 ± 2.34 |
| Methionine | 1.85 ± 0.49 | 1.80 ±0.60 | 1.45 ± 0.32 | 1.74 ± 0.49 |
| Threonine | 5.77 ± 1.60 | 6.07 ± 1.47 | 5.84 ±0.64 | 4.34 ± 0.57 |
| Phenylalanine | 2.00 ± 0.56 | 1.96 ±0.58 | 1.67 ± 0.28 | 1.52 ± 0.25 |
Values shown are mean ± SD (pmol/μg of protein) n=4 animal/group; mALD: mice with ALD; HMB: β-Hydroxy β-methyl butyrate; PF: pair-fed mice
Table 3.
Body composition and characteristics of mice.
| Characteristics | Pair-fed | mALD | Pair-fed+HMB | mALD+HMB |
|---|---|---|---|---|
| Initial body weight (g) | 19.38 ± 1.85 | 19.60 ± 1.01 | 19.38 ± 0.85 | 19.62 ±0.63 |
| Final body weight (g) | 20.83 ±2.18 | 19.85 ± 1.00 | 20.28 ± 1.29 | 19.83 ±0.47 |
| Liver weight (g) | 0.88 ±0.09 | 0.82 ±0.03 | 0.82 ±0.07 | 0.84 ± 0.03 |
| Liver/body weight ratio | 0.04 ±0.01 | 0.04 ±0.00 | 0.04 ±0.00 | 0.04 ± 0.00 |
| Liver triglycerides [mg/G liver) | 51.4 ±9.9 | 115.0 ± 11.3a | 42.3 ±9.0 | 105.1 ± 13.6b |
| ALT (IU/L) | 16.9 ±3.0 | 55.4 ± 22.0a | 16.3 ± 1.9 | 54.3 ± 13.0b |
| AST [IU/L) | 36.7 ±4.6 | 91.1 ±30.00a | 40.5 ±6.8 | 86.4 ± 12.3 |
ALT Alanine amino transferase; AST aspartate amino transferase; mALD mouse with alcoholic liver disease; PF pair-fed mice.
p<0.001 mALD-fed vs. pair-fed.
p<0.001 mALD+HMB vs. Pair-fed+HMB
We then tested if these signaling and metabolic perturbations with mitochondrial dysfunction caused dysregulated protein homeostasis and if HMB could reverse the functional consequences.
Ethanol exposure inhibits protein synthesis, increases autophagy and sarcopenic phenotype that are reversed by HMB
Consistent with our current data from multiomics analyses across myotube and muscle tissue responses, signaling and bioenergetic perturbations, ethanol lowered protein synthesis in myotubes and increased autophagy flux (Fig. 9A, 9B). Treatment with HMB alone increased the rate of protein synthesis and decreased autophagy flux in untreated myotubes as expected based on the mTORC1 responses to HMB published previously [35, 36, 40, 41]. Ethanol decreased protein synthesis and autophagy flux in myotubes that were reversed by HMB treatment. Since the muscle mass is maintained by a balance between protein synthesis and proteolysis, we then tested if phenotype abnormalities caused by ethanol were reversed by HMB treatment. Myotubes treated with ethanol had lower mean diameter than those treated with medium alone and HMB partially reversed this phenotype (Fig. 9C).
Fig. 9.

Skeletal muscle dysregulated proteostasis and ethanol-induced phenotype reversed by HMB. (A) Representative immuno- blots and densitometry (for the indicated conditions) for puromycin incorporation in C2C12 myotubes that were untreated or treated with 100 mM ethanol with or without 50 μM HMB. (B) Representative immunoblots and densitometry of LC3 lipidation in in differentiated C2C12 myotubes treated with 100 mM ethanol for 6h with and without 50 μM HMB. Chloroquine was used to determine autophagy flux. (C). Representative photomicrographs of C2C12 myotubes treated with and without ethanol and HMB. Mean diameter of at least 100 myotubes in each group. All data mean+SD from at least 3 biological replicates for myotubes and n=4 PF and n=6 mALD mice. *p<0.05; **p<0.01; ***p<0.001. EtOH: ethanol; HMB β-hydroxy-β-methyl butyrate; UnT: untreated controls; PF: pair-fed; mALD: mice with alcoholic liver disease.
Consistently, in mALD, muscle protein synthesis was lower as quantified by puromycin incorporation ex vivo and D5 phenylalanine incorporation in vivo and markers of autophagy were higher than that in PF mice (Fig. 10A–C). In PF mice, HMB alone increased the rate of muscle protein synthesis but did not alter LC3 lipidation. Treatment with HMB prevented these ethanol-induced changes in muscle protein synthesis and decreased autophagy markers. Phenotype also depends on body composition and muscle mass. Whole body weight was similar in all groups of mice. In vivo administration of HMB to PF mice resulted in no significant change in body weight, muscle mass or muscle fiber diameter that were similar to that of PF mice treated with vehicle alone (Table 3). Muscle mass and muscle fiber diameter in HMB-treated mALD was similar to that of PF mice (Fig. 10D, 10E).
Fig. 10.

Skeletal muscle responses to HMB in muscle from mice with ALD. (A). Representative immunoblots of incorporation of puromycin in ex vivo gastrocnemius muscle from PF mice and mALD treated with and without HMB. Densitometry of all blots in each lane. (B). Fractional and total synthesis rate of muscle protein in gastrocnemius muscle from PF mice and mALD above. (C). Representative immunoblots and densitometry of LC3 lipidation and Beclin 1 expression in mouse gastrocnemius muscle from mALD and PF mice with or without HMB. (D). Tissue weight (gm) of gastrocnemius and tibialis anterior muscles from mALD and PF mice treated with or without HMB. (E). Representative cross-sectional histological cryosections of gastrocnemius muscle oriented in the longitudinal direction from mALD and PF mice treated with or without HMB. All data expressed as mean±SD from n=4 PF mice and n= 6 mALD. * p<0.05; ** p<0.01, *** p<0.001. EtOH: ethanol; HMB β-hydroxy-β-methyl butyrate; UnT: untreated controls; PF: pair-fed; mALD: mouse model of ALD.
Discussion
The present studies show that ethanol causes skeletal muscle signaling perturbations, mitochondrial dysfunction, dysregulated proteostasis and consequent sarcopenic phenotype in validated preclinical models of ALD. We used a novel approach with a combination of unbiased data analyses to identify potential therapeutic agents complemented by experimental observations of ethanol responses in myotubes and in a validated mouse model of sarcopenia of ALD. We show that a multitargeting molecule, HMB, reversed ethanol-induced skeletal muscle molecular, metabolic, organelle function, bioenergetics and phenotype abnormalities. Novel observations include beneficial responses to HMB in ALD including mTORC1 signaling, mitochondrial oxidative function, protein synthesis and autophagy flux/markers with an increase in muscle mass. Consistent with previous publications, HMB treatment of myotubes and mice increased mTORC1 signaling and reduced autophagy [40, 41]. These data provide the mechanistic rationale and experimental evidence to support the use of HMB supplementation in human patients with sarcopenia of ALD.
Despite the high clinical significance of sarcopenia in ALD, there are no data on HMB in preclinical models or patients with ALD. Mechanistic studies on responses to HMB in myotubes, wild type and aged mice, as well as physiological studies in preclinical models of sepsis, cancer, immobilization, steroid use and carbon tetrachloride-induced liver disease demonstrate beneficial responses [10, 20, 40, 42–44]. In humans, HMB has been evaluated in healthy athletes, elderly subjects with sarcopenia, and in human patients with chronic obstructive lung disease, bronchiectasis, heart failure, renal failure and those undergoing orthopedic surgery with conflicting data on clinical outcomes [10, 13, 45–47]. In 2 published studies on models of liver disease, rats with carbon tetrachloride-induced cirrhosis and partial hepatectomy, HMB improved muscle ATP content and BCAA concentrations but did not increase muscle mass in either the control or cirrhotic/hepatectomized rats [20, 21]. These data are different from those observed in the present studies in models of sarcopenia in ALD and may be due to the direct and ongoing effect of carbon tetrachloride on the muscle, variable hepatic responses with carbon tetrachloride and duration and dose of HMB. Unlike the deleterious effects of HMB in the carbon tetrachloride cirrhosis rat model reported by others [20], in the present studies, muscle mass and signaling responses were higher in mALD treated with HMB. This may be due to species differences in responses between rats and mice, effect of carbon tetrachloride, or the longer duration of disease in the previous report [20]. Interestingly, even though muscle mass and fiber diameter were significantly lower in mALD compared to PF mice, whole body weights were not different in the 2 groups. These may be due to alterations in other compartments but are consistent with previous reports on mALD [2].
Unbiased approaches to identify perturbations in signaling responses have been extensively studied in primary sarcopenia of aging [48, 49]. However, similar data are limited in the skeletal muscle of secondary sarcopenia that occurs in chronic diseases including ALD. Using unbiased data including transcriptomics and proteomics in myotubes and skeletal muscle from mice with ALD and data from Open Targets, we observed that a number of previously reported molecular and organelle perturbations in preclinical models of sarcopenia of ALD [4–7, 50] are components of skeletal muscle protein homeostasis and are, therefore, potential molecular targets of HMB. Decreased protein synthesis and increased autophagic proteolysis are critical components that contribute to sarcopenia in ALD [5, 50]. Our unbiased data showed alterations in multiple pathways that have the potential to cause sarcopenia. These include impaired mTORC1 signaling, t-RNA charging and IGF-1 signaling pathways, decreased ribosomal components, mitochondrial oxidative phosphorylation and oxidative stress responses and increased senescence and IL-6 signaling that inhibit mRNA translation, protein synthesis and increase autophagic proteolysis.
Our experimental observations in vitro and in vivo show that ethanol treatment decreased phosphorylation of mTOR and mTORC1 kinase targets, P70S6 kinase and ribosomal S6 proteins. These are consistent with our unbiased data analyses, which showed that ethanol decreased expression of components of mTORC1 and P70S6K target pathways in myotubes, as well as previous reports on skeletal muscle responses in ALD [3]. Autophagy is inhibited by mTORC1 and ethanol inhibits mTORC1 signaling. Consistently, autophagy flux is increased in myotubes and expression of autophagy markers was higher in muscle from mice with ALD. These are similar to previous reports of increased muscle autophagy flux in ALD [6]. Our data that HMB reversed ethanol-induced mTORC1 signaling and autophagy markers was similar to reports by others that HMB increases mTORC1 signaling in skeletal muscle during stress [10, 35]. However, we did not observe significant changes in mTORC1 signaling or autophagy markers with HMB treatment in untreated myotubes or pair-fed mice suggesting that the beneficial effect of HMB was restricted to stressed cells and animals as reported previously by others [10, 42].
Protein synthesis and autophagy are sensitive to cellular ATP levels, as such, during cellular energy depletion, autophagy is activated [7]. In the present studies, ethanol-treated myotubes and muscle from mALD had decreased mitochondrial oxidative phosphorylation with defects in multiple ETC complexes including reduced ATP synthesis and lower cellular and muscle ATP. These observations were similar to previous reports on muscle mitochondrial dysfunction in ALD [7, 8]. Unaltered mitochondrial mass in response to ethanol was also consistent with our previous report [7]. Mitochondrial functional abnormalities without changes in mitochondrial mass have been reported previously with ethanol treatment as well as during hyperammonemia [7, 51]. However, our observations are different from that of others that HMB increases mitochondrial mass and oxygen consumption rates in myotubes [39, 41, 52], but there are very limited data on muscle mitochondrial biogenesis in disease or exposure to cellular stressors. Our studies show that muscle mitochondrial functional alterations and reduced ATP content in muscle from mice with ALD and ethanol-treated myotubes were reversed by HMB. These data were similar to those reported by others that mitochondrial oxidative function in intact myotubes was increased with HMB with increased mitochondrial biogenesis [39]. However, in the present studies, mitochondrial mass was not altered by HMB in myotubes or muscle tissue. Unaltered mitochondrial mass with HMB despite improvement in mitochondrial function in myotubes in the present study was different from that reported by others [52], and may be related to differences in the HMB molecule used (free) or the duration of myotubes in differentiation medium [39]. Potential mechanisms of beneficial responses to HMB in ALD in the present studies include restoration of mitochondrial intermediary metabolites and reversal of free radical generation, as was observed. Another potential mechanism by which HMB improves mitochondrial oxidative function and ETC function in response to different substrates may be related to changes in mitochondrial membrane composition since HMB is metabolized to cholesterol, a membrane constituent [53], and needs to be evaluated in future. Ethanol-induced cataplerosis or loss of TCA cycle intermediates [7] can explain the impaired mitochondrial respiration. Interestingly, despite an improvement in mitochondrial oxidative function, HMB did not consistently restore the cellular concentrations of TCA cycle intermediates in myotubes or muscle from mALD. However, in untreated myotubes and muscle from pair-fed mice, HMB increased the concentrations of some TCA cycle intermediates. Some amino acids, especially, some BCAA (isoleucine and valine), are physiological anaplerotic agents and restore decreased TCA cycle intermediates [54]. However, L-leucine is ketogenic and is not directly anaplerotic, and HMB, a leucine metabolite is not metabolized into precursors or intermediates in the TCA cycle intermediates during stress. The tissue concentrations of other anaplerotic amino acids also did not change and the mechanisms by which HMB alters the tissue concentrations of certain TCA cycle intermediates are likely to be indirect via regulating metabolism of anaplerotic molecules. Our observations that HMB was quantified only in the medium but not cell or muscle tissue lysates is different from reports by others who suggest that cellular uptake of HMB is mediated via yet unidentified transporters [55]. However, those investigators did not quantify HMB directly and instead measured radioactivity that maybe due to metabolites of HMB intracellularly. Rapid flux of HMB in skeletal muscle is another potential explanation for undetectable concentrations of HMB in tissue or cell lysates while it was measurable in the medium.
The parent molecule of HMB, L-leucine, is an essential amino acid that is a potent anabolic molecule with signaling properties which serves as a substrate for oxidative phosphorylation via generation of acetyl-CoA [15, 33, 38]. L-leucine has been extensively evaluated in preclinical and clinical studies including in ALD [3, 13, 35]. However, in ALD, oxidation of leucine is increased to generate an equimolar concentration of ammonia that is physiologically metabolized in the liver to non-toxic urea for excretion [14]. The initial step for leucine catabolism is catalyzed by BCAT that is expressed most abundantly in the skeletal muscle to generate the non-nitrogenous keto-isocaproate that is then metabolized in the hepatocyte mitochondria to form isovaleryl CoA, while a small proportion is converted to HMB in the cytosol by KIC dioxygenase [10, 14, 15, 38].
Even though we did not directly compare L-leucine to HMB, others have reported that muscle protein synthesis was increased with lower doses of HMB than that of L-leucine [13, 56]. However, when ammonia metabolism is perturbed as occurs in a number of chronic diseases including ALD and ethanol exposure [12], the resultant hyperammonemia causes skeletal muscle loss. Hence, non-nitrogenous anabolic molecules have the potential to be effective during hyperammonemia and could explain the favorable responses to HMB in ALD. It is also possible that HMB can alter the metabolism of its precursors and others have reported an increase in plasma leucine levels in septic rats [10, 42]. In contrast to these reports, we did not find any alteration in muscle leucine or BCAA levels in response to ethanol. Our observations are novel and are supported by previous data that plasma amino acids levels do not correlate with muscle levels of amino acids in liver disease [57, 58]. Interestingly muscle transcriptomics showed variable expression of BCAT, the key enzyme for transamination of leucine, and proteomics analyses did not show a significant change in either our cellular model or mALD. Even though we did not directly measure leucine fluxes in different tissues in response to ethanol, our transcriptomic and proteomic data suggest lower rates of leucine metabolism in our models of ALD that is consistent with previous reports of reduced leucine oxidation during ethanol exposure [17, 18].
Although the preparation of HMB, whether free acid or calcium salt, may have different kinetics, signaling and functional responses seem to be equivalent [22, 23] and our observations of a beneficial effect of the calcium salt support such an interpretation. Most clinical studies published to date have used the calcium salt with beneficial responses [10, 13, 45–47, 59, 60], and our current studies show that calcium HMB has the potential for rapid clinical translation to human studies (Fig. 11 (Graphical Abstract)).
Fig. 11.
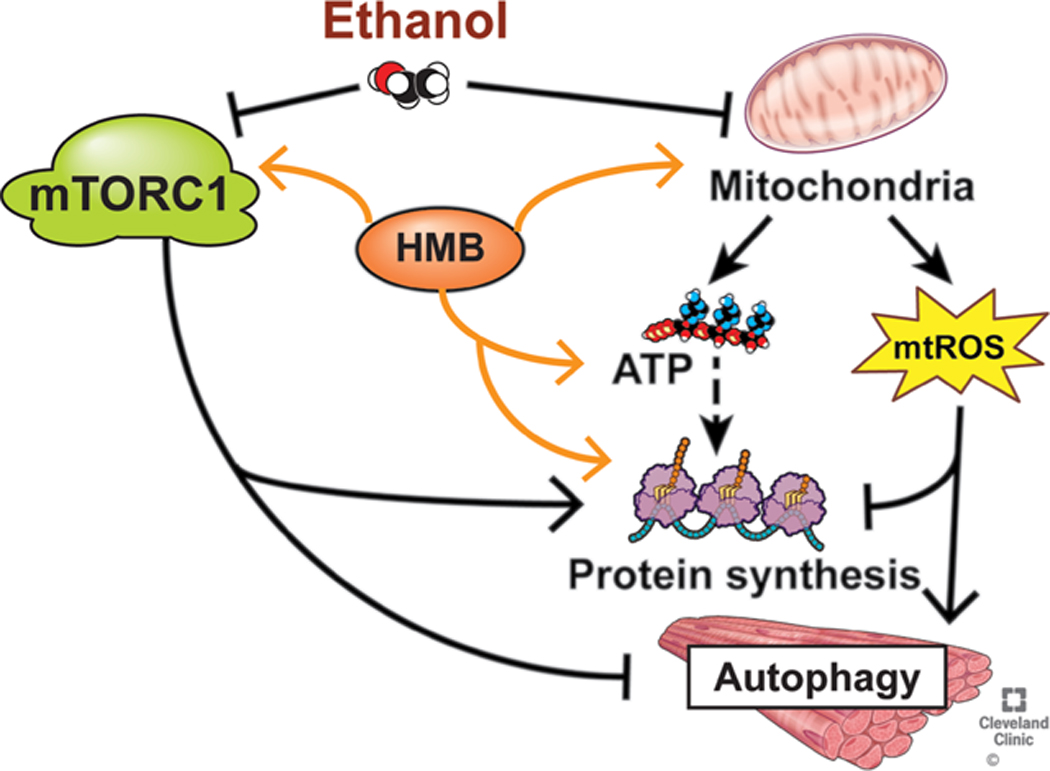
Ethanol induced perturbations in skeletal muscle that are responsive to HMB supplementation.
Our studies developed from previously published data complemented our unbiased approaches to identify HMB as a potential targeted therapeutic option were followed by experimental evidence of reversal of ethanol induced molecular, metabolic and functional perturbations in preclinical models lay the foundation for novel approaches identify responses to cellular stress and integrated mechanistic approaches to restore homeostasis. Our approach of leveraging potent bioinformatics tools to analyze large unbaised data and test potential therapeutic molecules in preclinical models has the potential for rapid and efficient development of novel, mechanism based therapies in other disorders also.
Supplementary Material
Acknowledgements
Author Contributions
SD: conceptualization, research design, supervision, project administration, funding acquisition, methodology, writing original draft, review and editing; LEN: Research design, methodology, review and editing; GD: Research design, data curation, investigation, validation, review and editing; SSS, AK, NW: Data curation, formal analysis, writing manuscript, review and editing, methodology, investigation; JS, SM, HAK, AB: Data curation, methodology, review and editing; MG, AA, XW, VP, VA, MRM, TAH: Data curation, review and editing.
Funding Sources
The following authors are funded by following support: Srinivasan Dasarathy partially supported by NIH grants R21 AR 071046; RO1 GM119174; RO1 DK113196; P50 AA024333; RO1 AA021890; 3U01AA026976 – 03S1; UO1 AA 026976; R56HL141744; UO1 DK061732; 5U01DK062470–17S2. Gangarao Davaluri partially supported by NIH grant R21 AR 071046. Laura Nagy was supported in part by NIH grants P50 AA024333; UO1 AA021890. Troy Hornberger was funded in part by NIH grant R01 AR057347. Nicole Welch was funded by NIH training grant T32 DK083251, K12HL141952 and by the American College of Gastroenterology Clinical Research Award. Amy Attaway was funded in part by NIH K12HL141952.
The Fusion Lumos instrument was purchased via an NIH shared instrument grant, 1S10OD023436–01 to perform the proteomics assays.
Statement of Ethics
All animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All national laws and regulations for these studies were also followed. Animal experiments conform to internationally accepted standards and have been approved by the Cleveland Clinic Institutional Animal Care and Use Committee.
Abbreviations
- ALD
alcohol related liver disease
- ATP
adenosine triphosphate
- BCAT
branched chain amino transferase
- BCKD
branched chain keto-dehydrogenase
- ETC
electron transport chain
- ETOH
ethanol
- FCCP
carbonyl cyanide-4-(tri fluoromethoxy) phenylhydrazone
- FSR
fractional muscle protein synthesis rate
- GC MS
gas chromatogram mass spectrometry
- HMB
β-hydroxy-βmethyl-butyrate
- IGF
insulin like growth factor
- IL
interleukin
- KIC
ketoisocaproic acid
- mALD
mouse model of alcoholic liver disease
- mTORC1
mammalian target of rapamycin complex 1
- OXPHOS
oxidative phosphorylation
- PF
pair fed
- TBARS
thiobarbituric acid reactive substances
- TCA
tricarboxylic acid
- TMPD
tetramethyl phenylene diamine
- TSR
total muscle protein synthesis rate
Footnotes
Disclosure Statement
The authors have no conflicts to declare.
References
- 1.Ferreira LLG, Andricopulo AD: ADMET modeling approaches in drug discovery. Drug Discov Today 2019;24:1157–1165. [DOI] [PubMed] [Google Scholar]
- 2.Davuluri G, Welch N, Sekar J, Gangadhariah M, Alsabbagh Alchirazi K, Mohan ML, et al. : Activated protein phosphatase 2A disrupts nutrient sensing balance between mTORC1 and AMPK causing sarcopenia in alcoholic liver disease. Hepatology 2020; DOI: 10.1002/hep.31524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasarathy J, McCullough AJ, Dasarathy S: Sarcopenia in Alcoholic Liver Disease: Clinical and Molecular Advances. Alcohol Clin Exp Res 2017;41:1419–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kimball SR, Lang CH: Mechanisms Underlying Muscle Protein Imbalance Induced by Alcohol. Annu Rev Nutr 2018;38:197–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steiner JL, Lang CH: Dysregulation of skeletal muscle protein metabolism by alcohol. Am J Physiol Endocrinol Metab 2015;308:E699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thapaliya S, Runkana A, McMullen MR, Nagy LE, McDonald C, Naga Prasad SV, et al. : Alcohol-induced autophagy contributes to loss in skeletal muscle mass. Autophagy 2014;10:677–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar A, Davuluri G, Welch N, Kim A, Gangadhariah M, Allawy A, et al. : Oxidative stress mediates ethanol-induced skeletal muscle mitochondrial dysfunction and dysregulated protein synthesis and autophagy. Free Radic Biol Med 2019;145:284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duplanty AA, Simon L, Molina PE: Chronic Binge Alcohol-Induced Dysregulation of Mitochondrial-Related Genes in Skeletal Muscle of Simian Immunodeficiency Virus-Infected Rhesus Macaques at End-Stage Disease. Alcohol Alcohol 2017;52:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kant S, Davuluri G, Alchirazi KA, Welch N, Heit C, Kumar A, et al. : Ethanol sensitizes skeletal muscle to ammonia-induced molecular perturbations. J Biol Chem 2019;294:7231–7244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holecek M: Beta-hydroxy-beta-methylbutyrate supplementation and skeletal muscle in healthy and muscle-wasting conditions. J Cachexia Sarcopenia Muscle 2017;8:529–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Holecek M: Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab (Lond) 2018;15:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dasarathy S, Hatzoglou M: Hyperammonemia and proteostasis in cirrhosis. Curr Opin Clin Nutr Metab Care 2018;21:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engelen M, Deutz NEP: Is beta-hydroxy beta-methylbutyrate an effective anabolic agent to improve outcome in older diseased populations? Curr Opin Clin Nutr Metab Care 2018;21:207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holecek M: The BCAA-BCKA cycle: its relation to alanine and glutamine synthesis and protein balance. Nutrition 2001;17:70. [DOI] [PubMed] [Google Scholar]
- 15.Suryawan A, Hawes JW, Harris RA, Shimomura Y, Jenkins AE, Hutson SM: A molecular model of human branched-chain amino acid metabolism. Am J Clin Nutr 1998;68:72–81. [DOI] [PubMed] [Google Scholar]
- 16.Van Koevering M, Nissen S: Oxidation of leucine and alpha-ketoisocaproate to beta-hydroxy-beta-methylbutyrate in vivo. Am J Physiol 1992;262:E27–31. [DOI] [PubMed] [Google Scholar]
- 17.Bernal CA, Vazquez JA, Adibi SA: Leucine metabolism during chronic ethanol consumption. Metabolism 1993;42:1084–1086. [DOI] [PubMed] [Google Scholar]
- 18.Avogaro A, Cibin M, Croatto T, Rizzo A, Gallimberti L, Tiengo A: Alcohol intake and withdrawal: effects on branched chain amino acids and alanine. Alcohol Clin Exp Res 1986;10:300–304. [DOI] [PubMed] [Google Scholar]
- 19.Mullen KD, Denne SC, McCullough AJ, Savin SM, Bruno D, Tavill AS, et al. : Leucine metabolism in stable cirrhosis. Hepatology 1986;6:622–630. [DOI] [PubMed] [Google Scholar]
- 20.Holecek M, Vodenicarovova M: Effects of beta-hydroxy-beta-methylbutyrate supplementation on skeletal muscle in healthy and cirrhotic rats. Int J Exp Pathol 2019;100:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Holecek M, Vodenicarovova M: Effects of beta-hydroxy-beta-methylbutyrate in partially hepatectomized rats. Physiol Res 2018;67:741–751. [DOI] [PubMed] [Google Scholar]
- 22.Shreeram S, Johns PW, Subramaniam S, Ramesh S, Vaidyanathan V, Puthan JK, et al. : The relative bioavailability of the calcium salt of beta-hydroxy-beta-methylbutyrate is greater than that of the free fatty acid form in rats. J Nutr 2014;144:1549–1555. [DOI] [PubMed] [Google Scholar]
- 23.Fuller JC Jr., Sharp RL, Angus HF, Baier SM, Rathmacher JA: Free acid gel form of beta-hydroxy-beta-methylbutyrate (HMB) improves HMB clearance from plasma in human subjects compared with the calcium HMB salt. Br J Nutr 2011;105:367–372. [DOI] [PubMed] [Google Scholar]
- 24.Kumar A, Davuluri G, Silva RNE, Engelen M, Ten Have GAM, Prayson R, et al. : Ammonia lowering reverses sarcopenia of cirrhosis by restoring skeletal muscle proteostasis. Hepatology 2017;65:2045–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, et al. : g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res 2019;47:W191–W198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.The Gene Ontology Consortium: The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 2019;47:D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M: New approach for understanding genome variations in KEGG. Nucleic Acids Res 2019;47:D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, et al. : TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res 2006;34:D108–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dasarathy S, McCullough AJ, Muc S, Schneyer A, Bennett CD, Dodig M, et al. : Sarcopenia associated with portosystemic shunting is reversed by follistatin. J Hepatol 2011;54:915–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garlick PJ, McNurlan MA, Preedy VR: A rapid and convenient technique for measuring the rate of protein synthesis in tissues by injection of [3H]phenylalanine. Biochem J 1980;192:719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goodman CA, Mabrey DM, Frey JW, Miu MH, Schmidt EK, Pierre P, et al. : Novel insights into the regulation of skeletal muscle protein synthesis as revealed by a new nonradioactive in vivo technique. FASEB J 2011;25:1028–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jha P, Wang X, Auwerx J: Analysis of Mitochondrial Respiratory Chain Supercomplexes Using Blue Native Polyacrylamide Gel Electrophoresis (BN-PAGE). Curr Protoc Mouse Biol 2016;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davuluri G, Krokowski D, Guan BJ, Kumar A, Thapaliya S, Singh D, et al. : Metabolic adaptation of skeletal muscle to hyperammonemia drives the beneficial effects of l-leucine in cirrhosis. J Hepatol 2016;65:929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carvalho-Silva D, Pierleoni A, Pignatelli M, Ong C, Fumis L, Karamanis N, et al. : Open Targets Platform: new developments and updates two years on. Nucleic Acids Res 2019;47:D1056–D1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suryawan A, Rudar M, Fiorotto ML, Davis TA: Differential regulation of mTORC1 activation by leucine and beta-hydroxy-beta-methylbutyrate in skeletal muscle of neonatal pigs. J Appl Physiol (1985) 2020;128:286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong Y, Zeng L, Deng J, Duan Y, Li F: beta-hydroxy-beta-methylbutyrate (HMB) improves mitochondrial function in myocytes through pathways involving PPARbeta/delta and CDK4. Nutrition 2019;60:217–226. [DOI] [PubMed] [Google Scholar]
- 37.Szczesniak KA, Ciecierska A, Ostaszewski P, Sadkowski T: Characterisation of equine satellite cell transcriptomic profile response to beta-hydroxy-beta-methylbutyrate (HMB). Br J Nutr 2016;116:1315–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutson SM: The case for regulating indispensable amino acid metabolism: the branched-chain alpha-keto acid dehydrogenase kinase-knockout mouse. Biochem J 2006;400:e1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He X, Duan Y, Yao K, Li F, Hou Y, Wu G, et al. : beta-Hydroxy-beta-methylbutyrate, mitochondrial biogenesis, and skeletal muscle health. Amino Acids 2016;48:653–664. [DOI] [PubMed] [Google Scholar]
- 40.Giron MD, Vilchez JD, Shreeram S, Salto R, Manzano M, Cabrera E, et al. : beta-Hydroxy-beta-methylbutyrate (HMB) normalizes dexamethasone-induced autophagy-lysosomal pathway in skeletal muscle. PLoS One 2015;10:e0117520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Standley RA, Distefano G, Pereira SL, Tian M, Kelly OJ, Coen PM, et al. : Effects of beta-hydroxy-beta-methylbutyrate on skeletal muscle mitochondrial content and dynamics, and lipids after 10 days of bed rest in older adults. J Appl Physiol (1985) 2017;123:1092–1100. [DOI] [PubMed] [Google Scholar]
- 42.Kovarik M, Muthny T, Sispera L, Holecek M: Effects of beta-hydroxy-beta-methylbutyrate treatment in different types of skeletal muscle of intact and septic rats. J Physiol Biochem 2010;66:311–319. [DOI] [PubMed] [Google Scholar]
- 43.Munroe M, Pincu Y, Merritt J, Cobert A, Brander R, Jensen T, et al. : Impact of beta-hydroxy beta-methylbutyrate (HMB) on age-related functional deficits in mice. Exp Gerontol 2017;87:57–66. [DOI] [PubMed] [Google Scholar]
- 44.Vallejo J, Spence M, Cheng AL, Brotto L, Edens NK, Garvey SM, et al. : Cellular and Physiological Effects of Dietary Supplementation with beta-Hydroxy-beta-Methylbutyrate (HMB) and beta-Alanine in Late Middle-Aged Mice. PLoS One 2016;11:e0150066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bear DE, Langan A, Dimidi E, Wandrag L, Harridge SDR, Hart N, et al. : beta-Hydroxy-beta-methylbutyrate and its impact on skeletal muscle mass and physical function in clinical practice: a systematic review and meta-analysis. Am J Clin Nutr 2019;109:1119–1132. [DOI] [PubMed] [Google Scholar]
- 46.Oktaviana J, Zanker J, Vogrin S, Duque G: The Effect of beta-hydroxy-beta-methylbutyrate (HMB) on Sarcopenia and Functional Frailty in Older Persons: A Systematic Review. J Nutr Health Aging 2019;23:145–150. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez-Martinez J, Santos-Lozano A, Garcia-Hermoso A, Sadarangani KP, Cristi-Montero C: Effects of beta-hydroxy-beta-methylbutyrate supplementation on strength and body composition in trained and competitive athletes: A meta-analysis of randomized controlled trials. J Sci Med Sport 2018;21:727–735. [DOI] [PubMed] [Google Scholar]
- 48.Giresi PG, Stevenson EJ, Theilhaber J, Koncarevic A, Parkington J, Fielding RA, et al. : Identification of a molecular signature of sarcopenia. Physiol Genomics 2005;21:253–263. [DOI] [PubMed] [Google Scholar]
- 49.Lin IH, Chang JL, Hua K, Huang WC, Hsu MT, Chen YF: Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet 2018;19:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fernandez-Sola J, Preedy VR, Lang CH, Gonzalez-Reimers E, Arno M, Lin JC, et al. : Molecular and cellular events in alcohol-induced muscle disease. Alcohol Clin Exp Res 2007;31:1953–1962. [DOI] [PubMed] [Google Scholar]
- 51.Davuluri G, Allawy A, Thapaliya S, Rennison JH, Singh D, Kumar A, et al. : Hyperammonaemia-induced skeletal muscle mitochondrial dysfunction results in cataplerosis and oxidative stress. J Physiol 2016;594:7341–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schnuck JK, Johnson MA, Gould LM, Gannon NP, Vaughan RA: Acute beta-Hydroxy-beta-Methyl Butyrate Suppresses Regulators of Mitochondrial Biogenesis and Lipid Oxidation While Increasing Lipid Content in Myotubes. Lipids 2016;51:1127–1136. [DOI] [PubMed] [Google Scholar]
- 53.Nissen S, Sharp RL, Panton L, Vukovich M, Trappe S, Fuller JC Jr.,: beta-hydroxy-beta-methylbutyrate (HMB) supplementation in humans is safe and may decrease cardiovascular risk factors. J Nutr 2000;130:1937–1945. [DOI] [PubMed] [Google Scholar]
- 54.Owen OE, Kalhan SC, Hanson RW: The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 2002;277:30409–30412. [DOI] [PubMed] [Google Scholar]
- 55.Ogura J, Sato T, Higuchi K, Bhutia YD, Babu E, Masuda M, et al. : Transport Mechanisms for the Nutritional Supplement beta-Hydroxy-beta-Methylbutyrate (HMB) in Mammalian Cells. Pharm Res 2019;36:84. [DOI] [PubMed] [Google Scholar]
- 56.Duan Y, Li F, Guo Q, Wang W, Zhang L, Wen C, et al. : beta-Hydroxy-beta-methyl Butyrate Is More Potent Than Leucine in Inhibiting Starvation-Induced Protein Degradation in C2C12 Myotubes. J Agric Food Chem 2018;66:170–176. [DOI] [PubMed] [Google Scholar]
- 57.Montanari A, Simoni I, Vallisa D, Trifiro A, Colla R, Abbiati R, et al. : Free amino acids in plasma and skeletal muscle of patients with liver cirrhosis. Hepatology 1988;8:1034–1039. [DOI] [PubMed] [Google Scholar]
- 58.Iob V, Coon WW, Sloan M: Free amino acids in liver, plasma, and muscle of patients with cirrhosis of the liver. J Surg Res 1967;7:41–43. [DOI] [PubMed] [Google Scholar]
- 59.Deutz NE, Pereira SL, Hays NP, Oliver JS, Edens NK, Evans CM, et al. : Effect of beta-hydroxy-beta-methylbutyrate (HMB) on lean body mass during 10 days of bed rest in older adults. Clin Nutr 2013;32:704–712. [DOI] [PubMed] [Google Scholar]
- 60.Wu H, Xia Y, Jiang J, Du H, Guo X, Liu X, et al. : Effect of beta-hydroxy-beta-methylbutyrate supplementation on muscle loss in older adults: a systematic review and meta-analysis. Arch Gerontol Geriatr 2015;61:168175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.