

SUMMARY
The transcription factors T-bet and Eomesodermin (Eomes) regulate CD8 T cell exhaustion through undefined mechanisms. Here, we show that the subcellular localization of T-bet and Eomes dictate their regulatory activity in exhausted T cells (TEXs). TEXs had a higher ratio of nuclear Eomes:T-bet than memory T cells (TMEMs) during chronic lymphocytic choriomeningitis virus (LCMV) infection in preclinical cancer models and in human tumors. Biochemically, T-bet and Eomes compete for the same DNA sequences, including the Pdcd1 T-box. High nuclear T-bet strongly represses Pdcd1 transcription in TMEM, whereas low nuclear T-bet in TEX leads to a dominant effect of Eomes that acts as a weaker repressor of Pdcd1. Blocking PD-1 signaling in TEXs increases nuclear T-bet, restoring stronger repression of Pdcd1, and driving T-bet-associated gene expression programs of chemotaxis, homing, and activation. These data identify a mechanism whereby the T-bet-Eomes axis regulates exhaustion through their nuclear localization, providing insights into how these transcription factors regulate TEX biology.
In brief
McLane et al. demonstrate that T-bet and Eomes expression contributes to exhaustion, but also their nuclear localization, and therefore functional activity, plays a key role. PD-1 blockade restores nuclear T-bet and promotes T cell homing and activation through direct competition with Eomes at gene promoters, such as Pdcd1.
Graphical Abstract
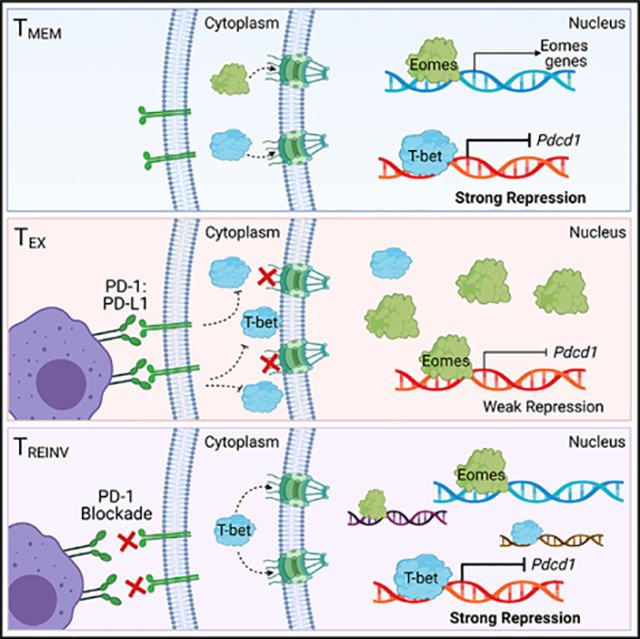
INTRODUCTION
CD8 T cell exhaustion is characterized by a loss of proliferative potential, decreased and modified effector function, high co-expression of inhibitory receptors (IRs), and an altered transcriptional network (Wherry and Kurachi, 2015). Initially defined during chronic LCMV infection (Zajac et al., 1998), T cell exhaustion has been described during multiple chronic infections, such as HIV (Buggert et al., 2014; Hersperger et al., 2011) and hepatitis C virus (Kurktschiev et al., 2014), as well as in preclinical cancer models and numerous human cancers (Baitsch et al., 2011; Curiel et al., 2003; Huang et al., 2017; Kim and Ahmed, 2010; Lee et al., 1999; Riches et al., 2013). Exhausted T cells (TEXs) have high expression of IRs, such as PD-1 (programmed cell death 1), that directly inhibit T cell activation by blocking T cell receptor (TCR) and/or co-stimulatory signaling. However, TEXs are not functionally inert, as therapeutically blocking IRs in the settings of chronic infections and cancer can augment both proliferation and effector function (Barber et al., 2006; Pauken and Wherry, 2015), although the mechanisms underlying this reinvigoration remain incompletely understood.
Underlying CD8 T cell differentiation and function are transcription factors, including the T-box family members T-bet and Eomesodermin (Eomes). Although some redundancy in genes regulated by T-bet and Eomes exists (Banerjee et al., 2010; Glimcher et al., 2004; Intlekofer et al., 2007; Pearce et al., 2003; Pipkin et al., 2010), during acute infection, T-bet is associated with effector function and terminal effector T cell subsets (Hersperger et al., 2011; Intlekofer et al., 2005, 2007, 2008; Joshi et al., 2007; Szabo et al., 2000) and Eomes is associated with memory T cell formation (Banerjee et al., 2010; Cui et al., 2009; Intlekofer et al., 2005; Paley et al., 2012; Pearce et al., 2003; Pipkin et al., 2010). The role of T-bet is primarily in effector T cells; however, this transcription factor is also expressed in memory T cells (TMEMs) in which, in addition to contributing to memory and effector gene regulation, T-bet acts as a transcriptional repressor of PD-1 and potentially other IRs (Kao et al., 2011). In TEXs, the function of T-bet and Eomes is partially distinct from that observed in TMEMs (Doering et al., 2012). TEXs are a heterogeneous population (Hashimoto et al., 2018), and the severity of exhaustion appears to be linked to Eomes expression (Paley et al., 2012), suggesting that high Eomes expression might positively regulate exhaustion. Moreover, despite the apparent opposing roles for T-bet and Eomes in TEXs (Paley et al., 2012), the majority of TEXs co-express both of these transcription factors. In fact, recent work identified further heterogeneity in TEX subsets (Beltra et al., 2020; Jadhav et al., 2019). For example, a four-stage developmental trajectory exists that encompasses a quiescent non-proliferative progenitor (TEXprog1), an activated progenitor (TEXprog2), an intermediate population with some effector activity and migratory capacity (TEXint), and a terminal population that is slightly more cytotoxic, but now non-recirculating (TEXterm) (Beltra et al., 2020). Notably, Eomes is expressed by the rare TEXprog1 but downregulated in TEXprog2 and TEXint only to rebound to high expression in TEXterm. In contrast, T-bet is low in the progenitor subsets but increases in the TEXint population before being lost again in the TEXterm. There is, however, some co-expression of these T-box transcription factors throughout and especially during the transitions between subsets. What mechanisms allow the effects of one of these transcription factors to dominate over the other to promote or antagonize exhaustion, however, is not clear.
Transcription factor activity can be regulated through multiple mechanisms including, but not limited to, subcellular localization (Lange et al., 2007). Disruption of transcription factor localization or perturbation of the regulatory signaling and/or nuclear import pathways that control nuclear trafficking is a common feature in human cancers and genetic disorders (McLane and Corbett, 2009). Recently, we showed that both T-bet and Eomes can be localized to different subcellular compartments in subsets of human CD8 T cells (McLane et al., 2013). Additionally, the nuclear localization of T-bet, but not Eomes, is driven by TCR activation, implying that the signaling mechanisms regulating the nuclear import of these transcription factors could have functional consequences. Because TEXs express both T-bet and Eomes but the function of these transcription factors appears distinct from TMEMs (Doering et al., 2012; Paley et al., 2012), it is possible that the subcellular localization of T-bet and/or Eomes could contribute to the development of different T cell states.
In this study, we sought to define the mechanisms regulating the function of T-bet and Eomes during T cell exhaustion. Using imaging flow cytometry, we show that TEXs from chronically LCMV-infected mice have low nuclear T-bet and high nuclear Eomes compared to those of TMEMs. Additionally, PD-1 expression directly correlated with the ratio of nuclear Eomes:T-bet. Furthermore, the relative relationship of high nuclear Eomes to T-bet was also observed in severely exhausted tumor infiltrating lymphocytes (TILs) from stage IV metastatic melanoma patients compared with CD8 T cells from the blood. Blocking the PD-1:PD-L1 pathway during chronic LCMV clone 13 infection or in the CT26 tumor model resulted in a lower nuclear Eomes:T-bet ratio likely due to an influx of nuclear T-bet. Moreover, in CT26 tumors, the Eomes:T-bet ratio appeared directly linked to tumor burden and disease progression. PD-1 blockade in chronic LCMV infection provoked changes in the expression of T-bet- and Eomes-associated genes involved in T cell chemotaxis, T cell activation, and exhaustion consistent with an altered nuclear ratio of Eomes to T-bet following blockade. Forcing T-bet into the nucleus during chronic LCMV infection restores the expression of key activation and effector molecules, such as KLRG1 and Ki67, suggesting that redistribution of T-bet to the nucleus drives an activated T cell phenotype. Biochemical analysis revealed that T-bet and Eomes can compete with each other for binding to T-box consensus sequences, providing a possible mechanism for the nuclear Eomes:T-bet ratio, rather than the absolute expression of either transcription factor, in regulating exhaustion. The same conserved enhancer region of the Pdcd1 locus bound by T-bet also was bound by Eomes in which this transcription factor functioned as a weak transcriptional repressor of Pdcd1. These data suggest a model in which T-bet is mainly localized to the cytoplasm in TEXs and blocking the PD-1 pathway results in T-bet nuclear localization. Although Eomes can repress Pdcd1 expression, this transcription factor appears to be a functionally weaker repressor that T-bet. Our data suggest that upon PD-1 pathway blockade, T-bet could either cooperate with or outcompete Eomes for binding to Pdcd1, resulting in stronger transcriptional repression. This T-box transcription factor interaction likely also occurs at other genes. Taken together, these data reveal a mechanism by which Eomes could partially antagonize the function of T-bet and potentially tune the program of TEXs through regulated nuclear localization and possible competition for DNA binding. These findings have implications for interpreting how T-bet and Eomes expression relate to features of T cell exhaustion and may suggest therapeutic opportunities based on regulating activity or localization of T-bet and Eomes.
RESULTS
Exhausted CD8 T cells have a high ratio of nuclear Eomes to T-bet that correlates with PD-1 expression
Compared to their well-defined functions during acute infection (Banerjee et al., 2010; Intlekofer et al., 2005, 2007; Joshi et al., 2007; Kaech et al., 2002; Pearce et al., 2003; Zhou et al., 2010), T-bet and Eomes appear to have disparate roles in TEXs during chronic infection (Buggert et al., 2014; Kao et al., 2011; Paley et al., 2012). How the function of these transcription factors changes in TEXs compared to TMEMs, however, is poorly understood. We have previously found that T-bet and Eomes can localize to the nucleus or cytoplasm in human TMEM subsets, suggesting the function of these transcription factors can be regulated by their subcellular distribution (McLane et al., 2013). Because T-bet can repress IR expression (Kao et al., 2011), but TEXs often express high IRs despite retaining T-bet expression, we hypothesized that T-bet might be mislocalized to the cytoplasm in TEXs, rendering this transcription factor incapable of effectively repressing IR gene expression. To test this hypothesis, we investigated the localization of T-bet and Eomes in LCMV-specific (H-2Db gp276+) CD8+ T cells from Armstrong-immune (TMEMs) and chronic clone 13-infected mice (TEXs) at day 30 post-infection (p.i.) by using both imaging cytometry (Figure 1) and confocal microscopy (Figures S1A and S1B). T-bet was observed in the nucleus and/or the cytoplasm of both TMEMs and TEXs (Figure 1A; Figure S1A).We next used imaging cytometry to quantify the nuclear and cytoplasmic localization of T-bet from TMEMs and TEXs. Compared to TMEMs, nuclear localization of T-bet in TEXs was significantly lower in both frequency and amount of T-bet within the nucleus, as measured by the median fluorescence intensity (MFI) of nuclear T-bet (Figure 1B). Additionally, the MFI of DAPI (Figure S1C), a DNA stain, and an unrelated transcription factor, NFAT2 (Figure S1D), showed little to no significant difference in TMEMs versus TEXs, suggesting that the observed differential localization of T-bet in T cell populations was not due to differences in the permeability of the nuclei or nuclear staining. Taken together, our data suggest that T-bet might be unable to efficiently repress Pdcd1 transcription or regulate other transcriptional features of exhaustion, due to its relative absence from the nucleus in TEXs.
Figure 1. Exhausted CD8 T cells have a high ratio of nuclear Eomes to T-bet that correlates with PD-1 expression during LCMV infection.

ImageStream analysis was performed on CD8+ T cells from Armstrong-immune (TMEM, blue)- or clone 13 (TEX, red)-infected mice at day 30 p.i. (A) Representative cell images acquired in IDEAS software from an Armstrong-immune mouse (TMEM, left) or chronic clone 13 mouse (TEX, right) are shown. Splenocytes were permeabilized and stained with T-bet (yellow). The location of the nucleus is indicated by DAPI (cyan).
(B) Representative ImageStream flow plots displaying T-bet localization in TMEMs or TEXs are shown (left). Bar graphs display the frequency and median fluorescence intensity (MFI) of nuclear T-bet in LCMV-specific H-2Db gp276+ CD8+ T cells (right).
(C) Representative cell images acquired in IDEAS software from an Armstrong-immune mouse (TMEM, left) or chronic clone 13 mouse (TEX, right) are shown. Splenocytes were permeabilized and stained with Eomes (magenta). The location of the nucleus is indicated by DAPI (cyan).
(D) Representative ImageStream flow plots displaying Eomes localization are shown (left). Bar graphs show the frequency and MFI of nuclear Eomes in LCMV-specific H-2Db gp276+ CD8+ T cells (right).
(E) The ratio of the MFI of nuclear Eomes:T-bet in LCMV-specific H-2Db gp276+ T-bet+ Eomes+ CD8 T cells is shown.
(F) A correlation plot displaying the ratio of the MFI of nuclear Eomes:T-bet versus MFI of PD-1 in LCMV-specific T-bet+ Eomes+ CD8+ T cells from TEXs is shown. A Pearson coefficient (r value) is displayed on the graph. All data are represented as mean ± SEM; p values were determined using the Student’s t test (***p < 0.0001, **p < 0.001, *p < 0.05). Each graph is representative of 3 or 4 independent experiments with 4–5 mice per experimental condition.
Eomes expression is highly upregulated in TEXs and strongly correlates with high IR expression (Paley et al., 2012), suggesting Eomes could be a positive regulator of T cell exhaustion. Therefore, we hypothesized that Eomes nuclear localization in TEXs might be elevated compared with that of TMEMs. Like T-bet, Eomes was detected in both the nuclear and cytoplasmic compartments of TMEMs and TEXs by both imaging cytometry (Figure 1C) and confocal microscopy (Figure S1B). In contrast to T-bet, quantification of nuclear Eomes revealed a significant increase in both the frequency and the MFI of nuclear Eomes in TEXs compared to TMEMs (Figure 1D), suggesting that the role of Eomes in TEXs is likely different from T-bet due to its high nuclear localization in TEXs compared to TMEMs. Taken together, these data demonstrate that the subcellular localization of T-bet and Eomes in LCMV-specific TEXs is altered compared to TMEMs. Furthermore, these data suggest that the loss of T-bet from, and gain of Eomes into, the nucleus may contribute to CD8 T cell exhaustion.
Previous studies have showed that the ratio of transcription factor pairs may contribute to CD8 T cell differentiation states (Kaech and Cui, 2012). In fact, the majority of virus-specific CD8 TMEMs and TEXs co-express some level of T-bet and Eomes (Paley et al., 2012; Beltra et al., 2020; Jadhav et al., 2019; Kaech and Cui, 2012). Based on the differential localization of T-bet and Eomes in TEXs, we posited that exhaustion was not simply defined by the overall relative amount of T-bet to Eomes expressed within a given cell (Paley et al., 2012), but rather by the ratio of T-bet and Eomes within the nucleus. Thus, we next investigated the ratio of nuclear Eomes to nuclear T-bet in TMEMs compared to TEXs. LCMV-specific CD8+ TMEMs, which express low levels of PD-1 (Paley et al., 2012), had a low nuclear Eomes:T-bet ratio (Figure 1E). In contrast, LCMV-specific CD8+ TEXs, which express high PD-1 (Paley et al., 2012), had a significantly higher nuclear Eomes:T-bet ratio than TMEMs. In LCMV-specific TEXs, the MFI of PD-1 directly correlated with the nuclear Eomes:T-bet ratio (Figure 1F), suggesting that the Eomes:T-bet ratio within the nucleus of CD8 T cells, rather than just the overall expression level of either transcription factor, might contribute to PD-1 expression and T cell exhaustion.
We extended these studies to examine the subcellular localization of T-bet and Eomes in CD8 T cells in the context of human cancer to investigate if differences in nuclear localization of T-bet and Eomes can be found beyond the LCMV model. Non-naive CD8 T cells from either peripheral blood or TILs from stage IV metastatic melanoma patients were analyzed by imaging flow cytometry. Based on the data above, we tested whether human CD8 T cells expressing high PD-1 had less nuclear T-bet than cells with lower PD-1 expression. Indeed, ImageStream analysis of non-naive PD1hi (top) and PD1lo (bottom) TIL showed that higher PD-1 expression correlated with cytoplasmic T-bet (Figure 2A, top), whereas lower PD-1 expression was associated with nuclear T-bet (Figure 2B, bottom). Moreover, the MFI of nuclear T-bet was significantly reduced, whereas the MFI of nuclear Eomes increased in CD8+ TIL compared with non-naive CD8 T cells in the blood (Figure 2B). Consequently, the ratio of nuclear Eomes:T-bet was significantly elevated in human TIL compared to blood (Figure 2C). Taken together, these data suggest that TEXs from chronic LCMV mice and human melanoma patients are characterized by a high ratio of nuclear Eomes compared to T-bet.
Figure 2. The ratio of nuclear Eomes:T-bet correlates with severe exhaustion in TIL from metastatic melanoma patients.

Peripheral blood and tumor-infiltrating lymphocytes were isolated from stage IV human metastatic melanoma patients and analyzed on the ImageStream. (A) Representative images of CD8+ (orange) TIL from two patients are shown. Cells were stained with PD-1 (blue), T-bet (yellow), and Eomes (magenta), and nuclei are indicated by DAPI (cyan).
(B) The bar graphs show the MFI of nuclear T-bet (left) and the MFI of nuclear Eomes (right) in non-naive CD8+ PBMC (gray) and TIL (light purple).
(C) The ratio of the MFI of nuclear Eomes:T-bet in non-naive CD8+ T-bet+ Eomes+ PBMC and TIL is displayed. Data are represented as mean ± SEM; p values were determined using the unpaired Mann-Whitney Student’s t test (***p < 0.0001; **p < 0.001). Bar graphs were generated from 6 blood samples and 9 TIL samples.
Blocking the PD-1 pathway re-balances the nuclear Eomes:T-bet ratio during chronic LCMV infection
Previous studies have shown that blocking the PD-1:PD-L1 pathway during chronic infections and cancer reinvigorates TEXs and promotes viral and tumor control (Barber et al., 2006; Curiel et al., 2003; Hirano et al., 2005; Huang et al., 2017; Iwai et al., 2002, 2005; Strome et al., 2003; Topalian et al., 2012). Additionally, we have previously shown that T-bet nuclear localization can be triggered by TCR signaling (McLane et al., 2013), suggesting that blocking PD-1, and thus enhancing TCR signaling, might result in the re-localization of T-bet to the nucleus in TEXs. To investigate the effects of PD-1 blockade in antigen-specific CD8 T cells, C57BL/6 mice were infected with LCMV clone 13 and the PD-1 pathway was blocked during chronic infection. In agreement with previous studies (Barber et al., 2006; Pauken et al., 2016), the number of LCMV-specific CD8 T cells increased following PD-1 blockade (data not shown). The majority of CD8 T cells from both control untreated (PBS) and αPD-L1-treated mice expressed some level of both T-bet and Eomes, suggesting additional levels of regulation for the role of these transcription factors beyond their expression following PD-1 pathway blockade (Figure 3A). Indeed, within the T-bet+ Eomes+ population following αPD-L1 therapy, the localization of T-bet became more nuclear and the MFI of nuclear T-bet increased, whereas Eomes localization remained largely unchanged (Figure 3B). Quantification of the ratio of nuclear Eomes:T-bet in LCMV-specific cells demonstrated that virus-specific CD8 T cells from αPDL1-treated mice had a significantly lower ratio of nuclear Eomes:T-bet than TEXs from untreated mice (Figure 3C). Changes in the Eomes:T-bet ratio were unlikely due to differences in viral titer alone, as viral load was not significantly different at day 35 p.i. in control PBS and αPD-L1 mice (Figure 3D). Note that the control of viral replication in this experimental design is consistent with the original studies of PD-1 pathway blockade (Barber et al., 2006), because at this time point even control mice had largely controlled systemic viral replication. Taken together, these data show that blocking the PD-1:PD-L1 interaction triggers T-bet nuclear localization, resulting in a decrease in the ratio of nuclear Eomes:T-bet. Moreover, these results suggest that an increase in nuclear T-bet could directly contribute to the reinvigoration of TEXs during chronic LCMV infection.
Figure 3. Blocking the PD-1 pathway re-balances the nuclear Eomes:T-bet ratio during chronic LCMV infection.

(A) LCMV-specific H-2Db gp276+ CD8 T cells from B6 control PBS (gray) or αPD-L1-treated mice (green) infected with LCMV clone 13. Representative flow plots show the co-expression of T-bet and Eomes in LCMV-specific CD8 T cells. The frequency of the T-bet+Eomes+ population is shown.
(B) Representative ImageStream flow plots displaying the subcellular distribution of T-bet (top) or Eomes (bottom) in LCMV-specific T-bet+ Eomes+ CD8 T cells from PBS-treated (gray) or αPD-L1-treated (green) mice are shown.
(C) The ratio of the MFI of nuclear Eomes:T-bet in LCMV-specific T-bet+ Eomes+ CD8 T cells is shown.
(D) The mean viral load of mice pre-PBS (day 15) and post-PBS (day 35) or αPD-L1 therapy is shown. Data are represented as mean ± SEM; p values were determined using the unpaired Mann-Whitney Student’s t test (***p < 0.0001). Bar graphs were generated from a single experiment with 4–5 mice per experimental condition. Each bar graph is representative of 3 independent experiments.
Blocking the PD-1 pathway in CT26 tumors re-balances the nuclear Eomes to T-bet ratio and correlates with tumor progression
To further investigate the role of T-box transcription factor nuclear localization in CD8 T cells in cancer, we next investigated the effects of PD-1 pathway blockade on T-bet and Eomes localization and the relationship to tumor growth and disease progression in the CT26 colon tumor model. Mice with CT26 tumors were treated every 3 days with αPD-L1 beginning at day 10 post-tumor cell injection, and tumor weights and TIL were analyzed at day 20 (Figure 4A). In agreement with previous studies (Duraiswamy et al., 2013; Sakuishi et al., 2010), αPD-L1-treated mice had significantly smaller tumors than control (PBS) mice, although a range of tumor weights was observed in both control and αPD-L1-treated groups (Figure 4B). Additionally, there was a trend in αPD-L1-treated mice toward more nuclear T-bet in CD8+ TIL than in PBS-treated mice (Figure 4C). Moreover, there was an inverse correlation between the frequency of TIL with nuclear T-bet and the tumor weight in αPD-L1-treated mice compared to untreated controls (Figure 4D), suggesting that re-localizing T-bet to the nucleus contributed to smaller tumor size. Although there was no significant difference in the ratio of nuclear Eomes:T-bet between treated and untreated mice (Figure 4E, left), there was a significant and direct correlation between the ratio of nuclear Eomes:T-bet and tumor weight in both control-PBS- and αPD-L1-treated mice (Figure 4E, right). Taken together, these data support the notion that high nuclear T-bet, and thus a lower nuclear Eomes:T-bet ratio, in CD8+ TIL contributed to a lower tumor burden and, potentially, enhanced control of disease.
Figure 4. Blocking the PD-1 pathway in CT26 tumors re-balances the nuclear Eomes-to-T-bet ratio and correlates with tumor progression.

(A) Experimental design for CT26 tumor studies.BALB/C mice were injected with CT26 tumor cells, and beginning at day 10 post-injection, mice were treated 4 times with control PBS or αPD-L1 every 3 days. Mice were sacrificed at day 20 post-tumor injection.
(B) Bar graph displaying the weight of CT26 tumors at day 20 post-tumor cell injection is shown for control PBS-treated (gray) or αPD-L1-treated (blue) mice.
(C) The frequency of CD8+ TIL that contain nuclear T-bet is shown.
(D) Correlation plots displaying the frequency ofCT26 TIL with nuclear T-bet compared to tumor weight are shown.
(E) A bar graph displaying the ratio of nuclear Eomes:T-bet in CT26 CD8+ TIL is shown (left). A correlation plot highlighting the ratio of the MFI of nuclear Eomes:T-bet in CD8+ TIL versus tumor weight in control PBS-treated mice (gray) compared to αPD-L1-treated mice (blue) is shown (right). Data are represented as mean ± SEM; p values were determined using the unpaired Mann-Whitney Student’s t test (**p < 0.001). Bar graphs were generated from a single experiment with 20–30 mice per experimental condition. For correlation graphs, a Pearson correlation analysis was performed, and p values are displayed on the graph.
Blocking the PD-1 pathway re-engages T-bet transcriptional circuits during chronic LCMV infection
A key prediction of changing the nuclear ratio of Eomes:T-bet following PD-1 blockade is that the expression of T-bet and Eomes target genes should also change. To investigate this possibility, we used existing RNA sequencing (RNA-seq) data obtained following PD-1 pathway blockade in LCMV-clone-13-infected mice (Pauken et al., 2016). We performed gene set enrichment analysis (GSEA) by using genes that are known to be regulated by T-bet and Eomes in TEXs (Doering et al., 2012) and asked whether αPD-L1 blockade-mediated changes in the nuclear Eomes:T-bet ratio correlated with changes in expression of these predicted T-bet and Eomes target genes. Leading-edge genes from GSEA were clustered using K means (described in STAR Methods), and heatmaps of these genes are shown in Figure 5A. T-bet can act as either a transcriptional activator or repressor (Szabo et al., 2000; Afkarian et al., 2002; Intlekofer et al., 2005; Doering et al., 2012), and as such, we identified T-bet-associated genes that either increased or decreased following PD-1 pathway blockade (Figure 5A, left). In addition, expression of IRs such as Lag-3 and PD-1, which are repressed by T-bet (Kao et al., 2011), are part of the T-bet-associated gene set, and as anticipated, Lag3 and Pdcd1 were transcriptionally repressed following PD-1 pathway blockade (Figure 5A, clusters 2 and 4). Of the four clusters of gene expression associated with T-bet, cluster 3, which included genes that increased following PD-1 blockade, was of particular interest given the expression of many genes involved in homing, migration, and T cell activation (Figure 5B). Furthermore, cluster 3 was strongly enriched for GO terms of T cell chemotaxis, regulation of interleukin-1 (IL-1) signaling, and response to virus (Figure 5B). The re-engagement of these genes is consistent with recent data identifying a TEXint subset that is increased following PD-1 pathway blockade and possesses homing and recirculation activity (Hudson et al., 2019; Zander et al., 2019; Beltra et al., 2020). The current data suggested that this biological activity is likely regulated by repositioning T-bet to the nucleus upon PD-1 pathway blockade. Like T-bet-associated genes, genes connected to Eomes in TEXs also changed following PD-1 pathway blockade, and the prominent cluster of genes that was de-repressed following PD-1 pathway blockade was enriched for GO terms involved in lymphocyte activation and antigen receptor signaling (Figures 5A and 5B). Eomes protein expression increases upon PD-1 pathway blockade (Pauken et al., 2016), and indeed, Eomes transcription increased (Figure 5A, right). To validate the transcriptional data, we next examined selected changes in protein expression of T-bet- and Eomes-associated target genes following PD-1 pathway blockade. The activation marker CD38, and proteins involved in homing and migration such as CCR5 and CCL5, increased in CD44+ PD-1+ CD8 T cells following PD-1 pathway blockade, whereas the IR CD244 (2B4) decreased (Figure 5C). Thus, these data not only provide an indication of the functional consequence of rebalancing the nuclear Eomes:T-bet ratio following PD-1 pathway blockade but also identify specific sets of transcriptional re-engagement that might have relevance for the therapeutic effect of PD-1 pathway blockade therapies. In particular, changes in T cell activation and signaling, as well as a set of chemotaxis, homing, and migration genes, may be of particular interest in immunotherapy for chronic infections and cancer.
Figure 5. Blocking the PD-1 pathway re-engages T-bet transcriptional circuits during chronic LCMV infection.

(A) Heatmaps display the raw expression values of chronic-only T-bet (left) or Eomes (right) gene neighbors from clone-13-infected mice following PBS (TEX) or αPD-L1 treatment. Genes were clustered by K-means.
(B) Gene Ontology pathway analysis was performed on the gene clusters from T-bet neighbors (top, cluster 3) or Eomes neighbors (bottom, cluster 4).
(C) Histograms from a representative clone-13-infected control (shaded gray) or αPD-L1-treated (green line) mouse show the MFI of candidate proteins in CD44+ PD-1+ CD8 T cells. Expression of candidate proteins in naive CD8 T cells are shown (dashed line). Histograms are representative of 5 mice per experimental condition.
Forcing T-bet to the nucleus engages an effector-like phenotype in TEXs
The data described above support a model in which PD-1 expression and T cell exhaustion are regulated, in part, by the ratio of nuclear Eomes:T-bet. Moreover, PD-1 pathway blockade appears to re-balance the nuclear ratio of Eomes:T-bet, and the increase in nuclear T-bet likely contributes to TEX reinvigoration in LCMV and CT26 models. To directly test the effects of increased nuclear T-bet on T cell activation, naive P14 TCR transgenic CD8 T cells specific for LCMV Db GP33–41 were retrovirally transduced with a recombinant version of T-bet fused to the estrogen receptor (ER) by using a GFP reporter to identify transduced cells. The T-bet-ER protein will be retained in the cytoplasm until treatment with tamoxifen that will cause translocation to the nucleus (Kao et al., 2011; Kurachi et al., 2019; unpublished data). T-bet-ERGFP-retroviral (RV)-transduced P14 cells were adoptively transferred into congenic LCMV-clone-13-infected mice (Figure 6A). Once chronic infection was established, mice were treated with tamoxifen for 5 days, and CD44+ P14 cells from tamoxifenversus control-treated mice were compared (Figure 6B). We previously demonstrated that enforced constitutive expression of WT T-bet by RV transduction during LCMV clone 13 infection repressed PD-1 expression (Kao et al., 2011). Here, we tested whether forcing more T-bet into the nucleus was also able to engage other T-bet-dependent gene expression activities. Following tamoxifen treatment, expression of several key T-bet target genes was increased at the protein level. Previous studies have shown that overexpressing T-bet during LCMV clone 13 infection results in increased KLRG1 expression (Kao et al., 2011). Indeed, expression of KLRG1, a marker of effector function, as well as CD39, was significantly increased upon tamoxifen treatment (Figure 6C). Notably, co-expression of CD39 and KLRG1 during LCMV clone 13 infection is characteristic of more effector-like activity (Beltra et al., 2020; Chen et al., 2019). Additionally, CD69, a marker of T cell activation, was upregulated following tamoxifen treatment (Figures 6D and 6E). Taken together, these data suggest that forcing T-bet into the nucleus in TEXs directly promotes gene (and protein) expression patterns consistent with the role of T-bet in other settings, including regulating effector genes. Thus, redistributing T-bet to the nucleus could contribute to some of the reinvigoration of TEXs following PD-1 pathway blockade.
Figure 6. Forcing T-bet to the nucleus engages an effector-like program in TEXs.

(A) Naive CD45.2+ recipient mice were infected with LCMV clone 13 on day 0. Concurrently, naive CD45.1+ CD45.2+ P14 cells were isolated, activated using anti-CD3/CD28/IL-2 for 24 h, and transduced with a MSCV-T-bet-ER-GFP retroviral vector. Transduced cells were then transferred into LCMV-clone-13-infected mice on day 1. Beginning on day 22, recipient mice were treated with tamoxifen or PBS (control) every day for 5 days. Splenocytes were harvested on day 32 post-infection and analyzed by flow cytometry.
(B) The frequency (left) and total cell number (right) of GFP+ non-naive (CD44+) P14 cells from tamoxifen-treated or control PBS-treated mice are shown.
(C) The frequency (left) and total cell number (right) of KLRG1+CD39+ effector-like cells within the GFP+ P14 splenocytes are shown.
(D) A representative histogram of CD69 expression within GFP+ P14 cells from control (black) or tamoxifen-treated (red) mice is shown (left). The frequency of CD69+ cells within the GFP+ P14 population is plotted (right). Data are represented as mean ± SEM; p values were determined using the unpaired Mann-Whitney Student’s t test (**p < 0.001). Bar graphs were generated from a single experiment with 4–5 mice per experimental condition and are representative of 2 independent experiments. Histograms are representative of 5 mice per experimental condition.
T-bet and Eomes compete for binding to a T-box consensus sequence
Thus far, in vivo data suggest a model in which CD8 T cell exhaustion and reinvigoration following PD-1 pathway blockade are, at least in part, the result of changes in T-bet and, to a lesser extent, Eomes nuclear localization. T-bet and Eomes share a highly homologous T-box DNA binding domain, and previous groups have shown that both factors can regulate an overlapping set of genes (Banerjee et al., 2010; Glimcher et al., 2004; Intlekofer et al., 2007; Pearce et al., 2003; Pipkin et al., 2010). The data described above show that the ratio of nuclear Eomes to T-bet differs between TMEMs, TEXs, and reinvigorated T cells (TREINVs). Because these transcription factors share DNA binding sequences, we hypothesized that T-bet and Eomes might directly compete for binding to specific regulatory elements of key genes relevant to exhaustion and modulate expression. To begin to address this possibility, we performed competition DNA binding experiments to interrogate if T-bet and Eomes can compete for binding to a T-box DNA sequence. To this end, we modified a commercially available T-bet-DNA binding ELISA (Active Motif) to detect binding of T-bet or Eomes to a canonical T-box sequence. Wild-type (WT) T-bet and Eomes showed strong binding to the canonical T-box sequence, whereas DNA-binding mutants of T-bet and Eomes lost their ability to bind (Figures S2A and S2B), demonstrating the specificity of this assay. The observed difference in binding of mutant T-bet and Eomes to the canonical T-box sequence was not due to differences in expression compared to WT T-bet and Eomes (Figure S2A).
To investigate whether T-bet can compete with Eomes (and vice versa), we tested whether DNA binding of one transcription factor was competitively inhibited by the other transcription factor. Thus, to test whether Eomes could compete T-bet off of DNA, T-bet was bound to the canonical T-box consensus sequence, and 293T lysate overexpressing Eomes (or control lysate) was added (Figure S2C). Upon addition on Eomes, T-bet binding was significantly reduced (Figure 7A, left) and replaced with Eomes (Figure 7A, right), suggesting that Eomes can compete with T-bet to bind a consensus T-box sequence. We also performed the reverse experiment in which, upon addition of T-bet, Eomes binding was also significantly reduced and T-bet binding was detected in place of Eomes (Figure 7B). Taken together, these data show that T-bet and Eomes can compete for binding to a consensus T-box binding site and suggest that competition between these two factors could occur at T-box consensus sites, regulating genes relevant for T cell exhaustion.
Figure 7. T-bet and Eomes compete for binding to and repressing Pdcd1 transcription.

(A) Immobilized T-bet bound to a T-box consensus sequence was competed with recombinant Eomes, and binding of both T-bet and Eomes was determined using an ELISA-based binding assay (described in STAR Methods and Figure S2B). The bar graph displays the fold change in binding of T-bet or Eomes following competition with Eomes+ lysate relative to each uncompleted protein.
(B) Immobilized Eomes bound to a T-box consensus sequence was competed with recombinant T-bet, and binding of both T-bet and Eomes was determined using an ELISA-based binding assay (described in STAR Methods and Figure S2B). The bar graph displays the fold change in binding of Eomes or T-bet following competition with T-bet+ lysate relative to each uncompleted protein.
(C) T-bet or Eomes binding to a consensus T-box sequence was competed with oligos corresponding to a canonical T-box half site sequence or the T-box sequences found in Pdcd1 or Il2. The bar graph displays the fold change in binding of Eomes or T-bet following competition with various T-box-containing oligos relative to each uncompleted protein.
(D) PCR amplification of immunoprecipitated DNA sequences in EL4 cells expressing exogenous T-bet or Eomes is shown. Ifng(positive) and Il4 (negative) regions were used as controls.
(E) Dual-luciferase reporter assays were performed using a reporter plasmid containing two conserved regions within the Pdcd1 promoter region (CR-B+C). A bar graph shows the luciferase activity observed in PMA/ionomycin-stimulated EL4 cells nucleofected with wild-type T-bet, Eomes, or co-nucleofected T-bet and Eomes plasmids. Data were normalized to EL4 cells containing the firefly luciferase reporter only.
(F) Endogenous PD-1 expression in unstimulated (left) or PMA/ionomycin-stimulated (right) EL4 cells is shown in T-bet+ (green), Eomes+ (blue), or T-bet+ Eomes+ (pink) cells relative to T-bet−, Eomes−, or T-bet−Eomes− cells (gray shaded). Representative histograms from one experiment are shown. Data in bar graphs are represented as mean ± SEM; p values were determined using a one-way ANOVA test (****p < 0.0001; ***p < 0.001, *p < 0.05).
Eomes binds to and weakly represses Pdcd1 transcription
T-bet has previously been shown to bind directly to and repress the transcription of Pdcd1 through a region upstream of the transcriptional start site containing a putative T-box binding site (Kao et al., 2011). Based on that data described above that T-bet and Eomes can compete for binding to a canonical T-box sequence (Figures 7A and 7B) and because high PD-1 expression in TEXs is associated with elevated nuclear Eomes (Figure 1), we hypothesized Eomes might act as a direct regulator of Pdcd1 expression. To test this hypothesis, we first asked if Eomes could bind to the Pdcd1 T-box sequence previously shown to be bound by T-bet (Kao et al., 2011). Thus, T-bet or Eomes were bound to immobilized consensus T-box DNA, and then DNA sequences containing T-box sites from different genes were tested for their ability to compete each transcription factor off the index consensus T-box site containing DNA (Figures S3A and S3B). As a proof of concept for this approach, T-bet and Eomes were competed with either a WT T-box or mutant T-box DNA sequence (Figure S3C). In both instances, T-bet or Eomes binding was lost when competed with the WT but not the mutant sequence. We next tested if Eomes could bind a previously characterized part of the Pdcd1 regulatory region (CR-C) containing a T-box consensus half-site that is bound by T-bet (Kao et al., 2011; Figure 7C). In agreement with chromatin immunoprecipitation data from Kao et al. (2011), T-bet binding was competed with an oligo encoding the Pdcd1 T-box half-site, confirming the ability of T-bet to bind to this Pdcd1 T-box sequence (Figure 7C, left). Eomes binding was also competed with the Pdcd1 half-site, suggesting that Eomes can also bind the same sequence as T-bet from Pdcd1 (Figure 7C, right). As controls, a consensus T-box half-site as well as the Il2 T-box sequence previously been shown to be bound by T-bet (Szabo et al., 2000) competed Eomes from the canonical full-site T-box sequence (Figure 7C). We also performed CUT&RUN (Skene et al., 2018) followed by PCR to test whether Eomes directly bound to the Pdcd1 promoter in EL4 cells. As controls, both T-bet and Eomes bound to the interferon gamma (Ifng) promoter, but not the Il4 promoter, as previously described (Cruz-Guilloty et al., 2009). Binding of Eomes (and T-bet) to the Pdcd1 T-box region was also detected in EL4 cells (Figure 7D). Taken together, these data indicate that Eomes can bind the same region of the Pdcd1 promoter bound by T-bet, thus providing a mechanism for Eomes to regulate the activity of T-bet and/or expression of Pdcd1 at this locus.
Because high Eomes expression in TEXs is associated with high PD-1 (Paley et al., 2012), we next hypothesized that Eomes might be a positive regulator of PD-1 expression. To test this hypothesis, we used a luciferase reporter assay with a construct containing the previously described conserved Pdcd1 T-box half-site (Pdcd1 CR-C+B) (Oestreich et al., 2008) cloned upstream of a luciferase reporter gene. T-bet and/or Eomes were exogenously expressed in EL4 cells containing the luciferase reporter construct, and luciferase activity was measured (Figure 7E, top). As previously shown (Kao et al., 2011), significant luciferase activity was observed following phorbol 12-myristate 13-acetate (PMA) and ionomycin treatment of EL4 cells containing the PD-1 CR-C+B reporter construct (Figure 7E, gray), and this activity was repressed upon exogenous T-bet expression (Figure 7E, green). Expression of exogenous Eomes also decreased luciferase activity, consistent with repression rather than activation (Figure 7E, blue). The magnitude of this Eomes repression, however, was significantly less than the repression mediated by T-bet, suggesting that T-bet may be a stronger repressor than Eomes. Co-expression of both T-bet and Eomes also decreased luciferase activity to the same level as T-bet, consistent with the notion that T-bet may be a more dominant repressor than Eomes, at least under these conditions of enforced high expression of both transcription factors.
We next investigated the effect of exogenously expressing T-bet and/or Eomes directly on PD-1 protein expression. Ectopic T-bet and Eomes protein expression was confirmed in EL4 cells by flow cytometry (Figure S3D). EL4 cells constitutively express PD-1 protein, and PD-1 expression can be further induced with PMA and ionomycin (Figure 7F, gray histograms; Figure S3E). Resting EL4 cells exogenously expressing T-bet, Eomes, or both T-bet and Eomes expressed significantly less PD-1 than control EL4 cells lacking enforced T-bet and/or Eomes expression (Figure 7F), confirming the role of T-bet and Eomes as negative regulators of Pdcd1 expression. In EL4 cells stimulated with PMA and ionomycin, PD-1 expression was reduced in T-bet+ and T-bet+ Eomes+ cells but not Eomes+ cells (Figure 7F, right), suggesting that when there is a strong transcriptional activation signal, T-bet can partially repress PD-1 expression but Eomes is less efficient. Expression of DNA-binding mutants of T-bet and Eomes did not alter the expression of PD-1 in EL4 cells (data not shown), confirming that these effects were due to the DNA binding activity of these transcription factors. These data, in combination with the luciferase reporter assay, reveal that Eomes is a transcriptional repressor of Pdcd1. Furthermore, these data suggest that T-bet may play a dominant role over Eomes in repressing Pdcd1 transcription because, when both factors are present and nuclear, there is less Pdcd1 transcription than when Eomes is exogenously expressed by itself. Moreover, our data suggest that when nuclear concentrations of T-bet increase, the weak repressive activity of Eomes may be overridden by the strong repressive activity of T-bet at genes such as Pdcd1. Such a T-box transcription factor competition model provides a potential mechanism for the benefit of increased T-bet, and nuclear re-localization of this transcription factor, during PD-1 pathway blockade-mediated reinvigoration of TEXs.
DISCUSSION
The T-box factors T-bet and Eomes control distinct transcriptional circuits in TMEMs and TEXs (Doering et al., 2012). T-bet and Eomes could be viewed as having potentially opposing roles in TEXs because T-bet negatively regulates the transcription of Pdcd1 and thus T cell exhaustion (Kao et al., 2011), whereas Eomes expression positively correlates with high IR expression and other features of more severe T cell exhaustion (Blackburn et al., 2009; Doering et al., 2012; Paley et al., 2012). More recent analyses found slightly elevated Eomes and low T-bet in the most quiescent TEX progenitors (Beltra et al., 2020). Following this population, there is an inversion of the Eomes:T-bet ratio with high T-bet in an intermediate TEX population and, finally, a loss of T-bet and final elevation of Eomes expression associated with terminal exhaustion (Beltra et al., 2020). Nevertheless, many TEXs co-express both transcription factors, and it is unclear what mechanisms allow T-bet and Eomes to foster versus antagonize exhaustion. In this study, we identified a key role for the differential subcellular localization of T-bet and Eomes as a mechanism for regulating T cell exhaustion. We show that a high ratio of Eomes:T-bet in the nucleus of TEXs correlates with PD-1 expression and exhaustion, likely due to the inability of T-bet to enter the nucleus to repress Pdcd1 transcription and regulate other T-bet dependent genes. Furthermore, modulating the relative nuclear Eomes:T-bet ratio by blocking the PD-1 pathway promoted T-bet nuclear localization (and perhaps Eomes cytoplasmic localization), re-balanced the relative nuclear Eomes: T-bet ratio, and decreased Pdcd1 transcription, potentially contributing to some aspects of TEX reinvigoration. The current studies may also have relevance for understanding subsets of TEXs. For example, in addition to TEX subsets defined based on PD-1 expression and T-bet and Eomes (Blackburn et al., 2009; Buggert et al., 2014; Paley et al., 2012), recent work has also identified subsets based on the expression of TCF-1, CXCR5, and/or Tim3 (He et al., 2016; Im et al., 2016; Utzschneider et al., 2016; Wu et al., 2016). Other recent studies defined four subsets of TEXs, and the developmental transitions between these subsets revealed roles for Eomes and T-bet at each developmental stage (Beltra et al., 2020). However, it has remained unclear how the activity of these two transcription factors was regulated in TEXs. The current studies suggest that, in addition to the overall amount of T-bet or Eomes protein detected by flow cytometry, the subcellular location is likely an important factor when considering the role of these transcription factors in the biology of TEXs. Indeed, our data suggest that not only does the localization of T-bet and Eomes affect T cell function but also the relative amounts of these factors within the nucleus, rather than total cellular protein levels, influence the program of genes expressed.
This study also provides a molecular mechanism underpinning the role of T-bet and Eomes in T cell exhaustion. T-bet and Eomes are members of the same family of transcription factors based on the high homology within their T-box DNA binding domain. The DNA binding domain coupled with the ability of many T-box family members to bind to consensus T-box sequences (Coll et al., 2002) suggest that T-bet and Eomes, when co-expressed, could act cooperatively or in competition with one another for DNA binding and transcriptional control. Indeed, our data show that Eomes binds to, and weakly contributes to, the transcriptional repression of Pdcd1 through the same regulatory region targeted by T-bet for repression. Moreover, competition DNA binding studies revealed that T-bet and Eomes compete for DNA binding at a T-box consensus sequence. Co-expression of T-bet and Eomes in EL4 cells resulted in stronger repression of PD-1 expression than Eomes alone, suggesting that T-bet has a dominant effect in controlling PD-1 expression when both factors are available.
The finding that T-bet is often in the cytoplasm rather than nucleus of TEXs suggested a key mechanism for the regulation of the function of T-bet and Eomes in TEXs. The sequestration of T-bet in the cytoplasm of TEXs limited the stronger repressive activity of T-bet at Pdcd1 and likely other genes. In contrast, Eomes accumulated in the nucleus to high levels in TEXs and was able to bind to the Pdcd1 promoter in situations in which T-bet was not present or present at low levels. Under these conditions, Eomes acted as only a weak repressor of Pdcd1 expression. Thus, the dominance of nuclear Eomes over nuclear T-bet was permissive to high expression of PD-1 by TEXs. Upon PD-1 blockade, however, likely due to increased TCR signaling, T-bet is able to reenter the nucleus and displace, or potentially cooperate with, Eomes at the Pdcd1 locus to mediate stronger transcriptional repression. Such an exchange is likely to occur at other genes regulated by T-bet and Eomes given the changes in expression of T-bet and Eomes-associated genes observed by GSEA when this nuclear Eomes:T-bet ratio changes following PD-1 pathway blockade.
Indeed, this re-engagement of T-bet-dependent transcriptional activity due to the influx of T-bet to the nucleus in reinvigorated TEXs could have implications for immunotherapy. For example, T-bet is connected to sets of genes involved in T cell homing and migration, in addition to effector function and IRs. The ability of PD-1 pathway blockade to re-engage this T-bet-dependent transcriptional activity could have benefits for tissue-localized infections or solid tumors. It is possible, therefore, that PD-1 pathway blockade might synergize with approaches that drive this set of T-bet-dependent homing and migration-related genes. Moreover, some of the prominent Eomes-connected genes increased following PD-1 blockade included not only Eomes itself but also genes involved in T cell activation and signaling, suggesting further re-wiring of T cell responses due to changes in the nuclear Eomes:T-bet ratio.
Although TEXs have been examined in many settings, the mechanisms underlying exhaustion and the reinvigoration of TEXs remain incompletely understood. The current study reveals that underpinning T cell dysfunction is the dysregulation of the transcription factors T-bet and Eomes that are critical for functional effector and memory T cells. These findings provide key insights into the transcriptional control of CD8 T cell exhaustion and TEX reinvigoration following therapeutic intervention and, furthermore, have direct implications for strategies aimed at TEX reinvigoration in both chronic disease and cancer.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, E. John Wherry (wherry@pennmedicine.upenn.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a complete Materials Transfer Agreement.
Data and code availability
Original source data for Figure 5 is available in Doering et al. (2012) and Pauken et al. (2016).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Models
For LCMV studies, female C57BL/6 (5–6 weeks old) were purchased from the National Cancer Institute or Charles River, depending on the experiment. For mouse tumor studies, female BALB/C mice (5–6 weeks old) were purchased from the National Cancer Institute. For all experiments, mice were sacrificed in a CO2 chamber followed by neck dislocation. All were used in accordance with the Institutional Animal Care and Use Committee guidelines for the University of Pennsylvania.
Human Subjects
Patients with stage III or IV melanoma were enrolled for immunotherapy treatment as previously described (Huang et al., 2017, 2019). Demographics of patients used in this study are listed in Table S1. Blood and tumor samples were collected under University of Pennsylvania Abramson Cancer Center’s melanoma research program tissue collection protocol UPCC 08607 and clinical trial UPCC 01615 in accordance with the Institutional Review Board. For the purposes of this study, only peripheral blood collected from patients before initiation of immunotherapy was analyzed. Tumor samples were procured from the operating room and processed the same day using manual dissociation into single cell suspension. Tumor samples were then frozen immediately using standard freeze media, and stored in liquid nitrogen.
Cell lines
The HEK293T cell line was provided by Warren Pear (University of Pennsylvania) and was maintained in RPMI 1640 (Corning) supplemented with 10% fetal bovine serum, 2mM L-glutamine, and 100U/mL penicillin/streptomycin.
EL4 cells were obtained from ATCC and were maintained in DMEM (Corning) with 4.5g/L glucose, L-glutamine and sodium pyruvate and supplemented with 10% fetal bovine serum, 100U/mL penicillin/streptomycin, and 50nM beta-mercaptoethanol.
CT26 tumor cells were obtained from ATCC and were maintained in RPMI 1640 (Corning) supplemented with 10% fetal bovine serum, 2mM L-glutamine, 100U/mL penicillin/streptomycin and 1% Glutamax.
METHOD DETAILS
Mice and Infections
All animal work was done in accordance with the Institute Animal Care and Use Guidelines for the University of Pennsylvania. LCMV strains were prepared and tittered as described previously (Kao et al., 2011). Female C57BL/6 (5–6 weeks old) were infected with LCMV Armstrong (2×05 PFU) by intraperitoneal injection (i.p.) or LCMV clone 13 (4×106 PFU) by intravenous injection (i.v.). Mice were bled and serum was collected to determine viral titers. Mice were euthanized at indicated time points post-infection. Spleens were harvested and splenocytes isolated for subsequent experiments. For some experiments, CD8 T cells were isolated using a negative selection kit per the manufacturer’s protocol (Stem Cell Technologies).
Flow Cytometry and Imaging Flow Cytometry
For indicated experiments, MHC class I H-2Db gp276 tetramers were conjugated and used as previously described to identify LCMV-specific CD8 T cells (Murali-Krishna et al., 1998; Wherry et al., 2003). For flow cytometry and imaging flow cytometry of mouse splenocytes or tumor infiltrating lymphocytes, cells were stained using a modified staining protocol (Heinen et al., 2014; Kurachi et al., 2017). For flow cytometry, cells were stained with LIVE/DEAD aqua blue (Invitrogen) for 15 min at 4°C. A cocktail of surface antibodies and tetramer, when applicable, was incubated for 25 min at 4°C. Cells were washed in FACS buffer and then fixed in a 2% formaldehyde solution for 15 min at 4°C. Cells were again washed twice in FACS buffer and permeabilized using the Foxp3 perm solution (eBioscience) for 30 min at 4°C. A cocktail of intracellular antibodies was added and incubated for 1 hr at 4°C. Cells were then fixed in 1% paraformaldehyde (PFA). Samples were analyzed by either flow cytometry or imaging flow cytometry.
For flow cytometry, 1,000,000 total events were acquired on a modified flow cytometer (LSRII, BD Immunocytometry Systems) equipped for the detection of 18 fluorescent parameters. Antibody capture beads (BD biosciences) were used for individual compensation controls for each fluorophore. Data was analyzed in FlowJo version 10.4.2 (TreeStar) and statistical analysis was performed using Prism Software (Version 7.0).
For imaging flow cytometry, cells were fixed and stained just before imaging with with 4’,6-Diamidino-2-Phenylindole, Dihydrochloride (5 μg/mL) for 5 min at room temperature. Samples were then imaged on an ImageStreamX (Amnis Corp). For each sample, 50,000 events were collected. Antibody capture beads (BD Biosciences) were used as individual compensation tubes for each fluorophore. Images were captured using a 60x lens with an extended depth of field upgrade through Inspire software (Amnis Corp). Nuclear and cytoplasmic T-bet and Eomes were defined using masking functions within IDEAS 5.0 as previously described (McLane et al., 2013; Figures S4A–S4C). Cells which harbored either exclusively nuclear or nuclear and cytoplasmic T-bet or Eomes were considered “nuclear” while cells with exclusive cytoplasmic T-bet or Eomes were scored as “cytoplasmic.”
Confocal Microscopy
Isolated CD8 T cells in suspension from d8 p.i. Armstrong and clone 13 mice were placed on coverslips coated with Celltak (Corning Inc.) and incubated at 37°C for 10 min. Cells were then fixed with 3.7% formaldehyde for 1 hour, permeabilized for 20 min in 0.2% Triton X-100, and blocked overnight at 4°C in 2% BSA. Immunolabeling was performed by incubating cells for 2 hours at 37°C with primary antibodies: 1:100 Alexa 488-conjugated mouse anti-T-bet (BD Biosciences) or 1:100 Alexa 488-conjugated rat anti-Eomes (Affymetrix eBioscience). After 3 washes in PBS, cells were stained with Hoechst dye for 30 minutes, washed twice in PBS, and finally once in ddH20. The coverslips were mounted onto slides with Fluoromount-G (Southern Biotech) and cells were imaged on a Leica TCS-SP5 inverted confocal microscope.
Human TIL Isolation and Staining
Patients with stage III or IV melanoma were enrolled for immunotherapy treatment as previously described (Huang et al., 2017). Blood and tumor samples were collected under University of Pennsylvania Abramson Cancer Center’s melanoma research program tissue collection protocol UPCC 08607 in accordance with the Institutional Review Board. For the purposes of this study, only peripheral blood collected from patients before initiation of immunotherapy was analyzed. Tumor samples were procured from the operating room and processed the same day using manual dissociation into single cell suspension. Tumor samples were then frozen immediately using standard freeze media, and stored in liquid nitrogen.
For imaging flow cytometry, cells were thawed in a 37°C water bath and washed in RPMI. Cells were then stained using a cocktail of cell surface and intracellular antibodies using the BD staining buffer kit according to the manufacturers protocol.
PD-1 pathway blockade
For chronic LCMV studies, female C57BL/6 mice (5–6 weeks old) were infected with clone 13 as described above. Mice were injected i.p. with either 200 μg αPD-L1 antibody (clone 10F.9G.2, Bio X Cell) or control PBS every 3 days starting at d22 for 5 total treatments as previously described (Barber et al., 2006). Spleens were harvested on d35 post-infection and splenocytes were stained and analyzed by flow cytometry and imaging flow cytometry.
For mouse tumor studies, female BALB/C mice (5–6 weeks old) were purchased from the National Cancer Institute. CT26 cells were maintained in RPMI1640 supplemented with 10% FCS, 1% Glutamax, and 1% penicillin/ streptomycin and maintained at 5% CO2. For primary tumor experiments, 2×105 CT26 cells/100 μL were injected subcutaneously into mice. Cohorts of tumor-bearing mice were then injected with phosphate buffered saline (PBS) or 200 μg of αPD-L1 (clone 10F.9G2, Bio X Cell) on d10, d13, d16, and d19. Established CT26 tumors were excised from mice on d20 post-tumor injection and processed for flow cytometry analysis as previously described (Knight et al., 2013). Briefly, tumors were digested with 1 mg/ml collagenase D and 0.02 mg/ml DNase I at 37°C. Digested tumors were passed through a cell strainer to prepare a single cell suspension. Cells were then stained for ImageStream analysis as described.
Transcriptional profiling of chronic T-bet and Eomes genes
The gene expression profiles of the top 150 chronic-only gene neighbors for both T-bet and Eomes (Doering et al., 2012) was determined using previously published transcriptional data from clone 13-infected untreated (exhausted) and αPD-L1-treated mice (Pauken et al., 2016). Genes were clustered into four optimal clusters by K-means using the pheatmap function from the R package pheatmap_1.0.8. Each of these clusters was then analyzed using metascape (Tripathi et al., 2015; http://metascape.org) for Gene Ontology Biological Process enrichment using meta-analysis for T-bet and Eomes genes separately. Heatmaps of expression values were created using the pheatmap function with default row-wise and column-wise clustering. Protein expression of candidates identified in the GO analysis was determined by flow cytometry in chronically-infected mice ± αPD-L1 therapy.
Retroviral transduction of splenocytes followed by tamoxifen treatment in clone 13-infected mice
Retroviral (RV) particles were produced in HEK293T cells with MSCV and pCL-Eco plasmids using Lipofectamine 3000. RV transduction was performed as described (Kurachi et al., 2017). Briefly, CD8+ T cells were purified from spleens of P14 mice using EasySep™ Mouse CD8+ T Cell Isolation Kit. Cells were stimulated with 100 U/mL recombinant human IL-2, 1 μg/mL anti-mouse CD3ε, and 5 μg/mL anti-mouse CD28 in RPMI-1640 medium with 10% fetal bovine serum (FBS), 10 mM HEPES, 100 μM non-essential amino acids (NEAA), 50 U/mL penicillin, 50 μg/mL streptomycin, and 50 μM β-mercaptoethanol. After 18–24 hr of in vitro stimulation, P14 cells were transduced with RV in the presence of polybrene (0.5 μg/ml) using spin infection (2,000 g for 1 hr at 32°C) followed by an incubation at 37°C for 6 hr. RV-transduced P14 cells were then adoptively transferred into recipient mice. These recipient mice were previously infected intravenously 24 hr prior to transfer with 4 × 106 PFU LCMV clone 13. Mice were administered 1mg of tamoxifen dissolved in an ethanol:Kolliphor (1:1) solution orally for four consecutive days starting at day 22 post-infection. Spleens were harvested at day 32 post-infection and RV-transduced cells (GFP+) were analyzed by flow cytometry.
DNA constructs and T-box alignments
A PD-1 reporter construct containing a regulatory region (PD-1 CR-B+C) upstream of the Pdcd1 transcriptional start site was cloned into the pGL3Basic vector (Promega) as previously described (Oestreich et al., 2008). T-bet and Eomes were cloned into pCMV-N-myc or pCMV-C-FLAG (Clontech) expression plasmids. DNA binding mutants of T-bet and Eomes were created (T-bet R163A R164A; Eomes R296A R297A) using a QuikChange site-directed mutagenesis kit per the manufacturer’s instructions (Agilent) (Figure S2A). Sequence alignments of the T-box family members were performed using MegAlign from DNAstar (Lasergene).
Immunoblotting
Exogenous expression of recombinant myc-tagged T-bet or FLAG-tagged Eomes in 293T cells was determined by immunoblotting. Briefly, transfected 293T cells were pelleted and washed with 1x PBS. Pellets were resuspending in RIPA buffer (Sigma Aldrich) supplemented with protease inhibitor cocktail (Sigma Aldrich) and incubated on ice for 30 min. Lysates were then cleared by centrifugation for 10 min at 4°C. Lysates were quantified using a Qubit protein assay kit (Thermo Fisher). 25 μg of total lysate was resolved on a 4%–12% NuPage Bis Tris gel (Invitrogen) and transferred to a nitrocellulose membrane with an iBlot (Invitrogen). The nitrocellulose membrane was then blocked in 5% milk (Lab Scientific) in TBST for 1 hr at RT. Primary antibodies were added to 5% milk in TBST and rocked overnight at 4°C. T-bet was detected using a 1:1000 dilution of a monoclonal α-myc antibody (Cell Signaling). Eomes was detected using a 1:1000 dilution of a monoclonal α-FLAG antibody. As a loading control, a polyclonal α-α-actin antibody was used at a 1:2000 dilution. Blots were then washed 3x for 15 min in TBST and secondary α-mouse or α-rabbit HRP antibodies were added to 5% milk in TBST and rocked for 1hr at RT. Blots were then washed 3x for 15 min in TBST and proteins were detected using the Ultra ECL detection reagent (KwikQuant).
DNA-protein binding ELISA
293T cells were transfected with wild-type or mutant T-bet and Eomes plasmids described above. After 48 hr, lysates were prepared in RIPA buffer and quantified using a Qubit (Thermo Fisher). Using the TransAM T-bet DNA binding ELISA kit (Active Motif), DNA binding of T-bet and Eomes was measured using a modified version of the manufacturers protocol (Figures S2B and S3A). For competition studies, 10 μg of lysate containing one transcription factor was allowed to bind to the plate for 1 hr at room temperature. Wells were then washed three times with wash buffer and 10 μg of lysate containing the other transcription factor was added to individual wells and allowed to bind for 1 hr at room temperature. Wells were seeded in duplicate. Recombinant T-bet and Eomes binding was detected using α–myc (T-bet) (9B10, Cell Signaling Technologies) or α–FLAG (Eomes) (M2, Sigma) monoclonal antibodies, respectively. For oligo competition studies, 40 pmol of competing oligos were added following addition of lysate to the wells. Protein binding was measured at OD450 nm on a BioTek Synergy HT plate reader. Results are plotted as fold change from the uncompeted lysate containing WT T-bet or Eomes. Oligonucleotide sequences used in competition studies are listed in Table S2.
Cleavage under targets and release using nuclease (Cut & Run) assay
To assess direct binding of Eomes to the Pdcd1 promoter, we used a modified version of Cut & Run (Skene et al., 2018) with a qPCR readout. EL4 cells were nucleofected using an Amaxa cell line Nucleofector kit L (Lonza) with either pCMV-myc T-bet or pCMV-Eomes FLAG plasmids. Following a 48 hr incubation to induce recombinant protein expression, 100,000 EL4 cells were used in the Cut&Run assay. For primary mouse cells, CD8+ splenocytes from d45 Armstrong (TMEM) or d45 clone 13 (TEX) mice were isolated and 10,000 cells were used in the Cut&Run assay. α-myc (Invitrogen) or α-FLAG (Invitrogen) antibodies were used to IP myc-T-bet or Eomes-FLAG proteins in EL4 cells. α-T-bet (4B10, eBioscience) or α-Eomes (Dan11mag, eBioscience) was used to IP T-bet or Eomes from primary mouse splenocytes. Immunoprecipitated DNA was subjected to two rounds of nested PCR using the following oligo pairs: I–forward 5′-actctaacatgccacaaaaccatag, reverse 5′-cttccagttttatacctgatcgaag (Cruz-Guilloty et al., 2009), Pdcd1–5’-ccttgctcctcaccacactgc, reverse 5′-cagagcagatcatgaggactg (Kao et al., 2011), and Il4–forward 5′-gagttaaagttgctgaaaccaagg, reverse 5′-attttccaattggtctgatttcac (Cruz-Guilloty et al., 2009).
Dual luciferase reporter assay
The dual luciferase reporter assay was performed as previously described (Beima et al., 2006). Briefly, EL4 cells were nucleofected using an Amaxa cell line Nucleofector Kit L (Lonza) with the pGL3-Pdcd1 CR-B+C-Firefly plasmid, control Renilla plasmid pRL-TK, and either pCMV-myc T-bet or pCMV-Eomes FLAG plasmids. Cells were rested overnight and stimulated with 50 ng/mL PMA and 2 μM ionomycin for 24 hr to induce expression of the reporter constructs. Cells were then collected, lysed, and the dual luciferase assay was performed according to the manufacturer’s instructions (Promega). Results were normalized to the expression of the Renilla luciferase control vector and plotted as fold change from empty vector alone.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all experiments shown, n (number of experimental subjects) is indicated in the Figure legends. Data are expressed as mean ± SEM from at least three independent experiments unless otherwise specified. Prism has been used for analysis. Comparison between two groups was done using unpaired two-tailed Student’s t test. Two-way ANOVA has been used for multiple comparisons. The appropriate statistical methods and P-values are indicated in the figure legends.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD279/PD-1 PE Cy7 | BioLegend | Cat# 109109; RRID: AB_572016 |
| Anti-human CD8 APC Cy7 | BD Biosciences | Cat# 557834; RRID: AB_396892 |
| Anti-human CD279/PD1 PE Cy7 | BioLegend | Cat# 329917; RRID: AB_2159325 |
| Anti-human Eomes AF647 | eBioscience | Custom |
| Anti-human CD197CCR7 FITC | BD Biosciences | Cat# 561271; RRID: AB_10561679 |
| Anti-human CD45RO ECD | Beckman Coulter | Cat# IM2712U; RRID: AB_10639537 |
| Anti-human CD3 BV510 | BD Biosciences | Cat# 563109; RRID: AB_2732053 |
| Anti-mouse CD62L BV605 | BD Biosciences | Cat# 563252; RRID: AB_2738098 |
| Anti-mouse CD223/Lag3 BV650 | BioLegend | Cat# 125227; RRID: AB_2687209 |
| Anti-mouse CD8 BV785 | BioLegend | Cat# 100749; RRID: AB_11218801 |
| Anti-mouse CD244/2B4 FITC | BD Biosciences | Cat# 553305; RRID: AB_394769 |
| Anti-mouse CD195/CCR5 PerCP eF710 | ThermoFisher | Cat# 46–1951-82; RRID: AB_11449119 |
| Anti-mouse CD38 A700 | ThermoFisher | Cat# 56–0381-82; RRID: AB_657740 |
| Anti-mouse CD45.2 APC Cy7 | BioLegend | Cat# 109823; RRID: AB_830788 |
| Anti-T-bet BV421 | BioLegend | Cat# 644815; RRID: AB_10896427 |
| Anti-CCL5 PE | BioLegend | Cat# 149103; RRID: AB_2564405 |
| Anti-mouse Eomes PE eF610 | ThermoFisher | Cat# 61-4875-82; RRID: AB_2574614 |
| Anti-T-bet Alexa Fluor 488 | BD Biosciences | Cat# 561266; RRID: AB_10562570 |
| Anti-mouse CD45.2 PE eF610 | ThermoFisher | Cat# 61-0454-82; 2574562 |
| Anti-mouse CD44 APC | BD Biosciences | Cat# 559250; RRID: AB_398661 |
| Anti-Mouse CD45.1 PE Cy5 | ThermoFisher | Cat# 15-0453-82; RRID: AB_468759 |
| Anti-Mouse CD45.2 APC eFluor780 | ThermoFisher | Cat# 47-0454-82; RRID: AB_1272175 |
| Anti-Mouse CD8 BV650 | BioLegend | Cat# 100742; RRID: AB_2563056 |
| Anti-Mouse KLRG1 BV605 | BioLegend | Cat# 138419; RRID: AB_2563357 |
| Anti-Mouse CD39 PE Cy7 | ThermoFisher | Cat# 25-0391-80; RRID: AB_1210767 |
| Anti-Mouse CD44 BV785 | BioLegend | Cat# 103059; RRID: AB_2571953 |
| Anti-Mouse CD69 PE Cy7 | BioLegend | Cat# 104512; RRID: AB_493564 |
| Mouse anti-myc | Cell signaling | Cat# 2276; RRID: AB_331783 |
| Mouse anti-FLAG | Sigma Aldrich | Cat# F3165; RRID: AB_259529 |
| Mouse anti-T-bet | ThermoFisher | Cat# 14-5825-82; RRID: AB_763634 |
| Mouse anti-Eomes | ThermoFisher | Cat# 14-4875-82; RRID: AB_11042577 |
| Rabbit anti-β-actin | Abcam | Cat# 8227; RRID: AB_2305186 |
| Anti-mouse Ig, κ/Negative control compensation particle set | BD Bioscience | Cat# 552843 |
| Anti-rat Ig, κ/Negative control compensation plus | BD Bioscience | Cat# 560499 |
| Chemicals, peptides, and recombinant proteins | ||
| 4’, 6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) | ThermoFisher | Cat# D1306 |
| LIVE/DEAD Aqua Dead Cell Stain | ThermoFisher | Cat# L34957 |
| Live/Dead Zombie NIR Dye | BioLegend | Cat# 423106 |
| Recombinant human IL-2 | NIH | N/A |
| Anti-Mouse CD3(145–2C11) | BioLegend | Cat# 100302; RRID:AB_312667 |
| Anti-Mouse CD28(37.51) | ThermoFisher | Cat# 16-0281-82; RRID:AB_468921 |
| EasySep Mouse CD8+ T Cell Isolation Kit | STEMCELL Technologies | Cat# 19853 |
| RPMI-1640 medium | Corning/Mediatech | Cat# 10-040-CV |
| DMEM medium | Corning/Mediatech | Cat# 10-017-CV |
| HI Fetal Bovine Serum | ThermoFisher | Cat# 26170-043 |
| HEPES | ThermoFisher | Cat# 15630080 |
| Non-Essential Amino Acids | ThermoFisher | Cat# 11140050 |
| Penicillin-Streptomycin | ThermoFisher | Cat# 15140122 |
| β-mercaptoethanol | Sigma-Aldrich | Cat# M6250-500ML |
| Opti-MEM | ThermoFisher | Cat# 31985088 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003-G |
| Lipofectamine 3000 Transfection Reagent | ThermoFisher | Cat# L3000001 |
| Digitonin | Millipore | Cat#300410-1GM |
| BioMag Plus Concanavalin A (10mL) | Bangs laboratories | Cat# BP531 |
| Complete, EDTA-free Protease Inhibitor Cocktail | Sigma | Cat# 4693132001 |
| Glycogen | Thermo | Cat# R0561 |
| Proteinase K | Denville Scientific | Cat# CB3201-5 |
| RNase A | Thermo | Cat# EN0531 |
| Spermidine | Sigma | Cat# 85558-1G |
| Fugene6 Transfection Reagent | Promega | Cat# E2691 |
| Phorbol 12-myristate 13-acetate (PMA) | Sigma Aldrich | Cat# 79346 |
| Ionomycin caldium salt | Sigma Aldrich | Cat# I3909 |
| RIPA buffer | Sigma Aldrich | Cat# R0278 |
| NuPAGE 4–12% Bis Tris protein gels | ThermoFisher | Cat# NP0321 |
| NuPAGE MOPS SDS running buffer (20x) | ThermoFisher | Cat# NP0001 |
| iBlot transfer stack, nitrocellulose mini | ThermoFisher | Cat# 1B301002 |
| Non-fat dry milk | Lab Scientific | Cat# M0842 |
| Cloned Pfu DNA polymerase AD | Agilent Technologies | Cat# 600357 |
| Ultra Digital-ECL substrate solution | KwikQuant | Cat# R1002 |
| Hoechst 33342 solution | ThermoFisher | Cat# 62249 |
| Bovine serum albumin | Sigma Aldrich | Cat# 05470 |
| Fluoromount-G mounting medium | Southern Biotech | Cat# 0100-01 |
| GlutaMAX | ThermoFisher | Cat# 35050061 |
| Collagenase D | Sigma Aldrich | Cat# 11088858001 |
| DNase I | Roche | Cat# 10104159001 |
| Tamoxifen | Sigma-aldrich | CAT# T5648-1G |
| in vivo rat anti-mouse PD-L1 | BioXCell | Cat# BE0101; RRID: AB_10949073 |
| Critical commercial assays | ||
| Ultra Digital-ECL substrate solution | KwikQuant | Cat# R1002 |
| QuikChange II site-directed mutagenesis it | Agilent Technologies | Cat# 200523 |
| Dual-luciferase reporter assay system | Promega | Cat# E1910 |
| Cell line nucleofector kit L | Lonza | Cat# VCA-1005 |
| Taq PCR master mix kit | QIAGEN | Cat# 201443 |
| TransAM T-bet DNA binding ELISA | Active Motif | Cat# 51396 |
| Qubit dsDNA BR assay kit | ThermoFisher | Cat# Q32850 |
| Qubit protein assay kit | ThermoFisher | Cat# Q33211 |
| Deposited data | ||
| RNaseq data | Pauken et al., 2016 | GSE86881 |
| Microarray data | Doering et al., 2012 | GSE41867 |
| Experimental models: Cell lines | ||
| LCMV Armstrong | Rafi Ahmed | Grew in-house |
| LCMV Clone 13 | Rafi Ahmed | Grew in-house |
| CT26 | ATCC | Cat# CRL-2638; RRID: CVCL_7256 |
| HEK293 cell line | ATCC | Cat# CRL-1573 |
| EL4 cell line | ATCC | Cat# TIB-39 |
| CT26.WT cell line | ATCC | Cat# CRL-2638 |
| Experimental models: Organisms/strains | ||
| C57BL/6 mice | National Cancer Institute | N/A |
| BALB/c mice | National Cancer Institute | N/A |
| C57BL/6 | Charles River | N/A |
| CD45.1+ C57BL/6 | Charles River | N/A |
| TCRα−; P14 TCRVα2Vβ8 | The Jackson Lab | Stock No. 37394-JAX |
| Oligonucleotides | ||
| See Table S1 | ||
| Recombinant DNA | ||
| pCMV-myc-N | Clontech | Cat# 631604 |
| pCMV-FLAG-C | Clontech | Cat# 635688 |
| pCMV-myc-T-bet (pLM243) | This study | N/A |
| pCMV-Eomes-FLAG (pLM258) | This study | N/A |
| pCMV-myc-T-bet R163A R164A (pLM263) | This study | N/A |
| pCMV-Eomes R296A R297A-FLAG (pLM268) | This study | N/A |
| pGL3-basic luciferase vector | Promega | |
| pGL3-Pdcd1 CR B+C | Oestreich et al., 2008 | |
| pRL Renilla luciferase control vector | Promega | Cat# E2231 |
| MSCV-T-bet-ER vector | This study | N/A |
| MSCV-GFP vector | This study | N/A |
| Software and algorithms | ||
| FlowJo v. 10.4.2 | FlowJo, LLC | https://www.flowjo.com |
| IDEAS v. 5.0 | Amnis Corp | |
| Graphpad Prism 7 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| SnapGene 4.1.9 | SnapGene | https://www.snapgene.com |
| pheatmap(R package) version 1.0.8 | R package | https://cran.r-project.org/web/packages/pheatmap/index.html |
| Metascape analysis | Gene Annotation and Analysis | http://metascape.org |
Highlights.
The relative amounts of nuclear T-bet and Eomes T cells partially define exhaustion
PD1 blockade increases nuclear T-bet and upregulates T cell activation and homing genes
T-bet and Eomes recognize and bind to the same T-box domain in the Pdcd1 promoter
Eomes is a weak transcriptional repressor of Pdcd1 in TEXs
ACKNOWLEDGMENTS
We would like to thank all the members of the Wherry lab for scientific input, technical advice, and comments on the manuscript. Tetramers were obtained from the NIH tetramer core. This work was supported by grants from the NIH to E.J.W. (AI105343, AI082630, AI112521, AI115712, AI117718, AI108545, and AI117950); X.X., L.M.S., and T.C.G. (P50-CA174523 and P01-CA114046), and B.D.F. (R56 AI125415), as well as the Tara Miller Foundation (L.M.S., R.K.A., G.C.K., T.C.G., and X.X.). S.F.N. is supported by an Australia NH&MRC C.J. Martin Fellowship (1111469) and the Mark Foundation Momentum Fellowship. Confocal imaging studies were performed in the Penn Vet Imaging Core, which is supported by S10OD021633 and S10RR027128. E.J.W. is a member of the Parker Institute for Cancer Immunotherapy, which supported the UPenn Cancer Immunotherapy Program. Graphical abstract was created using Biorender.com.
Footnotes
DECLARATION OF INTERESTS
E.J.W. has consulting agreements with and/or is on the scientific advisory board for Merck, Elstar, Janssen, Related Sciences, Synthekine, and Surface Oncology. E.J.W. is a founder of Surface Oncology and Arsenal Biosciences. E.J.W. has a patent licensing agreement on the PD-1 pathway with Roche/Genentech.
INCLUSION AND DIVERSITY
We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109120.
REFERENCES
- Afkarian M, Sedy JR, Yang J, Jacobson NG, Cereb N, Yang SY, Murphy TL, and Murphy KM (2002). T-bet is a STAT1-induced regulator of IL-12R expression in naïve CD4+ T cells. Nat. Immunol 3, 549–557. [DOI] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. (2011). Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest 121, 2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, and Reiner SL (2010). Cutting edge: The transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol 185, 4988–4992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, and Ahmed R (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687. [DOI] [PubMed] [Google Scholar]
- Beima KM, Miazgowicz MM, Lewis MD, Yan PS, Huang TH, and Weinmann AS (2006). T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J. Biol. Chem 281, 11992–12000. [DOI] [PubMed] [Google Scholar]
- Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity 52, 825–841.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, and Wherry EJ (2009). Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol 10, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buggert M, Tauriainen J, Yamamoto T, Frederiksen J, Ivarsson MA, Michaëlsson J, Lund O, Hejdeman B, Jansson M, Sönnerborg A, et al. (2014). T-bet and Eomes are differentially linked to the exhausted phenotype of CD8+ T cells in HIV infection. PLoS Pathog 10, e1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, Johnson J, Staupe RP, Bengsch B, Xu C, et al. (2019). TCR-1-centered transcriptional network drives an effector versus exhausted CD8 cell-fate decision. Immunity 51, 840–855.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll M, Seidman JG, and Müller CW (2002). Structure of the DNA-bound T-box domain of human TBX3, a transcription factor responsible for ulnarmammary syndrome. Structure 10, 343–356. [DOI] [PubMed] [Google Scholar]
- Cruz-Guilloty F, Pipkin ME, Djuretic IM, Levanon D, Lotem J, Lichtenheld MG, Groner Y, and Rao A (2009). Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med 206, 51–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W, Joshi NS, Jiang A, and Kaech SM (2009). Effects of Signal 3 during CD8 T cell priming: Bystander production of IL-12 enhances effector T cell expansion but promotes terminal differentiation. Vaccine 27, 2177–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC, et al. (2003). Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med 9, 562–567. [DOI] [PubMed] [Google Scholar]
- Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, and Wherry EJ (2012). Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity 37, 1130–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duraiswamy J, Freeman GJ, and Coukos G (2013). Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 73, 6900–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimcher LH, Townsend MJ, Sullivan BM, and Lord GM (2004). Recent developments in the transcriptional regulation of cytolytic effector cells. Nat. Rev. Immunol 4, 900–911. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, Araki K, and Ahmed R (2018). CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med 69, 301–318. [DOI] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. (2016). Follicular CXCR5- expressing CD8(+) T cells curtail chronic viral infection. Nature 537, 412–428. [DOI] [PubMed] [Google Scholar]
- Heinen AP, Wanke F, Moos S, Attig S, Luche H, Pal PP, Budisa N, Fehling HJ, Waisman A, and Kurschus FC (2014). Improved method to retain cytosolic reporter protein fluorescence while staining for nuclear proteins. Cytometry A 85, 621–627. [DOI] [PubMed] [Google Scholar]
- Hersperger AR, Martin JN, Shin LY, Sheth PM, Kovacs CM, Cosma GL, Makedonas G, Pereyra F, Walker BD, Kaul R, et al. (2011). Increased HIV-specific CD8+ T-cell cytotoxic potential in HIV elite controllers is associated with T-bet expression. Blood 117, 3799–3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. (2005). Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res 65, 1089–1096. [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Orlowski RJ, Xu X, Mick R, George SM, Yan PK, Manne S, Kraya AA, Wubbenhorst B, Dorfman L, et al. (2019). A single dose of neoadjuvant PD-1 blockade predicts clinical outcomes in resectable melanoma. Nat. Med 25, 454–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1+ Stem-like CD8+ T cells during chronic infection. Immunity 51, 1043–1058.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, et al. (2005). Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol 6, 1236–1244. [DOI] [PubMed] [Google Scholar]
- Intlekofer AM, Takemoto N, Kao C, Banerjee A, Schambach F, Northrop JK, Shen H, Wherry EJ, and Reiner SL (2007). Requirement for T-bet in the aberrant differentiation of unhelped memory CD8+ T cells. J. Exp. Med 204, 2015–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Banerjee A, Takemoto N, Gordon SM, Dejong CS, Shin H, Hunter CA, Wherry EJ, Lindsten T, and Reiner SL (2008). Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, and Minato N (2002). Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 99, 12293–12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Terawaki S, and Honjo T (2005). PD-1 blockade inhibits hematogenous spread of poorly immunogenic tumor cells by enhanced recruitment of effector T cells. Int. Immunol 17, 133–144. [DOI] [PubMed] [Google Scholar]
- Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, Leonard WJ, Greenleaf WJ, Ahmed R, and Goronzy JJ (2019). Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl. Acad. Sci. USA 116, 14113–14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, and Cui W (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol 12, 749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech SM, Hemby S, Kersh E, and Ahmed R (2002). Molecular and functional profiling of memory CD8 T cell differentiation. Cell 111, 837–851. [DOI] [PubMed] [Google Scholar]
- Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, and Wherry EJ (2011). Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol 12, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim PS, and Ahmed R (2010). Features of responding T cells in cancer and chronic infection. Curr. Opin. Immunol 22, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight DA, Ngiow SF, Li M, Parmenter T, Mok S, Cass A, Haynes NM, Kinross K, Yagita H, Koya RC, et al. (2013). Host immunity contributes to the anti-melanoma activity of BRAF inhibitors. J. Clin. Invest 123, 1371–1381. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kurachi M, Kurachi J, Chen Z, Johnson J, Khan O, Bengsch B, Stelekati E, Attanasio J, McLane LM, Tomura M, et al. (2017). Optimized retroviral transduction of mouse T cells for in vivo assessment of gene function. Nat. Protoc 12, 1980–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Ngiow SF, Kurachi J, Chen Z, and Wherry EJ (2019). Hidden caveat of inducible cre recombinase. Immunity 51, 591–592. [DOI] [PubMed] [Google Scholar]
- Kurktschiev PD, Raziorrouh B, Schraut W, Backmund M, Wächtler M, Wendtner CM, Bengsch B, Thimme R, Denk G, Zachoval R, et al. (2014). Dysfunctional CD8+ T cells in hepatitis B and C are characterized by a lack of antigen-specific T-bet induction. J. Exp. Med 211, 2047–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange A, Mills RE, Lange CJ, Stewart M, Devine SE, and Corbett AH (2007). Classical nuclear localization signals: definition, function, and interaction with importin alpha. J. Biol. Chem 282, 5101–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS, Johnson D, Swetter S, Thompson J, Greenberg PD, et al. (1999). Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat. Med 5, 677–685. [DOI] [PubMed] [Google Scholar]
- McLane LM, and Corbett AH (2009). Nuclear localization signals and human disease. IUBMB Life 61, 697–706. [DOI] [PubMed] [Google Scholar]
- McLane LM, Banerjee PP, Cosma GL, Makedonas G, Wherry EJ, Orange JS, and Betts MR (2013). Differential localization of T-bet and Eomes in CD8 T cell memory populations. J. Immunol 190, 3207–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K, Altman JD, Suresh M, Sourdive D, Zajac A, and Ahmed R (1998). In vivo dynamics of anti-viral CD8 T cell responses to different epitopes. An evaluation of bystander activation in primary and secondary responses to viral infection. Adv. Exp. Med. Biol 452, 123–142. [DOI] [PubMed] [Google Scholar]
- Oestreich KJ, Yoon H, Ahmed R, and Boss JM (2008). NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol 181, 4832–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, and Wherry EJ (2012). Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, and Wherry EJ (2015). Overcoming T cell exhaustion in infection and cancer. Trends Immunol 36, 265–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, Drake AM, Chen Z, Sen DR, Kurachi M, et al. (2016). Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science 354, 1160–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, et al. (2003). Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302, 1041–1043. [DOI] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, and Rao A (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, Ramsay AG, and Gribben JG (2013). T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood 121, 1612–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J. Exp. Med 207, 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skene PJ, Henikoff JG, and Henikoff S (2018). Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc 13, 1006–1019. [DOI] [PubMed] [Google Scholar]
- Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W, et al. (2003). B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res 63, 6501–6505. [PubMed] [Google Scholar]
- Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, and Glimcher LH (2000). A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669. [DOI] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, and Pardoll DM (2012). Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr. Opin. Immunol 24, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, and Kurachi M (2015). Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol 15, 486–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol 77, 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1, eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac AJ, Blattman JN, Murali-Krishna K, Sourdive DJ, Suresh M, Altman JD, and Ahmed R (1998). Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med 188, 2205–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, and Cui W (2019). CD4+ T cell help is required for the formation of a cytolytic CD8+ T cell subset that protects against chronic infection and cnacer. Immunity 51, 1028–1042.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH (2010). Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original source data for Figure 5 is available in Doering et al. (2012) and Pauken et al. (2016).