

Polymer-drug conjugates are developed for the topical ratiometric delivery of a synergistic drug pair to treat skin cancers.
Abstract
Skin cancer is one of the most common types of cancer in the United States and worldwide. Topical products are effective for treating cancerous skin lesions when surgery is not feasible. However, current topical products induce severe irritation, light-sensitivity, burning, scaling, and inflammation. Using hyaluronic acid (HA), we engineered clinically translatable polymer-drug conjugates of doxorubicin and camptothecin termed, DOxorubicin and Camptothecin Tailored at Optimal Ratios (DOCTOR) for topical treatment of skin cancers. When compared to the clinical standard, Efudex, DOCTOR exhibited high cancer-cell killing specificity with superior safety to healthy skin cells. In vivo studies confirmed its efficacy in treating cancerous lesions without irritation or systemic absorption. When tested on patient-derived primary cells and live-skin explants, DOCTOR killed the cancer with a selectivity as high as 21-fold over healthy skin tissue from the same donor. Collectively, DOCTOR provides a safe and potent option for treating skin cancer in the clinic.
INTRODUCTION
In the United States, more than 5.4 million people are diagnosed with nonmelanoma skin cancers (NMSCs) every year (1). Increasing trends in its incidence in North America, Europe, Australia, and the Asia-Pacific region indicate the growing scope of this global epidemic (2, 3). Basal cell carcinoma and cutaneous squamous cell carcinoma (cSCC) constitute 99% of all NMSCs. Precancerous skin lesions such as actinic keratosis (AK) also pose a major challenge, with 58 million people affected in the United States (4). NMSCs and AKs are typically removed by surgical excision, cryotherapy, curettage, and electrodesiccation or Mohs surgery (5, 6). While effective for highly localized cancers, these techniques are not suitable for multiple lesions or those located at anatomically sensitive areas (6). Extended healing time of surgical wounds is also a challenge (5).
Current topical products for treating NMSCs or AKs include treatments such as cryotherapy and photodynamic therapy or topical chemotherapeutics such as 5-fluorouracil (5-FU; Efudex). Procedures are limited by cost, complexity, and, in case of photodynamic therapy, light sensitivity (7). Besides, the current topical products are effective in treating superficial skin lesions, but not the deeper regions, resulting in appearance of new lesions or progression to distant sites. Topical products such as Efudex are known to cause inflammation, swelling, and scaling when applied on the skin (8–10). Some inflammation induced by Efudex may be helpful in generating an immune response (11). However, the high inflammation induced by Efudex is also a deterrent in its use by patients, especially when multiple applications are required for an effective response. At the current prescribed doses for Efudex, many patients opt out of topical treatment because of side effects, thus leading to disease mismanagement.
Here, we report a new topical treatment based on a drug combination—doxorubicin (DOX) and camptothecin (CPT)—specifically designed to improve selectivity toward skin cancers. We sought to assess whether efficacy can be obtained in the absence of inflammation. This combination, DOxorubicin and Camptothecin Tailored at Optimal Ratios (DOCTOR), was delivered via topical applications using a hyaluronic acid (HA) conjugate. Synergistic action of DOX and CPT has been extensively studied in the literature (12–14). CPT and DOX are inhibitors of topoisomerase I (TOP I) and topoisomerase II (TOP II), which are critical for DNA transcription and cell replication (12). The levels of TOP I enzymes are relatively high in cancer tissues including SCCs compared to the adjacent healthy tissues (15–17). However, the clinical utility of DOX-CPT combination has never translated to the clinic. This is primarily due to the poor solubility of CPT, and for that reason, the free drug combination was not tested in this study because it is not a translationally viable form of formulation (18, 19). DOX and CPT have highly distinct hydrophobicities and solubilities, which lead to differences in skin permeabilities. This changes ratios of the two drugs delivered in the skin and, therefore, the difficulty in maintaining the pair’s synergy in vivo. This makes topical synergistic ratiometric dosing of free drugs challenging and a direct comparison to their conjugated counterparts difficult. By conjugating DOX and CPT to the HA polymer, we demonstrate the successful retention of this pair’s safety and synergistic potency. By using the polymer conjugate, we overcome the limitations faced by free drugs that hinder their translation to the clinic.
Conjugation of DOX and CPT to HA led to the formation of worm-like micelles (DOCTOR) that delivered both drugs deep into the skin. Studies performed in multiple cell lines in vitro, ultraviolet (UV)–induced mouse tumor models in vivo, and patient-derived healthy and cSCC biopsies confirm the anticancer activity and safety of DOCTOR in treating skin cancer.
RESULTS
Synthesis of HA-DOX, HA-CPT, and DOCTOR
Free DOX (Table 1) and free CPT (Table 1) were covalently linked to HA (Table 1) to obtain single-drug conjugates (HA-DOX and HA-CPT; Table 1) or the dual-drug conjugates (DOCTOR; Table 1) incorporated with DOX:CPT at three molar ratios of R2, R5, and R15 (R = molar ratio). Drug incorporation on HA was confirmed via Fourier transform infrared (FTIR) spectroscopy and nuclear magnetic resonance (NMR) spectroscopy. The covalent conjugation of each drug to HA was confirmed via the formation of amide and ester bonds (Fig. 1A and fig. S2, A to D). Native HA is a linear hydrophilic polymer, which exists in an extended confirmation in the solution. Conjugation of DOX and CPT imparts hydrophobicity to HA, thus leading to its self-assembly into micelles (Fig. 1, B and C, and fig. S2F). The amount of DOX and CPT incorporated in DOCTOR was quantified using fluorescence spectra specific to each drug molecule.
Table 1. List of relevant abbreviations.
|
DOCTOR (DOX-HA-CPT) |
DOxorubicin and Camptothecin Tailored at Optimal Ratios |
| HA | Hyaluronic acid |
| DOX | Free doxorubicin |
| CPT | Free camptothecin |
| HA-DOX | HA-conjugated doxorubicin |
| HA-CPT | HA-conjugated camptothecin |
| SCC | Squamous cell carcinoma |
Fig. 1. Physical characterization of DOCTOR.
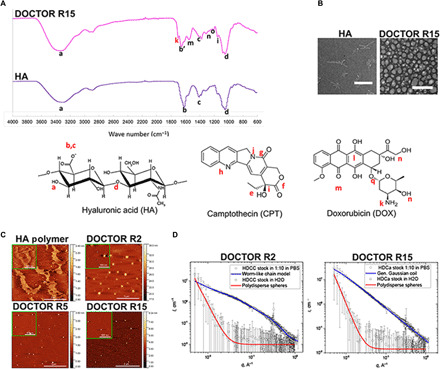
(A) Differences in FTIR spectra of native HA and DOCTOR confirmed the covalent linkage of DOX and CPT to the HA polymer backbone. (B) TEM imaging revealed the transition from fibrillar morphology for native HA to a micellar appearance for DOCTOR (scale bar, 500 nm). (C) Topographic AFM images, 5 μm by 5 μm in size (inset: 1 μm by 1 μm), of (top row) native HA and DOCTOR R2 and (bottom row) DOCTOR R5 and R15 revealed a fibrillar morphology for native HA and a particulate structure with average mean sizes of 146, 63.5, and 72 nm for R2, R5, and R15, respectively. The adjacent color scale represents the height (z value) for the 5 μm–by–5 μm frames, respectively. (D) SAXS curves for DOCTOR in DI water and PBS obtained via SAXS analysis. Weak or negligible scattering was observed for the conjugates in water (red trace). The scattering intensity is almost flat at the intermediate and high q values and shows a rise fitted by the model of polydisperse hard spheres at low values of scattering vector q < 0.05 A−1. In PBS (blue trace), the scattering intensity increased by two orders of magnitude and is fitted by worm-like chain or generalized Gaussian coil model described in Materials.
The FTIR spectra for DOCTOR show the presence of signature peaks “a” (─OH and ─NH groups at 3309 cm−1 from HA), “b and c” (asymmetric vibration of COO− at 1613 and 1400 cm−1 from HA), and “d” (C─O─C hemiacetalic saccharide linkages at 1030 cm−1 from HA), indicating the intact polysaccharide structure of the HA backbone (Fig. 1A and fig. S2A). On conjugation of CPT with HA, the characteristic peaks “i” [C─C(═O)─O stretching at 1150 cm−1] and “j” (contribution from the four adjacent hydrogen bonds on hetero-aromatic nucleus at 767 cm−1) remain unaffected (Fig. 1A and fig. S2A). Furthermore, the formation of ester bond between HA and CPT is evident from the shift of the carbonyl peak at 1613 cm−1 (b) to 1663.8 cm−1 (b′) and the disappearance of peak “e” (─OH groups on CPT; fig. S2A). As a control, physically mixed CPT and HA yielded just the signature peaks a, b, c, and d from the polymer, with no peaks from CPT in the FTIR spectrum of purified product (fig. S2B). The FTIR spectrum of HA-DOX conjugate revealed close to complete disappearance of the peak corresponding to primary amines at 1730 cm−1 (k), indicating its reaction with carboxyl groups of HA to form amide bonds (fig. S2A). For the control sample made via the physical mixing of HA and DOX, the peak “k” still existed for the final purified product. This is likely due to the presence of small amounts of residual DOX physically attached to HA (fig. S2C). The FTIR spectrum for DOX-HA-CPT confirmed the presence of signatures for bonds, namely, the amide and ester bonds formed following the conjugation of HA to CPT and DOX (Fig. 1A).
1H NMR analysis of the HA conjugates indicated the presence of drug, although not quantifiable because of its low peak signal (fig. S2D). The signature peaks for HA were present in all conjugates when compared to the HA control (fig. S2D). The aromatic H─C bonds of free CPT—a, b, c, d, and e—were visible in the HA-CPT conjugate (fig. S2D). These peaks were slightly shifted because of the D2O solvent used for HA-CPT in contrast to the dimethyl sulfoxide (DMSO)–d6 solvent used for free CPT. These peaks, however, were not visible in the dual-drug conjugate R15 where DOX:CPT molar ratio is 15:1. We attribute this to the low CPT content in the dual conjugate with respect to HA-CPT. The aromatic peaks—f, g, and h—of DOX were visible as a merged peak around 7.6 parts per million in both HA-DOX and DOCTOR R15 (fig. S2D). The low signal intensity of the aromatic peaks for individual drugs can be attributed likely to the existence of drugs in a self-assembled form when incorporated on HA. This is consistent with previous reports of strong attenuations or complete disappearance of the drugs’ aromatic signals, which remains buried within the core of self-assembled HA conjugate aggregates (20–23). The presence of strong signals for HA and the low aromatic signals for the drugs during NMR analysis indicate the formation of HA self-assembled structures in the aqueous solution.
The formation of different covalent bonds for DOX and CPT is further supported by the differences in in vitro release rates for both drugs. While a slow and steady release was observed for DOX [10.93 ± 0.33 weight % (wt %) DOX], approximately 70.17 ± 2 wt % CPT was released at the end of 5 days in physiological buffer conditions at 37°C (fig. S2E).
There is extensive evidence in the literature that supports validating the formation of amide and ester bonds in HA conjugates using FTIR spectroscopy (24, 25). FTIR analysis of HA describes the peak around 3300 cm−1 to be associated with the intra- and intermolecular stretching vibration of the ─OH group (24). The technique may not confirm the complete disappearance of ─OH peaks because only a fraction of ─OH groups from HA get conjugated to CPT. However, there is a clear and notable difference between the ─OH peak regions of HA, CPT, and HA-CPT, indicating interaction between HA and CPT molecules via ─OH. Furthermore, there are no other functional groups like amine on CPT to conjugate to HA. Neither is there considerable physical adsorption of CPT [as evident from HA-CPT without N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) spectrum; fig. S2B]. Thus, ester formation seems to be the only obvious or possible route for the EDC reaction to proceed. Note also that the hydroxyl groups on DOX can lead to esterification. However, in the presence of EDC, amide formation (via strongly nucleophilic amine group) is favored more over ester formation (via hydroxyl group). Competitive ester formation would require addition of an accelerating reagent like 4-(dimethylamino)pyridine (DMAP) (26, 27). Amide bond formation is evident from the complete disappearance of the peak corresponding to primary amines at 1730 cm−1 (k) in the HA-DOX FTIR spectrum, as well as the slow release of DOX from DOCTOR in in vitro release study.
Morphology and size of DOCTOR was further confirmed by transmission electron microscopy (TEM) (Fig. 1B), atomic force microscopy (AFM) (Fig. 1C), and nanoparticle tracking analysis (NTA) (fig. S2G). Native HA had a fibrillar morphology, whereas DOCTOR exhibited a particulate structure. The average mean sizes of DOCTOR were approximately 146, 63.5, and 72 nm for R2, R5, and R15, respectively (table S2A). These numbers also matched those measured via NTA in phosphate-buffered saline (PBS; pH 7.4) (fig. S2G). Furthermore, small-angle x-ray scattering (SAXS) was performed to determine the structure of DOCTOR in deionized (DI) water and PBS. For SAXS, DOCTOR was diluted 10 times in PBS to simulate the skin’s internal salinity that the conjugates would encounter following penetration. SAXS revealed a weak or negligible scattering for samples in water (Fig. 1D, red trace). However, the scattering intensity increased by two orders of magnitude in PBS, indicating a particulate shape for DOCTOR (Fig. 1D, blue trace). A similar trend was observed for the single-drug conjugates as well (fig. S2H). The zeta potential values changed from highly negative for blank HA to within the neutral range for DOCTOR in PBS (table S2C).
Efficacy of DOCTOR against cancerous keratinocytes: Role of synergy
DOCTOR was highly effective against the human SCC cell line (A431). The median inhibitory concentration (IC50) dose of DOX in DOCTOR “R2,” DOCTOR “R5,” and DOCTOR “R15” was approximately 140-fold, 60-fold, and 17-fold lower, respectively, than the IC50 dose of DOX in HA-DOX (Fig. 2A and table S3A). Similarly, the IC50 dose of CPT in DOCTOR R2, DOCTOR R5, and DOCTOR R15 was approximately sevenfold, eightfold, and sixfold lower, respectively, than the IC50 dose of CPT in HA-CPT (table S3A). A synergistic interaction was confirmed for DOCTOR at all ratios based on combination indices (CIs) estimated from the IC50 values of single- and dual-drug treatments. A value of CI < 1 indicates synergism, CI = 1 suggests an additive effect, and CI > 1 suggests antagonism. Because the CI values for DOCTOR at all ratios were <<1, it indicated a highly synergistic interaction between the drug pair when incorporated onto HA (table S3A).
Fig. 2. DOCTOR exhibits increased synergistic potency against cancerous (A431) human keratinocytes with less toxicity to the healthy cells.
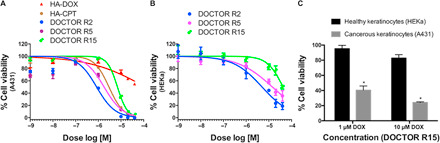
(A) When treated with A431, the IC50 dose of DOX in formulations DOCTOR R2, DOCTOR R5, and DOCTOR R15 were approximately 140-fold, 60-fold, and 17-fold lower than the IC50 dose of DOX in HA-DOX (table S3A). (B) Among the three different ratios tested, the molar ratio of DOX:CPT R15 was identified to be the least toxic to HEKa cells (table S3B). (C) When treated with DOCTOR R15, approximately 60 and 76% of A431 cells were killed while affecting only ~5 and ~17% of HEKa cells. Error bars represent means ± SEM (n = 3). Significantly different compared to the HEKa cells: *P < 0.05.
DOCTOR is well tolerated by healthy human keratinocytes
DOCTOR was less toxic to healthy human epidermal keratinocytes (HEKa) compared to the cancerous (A431) human keratinocytes. The IC50 doses of DOX in DOCTOR R2, DOCTOR R5, and DOCTOR R15 were approximately 3.6-fold, 3.5-fold, and 7-fold higher, respectively, in HEKa compared to A431 (Fig. 2B and table S3B). Among the three different ratios tested, the molar ratio of DOX:CPT R15 was identified to be the least toxic to HEKa cells (Fig. 2B and table S3B). DOCTOR R15 exhibited high selectivity toward A431. At a dose of 10 μM DOX, delivered as DOCTOR R15, 76% of A431 cells were killed while affecting only ~17% of HEKa cells. Similar observations were made at other doses of DOCTOR R15 (Fig. 2C).
DOCTOR delivers DOX and CPT inside the skin
The ability of DOCTOR to deliver DOX and CPT into the skin was assessed ex vivo using a porcine skin model where the formulation was applied topically and left undisturbed for 24 hours. DOCTOR R15 was used for these studies at a dose of 0.5 mg/ml or 920 μM DOX in DOCTOR. When compared with the negative HA control–treated skin (fig. S4A, top), confocal microscopy imaging revealed bright fluorescent signals for DOX and CPT, suggesting that DOCTOR R15 can deliver the drugs within the epidermis and dermis (Fig. 3A). The amounts of DOX and CPT delivered into the skin were quantified using the tape-stripping method (Fig. 3B and table S4). Quantification via the tape-stripping method revealed the amounts of DOX and CPT delivered into the skin and the pair’s molar ratio. Approximately 18 ± 7.0% of the applied DOX and 8 ± 3.0% of the applied CPT were measured in the combined layers of the lower stratum corneum and the epidermis, resulting in a DOX:CPT molar ratio of 27 (Fig. 3B and table S4). For the dermis, approximately 2 ± 0.7% of the applied DOX and 2 ± 0.8% of the applied CPT were detected, resulting in a DOX:CPT molar ratio of 15 (Fig. 3B and table S4). Separately, in an in vitro cell uptake study, confocal imaging revealed that DOCTOR enabled simultaneous internalization of DOX and CPT in A431 cells (fig. S4B). Intracellular colocalization of the drug pair was confirmed by the cyan signal (fig. S4B, white arrow). This indicates that DOCTOR is able to deliver both the drugs deep into the skin and further inside the cells to induce synergistic potency.
Fig. 3. DOCTOR delivers the drug pair inside the tissue without causing skin inflammation.
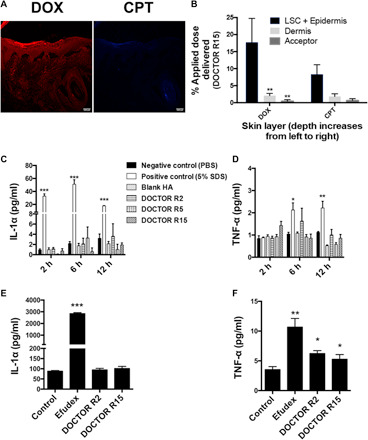
(A) Confocal microscopy imaging and (B) quantification via tape stripping revealed penetration across the stratum corneum and deposition of DOX (red channel) and CPT (blue channel) within the epidermis and dermis of porcine skin ex vivo (integration time, 1.58 μs; scale bar, 100 μm). Error bars represent means ± SEM (n = 3). Significantly different compared to the DOX readings in lower SC (LSC) + epidermis: **P < 0.01. (C and D) DOCTOR induced negligible inflammation on human skin. Levels of (C) IL-1α and (D) TNF-α were assessed on a MatTek EpiDerm human skin equivalent model. PBS and 5% SDS were used as negative and positive controls, respectively. Error bars represent means ± SEM (n = 3). Significantly different compared to the negative control: *P < 0.05, **P < 0.01, and ***P < 0.001. (E and F) DOCTOR induced negligible skin inflammation in vivo. Levels of (E) IL-1α and (F) TNF-α were assessed in mice during a 2-week repeat dose study with DOCTOR. Efudex was used as the clinical comparator, and PBS was used as the negative control. Error bars represent means ± SEM (n = 5). Significantly different compared to the negative control: *P < 0.05, **P < 0.01, and ***P < 0.0001.
Tolerability of DOCTOR
Using reconstructed human epidermis (EpiDerm) as a model, the tolerability of DOCTOR was assessed. SDS (5%) was used as a positive irritation control (28). Tolerability was assessed via release of inflammatory cytokines [interleukin-1α (IL-1α) and tumor necrosis factor–α (TNF-α)]. All three drug ratios R2, R5, and R15 were tested in EpiDerm. Native HA and all DOCTOR compositions induced IL-1α and TNF-α at levels similar to that of PBS (Fig. 3, C and D). Furthermore, the in vivo tolerability of DOCTOR was tested in immunocompetent SKH1-E hairless mice over a 2-week repeat dose study (five times a week for 2 weeks). DI water (pH 5.0) was used as a negative control, and Efudex (5% fluorouracil) was the positive control. R5 was not used in these studies in anticipation that its behavior is not distinctive from R2 or R15 and thus unlikely to provide benefits and insights offered by either. DOCTOR R2 and R15 were dissolved in DI water (pH 5.0) and applied at a dose of 0.006 mg/ml or 11 μM DOX (3 μg/week) in DOCTOR. The CPT doses were 0.0012 mg/ml or 3.5 μM CPT in DOCTOR R2 and 0.000164 mg/ml or 0.47 μM CPT in DOCTOR 15. The dose for Efudex (160 μg/week), the clinical comparator, was determined on the basis of a previous study (29). Efudex induced high levels of IL-1α and TNF-α compared to the PBS negative control, whereas DOCTOR R2 and DOCTOR R15 induced cytokine production comparable to PBS, suggesting that the DOCTOR formulations cause negligible skin inflammation compared to Efudex (Fig. 3, E and F). Furthermore, systemic absorption of DOX and CPT after topical application of DOCTOR was assessed in vivo. No DOX was detected, and less than 1% of the applied CPT dose was detected in the plasma at the end of 10 topical applications over 2 weeks (fig. S5A).
In vivo efficacy
In vivo efficacy of DOCTOR was tested in a UV-induced model of spontaneous cSCC in immunocompetent SKH1-E mice. Mice were topically treated three times a week over a period of 90 days with (i) blank HA control (100 mg/kg) prepared in DI water (pH 5.0) or (ii) DOCTOR R15 at a dose of 0.003 mg/ml or 5.5 μM DOX and 0.000082 mg/ml or 0.235 μM CPT in DOCTOR after dissolving in DI water (pH 5.0). R15 was selected for these experiments based on better safety profile in HEKa cells. Compared to the group treated with blank HA, a considerable delay in tumor progression was observed in mice when treated with DOCTOR R15 (Fig. 4, A and C). In addition, treatment with DOCTOR R15 significantly increased mean survival by 65% (P < 0.0001) compared to the HA control group (Fig. 4B). Skin biopsies from mice treated with DOCTOR were assessed to identify the mechanism of DOCTOR-induced cSCC cell death. A positive nuclear and cytoplasmic immunostaining was observed for cleaved caspase-3 in DOCTOR R15–treated cSCC tumor biopsies, indicative of apoptotic cell death (Fig. 4D). No apoptotic cell death signals were observed in biopsy samples from mice treated with the HA control (Fig. 4D).
Fig. 4. In vivo efficacy.
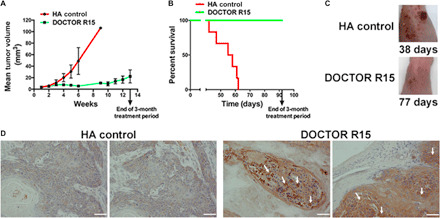
(A to C) DOCTOR prevents progression of cSCC lesions and enhanced survival by 65% (P < 0.0001) at extremely low doses (0.003 mg/ml or 0.016 mg/kg) of DOX in DOCTOR. Error bars represent means ± SEM (n = 9). (D) cSCC tumors showing nuclear and cytoplasmic immunostaining (golden brown) for cleaved caspase-3 depicting apoptotic tumor cell death on treatment with DOCTOR R15 (scale bar, 50 μm). Photo credit: (C) B.R.S. and K.Y.T., H. Lee Moffitt Cancer Center and Research Institute; V.K., Harvard University; (D) V.K., Harvard University.
Safety and anticancer activity in patient-derived primary cells and live skin explants
The potential clinical safety and anticancer activity of DOCTOR in treating cSCC was further tested on normal and cSCC skin tissues obtained from patients. The translation of ratios that worked best in in vitro and in vivo mouse experiments to human samples is not obvious, given the biological and physical differences in cells, animal tissues, and human tissues. Hence, we tested all three ratios on primary patient cells obtained from tumor lesions and corresponding normal controls. In these studies, R2 showed the highest level of specificity among all ratios tested [fig. S6A (A to C)]. Hence, R2 was used for further testing with human cancer specimens.
While more than 80 to 90% viability was observed for DOCTOR-exposed primary cells isolated from the normal skin adjacent to the cSCC tumor lesion, DOCTOR reduced the viability of primary cSCC cells isolated from the lesion to as low as 30% [Fig. 5A and fig. S6A (A to C)]. Patient donor 3 displayed high sensitivity to the treatment [fig. S6A (C and F)] at half the dose deemed highly effective for the other two patient samples [fig. S6A (D and E)]. These variations are critical and underscore the interindividual differences in response to therapy in the clinic. The variations could possibly be attributed to factors that are inherited or acquired. Furthermore, DOCTOR revealed an increasingly high potency and safety when compared against the single-drug conjugates [fig. S6A (D to F)]. This highlights the synergistic interaction between DOX and CPT, thereby making the case for clinical use of DOCTOR against cutaneous cancers. Patient-derived skin explants from normal and lesional skin have the ability to stay viable and preserve their metabolic and proliferative capacity for several days in culture. When DOCTOR was tested on patient-derived live tissue explants, the proliferative activity of normal skin to the matched cSCC tissue was approximately 3.6-fold higher for patient 4, 21-fold higher for patient 5, and 5.4-fold higher for patient 6 over 5-FU or Efudex, the clinical comparator (Fig. 5, B and C, and fig. S6B). Together, these data indicate the therapeutic potential of DOCTOR in eliminating cSCC lesions without harming the healthy skin tissue.
Fig. 5. Safety and anticancer activity in patient-derived primary cells and live skin explants.
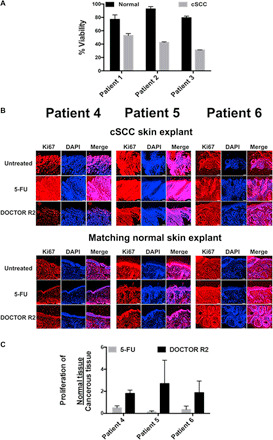
(A) When tested on primary cultures of patient-derived cells, DOCTOR induced a maximum of 70% cSCC cell death, while more than 80 to 90% of adjacent noncancerous skin cells still remained viable. (B) When tested with live cSCC skin and adjacent noncancerous live skin explants, DOCTOR did not affect the proliferation of normal skin cells but rather inhibited the growth of cSCC cells in contrast to treatment with the clinical comparator 5-FU (Efudex) (scale bar, 100 μm). The proliferation activity of cSCC and noncancerous cells was measured by Ki67 staining (red). The number of Ki67-positive cells in the treated explant was normalized to the number of cells in the untreated control for each patient tissue. Quantitative image analysis was performed using ImageJ software on three separate tissue sections per sample after “threshold”-based reduction of background noise. Positive “red” signal was quantified in the entire tissue section for the cSCC samples and in the epidermis only for the matched normal tissues. All samples were stained at the same time under the same conditions to keep the background signal (false-positive counts) similar across all samples. An increase in cSCC proliferation activity was observed for untreated and 5-FU–treated skin explants. (C) The proliferation of matched healthy skin tissue:cSCC skin tissues was calculated on the basis of quantified proliferation activity for adjacent noncancerous tissue over the matching cSCC tissue. No significant difference compared to the treatment control 5-FU, P > 0.05.
DISCUSSION
Previously, HA conjugates have been used for the localized and systemic therapies of solid tumors (22, 30). Here, we sought to develop a translatable formulation of CPT and DOX for the topical treatment of skin cancers. The choice of this combination was motivated by previous reports on success of this pair for the treatment of other cancers (12–14). However, the efficacy and safety of this formulation is strongly dependent on the molar ratio of the two drugs. This makes the drug pair’s formulation process challenging because a simple topical formulation comprising DOX and CPT is unlikely to maintain the desired ratios in the skin because of differences in their molecular properties that lead to differences in their solubility and skin permeation rates. We sought to address this challenge by conjugating DOX and CPT to HA, which fixes its ratio. However, in this process, the small molecular drugs are converted into a macromolecule. We thus sought to assess whether this macromolecular drug exhibits the necessary efficacy/safety profile in vitro, in vivo, and in patient-derived samples.
In vitro toxicity results revealed an increased potency for DOX and CPT in DOCTOR compared to the single-drug conjugates (HA-DOX or HA-CPT) (Fig. 2A and table S3A) with CI values calculated to be much less than 1. Conjugating DOX to HA increased its IC50 dose by 163-fold when compared to its nonconjugated form, the free DOX (table S3A). In case of CPT, conjugating the drug to HA increased its IC50 dose by fivefold when compared to free CPT (table S3A). We attribute this to the differences in drug availability due to bond hydrolysis and probable differences in its uptake mechanism. While free drugs permeate across the membrane directly, the conjugates are likely to be internalized via endocytosis. This was confirmed via confocal microscopy analysis, which revealed intracellular colocalization of the drug pair (fig. S4B, white arrow). FTIR and NMR spectroscopy analysis revealed that CPT was chemically conjugated to HA via an ester bond, while DOX was incorporated with an amide bond (Fig. 1A and fig. S2, A to D). Thus, the differences in drug availability are supported by the fact that CPT release was faster on account of ester hydrolysis from HA, while DOX release was slower because of amide hydrolysis from HA (fig. S2E). However, the differences in the release rates did not compromise their synergistic anticancer activity, as suggested by the CI values being increasingly less than 1 (table S3A).
DOCTOR exhibited minimal toxicity to the primary human epidermal keratinocytes in vitro (Fig. 2C and table S3B) when compared to the cancerous A431 cells. DOCTOR R15 was identified to be the least toxic to the HEKa cells, followed by DOCTOR R5 and DOCTOR R15 (Fig. 2B and table S3B). The safety of DOCTOR was also demonstrated with primary patient-derived normal keratinocytes in vitro (Fig. 5A and fig. S6A) and with patient-derived normal live skin explants ex vivo (Fig. 5, B and C). The lack of toxicity to healthy cells was also evident from the absence of skin inflammation when tested with three-dimensional (3D) human EpiDerm model ex vivo and with immunocompetent SKH1-E mice in vivo (Fig. 3, C to F). IL-1α is known to induce a considerable proinflammatory effect in the skin. When stimulated by large amounts of TNF-α, the keratinocytes secrete IL-1α. An imbalance in the levels of IL-1α results in an onset of skin inflammation (31, 32). Although treatment with DOCTOR did stimulate the production of TNF-α (Fig. 3F), its levels were not sufficient enough to increase the production of IL-1α, the endogenous marker for skin inflammation (Fig. 3E).
Thus, DOCTOR’s safety was validated across multiple systems including primary human epidermal keratinocytes in vitro, primary patient-derived normal skin cells in vitro, and immunocompetent SKH1-E mice in vivo and with the 3D human EpiDerm model and patient-derived normal skin explants ex vivo. We attribute this specific anticancer activity seen across multiple systems primarily to CPT, a TOP I enzyme inhibitor. As stated previously, the levels of TOP I enzymes are relatively high in cancer tissues including SCCs compared to the adjacent healthy tissue. This render “TOP I” as a highly specific and attractive anticancer target for its inhibitors such as CPT (15–17). In addition, HA is known to bind specifically to CD44, a cell surface receptor that has elevated expression in many cancers including the skin. This could possibly support a highly specific uptake for the conjugates (33, 34).
TEM (Fig. 1B), AFM (Fig. 1C), and NTA (fig. S2G) revealed a polydisperse spherical morphology for DOCTOR in PBS with a mean size ranging from ∼60 to 140 nm (table S2A). However, SAXS revealed a weak or negligible scattering from the conjugates when dissolved in water at 10 mg/ml (Fig. 1D, red trace). While the scattering intensity is almost flat in the intermediate and at increasing q values, the upturn at the low q values (>0.05 Å) indicate the presence of a small fraction of aggregates in the range of 1 to 100 nm (table S2B). Thus, DOCTOR exists as monomers or molecularly dissolved short moieties that coexist with a small fraction of the 100 Å–sized aggregates in water. To simulate the skin’s internal salinity that the conjugates would be exposed following penetration, the samples were further diluted 10 times in PBS. The scattering intensity unexpectedly rose by two orders of magnitude (Fig. 1D and fig. S2G, blue trace). This implied an enhanced aggregation of the molecularly dissolved monomers in the presence of salt. In water, the electrostatic repulsion and the hydrophilic nature of HA enable DOCTOR to exist as molecularly dissolved entities. Salt mitigates these interactions by shielding the charges and promotes self-assembly via the hydrophobic drugs covalently linked on these polymers. To assess the potential impact on DOCTOR’s microstructure upon dilution in PBS, it is best if the effect of salt and concentration is studied separately, that is, (i) compare the data at high concentration with or without PBS and (ii) compare the data at high and low concentration in PBS. A detailed mapping on the effect of such conditions on microstructure could be performed in future studies. SAXS data analysis revealed the nanoparticle diameter and Kuhn length to be at a maximum value of 3.6 and 115 Å, respectively, for DOCTOR R15 (table S2B). Thus, SAXS confirms the existence of self-assembled HA polymeric nanoparticles supported by the interplay of three important molecular forces: electrostatic repulsion, hydrophilicity, and hydrophobicity (35, 36).
Results from ex vivo porcine skin permeation studies revealed that a total of approximately 20% of the applied dose of DOX in DOCTOR R15 (DOX:CPT molar ratio, 15:1) and 10% of the applied dose of CPT in DOCTOR R15 (DOX:CPT molar ratio, 15:1) was detected (Fig. 3B and table S4) within the tissue. The values represent the combination of conjugated and released drugs, thereby contributing to the robust fluorescent signals observed via confocal microscopy imaging of the tissue cross section (Fig. 3A and fig. S4A). In vitro drug release studies indicate that the release rates of DOX and CPT from the conjugate under physiological conditions are slow and steady. At the end of 5 days in physiological buffer conditions at 37°C, approximately 10.93 ± 0.33 wt % DOX and 70.17 ± 2 wt % CPT were released (fig. S2E). Hence, we hypothesize that the conjugates that permeate into the skin undergo hydrolysis to release drugs. This results in differences in the amounts of DOX and CPT detected and the pair’s molar ratios in the skin layers (table S4).
When tested in UVB-exposed SKH1-E hairless mice, a highly aggressive and immunocompetent model of cSCC, topically applied DOCTOR reduced tumor growth and improved survival significantly (P < 0.0001) with no irritation or inflammation. In certain instances, DOCTOR eliminated full-size cSCC tumors in mice by inducing apoptotic tumor cell death (fig. S5B). It is quite remarkable that this efficacy was achieved at a low dose of DOX (0.003 mg/ml). By directly administering drugs to the pathological site, DOCTOR can avoid potential adverse effects associated with systemic toxicity. It also has an increased benefit-risk ratio against current clinical comparator, Efudex, which reduces cSCC lesions in humans. However, the challenge with this treatment is that it induces irritation and, thus, poor patient compliance. Hence, we focused comparison of DOCTOR to Efudex with respect to safety (Fig. 3, E and F).
Free drug combination was not tested in this study because it is not a translationally viable form of a formulation. Highly distinct hydrophobicities and solubilities of DOX and CPT lead to differences in skin permeabilities, thereby changing ratios of the two drugs delivered in the skin. Consequently, topical synergistic ratiometric dosing of free drugs is a challenge, and their comparison to conjugated counterparts is difficult. Instead, the focus was on comparing DOCTOR to single-drug conjugates and to 5-FU, the current clinical standard for treating AKs and cSCCs. Thus, conjugating a potent drug pair such as DOX and CPT to the HA backbone at precise molar ratios ensures topical delivery and accumulation of both the drugs at the target for enhanced therapeutic efficacy and improved prognosis in localized and metastatic skin cancer. It eliminates painful surgery, nonspecific systemic injections, and highly invasive treatment methods for prevention and treatment of skin cancers. This improves patient compliance. Upon further research on efficacy and safety, these polymer drug conjugates may open new opportunities for treating skin neoplasms and precancerous lesions.
MATERIALS
CPT and DMAP were purchased from Sigma-Aldrich (St. Louis, MO, USA). EDC was purchased from Life Technologies, USA. DOX-HCl was obtained from LC Laboratories (Woburn, MA, USA). HA of 50-kDa molecular weight (MW) was purchased from Creative PEGWorks (Winston Salem, NC, USA). CellTiter-Blue and Hoechst were purchased from Life Technologies, USA. Human SCCs (A431) and primary adult epidermal keratinocytes (HEKa) were purchased from the American Type Culture Collection (ATCC; Manassas, VA, USA). A431 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS). HEKa cells were maintained in dermal cell basal medium and components of the keratinocyte growth kits purchased from ATCC. The cells were cultured according to instructions provided by ATCC. The T cell lymphoma cell line HUT78: CTCL was gifted by D. Weinstock (Department of Medical Oncology, Dana-Farber Cancer Institute and Harvard Medical School). The normal B lymphoblast cell line HCC2218 BL was purchased from ATCC (Manassas, VA, USA). Both cell lines were cultured in RPMI 1640 medium (Life Technologies) supplemented with 20% FBS and were maintained at 37°C under a humidified atmosphere of 95% air and 5% CO2. All cells were maintained at 37°C under a humidified atmosphere of 95% air and 5% CO2. Sephadex G-25 PD-10 columns were obtained from GE Healthcare Life Sciences (Piscataway, NJ, USA), and dialysis cassettes with 3500 MW cutoff (MWCO) were obtained from Life Technologies, USA. All other chemicals used for this study were obtained from Fisher Scientific and were the highest possible grade commercially available.
METHODS
Synthesis of DOCTOR
DOX and CPT were conjugated to HA polymer via nucleophilic acyl substitution reactions. Briefly, 100 mg of 50-kDa-MW HA was dissolved in a 2-ml mixture of 1:1 DI water/DMSO at 40°C. To this mixture, DMAP and EDC were added at a molar ratio of 1:1 relative to the HA monomers. The activation was allowed to continue for 30 min under stirring. For the synthesis of DOX-HA or CPT-HA, the drugs were added dropwise into the reaction mixture at molar ratios of 0.4:1 and 0.2:1, respectively. For the dual-drug conjugates, DOX was initially added to the polymer at varying amounts and durations depending on the ratio synthesized, followed by reacting CPT in a similar manner. All conditions have been summarized in table S1. Subsequently, the drug-incorporated particles were purified by size exclusion chromatography via Sephadex G-25 PD-10 desalting columns (5000-MW exclusion limit) followed by overnight dialysis (3500 MWCO) against DI water. The samples were then lyophilized, stored at 4°C, and reconstituted in PBS before use. Concentrations of DOX and CPT were measured using their distinct fluorescence spectra at excitation (Ex)/emission (Em) 470/590 and 370/448 nm, respectively.
Physical characterization
FTIR characterization
To establish the covalent conjugation of DOX and CPT with HA, infrared spectra of DOCTOR were collected using a Nicolet iS10 FTIR spectrometer (Thermo Fisher Scientific, Waltham, MA) in the range of 400 to 4000 cm−1 with a spectral resolution of 4 cm−1. The lyophilized samples were placed on the crystal surface of a single reflection diamond attenuated total reflection (ATR) device, and a 32-scan interferogram was recorded for each of them. The absorbance spectra were processed for baseline, atmospheric, and ATR correction using Thermo Scientific OMNIC Specta software, before analyzing the peaks.
NMR characterization
NMR samples were prepared by dissolving the HA conjugates at a concentration of 10 mg/ml in D2O. HA (50 kDa) and EDC-modified HA (50 kDa) were included as controls. The free DOX control was prepared in D2O and free CPT control in DMSO-d6.
In vitro release studies
To study the release rate of DOX and CPT from single- or dual-drug conjugates in physiological buffer conditions, lyophilized DOCTOR was resuspended in PBS (pH 7.4) at a dose of 1 mg/ml DOX in DOCTOR R15 and incubated in Slide-A-Lyzer MINI dialysis devices (10,000 MWCO). The Slide-A-Lyzer MINI dialysis devices were then inserted into microcentrifuge tubes with 1 ml of PBS for up to 3 days. At the indicated time points, release medium in the microcentrifuge tubes was collected and analyzed by reading the drugs’ concentration via fluorescence using a Tecan plate reader. The cumulative release was calculated by dividing the amount of drug released each day with the total mass initially loaded. All measurements were carried out in triplicate, and the results were indicated as means ± SE.
Size and surface charge measurement
The size of HA-DOX, HA-CPT, and DOCTOR R2, R5, and R15 in physiological buffer conditions (PBS, pH 7.4) was measured using the NanoSight LM10 System (NanoSight, Amesbury, UK) supplemented with a fast video capture and NTA (NTA 2.3) software. The samples were measured at room temperature by capturing videos set at a recording time of 30 s, each with manual shutter and gain adjustments. The images were then processed using the NTA 2.3 software, and size was recorded. Measurements were made in triplicates for each sample following instrument recalibration. Zeta potential of the particles was measured in PBS (pH 7.4) using the Zetasizer Nano ZS (Malvern Instruments, Westborough, MA).
AFM characterization
Sample preparation
HA-DOX, HA-CPT, and DOCTOR were diluted to a concentration of 2 μg/ml using DI water. The dilute solutions were stirred using a tube revolver for 30 min at room temperature, with intermittent vortexing to ensure mixing at the molecular scale. Just before imaging, 4 μl of the solution was dropped on a freshly cleaved mica surface and allowed to dry for 15 min at room temperature. This procedure was used to prepare all samples.
AFM measurement
The structure of the polymer conjugates was studied with AFM using Cypher microscope (Asylum Research, Santa Barbara, CA), operated in tapping mode at ambient conditions. Silicon cantilevers having chromium/gold (Cr/Au) coating, with resonance frequencies between 44 and 95 kHz and spring constant in the range of 0.3 to 4.8 N/m and 9 ± 2–mm uncoated silicon tip (AC240TSA-R3, Asylum Research, Santa Barbara, CA), were used for the dry imaging in air. Depending on the frame size, the scan rate was set in the 0.7- to 1.0-Hz range. The images were processed and analyzed using Gwyddion 2.47 software (n = 5).
SAXS analysis
SAXS experiments help to shed light on the nanoparticles’ architecture and dimensions. A flow-cell setup was used for SAXS measurements at the LiX-16-ID beamline at the National Synchrotron Light Source II of the Brookhaven National Laboratory (Upton, NY). A series of scattering images were recorded each with a 1-s exposure for both polymer and buffer samples. All scattering curves were radially averaged and inspected for possible radiation damage. Final scattering intensity was reported after proper buffer subtraction. The x-ray energy was 13 keV.
Form factors used depend on the sample and solvent and are described below. Because of low concentration of nanoparticles in solution, interparticle interactions are neglected [structure factor S(q) = 1]. The scattered intensity curves were fitted using SASfit software (37).
Polydisperse hard sphere model
The spherical shell form factor has the following form
| (1) |
where R is the radius of sphere and ΔSLD is the difference in scattering length densities between particle and solvent.
To account for nanoparticle polydispersity, a Schulz-Zimm distribution of R with polydispersity parameter σ was included in the following way
| (2) |
where . Because the polydispersity parameter σ and radius of sphere are correlated parameters, the σ value was set to 0.3 for all fitting procedures.
Generalized Gaussian coil model
The form factor of generalized Gaussian coil has the following form
| (3) |
Here, and – Gamma function. The fitting parameters for this model are Rg (gyration radius) and ν (Flory exponent).
Worm-like chain model
The form factor of a worm-like chain with contour length L, Kuhn length A, and diameter d has been described previously (38).
In vitro toxicity analysis
The in vitro toxicity of free drugs, HA-DOX, HA-CPT, and DOCTOR (R2, R5, and R15) was evaluated with the cancerous (A431) keratinocytes. The cells were seeded at a density of 5000 cells/100 μl of medium in a 96-well cell culture plate and allowed to adhere for 18 to 20 hours at 37°C in 5% CO2 atmosphere. Medium was then replaced with fresh medium containing the drugs. For this, lyophilized DOCTOR was dissolved in DI water (pH 5.0) and then serially diluted in cell culture medium to obtain the doses for the toxicity assays. The toxicity assays were carried out in the “log” cellular-growing phase of cells during which the cells were subjected to each treatment at a range of concentrations for 48 hours. Cell viability was measured by CellTiter-Blue Viability Assay and expressed as the percentage of viable cells relative to the survival of untreated cells (defined as the maximum cell viability). The CI was then estimated from the dose-response data of single- and dual-drug conjugate drug treatments. A value of CI less than 1 indicates synergism, CI = 1 indicates additive effect, and CI > 1 indicates antagonism. The further a CI value is from 1, the more pronounced is the drug interaction, i.e., synergism or antagonism.
In a separate study, an in vitro toxicity assay was set up as described above to evaluate the effect of DOCTOR on healthy keratinocytes versus the cancerous keratinocytes. The cells were exposed to DOCTOR at a range of drug concentrations for 18 hours. Subsequently, the treated cells were washed with fresh medium at least two times and left for further incubation up to 48 hours. Cell viability was once again measured by CellTiter-Blue viability assay and was expressed as the percentage of viable cells relative to the survival of untreated cells defined as the maximum cell viability.
Ex vivo porcine skin permeation study
Visualizing dermal delivery
The ability of DOCTOR to deliver DOX and CPT inside the skin was assessed via ex vivo porcine skin permeation studies using protocols reported previously (39). Briefly, full-thickness porcine skin samples (Lampire Biological Laboratories, Pipersville, PA) stored at −80°C were defrosted, and hair was trimmed before use. The pieces were washed with PBS (pH 7.4), and resistivity was measured to ensure that samples with an intact barrier were used for the study. The permeation rates were assessed in Franz diffusion cells (penetration area, 1.77 cm2; receptor volume, 12.0 ml) as described here. The receptor compartment was filled with PBS (pH 7.4), and skin samples were mounted with the stratum corneum (SC) facing up. Care was taken to ensure that the donor compartment was dry and left open to air for 30 min. Extra caution was taken to remove air bubbles between the skin’s base and the receptor solution. Lyophilized DOCTOR was dissolved in DI water (pH 5.0) and then diluted in the same solvent to obtain a dose of 0.5 mg/ml or 920 μM DOX in DOCTOR. This formulation (200 μl) was applied topically on the skin surface inside the donor compartment and incubated for 24 hours at 37°C under occlusive conditions with moderate stirring (200 rpm) in the acceptor compartment. The controls and treatments for the study were assessed in triplicates. At 24 hours, the skin surface was washed at least five times with PBS (pH 7.4) to remove any excess drugs. The skin samples were then cryosectioned for confocal microscopy analysis or subjected to tape-stripping studies for drug quantification purposes.
Measuring dermal delivery
For quantifying skin permeation of the applied DOCTOR, the skin sections were retrieved after incubation for 24 hours and rinsed with PBS. Each section was then subjected to the tape-stripping method as described. For this, an adhesive tape (Scotch Transparent Tape, 3M Corporate, St. Paul, MN) was used to separate the upper stratum corneum from the lower stratum corneum and the epidermis. Ten consecutive tape strips were performed to remove the upper stratum corneum. The lower stratum corneum combined with the epidermis of each skin section was then separated from the dermis with a sterile surgical scalpel and placed in the same glass vial. The dermis obtained from each section was then cut into small pieces and placed in different glass vials. For drug extraction, each separated skin layer was incubated with 3 ml of 50% methanol/PBS mixture. To determine the amount of drug that passed completely through the length of the skin, 3 ml of the acceptor chamber solution was mixed with 3 ml of methanol/PBS. All vials were shaken overnight at room temperature. The solutions were then centrifuged to remove the skin remnants. The supernatants collected were used to measure the concentration of DOX and CPT using their distinct fluorescence spectra at Ex/Em 470/590 and 370/448 nm, respectively.
Cell uptake study
For visualizing DOX (479Ex/590Em) and CPT (370Ex/450Em) uptake with its nuclear colocalization, the cells were seeded at 5000 cells/100 μl and allowed to adhere overnight. Medium was then replaced with fresh medium containing a dose of 0.06 mg/ml or 110.4 μM DOX in DOCTOR R15. The cells were incubated for 3 hours at 37°C and 5% CO2 atmosphere. Following incubation, the cells were washed three times with warm PBS and nucleus was stained with 5 μM solution of SYTO 17 (621Ex/634Em) for 10 min at 37°C and 5% CO2. The cells were washed once again with warm PBS and fixed with ice-cold methanol for 10 min. The fixed cells were imaged immediately under Cell Discover, and z-stacks of 10 μm were captured and averaged.
Testing for inflammation with EpiDerm human skin model
The irritation or inflammation potential of DOCTOR was assessed on a MatTek EpiDerm human skin model (MatTek Corporation) by measuring the release of inflammatory cytokines, including IL-1α and TNF-α. Briefly, EpiDerm tissue inserts were incubated overnight in medium. The tissue inserts were then transferred into new six-well plates with fresh medium. PBS (100 μl; negative control), 5% SDS (positive control), blank HA, and DOCTOR (R2, R5, and R15 at a dose of 0.0027 mg/ml or 5 μM DOX in DOCTOR) were dosed on top of the inserts. At predetermined time points (2, 6, and 12 hours), 200 μl of medium was collected and equivalent volume of medium was added. Cytokine concentrations were measured by enzyme-linked immunosorbent assay (ELISA) according to protocols provided by the manufacturer.
Evaluating tolerability and systemic absorption in vivo
Female SKH1-E hairless mice (4 to 6 weeks of age; five per group) obtained from Charles River Laboratories received topically PBS, Efudex (160 μg/week; 5% fluorouracil), and DOCTOR R2 or R15 at a dose of 0.006 mg/ml or 11 μM DOX (3 μg/week) in DOCTOR after dissolving in DI water (pH 5.0). The CPT doses were 0.0012 mg/ml or 3.5 μM CPT in DOCTOR R2 and 0.000164 mg/ml or 0.47 μM CPT in DOCTOR 15. The treatments were evenly applied with a cotton applicator to the back skin of the mouse five times a week over 2 weeks and allowed to dry following each application. Any change in the body weight of mice in all treatment groups was monitored throughout the study. Animals that exhibit any signs of anaphylaxis or drug toxicity, including but not limited to respiratory distress, pale or cyanotic skin, lack of grooming, sluggishness, ruffled fur and hunched posture, and acute weight loss greater than 10%, were euthanized in accordance with Harvard University’s Institutional Animal Care and Use Committee (IACUC)–approved protocol. At the end of the study, blood was collected via cardiac puncture in heparin-coated collection tubes. Following centrifugation, plasma was obtained and drugs were extracted following a previous procedure to measure drug (DOX and CPT) and cytokine concentrations (40). Plasma levels of DOX and CPT were measured at their respective fluorescence spectra as described earlier. Cytokine concentrations in the plasma were measured using ELISA according to protocols provided by the manufacturer.
Evaluating cSCC therapeutic efficacy of DOCTOR in vivo
In vivo efficacy of DOCTOR was evaluated in SKH1-E hairless mice (12 weeks of age; nine per group) obtained from Charles River Laboratories. Following the protocols described previously, female mice were exposed to UVB (12.5 kJ/m2) weekly (total divided over three doses Monday, Wednesday, and Friday) for 90 days using an Oriel solar simulator (Newport) (41, 42). Once the mice developed single cSCC lesions that measured 4 mm in diameter, they were randomly enrolled to receive blank HA control (100 mg/kg) or DOCTOR R15 (100 mg/kg) at a dose of 0.003 mg/ml or 5.5 μM DOX and 0.000082 mg/ml or 0.235 μM CPT in DOCTOR.
For the treatments, lyophilized DOCTOR was dissolved in DI water (pH 5.0) at the required dose and 100 μl of this solution was applied dropwise all over the dorsal side of the mouse. The drops were then spread evenly using a cotton applicator to the animal’s back skin three times a week and allowed to dry following application. No other vehicle was used to solubilize DOCTOR. Mice were euthanized once the tumor reached its endpoint size (100 mm3) or until completion of the treatment for 3 months. All animal studies were conducted under the guidelines of University of South Florida IACUC-approved protocol (IACUC IS00002374).
Assessing safety and efficacy of DOCTOR with patient-derived skin tissues
The safety and efficacy of DOCTOR was evaluated with samples from normal skin and cSCC lesional skin. Human epidermal keratinocytes were isolated from human skin tissue samples obtained during abdominoplasty procedures at Massachusetts General Hospital, Boston, MA with Institutional Review Board (IRB) approval for discarded human tissue (MGH 2008P001742). The safety and efficacy of DOCTOR was further evaluated with samples from normal skin and cSCC lesional skin taken from patients during tumor resection surgery, in agreement with the guidelines of the IRB at Massachusetts General Hospital, Boston, MA (MGH 2018P003156). Primary cell cultures were isolated as described previously and used in passages 1 to 3 for viability assessment by alamarBlue Cell Viability Reagent (Thermo Fisher Scientific, catalog no. DAL1025) (43). For tissue explant studies, the skin samples were placed dermal side down onto membranes of transwell inserts (Costar) for culturing in growth medium DMEM containing 1% l-arginine, 10% human serum (Gemini Bio Products), and antibiotics/antimycotics. The lesion surface was kept in contact with the air and treated with 0.07 mg/ml or 500 μM 5-FU or a dose of 0.3 mg/ml or 500 μM DOX in DOCTOR R2 for 48 hours. To ensure topical application only on the lesion surface, they were sealed with semisoft agar. For quantification of cellular proliferation, frozen sections were mounted in OCT (optimal cutting temperature) embedding compound and frozen at −80°C and subsequently stained with anti-Ki67 antibody (anti-Ki67, Abcam, ab16667, 1:200). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen), and the slides were mounted in ProLong Gold Antifade Mount (Invitrogen). For precise counting of cells, ImageJ was used as described in the caption for Fig. 5C.
Statistical analysis
All experiments were carried out in triplicates, and results are indicated as means ± SEM unless otherwise indicated. One-way analysis of variance (ANOVA) and two-way ANOVA were performed using GraphPad Prism nonlinear regression software (GraphPad Software Inc.). Results are depicted as averages ± SEM. A P value of <0.05 was considered significant.
Acknowledgments
This research used resources (Beamline 16-ID) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract no. DE-SC0012704. We would like to thank L. Yang (NSLS-II) for help and support with the SAXS experiments. We would also like to acknowledge A. Clocchiatti (MGH) for technical assistance with the image analysis and Y. Gao (Harvard) for suggestions offered during the conceptual stages of this project. Funding: This work was financially supported by the Wyss Institute and School of Engineering and Applied Sciences at Harvard University. K.Y.T. acknowledges that this work was additionally funded by the Donald A. Adam Melanoma and Skin Cancer Center of Excellence at the Moffitt Cancer Center. Author contributions: V.K. and S.M. designed the experiments and wrote and edited the manuscript. V.K., K.P., A.S., S.P., Z.Z., S.K.F., K.T., B.R.S., O.L., S.B., A.P., and D.V. conducted and analyzed the experiments. V.K. and A.S. contributed to FTIR, TEM, and AFM studies and subsequent analyses. V.K., S.P., O.L., S.B., and D.V. contributed to formulation studies and subsequent analyses. V.K. and S.K.F. contributed to SAXS studies and subsequent analyses. V.K., B.R.S., and K.Y.T. contributed to animal studies and subsequent analyses. V.K., K.T., A.M., and S.M. contributed to patient sample studies and subsequent analyses. Competing interests: V.K., D.V., and S.M. are inventors on patent applications that cover some aspects of the polymer drug conjugates described in this manuscript (owned and managed by University of California, Santa Barbara and Harvard University). The remaining authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabe6627/DC1
REFERENCES AND NOTES
- 1.American Academy of Dermatology, “Incidence Rate—Skin Cancer” (American Academy of Dermatology Association, 2021); www.aad.org/media/stats-skin-cancer.
- 2.Apalla Z., Lallas A., Sotiriou E., Lazaridou E., Ioannides D., Epidemiological trends in skin cancer. Dermatol. Pract. Concept. 7, 1–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sari E., Non-metastatic non-melanoma skin cancers: Our 3 years of clinical experiences. World J. Plast Surg. 6, 305–312 (2017). [PMC free article] [PubMed] [Google Scholar]
- 4.Barton M. K., Nicotinamide found to reduce the rate of nonmelanoma skin cancers in high-risk patients. CA Cancer J. Clin. 66, 91–92 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Christensen S. R., Recent advances in field cancerization and management of multiple cutaneous squamous cell carcinomas. F1000Res 7, F1000 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krishnan V., Mitragotri S., Nanoparticles for topical drug delivery: Potential for skin cancer treatment. Adv. Drug Deliv. Rev. 153, 87–108 (2020). [DOI] [PubMed] [Google Scholar]
- 7.Neidecker M. V., Davis-Ajami M. L., Balkrishnan R., Feldman S. R., Pharmacoeconomic considerations in treating actinic keratosis. Pharmacoeconomics 27, 451–464 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Johnson M. R., Hageboutros A., Wang K., High L., Smith J. B., Diasio R. B., Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin. Cancer Res. 5, 2006–2011 (1999). [PubMed] [Google Scholar]
- 9.Loyal J., Romano M., Pierson J. C., Exuberant inflammatory reaction to occlusion of topical 5-fluorouracil (FU) under a continuous positive airway pressure (CPAP) mask: A warning to dermatologists and patients. JAAD Case Rep. 2, 278–280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cohen P. R., Topical application of 5-fluorouracil 5 percent cream associated with severe neutropenia: Discussion of a case and review of systemic reactions after topical treatment with 5-fluorouracil. Dermatol. Online J. 24, 13030 (2018). [PubMed] [Google Scholar]
- 11.Jury C. S., Ramraka-Jones V. S., Gudi V., Herd R. M., A randomized trial of topical 5% 5-fluorouracil (EfudixR cream) in the treatment of actinic keratoses comparing daily with weekly treatment. Br. J. Dermatol. 153, 808–810 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Pavillard V., Kherfellah D., Richard S., Robert J., Montaudon D., Effects of the combination of camptothecin and doxorubicin or etoposide on rat glioma cells and camptothecin-resistant variants. Br. J. Cancer 85, 1077–1083 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krauze M. T., Noble C. O., Kawaguchi T., Drummond D., Kirpotin D. B., Yamashita Y., Kullberg E., Forsayeth J., Park J. W., Bankiewicz K. S., Convection-enhanced delivery of nanoliposomal CPT-11 (irinotecan) and PEGylated liposomal doxorubicin (Doxil) in rodent intracranial brain tumor xenografts. Neuro Oncol. 9, 393–403 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li J., Hu Z. E., Yang X. L., Wu W. X., Xing X., Gu B., Liu Y. H., Wang N., Yu X. Q., GSH/pH dual-responsive biodegradable camptothecin polymeric prodrugs combined with doxorubicin for synergistic anticancer efficiency. Biomater. Sci. 7, 3277–3286 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Husain I., Mohler J. L., Seigler H. F., Besterman J. M., Elevation of topoisomerase I messenger RNA, protein, and catalytic activity in human tumors: Demonstration of tumor-type specificity and implications for cancer chemotherapy. Cancer Res. 54, 539–546 (1994). [PubMed] [Google Scholar]
- 16.Masin J. S., Berger S. J., Setrakian S., Stepnick D. W., Berger N. A., Topoisomerase I activity in squamous cell carcinoma of the head and neck. Laryngoscope 105, 1191–1196 (1995). [DOI] [PubMed] [Google Scholar]
- 17.Bronstein I. B., Vorobyev S., Timofeev A., Jolles C. J., Alder S. L., Holden J. A., Elevations of DNA topoisomerase I catalytic activity and immunoprotein in human malignancies. Oncol. Res. 8, 17–25 (1996). [PubMed] [Google Scholar]
- 18.Scott D. O., Bindra D. S., Stella V. J., Plasma pharmacokinetics of lactone and carboxylate forms of 20(S)-camptothecin in anesthetized rats. Pharm. Res. 10, 1451–1457 (1993). [DOI] [PubMed] [Google Scholar]
- 19.Gabr A., Kuin A., Aalders M., El-Gawly H., Smets L. A., Cellular pharmacokinetics and cytotoxicity of camptothecin and topotecan at normal and acidic pH. Cancer Res. 57, 4811–4816 (1997). [PubMed] [Google Scholar]
- 20.Shi Y., van Steenbergen M. J., Teunissen E. A., Novo L., Gradmann S., Baldus M., van Nostrum C. F., Hennink W. E., Π-π stacking increases the stability and loading capacity of thermosensitive polymeric micelles for chemotherapeutic drugs. Biomacromolecules 14, 1826–1837 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Oommen O. P., Garousi J., Sloff M., Varghese O. P., Tailored doxorubicin-hyaluronan conjugate as a potent anticancer glyco-drug: An alternative to prodrug approach. Macromol. Biosci. 14, 327–333 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Vogus D. R., Evans M. A., Pusuluri A., Barajas A., Zhang M., Krishnan V., Nowak M., Menegatti S., Helgeson M. E., Squires T. M., Mitragotri S., A hyaluronic acid conjugate engineered to synergistically and sequentially deliver gemcitabine and doxorubicin to treat triple negative breast cancer. J. Control. Release 267, 191–202 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Liang Y., Sun Y., Fu X., Lin Y., Meng Z., Meng Y., Niu J., Lai Y., Sun Y., The effect of π-conjugation on the self-assembly of micelles and controlled cargo release. Artif. Cells Nanomed. Biotechnol. 48, 525–532 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Vasi A. M., Popa M. I., Butnaru M., Dodi G., Verestiuc L., Chemical functionalization of hyaluronic acid for drug delivery applications. Korean J. Couns. Psychother. 38, 177–185 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Liu R., Zhang J., Zhao C., Duan X., McClements D. J., Liu X., Liu F., Formation and characterization of lactoferrin-hyaluronic acid conjugates and their effects on the storage stability of sesamol emulsions. Molecules 23, 3291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Organic Chemistry Portal (Stream Chemicals), vol. 2020; https://www.organic-chemistry.org/namedreactions/steglich-esterification.shtm.
- 27.Neises B., Steglich W., Simple method for the esterification of carboxylic acids. Angew. Chem. Int. Ed. Engl. 17, 522–524 (1978). [Google Scholar]
- 28.Groeber F., Schober L., Schmid F. F., Traube A., Kolbus-Hernandez S., Daton K., Hoffmann S., Petersohn D., Schafer-Korting M., Walles H., Mewes K. R., Catch-up validation study of an in vitro skin irritation test method based on an open source reconstructed epidermis (phase II). Toxicol. In Vitro 36, 254–261 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Pommergaard H. C., Burcharth J., Rosenberg J., Raskov H., Topical combination of diclofenac, calcipotriol, and difluoromethylornithine has beneficial effects comparable to 5-fluorouracil for the treatment of non-melanoma skin cancer in mice. J. Chemother. 26, 105–110 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Cai S., Thati S., Bagby T. R., Diab H. M., Davies N. M., Cohen M. S., Forrest M. L., Localized doxorubicin chemotherapy with a biopolymeric nanocarrier improves survival and reduces toxicity in xenografts of human breast cancer. J. Control. Release 146, 212–218 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Corsini E., Bruccoleri A., Marinovich M., Galli C. L., Endogenous interleukin-1 alpha associated with skin irritation induced by tributyltin. Toxicol. Appl. Pharmacol. 138, 268–274 (1996). [DOI] [PubMed] [Google Scholar]
- 32.Bou-Dargham M. J., Khamis Z. I., Cognetta A. B., Sang Q. A., The role of interleukin-1 in inflammatory and malignant human skin diseases and the rationale for targeting interleukin-1 alpha. Med. Res. Rev. 37, 180–216 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Journo-Gershfeld G., Kapp D., Shamay Y., Kopecek J., David A., Hyaluronan oligomers-HPMA copolymer conjugates for targeting paclitaxel to CD44-overexpressing ovarian carcinoma. Pharm. Res. 29, 1121–1133 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Platt V. M., Szoka F. C. Jr., Anticancer therapeutics: Targeting macromolecules and nanocarriers to hyaluronan or CD44, a hyaluronan receptor. Mol. Pharm. 5, 474–486 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grzelczak M., Vermant J., Furst E. M., Liz-Marzan L. M., Directed self-assembly of nanoparticles. ACS Nano 4, 3591–3605 (2010). [DOI] [PubMed] [Google Scholar]
- 36.Blahova M., Filippov S. K., Kovacik L., Horsky J., Hybelbauerova S., Syrova Z., Krizek T., Jindrich J., Synthesis and supramolecular properties of regioisomers of mononaphthylallyl derivatives of γ-cyclodextrin. Beilstein J. Org. Chem. 13, 2509–2520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bressler I., Kohlbrecher J., Thunemann A. F., SASfit: A tool for small-angle scattering data analysis using a library of analytical expressions. J. Appl. Cryst. 48, 1587–1598 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pedersen I. S., Schurtenberger P., Scattering functions of semiflexible polymers with and without excluded volume effects. Macromolecules 29, 7602–7612 (1996). [Google Scholar]
- 39.Banerjee A., Ibsen K., Iwao Y., Zakrewsky M., Mitragotri S., Transdermal protein delivery using choline and geranate (CAGE) deep eutectic solvent. Adv. Healthc. Mater. 6, (2017). [DOI] [PubMed] [Google Scholar]
- 40.Pusuluri A., Krishnan V., Lensch V., Sarode A., Bunyan E., Vogus D. R., Menegatti S., Soh H. T., Mitragotri S., Treating tumors at low drug doses using an aptamer-peptide synergistic drug conjugate. Angew. Chem. Int. Ed. Engl. 58, 1437–1441 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Vin H., Ojeda S. S., Ching G., Leung M. L., Chitsazzadeh V., Dwyer D. W., Adelmann C. H., Restrepo M., Richards K. N., Stewart L. R., Du L., Ferguson S. B., Chakravarti D., Ehrenreiter K., Baccarini M., Ruggieri R., Curry J. L., Kim K. B., Ciurea A. M., Duvic M., Prieto V. G., Ullrich S. E., Dalby K. N., Flores E. R., Tsai K. Y., BRAF inhibitors suppress apoptosis through off-target inhibition of JNK signaling. eLife 2, e00969 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chitsazzadeh V., Coarfa C., Drummond J. A., Nguyen T., Joseph A., Chilukuri S., Charpiot E., Adelmann C. H., Ching G., Nguyen T. N., Nicholas C., Thomas V. D., Migden M., MacFarlane D., Thompson E., Shen J., Takata Y., McNiece K., Polansky M. A., Abbas H. A., Rajapakshe K., Gower A., Spira A., Covington K. R., Xiao W., Gunaratne P., Pickering C., Frederick M., Myers J. N., Shen L., Yao H., Su X., Rapini R. P., Wheeler D. A., Hawk E. T., Flores E. R., Tsai K. Y., Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 7, 12601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neel V. A., Todorova K., Wang J., Kwon E., Kang M., Liu Q., Gray N., Lee S. W., Mandinova A., Sustained Akt activity is required to maintain cell viability in seborrheic keratosis, a benign epithelial tumor. J. Invest. Dermatol. 136, 696–705 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabe6627/DC1