

P. aeruginosa flagella are recorded across the entire biofilm life cycle as likely biofilm structural scaffolds.
Abstract
The opportunistic pathogen, Pseudomonas aeruginosa, a flagellated bacterium, is one of the top model organisms for biofilm studies. To elucidate the location of bacterial flagella throughout the biofilm life cycle, we developed a new flagella biotracking tool. Bacterial flagella were site-specifically labeled via genetic code expansion. This enabled us to track bacterial flagella during biofilm maturation. Live flagella imaging revealed the presence and synthesis of flagella throughout the biofilm life cycle. To study the possible role of flagella in a biofilm, we produced a flagella knockout strain and compared its biofilm to that of the wild-type strain. Results showed a one order of magnitude stronger biofilm structure in the wild type in comparison with the flagella knockout strain. This suggests a possible structural role for flagella in a biofilm, conceivably as a scaffold. Our findings suggest a new model for biofilm maturation dynamic which underscores the importance of direct evidence from within the biofilm.
INTRODUCTION
Pseudomonas aeruginosa is a well-studied opportunistic pathogen (1). Despite the fact that quorum sensing and biofilm formation have been studied for the past 40 years, previously unknown information is constantly being revealed (2, 3). Biofilms provide a more tolerant form of existence for bacteria than their planktonic forms, proffering them with protection from possible stressors. They, therefore, have been intensely studied for their complexity and the mechanisms involved in their life cycle (4, 5). Any previously unreported information emerging from these studies is crucial, allowing the development of new and diverse strategies to resist infections.
The biofilm life cycle is composed of several commonly reported steps (1, 6–8). Initially, planktonic bacteria propel themselves to a proximal surface, followed by an irreversible attachment to the surface. Once attachment is established, exopolymeric substances are secreted from within the cells to generate a matrix of a supporting microenvironment for the dividing cells and to initiate formation of microcolonies. Next, mushroom-like structures start to emerge. Last, the cells secrete enzymes to digest the exopolymeric substances at the center of the grown mushroom-like structures where the newly flagellated cells are released in a planktonic form to attach to new exposed surfaces.
Flagella, the bacterial rotor with its unique structure, are therefore an inseparable part of biofilm research (9). Several reports have expressed a consensus regarding the importance of flagella in biofilm formation, specifically in its initial formation (10). However, reports regarding the presence of flagella during biofilm maturation are still somewhat contradictory. While some suggest lack of flagella during biofilm maturation right until the start of the detachment process, it was mainly concluded through observations of down-regulation of flagella-related genes in bulk populations or through light transmission microscopy or scanning electron microscopy (SEM) (5, 7, 8, 11–15). On the other hand, others suggest continued flagella synthesis during biofilm maturation (16–19). Therefore, we have found it necessary to assess the presence and possible role of flagella in biofilms. Direct imaging of flagella inside a biofilm, which was not achieved before, may thus serve as a tool to shed a better light on their role in biofilm formation and maintenance and provide an improved understanding of what occurs during the biofilm life cycle.
To date, live-cell imaging can be obtained through different approaches; however, genetic code expansion, the reassignment of codons and incorporation of unnatural amino acids (Uaas) into proteins (20), displays advantages over other methodologies and gains increased exposure and momentum (21–24). Genetic code expansion systems are being constantly improved, expanded, and adapted to a growing number of organisms (25–29). Genetic code expansion uses an orthogonal translation system (OTS) for the incorporation of Uaas. The OTS, composed of an orthogonal transfer RNA (tRNA) and orthogonal tRNA synthetase, has no cross-reactivity with the target organism’s translation system. There are several different OTSs that are used in different organisms, and each OTS has compatibility to specific Uaa family (30). Uaa incorporation into a protein occurs using a reassigned codon, most commonly a stop codon. Suppression of the stop codon with an orthogonal tRNA anticodon results in the site-specific incorporation of a Uaa into a protein.
Genetic code expansion presents advantages when used for imaging purposes, for example, the incorporation of a Uaa may alleviate the need for large and bulky labeling agents, such as fluorescent proteins or antibodies. Moreover, a protein with a site-specifically incorporated Uaa can be labeled using bioorthogonal chemistry and serve as a specific reporter inside cells. Incorporating Uaa into P. aeruginosa flagellum enables live-cell imaging of flagella inside the complex environment of a biofilm. Despite the fact that P. aeruginosa’s flagella imaging using genetic code expansion was recently achieved by Zheng et al. (31), the used OTS was yet to be proven orthogonal, and the methodology was not demonstrated inside a growing biofilm. Here, we provide a detailed account of the regulatory components and sequences needed for genetic code expansion in P. aeruginosa. We have designed a genetic code expansion system for flagella labeling and biotracking in a live and growing biofilm that revealed novel information regarding the biofilm life cycle.
RESULTS
Genetic code expansion of P. aeruginosa
Aiming for optimized Uaa incorporation into P. aeruginosa proteins, a new plasmid was constructed (Fig. 1A), resulting in a plasmid harboring an OTS, as well as a reporter gene [green fluorescent protein (gfp)] for system validation. An OTS, composed of a tRNA and tRNA synthetase pair, could be considered orthogonal if it does not interact with native translational components (30). Methanosarcina mazei pyrrolysyl OTS (MmPyl OTS) (32) has been previously found to be orthogonal in several organisms, including Gram-negative bacteria, and was therefore our choice. Combined with MmPyl OTS, a gfp reporter gene encoded for Uaa incorporation.
Fig. 1. Genetic code expansion system of P. aeruginosa using GFP.
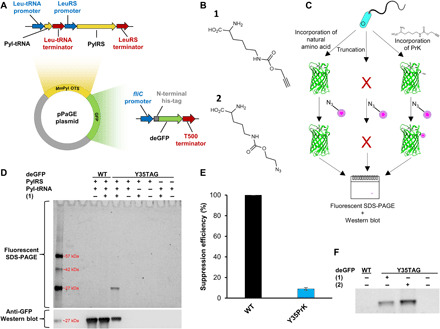
(A) Plasmid map of pPaGE. (B) Uaas used in this study, propargyl-l-lysine (PrK) (1) and azido-carboxy-lysine (AzCK) (2). (C) Schematic representation of possible translation outcomes for mid-gene TAG mutants. (D) Lysed cells following a click reaction to tetramethylrhodamine (TAMRA)–azide were analyzed through fluorescent SDS–polyacrylamide gel electrophoresis (PAGE) and anti-GFP Western blot analyses. (E) Calculated suppression efficiency of Y35PrK GFP mutant. Error bar represents SD between five different calculated suppression efficiencies. Each suppression efficiency was calculated from a separate set of wild-type (WT) GFP, Y35PrK GFP, and native PAO1. (F) Fluorescent gel of lysed samples followed by a click reaction between either TAMRA azide to (1) or TAMRA alkyne to (2) analyzed through fluorescent SDS-PAGE (full-length gel is available as fig. S3C).
The P. aeruginosa genetic code expansion plasmid (pPaGE) was assembled using P. aeruginosa endogenous promoters and terminators, to maintain physiologically relevant expression levels of the exogenous OTS. We thus identified the most abundant codon in P. aeruginosa PAO1 genome (33), which was found to be the CUG codon encoding for leucine (Leu). The native promoters and terminators of Leu-tRNA and leucyl-tRNA synthetase were assigned as the upstream and downstream regions of pyrrolysyl tRNA (Pyl-tRNA) and Pyl-tRNA synthetase (PylRS), respectively. The fliC endogenous promoter for flagellin expression was chosen to serve as a promoter for the GFP reporter in the pPaGE plasmid. Toxicity tests for the Uaas, propargyl-l-lysine (PrK) and azido-carboxy-lysine (AzCK) (Fig. 1B, 1 and 2, respectively), were performed and did not hamper bacterial growth rates in a notable manner (fig. S1).
We reassigned the TAG stop codon for site-specific incorporation of Uaas and introduced a TAG mutation to the gfp reporter gene at its 35th position. MmPyl OTS orthogonality in P. aeruginosa was tested next. Several protein elongation scenarios for a premature TAG introduction to GFP were considered and tested, as well as the possibility of nonspecific incorporation of a Uaa into the host organism proteome (Fig. 1, C and D). For that purpose, pPaGE variants containing partial OTS were generated by the removal of Pyl-tRNA or PylRS. Cells were grown, lysed, and analyzed using fluorescent chemical conjugation and anti-GFP Western blot analyses (Fig. 1D). To verify the incorporation of PrK into GFP only, a click reaction using Cu(I)-catalyzed azide-alkyne cycloaddition (CuAAC) (34) to an azide-bearing fluorophore tetramethylrhodamine (TAMRA) azide was conducted. Since there are no endogenous alkynes in bacteria, this experiment was meant to reveal whether there exists any PrK that was misincorporated in response to the TAG stop codon in the bacterial proteome. Another possibility that needed to be ruled out was whether PylRS can aminoacylate endogenous tRNAs, where PrK would have been incorporated into random locations in the genome. Both scenarios, whether to occur, will result in nonspecific fluorescent labeling of endogenous proteins. On the other hand, if Pyl-tRNA is not orthogonal and is aminoacylated by the host organism’s tRNA synthetases by natural amino acids, then full-length GFP may still be synthesized and observed in the Western blot.
When examining all protein expression scenarios, as can be seen in Fig. 1D, we did not observe fluorescent labeling or GFP expression in the presence of the partial OTS variants. This directly proved MmPyl OTS’s orthogonality. Following the establishment that our system is indeed orthogonal, we were then able to analyze proper Uaa incorporation into GFP. Wild-type (WT) GFP expression was observed either in the presence or absence of PrK and could be seen only in the Western blot analysis and not in the fluorescent gel. This indicated that there was no incorporation of PrK into WT GFP. When OTS was present along with PrK, a fully elongated GFP was detected in the Y35TAG Western blot analysis. In addition, a clear fluorescent labeling corresponding to GFP in size was visible. This indicated that the reporter gene was able to not only synthesized but also be verified the presence of PrK inside the protein. When those cells were grown in the absence of PrK, no GFP expression was observed, signifying that the expressed protein was not a result of a read-through event. Final validation was performed using electrospray mass spectrometry (fig. S2), where the deconvoluted mass corresponded to GFP molecular weight with PrK at position 35, replacing a tyrosine. Thus, it was concluded that Uaa was successfully incorporated and encoded for, in a protein expressed in P. aeruginosa.
Suppression efficiency of Uaa incorporation into GFP was calculated to be only below 10% (Fig. 1E). However, it is important to note the context effects (35) at play in this methodology that may alter incorporation yields and suppression efficiency. For example, using the fliC promoter may impair incorporation into GFP but could be found beneficial for Uaa incorporation into a flagellin. To further establish the generated system following genetic code expansion with PrK, AzCK was also incorporated into GFP; moreover, AzCK has the ability to undergo a Cu-free click reaction (34), which makes it useful for future in vivo applications. AzCK was detected, when clicked to an alkyne-containing fluorophore (Fig. 1F). Thus, genetic code expansion was successfully established in P. aeruginosa using two different Uaas. A Cu-free click reaction was also attempted for Y35AzCK GFP (fig. S3). Using Cyanine5–dibenzocyclooctyne (DBCO) fluorophore, a fluorescent band was detected only for a TAG mutant in the presence of AzCK. However, nonspecific labeling was also observed in P. aeruginosa proteome and was detected both by SDS–polyacrylamide gel electrophoresis (PAGE) and by fluorescent microscopy. Because of this limitation, together with the fact that P. aeruginosa demonstrated resilience to the Cu(I) levels used in the Cu(I)-catalyzed click reaction, Cu(I)-catalyzed click reaction was chosen for further study.
Flagella “biotracking” in a biofilm
Flagella labeling was demonstrated before mainly using specific antibodies (36) or through cysteine knock-ins (37). However, antibodies are large proteins that may be difficult to transport across membranes and, in crowded environments, are less specific (38). Maleimide reactions lack selectivity and often result in nonspecific labeling. Fluorescent labeling using a Uaa uses a small molecule with the ability for biorthogonal conjugation, rendering labeling extremely specific and selective. To generate a flagella biotracking tool, we incorporated a Uaa into the endogenous flagellin gene. The flagellum is a complex machinery composed of several genes and proteins that generate filaments extending up to 10 μm in length in P. aeruginosa (39–41). Although assembled by a consortium of proteins, the flagellin protein encoded by the fliC gene is the repeating monomer subunit giving the flagellum its high aspect ratio. We decided to introduce modifications mainly in the D3 domain of flagellin, facing outward in the cylindrical structure of flagellum, thus minimizing possible interference with filament assembly (40). We have modulated the P. aeruginosa flagellum that is composed of 41 subunits [based on Protein Data Bank (PDB) accession number 5WK5] with PrK incorporated at flagellin’s 248th position instead of threonine (Fig. 2A).
Fig. 2. Uaa incorporation into flagellin and inoculated flagella survival in a biofilm.
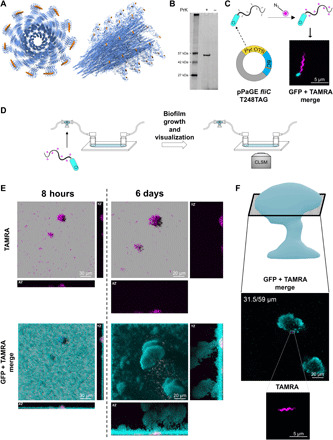
(A) Theoretical model of P. aeruginosa flagella filament with incorporated PrK at the D3 domain (based on PDB accession number 5WK5). Left: A top view. Right: A side view. Uaa is an orange sphere. (B) Fluorescent SDS-PAGE examining PrK incorporation into flagellin protein. Samples were lysed, and insoluble proteins fraction underwent CuAAC click reaction to an azide-containing fluorescent dye (TAMRA azide). (C) Predicted system functionality and resulting fluorescent imaging using confocal laser scanning microscopy (CLSM). PAO1 encoding genomic GFP (gGFP) reporter (cyan) were transformed with mutant flagella plasmid incorporating PrK into exogenous flagellin, following flagella fluorescent labeling using click reaction with TAMRA azide (magenta). Planktonic bacteria were imaged with incorporated PrK in the flagellum. (D) Schematic representation of the flow cell and experimental setup for biotracking of inoculated flagella throughout biofilm maturation. Flagella with a Uaa were fluorescently labeled (click reaction with TAMRA azide) before bacterial injection into the flow cell. Continuous biofilm growth and the TAMRA azide–labeled inoculated flagella were monitored for up to 6 days using CLSM. (E) Flagella labeled before flow cell injection, present since initial biofilm establishment, were monitored inside biofilm’s three-dimensional (3D) structure over time using CLSM (image planes are indicted in the figure). (F) Flagella labeled before flow cell injection located in a mature biofilm at mid-height after 6 days of growth.
We reasoned that replacing the native flagellin copy in the genome of P. aeruginosa with an incorporated Uaa may result in a short flagellum, leading to artifacts, as Uaa incorporation is a slower translational process than native amino acid incorporation (42). Hence, we chose an integrative approach: While encoding for a Uaa incorporation into the fliC gene, we still retained native flagellin in the genome for a hybrid assembly into a single unified flagellum. The integrated flagellum was thus predicted to have fewer chemical anchor points for biolabeling. Despite not having every monomer subunit carry a Uaa, the microfiber was still labeled. Using the fliC promoter, the downstream sequence from the inserted fliC gene was also chosen as the native flagellin terminator; hence, the sequence was completely identical to the native genomic sequence apart from the premature TAG stop codon mutation (Fig. 2C). The new plasmid was assembled and served for flagella genetic code expansion from that point onward.
pPaGE fliC was transformed into P. aeruginosa and resulted in PrK incorporation into flagellin monomers (Fig. 2B). It was also established that exogenous flagellin expression with PrK maintained a singular flagellum synthesis per individual cell and did not impair flagella functionality (fig. S4). Next, the system was used for live-cell imaging using a click reaction. pPaGE fliC plasmid was inserted into P. aeruginosa PAO1 genomic GFP (gGFP) strain, carrying a gGFP reporter gene for the convenience of whole-cell detection. Using a confocal laser scanning microscope (CLSM), successfully labeled flagella were observed including its unique wave-like morphology features indicating a repetitive occurrence of the incorporated label (Fig. 2C, magenta). No signal was observed when expression was attempted in the absence of PrK (fig. S5A).
With this labeling tool, it was now possible to “biotrack” flagella inside a biofilm. We wanted to test whether flagella-initiating biofilm formation and surface attachment are maintained in the biofilm or are degraded as the biofilm matures since motility is not needed any longer. We have thus labeled PrK containing planktonic flagella, before biofilm inoculation, followed by its tracking inside the developing biofilm over time. With the following experimental setup (Fig. 2D), P. aeruginosa expressing gGFP, harboring the pPaGE fliC plasmid, was grown in the presence of PrK. These planktonic bacteria incorporated PrK into flagellin and were fluorescently labeled by a click reaction, followed by growth in a flow cell. Biofilm was grown for up to 6 days (in the absence of PrK), while prelabeled, inoculated flagella were monitored inside the biofilm every couple of hours. In this scenario, any signal was assigned to labeled flagella that were synthesized before biofilm inoculation. Labeled flagella, with their distinct morphology, were clearly observed after initial surface attachment and growth in the flow cell either as singles or as clusters of different sizes (Fig. 2E). This is the first report demonstrating live imaging of the emergence of flagella clusters in a biofilm.
Flagella biotracking for up to 6 days of biofilm growth enabled us to provide evidence regarding changes in the state of inoculated flagella in a biofilm. Computational semiquantification of labeled flagella biovolume allowed comparison between the signal in a mature biofilm and the signal in a young biofilm. The result was a signal decrease from 100 to 97.24%, with an SD of 18.78%. On the basis of these calculated values, some flagella signal has diminished, but its majority remained intact. It is important to note the limitations of this method, apart from observed movement of some flagella in/out of an examined field; the fluorescent signal weakens and bleaches in time as the same volume was exposed to excitation for a long time. We believe that these circumstances are the reason for the large SD. We could demonstrate in a direct manner that despite continued biofilm growth to heights of above 59 μm, most inoculated flagella remained approximately at the bottom of the biofilm (up to 13 μm; Fig. 2E). That means that most inoculated flagella are not metabolized or recycled within the time frame of the experiment.
While most inoculated flagella seemed to remain at the bottom of the biofilm, rarely, we observed an inoculated flagellum in higher sections of a growing biofilm, at around 31.5 μm (Fig. 2F). Because of imaging limitations, it is hard to determine whether the observed flagella are still attached to a cell. Individual flagella that are observed in the biofilm’s mid-height were possibly positioned there because of a response to bacterial signaling or by detachment, followed by reattachment elsewhere. Hence, this has led us to believe that most of the observed flagella are indeed attached to bacterial cells.
Newly synthesized flagella in the biofilm life cycle
After locating labeled inoculated flagella in the biofilm, we wanted to ascertain whether we could track newly synthesized flagella in the biofilm and, if so, when and where will they appear. The biofilm life cycle is divided into several stages where planktonic cells transform and grow together into mushroom-like structures. Throughout the biofilm’s growth, flagella synthesis is mainly believed to be reinitiated during the final step of dispersion from the mature biofilm (5, 7, 8, 13–15). We therefore set out to localize newly synthesized flagella at different biofilm growth stages. This time, the media supply into the flow cell contained PrK throughout the entire growth for continued Uaa incorporation inside the growing biofilm for 2, 4, or 6 days. During the different time points, a secondary flagella labeling was performed inside the flow cell chamber using the same fluorophore as in the first labeling step (Fig. 3A).
Fig. 3. Newly synthesized flagella during biofilm life cycle and maturation.
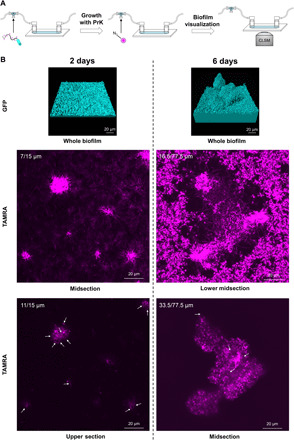
(A) Schematic representation of the experimental setup identifying newly synthesized flagella within different stages of biofilm growth. Flagella with incorporated PrK were fluorescently labeled with TAMRA azide before injection and used as bacterial inoculation for continuous biofilm growth for up to 6 days. Flow cell medium was supplemented with PrK. A secondary click reaction to TAMRA azide was performed inside a flow cell chamber for visualization of newly synthesized flagella. Secondary labeling was performed at different biofilm’s growth time points of 2 and 6 days and visualized using CLSM. Bacteria expressing GFP were labeled in cyan, and TAMRA azide–conjugated flagella were labeled in magenta. (B) CLSM imaging from within the biofilm at different growth stages. For each time point during biofilm life cycle, biofilm was imaged (bacteria’s GFP in biofilms presented as biovolume using Imaris software), and TAMRA azide–conjugated flagella were documented in magenta at different heights of the biofilm (cross sections from within the biofilm). White arrows indicate harder-to-detect flagella.
Large quantities of fluorophore were trapped inside the viscous exopolymeric matrix of the biofilm (6) and appeared as a bulk cloud-like signal. The cloud-like fluorescent signal also appeared when a biofilm that was grown in the absence of PrK was labeled; however, flagella morphology was not apparent (fig. S5B). Possible explanations are either by trapped free fluorophore or, in the case of growth with PrK, by a trapped PrK to which the fluorophore conjugated. However, for that reason, conclusions were made on the basis of clear and abundant flagella morphology only. A mature 6-day-old biofilm had abundant and noticeable flagella at mid-height (33.5 μm) of the 77.5-μm maximal height (Fig. 3B). Flagella did not appear at the top of the mature biofilm, while many flagella were observed at the lower and middle sections of the grown mushroom-like structure. Aiming to identify the stage at which newly synthesized flagella are being made, younger biofilm forms were also examined at the same manner. A 4-day-old biofilm had flagella at the base of the biofilm and at the height of approximately 22 μm of 30.5 μm in total. In a younger biofilm (2 days old), flagella were also located throughout the entire biofilm up to the height of 15 μm on average. These results not only confirmed our prediction of being able to detect flagella in the biofilm through its unique shape but also shed light regarding the constant flagella synthesis and localization inside a growing biofilm.
Cells may detach from a mature biofilm by several ways: through erosion, bulk detachment, or by planktonic release (8). To ascertain the possible changes occurring in detaching cells, the flow cells’ effluent was collected before whole biofilm labeling and was labeled using a second different fluorophore to distinguish between inoculated flagella and newly synthesized flagella (Fig. 4A). Biomass was collected into chilled tubes immediately upon their exit from the flow cells. Thus, we could assume that any flagellum that was observed was synthesized inside the biofilm. Looking at the double-labeled effluent of a young biofilm, new flagella were clear and abundant. This meant that cells are frequently flagellated before leaving the biofilm even in a young biofilm (Fig. 4B).
Fig. 4. Flagellated cells are continually dispersed in the biofilm life cycle.
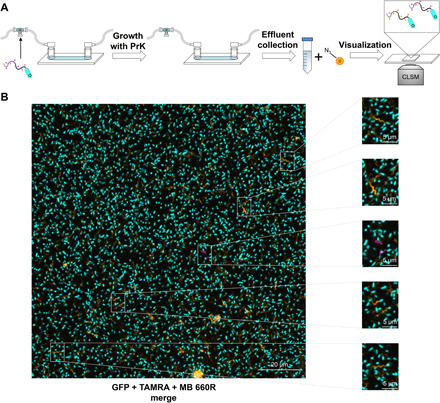
(A) Schematic representation of the experimental setup meant to identify flagellated dispersed cells from the biofilm. Flagella with incorporated PrK were fluorescently labeled with TAMRA azide before injection (magenta) and used as bacterial inoculation for continuous biofilm growth in the presence of PrK for up to 6 days. Effluent was collected at different time points, labeled with a second click reaction to a different fluorophore MB 660R azide (orange), and visualized using CLSM. (B) CLSM imaging of dispersed flagellated cells from a 2-day-old biofilm. Bacteria’s GFP was labeled in cyan, inoculated TAMRA azide–conjugated flagella were labeled in magenta, and new MB 660R–conjugated flagella were labeled in orange.
Some rare cases of labeled inoculated flagella used for biofilm initiation were observed as well (Fig. 4B). Although these observations were rare, indicating that this may be a statistical error if analyzed quantitatively, the image was clear and reproducible. As mentioned previously, it is difficult to detect whether the flagella that were observed in the biofilm were connected to cells. Despite this, we showed that labeled inoculated flagella were still attached to cells. This served as further support for our initial hypothesis, suggesting that most of flagella were indeed still connected to a cell with some level of movement inside the biofilm.
All the information gathered in this work enabled us to portray an updated model for P. aeruginosa biofilm life cycle (Fig. 5). Flagella used for biofilm’s inoculation were maintained throughout the biofilm life cycle at lower levels and displayed a level of viability or deliberate movement. Flagella synthesis was present in the entire life cycle, and newly synthesized flagella were located until midlevel of grown biofilm structures. Moreover, flagellated cells were continuously leaving the biofilm.
Fig. 5. Updated biofilm life cycle model based on the presented direct imaging discoveries.
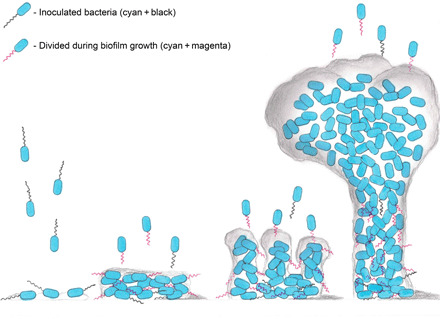
Previously unreported discoveries include inoculated flagella presence throughout the entire life cycle (flagella in black), as well as some level of movement to higher layers in a mature biofilm. Moreover, newly synthesized flagella are present throughout all the different stages of the biofilm life cycle (flagella in magenta). All observed flagella were located at a maximum height of no more than the midlevel of a mature mushroom-like structure. Last, flagellated cells were spotted leaving the biofilm in a continuous manner.
The possible structural importance of flagella in a biofilm
Since we could learn that flagella are being continuously synthesized within a growing biofilm, we asked ourselves whether they might play a role in a biofilm mechanical structure. Considering that flagella synthesis is energetically costly (43) and that cells within the biofilm are mainly nonmotile, there must be an explanation as to why the flagella are formed in cells inside the biofilm. We observed a visible grid-like appearance of the flagella in the lower part of the biofilm (Fig. 6A). In addition, we observed several bacteria assembling on a single flagellum, as shown in Fig. 6B. These observations seemed to be of importance as the structure resembled a structural scaffold that supports a substantial architecture. The idea of the role of flagella as a mechanical support in a biofilm has been previously suggested in Escherichia coli macrocolony biofilms but was yet to be demonstrated (44). An additional observation to support the possible spatial contribution of flagella is the observation of Geobacter sulfurreducens electrochemical chamber biofilm, using their flagella for cytochromes’ spatial arrangement (45). To investigate this possibility, we constructed a P. aeruginosa–flagella knockout (KO) strain.
Fig. 6. Flagella as possible physical support in a biofilm microenvironment.
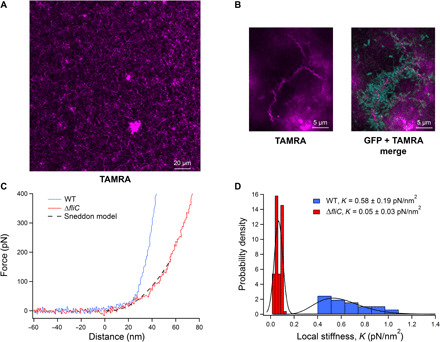
(A) CLSM imaging of a cross section located in the lower third of a 4-day-old biofilm with numerous TAMRA azide–labeled flagella after a click reaction (magenta) arranged in a grid-like structure. (B) CLSM imaging of bacterial assembly on flagella in a growing biofilm. gGFP bacteria appear attached to TAMRA azide–labeled flagella. (C) Nanoindentation force-distance curves of the WT biofilm (blue) and a ΔfliC biofilm (red). The ΔfliC curve is fitted with the Sneddon model (dashed black line), from which K, the local stiffness modulus, was estimated. (D) Local stiffness probability density distributions of WT biofilm (blue) and ΔfliC biofilm (red). The distributions were fitted by relevant statistical models (black line), from which their means and SDs were calculated.
Using CRISPR-Cas9 for genome editing based on Streptococcus pyogenes Cas9, which was adapted for P. aeruginosa genome editing (46), we generated a substrain lacking the ability to produce flagella. This was carried out by deleting the flagellin gene (ΔfliC) from the P. aeruginosa genome. Transmission electron microscope (TEM) imaging and the loss of its swarming abilities validated that the new strain does not express flagella (fig. S6). When pPaGE fliC T248TAG was introduced into ΔfliC but grown in the absence of Uaa, no swarming was observed; however, in the presence of PrK, a rescue effect was identified by regaining swarming activity albeit less robust than that of WT (fig. S6). Since Uaa incorporation during protein translation is much slower than natural amino acid incorporation (42), it is reasonable that the rescued swarming demonstrated lower functionality than WT swarming.
Next, we have attempted to differentiate between WT- and ΔfliC-based biofilms. We hypothesized that mushroom-like structures will be affected by the absence of flagella. We could not detect any visible morphological/architectural differences between the two biofilms using confocal microscopy (fig. S7). However, high-resolution SEM (HR-SEM) revealed noticeable visual difference between the two biofilms (fig. S8). In the WT biofilm, we could notice a web of fibers between the cells, while the ΔfliC biofilm cells were much more crowded with notably less fibers. Since multiple string-like fibers were present in the biofilm and represented the complex environment surrounding the cells (sugars, proteins, DNA, etc.), it was not possible to use regular microscopic methods to determine morphologically which elements are flagella and which are not. Therefore, further comparison was performed using atomic force microscopy (AFM) imaging on WT and ΔfliC biofilms (fig. S9), where flagella were easily recognized within the WT biofilm only. These results serve as further validation of the consistent presence of flagella in biofilm.
Hypothesizing that flagella contribute to the strength of the biofilm’s structure, we decided to inspect the biofilm’s mechanical strength and stiffness. A biofilm’s mechanical strength is important to its survival. This characteristic is being actively studied to better understand the contribution of different elements to the exopolymeric environment or to measure biofilm’s resilience following different treatments (47–49). For that purpose, we used AFM nanoindentation measurements on the two variants’ mature biofilms. If the hypothesis is correct and the flagella take part in the biofilms’ structural strength, then a biofilm lacking flagella should be mechanically weaker. Figure 6C shows examples of WT and ΔfliC (marked as blue and red, respectively) biofilm force-distance traces obtained from the nanoindentation measurements. Each indentation curve demonstrates specific elastic response. There is a clear difference in the elastic response curvatures for the two biofilms, indicating lower local stiffness for the ΔfliC biofilm. Fitting the curves with the Sneddon model allowed the estimation of local elastic stiffness, K, as illustrated for the ΔfliC biofilm (Fig. 6C, black dashed line).
We analyzed 100 force-distance curves collected for each of the biofilms, enabling the estimation of local stiffness and verification that the biofilms did not undergo irreversible deformation. The local stiffness, K, was collated into probability density functions that interestingly displayed two spreading behaviors. While the stiffness of the ΔfliC displayed a narrow normal distribution, the stiffness of the WT biofilm showed a wide heavy tail distribution. This means that the stiffness values of the WT biofilm spread over a wider range compared to those of the ΔfliC biofilm, with stiffness values that can get considerably high. For this reason, the ΔfliC K probability distribution was fitted with a normal (Gaussian) distribution, and the WT K was fitted with gamma distribution, to properly assess their collective values, i.e., means and SDs (Fig. 6D). Local stiffness value estimated for WT biofilm was one order of magnitude higher than ΔfliC biofilm’s local stiffness. This proved that the ΔfliC biofilm is weaker in terms of physical strength (KWT = 0.58 ± 0.19 pN/nm2 versus KΔfliC = 0.05 ± 0.03 pN/nm2). This result implied that flagella may take a crucial part in the biofilm mechanical strength, much similar to scaffolds in a building construction.
DISCUSSION
Biofilm studies are of great importance for basic microbiological understanding and for medical applications. Despite being studied for over 40 years, there are still many pieces missing in the complex biofilm puzzle. One of the reasons for this great mystery is its complexity and the challenges in its manipulation. Previous research conducted on the role of flagella in biofilm was mainly performed using bulk population analysis (12, 16, 50, 51) and comparison between different modified strains (9, 11, 13, 17–19, 36, 44). While some demonstrated the relevance of flagella in the biofilm (9, 16–19, 44), not only at the attachment stage or dispersion stage but also during its maturation, others documented gene expression down-regulation (11, 12, 51). This resulted in inconsistent biofilm life cycle models when it comes to flagella localization (5–8, 10, 14, 15, 18, 52). Direct evidence from within the biofilm provides an important tool to tackle this challenge. The work presented here has focused on spatial and temporal localization of flagella in the biofilm, providing direct visualization.
A designated genetic code expansion system was developed for direct flagella labeling in P. aeruginosa (Fig. 1). Using GFP reporter protein, orthogonality establishment for MmPyl OTS in P. aeruginosa was achieved (Fig. 1D), and suppression efficiency was calculated to be only below 10% (Fig. 1E). The developed system was later used for direct flagella tracking and localization throughout the biofilm life cycle. Using an integrative approach, Uaa-containing flagella monomers were combined with native monomers for the creation of a functional microstructure capable of fluorescent labeling (Fig. 2C). Examination of different biofilm life cycle models led us to try and better understand the dynamics of flagella in the different biofilm growth stages, assuming that they would either be degraded or be maintained. Direct imaging revealed inoculated flagella persistence during initial biofilm growth and maturation (Fig. 2E), meaning that, for the most part, it would not degrade despite the prolonged lack of motility. While mainly remaining at the lower regions, on rare occasions, inoculated flagella movement toward higher regions within a mature biofilm was revealed (Fig. 2F). This result could serve as indication for the viability of flagella inside the biofilm and throughout its growth.
Next, we wanted to use our generated system to localize flagella synthesis inside a mature biofilm, right before the dispersion stage. We expected to locate some newly synthesized flagella only at a fully grown biofilm. However, to our surprise, we directly demonstrated that flagella are present in the biofilm throughout its entire life cycle and are constantly being synthesized in the lower and middle layers (Fig. 3B). We did not detect flagella any higher than the midsection of mushroom-like structures found in a grown biofilm, yet numerous flagella were constantly located at lower regions. Consistently seeing numerous flagella inside the biofilm throughout its whole life cycle was unexpected. This discrepancy provides important evidence for the importance of direct flagella visualization from within the biofilm, at an appropriate spatiotemporal resolution. The notion that flagella exist at the top of microcolonies in young biofilms, to allow flagella-mediated movement in between microcolonies, or the flagella’s part in biofilm architecture was previously suggested (17, 18). Direct visualization of flagella at the top of microcolonies of a 2-day-old biofilm, as well as a grown, flagella-embedded biofilm, serves as further support for these reports.
The final aspect we examined as far as flagella’s observation in the biofilm life cycle was that of detached cells. Expecting to locate detached flagellated cells only from a mature biofilm, we were surprised to find that flagellated cells constantly left the biofilm, carrying either newly synthesized flagella or, on rare occasions, inoculated flagella (Fig. 4). Finding continued flagellated dispersion is novel information regarding the biofilm life cycle model and reflects the observation that the biofilm is filled with flagella, as was found in this study. From a clinical aspect, mature biofilms are prone to planktonic dispersion at times, causing exacerbations in chronic infections and afflicting new environment within the host (53). Usually, these exacerbations are in need of antibiotics treatment but are only taken under consideration in case of a mature biofilm. Hence, continuous planktonic cells release could affect the clinical view of possible treatments for chronic “biofilm-infected” patients.
The information gathered in this paper could not have been quantified, as there are at least three degrees of freedom to any possible calculation involving the presented system. However, direct observations made here were reproducible and indisputable. The combination of our direct imaging findings led us to create an updated biofilm life cycle model that includes a more accurate dynamics of flagella within the biofilm.
Following the unexpected discovery of constant synthesis of flagella inside the biofilm, we were in search of a possible explanation, especially as flagella synthesis is highly energy consuming. The detection of scaffold-like structures of flagella inside the biofilm (Fig. 6, A and B) resembled mechanical support needed for architectural buildings and led us to inspect the biofilm with regard to the analysis of biofilm’s physical strength. Our hypothesis was strengthened when native biofilm displayed higher physical strength compared to a biofilm lacking flagella (Fig. 6, C and D). Accordingly, we concluded that flagella could play an important role in providing mechanical and physical support of the biofilm. However, further research is needed to clarify the exact details behind this previously undiscovered role of flagella in a biofilm setting using versatile experimental platforms. For example, a reconstituted flagella strain could be generated (ΔfliC strain with exogenous expression of flagellin, WT, or genetically expended copies) for further strength measurements. Such an experiment may reveal a “rescue” effect for the ΔfliC strain–reduced biofilm strength.
To exemplify the importance of the findings of biofilm’s strength, we returned to the literature and found that in some cases, P. aeruginosa strains isolated from patients with cystic fibrosis lack flagella because of various mutations in flagella synthesis–involved genes (54). It was found that cystic fibrosis isolates occasionally develop differently to evade the human immune system that mainly targets bacterial flagella. Recently, Harrison et al. (54) revealed the connection between the loss of flagellum and overexpression of exopolysaccharides in biofilms created from cystic fibrosis isolates. The nanoindentation results presented here can elucidate further details on this fascinating mechanism, as it is possible that bacteria produce more exopolysaccharides in the attempt to compensate for the loss of flagella and its mechanical support. In addition, if flagella indeed contribute to the biofilm’s strength, then future biofilm treatment approaches may potentially target flagella, to weaken the biofilm’s structure and improve antibiotics penetration. Such a strategy may then be followed by an effective antibiotic treatment, which would otherwise be less efficient in a strong and fully functional biofilm, and give way to future studies in clinical/environmental biofilms.
We do recognize the existing obstacles at play; however, the work presented herein strongly emphasizes the need for additional direct evidence of occurrences within the biofilm environment. Direct imaging can thus serve as a window for new research venues. We posit that the previously unreported knowledge afforded by this novel model, and approach that uses genetic code expanded strains as presented here will serve in tackling clinical/environmental biofilm research and envision multiple other new studies in regard to P. aeruginosa.
MATERIALS AND METHODS
Reagents
PrK [(S)-2-amino-6-((prop-2-ynyloxy)carbonylamino)hexanoic acid] and AzCK [(S)-2-amino-6-((2-azidoethoxy)carbonylamino)hexanoic acid] were both synthesized according to a protocol reported by Nguyen et al. (55). Tris(3-hydroxypropyltriazolylmethyl)amine (THPTA), TAMRA azide, and TAMRA alkyne were purchased from Sigma-Aldrich (Rehovot, Israel). MB 660R azide was a donation from Click Chemistry Tools (Scottsdale, AZ, USA). All restriction enzymes were purchased from Thermo Fisher Scientific (Waltham, MA, USA), while all DNA oligonucleotides were obtained from Syntezza Bioscience (Jerusalem, Israel).
Plasmid construction
All plasmids for initial method establishment were constructed by a standard yeast assembly protocol (25). Final plasmid for flagellin genetic code expansion was constructed using standard Gibson assembly. All plasmids inserted into E. coli underwent standard heat-shock transformation protocol. All plasmids inserted into P. aeruginosa underwent standard electroporation protocol. All primers are available in table S1.
The upstream and downstream regions from P. aeruginosa’s leucyl translational system were chosen for OTS expression. Upstream and downstream regions of leucyl tRNA “flanked” Pyl-tRNA, as well as for the upstream and downstream regions of leucyl-tRNA synthetase that flanked PylRS. The dual emission GFP (deGFP) reporter protein (a variant designed for better expression in in vitro systems but also works well in in vivo systems) was also chosen to have a PAO1 endogenous promoter, the flagellin native promoter (fliC promoter). It was chosen considering fliC gene was designed for Uaa incorporation after deGFP.
The initial construct (pPaGE Pyl TAG fliC prom gfp WT NHis) was assembled in two stages. First, pMRP9-1 backbone was amplified using primers 1 and 2 (table S1), without the deGFP gene, and was assembled with the 2μ origin and URA3 selectivity gene for yeast. Second, the new vector after restriction with Bam HI was assembled with seven other polymerase chain reaction (PCR)–amplified parts, containing the OTS, gfp expression gene, and necessary promoters and terminators (primers 3 to 16; table S1). Endogenous regions of P. aeruginosa were amplified from PAO1 genome. N-terminal his-tag gfp and its T500 terminator were amplified from the pBEST plasmid (32). PylRS was amplified from pEVOL-Pyl plasmid described in previous work (32). Pyl-tRNA was assembled through primers 4 and 5 homology (table S1) during the yeast assembly. The pPaGE Pyl TAG fliC prom gfp Y35TAG NHis construct was assembled in the same manner, only with a mutant deGFP amplified from the pBEST plasmid.
Variant for orthogonality testing of Pyl tRNA was generated through standard DNA collapse using Hind III. Hind III restriction was followed by self-ligation of the plasmid without the PylRS gene. During the initial construct generation, a deletion construct without Pyl tRNA was also created. This variant was used for orthogonality testing of PylRS.
A construct containing the fliC gene with a TAG mutation (pPaGE Pyl TAG fliC T248TAG) was assembled through Gibson assembly. pPaGE was restricted with Nco I for vector generation without the gfp gene and T500 terminator. fliC gene had the TAG mutation installation as part of the assembly, by two pieces amplification. The gene, together with its downstream sequence, was amplified from the PAO1 genome using primers 17 ans 20 and 19 and 18 (table S1).
Viability assay
Bacterial liquid culture in LB Miller, after 24 hours of growth at 37°C, was diluted 1:100. The diluted culture was placed at 37°C. Every 1 hour, a duplicate was measured for optical density at a wavelength of 600 nm (OD600) using a Synergy HT plate reader (BioTek, Winooski, VT, USA). After 17 hours of measurements, when the cells reached growth plateau, the culture was left for incubation for another 10 hours, when a final measurement was taken. When needed, the culture was supplemented with final concentration of 1 mM Uaa (optimal concentration found is shown in fig. S10). In case of plasmid containing bacteria, growth medium was supplemented with carbenicillin (300 μg/ml).
Suppression efficiency
Five separate sets of liquid cultures were grown at 37°C for 24 hours. Each set was tested for GFP expression and was composed of a native strain of P. aeruginosa, WT GFP, and Y35TAG GFP with PrK. Following growth, each sample, from each set, was tested in triplicates for OD600 and GFP fluorescence using a Synergy HT plate reader (BioTek, Winooski, VT, USA). Each sample’s fluorescence was divided by the OD600 value. Values of WT and Y35TAG with PrK were normalized to the native strain’s value of fluorescence/OD600. Last, the suppression efficiency of each set was determined through the value of normalized fluorescence/OD600 of mutant divided by the value of normalized fluorescence/OD600 of WT. Error bar in Fig. 1E could only be calculated for mutant expression (as WT is always 100% by definition), representing the SD between five different suppression efficiencies values calculated.
Protein expression in PAO1 and cells lysis
Culture growth
PAO1 harboring pPaGE variants were grown in LB Miller with carbenicillin (300 μg/ml) at 37°C for 24 hours. After growth, the cultures were diluted 1:100 for another 24 hours of growth. In case of needed Uaa addition, the culture was supplemented with a final concentration of 1 mM Uaa.
Lysis
Following 24 hours of growth, 1 ml of culture was sedimented and resuspended with 1 ml of 100 mM phosphate buffer (PB) (pH 7). The cells were once again sedimented and were resuspended with 100 μl of lysis solution composed of 90% (v/v) PB, 10% (v/v) BugBuster 10X protein extraction reagent (Merck, Billerica, MA, USA), Turbonuclease (Sigma-Aldrich, St. Louis, MO, USA), lysozyme (Sigma-Aldrich, Rehovot, Israel), and protease inhibitor (Merck, Darmstadt, Germany). Cells were incubated with the lysis solution for 30 min at room temperature while shaking, followed by 4°C centrifugation at 10,000g for 10 min. Supernatant containing soluble protein fraction was taken for further analysis. Lysate was used for downstream analyses in the form of click reaction, SDS-PAGE, fluorescent imaging, and Western blot.
Cu-catalyzed click reaction followed by SDS-PAGE, fluorescent imaging, and Western blot analyses
CuAAC click reaction (34) was performed on lysates at a final volume of 50 μl. A fluorophore (with either an azide moiety or an alkyne moiety, according to necessity) was added to a concentration of 50 μM, while THPTA, sodium ascorbate, and CuCl2 were added to final concentration of 1.2, 2.5, and 200 μM, respectively. A volume of 20 μl of cell lysate was added to the reaction, followed by 1 hour of incubation at room temperature with shaking. Clicked samples were examined through SDS-PAGE 4 to 20% ExpressPlus protein gel (GenScript, Nanjing, China). Fluorescent SDS-PAGE images were obtained through an ImageQuant LAS 4000 imager (Fujifilm, Tokyo, Japan), using green light (520-nm Epi) and a 575-nm Cy3 detection filter. Next, when Western blot was needed, SDS-PAGE was transferred to a membrane (Bio-Rad, Hercules, CA, USA) through eBlot protein transfer system (GenScript, Nanjing, China). Using goat T-19 anti-GFP antibody (sc-5384, Santa Cruz Biotechnology, CA, USA) (56) as a primary antibody and donkey anti-goat IgG-HRP (sc-2020, Santa Cruz Biotechnology, CA, USA) (57) as a secondary antibody, standard Western blot protocol was performed. Chemiluminescence imaging was performed using an ImageQuant LAS 4000 imager (Fujifilm, Tokyo, Japan).
All protein gels were loaded with a protein ladder (PM2700 ExcelBand 3-color). A self-made fluorescent protein ladder was also added for Cy3 fluorescent imaging. The fluorescent ladder was assembled from Uaas incorporated in a purified deGFP of ~27 kDa, a purified alcohol dehydrogenase of ~42 kDa, and a purified Cu eflux oxidase of ~57 kDa that were subsequently labeled by a click reaction to a TAMRA fluorophore.
Cu-free click reaction
Twenty-four microliters of PB were added to 25 μl of cell lysates followed by the addition of Cyanine5-DBCO added at 1 μl of dye to achieve a final concentration of 50 μM and a final reaction volume of 50 μl. After 2 hours of incubation at 37°C, 20 μl from the reaction were loaded into SDS-PAGE. The fluorescent gels were observed using Typhoon FLA 9500 imager (GE Healthcare Life Sciences, Uppsala, Sweden). Measurements were performed using a 635-nm laser for excitation and a 665-nm barrier filter for emission detection, matching Cyanine5 detection wavelengths.
For whole-cell reaction, 25 μl of grown liquid culture were pelleted and resuspended with 49 μl of fresh LB. Cyanine5-DBCO was added at 1 μl of dye to achieve a final concentration of 50 μM and a reaction final volume of 50 μl. After 2 hours of incubation at 37°C, the reaction was washed five times with 100 mM PB. Following washes, cells were resuspended in 40 μl of 100 mM PB. Confocal images were acquired using Zeiss LSM880 system (Zena, Germany). Plan-Apochromat 63×/1.4 Oil DIC M27 objective was used. Red channel for Cyanine5-DBCO detection was imaged using 633-nm HeNe laser (usually in the range of 8 to 15%) with emission filter bandpass (BP) 638-755.
Protein purification and mass spectrometry
A 200-ml culture of the P. aeruginosa Y35TAG GFP mutant, in the presence of PrK, was grown at 37°C for 24 hours. Using standard needle sonication, culture was lysed and purified using IMAC (Novagene, Madison, WI, USA) according to the manufacturer’s guidelines. Elution fraction was concentrated using Vivaspin 6, 10,000 MWCO polyethersulfone (Sartorius, Goettingen, Germany), and the concentrated fraction was analyzed by liquid chromatography mass spectrometry (Finnigan Surveyor Autosampler Plus/LCQ Fleet, Thermo Fisher Scientific, Waltham, MA, USA).
Theoretical model of P. aeruginosa flagella
A monomer model of P. aeruginosa flagellin was obtained through SWISS-MODEL (58), and a PrK residue was incorporated in position 248 (PyMOL Molecular Graphics System, version 2.0, Schrӧdinger LLC). PDB file 5WK5, containing 41 subunits of the P. aeruginosa’s filament core (no outer protein structure of D2 and/or D3), was used as alignment reference for 41 SWISS-MODEL–generated monomers with PrK (PyMOL Molecular Graphics System, version 2.0, Schrӧdinger LLC). The final product is a theoretical model of a flagella filament, consisting of 41 P. aeruginosa flagellin proteins, all of which contain PrK in the 248th position.
Live-cell click reaction and flow cell construction
pPaGE harboring fliC with TAG mutation at the 248th site was electroporated into PAO1 gGFP strain (a strain containing GFP expression gene in the genome). Liquid culture in the presence of PrK was grown at 37°C for 24 hours. Grown culture at the volume of 200 μl was centrifuged and resuspended with 20 μl of 100 mM PB (pH 7). Click reagents were added to a final reaction volume of 50 μl. Azide-containing 545 fluorophore was added to a concentration of 50 μM, while THPTA, sodium ascorbate, and CuCl2 were added to a final concentration of 1.2, 2.5, and 200 μM, respectively. Following 40 min of incubation at room temperature with shaking, cells were washed three times with 1 ml of 100 mM PB (pH 7) and were brought to a value of OD600 of 0.1. A flow cell system was constructed as described before (59), using the labeled bacteria as inoculation for growth. Following 1 hour of static attachment in the flow cell at 30°C, AB minimal growth medium at a temperature of 37°C was supplied at a rate of 4 ml/hour. The preparation of 1 liter of AB minimal medium included 2 g of (NH4)2SO4, 6 g of Na2HPO4, and 3 g of KH2PO4 dissolved in 1 liter of distilled water and autoclaved. Media “activation” was performed before every new flow cell by adding 1 ml of 0.1 M CaCl2 (sterile), 1 ml of 1 M MgCl2 (sterile), and 1 ml of 0.003 M FeCl3 (sterile). Flow cells were grown for up to 6 days, while imaging for different experimental procedures was performed using CLSM. All mentioned experimental procedures were repeated for a minimum number of five different flow cells. For each individual experiment, general impression was tested for being even throughout the flow cell, and at least three fields were captured with resulted layout.
Detailed settings for different flow cell experiments are as follows. For preinoculation-labeled flagella tracking, flow cell was constructed as described and was imaged using CLSM at two different channels (488 at cyan for bacteria’s GFP and 545 at magenta for preinoculation TAMRA azide–labeled flagella through a click reaction) at specific locations every several hours for up to 6 days.
For effluent flagella tracking, flow cell was constructed as described, while AB minimal medium was supplemented with PrK for continuous incorporation. Effluent of ~15 ml was collected into ice following 2, 4, or 6 days of growth. Effluent total volume was pelleted down and resuspended with 20 μl of 100 mM PB (pH 7). Click reagents were added to a final reaction volume of 50 μl. Azide-containing 660 fluorophore was added to a concentration of 50 μM, while THPTA, sodium ascorbate, and CuCl2 were added to a final concentration of 1.2, 2.5, and 200 μM, respectively. Following 40 min of incubation at room temperature with shaking, cells were washed two times with 1 ml of 100 mM PB (pH 7) and were lastly resuspended with ~50 μl of 100 mM PB (pH 7). Labeled effluent (5 μl onto a glass slide) was imaged using CLSM at three different channels (488 at cyan for bacteria’s GFP, 545 at magenta for preinoculation clicked TAMRA azide–labeled flagella, and 660 at orange for newly synthesized MB 660R azide clicked flagella).
For newly synthesized labeled flagella tracking within a biofilm, flow cell was constructed as described and grown for 2, 4, or 6 days, while AB minimal medium was supplemented with PrK for continuous incorporation. Click reagents solution was prepared for a final reaction volume of 500 μl. Azide-containing 545 fluorophore was added to a concentration of 50 μM, while THPTA, sodium ascorbate, and CuCl2 were added to a final concentration of 1.2, 2.5, and 200 μM, respectively. The click mixture was slowly injected into the flow cell chamber and remained stationary for 20 min at room temperature, followed by intensive wash with growth medium at a rate of 16 ml/hour at room temperature. Flow cell was imaged using CLSM at two different channels (488 at cyan for bacteria’s GFP and 545 at magenta for clicked TAMRA azide–labeled flagella).
Confocal microscope settings and image analysis
Confocal images were acquired using Zeiss LSM880 system (Zena, Germany). Plan-Apochromat 63×/1.4 Oil DIC M27 or Plan-Apochromat 40×/1.3 Oil DIC M27 objective were used. Cyan channel for GFP detection was imaged using 488-nm argon laser (usually in the range of 5 to 15%) with emission filter BP 493-556. Magenta channel for TAMRA azide–conjugated flagella detection was imaged using 561-nm diode-pumped solid-state laser (usually in the range of 2 to 4%) with emission filter BP 570-624. Orange channel for MB 660R azide–conjugated flagella detection was imaged using 633-nm HeNe laser (usually in the range of 8 to 15%) with emission filter BP 638-755. Scanning resolution for all flow cells was 1024 by 1024. Scanning resolution for effluent planktonic cells was 2048 by 2048. Image analysis was performed using either ImageJ software (National Institutes of Health, USA) or Imaris software (Bitplane AG, Zürich, Switzerland). Despite channel modifications in image analysis, a gamma value of 1.00 was strictly conserved in all images and analysis. In Fig. 2C, all nine Z planes were stacked, and channels were merged (488 and 545), and a smooth filter was used (ImageJ). In Fig. 2E, three-dimensional (3D) digital visualization of 545 alone or merged channels (488 and 545) were at 6000 by 6000 dimensions and 900 dpi (dots per inch) using the “shadow” function (Imaris). In Fig. 2F, of 118 Z planes, 61 to 65 were stacked and 488 and 545 channels were merged (ImageJ). TAMRA channel biovolume was computed for young and mature biofilms at specific locations. A value of biovolume (volume per area) was calculated for young biofilm and mature biofilm using six different experimental fields. To assess changes in TAMRA azide–conjugated flagella signal, a percentage ratio was calculated for each location by dividing the mature biofilm value with the young biofilm value. In Fig. 3B, digital biovolume was saved as 6000 by 6000 and 900 dpi. Two-day midsection was stacked from 13 to 16 Z planes. Upper section was stacked from 20 to 25 Z planes. Lower midsection was stacked from 27 to 39 Z planes. Midsection was stacked from 66 to 68 Z planes (Imaris). In Fig. 4B, of 13 Z planes, image was stacked using 4 to 9 Z planes and saved as individual channels (488/545/660) or a merged image of all channels (ImageJ). In Fig. 6A, of 39 Z planes, the 14 plane was presented only in the 545 channel (Imaris). In Fig. 6B, Z planes of 23 to 26 were stacked and saved as individual 545 channel or a merged image of 488 and 545 channels (Imaris).
P. aeruginosa PAO1 gGFP ΔfliC strain generation
For the creation of a KO strain to flagella filament, a CRISPR-Cas9–based platform adapted to P. aeruginosa (46) was used, and a PAO1 gGFP ΔfliC was generated. Of a two-plasmid system, pCasPA plasmid was used as is, while a relevant pACRISPR plasmid had to be constructed using Gibson assembly to contain guide RNA (gRNA) and complementary homology region to the genome. The gRNA used was chosen on the basis of CHOPCHOP (60) and complemented sites 168 to 174 in fliC gene. Homology region was chosen as 500–base pair (bp) upstream to the ATG codon of fliC, 30 bp from the end of the fliC gene (including stop codon), and 500-bp downstream to the stop codon of fliC, meaning deletion of 1437 bp of the fliC gene. Gibson assembly for pACRISPR with relevant gRNA and homology was performed using primers 21 to 28 (table S1), following standard assembly and plasmid sequence verification. pCasPA was electroporated into PAO1 gGFP strain using standard protocol and grown on tetracycline (100 μg/ml) selective plate. After first plasmid insertion, PAO1 gGFP pCasPA was grown over night at 37°C and was added with l-arabinose to a final concentration of 2 mg/ml for 2 hours of incubation at 37°C (targeted for Cas9 inductive protein expression). Cells then were prepared for electroporation using standard protocol. pACRISPR targeted for fliC deletion was electroporated into PAO1 gGFP pCasPA cells prepared in advance and grown on tetracycline (100 μg/ml) and carbenicillin (150 μg/ml). Colonies were screened for genome segment modification, and chosen colonies were cured from both plasmids by plating on sucrose 5% plates. Chosen colony was isolated, tested for positive plasmid curing, and tested by PCR for positive deletion (primers 29 and 30; table S1). PCR segment was purified using NucleoSpin gel and PCR clean-up (MACHEREY-NAGEL, Germany) and underwent Sanger sequencing with primer 31 (table S1).
Strains comparison
WT (PAO1 gGFP) and KO (PAO1 gGFP ΔfliC) strains were compared in a series of experiments including flow cells, TEM, swarming assays, HR-SEM, dual-beam HR-SEM, AFM imaging, and AFM nanoindentation.
WT/KO strains flow cell construction
WT (PAO1 gGFP) or KO (PAO1 gGFP ΔfliC) strain stationary grown liquid culture was diluted to OD600 of 0.1, and cells were inoculated in a flow cell as described before. After 1 hour of attachment at 30°C, 37°C heated AB minimal medium at a rate of 4 ml/hour were supplemented for 5 days of growth. Flow cells were examined for biofilm structure and 3D maturation using CLSM. Biovolume was produce using Imaris software analysis.
Transmission electron microscopy
Grown bacterial cultures of P. aeruginosa PAO1 gGFP WT/ΔfliC strains were diluted 1:20. A carbon type B TEM grid was prepared using plasma cleaner PDC-32G (Harrick Plasma, Ithaca, NY, USA) for 30 s. TEM grids were loaded with 2 μl of the diluted liquid culture, while excess liquid was dried using filter paper. Prepared grids were examined in FEI Tecnai T12 G2 TWIN TEM operating at 120 kV.
Swarming assay
P. aeruginosa PAO1 gGFP WT/ΔfliC strains were grown overnight in LB Miller medium. The next day, the bacteria were diluted 1:100 into fresh M9 medium and were grown to mid-log phase. The swarming plates were prepared using M9, solidified with 0.7% (w/v) Difco agar. Following bacteria refreshment, 2 μl of the inoculums were placed in the middle of the plates, enabling assessment of surface coverage after 24 hours of growth at 37°C.
Static biofilm growth
Round glass coverslips, 15 mm in diameter (Ted Pella Inc., Redding, CA, USA), were incubated with 8% HCl for 1 hour at room temperature and underwent autoclave sterilization in advance to biofilm growth. WT (PAO1 gGFP) or KO (PAO1 gGFP ΔfliC) was refreshed by taking 30 μl of stationary grown culture into 3 ml of LB Miller for 3 hours at 37°C with agitation. Reaching an OD600 of ~0.2, 20 μl of refreshed culture were taken into 2 ml of LB Lennox in a 12-well plate well. Treated glass coverslip was also placed in the well. Plate was incubated at 37°C for 16 hours. Wells surrounding the sample were filled with double-distilled water to keep high moisture during growth.
High-resolution scanning electron microscopy
Following static biofilm growth, liquid was aspirated and the glass coverslip underwent fixation (2.5% glutaraldehyde, 2% paraformaldehyde, and 0.2 M PB) for 15 min. Samples were washed twice with phosphate-buffered saline (PBS), 10 min each time, and were dehydrated using increasing ethanol concentrations before an addition of hexamethyldisilazane/ethanol solution in increasing concentrations. Last, all liquids were removed, and samples were dried in a chemical hood. Fixed glasses were coated in Cr using Quorum Q150T-ES sputter and examined in Verios 460L Thermo Fisher Scientific scanning electron microscope operating at 3.00 kV.
Dual-beam HR-SEM
For an inside look at a biofilm and not only outer-rim observations, the same glasses coated with Cr that were used for HR-SEM were also analyzed with dual-beam HR-SEM. Areas meant for examination were locally coated with Pt and were sliced using focused ion beam. Biofilm’s inside was examined in Helios G4 UC Thermo Fisher Scientific scanning electron microscope operating at 5.00 kV.
AFM imaging
Following static biofilm growth, the well was washed twice with PBS and left to dry in a hood. The dry samples were imaged on a MFP-3D-Bio (Oxford Instruments Asylum Research, Santa Barbara, CA, USA) in AC mode (“tapping mode”) in air, using an AC240BSA probe (Olympus) at room temperature. Imaging rate was of 0.5 Hz, with parameters of 512 scan lines and 1024 scan points.
AFM nanoindentation
Local stiffness modulus, K, of the two different biofilms was measured with nanoindentation experiments performed on a Luigs & Neumann Ltd. AFM. The biofilm samples were grown on a clean glass coverslip as was mentioned before. The grown biofilm on top the coverslip was gently washed with PBS and semidried in open air. The semidried biofilm was rehydrated when positioned above the cantilever and sealed a chamber filled with PBS. The indentation was performed with pyramidal silicon nitride cantilevers (V-shaped MLCT, Bruker) with a measured mean spring constant of 0.01 N/m. The normal spring constants of the cantilevers were determined before each measurement using the equipartition theorem (61). In a nanoindentation measurement, force-distance curves are collected by approaching the cantilever tip toward the surface of the biofilm sample at a rate of 400 nm/s and an indentation depth amplitude of 500 nm. Corresponding to the compliance of the sample, the cantilever deflects proportionally to the compliance of the sample. The curvature of this response region was fitted using the Sneddon model (62, 63), a contact mechanics model for a conical sharp probe (as the one used), to the force-distance curves
where v is Poisson ratio, taken as 0.5 (typical value for incompressible materials); δ is the indentation length coordinate; θ is the cone half angle (face angle) of the AFM probe, taken as the manufacturer’s nominal value of 29.1°; and K is the local stiffness modulus. The indentation was performed at random locations across the surface of each biofilm sample. The measurement sets were repeated three times for each biofilm sample, where each set was composed of 100 force-distance traces. All measurements were carried out under PBS buffer [150 mM NaCl and 20 mM PBS (pH 7.2)] at room temperature. All data were recorded and analyzed using custom software written in Igor Pro 6.37 (WaveMetrics).
From 100 collected values of K for each biofilm, we constructed their probability distributions and fitted them with the relevant statistical model. The Freedman-Diaconis rule was used as criterion for setting the bin size of the distributions (64). The stiffness distribution of the WT biofilm was fitted with a Gaussian distribution, ϕ(K) = (σ(2π)½)−1exp{−½[(K − μ)/σ]2}, and the stiffness distribution of the ΔfliC biofilm was fitted with the gamma distribution, which is given by ϕ(K) = (βα/Γ(α))Kα − 1e–βK. The latter is highly useful to describe the distribution of a random variable that does not normally distribute. The mean is calculated by μ and α/β (for the normal and gamma distributions, respectively), and the variance (from which the SD is calculated by taking its root) by σ2 and α/β2 (for the normal and gamma distributions, respectively).
Acknowledgments
E. Banin is acknowledged for useful discussions. We would like to thank the Ilse Katz Institute for Nanoscale Science and Technology Shred Resource Facility for technical contribution in image acquisition with Zeiss LSM880 Airyscan (U. Hadad), Verios 460L Thermo Fisher Scientific HR-SEM (E. Nativ-Roth), Helios G4 UC Thermo Fisher Scientific dual-beam HR-SEM (N. Maman), and MFP-3D-Bio AFM (J. Jopp). A. Bakhrat’s assistance in genetic engineering is acknowledged. Y. Ozer and I. Algov’s graphical assistance is acknowledged. E. K. Winter is acknowledged for writing assistance. MB 660R azide was a donation from Click Chemistry Tools (Scottsdale, AZ, USA). Funding: We thank the Kreitmann School for graduate students for a Ph.D. fellowship (to E.O., A.B., and K.Y.) and the Ben-Gurion University for a continued support of our research (to L.A. and A.K.). We include all funding sources, including grant numbers and funding agencies. Author contributions: E.O. and K.Y. share equal contribution to this paper. E.O. conceived, performed, and analyzed experimentations; established genetic code expansion system; and authored the manuscript. K.Y. conceived, performed, and analyzed all experimentations and coauthored the manuscript. E.C. performed nanoindentation experiments. A.B. synthesized Uaas used in this research. M.M.M. perceived and advised with P. aeruginosa biofilm experiments. R.B. supervised, analyzed, and wrote the manuscript regarding AFM measurements. A.K. conceived experiments, supervised, provided research facilities, and edited the manuscript. L.A. conceived experiments, supervised the research, provided facilities, written, and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional experimental results and control experiments, primers, gene sequences, and plasmid maps are available in the Supplementary Materials. pPaGE plasmid is available through Addgene with the following accession number and link: www.addgene.org/160089/. Enlarged and high-resolution images are available for download at: https://figshare.com/articles/figure/An_inside_look_at_a_biofilm_Pseudomonas_aeruginosa_flagella_bio-tracking_Appendix_docx/13640777. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg8581/DC1
REFERENCES AND NOTES
- 1.Lee K., Yoon S. S., Pseudomonas aeruginosa biofilm, a programmed bacterial life for fitness. J. Microbiol. Biotechnol. 27, 1053–1064 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Papenfort K., Bassler B. L., Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Høiby N., A short history of microbial biofilms and biofilm infections. Apmis. 125, 272–275 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Parsek M. R., Greenberg E. P., Sociomicrobiology: The connections between quorum sensing and biofilms. Trends Microbiol. 13, 27–33 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Lappin-Scott H., Burton S., Stoodley P., Revealing a world of biofilms-the pioneering research of Bill Costerton. Nat. Rev. Microbiol. 12, 781–787 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Maunders E., Welch M., Matrix exopolysaccharides; the sticky side of biofilm formation. FEMS Microbiol. Lett. 364, fnx120 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDougald D., Rice S. A., Barraud N., Steinberg P. D., Kjelleberg S., Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 10, 39–50 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Petrova O. E., Sauer K., Escaping the biofilm in more than one way: Desorption, detachment or dispersion. Curr. Opin. Microbiol. 30, 67–78 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barken K. B., Pamp S. J., Yang L., Gjermansen M., Bertrand J. J., Klausen M., Givskov M., Whitchurch C. B., Engel J. N., Tolker-Nielsen T., Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 10, 2331–2343 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Belas R., Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 22, 517–527 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Baraquet C., Harwood C. S., Cyclic diguanosine monophosphate represses bacterial flagella synthesis by interacting with the Walker a motif of the enhancer-binding protein FleQ. Proc. Natl. Acad. Sci. U.S.A. 110, 18478–18483 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Whiteley M., Bangera M. G., Bumgarner R. E., Parsek M. R., Teitzel G. M., Lory S., Greenberg E. P., Gene expression in Pseudomonas aeruginosa biofilms. Nature 413, 860–864 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Sauer K., Camper A. K., Ehrlich G. D., Costerton J. W., Davies D. G., Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 184, 1140–1154 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stoodley P., Sauer K., Davies D. G., Costerton J. W., Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 56, 187–209 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Watnick P., Kolter R., Biofilm, city of microbes. J. Bacteriol. 182, 2675–2679 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heacock-Kang Y., Sun Z., Zarzycki-Siek J., McMillan I. A., Norris M. H., Bluhm A. P., Cabanas D., Fogen D., Vo H., Donachie S. P., Borlee B. R., Sibley C. D., Lewenza S., Schurr M. J., Schweizer H. P., Hoang T. T., Spatial transcriptomes within the Pseudomonas aeruginosa biofilm architecture. Mol. Microbiol. 106, 976–985 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolker-Nielsen T., Brinch U. C., Ragas P. C., Andersen J. B., Jacobsen C. S., Molin S., Development and dynamics of Pseudomonas sp. biofilms. J. Bacteriol. 182, 6482–6489 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klausen M., Heydorn A., Ragas P., Lambertsen L., Aaes-Jørgensen A., Molin S., Tolker-Nielsen T., Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 48, 1511–1524 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Blair K. M., Turner L., Winkelman J. T., Berg H. C., Kearns D. B., A molecular clutch disables flagella in the Bacillus subtilis biofilm. Science 320, 1636–1638 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Lemke E. A., The exploding genetic code. ChemBioChem. 15, 1691–1694 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Cohen M., Ozer E., Kushmaro A., Alfonta L., Cellular localization of cytochrome bd in cyanobacteria using genetic code expansion. Biotechnol. Bioeng. 117, 523–530 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Aloush N., Schvartz T., König A. I., Cohen S., Brozgol E., Tam B., Nachmias D., Ben-David O., Garini Y., Elia N., Arbely E., Live cell imaging of bioorthogonally labelled proteins generated with a single pyrrolysine tRNA gene. Sci. Rep. 8, 14527 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kipper K., Lundius E. G., Ćurić V., Nikić I., Wiessler M., Lemke E. A., Elf J., Application of noncanonical amino acids for protein labeling in a genomically recoded Escherichia coli. ACS Synth. Biol. 6, 233–255 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Zhang J., Yan S., He Z., Ding C., Zhai T., Chen Y., Li H., Yang G., Zhou X., Wang P., Small unnatural amino acid carried raman tag for molecular imaging of genetically targeted proteins. J. Phys. Chem. Lett. 9, 4679–4685 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Chemla Y., Friedman M., Heltberg M., Bakhrat A., Nagar E., Schwarz R., Jensen M. H., Alfonta L., Expanding the genetic code of a photoautotrophic organism. Biochemistry 56, 2161–2165 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Gan Q., Lehman B. P., Bobik T. A., Fan C., Expanding the genetic code of Salmonella with non-canonical amino acids. Sci. Rep. 6, 39920–39926 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bianco A., Townsley F. M., Greiss S., Lang K., Chin J. W., Expanding the genetic code of Drosophila melanogaster. Nat. Chem. Biol. 8, 748–750 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Ernst R. J., Krogager T. P., Maywood E. S., Zanchi R., Beránek V., Elliott T. S., Barry N. P., Hastings M. H., Chin J. W., Genetic code expansion in the mouse brain. Nat. Chem. Biol. 12, 776–778 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ozer E., Alfonta L., Genetic code expansion of Vibrio natriegens. Front. Bioeng. Biotechnol. 9, 594429 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cervettini D., Tang S., Fried S. D., Willis J. C. W., Funke L. F. H., Colwell L. J., Chin J. W., Rapid discovery and evolution of orthogonal aminoacyl-tRNA synthetase–tRNA pairs. Nat. Biotechnol. 38, 989–999 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zheng H., Lin S., Chen P. R., Genetically encoded protein labeling and crosslinking in living Pseudomonas aeruginosa. Bioorganic Med. Chem. 28, 115545 (2020). [DOI] [PubMed] [Google Scholar]
- 32.Ozer E., Chemla Y., Schlesinger O., Aviram H. Y., Riven I., Haran G., Alfonta L., In vitro suppression of two different stop codons. Biotechnol. Bioeng. 114, 1065–1073 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Nakamura Y., Gojobori T., Ikemura T., Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 28, 292 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh M. S., Chowdhury S., Koley S., Advances of azide-alkyne cycloaddition-click chemistry over the recent decade. Tetrahedron 72, 5257–5283 (2016). [Google Scholar]
- 35.Chemla Y., Ozer E., Algov I., Alfonta L., Context effects of genetic code expansion by stop codon suppression. Curr. Opin. Chem. Biol. 46, 146–155 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Schniederberend M., Williams J. F., Shine E., Shen C., Jain R., Emonet T., Kazmierczak B. I., Modulation of flagellar rotation in surface-attached bacteria: A pathway for rapid surface-sensing after flagellar attachment. PLOS Pathog. 15, e1008149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Renault T. T., Abraham A. O., Bergmiller T., Paradis G., Rainville S., Charpentier E., Guet C. C., Tu Y., Namba K., Keener J. P., Minamino T., Erhardt M., Bacterial flagella grow through an injection-diffusion mechanism. eLife 6, e23136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slastnikova T. A., Ulasov A. V., Rosenkranz A. A., Sobolev A. S., Targeted intracellular delivery of antibodies: The state of the art. Front. Pharmacol. 9, 1–21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chaban B., Hughes H. V., Beeby M., The flagellum in bacterial pathogens: For motility and a whole lot more. Semin. Cell Dev. Biol. 46, 91–103 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Minamino T., Namba K., Self-assembly and type III protein export of the bacterial flagellum. J. Mol. Microbiol. Biotechnol. 7, 5–17 (2004). [DOI] [PubMed] [Google Scholar]
- 41.N. Dasgupta, S. K. Arora, R. Ramphal, in Pseudomonas (Springer US, 2004), pp. 675–698. [Google Scholar]
- 42.Schlesinger O., Chemla Y., Heltberg M., Ozer E., Marshall R., Noireaux V., Jensen M. H., Alfonta L., Tuning of recombinant protein expression in Escherichia coli by manipulating transcription, translation initiation rates, and incorporation of noncanonical amino acids. ACS Synth. Biol. 6, 1076–1085 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Zhao K., Liu M., Burgess R. R., Adaptation in bacterial flagellar and motility systems: From regulon members to’foraging’-like behavior in E. coli. Nucleic Acids Res. 35, 4441–4452 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serra D. O., Richter A. M., Klauck G., Mika F., Hengge R., Microanatomy at cellular resolution and spatial order of physiological differentiation in a bacterial biofilm. MBio 4, 10.1128/mBio.00103-13, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu X., Zhuo S., Jing X., Yuan Y., Rensing C., Zhou S., Flagella act as Geobacter biofilm scaffolds to stabilize biofilm and facilitate extracellular electron transfer. Biosens. Bioelectron. 146, 111748 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Chen W., Zhang Y., Zhang Y., Pi Y., Gu T., Song L., Wang Y., Ji Q., CRISPR/Cas9-based genome editing in Pseudomonas aeruginosa and cytidine deaminase-mediated base editing in pseudomonas species. iScience 6, 222–231 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tallawi M., Opitz M., Lieleg O., Modulation of the mechanical properties of bacterial biofilms in response to environmental challenges. Biomater. Sci. 5, 887–900 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Baniasadi M., Xu Z., Gandee L., Du Y., Lu H., Zimmern P., Minary-Jolandan M., Nanoindentation of Pseudomonas aeruginosa bacterial biofilm using atomic force microscopy. Mater. Res. Express. 1, 045411 (2014). [Google Scholar]
- 49.Zeng G., Vad B. S., Dueholm M. S., Christiansen G., Nilsson M., Tolker-Nielsen T., Nielsen P. H., Meyer R. L., Otzen D. E., Functional bacterial amyloid increases Pseudomonas biofilm hydrophobicity and stiffness. Front. Microbiol. 6, 1099 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morgan R., Kohn S., Hwang S.-H., Hassett D. J., Sauer K., BdlA, a chemotaxis regulator essential for biofilm dispersion in Pseudomonas aeruginosa. J. Bacteriol. 188, 7335–7343 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tart A. H., Wolfgang M. C., Wozniak D. J., The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ. J. Bacteriol. 187, 7955–7962 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hobley L., Harkins C., MacPhee C. E., Stanley-Wall N. R., Giving structure to the biofilm matrix: An overview of individual strategies and emerging common themes. FEMS Microbiol. Rev. 39, 649–669 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H., Moser C., Wang H. Z., Høiby N., Song Z. J., Strategies for combating bacterial biofilm infections. Int. J. Oral Sci. 7, 1–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harrison J. J., Almblad H., Irie Y., Wolter D. J., Eggleston H. C., Randall T. E., Kitzman J. O., Stackhouse B., Emerson J. C., Mcnamara S., Larsen T. J., Shendure J., Hoffman L. R., Wozniak D. J., Parsek M. R., Elevated exopolysaccharide levels in Pseudomonas aeruginosa flagellar mutants have implications for biofilm growth and chronic infections. PLOS Genet. 16, e1008848 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen D. P., Lusic H., Neumann H., Kapadnis P. B., Deiters A., Chin J. W., Genetic encoding and labeling of aliphatic azides and alkynes in recombinant proteins via a pyrrolysyl-tRNA synthetase/tRNACUA pair and click chemistry. J. Am. Chem. Soc. 131, 8720–8721 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Ketema M., Secades P., Kreft M., Nahidiazar L., Janssen H., Jalink K., De Pereda J. M., Sonnenberg A., The rod domain is not essential for the function of plectin in maintaining tissue integrity. Mol. Biol. Cell 26, 2402–2417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dunphy G., Flannery S. M., Almine J. F., Connolly D. J., Paulus C., Jønsson K. L., Jakobsen M. R., Nevels M. M., Bowie A. G., Unterholzner L., Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-κB signaling after nuclear DNA damage. Mol. Cell 71, 745–760.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., De Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T., SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yaniv K., Golberg K., Kramarsky-Winter E., Marks R., Pushkarev A., Béjà O., Kushmaro A., Functional marine metagenomic screening for anti-quorum sensing and anti-biofilm activity. Biofouling 33, 1–13 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Labun K., Montague T. G., Krause M., Torres Cleuren Y. N., Tjeldnes H., Valen E., CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 47, W171–W174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hutter J. L., Bechhoefer J., Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 64, 1868–1873 (1993). [Google Scholar]
- 62.Sneddon I. N., The relation between load and penetration in the axisymmetric boussinesq problem for a punch of arbitrary profile. Int. J. Eng. Sci. 3, 47–57 (1965). [Google Scholar]
- 63.Krieg M., Fläschner G., Alsteens D., Gaub B. M., Roos W. H., Wuite G. J. L., Gaub H. E., Gerber C., Dufrêne Y. F., Müller D. J., Atomic force microscopy-based mechanobiology. Nat. Rev. Phys. 1, 41–57 (2019). [Google Scholar]
- 64.Freedman D., Diaconis P., On the histogram as a density estimator:L2 theory. Z. Wahrscheinlichkeit. 57, 453–476 (1981). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/24/eabg8581/DC1