

Abstract
The disease outbreak of Coronavirus disease-19 (COVID-19), caused by the novel SARS-CoV-2 virus, remains a public health concern. COVID-19 is spreading rapidly with a high mortality rate due to unavailability of effective treatment or vaccine for the disease. The high rate of mutation and recombination in SARS-CoV2 makes it difficult for scientist to develop specific anti-CoV2 drugs and vaccines. SARS-CoV-2-Mpro cleaves the viral polyprotein to produce a variety of non-structural proteins, but in human host it also cleaves the nuclear transcription factor kappa B (NF-κB) essential modulator (NEMO), which suppresses the activation of the NF-κB pathway and weakens the immune response. Since the main protease (Mpro) is required for viral gene expression and replication, it is a promising target for antagonists to treat novel coronavirus disease and discovery of high resolution crystal structure of SARS-CoV-2-Mpro provide an opportunity for in silico identification of its possible inhibitors. In this study we intend to find novel and potential Mpro inhibitors from around 1830 chemically diverse and therapeutically important secondary metabolites available in the MeFSAT database by performing molecular docking against the Mpro structure of SARS-CoV-2 (PDB ID: 6LZE). After ADMET (absorption, distribution, metabolism, excretion, and toxicity) profile and binding energy calculation through MM-GBSA for top five hits, Sterenin M was proposed as a SARS-CoV2-Mpro inhibitor with validation of molecular dynamics (MD) simulation study. Sterenin M seems to have the potential to be a promising ligand against SARS-CoV-2, and thus it requires further validation by in vitro and in vivo studies.
Keywords: SARS-CoV2, Main protease (Mpro), MeFSAT database, Free energy calculation, Computational drug prediction
Graphical abstract

1. Introduction
The whole world is suffering from current unique and dynamic coronavirus disease 2019 (COVID-19) pandemic, due to the global outbreak of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). SARS-CoV-2 emerged in late December 2019 in Wuhan, China's Hubei province, and then spread across the globe, affecting millions of people [1]. In massive populations, it primarily causes respiratory or gastrointestinal sickness with symptoms like fever, pneumonia, diarrhoea vomiting and shortness of breath that needs urgent intensive care [2]. As of April 7, 2021, more than 132 million confirmed cases including 2,904,013 deaths from all around the world had been reported to World Health Organization (WHO) and the number of incidents and deaths increasing due to the unavailability of clinically effective therapies to prevent SARS-CoV-2 infection and available treatment is restricted to symptom-relieving drugs. It is a challenging task for both pharmacology and medical science researchers to develop effective treatments, such as a vaccine and a small molecular drug to paralyse COVID-19 outbreak.
SARS-CoV-2 is enveloped, positive stranded RNA virus belongs to the Betacoronavirus genus classified under Coronaviridae family including severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) [3]. The receptor binding domain (RBD) of spike protein present on SARS-CoV-2 interacts with the receptor on the eukaryotic membrane surface called human angiotensin-converting enzyme (ACE-2), to hijacks the host cell's molecular machinery and produce more copies (Fig. 1 ). After acid-dependent proteolytic cleavage of the S-protein and fusion of the host cell membrane with viral membrane, the virus can enter the cytosol of the host cell and release its RNA with utmost priority of its own genome replication [4]. SARS-CoV-2 genome contains two open reading frames 1a and 1b (ORF1a and ORF1b) genes that encodes two overlapping replicase proteins pp1a and pp1b. Main protease (Mpro) and Papain-like protease (PLpro) are two major viral enzymes that acted upon pp1a and pp1b polyproteins to generate 16 non-structural proteins (NSPs) involved in viral replication, transcription and packing of the virus nuclear material into the capsid protein coat [5]. Viral proteins such as S-protein, Mpro, PLpro, and RNA dependent RNA polymerase are significant targets for antiviral drug development due to their engagement in the biochemical events and/or control on the replication cycle of the virus [6]. Of all viral target proteins, Mpro also known as 3-Chymotrypsin like protease (3-CLpro) has received a lot of attention from the researchers around the globe due to its vital role in the processing of polyproteins and controlling viral replication and transcription of SARS-CoV-2 genome. Additionally, Mpro of two CoVs infecting pigs have been found to responsible for suppressing the host's immune system [7]. They act as IFN antagonist by cleaving the nuclear transcription factor kappa B (NF-κB) essential modulator (NEMO), which is essential for activation of NF-κB pathway [8]. It has been reported that, the Mpro of PDCoV also impairs the JAK-STAT pathway [9]. As a result, developing quintessential enzyme Mpro specific inhibitors would be crucial for blocking viral replication and preventing host immune system suppression without any toxicity due to unavailability of Mpro human analogue.
Fig. 1.

Illustration depicting the life cycle of SARS-CoV-2 and function of Mpro as protease to produce non-structural proteins involved in viral replication and transcription.
In order to develop potential SARS-CoV-2 Mpro blocker, two compounds 11a and 11b have been designed based on the knowledge of active residue of the binding site and developed synthetically which showed 100% and 96% inhibition activity at 1 μM, respectively [10]. Other wide-spectrum Mpro inhibitors, such as N1, N3, and N9, have been also synthesised to track down various corona viruses [11,12]. Since these compounds are synthetically designed and need to cross barrier of substantial clinical trials before coming to any conclusion, natural molecules as lead compounds have a direct path to prove their competence as Mpro inhibitors. Nowadays, incremental advancement of computer hardware and software technology has increased the odds of discovering new drugs from metagenomic databases of natural small molecules. Number of studies have been employed the use of advance computational tools including docking, molecular dynamics simulation, and combination of developed in silico methods for providing natural compounds as potential lead molecule against SARS-CoV-2 virus [[13], [14], [15], [16], [17]].
This study was conducted to discover potential Mpro inhibitor from around 1830 chemically diverse and therapeutically important secondary metabolites available in the Medicinal Fungi Secondary metabolites And Therapeutics (MeFSAT) database through performing molecular docking against the Mpro structure of SARS-CoV-2 (PDB ID: 6LZE). At the next step ADMET (drug absorption, distribution, metabolism, excretion, and toxicity) profile and binding energy calculation through MM-GBSA was performed for top-5 hit ligands and five known Mpro inhibitors. Finally, a 100-ns MD simulation was conducted to evaluate the nature of the ligand-target interaction under simulated physiological conditions for the most compatible drug-like molecule, Sterenin M (MSID001413), which can be used in the pursuit of truly required medication of COVID-19.
2. Materials and methods
2.1. Preparation of protein
The X-ray crystal structure of SARS-CoV-2 main protease (Mpro) in complex with ~{N}-[(2~{S})-3-cyclohexyl-1-oxidanylidene-1-[[(2~{S})-1-oxidanylidene-3-[(3~{S})-2-oxidanylidenepyrrolidin-3-yl]propan-2-yl]amino]propan-2-yl]-1~{H}-indole-2-carboxamide (11a) inhibitor was imported from the Protein Data Bank (PDB ID: 6LZE; 1.50 Å resolution) as the receptor for molecular docking study [10]. The Protein Preparation Wizard (PrepWizard) available in Maestro was used for the preparation and minimization of the energy of protein [18]. In which, crystallographic water molecules and attached ligands were removed, followed by adding missing hydrogen and/or side chain atoms, and proper charges and protonation states being assigned to acidic and basic amino acid residues at pH 7.0 [19]. Finally, the pre-processed protein structure was minimized using the OPLS-2005 force-field with a root mean square deviation (RMSD) cut-off value of 0.30. Protein minimization process was applied to relieved the steric clashes among the residues caused by the addition of hydrogen atoms [20].
2.2. Ligand preparation
The MeFSAT database containing chemically diverse secondary metabolites of solely fungal origin, was used to find natural product-based drug leads [21]. The LigPrep module of Maestro was used to prepare 1830 compounds retrieved from MeFSAT as well as the five known Mpro blockers (N1, N3, N9, 11a and 11b) [[10], [11], [12]]. All ligands were prepared by adding hydrogen atoms, assigning proper bond orders, adjusting bond lengths and angles, stereo chemistries, and ring conformations. Subsequently, Epik ionization tool was utilized to set ionization state at the neutral pH 7.0 [22]. Using the OPLS-2005 force-field, partial charges were applied to the structures, preceded by energy minimization process.
2.3. Receptor grid generation and molecular docking
A grid box [co-ordinates X −10.86, Y 11.94, Z 693.32, 10 X 15 X 15] was generated around the protein (PDB: 6LZE) by selecting the co-crystallized inhibitory peptide ligand 11a in Receptor Grid Generation tool of Glide in Maestro. All docking calculations for the prepared ligand molecules were performed in Glide's extra precision (XP) mode using default settings. Prior to docking of all ligands, Glide's XP mode was used to redock the co-crystallized ligand 11a (Pubchem ID: 145343771) into the active site of the protein to ensure the reliability of docking protocol [23]. Following Glide's best pose based on energy, post docking minimization was performed. They were then exported and analysed in the BIOVIA Discovery Studio (DS) visualizer to explore how the functional groups of ligands interact with the amino acids in the binding site of the protein to form hydrogen bonds and hydrophobic interactions. Among all docked molecules, top five metabolites with the highest binding affinity and the most interactions with amino acids that interact with the essential residues that form the catalytic dyad HIS41 and/or CYS145 of SARS-CoV2-Mpro were chosen for further examination.
2.4. ADMET properties assessment
All drug molecules with therapeutic potential against specific disease have to go through preclinical and clinical trials for the evaluation of absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties. To be a good new drug candidate, there must be fine balancing between drug-likeness and ADMET profiling [24]. Early drug-likeness prediction and ADMET of possible drug molecules can thus help to prevent expensive late-stage drug failure in the drug development phase and therefore, the ADMET properties of the considered compounds were calculated using the pkCSM - pharmacokinetics server (http://biosig.unimelb.edu.au/pkcsm/) by providing SMILES (Simplified Molecule Input Line Entry Specification) of the compounds downloaded from PubChem to generate physiochemical and pharmacological properties [25]. It computed in vivo Absorption parameters like; Water solubility (SK atomic types, mg/L), Caco2 cell permeability (Human colorectal carcinoma), Human Intestinal Absorption (HIA, %), P-glycoprotein inhibition and skin permeability (logKp, cm/hour). The metabolic parameters were determined using in vivo Cytochrome P450 2C19, Cytochrome P450 2C9, Cytochrome P450 2D6, and Cytochrome P450 3A4 inhibition, as well as in vivo Cytochrome P450 2D6 and in vivo Cytochrome P450 3A4 substrate. The penetration of the Blood-Brain Barrier (BBB), Lipinski's Rule (Rule of Five), and CNS permeability were all employed to determine distribution property. Excretion is another crucial factor, with many drugs being removed from clinical trials due to inadequate renal clearance and thus, Total Renal Clearance and Renal OCT2 Substrate were applied in this analysis to ascertain the excretion performance of selected metabolites. To determine the toxicity of the compounds under investigation, a variety of important endpoints were used, including the Ames test, a 2-year carcinogenicity bioassay in mouse and rat, and an in vivo Ames test result in the TA100 strain (Metabolic activation by rat liver homogenate). In the era of green chemistry, concerns about potential ecotoxic effects of pharmaceuticals prompted us to investigate their toxicity against major organisms (Tetrahymena pyriformis and fat-head minnow) for ecotoxicity assessment in this study.
2.5. Binding free energy calculations
The relative binding free energies of docked complexes of top five hits and five known inhibitor molecules with Mpro were evaluated using the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method available in Prime wizard of Maestro. These protein-ligand complexes were used as inputs for calculating the binding free energy using the OPLS-2005 force field. The following equation was used to calculate the binding free energy ΔGbind:
Where, E_complex is the free energy of minimized protein-ligand complex, E_protein is the free energy of minimized protein only, while E_ligand is the free energy of minimized ligand.
2.6. Molecular dynamics simulation
Molecular Dynamics (MD) simulation is considered as the most significant tool for understanding the nature of the fundamental structure and function of biological macromolecules [26]. This approach aids in determining the underlying dynamics and how they relate to the biomolecular activity of enzymes. MD simulation analysis was conducted for SARS-CoV2-Mpro-11a and SARS-CoV2-Mpro-Sterenin M complexes in two sets of experiments using Desmond, where the former was taken as control [27]. When compared to other metabolites in the MeFSAT database and known Mpro blockers, Sterenin M has shown important interactions with amino acid residues with strong docking score as well as have good ADMET profile. To allow complex relaxation, both complexes were prepared using a protein preparation wizard prior to MD simulation. The following requirements are fulfilled: adding hydrogens, assigning bond orders, filling in missing amino acid side chains and loops with hydrogen-bond assignment optimization, and sampling water orientations (pH 7.0). The simulation system was built using the TIP3P solvent model, and the boundaries were defined using an orthorhombic box shape with a dimension of 10 Å × 10 Å × 10 Å and filled with water molecules. Then, sodium and calcium ions were added as counter ions to neutralize charges of the system. The MD was performed with the NPT (constant Number of particles, Pressure, and Temperature) ensemble, for 100ns with 300 K and 1.01 bar, constant volume, Smooth Particle Mesh Ewald (PME) method. A simple point charge solvent model was set to measure the trajectory. On completion of simulation, Simulation Interaction Diagram wizard was used to sketched plots and figures of Ligand–protein interaction profiles, root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), for Ligand and Protein changes.
3. Results
3.1. Molecular docking studies
The prepared 3D molecular structure of co-crystallized inhibitor 11a was docked into the binding site of viral protease after defining the grid box using Glide's Receptor Grid Generation tool in Maestro. The docked orientation of 11a was compared with the crystallized orientation. The identical docked orientation represented in Fig. 2 , which indicated that the docking protocol could be reliable for the final docking studies of the selected compounds against the SARS-CoV2-Mpro. Analysis of the co-crystallized inhibitor (11a) revealed its molecular interaction with the binding site of viral protease SARS-CoV-Mpro. The synthetically developed molecule 11a interacted through hydrogen bonds with residues Cys145, Gly143, His163, Phe140, Glu166 and through alkyl, Pi-Sulfur and van der Waals interactions with Asn142, Ser144, Met49, His41, Pro52, Cys44, Tyr54, Arg188, Asp187, Leu167, Gln189, Gln192, Thr190, Ala191, Pro168, Met165, His164, His172 and Leu141 of SARS-CoV2-Mpro catalytic site.
Fig. 2.

Orientation and position of 11a in the binding cleft of Mpro (PDB ID: 6LZE) of SARS CoV-2 is shown in 3D representation where ligand (11a) in cyan blue is representing co-crystallized orientation and in maroon is the orientation of same ligand obtained after performing docking.
Total 1830 fungal metabolites obtained from MeFSAT database and five known Mpro inhibitors were considered for docking analysis using Glide XP lead optimization protocol of Schrödinger package. On the competition of docking, total of five fungal metabolites, namely Poh 3, Epi-phelligrin A, Sterenin M, Termitomycamide B and Enokipodin D showed effective binding with HIS41 and/or CYS145 of catalytic dyad along with multiple interactions with other amino acid residues in active site of SARS-CoV2-Mpro and showed binding energy close to known Mpro inhibitors were selected. The Glide Score and interacting amino acid residues of the selected lead molecules and five Mpro antagonists presented in Table 1 .
Table 1.
Docking scores and the contributing binding residues of known Mpro inhibitors and selected top five fungal metabolites generated using XP docking.
| Compounds |
Glide Score (Kcal/mol) |
Contributing Binding Residues |
|---|---|---|
| Known inhibitors of Mpro | ||
| N1 | −8.255 | PHE140, LEU141, CYS145, HIS172, SER144, HIS163, ASN142, GLY143, LRU27, THR25, THR26, HIS164, MET49, GLU166, MET165, GLN189, LEU167, THR190, GLN192, ALA191, PRO168, ARG188, PRO52, ASP187, TYR54, HIS41, THR45, CYS44, VAL42 |
| 11b | −8.158 | PHE140, HIS163, HIS172, ASP187, TYR54, CYS44, MET49, HIS41, ARG188, GLN189, GLN192, THR190, PRO168, LEU167, MET165, HIS164, GLU166, ASN142, GLY143, SER144, CYS145, LEU141 |
| 11a | −7.857 | PHE140, ASN142, SER144, GLY143, CYS145, MET49, HIS41, PRO52, CYS44, TYR54, ARG188, ASP187, LEU167, GLN189, GLN192, THR190, ALA191, PRO168, MET165, HIS164, HIS172, HIS163, LEU141, GLU166 |
| N9 | −5.861 | PHE140, CYS145, SER144, LEU27, ASN142, HIS163, LEU141, GLY170, LEU167, PRO168, GLU166, GLN189, MET49, MET165, HIS164, HIS41, GLY143 |
| N3 |
−5.482 |
PHE140, HIS172, HIS163, SER144, HIS164, MET165, GLU166, GLY170, LEU167, PRO168, THR190, GLN189, GLN192, TYR54, PRO52, HIS41, CYS44, ASP187, ARG188, MET49, LEU27, THR25, THR26, CYS145, GLY143, LEU141, ASN142 |
|
Top 5 compounds from MeFSAT Database | ||
| Poh 3 | −8.779 | THR169, GLY170, HIS172, PHE140, LEU141, SER139, GLY138, GLY145, LYS137, ILE136, VAL171, GLU166, GLN189, THR190, GLN192 |
| Epi-phelligrin A | −8.632 | PRO168, LEU167, GLN192, ARG188, VAL186, MET49, HIS41, THR25, THR45, CYS44, MET165, GLU166, THR190, GLN189 |
| Sterenin M | −8.431 | GLU166, LEU141, HIS172, PHE140, SER144, HIS163, CYS145, GLY143, ASN142, THR26, LEU27, THR25, THR24, HIS41, CYS44, TYR54, ASP187, ARG188, MET49, GLN189, HIS164, MET165 |
| Termitomycamide B | −6.694 | CYS44, THR25, THR26, LEU27, CYS145, HIS163, SER144, LEU141, PHE140, HIS172, GLY143, ASN142, VAL42, PRO52, TYR54, VAL186, LEU167, MET165, PRO168 |
| Enokipodin D | −5.645 | PRO168, MET165, HIS41, MET49, TYR54, VAL186, HIS164, GLN189, THR190, GLN192 |
All five known Mpro inhibitors, N1, 11b, 11a, N9 and N3 interact with both the residues Cys145 and His41 of catalytic dyad along with important residues of active site that are Glu166, Phe140, His163, Gly143, His164, Met165 with binding energy ranging from −5.482 to −8.255 kcal/mol. Interactions of Mpro of SARS-CoV-2 with known inhibitors are shown in Fig. 3 and with five fungal metabolites are shown in Fig. 4 . Among the five fungal metabolites, only Sterenin M interacts with both residues of catalytic dyad, Cys145 with Pi-Sulfur bond and His41 with hydrogen bond as well as alkyl and Pi-alkyl interaction, while Poh 3 and Termitomycamide B interacts with only Cys145 and Epi-phelligrin A and Enokipodin D interacts with only His41. Furthermore, Sterenin M also interact by making hydrogen bonds with the key amino acids of Mpro Gly143, His163, Phe140 and Glu166 in same fashion as done by 11a and 11b. The structural features of all selected known Mpro inhibitors and these selected fungal metabolites are presented in Table 2 .
Fig. 3.

Interaction profile of five known Mpro inhibitors docked with SARS-CoV2-Mpro.
Fig. 4.

Interaction profile of selected five fungal metabolites from MeFSAT database docked with SARS-CoV2-Mpro.
Table 2.
Structures and chemical properties of known Mpro inhibitors and selected fungal metabolites.
| Compound | Structure | Molecular Weight | LogP | #Rotatable Bonds | #Acceptors | #Donors | Surface Area |
|---|---|---|---|---|---|---|---|
| N1 | 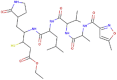 |
652.815 | 1.03562 | 17 | 10 | 6 | 269.231 |
| 11b | 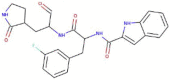 |
464.497 | 1.858 | 9 | 4 | 4 | 194.849 |
| 11a | 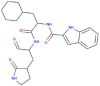 |
452.555 | 2.4466 | 9 | 4 | 4 | 192.810 |
| N9 | 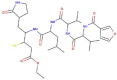 |
665.854 | 1.9683 | 18 | 9 | 6 | 276.425 |
| N3 |
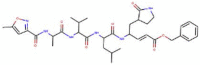 |
680.803 |
2.08362 |
17 |
9 |
5 |
286.079 |
| Poh 3 | 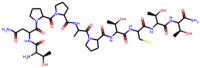 |
1001.131 | −7.9709 | 23 | 17 | 14 | 403.027 |
| Epi-phelligrin A | 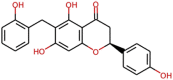 |
378.38 | 3.8063 | 3 | 6 | 4 | 160.452 |
| Sterenin M | 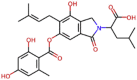 |
497.544 | 4.29482 | 8 | 7 | 4 | 208.802 |
| Termitomycamide B | 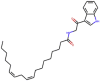 |
436.64 | 7.2803 | 17 | 2 | 2 | 193.115 |
| Enokipodin D | 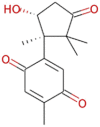 |
262.305 | 1.3771 | 1 | 4 | 1 | 111.736 |
3.2. ADMET properties assessment
Absorption, Distribution, Metabolism, Excretion, and Toxicity plays a significant role in medicinal chemistry. All the ADMET properties of the five known Mpro inhibitors and top five selected compounds are represented in Table 3 . The efficacy of a selected compound as an oral drug was determined using different models to measure property of absorption in which CaCO2 permeability and intestinal absorption were tested. In present study all the screened compounds show the CaCO2 permeability values in positive integer with exception of Poh 3, Epi-phelligrin A, and Sterenin M. Furthermore, Poh 3 demonstrated its intestinal absorption inability with 0%, while other screened compounds demonstrated absorption at greater than 40%, which is considered as reasonable absorption. The next crucial variable in absorption is skin permeability, and all of the substances under consideration have permeability values of less than −2.5 log Kp, suggesting poor permeability. P-glycoprotein is a component of the ATP-binding cassette (ABC) transporter, which is required for effective molecular transport across cell membranes. All screened compounds were tested as P-glycoprotein substrates as well as inhibitors of P-glycoprotein I and II. Except for Enokipodin D, all of the compounds were discovered to be substrates, implying that they can move through the cell membrane through the ABC transporter. Besides this, Poh 3 and Sterenin M were found to be ineffective as inhibitors for P-glycoprotein I and II transporters, implying that they are unable to inhibit both of these drug efflux pumps. Four different assays, namely fraction unbound, Volume of distribution (VDss), BBB permeability, and central nervous system (CNS) permeability were used to determine the distribution of the compounds in the body. The VDss assay is used to determine the total amount of medications required for uniform drug distribution in the bloodstream, and a value of less than −0.15 log is considered negative, while values greater than 0.45 log is interpreted good diffusion. However, 11a, 11b and Termitomycamide B show strong VDss values indicating faster diffusion in blood, while other compounds have low distribution capacity. The permeability of the Blood-Brain Barrier (BBB) determines a compound's ability to move to the brain. They will move BBB if the logBB values are greater than 0.3. None of the screened compounds have a logBB value greater than 0.3, implying that none of them would be able to cross BBB. Metabolism of the test drugs in the body was assessed using seven different cytochrome models. All the compounds were assessed for their ability to serve as a substrate for CYP2D6 and CYP3A4, as well as their inhibition of CYP1A2, CYP2C19, CYP2D6, CYP2C9, and CYP3A4. Out of all screened compounds, Poh 3, Sterenin M, and Enokipodin D predicted to remain inactive for CYP1A2, CYP2C19, CYP2D6, CYP2C9, and CYP3A4 inhibition. All of the screened compounds had different total clearance rates, and none of them appeared as a substrate for organic cation transporter 2 (OCT2). Furthermore, none of them predicted AMES toxicity, implying that these compounds are neither carcinogenic nor mutagenic. Skin sensitisation was not seen in any of the screened compounds but only Epi-phelligrin A, Sterenin M, and Enokipodin D tested negative for hepatotoxicity. The toxic effect of all screened compounds on T. pyriformis and fat-head Minnow are shown in Table 3 along with other essential ADMET properties.
Table 3.
ADMET properties of known Mpro inhibitors and selected fungal metabolites.
| Property | Model Name | Predicted Value |
Unit | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N1 | 11b | 11a | N9 | N3 | Poh 3 | Epi-phelligrin A | Sterenin M | Termitomycamide B | Enokipodin D | |||
| Absorption | Water solubility | −3.301 | −3.568 | −3.548 | −3.129 | −4.144 | −2.764 | −4.081 | −3.002 | −6.802 | −2.124 | Numeric (log mol/L) |
| Caco2 permeability | 0.522 | 0.248 | 0.454 | 0.573 | 0.639 | −0.822 | −0.11 | −0.712 | 0.151 | 0.504 | Numeric (log Papp in 10−6 cm/s) | |
| Intestinal absorption (human) | 41.769 | 74.619 | 75.471 | 49.568 | 57.884 | 0 | 87.22 | 52.154 | 88.452 | 97.425 | Numeric (% Absorbed) | |
| Skin Permeability | −2.741 | −2.749 | −2.889 | −2.737 | −2.734 | −2.735 | −2.735 | −2.735 | −2.721 | −3.712 | Numeric (log Kp) | |
| P-glycoprotein substrate | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | No | Categorical (Yes/No) | |
| P-glycoprotein I inhibitor | Yes | Yes | Yes | Yes | Yes | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| P-glycoprotein II inhibitor | No | Yes | Yes | No | Yes | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| Distribution | VDss (human) | −0.942 | 0.626 | 0.667 | −0.684 | −0.762 | −1.188 | −1.091 | −0.749 | 0.87 | −0.149 | Numeric (log L/kg) |
| Fraction unbound (human) | 0.271 | 0 | 0.093 | 0.282 | 0.067 | 0.589 | 0 | 0.031 | 0 | 0.511 | Numeric (Fu) | |
| BBB permeability | −1.614 | −0.813 | −0.587 | −1.438 | −1.261 | −2.193 | −0.976 | −1.528 | −0.324 | −0.081 | Numeric (log BB) | |
| CNS permeability | −4.24 | −3.287 | −3.111 | −3.807 | −3.568 | −5.93 | −3.013 | −3.274 | −2.483 | −2.905 | Numeric (log PS) | |
| Metabolism | CYP2D6 substrate | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) |
| CYP3A4 substrate | Yes | Yes | Yes | Yes | Yes | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| CYP1A2 inhibitor | No | No | No | No | No | No | Yes | No | No | No | Categorical (Yes/No) | |
| CYP2C19 inhibitor | No | No | No | No | No | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| CYP2C9 inhibitor | No | Yes | No | No | No | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| CYP2D6 inhibitor | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) | |
| CYP3A4 inhibitor | No | Yes | Yes | Yes | Yes | No | Yes | No | Yes | No | Categorical (Yes/No) | |
| Excretion | Total Clearance | −0.162 | 0.505 | 0.696 | 0.058 | 0.653 | 0.788 | 0.084 | 0.3 | 1.697 | 0.233 | Numeric (log ml/min/kg) |
| Renal OCT2 substrate | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) | |
| Toxicity | AMES toxicity | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) |
| Max. tolerated dose (human) | 0.87 | −0.669 | −0.527 | 0.47 | −0.348 | 0.618 | 0.249 | 0.419 | −0.243 | 0.359 | Numeric (log mg/kg/day) | |
| hERG I inhibitor | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) | |
| hERG II inhibitor | No | Yes | No | Yes | Yes | Yes | Yes | No | Yes | No | Categorical (Yes/No) | |
| Oral Rat Acute Toxicity (LD50) | 2.218 | 2.345 | 1.911 | 3.387 | 3.63 | 2.633 | 2.183 | 2.622 | 2.43 | 2.005 | Numeric (mol/kg) | |
| Oral Rat Chronic Toxicity (LOAEL) | 1.382 | 1.844 | 1.021 | 1.392 | 3.935 | 2.873 | 2.918 | 2.772 | 2.901 | 2.437 | Numeric (log mg/kg_bw/day) | |
| Hepatotoxicity | Yes | Yes | Yes | Yes | Yes | Yes | No | No | Yes | No | Categorical (Yes/No) | |
| Skin Sensitisation | No | No | No | No | No | No | No | No | No | No | Categorical (Yes/No) | |
| T. pyriformis toxicity | 0.285 | 0.317 | 0.356 | 0.285 | 0.285 | 0.285 | 0.29 | 0.285 | 0.382 | 0.47 | Numeric (log ug/L) | |
| Minnow toxicity | 4.519 | 3.463 | 2.071 | 4.62 | 4.136 | 12.433 | −0.412 | 1.892 | −1.909 | 2.127 | Numeric (log mM) | |
3.3. Binding free energy calculations
Binding energy is the energy released (ΔGBind) as a result of bond formation, or rather the interaction of the ligand with the protein, and it modifies the energies of both the free receptor and the ligand; additionally, these energies have a direct effect on the stability of the receptor-ligand complex. Negative values of free energy mean that a system is more stable. Table 4 entails the binding free energy change profiles of the top five fungal metabolites from the MeFSAT database in comparison with five known Mpro inhibitors. Because the ΔGBind for all three instances is less than −60.00 kcal/mol, the interaction of three control molecules, 11a, 11b, and N3, with the SARS-CoV2-Mpro is thought to occur highly spontaneously. Among the fungal metabolites, Sterenin M is the next best ligand in terms of binding free energy change, with ΔGBind equal to −49.57 kcal/mol. Other screened compounds with ΔGBind greater than −40.0 kcal/mol have low spontaneous interactions with Mpro, however this does not necessarily imply that the interaction is unfavourable; the value in the negative dictates the interaction to occur when offered a chance. Addition to the total energy, other parameters of the MM/GBSA profile including Hydrogen-bonding correction, Coulomb energy, Lipophilic energy, Pi-pi packing correction and van der Waals energy is also provided in Table 4.
Table 4.
MM/GBSA binding free energy change profiles of known Mpro inhibitors and selected top five fungal metabolites with SARS-CoV2-Mpro docked complexes.
| Ligand | ΔGBind (Kcal/mol) | ΔGCoulomb (Kcal/mol) | ΔGHbond (Kcal/mol) | ΔGLipo (Kcal/mol) | ΔGPacking (Kcal/mol) | ΔGvdW (Kcal/mol) |
|---|---|---|---|---|---|---|
| Known Mpro inhibitors interacting with SARS-CoV2-Mpro | ||||||
| N1 | −35.02 | −32.35 | −4.62 | −17.29 | −0.49 | −64.78 |
| 11b | −65.58 | −49.74 | −3.29 | −14.46 | −4.60 | −49.15 |
| 11a | −60.92 | −42.28 | −3.17 | −16.51 | −2.93 | −57.49 |
| N9 | −36.31 | −40.27 | −4.02 | −12.25 | 0 | −44.89 |
| N3 | −65.95 | −44.61 | −3.83 | −16.15 | −0.77 | −70.46 |
| Top five fungal metabolites interacting with SARS-CoV2-Mpro | ||||||
| Poh 3 | −28.28 | −37.25 | −7.09 | −14.52 | 0 | −54.88 |
| Epi-phelligrin A | −35.71 | −18.48 | −3.29 | −11.17 | −2.37 | −33.34 |
| Sterenin M | −49.57 | −39.75 | −3.71 | −10.71 | −2.24 | −49.98 |
| Termitomycamide B | −39.62 | −32.31 | −1.96 | −17.02 | −2.79 | −50.23 |
| Enokipodin D | −34.52 | −15.43 | −0.83 | −09.17 | 0 | −31.14 |
Note, meaning of abbreviations used in the table are as follows.
Coulomb—Coulomb energy.
Hbond—Hydrogen-bonding correction.
Lipo—Lipophilic energy.
Packing—Pi-Pi packing correction.
vdW—Van der Waals energy.
3.4. Molecular dynamics simulation
Sterenin M was chosen as the best hit based on interactions with important residues of binding site, docking score, ADMET and MM/GBSA profiles. The interaction profile of Sterenin M was compared to that of a known Mpro inhibitor, 11a, using 100ns MD simulations. In two sets of experiments using Desmond, MD simulation analysis for SARS-CoV2-Mpro-11a as control set and SARS-CoV2-Mpro-Sterenin M as test set was performed. Following the MD simulations, the Root Mean Square Deviation (RMSD) evaluation was carried out, which reflects the changes in the state of specific atoms with reference to their initial state. The docked pose of the protein and ligand in the complex serves as the reference frame, and the movement for this original orientation throughout MD simulation is assessed by aligning all of the protein frames in terms of the time. The RMSD movements in the protein portions are depicted in Fig. 5 on left Y-axis. Examining the RMSD of the protein during the simulation will indicate its integral 3D structural movement on a graph. The equilibration of the simulation can be demonstrated using RMSD analysis — Its conformational alterations at the end of the process are focused on a thermal energetically stable configuration. For tiny, globular proteins, variations in the range of 1–4 Å are completely reasonable. However, as the size of the protein becomes larger, this value range broadens. The SARS-CoV2-Mpro-11a complex (Fig. 5a) has a protein backbone with an RMSD of less than 2.5; however, the SARS-CoV2-Mpro-Sterenin M complex (Fig. 5b) has an RMSD of less than 4.0. Ligand RMSD (right Y-axis, plots of Fig. 5) reflects the ligand posture stability in respect to its docked position in the protein's binding cleft. The RMSD of a ligand for the backbone of a protein is referred to as the ‘Lig fit Prot’. For this, values slightly bigger than the protein's RMSD are acceptable but, if the observed values are significantly bigger than the protein's RMSD, the ligand is more likely to propose a stable position other than the native posture. For SARS-CoV2-Mpro-11a (Fig. 5a), the Lig fit Prot stays around 3.5 Å. The Lig fit Prot value for SARS-CoV2-Mpro-Sterenin M (Fig. 5b) remains below 4.8. The Root Mean Square Fluctuation (RMSF) is being used to depict restricted shifts in the protein chain (Fig. 6 ). The protein areas that differ the most in the simulation are shown by the peaks in the graph. Protein tails (N- and C-terminal) normally shift more than the rest of the protein. Alpha helices and beta strands, for example, are more rigid and inflexible than unstructured sections of proteins, and so fluctuate differently than loop-forming areas. The red and blue foundations, respectively, feature alpha-helical and beta-strand regions. These districts are distinguished by helices or strands that last for more than 70% of the re-enactment. Green-hued vertical bars separate protein deposits that come into contact with the ligand. The RMSF of the protein can also be connected to the exploratory x-beam B-factor (right Y-hub). Balanced correspondence should not be common because of the difference between the RMSF and B-factor definitions. Regardless, the reproduction findings should be consistent with the crystallographic data. The protein interacts with both ligands in complexes SARS-CoV2-Mpro-11a (Fig. 6a) and SARS-CoV2-Mpro-Sterenin M (Fig. 6b), along with the RMSF and B-factor definitions are comparable.
Fig. 5.

MD simulation Protein-ligand interaction root-mean-square deviation (RMSD) profile of (a) SARS-CoV2-Mpro-11a (b) SARS-CoV2-Mpro-Sterenin M.
Fig. 6.

MD simulation Protein-ligand interaction root-mean-square fluctuation (RMSF) profile of (a) SARS-CoV2-Mpro-11a (b) SARS-CoV2-Mpro-Sterenin M.
Throughout the simulation, interactions between proteins and ligands can be observed. The interactions in SARS-CoV2-Mpro-11a complex and the SARS-CoV2-Mpro-Sternine M complex can be characterised and summarised, as illustrated in Fig. 7 . The four types of protein-ligand interactions include hydrogen bonds, hydrophobic interactions, ionic interactions, and water bridge interactions. The ‘Simulation Interactions Diagram’ board can be used to examine the explicit subtypes of each relation form. The stacked bar outlines are standardised in the direction: for example, an estimate of 0.8 suggests that collaboration will be maintained for 80% of the simulation time. Quality scores exceeding 1.0 are possible because some protein build-up can create numerous connections of the same subtype with the ligand. The docking results for 11a are confirmed in Fig. 7a, where it interacts with Phe140, Gly143, Cys145, His163 and Glu166. Similarly, a very strong interaction by Sterenin M is represented in Fig. 7b, where it interacts with same amino acids which are Phe140, Gly143, Cys145, His163 and Glu166. A schematic portrayal of the interactions and contacts (Hydrogen bonds, Hydrophobic, Ionic, Water spans) is shown in Fig. 8 a for the SARS-CoV2-Mpro-11a complex and Fig. 8b for the SARS-CoV2-Mpro-Sterenine M complex. These diagrams depict how the ligand interacts with the deposits in each direction. The scale on one side of the map, which is illustrated by a darker shade of orange, shows that a few residues make multiple overt interactions with the ligand. The plots support the docking results, indicating that the associations proposed to form between ligand and amino acids during docking are generated with same amino acids during simulations.
Fig. 7.

Protein-Ligand interaction profile of crucial interacting amino acids during the course of MD simulation of (a) SARS-CoV2-Mpro-11a complex (b) SARS-CoV2-Mpro-Sterenin M complex.
Fig. 8.

Timeline representation of the interactions of ligand with amino acids for the complex (a) SARS-CoV2-Mpro-11a (b) SARS-CoV2-Mpro-Sterenin M.
4. Discussion
Coronaviruses (CoVs) are a large virus group that belong to zoonotic viruses. They are widely spread among humans and other animals and have the potential to cause global epidemics and pandemics [28]. The fact that CoVs are virions with crown-like projections contributes to their explanation and are identical in terms of organization and genomic expression with at least four structural proteins, namely S-protein, E-protein, M-protein, and N-protein and 16 non-structural proteins. AlphaCoV, betaCoV, gammaCoV, and deltaCoV are the four genera of CoVs. Among them alphaCoVs and betaCoVs are known to infect humans. To date, seven human CoVs namely 229E (alfaCoV), NL63 (alfaCoV), OC43 (betaCoV), HKU1 (betaCoV), MERS-CoV (betaCoV, which causes MERS), SARS-CoV (betaCoV, which causes SARS) and 2019-nCoV/SARS-CoV-2 have been identified. Other members of coronavirinae subfamily, namely Porcine Transmissible Gastroenteritis Virus (TGEV), Bovine Coronavirus (BCV), Avian Infectious Bronchitis Virus (IBV), Feline Infectious Peritonitis Virus (FIPV), Canine Coronavirus (CCoV), Porcine Hemagglutinating Encephalomyelitis Virus (HEV), and Turkey Coronavirus are unable to infect humans due to lack of the necessary S-proteins for cellular entry [1,3]. S-proteins are host-specific, so cross transmission is rare. However, it has been recorded on occasion, such as when SARS-CoV (2003) was transmitted from bats to humans and MERS-CoV (2005) was transmitted from camels [29,30]. SARS-CoV2, a member of the Covid community, has recently been identified as an infectious agent in humans due to cross infectivity. Since it has infected large populations, there is an unmet need to vaccinate the majority of the world's population in order to prevent the virus from spreading, and scientists from all over the world are working on developing vaccines.
In addition to vaccine development, scientists have focused their efforts and funds on finding molecules that can combat the SARS-CoV2 virus. Computational methods such as molecular docking and MD simulations have been used extensively to identify such compounds. A wide range of metabolites from various natural sources such as plants, bacteria and fungi have been investigated to check its potential as inhibitor of virus essential proteins. For instance, molecular docking and simulation studies of the fungal metabolite, Flaviolin indicate that it interacts with PLpro and Mpro proteases and can inhibit key processes for completion of SARS-CoV2 life cycle [15,31]. Similarly, a plant metabolite Lepidine E along with a polyphenol Hispidin found in a variety of plants and fungi has been tested against Mpro of the SARS-CoV2 virus [32]. It is noteworthy that there are numerous other reports on drug repurposing approach with expectation that one of available drug may interfere with the life cycle of the virus and restrict it [33,34]. Following a thorough investigation, two FDA-approved molecules, ABBV744 and Onalespibas, have been recently recommended as possible inhibitors of SARSCoV2 main protease [34]. Computational methods have proven to be extremely useful in a number of other areas of life science [[35], [36], [37]]. Molecular docking and MD simulations are the central strategies for the in silico analysis in this approach, and many lead compounds are recently revealed to have the potential to influence the biochemistry and life cycle of SARS-CoV2.
Several attempts have been made to discover Mpro inhibitors. In a virtual screening of ~8793 natural compounds carried out by Abdallah and colleagues, they found that Naringenin interacts with Mpro, and it has moderate anti-SARS-CoV-2 activity at non-cytotoxic micromolar concentrations with a significant selectivity index in an in vitro assay [38]. In another study, Ahmed and collaborators targeted 1397 FDA-approved antiviral and anti-infection agents to find inhibitors for Mpro, their best compound was Amikacin which made hydrogen bonds with crucial residues Phe140, Cys145 and Glu166 [39]. Under current study the fungal metabolite, Sterenin M showed Pi-Sulfur bond with Cys145 as well as hydrogen bond interaction to crucial residues Phe140, Gly143, His163 and Glu166. Besides, their research did not made use of any known Mpro inhibitor as control to compare their results. Efforts are also made to repurposing 10,254 drugs from DrugBank as Mpro inhibitors using QSAR model followed by docking and MD simulation [40]. In a review on recent progress in the drug development targeting SARS-CoV-2 main protease done by Cui and colleagues recognized published inhibitors for Mpro such as disulfiram, carmofur, Ebselen, α-ketoamide 13b, peptidomimetic inhibitor N3, peptidomimetic aldehydes 11a/11b, shikonin, tideglusib, PX-12, GC-376, TDZD-8, these were either discovered by ab initio designed drugs or using drug repurposing approaches. According to the same review, 11a and 11b are the best inhibitors for SARS-CoV2-Mpro discovered to date, with strong antiviral activity [41]. This supports our choice to use 11a as the reference inhibitor in our MD simulation study. With the restricted ability to work with SARS-CoV2 due to the requirement of a Biosafety Level 4 (BSL4) arrangement, more in silico exploration using docking and MD simulations is becoming the backbone of the drug discovery.
For millennia, medicinal fungi have been used to treat human illnesses in traditional remedies. Fungi are abundant in secondary metabolites, which provide a valuable and diverse chemical resource of natural products with potential bioactivity. Alexander Fleming's discovery of penicillin from fungus Penicillium notatum in 1928 is the classical example of bioactive fungal metabolite [42]. Since then, fungal metabolites have been widely used in pharmaceuticals, agriculture, and food industries. Even So, fungal metabolites are poorly explored in contrast to other natural compounds and there is a significant chance that few of them interact with Mpro. Based on this underlying principle MeFSAT database containing 1830 chemically diverse therapeutic metabolites were used in pursuing of this research. In present study, Sterenin M was identified as the lead fungal metabolite based on the result of molecular docking, ADMET profile, MM/GBSA profile and MD simulation. Sterenin M is an isoprenylated depside, which was first isolated in the year of 2014 from a culture of the mushroom Stereum hirsutum [43]. In view of unavailability of medication for corona virus infection, present study proposes Sternine M as the lead molecule that interact with SARS-CoV2-Mpro catalytic dyad Cys145 and His41 to inhibit its function and can be beneficial in forthcoming in vitro and in vivo studies for COVID-19 therapeutics.
Funding sources
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We acknowledge University Grants Commission (UGC), New Delhi, India for providing the fellowship for the award of ‘CSIR-NET Junior Research Fellowship (JRF)’ to J.P. Authors are thankful to Department of Chemistry, School of Sciences, Gujarat University, for allowing to use license version of Schrodinger suit and Department of Microbiology and Biotechnology, School of Sciences, Gujarat University, DST-FIST Sponsored Department, for providing necessary facilities to perform experiments. We acknowledge GSBTM, DST, Government of Gujarat for providing Bioinformatics Node facility and Finishing School support. We acknowledge GUJCOST, DST, Government of Gujarat for Super-computing facility provision.
References
- 1.Gabutti G., d'Anchera E., Sandri F., Savio M., Stefanati A. Coronavirus: update related to the current outbreak of COVID-19. Infect. Dis. Ther. 2020;9:241–253. doi: 10.1007/s40121-020-00295-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jutzeler C.R., Bourguignon L., Weis C.V., Tong B., Wong C., Rieck B., et al. Comorbidities, clinical signs and symptoms, laboratory findings, imaging features, treatment strategies, and outcomes in adult and pediatric patients with COVID-19: a systematic review and meta-analysis. Trav. Med. Infect. Dis. 2020;37 doi: 10.1016/j.tmaid.2020.101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shukla A., Parmar P., Kapoor G., Goswami D., Jha C.K., Patel B., et al. Curse of La Corona: unravelling the scientific and psychological conundrums of the 21st century pandemic. Mol. Divers. 2021 doi: 10.1007/s11030-020-10167-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sayed A.M., Khattab A.R., AboulMagd A.M., Hassan H.M., Rateb M.E., Zaid H., et al. Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale. RSC Adv. 2020;10:19790–19802. doi: 10.1039/d0ra04199h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amin S.A., Banerjee S., Ghosh K., Gayen S., Jha T. Protease targeted COVID-19 drug discovery and its challenges: insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Bioorg. Med. Chem. 2021;29:337–339. doi: 10.1016/j.bmc.2020.115860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui W., Yang K., Yang H. Recent progress in the drug development targeting SARS-CoV-2 main protease as treatment for COVID-19. Front Mol Biosci. 2020;7:1–10. doi: 10.3389/fmolb.2020.616341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lei J., Hilgenfeld R. RNA-virus proteases counteracting host innate immunity. FEBS Lett. 2017;591:3190–3210. doi: 10.1002/1873-3468.12827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai D., Wang A., Cao Y., Zhou K., Mao Z., Dong X., et al. Bioactive dibenzo-α-pyrone derivatives from the endophytic fungus rhizopycnis vagum Nitaf22. J. Nat. Prod. 2016;79:2022–2031. doi: 10.1021/acs.jnatprod.6b00327. [DOI] [PubMed] [Google Scholar]
- 9.Zhu X., Wang D., Zhou J., Pan T., Chen J. Porcine deltacoronavirus nsp5 antagonizes type I interferon signaling by cleaving STAT2. J. Virol. 2017;91:1–14. doi: 10.1128/JVI.00003-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. 80- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020 doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 12.Yang H., Xie W., Xue X., Yang K., Ma J., Liang W., et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005;3:1–11. doi: 10.1371/journal.pbio.0030324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel C.N., Goswami D., Jaiswal D.G., Parmar R.M., Solanki H.A., Pandya H.A. Journal of Molecular Graphics and Modelling Pinpointing the potential hits for hindering interaction of SARS-CoV-2 S-protein with ACE2 from the pool of antiviral phytochemicals utilizing molecular docking and molecular dynamics ( MD ) simulations. J. Mol. Graph. Model. 2021;105 doi: 10.1016/j.jmgm.2021.107874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patel C.N., Kumar S.P., Pandya H.A., Rawal R.M. Identification of potential inhibitors of coronavirus hemagglutinin-esterase using molecular docking, molecular dynamics simulation and binding free energy calculation. Mol. Divers. 2021;25:421–433. doi: 10.1007/s11030-020-10135-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rao P., Patel R., Shukla A., Parmar P., Rawal R.M., Saraf M. Identifying structural – functional analogue of GRL0617 , the only well - established inhibitor for papain - like protease ( PLpro ) of SARS - CoV2 from the pool of fungal metabolites using docking and molecular dynamics simulation. Mol. Divers. 2021 doi: 10.1007/s11030-021-10220-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Basu A., Sarkar A., Maulik U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep. 2020;10:1–15. doi: 10.1038/s41598-020-74715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar B.K., Faheem, Sekhar K.V.G.C., Ojha R., Prajapati V.K., Pai A., et al. Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases. J. Biomol. Struct. Dyn. 2020:1–24. doi: 10.1080/07391102.2020.1824814. 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 19.Sastry G.M., Adzhigirey M., Sherman W. Protein and ligand preparation : parameters , protocols , and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013;27:221–234. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 20.Shivakumar D., Williams J., Wu Y., Damm W., Shelley J., Sherman W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the. J. Chem. Theor. Comput. 2010;6:1509–1519. doi: 10.1021/ct900587b. [DOI] [PubMed] [Google Scholar]
- 21.Sahoo A.K., Kumaravel K., Mohanraj K., Samal A. MeFSAT : a curated natural product database speci fi c to secondary metabolites of medicinal fungi †. RSC Adv. 2021;11:2596–2607. doi: 10.1039/d0ra10322e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shelley J.C., Cholleti A., Frye L.L., Greenwood J.R., Timlin M.R., Uchimaya M. Epik: a software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007;21:681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 23.Halgren T.A., Murphy R.B., Friesner R.A., Beard H.S., Frye L.L., Pollard W.T., et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 24.Kar S., Leszczynski J. Open access in silico tools to predict the ADMET profiling of drug candidates. Expet Opin. Drug Discov. 2020;15:1473–1487. doi: 10.1080/17460441.2020.1798926. [DOI] [PubMed] [Google Scholar]
- 25.Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karplus M., McCammon J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002;9:646–652. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- 27.Bowers K., Chow D., Xu H., RD-SP . 2006. Undefined. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. n.D. [Google Scholar]
- 28.St John S.E., Tomar S., Stauffer S.R., Mesecar A.D. Targeting zoonotic viruses: structure-based inhibition of the 3C-like protease from bat coronavirus HKU4 - the likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS) Bioorg. Med. Chem. 2015;23:6036–6048. doi: 10.1016/j.bmc.2015.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., et al. The newly emerged SARS-Like coronavirus HCoV-EMC also has an “Achilles'’ heel": current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4:248–250. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang H., Bartlam M., Rao Z. Drug design targeting the main protease, the achilles heel of coronaviruses. Curr. Pharmaceut. Des. 2006;12:4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 31.Rao P., Shukla A., Parmar P., Rawal R.M., Patel B.V., Saraf M., et al. Proposing a fungal metabolite-flaviolin as a potential inhibitor of 3CLpro of novel coronavirus SARS-CoV-2 identified using docking and molecular dynamics. J. Biomol. Struct. Dyn. 2020:1–13. doi: 10.1080/07391102.2020.1813202. 0. [DOI] [PubMed] [Google Scholar]
- 32.Serseg T., Benarous K., Yousfi M. Hispidin and Lepidine E: two natural compounds and folic acid as potential inhibitors of 2019-novel coronavirus main protease (2019-nCoVMpro), molecular docking and SAR study. 2020. ArXiv. [DOI] [PubMed]
- 33.Cavasotto C.N., Di Filippo J.I. In silico drug repurposing for COVID-19: targeting SARS-CoV-2 proteins through docking and consensus ranking. Mol Inform. 2021;40:1–8. doi: 10.1002/minf.202000115. [DOI] [PubMed] [Google Scholar]
- 34.Fakhar Z., Khan S., AlOmar S.Y., Alkhuriji A., Ahmad A. ABBV-744 as a potential inhibitor of SARS-CoV-2 main protease enzyme against COVID-19. Sci. Rep. 2021;11:1–15. doi: 10.1038/s41598-020-79918-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pandya P.N., Kumar S.P., Bhadresha K., Patel C.N., Patel S.K., Rawal R.M., et al. Identification of promising compounds from curry tree with cyclooxygenase inhibitory potential using a combination of machine learning, molecular docking, dynamics simulations and binding free energy calculations. Mol. Simulat. 2020;46:812–822. doi: 10.1080/08927022.2020.1764552. [DOI] [Google Scholar]
- 36.Parmar P., Shukla A., Rao P., Saraf M., Patel B., Goswami D. The rise of gingerol as anti-QS molecule: darkest episode in the LuxR-mediated bioluminescence saga. Bioorg. Chem. 2020;99:103823. doi: 10.1016/j.bioorg.2020.103823. [DOI] [PubMed] [Google Scholar]
- 37.Patel S., Waghela B., Shah K., Vaidya F., Mirza S., Patel S., et al. Silibinin, A natural blend in polytherapy formulation for targeting Cd44v6 expressing colon cancer stem cells. Sci. Rep. 2018;8:1–13. doi: 10.1038/s41598-018-35069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abdallah H.M., El-Halawany A.M., Sirwi A., El-Araby A.M., Mohamed G.A., Ibrahim S.R.M., et al. Repurposing of some natural product isolates as SARS-COV-2 main protease inhibitors via in vitro cell free and cell-based antiviral assessments and molecular modeling approaches. Pharmaceuticals. 2021;14:213. doi: 10.3390/ph14030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahmed M.Z., Zia Q., Haque A., Alqahtani A.S., Almarfadi O.M., Banawas S., et al. FDA-approved antiviral and anti-infection agents as potential inhibitors of SARS-CoV-2 main protease: an in silico drug repurposing study. J Infect Public Health. 2021;14:611–619. doi: 10.1016/j.jiph.2021.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tejera E., Munteanu C.R., López-Cortés A., Cabrera-Andrade A., Pérez-Castillo Y. Drugs repurposing using QSAR, docking and molecular dynamics for possible inhibitors of the SARS-CoV-2 Mpro protease. Molecules. 2020;25:5172. doi: 10.3390/molecules25215172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui W., Yang K., Yang H. Recent progress in the drug development targeting SARS-CoV-2 main protease as treatment for COVID-19. Front Mol Biosci. 2020;7:1–10. doi: 10.3389/fmolb.2020.616341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fleming A. The discovery of penicillin. Br. Med. J. 1955;1:711. doi: 10.1136/bmj.1.4915.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang B.T., Qi Q.Y., Ma K., Pei Y.F., Han J.J., Xu W., et al. Depside α -glucosidase inhibitors from a culture of the mushroom Stereum hirsutum. Planta Med. 2014;80:918–924. doi: 10.1055/s-0034-1382828. [DOI] [PubMed] [Google Scholar]