

Abstract
INTRODUCTION:
Machine learning models were used to discover novel disease trajectories for autosomal dominant Alzheimer’s disease.
METHODS:
Longitudinal structural MRI, amyloid PET, and fluorodeoxyglucose PET were acquired in 131 mutation carriers and 74 non-carriers from the Dominantly Inherited Alzheimer Network; the groups were matched for age, education, sex, and apolipoprotein ε4 (APOE ε4). A deep neural network was trained to predict disease progression for each modality. Relief algorithms identified the strongest predictors of mutation status.
RESULTS:
The Relief algorithm identified the caudate, cingulate, and precuneus as the strongest predictors among all modalities. The model yielded accurate results for predicting future Pittsburgh Compound-B (R2=0.95), fluorodeoxyglucose (R2=0.93), and atrophy (R2=0.95) in mutation carriers compared to non-carriers.
DISCUSSION:
Results suggest a sigmoidal trajectory for amyloid, a biphasic response for metabolism, and a gradual decrease in volume, with disease progression primarily in subcortical, middle frontal, and posterior parietal regions.
Keywords: Autosomal dominant Alzheimer disease (ADAD), Machine learning, Pittsburgh compound B (PiB), Fluorodeoxyglucose (FDG), Magnetic resonance imaging (MRI)
1. INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia, accounting for 60%–70% of the 50 million dementia cases worldwide [1]. AD leads to slow cognitive decline, behavioral and psychiatric disorders, and impairments in functional status. Pathological features of AD include the accumulation of amyloid-beta (Aβ) plaques, neurofibrillary tau tangles, and neuronal/synaptic losses that correspond with atrophy and decreased glucose metabolism [2]. The most common form of AD occurs in older age and is known as late-onset Alzheimer’s disease (LOAD). Autosomal dominant Alzheimer’s disease (ADAD) accounts for less than 1% of all AD cases and is caused by pathogenic mutations in amyloid precursor protein (APP), presenilin 1 (PSEN1), or presenilin 2 (PSEN2) genes that lead to early increases in Aβ deposition in the brain, which, in turn, is hypothesized to initiate a cascade that causes cognitive decline [3,4]. The age of onset of cognitive impairment in ADAD mutation carriers (MC) is earlier than LOAD and remains fairly consistent within a family, allowing for calculation of the estimated age of symptom onset (EAO) [5].
Multiple neuroimaging methods have been used to evaluate in vivo changes in the brain due to AD. [11C]Pittsburgh Compound-B (PiB) has high affinity for Aβ plaques, with distributions similar to those seen at autopsy [6]. PiB PET has been employed in ADAD to identify amyloid deposition, with amyloid deposition identified more than 20 years prior to EAO in MC [7–10]. Studies have also shown increases in PiB retention in MC are associated with a worsening cognitive performance, a decrease in glucose metabolism, and a decrease in hippocampal volume [7,11].
[18F]Fluorodeoxyglucose (FDG) uptake reflects glucose metabolism and has shown promise in discriminating symptomatic MCs from cognitively normal, mutation-negative non-carriers (NC) [6,9,10]. In ADAD, studies have shown FDG uptake in MCs is decreased in the precuneus and is inversely correlated with PiB binding. Marked decreases in glucose metabolism occur approximately 5–10 years before EAO in MCs [10,12].
Structural MRI provides a method to evaluate regional volumetric changes in neurodegeneration that occur with disease progression [13]. MRI can reveal regional brain atrophy, which is a characteristic feature of neurodegeneration due to synaptic losses [14]. ADAD is characterized by progressive atrophy that manifests as changes initially in the temporal lobes and subcortical regions with eventual spread to other regions. Observed changes in atrophy are related to the spread of neurofibrillary tangles in AD [14].
Machine learning (ML) is a branch of artificial intelligence that can learn to extract patterns from existing data to predict future events [15]. Advances in ML offer promise for a number of applications, including medical imaging and predictive analytics [15,16]. Compared to traditional statistics that provide primarily group-level results, ML algorithms can predict clinical outcomes at the individual level and could enable personalized treatments that provide targeted care for patients [17]. Although a number of studies have applied ML to neuroimaging measures to study LOAD [18–22], few studies to date have applied these techniques to ADAD. Because time until conversion to symptomatic impairment can be estimated with EAO, ADAD provides a unique opportunity for ML to model the progression of the disease and provide decision support to evaluate therapies currently being investigated in the Dominantly Inherited Alzheimer Network (DIAN) Trials Unit (DIAN-TU).
In this longitudinal study, we used artificial neural networks (ANNs) to evaluate progression to cognitive impairment using multimodal neuroimaging biomarkers. Specifically, within a cohort of MCs (n = 131) and NCs (n = 74), we used ANNs to investigate: (1) changes in Aβ deposition (using PiB), (2) changes in glucose metabolism (using FDG), and (3) brain atrophy (using structural MRI) as a function of aging in relation to EAO. Further, we utilized feature selection to identify regions that were the strongest discriminators of mutation status for each modality. We then performed Monte Carlo simulations to identify cutoffs for the identified regions. This data-driven approach provides an opportunity to discover novel mechanisms and disease trajectories specific for ADAD.
2. METHODS
2.1. Participants
One hundred thirty-one MCs with mutations in PSEN1, PSEN2, or APP and 74 healthy, mutation-negative NCs were recruited from sites participating in the DIAN study. Participants from the 12th data freeze with genetic, clinical, and longitudinal neuroimaging data that passed quality control procedures were included. The Washington University Institutional Review Board provided supervisory review and human subjects’ approval. Participants provided written, informed consent or assent with proxy consent. All study procedures were approved by the Washington University Human Research Protection Office and the institutional review boards of the participating sites.
2.2. Clinical Classification
The CDR® Dementia Staging Instrument was used to assess dementia status at each clinical assessment [23]. A participant’s EAO was calculated at each visit on the basis of the participant’s current age relative to the family mutation–specific expected age at onset of dementia symptoms [5]. Parental age at first progressive cognitive decline was used if the mutation-specific EAO was unknown. EAO was calculated identically for both MCs and NCs. All clinical evaluators were blinded to the mutation status of participants. The presence/absence and type of mutation were determined using polymerase chain reaction amplification followed by Sanger sequencing [7].
2.3. MRI Acquisition and Processing
MRI was performed using the Alzheimer’s Disease Neuroimaging Initiative protocol (ADNI) [24]. Sites used a 3T scanner that passed regular quality control assessments. The ADNI Imaging Core screened images for compliance. T1 weighted images at 1.1 x 1.1 x 1.2 mm voxel resolution were acquired for participants. FreeSurfer 5.3 [25,26] was used to perform volumetric segmentation, cortical surface reconstruction, and to define cortical and subcortical regions of interest (ROIs). Segmentations were inspected and edited as needed by members of the DIAN Imaging Core. A regression approach was used to correct subcortical volumes for intracranial volumes. Volumetric measures were averaged across hemispheres. FreeSurfer-defined cortical and subcortical ROIs (44 total) were used for regional processing of PET data. The FreeSurfer-defined ROIs were derived from the Deskian/Killiany atlas [27] for segmentation. These are standard regions used for volumetric analyses.
2.4. PET Acquisition and Processing
Amyloid PET was performed using a bolus injection of PiB. Data from the 40–70-minute post-injection timeframe were converted to regional standardized uptake value ratios (SUVRs) relative to the cerebellar gray matter using FreeSurfer-derived ROIs (PET Unified Pipeline) [28]. Glucose metabolism imaging was performed with a single bolus injection of FDG. A 30-minute dynamic acquisition beginning 30 minutes post-injection was acquired. The last 20 minutes of each FDG scan were converted to SUVRs using the cerebellar gray matter as a reference region. All PET data were partial volume corrected using a regional spread function technique [29,30]. PET images were aligned to the T1 image processed using FreeSurfer. PET scanner–specific filters were applied to account for differences in spatial resolution and to achieve a common resolution (8 mm) [31]. The DIAN imaging core performed quality control checks on the PET Unified Pipeline processing.
2.5. Machine Learning and Statistical Analyses
ML analyses were performed in MATLAB R2018b. Deep feedforward ANNs were trained for each of the neuroimaging modalities. Feedforward ANNs map an input to an output by composing sets of smaller functions laid out as a directed acyclic graph [32]. The feasibility of these networks is based on the Universal Approximation Theorem, which states a neural network with a single hidden layer contains a finite set of artificial neurons that approximate continuous functions on subsets of Rn [33].
Our ANNs contained 4 hidden layers with 10 artificial neurons in each layer. The network architecture was decided based on design methodologies [34,35], incremental pruning, and cross-validation. Further details on model design and validation can be found in supplementary material (Methods- Machine Learning Model Design). An ANN was trained to output all ROIs for each modality. Input to the models included age, sex, APOE ε4 status, mutation status, the amount of time in the future to predict, and the given imaging variables (MRI or SUVR) for 44 FreeSurfer ROIs. A complete list of the ROIs can be found in Supplementary Table 1. The output of each model corresponded to the ROI values at a time point in the future. Rates of change were calculated by subtracting scans at time point N by the scan at time point N-1. Rates were then divided by the number of months between the scans to obtain a normalized rate of change. The mean time between scans was 2.6 years (±1.4). If a participant had more than 2 scanning sessions, all possible combinations were evaluated. Using the first time point, data were projected into the future by iteratively adding the normalized rate of change, and these data were used for training. For each point, the rate of change was used to project the data ± 3 years from the current age. Data were projected into the future and the past to avoid biasing the model to later phases of the disease. We chose this window based on previous work [10], which showed the biomarkers’ rate of change is not constant along the disease continuum.
Predictive features of mutation status were ranked according to importance using a Relief algorithm [36]. Relief algorithms detect conditional dependencies between attributes using a nearest neighbor approach, with features ranked by estimating how well their values distinguish between proximal comparisons. Further, cutoff points for PiB, FDG, and brain volumetrics were identified based on the likelihood of the values generated by Monte Carlo model simulations. The simulations generated an equal number (by mutation status) of random sample points from the multivariate distribution defined by the mean and covariance matrix of the data given a specific mutation status, age, and EAO range [37].
We also trained a linear regression model to compare the results to our ANN. This comparative analysis was performed due to recent research suggesting that, in some cases, linear models can outperform nonlinear models [38]. When training the regression model, all methods previously described for training the ANN were applied. Each biomarker was modeled separately, and the models were trained using 5-fold cross-validation. Cross-validation was performed at the participant level, and all results reported were derived by combining the test data results from each of the 5 folds of cross-validation. In addition, the input to the regression model was the same as the ANN, but the only output considered was the precuneus. We chose the precuneus as it is highly predictive and heavily involved in disease progression in ADAD [10], making it optimal for comparison. Further, a multivariate linear regression was performed which derived brain regions in the same manner as the ANN. The regression model utilized ordinary multivariate normal maximum likelihood estimation with the full variance-covariance matrix and constant, linear, and interaction terms. We also performed the zero rule algorithm on the data to compare baseline predictability using the mean of the output values observed in the training data compared to the testing data.
3. RESULTS
3.1. Demographics of the Cohort
Detailed demographics are presented in Table 1. Participants were matched for age, sex, and education.
Table 1.
Demographics of participants
| Mutation carriers (MC) | Mutation-negative non-carriers (NC) | p Values | |
|---|---|---|---|
| N | 131 | 74 | |
| Age (years) ± SD | 39.2 ± 10.6 | 39.3 ± 10.2 | .95 |
| Sex (% Male) | 40% | 35% | .72 |
| Education (years) ± SD | 14.3 ± 2.7 | 15.1 ± 2.6 | .06 |
| APOE ε4 (% carriers) | 39% | 36% | .81 |
| EAO (years) ± SD | 46.3 ± 6.8 | 48.1 ± 5.7 | .90 |
Abbreviations: SD, standard deviation; APOE ε4, apolipoprotein ε4; EAO, estimated age of symptom onset.
3.2. PiB
The Relief algorithm identified the nucleus accumbens, caudate, precuneus, anterior cingulate, pallidum, putamen, and middle frontal regions as strong predictors of mutation status. The ANN was able to predict the future PiB values with an average R2 value of 0.95 and RMSE of 0.2. Figure 1 depicts results for the 4 best-predicted ROIs. The algorithm was able to accurately estimate the values in both MCs and NCs, with the NCs having lower SUVRs compared to the MCs. Supplementary Figure 1 shows the model predictions for MCs based on distance from EAO for PiB. Two relatively distinct clouds were seen for PiB, with lower SUVRs seen at greater distances from EAO, while MCs closer to EAO had elevated PiB SUVRs.
Figure 1.
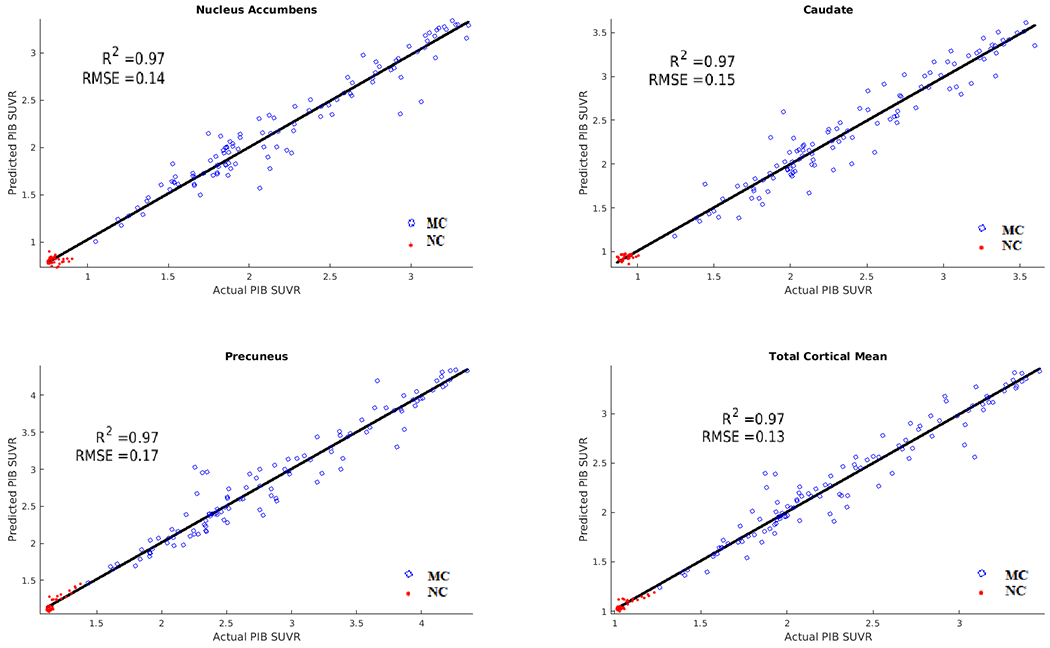
Results of Pittsburgh Compound-B (PiB) predictions for mutation carriers (MC) (blue) and non-carriers (NC) (red). Correlation and RMSE of predicted versus actual values. The ANN was able to predict future PiB values with an average R2 of 0.95 and RMSE of 0.2 in both MCs and NCs.
3.3. FDG
The strongest predictors of mutation status with respect to metabolism were the pericalcarine, caudate, precuneus, fusiform, anterior cingulate, insula, and transverse temporal regions. The ANN was able to predict future FDG values with an R2 value of 0.93 and RMSE of 0.02 in both groups. Figure 2 depicts results for the 4 best-predicted ROIs. The algorithm showed a trend of MCs having lower future FDG values than NCs. Supplementary Figure 2 shows the model predictions for MCs based on distance from EAO for FDG. Two clouds are seen for FDG, with higher SUVRs seen at greater distances from EAO, while MCs closer to EAO had lower FDG SUVRs.
Figure 2.
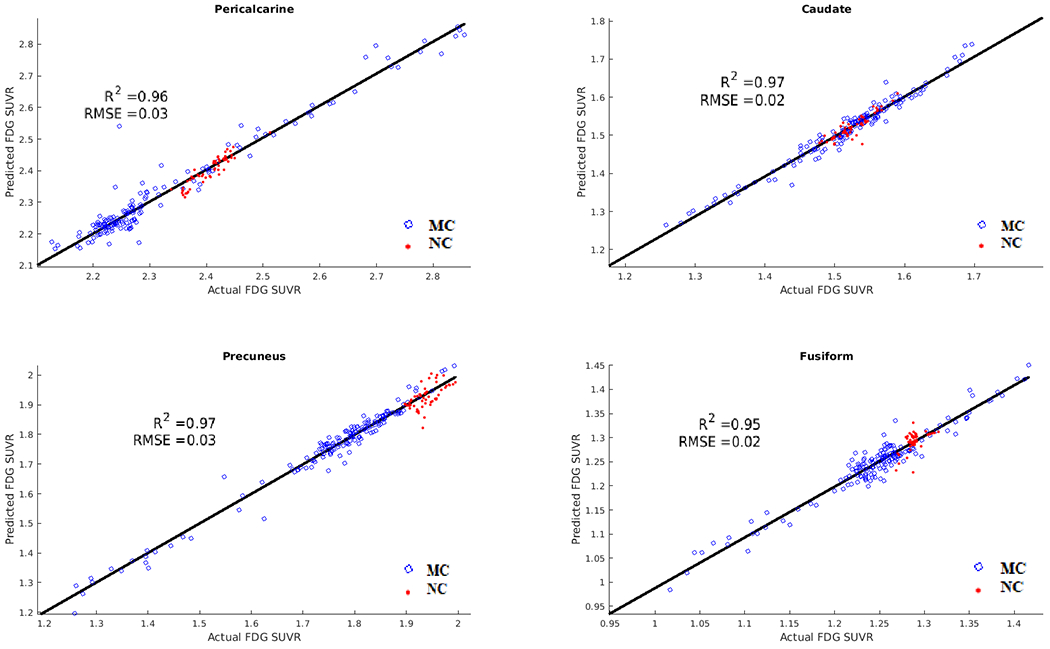
Results of fluorodeoxyglucose (FDG) predictions for mutation carriers (MC) (blue) and non-carriers (NC) (red) in select ROIs. Correlation and root mean squared error (RMSE) of predicted versus actual values. The ANN was able to predict future FDG values with an average R2 of 0.93 and RMSE of 0.02 in MCs and NCs, with MCs showing trends of lower predicted FDG values than NCs.
3.4. Volume
The strongest predictors of mutation status with respect to brain atrophy were seen in the nucleus accumbens, pericalcarine, caudate, precuneus, anterior cingulate, insula, entorhinal cortex, pallidum, and transverse temporal regions. The ANN was able to predict changes in brain volumes with an average R2 value of 0.95. Figure 3 depicts results for the 4 best-predicted regions. The algorithm showed a general trend of MCs having more brain atrophy than NCs. Supplementary Figure 3 shows the model predictions for MCs as a function of distance from EAO for brain volumes.
Figure 3.
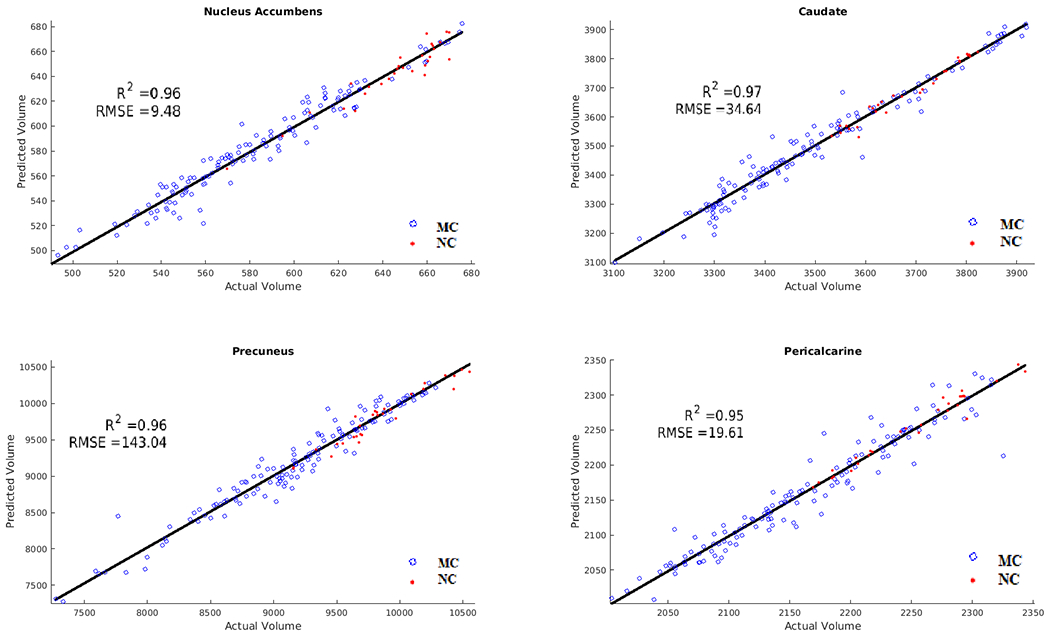
Results of brain volumetric predictions for mutation carriers (MC) (blue) and non-carriers (NC) (red). Correlation and root mean squared error (RMSE) of predicted versus actual values. The ANN was able to predict changes in brain volumes with an average R2 value of 0.95 and showed a general trend of MCs having more brain atrophy than NCs.
3.5. Simulations
Using the trained models, amyloid accumulation, changes in metabolism, and brain atrophy were simulated for MCs and NCs (Figure 4, top). Consistent with previous work, the models showed that in the MC group, the earliest changes are in amyloid deposition, which follows a sigmoidal trajectory and continues to accumulate past EAO. A biphasic response was seen for metabolism, with changes occurring earlier than expected, and progressive decline was observed in atrophy throughout the course of the disease, with the greatest changes occurring just prior to EAO. The NC groups showed little change over time for all modalities.
Figure 4.
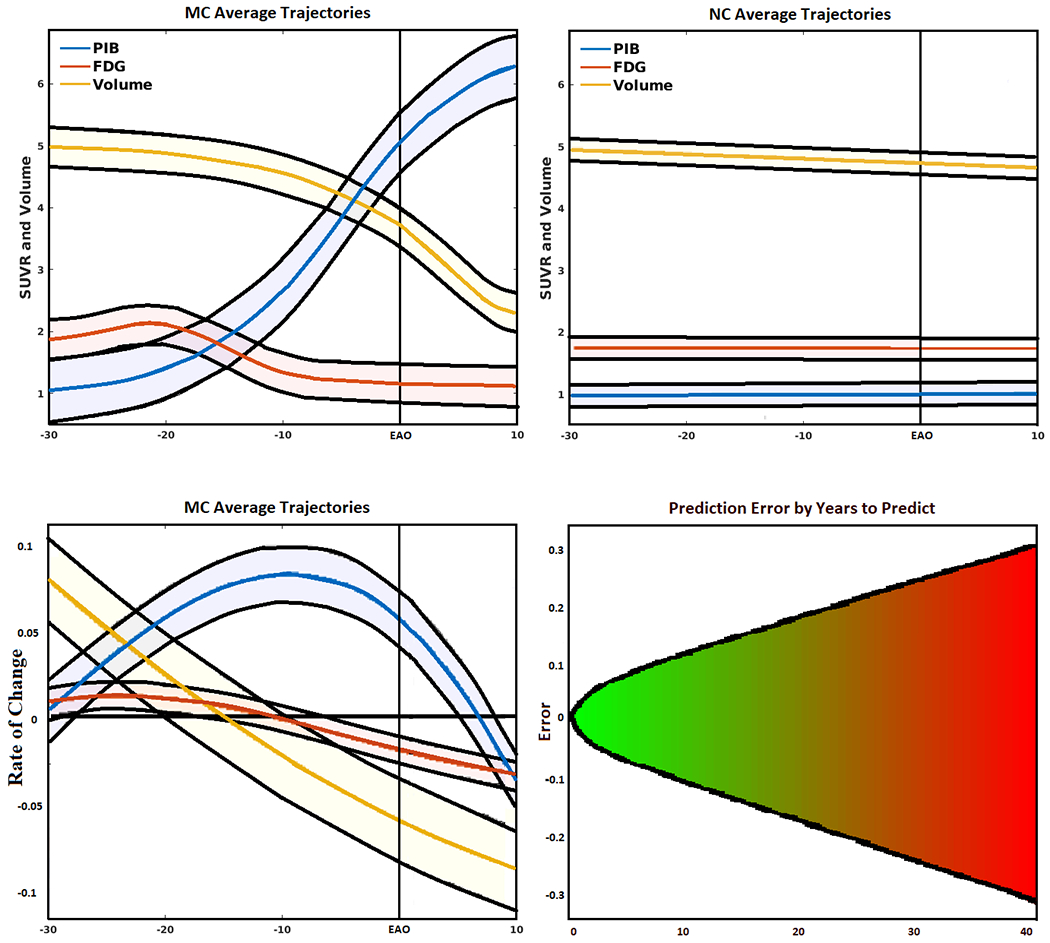
(Top left) Simulated biomarker evolution for total mean cortical and subcortical Pittsburgh Compound-B (PiB), total mean cortical and subcortical fluorodeoxyglucose (FDG), and total gray matter volume (scaled to a common interval) derived from the artificial neural network (ANN) in mutation carriers (MC). Shaded region indicates model variability, with EAO marked by perpendicular line. (Top right) Simulated biomarker evolution for total mean cortical and subcortical PiB, total mean cortical and subcortical FDG, and total gray matter volume (scaled to a common interval) derived from the ANN in mutation non-carriers (NC). (Bottom left) Normalized biomarker rate of change for mean PiB, mean FDG, and total gray matter volume (scaled to a common interval) fit to a polynomial curve showing 95% confidence interval. (Bottom right) Mean absolute error of predicted (normalized) biomarker values given the amount of time in the future to predict, fit with a 2-degree polynomial curve projected into the future. Errors increased linearly with an increase in the amount of time in the future to predict.
We fitted a polynomial curve to the normalized rates of change for each of the neuroimaging biomarkers (Figure 4, bottom left). Consistent with the models, amyloid showed an inverted U shape, with increases occurring early in the disease, and subsequently followed by a gradual decline in rate of PiB accumulation. FDG showed a slight increase in the early stages, followed by a gradual decrease when the distance from EAO approached 0. Finally, brain volumetrics showed a gradual increase in the rate of decline throughout progression to EAO.
Figure 4 (bottom right) shows the normalized models errors based on years to predict (e.g., the error for a participant’s PET/MRI values predicted 1 year in the future versus the error for predicting 5 years in the future). A 2 degree polynomial curve was fit to the error data, which showed a predominantly linear increase with increasing number of years to predict. The fit lines were projected into the future for up to 40 years. The plot shows that the model maintains a mean absolute error less than 0.1 up to 10 years in the future. The individual biomarkers showed similar trends, only at different scales.
Supplementary Figures 4–6 display the results of the Monte Carlo model simulations for each of the highly predictive regions for each modality. Larger values on the y-axis represent a greater likelihood of producing a given value. For PiB, clear cut-points were observed between MCs and NCs with nearly 100% specificity. Cut-points were 1.17 for the nucleus accumbens, 1.3 for the caudate, 1.4 for the precuneus, and 1.2 SUVR for total cortical mean. For FDG, the cut-points were less defined for some regions. Cut-points for the anterior cingulate, caudate, precuneus, and total cortical mean ranged from 1.4–1.825 SUVR. The model simulations indicate MCs had a trend for decreased FDG in each of these ROIs, as well as a biphasic response in the caudate and anterior cingulate. For brain volumes, MCs had greater atrophy than NCs. Cutoffs were identified for the nucleus accumbens (550 mm3), caudate (3300 mm3), precuneus (8500 mm3), and total gray matter (575,000 mm3).
3.6. Alternative Analysis Methods
Supplementary Figure 8 displays the error histograms [probabilities of errors (actual-predicted)] for the ANN versus the regression model for PiB in the precuneus. Although both models performed very well, the performance obtained through regression was lower than that obtained through the ANN. The ANN’s error probability distribution was highly clustered around 0 (RMSE = 0.17), whereas the regression model showed greater dispersion (RMSE = 0.28), indicating a greater likelihood of making a larger error compared to the ANN. Similar results were seen using FDG and volumetric data. Whole brain average RMSE for the ANN, multivariate linear regression, and zero rule algorithm are provided at the bottom of supplementary table 1. As expected, the ANN showed lower RMSE compared to multivariate linear regression and the zero rule algorithm for all modalities.
4. DISCUSSION
Our models yielded high accuracy in predicting amyloid accumulation, changes in metabolism, and brain atrophy in ADAD. The Relief algorithm identified both subcortical (caudate) and cortical (precuneus and anterior cingulate) ROIs as the strongest predictors of mutation status. Figure 5 displays the strongest predictors for each modality. For amyloid PET, which is believed to reflect the earliest changes in ADAD, changes were primarily seen within subcortical (pallidum, nucleus accumbens, caudate, putamen, and entorhinal) compared to cortical regions (middle frontal, anterior cingulate, and precuneus). For changes in metabolism measured by FDG, which reflect changes later in the disease process compared to amyloid, more cortical (insula, fusiform, middle frontal, precuneus, anterior cingulate, pericalcarine, and transverse temporal) rather than subcortical (caudate) regions were involved.
Figure 5.
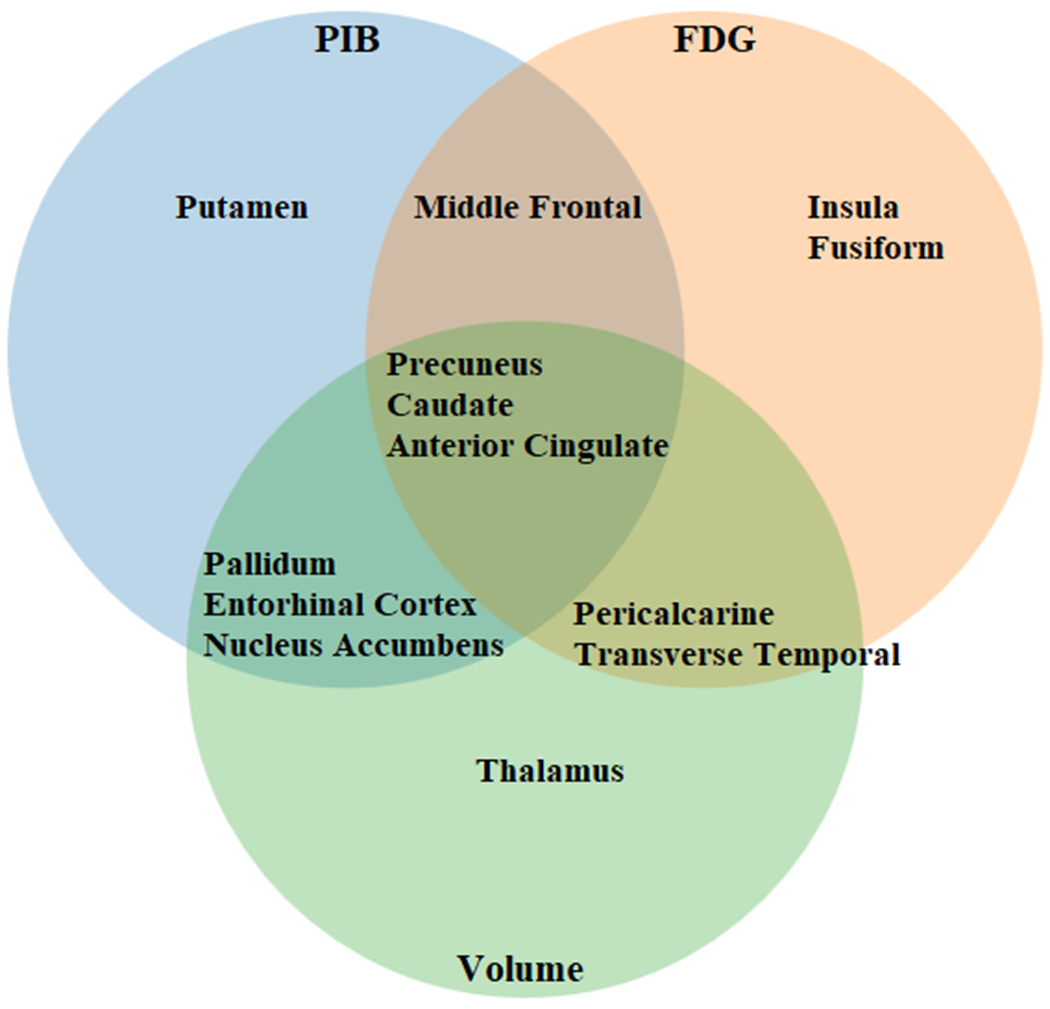
Strongest predictors of mutation carrier (MC) status for autosomal dominant Alzheimer’s disease (ADAD) as identified by Relief algorithms. The strongest predictors across all modalities were the precuneus, caudate, and anterior cingulate. Changes in amyloid PET (PiB, blue circle) were primarily seen within subcortical regions. Changes in metabolism (FDG, orange circle) showed more cortical involvement. Volumetric changes (Volume, green circle) showed both cortical and subcortical involvement.
For changes that occur late in the disease process due to atrophy, both cortical (precuneus, anterior cingulate, pericalcarine, transverse temporal) and subcortical (caudate, pallidum, nucleus accumbens, entorhinal, thalamus) regions were affected. This suggests that the disease may start within subcortical areas and quickly involve additional subcortical and cortical regions. Overall, these analyses point to multiple hubs being affected early in the disease process, followed by spread to other brain regions (Supplementary Figure 7). Supplementary Table 1 lists the RMSE for the individual ROIs for each of the 3 biomarkers, as well as the mean overall RMSE of the models compared to the zero rule algorithm and multivariate linear regression.
In the amyloid analysis, the model achieved 0.95 R2 and 0.2 RMSE (see Figure 1). The model showed PiB uptake was greater in MCs compared to NCs for most regions. Our results also confirm that the presence of amyloid alone is insufficient for conversion to symptomatic AD. The simulated trajectory for mean cortical amyloid accumulation (see Figure 4, top left) showed deposition started to occur approximately 15–20 years before EAO. These results are consistent with other studies that focused on global and regional amyloid deposition [7,11]. Our model indicates a sigmoidal trajectory of accumulation for amyloid, with a slow increase 20–30 years from EAO, an abrupt increase 0–15 years from EAO, and slowing to an eventual decline after EAO. This is consistent with what has been hypothesized to occur in LOAD [39].
As a point of reference, we calculated the normalized rates of change for all mutation-positive participants (see Figure 4, bottom left). The normalized rate of amyloid deposition shows a consistent increase from roughly 10 years prior to EAO followed by slowing in the rate of accumulation after EAO. Only after EAO does the rate of accumulation diminish, which is consistent with the sigmoidal model trajectory.
With regard to metabolism, our model yielded 0.93 R2 and 0.02 RMSE. Although the MCs had greater decreases in FDG for most brain regions, the separation between the 2 groups was not as well defined compared to PiB. This is likely because the rate and amount of change are less extreme compared to amyloid (see Figure 4, top left). Our model indicates metabolism did not decrease below a baseline until 10 years before symptom onset and continued to decline after EAO. These results are consistent with the normalized rate of change (see Figure 4, bottom left). The rate of metabolism did not decline below baseline until 10 years prior to EAO, followed by a steady decline.
An uptick in metabolic activity was observed in the early stages of amyloid accumulation and did not begin to decrease until amyloid significantly increased. This was observed in the simulated trajectory and the normalized rate of change. Similar results were observed within the precuneus in a cross-sectional analysis [7]. Rate of change analysis revealed this primarily occurs in the basal ganglia. Because the basal ganglia show the least toxic response to amyloid deposition [40–42], these transient increases may be prominent because these neurons mount a compensatory response preceding significant amyloid accumulation [43–47]. However, at a point, the brain is no longer able to buffer changes when amyloid deposition becomes significant.
Our model showed total gray matter volume slightly declined during the early stages of ADAD, followed by a dramatic decrease 5–10 years prior to EAO. The decrease in volume occurred when metabolism was decreased and amyloid had accumulated. Volumetrics continued to decline even after EAO. The model was able to predict volumes with an R2 of 0.95.
These findings have clinical importance for the care of people with ADAD in the context of amyloid, metabolism, and atrophy. Using feature selection methods, we have identified brain regions that are both common amongst modalities as well as unique for each modality (Figure 5). Specifically, we have shown the precuneus, caudate, and anterior cingulate are strong predictors of mutation status among all modalities. These findings are significant for multimodal imaging studies and clinical trials whose goal is to assess the overall impact of a therapy. Further, the fact that we have identified regions that are unique to each modality suggest a complex set of evolving interactions that are not localized to a small set of brain regions. Our models also suggest a complex disease progression that goes beyond a linear or sigmoidal pattern that has been hypothesized for LOAD (Figure 4). We have identified a biphasic response in metabolism, where hypermetabolism is seen very early in the disease process. Future studies should investigate this phenomenon, as previous studies have primarily focused on hypometabolism that occurs later in the disease process.
We also extend the literature by establishing clinically useful algorithms for modeling the progression of ADAD, and show the utility of ML in developing diagnostic and predictive tests. A major deficiency in AD clinical research is the problem of individual predictability versus group level differences. ML is ideal for research aimed at discovering patterns in high dimensional data that are believed to underlie complex clinical phenotypes that go beyond group level results. This is especially relevant for diseases such as ADAD and LOAD, which show chronic progression over long periods of time, as well as variability in terms of symptoms, risk factors, and progression. Our models were trained on the largest available ADAD data set, and are able to accurately forecast disease progression several years into the future at any stage of the disease. Inputting a patients unique demographics and imaging variables will yield trajectories that are specific to that individual. Further, by simulating our trained models, we are able to identify trajectories and cutoff values unique to each brain region which best discriminate MCs from NCs. Because our models have been trained on a variety of demographics, one can easily generate values that are specific to a given sex, APOE ε4 status, and education level. Utilization of these models provides the opportunity to expedite clinical trials and provide precision medicine tailored to a patient based on his/her unique set of demographics, disease subtype, and treatment response. As we have shown, while both linear regression and our ANN performed well in predicting disease progression, the ANN had a lower error rate. More accurate models could lead to better decision-making and improved efficiency of research, and accurate identification of participants whose progression patterns differ from model predictions could allow for decision support in evaluating the effects of specific therapies in clinical trials.
Limitations and future work for this study are detailed as follows. Data leakage, which refers to the use of test data in any part of the training process, is a major concern in the AD field [48], and is difficult to address due to the limited number of samples. This is especially relevant in the context of ADAD. While we utilize the largest available data set, the fact that ADAD only accounts for a small portion of the total AD cases restricts the number of available of samples. Still, choosing the proper set of hyperparameters in the context of deep neural networks is a difficult task. However, the performance and flexibility of these models crucially depends on how these parameters are set [49]. In our analysis, the number of layers and number of artificial neurons in each layer were identified by testing multiple network architectures within our data (see supplemental). Measures were taken to ensure that the partitioning of the data for 5 fold cross validation in this process did not match the data partitions used for the 5 fold cross validation for the final analysis. We also ensured the models trained in the network architecture identification process were discarded after the fact, and all subsequent models generated in the main analysis had randomly initialized weights. However, because the same data set was used to identify the size of the network as well to perform the main analysis, we acknowledge that this could be a source of data leakage, which could lead to an underestimation of errors. Future work will involve further validation and testing of the proposed models. Specifically, conducting blinded out of sample testing on newly acquired data from the DIAN study is needed to ensure issues such as data leakage and overfitting do not influence the model results. Further, alternative network models will need to be considered. As more longitudinal time points are acquired for participants in DIAN, time series specific networks, such as long short term memory networks may be more appropriate. Lastly, alternative forms of feature selection should be considered to investigate the relationships between biomarkers and brain regions.
4.1. Conclusion
To provide targeted treatment to persons with ADAD, novel methods are needed to model disease trajectories. We have shown ANNs can accurately forecast amyloid accumulation, changes in glucose metabolism, and brain atrophy. Using feature extraction methods, we identified the strongest predictors of mutation status over 44 brain regions. Our results show a sigmoidal progression of amyloid accumulation, a biphasic response to glucose metabolism, and a gradual increase in brain atrophy in MCs compared to NCs. Our models indicate disease progression is primarily in subcortical regions, followed by cortical involvement within anterior and posterior portions of the brain.
Supplementary Material
5.1. Acknowledgements
We would like to acknowledge the participants and their families, without whom these studies would not be possible. We additionally thank all of the participating researchers and coordinators in the DIAN (https://dian.wustl.edu/our-research/observational-study/dian-observational-study-sites/) who support the studies. DIAN ClinicalTrials.gov identifier: NCT00869817.
5.2. Funding
This research was funded by the National Institutes of Health (NIH) [grant numbers K01AG053474, R01AG052550, UFAG 032438, UL1TR000448, P30NS098577, R01EB009352, P50AG05131, U01AG042791, U01AG042791-S1 (FNIH and Accelerating Medicines Partnership), R1AG046179]; the German Center for Neurodegenerative Diseases (DZNE); the National Institute for Health Research (NIHR) Queen Square Dementia Biomedical Research Centre; and the Medical Research Council Dementias Platform UK [grant numbers MR/L023784/1, MR/009076/1, BrightFocus Foundation A2018817F, Alzheimer Association International Research Grant Program #AARFD-20-681815, NSF DMS 156243, and an anonymous organization.
We acknowledge the support of Fred Simmons and Olga Mohan, the Barnes-Jewish Hospital Foundation, the Charles F. and Joanne Knight Alzheimer Research Initiative, the Hope Center for Neurological Disorders, the Mallinckrodt Institute of Radiology, the Paula and Rodger O. Riney fund, and the Daniel J. Brennan fund. Computations were performed using the facilities of the Washington University Center for High Performance Computing, which were partially funded by the NIH [grant numbers 1S10RR022984–01A1, 1S10OD018091–01].
5.3. Competing Interests
The authors declare no conflicts of interest. Anne Fagan received research funding from the National Institute on Aging of the National Institutes of Health, Biogen, Centene, Fujirebio, and Roche Diagnostics. She is a member of the scientific advisory boards for Roche Diagnostics, Genentech, and AbbVie and also consults for Araclon/Grifols, DiademRes, DiamiR, and Otsuka. Carlos Cruchaga receives research support from Biogen, EISAI, Alector, and Parabon. The funders of the study had no role in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. Dr. Cruchaga is also a member of the advisory board of ADx Healthcare, Halia Therapeutics, and Vivid Genomics. Jasmeer P. Chhatwal served on the medical advisory board for Otsuka Pharmaceuticals. Johannes Levin reports speaker’s fees from Bayer Vital, speaker’s fees from Willi Gross Foundation, consulting fees from Axon Neuroscience, consulting fees from Ionis Pharmaceuticals, author fees from Thieme medical publishers and W. Kohlhammer GmbH medical publishers, compensation for work as part-time CMO from MODAG GmbH, and non-financial support from AbbVie outside the submitted work. John Morris is funded by NIH grants [numbers P50AG005681, P01AG003991, P01AG026276, UF1AG032438]. Dr. Jack serves on an independent data monitoring board for Roche and has served as a speaker for Eisai, but he receives no personal compensation from any commercial entity. He receives research support from NIH and the Alexander Family Alzheimer Disease Research Professorship of the Mayo Clinic. Eric McDade is involved in a clinical trial on AV-1451 sponsored by Avid and serves on a data safety monitoring committee for Eli-Lilly. Randall Bateman is on the scientific advisory board of C2N Diagnostics and reports research support from AbbVie, Biogen, Eisai, Eli Lilly, Co/Avid Radiopharmaceuticals, Roche, Janssen, and United Neuroscience.
Abbreviations.
- Aβ
Amyloid beta
- ADAD
autosomal dominant Alzheimer disease
- ANN
artificial neural networks
- DIAN
Dominantly Inherited Alzheimer Network
- DIAN-TU
Dominantly Inherited Alzheimer Network Trials Unit
- EAO
expected age of symptom onset
- EYO
estimated years to symptomatic onset
- FDG
[18F]Fluorodeoxyglucose
- MC
mutation carrier
- ML
machine learning
- NC
non-carrier
- PiB
[11C]Pittsburgh Compound-B
- RMSE
root mean squared error
- ROIs
regions of interest
- SUVRs
standardized uptake value ratios
Appendix
Consortia: Dominantly Inherited Alzheimer Network
Ricardo Allegri, Randall J. Bateman, Jacob Bechara, Tammie L.S. Benzinger, Sarah Berman, Courtney Bodge, Susan Brandon, William Brooks, Jill Buck, Virginia Buckles, Sochenda Chea, Jasmeer Chhatwal, Patricio Chrem, Helena Chui, Jake Cinco, Clifford R Jack Jr, Carlos Cruchaga, Tamara Donahue, Jane Douglas, Noelia Edigo, Nilufer Erekin-Taner, Anne Fagan, Martin R. Farlow, Nick C. Fox, Colleen Fitzpatrick, Gigi Flynn,Erin Franklin, Hisako Fujii, Douglas Galasko, Cortaiga Gant, Samantha Gardener, Bernardino Ghetti, Alison Goate, Jill Goldman, Brian Gordon, Neill Graff-Radford, Julia Gray, Alexander Groves, Jason Hassenstab, Laura Hoechst-Swisher, David Holtzman, Russ Hornbeck, Siri Houeland DiBari, Takeshi Ikeuchi, Snezana Ikonomovic, Gina Jerome, Mathias Jucker, Celeste Karch, Kensaku Kasuga, Takeshi Kawarabayashi, William Klunk, Robert Koeppe, Elke Kuder-Buletta, Christoph Laske, Jae-Hong Lee, Johannes Levin, Ralph Martins, Neal Scott Mason, Ralph Martins, Colin L. Masters, Denise Maue-Dreyfus, Eric McDade, Hiroshi Mori, John C. Morris, Akem Nagamatsu, Katie Neimeyer, James M.Noble, Joanne Norton, Richard J. Perrin, Marc Raichle, Alan Renton, John Ringman, Jee Hoon Roh, Stephen Salloway, Peter R. Schofield, Hiroyuki Shimada, Wendy Sigurdson, Hamid Sohrabi, Paige Sparks, Kazushi Suzuki, Kevin Taddei, Peter Wang, Chengjie Xiong, Xiong Xu
6. REFERENCES
- [1].World Health Organization. Dementia Fact sheet. WHO; 2017;17:751–60. 10.1063/1.3590158. [DOI] [Google Scholar]
- [2].Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med 2011;1:6189. 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bateman RJ, Aisen PS, De Strooper B, Fox NC, Lemere CA, Ringman JM, et al. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Res Ther 2011;3:1–13. 10.1186/alzrt59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schindler SE, Fagan AM. Autosomal dominant Alzheimer disease: A unique resource to study CSF biomarker changes in preclinical AD. Front Neurol 2015;6:142. 10.3389/fneur.2015.00142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ryman DC, Acosta-Baena N, Aisen PS, Bird T, Danek A, Fox NC, et al. Symptom onset in autosomal dominant Alzheimer disease: a systematic review and meta-analysis. Neurology 2014;83:253–60. 10.1212/WNL.0000000000000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Marcus C, Mena E, Subramaniam RM. Brain PET in the diagnosis of Alzheimer’s disease. Clin Nucl Med 2014;39:413. 10.1097/RLU.0000000000000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 2012;367:795–804. 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yau WYW, Tudorascu DL, McDade EM, Ikonomovic S, James JA, Minhas D, et al. Longitudinal assessment of neuroimaging and clinical markers in autosomal dominant Alzheimer’s disease: A prospective cohort study. Lancet Neurol 2015;14:804–13. 10.1016/S1474-4422(15)00135-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 2018;91:1295–306. 10.1212/WNL.0000000000006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer’s disease: a longitudinal study. Lancet Neurol 2018;17:241–50. 10.1016/S1474-4422(18)30028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oxtoby NP, Young AL, Cash DM, Benzinger TLS, Fagan AM, Morris JC, et al. Data-driven models of dominantly-inherited Alzheimer’s disease progression. Brain 2018;141:1529–44. 10.1093/brain/awy050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yan L, Liu CY, Wong KP, Huang SC, Mack WJ, Jann K, et al. Regional association of pCASL-MRI with FDG-PET and PiB-PET in people at risk for autosomal dominant Alzheimer’s disease. NeuroImage Clin 2018;17:751–60. 10.1016/j.nicl.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lowe VJ, Lundt E, Knopman D, Senjem ML, Gunter JL, Schwarz CG, et al. Comparison of [18F]Flutemetamol and [11C]Pittsburgh Compound-B in cognitively normal young, cognitively normal elderly, and Alzheimer’s disease dementia individuals. NeuroImage Clin 2017;16:295–302. 10.1016/j.nicl.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Johnson KA, Fox NC, Sperling RA, Klunk WE. Brain imaging in Alzheimer disease. Cold Spring Harb Perspect Med 2012;2:6213. 10.1101/cshperspect.a006213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Choy G, Khalilzadeh O, Michalski M, Do S, Samir AE, Pianykh OS, et al. Current Applications and Future Impact of Machine Learning in Radiology. Radiology 2018;288:318–28. 10.1148/radiol.2018171820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Erickson BJ, Korfiatis P, Akkus Z, Kline TL. Machine Learning for Medical Imaging. RadioGraphics 2017;37:505–15. 10.1148/rg.2017160130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Passos IC, Mwangi B, Kapczinski F. Big data analytics and machine learning: 2015 and beyond. The Lancet Psychiatry 2016;3:13–5. 10.1016/S2215-0366(15)00549-0. [DOI] [PubMed] [Google Scholar]
- [18].Ding Y, Sohn JH, Kawczynski MG, Trivedi H, Harnish R, Jenkins NW, et al. A Deep Learning Model to Predict a Diagnosis of Alzheimer Disease by Using 18 F-FDG PET of the Brain. Radiology 2018;290:456–64. 10.1148/radiol.2018180958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Moradi E, Pepe A, Gaser C, Huttunen H, Tohka J. Machine learning framework for early MRI-based Alzheimer’s conversion prediction in MCI subjects. Neuroimage 2015;104:398–412. 10.1016/j.neuroimage.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Khazaee A, Ebrahimzadeh A, Babajani-Feremi A. Application of advanced machine learning methods on resting-state fMRI network for identification of mild cognitive impairment and Alzheimer’s disease. Brain Imaging Behav 2016;10:799–817. 10.1007/s11682-015-9448-7. [DOI] [PubMed] [Google Scholar]
- [21].Collij LE, Heeman F, Kuijer JPA, Ossenkoppele R, Benedictus MR, Möller C, et al. Application of Machine Learning to Arterial Spin Labeling in Mild Cognitive Impairment and Alzheimer Disease. Radiology 2016;281:865–75. 10.1148/radiol.2016152703. [DOI] [PubMed] [Google Scholar]
- [22].Youssofzadeh V, McGuinness B, Maguire L, Wong-Lin K. Multi-kernel learning with dartel improves combined MRI-PET classification of Alzheimer’s disease in AIBL data: Group and individual analyses. Front Hum Neurosci 2017;11:380. 10.3389/fnhum.2017.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Morris JC. The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 2012;41:1588–92. 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- [24].Jack CR, Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, et al. Update on the Magnetic Resonance Imaging core of the Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement 2010;6:212–20. 10.1016/j.jalz.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fischl B FreeSurfer. Neuroimage 2012;62:774–81. 10.1016/j.neuroimage.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fischl B, Dale AM. Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proc Natl Acad Sci U S A 2000;97:11050–5. 10.1073/pnas.200033797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage 2006. 10.1016/j.neuroimage.2006.01.021. [DOI] [PubMed] [Google Scholar]
- [28].Su Y, D’Angelo GM, Vlassenko AG, Zhou G, Snyder AZ, Marcus DS, et al. Quantitative analysis of PiB-PET with FreeSurfer ROIs. PLoS One 2013;8:73377. 10.1371/journal.pone.0073377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Su Y, Blazey TM, Snyder AZ, Raichle ME, Marcus DS, Ances BM, et al. Partial volume correction in quantitative amyloid imaging. Neuroimage 2015;107:55–64. 10.1016/j.neuroimage.2014.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Rousset OG, Ma Y, Evans AC. Correction for partial volume effects in PET: principle and validation. J Nucl Med 1998;39:904–11. [PubMed] [Google Scholar]
- [31].Joshi A, Koeppe RA, Fessler JA. Reducing between scanner differences in multi-center PET studies. Neuroimage 2009;46:154–9. 10.1016/j.neuroimage.2009.01.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Goodfellow I, Bengio Y, Courville A. Deep Learning. MIT Press; 2016. 10.1038/nmeth.3707. [DOI] [Google Scholar]
- [33].Hornik K Approximation capabilities of multilayer feedforward networks. Neural Networks 1991;4:251–7. 10.1016/0893-6080(91)90009-T. [DOI] [Google Scholar]
- [34].Hagan MT, Demuth HB, Beale MH. Neural Network Design. Bost Massachusetts PWS 1995. 10.1007/1-84628-303-5. [DOI] [Google Scholar]
- [35].Heaton J Programming Neural Networks in Java. Java Dev J 2002. [Google Scholar]
- [36].Kira K, Rendell LA. A practical approach to feature selection. Proc. ninth Int. Work. Mach. Learn, 1992, p. 249–56. 10.1016/S0031-3203(01)00046-2. [DOI] [Google Scholar]
- [37].Kotz S, Balakrishnan N, Johnson NL. Continuous Multivariate Distributions, Models and Applications: Second Edition. 2005. 10.1002/9780471722069. [DOI] [Google Scholar]
- [38].Schulz M-A, Yeo BTT, Vogelstein JT, Mourao- J, Kather JN, Kording K, et al. Deep learning for brains?: Different linear and nonlinear scaling in UK Biobank brain images vs. machine-learning datasets. BioRxiv 2019. 10.1101/757054. [DOI] [Google Scholar]
- [39].Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 2010;9:119–28. 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, et al. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci 2007. 10.1523/JNEUROSCI.0730-07.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Suenaga T, Hirano A, Llena JF, Yen SH, Dickson DW. Modified Bielschowsky stain and immunohistochemical studies on striatal plaques in Alzheimer’s disease. Acta Neuropathol 1990. 10.1007/BF00294646. [DOI] [PubMed] [Google Scholar]
- [42].Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid β protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathol Appl Neurobiol 1997. 10.1111/j.1365-2990.1997.tb01302.x. [DOI] [PubMed] [Google Scholar]
- [43].Busche MA, Konnerth A. Neuronal hyperactivity - A key defect in Alzheimer’s disease? BioEssays 2015;37:624–32. 10.1002/bies.201500004. [DOI] [PubMed] [Google Scholar]
- [44].Palop JJ, Mucke L. Epilepsy and cognitive impairments in alzheimer disease. Arch Neurol 2009;66:435–40. 10.1001/archneurol.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Busche MA, Konnerth A. Impairments of neural circuit function in Alzheimer’s disease. Philos Trans R Soc B Biol Sci 2016;371:20150429. 10.1098/rstb.2015.0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Stargardt A, Swaab DF, Bossers K. The storm before the quiet: Neuronal hyperactivity and Aβ in the presymptomatic stages of Alzheimer’s disease. Neurobiol Aging 2015;36:1–11. 10.1016/j.neurobiolaging.2014.08.014. [DOI] [PubMed] [Google Scholar]
- [47].Bero AW, Bauer AQ, Stewart FR, White BR, Cirrito JR, Raichle ME, et al. Bidirectional relationship between functional connectivity and amyloid-β deposition in mouse brain. J Neurosci 2012;32:4334–40. 10.1523/JNEUROSCI.5845-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wen J, Sutre ET, Samper-gonzález J, Bottani S, Dormont D, Durrleman S, et al. How serious is data leakage in deep learning studies on Alzheimer’s disease classification? Organ. Hum. Brain Mapp, 2019. [Google Scholar]
- [49].Mendoza H, Klein A, Feurer M, Springenberg JT, Hutter F. Towards automatically-tuned neural networks. Work. Autom. Mach. Learn, 2016. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.