

Abstract
Organelles cooperate with each other to control cellular homeostasis and cell functions by forming close connections through membrane contact sites. Important contacts are present between the endoplasmic reticulum (ER), the main intracellular Ca2+-storage organelle, and the mitochondria, the organelle not only responsible for the majority of cellular ATP production but also for switching on cell death processes. Several Ca2+-transport systems focalize at these contact sites, thereby enabling the efficient transmission of Ca2+ signals from the ER towards mitochondria. This provides a tight control of mitochondrial functions at the microdomain level. Here, we discuss how ER-mitochondrial Ca2+ transfers support cell function and how their dysregulation underlie, drive or contribute to pathogenesis and pathophysiology with major focus on cancer and neurodegeneration, but also with attention for other diseases such as diabetes and rare genetic diseases.
Keywords: Ca2+ signaling, MAMs, contact sites, cancer, neurodegeneration, genetic diseases
Inter-organellar Ca2+ dynamics at membrane contact sites as a critical process underlying cell function
Organellar function is essential for cellular homeostasis and physiology. Organelles do not function as isolated entities. Instead, their function is impacted by other organelles via membrane contact sites. These junctions enable the formation of microdomains that comprise different cellular functions by hosting signaling complexes and enabling distinct processes, including Ca2+ signaling [1]. The role of membrane contact sites and their signaling functions in cell biology and physiology is a highly timely topic that has attracted widespread attention and interest from several disciplines [2].
Ca2+ transfer between endoplasmic reticulum (ER) and mitochondria is enabled by Ca2+-transport systems that reside at mitochondria-associated ER membranes (MAMs). This Ca2 transport controls cell biological processes such as mitochondrial metabolism, autophagy and cell death. Recent research revealed that several proteins whose dysfunction is associated with pathogenesis are located at MAMs and directly impact ER-mitochondrial Ca2+ transfers. These findings advanced the novel concept that disease processes are linked to dysregulated subcellular signaling events at ER-mitochondrial contact sites.
Here, we discuss recent advances in deciphering molecular mechanisms underlying Ca2+ signaling dysregulation at the ER-mitochondrial interface and their contribution to pathogenesis. After explaining basics and principles of ER-mitochondrial Ca2+ transfer, we focus on different diseases for which novel insights in ER-mitochondrial Ca2+ dysfunction have recently emerged, including not only common diseases such as cancer, neurodegenerative diseases and diabetes, but also rare genetic disorders such as Wolfram syndrome and polycystic kidney disease. It will become clear that ER-mitochondrial contact sites form a central hub hosting a growing list of disease-linked proteins that tightly control ER-mitochondrial Ca2+ transfer and thus cellular health. Further exploration of this field may lead to new strategies to tackle these diseases.
Basic players and principles of Ca2+ signaling at MAMs
The ER, the main intracellular Ca2+ store, forms areas of close contact (10–80 nm) with the outer mitochondrial membrane (OMM), termed MAMs (Figure 1, Key figure), enabling efficient mitochondrial Ca2+ transfer [1]. Various proteins control ER-mitochondrial apposition, including inositol 1,4,5 trisphosphate (IP3) receptors (IP3Rs) [3], Mitofusin 2 (Mfn2) [4, 5] and the vesicle-associated membrane protein-associated protein B (VAPB) - protein tyrosine phosphatase interacting protein 51 (PTPIP51) pair [6]. Close ER-mitochondrial connections overcome the relatively low affinity for mitochondrial Ca2+ uptake by establishing a microdomain, where Ca2+ concentrations can rise >10 fold higher than those in the bulk cytosol [7, 8]. The mitochondrial matrix accumulates Ca2+ via voltage-dependent anion channels (VDACs) at the OMM and mitochondrial Ca2+ uniporter (MCU) complexes at the inner mitochondrial membrane (IMM). This process is driven by the negative mitochondrial potential (−180 mV) [9].
Figure 1, key figure: Schematic overview of basic MAM composition and functions.
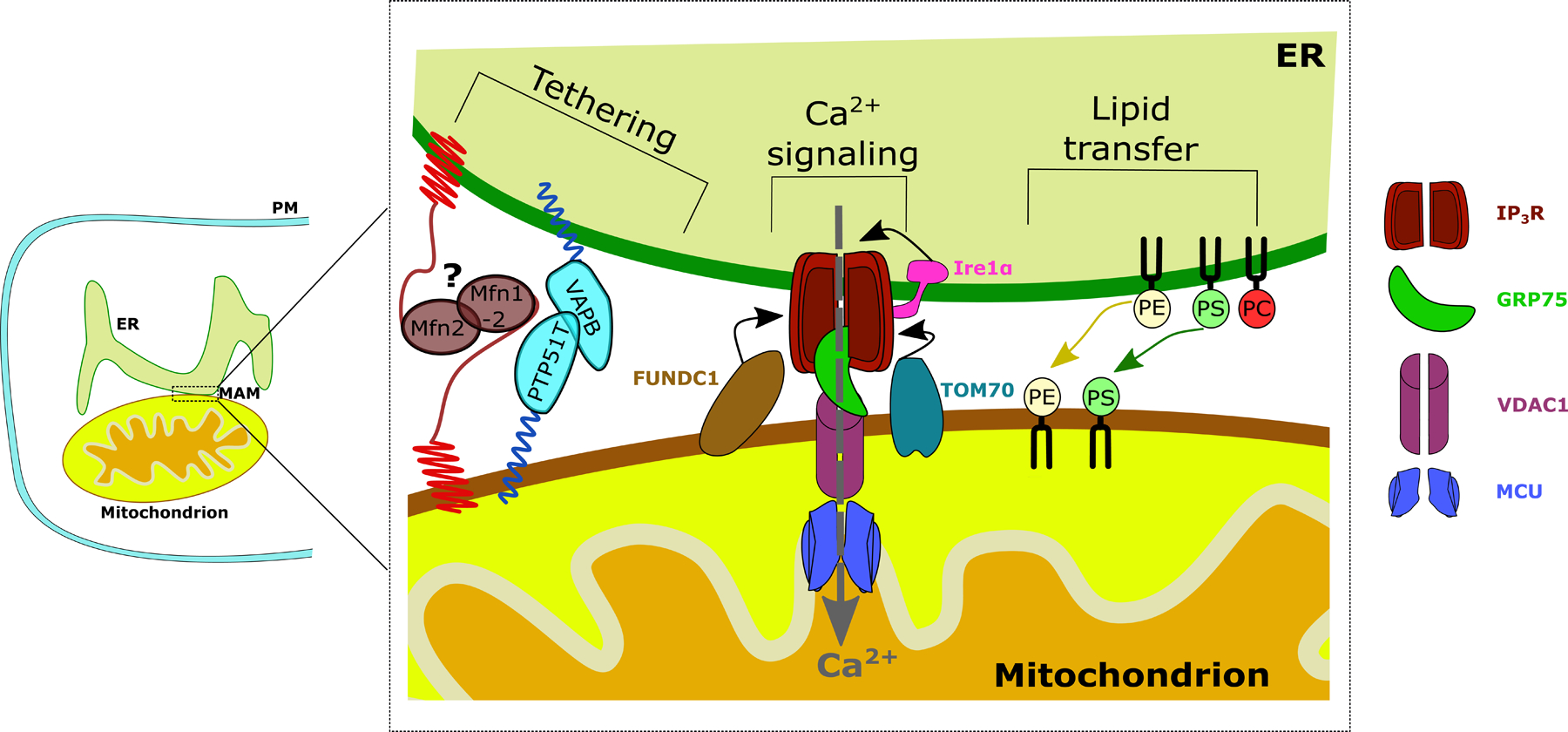
Structural MAM proteins function as physical MAM tethers such as VAPB. Another modulator of ER-mitochondrial tethering is Mfn1/2. Its role as a tether is debated, which is why it is annotated with a question mark. One of the main functions of the MAMs is ER to mitochondrial Ca2+ transfer through the IP3R-VDAC1-MCU axis, stabilized by GRP75. Recently, a non-canonical role for Ire1α was elucidated where Ire1α recruits IP3R to the MAMs. Both TOM70 and FUNDC1 recruit IP3R to the MAMs, via either direct or indirect interactions. Additionally, the synthesis and transfer of lipids from the ER to the mitochondria takes place at the MAMs. For simplicity reasons, only the most abundant membrane lipids are depicted. PM: Plasma membrane, ER: Endoplasmic reticulum, MAM: Mitochondria-associated ER membrane, Ca2+: Calcium ion, Mfn: mitofusin, VAPB: Vesicle-associated membrane protein-associated protein B, PTPIP51: Protein tyrosine phosphatase interacting protein 51, Ire1α: Inositol-requiring enzyme 1 α, FUNCD1: FUN14 domain containing protein 1, TOM70: Translocase of the outer mitochondrial membrane 70, IP3R: Inositol 1,4,5-trisphosphate receptor type 1, GRP75: Glucose-regulated protein 75, VDAC1: Voltage dependent anion channel 1, MCU: Mitochondrial Ca2+ uniporter, PE: phosphatidylethanolamine, PS: phosphatidylserine, PC: phosphatidylcholine.
Key mediators of ER-to-mitochondria Ca2+ transport are IP3Rs, tetrameric IP3-gated, Ca2+-release channels. IP3Rs are tightly controlled by various factors, including accessory proteins. The three IP3R isoforms display distinct IP3 affinity and regulation, thereby finetuning ER Ca2+ release [10]. IP3Rs also integrate other signals such as reactive oxygen species (ROS) [11]. ROS directly oxidizes cysteine residues in IP3Rs, thereby augmenting Ca2+ fluxes [12]. IP3R oxidation is also promoted by the MAM-resident ER oxidase 1 α (ERO1α), an enzyme responsible for forming disulfide bonds at the ER [13].
To facilitate ER-mitochondrial Ca2+ exchange, IP3Rs reside in a macromolecular complex, bringing it in close apposition to VDAC1 [14]. Glucose-regulated protein 75 (GRP75), amongst other proteins, functions as the molecular bridge connecting VDAC1 and IP3Rs [15]. Although all IP3R isoforms can tether ER and mitochondria, IP3R2 appeared the most efficient at transferring Ca2+ to mitochondria [3]. A pore-dead IP3R mutant revealed that the IP3R’s tethering function is independent of its channel activity [3]. In this work, the IP3R target responsible for mitochondrial tethering was not identified. Besides GRP75/VDAC1, other proteins may associate with IP3Rs and contribute to ER-mitochondrial tethering. In cardiac cells, FUN14 domain-containing protein 1 (FUNDC1), a mitochondrial quality-control protein, contributes to MAM formation by binding IP3R2 [16]. Besides FUNDC1, inositol-requiring enzyme 1 α (Ire1α), the ER-stress transducer involved in the unfolded protein response (UPR) [17], and translocase of the OMM 70 (TOM70) [18], bind IP3Rs and recruit them to ER-mitochondrial contact sites. IP3R recruitment to the MAMs by Ire1α is mediated through Ire1α’s C-terminal domain that binds IP3Rs and is independent from Ire1α’s UPR-related RNAse and kinase activities [17].
Effects of ER-mitochondrial Ca2+ fluxes on cell fate
ER-mitochondrial Ca2+ fluxes determine cell fate. Low-level, rhythmic Ca2+ oscillations provide the mitochondria with adequate Ca2+ levels, thereby stimulating mitochondrial ATP production and sustaining survival. The increase in bioenergetic output is driven by the Ca2+ dependence of metabolic enzyme activity such as pyruvate-, isocitrate- and α-ketoglutarate dehydrogenase [9]. Conversely, insufficient ER-mitochondrial Ca2+ flux, either by inhibition or suppression of IP3Rs [19], or by impaired recruitment to the MAMs [17, 18], impairs mitochondrial bioenergetics and increases the AMP/ATP ratio, thereby activating AMP-activated protein kinase, a driver of autophagic flux, and slowing cell proliferation. In the heart, the FUNDC1-IP3R2 axis maintains mitochondrial Ca2+ dynamics and metabolism, which underlies cardiac function in vivo [16].
In contrast, persistent mitochondrial Ca2+ accumulation adversely affects cell survival [20]. Beyond a certain threshold, mitochondrial Ca2+ accumulation will trigger mitochondrial permeability transition pore (mPTP) opening, a process rendering the IMM permeable, causing mitochondrial dysfunction and ultimately cell death [21]. Ca2+ may trigger mPTP opening directly [22], but also indirectly through binding to the mitochondrial lipid cardiolipin. Cardiolipin subsequently dissociates from complex II, thereby provoking its disassembly and unrestricted activity, excessive ROS production and mPTP opening [23].
Given the disparate consequences of ER-mitochondrial Ca2+ transfer, this process is tightly and dynamically regulated. For example, during cell death, SUMOylation of dynamin-related protein 1 (Drp1) is stabilizing the ER-mitochondrial contact sites, thereby facilitating ER-mitochondrial Ca2+ transfer [24]. This drives cell death through cristae remodeling following the disassembly of mitochondrial-dynamin like GTPase (Opa1) oligomers and subsequent cytochrome c release through Bax/Bak pores.
Also during early phases of ER stress, IP3R function is suppressed upon losing the interaction with immunoglobulin protein (BiP/GRP78) and recruiting ER-resident protein 44 [25]. During prolonged ER stress, however, ERO1α expression is induced, thereby hyperoxidizing IP3Rs and augmenting Ca2+ release [26]. These mechanisms together with the UPR sensor Ire1α may enable a mechanistic link between ER proteostasis and cellular functions, like mitochondrial metabolism and cell death, through IP3R modulation at the level of the MAMs.
Hypoxia is another cellular stress trigger underlying cell damage in ischemia/reperfusion but also contributes to the protective actions of cardiac ischemic pre-conditioning, whereby short ischemic periods enable the heart to cope with subsequent ischemic insults. During hypoxia, FUNDC1 accumulates at the MAMs integrating a concerted action of calnexin and Drp1 to drive mitochondrial fission and mitophagy [27]. In blood platelets, FUNDC1-mediated mitophagy could underly the protective effect of ischemic pre-conditioning against ischemia/reperfusion-induced cardiac damage, thus inhibiting platelet activation [28].
Deranged ER-mitochondrial Ca2+ signaling in disease
Cancer
Cancer cells remodel their Ca2+ signaling machinery, thereby contributing to cancer hallmarks [20]. Part of this remodeling is due to loss of tumor suppressor function and increased activity/upregulation of oncogene products [20]. Again, Ca2+ plays a dual role, while ER-mitochondrial Ca2+ fluxes may be dampened in cancer cells to evade cell death, enhanced Ca2+ signaling may promote metabolism and migration [20]. IP3Rs, thus, can act both in a pro-oncogenic and tumor-suppressive manner. The cancer-specific mechanisms discussed below are summarized graphically in Figure 2.
Figure 2: A graphic summary of targets in cancer cells altering IP3R-mediated ER-mitochondrial Ca2+ fluxes.
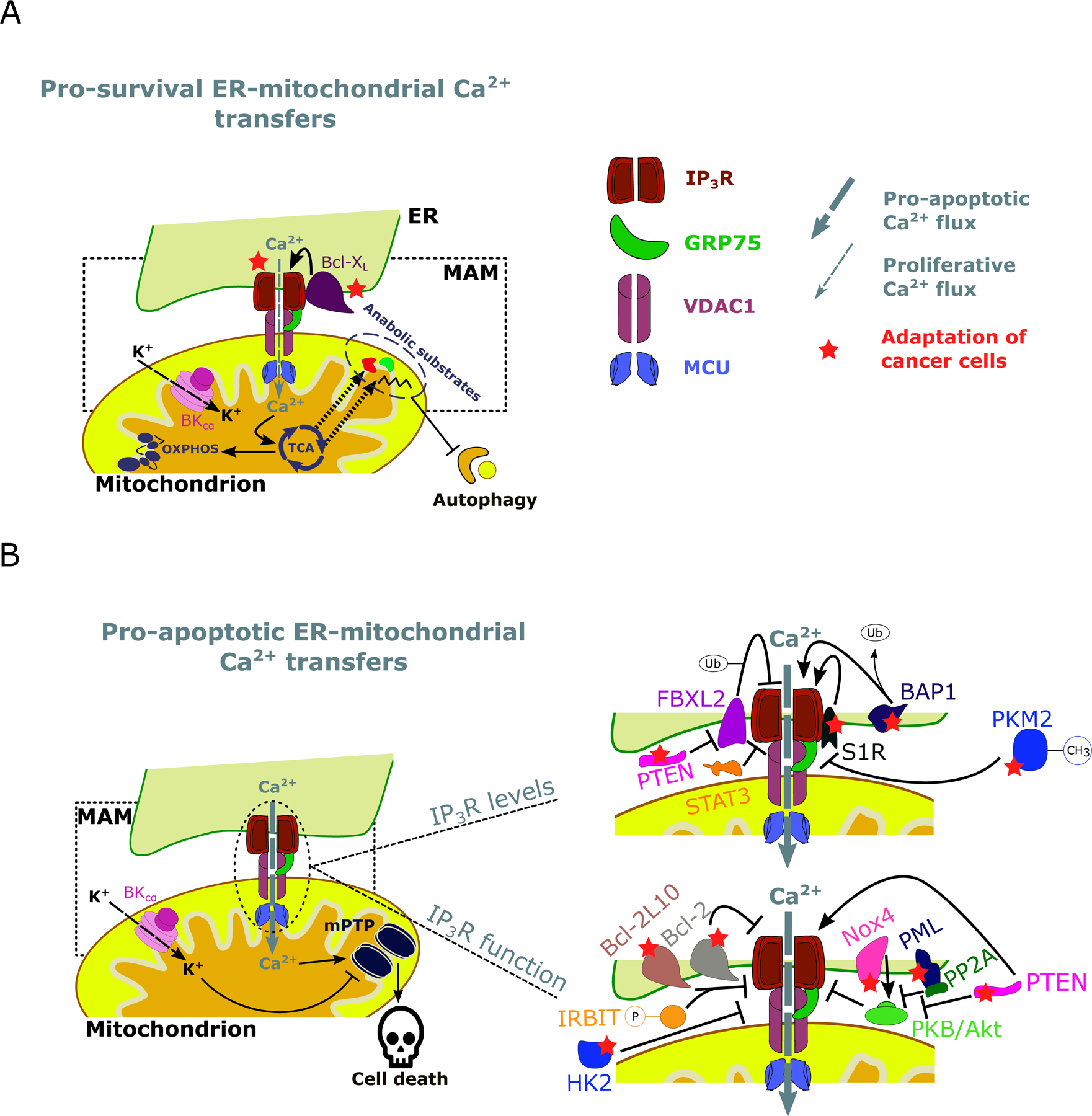
A: Cancer cells are addicted to pro-survival ER-mitochondrial Ca2+ transfers, which promote the mitochondrial metabolism through Ca2+-dependent stimulation of TCA and OXPHOS and suppress autophagic flux. This not only serves for ATP production but also for anabolic pathways through the production of intermediate substrates to synthesize nucleotides. IP3R function can be enhanced by anti-apoptotic/pro-survival proteins such as Bcl-XL. B: Cancer cells also avoid pro-apoptotic ER-mitochondrial Ca2+ transfers. Excessive Ca2+ levels in the mitochondrial matrix lead to cell death via opening of the mPTP. Cancer cells can circumvent this by i. decreasing IP3R levels or ii. modulate IP3R function. PTEN competes with FBXL2, which can mark IP3R for degradation by ubiquitination. Similarly, the oncogenic transcription factor STAT3 facilitates IP3R degradation at the MAMs. Conversely, BAP1 interacts with and deubiquitinates IP3R. Additionally, S1R binds and stabilizes IP3R. Methylated PKM2 was observed to interact with IP3R in the MAMs and to promote IP3R downregulation. Note that the methylations of PKM2 are more complex than the single methyl group depicted here. PTEN negatively regulates PKB/Akt-dependent phosphorylation of IP3Rs, this way promoting pro-apoptotic Ca2+ signaling in the MAMs. Additionally, Nox4 promotes PKB/Akt-mediated inhibitory IP3R phosphorylation. Also, PML recruits PP2A to the MAMs, where it counteracts PKB/Akt activity. Moreover, several anti-apoptotic Bcl-2 proteins can inhibit IP3R, either directly or in cooperation with phosphorylated IRBIT (as is the case for Bcl-2L10). Finally, HK2 negatively modulates pro-apoptotic IP3R-driven Ca2+ fluxes. ER: Endoplasmic reticulum, MAM: Mitochondria-associated ER membrane, TCA: Tricarboxylic acid cycle, OXPHOS: oxidative phosphorylation, Ca2+: Calcium ion, K+: Potassium ion, IP3R: Inositol 1,4,5-trisphosphate receptor type 1, GRP75: Glucose-regulated protein 75, VDAC1: Voltage dependent anion channel 1, MCU: Mitochondrial Ca2+ uniporter, BKCα: Calcium-activated potassium channel, Bcl-XL: B-cell lymphoma extra-large, mPTP: Mitochondrial permeability transition pore, FBXL2: F-box/LRR-repeat protein 2, Bap1: Ubiquitin carboxyl-terminal hydrolase BAP1, PTEN: Phosphatase and tensin homolog, STAT3: Signal transducer and activator of transcription 3, S1R: Sigma-1 receptor, Bcl-2: B-cell lymphoma 2, Bcl-2L10: Bcl-2 like protein 10, Nox4: NADPH oxidase 4, PML: Promyelocytic leukemia protein, PP2A: Protein phosphatase 2A, IRBIT: IP3R binding protein released with IP3, PKB/Akt: Protein kinase B, HK2: Hexokinase 2, Ub: Ubiquitinylations, P: Phosphorylations, CH3: Methylations.
Several cancer cell types appear to be addicted to ER-mitochondrial Ca2+ signaling for their survival [29–31]. Suppressing IP3R function has distinct effects in cancer cell models. Immortalized human MCF-7 breast-cancer cells, for example, display excessive autophagy leading to autophagic cell death after IP3R suppression [31]. However, driving ATP metabolism is not the only function of ER-mitochondrial Ca2+ transfer. In a variety of human cancer cell models (including several tumorigenic cancer cells and oncogene-transformed primary fibroblasts), IP3R inhibition resulted in impaired mitochondrial metabolism and shortage of nucleotides that are essential building blocks for the cell to undergo mitosis [30]. In combination with dysregulated cell cycle checkpoints, this leads to mitotic catastrophe. Also oxidative phosphorylation (OXPHOS) deficient cancer cells need ER-mitochondrial Ca2+ fluxes for survival, thereby preventing NAD+-driven autophagy [32]. Yet, DT40 cells (a chicken cell line derived from bursal lymphoma) and HeLa cells (a human cell line derived from cervival cancers) lacking all three IP3Rs isoforms have been generated and can survive [3, 33]. This indicates that the requirement of IP3Rs for cancer cell survival might be cancer-type dependent and/or that prolonged loss of IP3R function might be compensated by other mechanisms.
Evidence is emerging that elevated IP3R-expression levels promote cancer hallmarks. Immortalized human colorectal and non-small cell lung cancer (NSCLC) cells display increased IP3R3-protein levels [34, 35]. In human MCF-7 breast cancer cells, IP3R3 drives cell proliferation in concert with large conductance Ca2+-activated potassium channel subunit α (BKCa) channels [29]. In primary human glioblastoma cells, IP3R3 sustains cellular mobility, thereby driving cancer cell invasion [36]. Oncogenic proteins can also promote IP3R function to sustain cell survival. For instance, the anti-apoptotic protein Bcl-XL can sensitize IP3Rs to IP3, stimulating cell survival by enabling ER-mitochondrial Ca2+ transfer [37].
Not only do IP3Rs sustain cancer hallmarks but IP3R-mediated Ca2+ fluxes can also drive cell death in cancer cells that therefore often display suppressed IP3R function and ER-mitochondrial Ca2+ fluxes. For example, protein kinase B (PKB/Akt), a cell-growth promoting kinase, binds, phosphorylates and inhibits IP3Rs, conferring resistance against Ca2+-dependent pro-apoptotic stimuli in multiple cancer model cell lines, such as immortalized human glioblastoma cells [38, 39].
Several proteins appear to interfere with PKB/Akt-dependent modulation of IP3R function to control cell survival. For instance, the tumor suppressor promyelocytic leukemia protein is key to recruit protein phosphatase 2 A to the MAMs, where it dephosphorylates PKB/Akt and prevents subsequent IP3R inhibition [40]. More recently, NADPH oxidase 4 (Nox4) was found to be enriched at the MAMs during cellular stress, where it promotes PKB/Akt-mediated phosphorylation of IP3Rs through MAM-confined local redox signaling [41]. Phosphatase and tensin homolog (PTEN) is a tumor suppressor and an inhibitor of PKB/Akt. Immortalized human glioblastoma cells lacking PTEN, displayed IP3R1 hyperphosphorylation and decreased sensitivity to apoptotic stimuli [39]. Additionally, a subpopulation of PTEN proteins is present at ER membrane and MAMs [42]. There, PTEN functions as a protein phosphatase counteracting IP3R phosphorylation by PKB/Akt and increasing the susceptibility of cells to undergo Ca2+-dependent apoptosis [42]. PTEN also supports IP3R function outside its phosphatase function: it was found to compete with F-box/LRR protein 2, a ubiquitin ligase marking IP3R3 for degradation. Hence, lack of PTEN favors IP3R3 ubiquitination, thereby downregulating IP3R3 and increasing resistance against apoptotic stimuli [43]. Indeed, in human prostate cancer tissue, PTEN and IP3R3 levels directly correlate [43]. Similarly, BRCA1-associated protein 1 (BAP1), a tumor suppressor gene product, interacts with IP3R3, enabling its deubiquitination and stabilization. Thus, BAP1 promotes pro-apoptotic IP3R3-mediated Ca2+ fluxes, whereby BAP1 deficiency (BAP1+/−) in primary human fibroblasts and human multiple myeloma cells suppresses IP3R3 function [44].
Other proteins affecting IP3R levels include the ER-targeted chaperone sigma 1 receptor (S1R) [45], STAT3 [46] and methylated pyruvate kinase M2 (PKM2) [47]. Depletion of Ca2+ in the ER activates S1R to dissociate from BiP/GRP78 and to bind and stabilize IP3R3 [45]. It appears that breast, prostate and lung cancer cells in murine xenograft models are susceptible to S1R inhibitors, which reduced tumor growth in vitro and in vivo [48]. STAT3 is an oncogenic transcription factor that also displays an extra-nuclear function at the MAMs, where it downregulates IP3R3 and contributes to the cell-death resistance of basal-like mammary tumors [46]. Methylated PKM2 also downregulates IP3Rs, whereby inhibiting PKM2 methylation compromises in vivo tumor growth of human breast cancer cells xenografted in mice [47].
Members of the B-cell lymphoma 2 (Bcl-2) protein family also reside at MAMs [49], thereby exerting anti-apoptotic functions by preventing ER-mitochondrial Ca2+ transfer in cancer. Anti-apoptotic Bcl-2 is upregulated in several cancer types and directly targets IP3Rs, thereby preventing pro-apoptotic Ca2+ release [50].
Peptides that dislodge Bcl-2 from IP3Rs in cancer cells provoke cell death and/or augmented cell death elicited by other therapeutics. In human diffuse large B-cell lymphoma and primary chronic lymphocytic leukemia cells, these peptides elicit pro-apoptotic Ca2+ signaling [51], resulting in mitochondrial Ca2+ overload and mPTP opening [52]. In ovarian cancer cells, such peptides augment the cytotoxic effects of cisplatin by enhancing cisplatin-induced ER-mitochondrial Ca2+ fluxes [53].
Also, Bcl-2 homolog NRH/BCL-2L10, a proto-oncogene product upregulated in primary human breast cancer cells [54], associates with and inhibits IP3Rs [55]. Additionally, peptides that disrupt IP3R/Bcl-2L10 complexes inhibit the growth of immortalized human breast cancer cells [56]. Bcl-2L10 also cooperates with IRBIT, a protein that, when phosphorylated, binds and inhibits IP3Rs [57]. Together, Bcl-2L10 and phosphorylated IRBIT inhibit IP3Rs at the MAMs. Under cell stress, IRBIT is dephosphorylated and acts as an inhibitor of Bcl-2L10 that promotes ER-mitochondrial Ca2+ fluxes and apoptosis [57]. IP3Rs are not only modulated by anti-apoptotic Bcl-2-protein family members, but also pro-apoptotic members such as Bcl-2-related ovarian killer (Bok) can bind IP3Rs to protect them from proteolytic degradation [58]. Primary human NSCLC cells downregulate Bok, rendering IP3Rs more prone to degradation [59].
The ER Ca2+-store content indirectly controls IP3R function. Certain tumor suppressors increase apoptotic susceptibility via ER-mitochondrial Ca2+ fluxes by augmenting the ER Ca2+-filling state. The tumor suppressor p53 accumulates at ER-mitochondrial contact sites in cells exposed to chemical stresses and chemotherapeutic agents [60]. At the ER, p53 enhances the activity of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), thereby elevating ER Ca2+ levels and boosting pro-apoptotic IP3R-mediated Ca2+ fluxes [60]. In cells lacking p53, SERCA activity is not stimulated, underlying cell-death resistance to chemotherapeutics [60].
Surely, ER-mitochondrial Ca2+ fluxes are not only determined by IP3R function. The mitochondrial gateways for Ca2+, VDAC1 and MCU, are also dysregulated in cancer. The metabolic enzyme hexokinase 2, a VDAC1 partner, was observed to impact cell fate at the MAMs [61]. Displacement of hexokinase 2 from the MAMs with a peptide resulted in mitochondrial Ca2+ overload and subsequent cell death in primary human leukemia B-cells [61]. In addition to this, Bcl-2, Bcl-XL and Mcl-1 can also target VDAC1 in the MAMs and inhibit its ability to shuttle Ca2+ into the mitochondria, conferring apoptotic protection [49, 62]. Yet, anti-apoptotic Bcl-XL and Mcl-1 have also been reported to enhance basal Ca2+ uptake into the mitochondria through VDAC1, thereby promoting mitochondrial metabolism [63, 64]. In several human NSCLC cell lines, boosting VDAC1-mediated mitochondrial Ca2+ uptake and subsequent ROS production through Mcl-1 sustained cancer cell migration [64].
Finally, the amount of Ca2+ reaching the mitochondria will be dampened by local SERCA pumps, whose activity is counteracted by thioredoxin-related transmembrane protein 1 (TMX1), a MAM-localized redox-sensitive oxidoreductase. Low TMX1-protein levels allowed for high SERCA activity, thereby reducing mitochondrial Ca2+ transfer and promoting tumor growth in xenografts of mice injected with human melanoma cells [65]. Other work, however, indicated that several human melanoma cell lines are addicted to high TMX1 (and TMX3) levels, counteracting Nox4-mediated ROS production at the ER-mitochondrial interface. In melanoma cells with high TMX1/TMX3 levels, the Ca2+/calmodulin-dependent phosphatase calcineurin is activated, thereby driving the NFAT pathway and subsequent gene expression involved in cancer cell proliferation and migration [66]. In melanoma cells with low TMX1/TMX3 levels, mitochondrial Ca2+ uptake and Nox4 activity are increased, thereby generating excessive ROS that impairs melanoma growth and migration by oxidation and inhibition of calcineurin. This seemingly opposing role for TMX1 in cancer may reflect the importance of the specific cellular context.
Neurodegeneration
The dysregulation of IP3R-mediated Ca2+-release at ER-mitochondrial contact sites is emerging as a key feature in the pathogenesis of a variety of neurodegenerative diseases. Here, we will focus on Parkinson’s disease (PD) and Alzheimer’s disease (AD) not only because they are the most prevalent neurodegenerative diseases but especially because several PD- and AD-linked proteins reside and function at the MAMs (Figure 3).
Figure 3: A summary of alterations in MAMs for two neurodegenerative diseases: PD and AD.
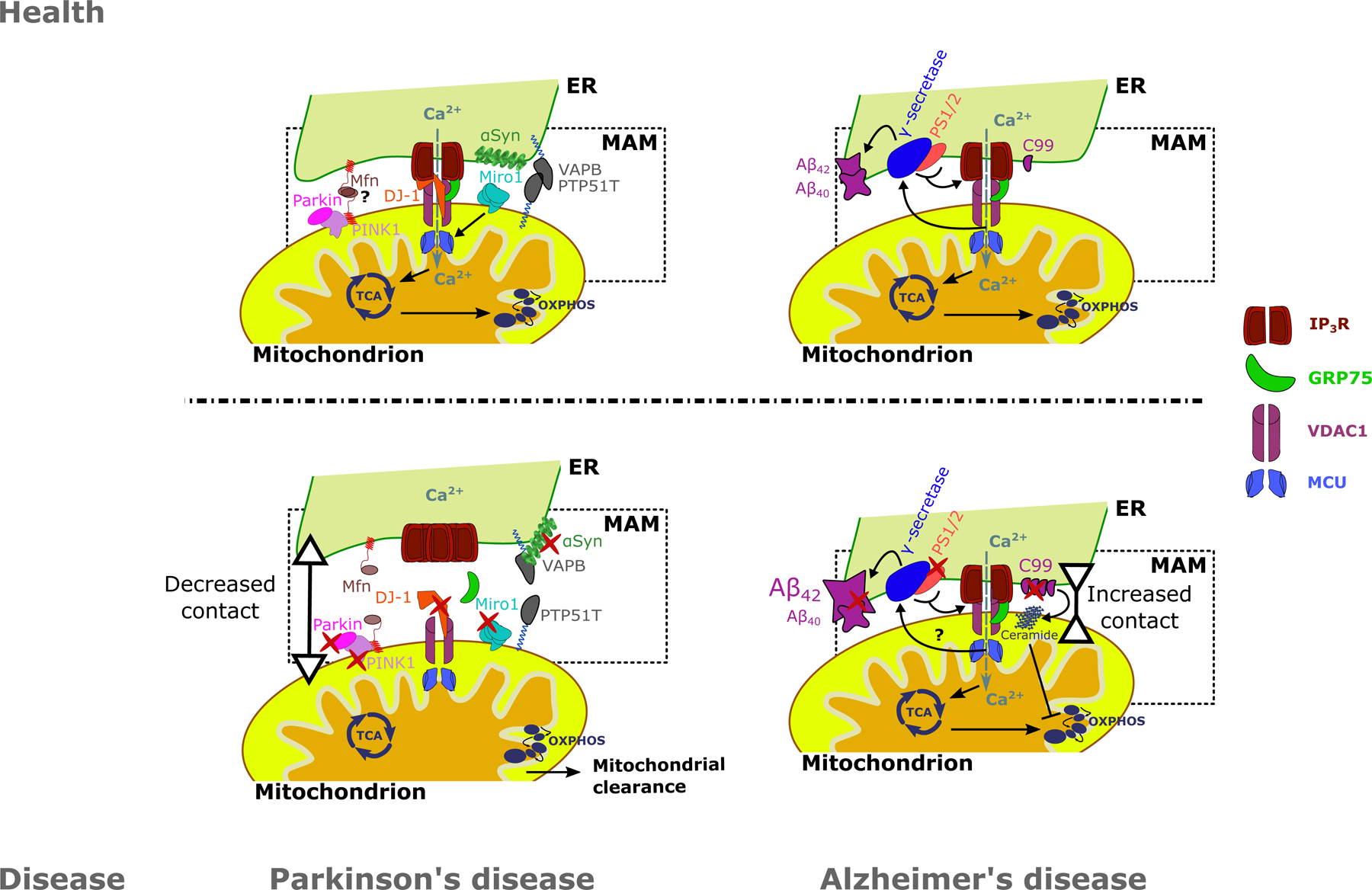
A: DJ-1, a protein associated with familial forms of PD, is a vital component of the IP3R-VDAC1-GRP75 axis in the MAMs. Loss of function of DJ-1 results in decreased ER-mitochondrial Ca2+ transfer, ER-mitochondrial contact sites and upregulation and aggregation of IP3R. PD-related proteins PINK1 and Parkin have been implicated in the tethering of ER to mitochondria. Moreover, Miro1 positively regulates mitochondrial Ca2+ uptake by MCU and mutations in Miro1 result in decreased ER-mitochondrial contact. Additionally, αSyn, of which its aggregated form is a hallmark of PD, is also present in the MAMs. Both WT and mutant αSyn interact with the VAPB-PTPIP51 MAM tethering proteins. B: AD is characterized by the deposition of Aβ plaques due to aberrant APP processing. Processing of APP is performed by the γ-secretase complex, of which PS1 and PS2 are components. APP and PS1–2 are particularly enriched in the MAMs. Mutations in APP, PS1–2 can increase ER-mitochondrial contact. Additionally, mutant PS1–2 are able to sensitize IP3Rs to IP3, increasing ER-mitochondrial Ca2+ transfer, which in turn enhances APP processing. C99, a product of APP processing, is enriched in the MAMs in AD patients cells, where they enhance the accumulation of ceramide in the mitochondria. In turn, ceramides inhibit mitochondrial respiration. Ultimately these changes lead to an increased Aβ42/Aβ40 ratio in AD patient cells. Red marks indicate alterations on the protein level in neurodegeneration, leading to decreased ER-mitochondrial contact in PD and increased ER-mitochondrial contact in AD. Grey arrows indicate Ca2+ fluxes. Question marks signify processes that are still uncertain. ER: Endoplasmic reticulum, MAM: Mitochondria-associated ER membrane, TCA: Tricarboxylic acid cycle, OXPHOS: oxidative phosphorylation, Ca2+: Calcium ion, IP3R: Inositol 1,4,5-trisphosphate receptor type 1, GRP75: Glucose-regulated protein 75, VDAC1: Voltage dependent anion channel 1, MCU: Mitochondrial Ca2+ uniporter, Mfn: Mitofusins, PINK1: PTEN-induced kinase 1, DJ-1: Protein deglycase DJ-1, Miro1: Mitochondrial Rho GTPase 1, VAPB: Vesicle-associated membrane protein-associated protein B/C, PTPIP51: Protein tyrosine phosphatase interacting protein 51, αSyn: α-synuclein, Aβ42/40: 42/40 amino acid proteolytic product of amyloid precursor protein, PS1/2: Presenilin 1 / 2, C99: C99 fragment of amyloid precursor protein.
Parkinson’s disease
PD is a progressive neurodegenerative disease caused by loss of neurons, particularly in the substantia nigra pars compacta, resulting in tremor, shuffling gait and muscle rigidity [67]. α-Synuclein, the main constituent of the intraneuronal Lewy bodies found in PD, is localized at the MAMs, whereas a pathogenic mutant form of α-synuclein (A30P) showed a decreased MAM-localization [68]. More recently, α-synuclein, as well as its pathogenic mutants (A30P and A53T), were shown to bind to the mitochondrial tethering protein VAPB, thereby hindering its association with the ER-localized PTPIP51 and disrupting ER-mitochondrial contact sites [69].
Moreover, PTEN-induced kinase 1 (PINK1) and Parkin, mitophagy proteins associated with recessive familial forms of PD, contribute to ER-mitochondria tethering [70]. For example, the ER-mitochondrial interface is perturbed in Parkin knockout mice and patients with Parkin mutations leading to aberrant ER-to-mitochondria Ca2+ transfers [70]. Work in Caenorhabditis elegans demonstrated that mutant PINK1 reduced mitochondrial toxicity upon exposure to rotenone and paraquat, two environmental risk factors associated with sporadic PD [71]. Exposure to these compounds effectively increased PINK1 expression, while alterations at the ultrastructural level in the MAMs were observed.
Additionally, mitochondrial rho GTPase 1 (Miro1), an OMM protein involved in mitochondrial mobility [72], regulates mitochondrial Ca2+ uptake through MCU [73] and has been implicated in PD due to its interaction with both PINK1 and Parkin [74]. Miro proteins can function as a Ca2+‐dependent docking site for Parkin recruitment [74]. Mutations in the Ca2+-binding EF-hand motif and GTPase domain of Miro1 have recently been identified in sporadic PD patients [75]. The Miro1 mutations led to a decrease in ER-mitochondrial contact sites, resulting in a reduced cytosolic Ca2+ buffering capacity of mitochondria in fibroblasts. Additionally, these mutations likely increase the rate of mitophagy [75].
Another PD-related protein with a strong link to MAMs is Parkinson’s disease protein 7 (Park7/DJ-1), a protein that has been implicated in several cellular processes, including Ca2+ homeostasis [76]. Loss of DJ-1 function causes a recessive-inherited form of PD [77]. DJ-1 is vital for mitochondrial health and regulates ER-mitochondrial tethering [78, 79]. Of note, DJ-1 has been identified as a vital component of the IP3R-GRP75-VDAC1 complex mediating ER- to-mitochondrial Ca2+ transfer [79]. Loss of function of DJ-1 resulted in a disruption of the IP3R-GRP75-VDAC1 complex and a decrease in mitochondrial Ca2+ levels upon IP3R stimulation. Additionally, DJ-1 ablation also led to an increase in IP3R3 levels at ER-mitochondrial contact sites, suggesting IP3R3 aggregates upon losing its association with the IP3R3-GRP75-VDAC1 complex [79].
Recently, VAPB and PTPIP51 were found to localize and interact at ER-mitochondrial contact sites in neuronal synapses, where they sustain pre- and post-synaptic activity [80]. Moreover, induction of synaptic activity increased VAPB-PTPIP51 interaction and promoted ER-mitochondrial contacts [80]. Conversely, loss of VAPB and PTPIP51 decreased the number of active dendritic spines, identified through apposition with the pre-synaptic marker synaptophysin, and reduced synaptic vesicle exocytosis [80]. Therefore, it is possible that loss of neuronal synapse function, a chief feature of PD and other neurodegenerative diseases, is intimately linked with loss of ER-mitochondrial tethering [80].
Alzheimer’s disease
Worldwide, AD is the most prevalent neurodegenerative disease and is the most common cause of dementia. The main pathophysiological mechanism is the formation of intracellular neurofibrillary tangles consisting of aggregated hyperphosphorylated tau and extracellular plaques composed of amyloid β (Aβ), resulting in neuronal atrophy. During the formation of senile plaques, Aβ is cleaved from the amyloid precursor protein (APP) by the γ-secretase complex, containing presenilin 1 (PS1) or presenilin 2 (PS2) as catalytic subunits [81].
Both APP and the γ-secretase complex have been reported to reside in MAM fractions [82, 83], while MAM-localized γ-secretase is active in vitro [82]. When exposing primary hippocampal neurons to Aβ, an upregulation of MAM-associated proteins is triggered, including IP3R3 and VDAC1, and the number of ER-mitochondrial contact sites is increased [84]. In SH-SY5Y cells, exposure to Aβ led to an increase in ER-mitochondrial Ca2+ fluxes [84], while the overexpression of a familial AD mutant form of APP also increased ER-mitochondrial contacts [82]. Moreover, the C99 protein, a product derived from β-secretase-cleaved APP, was found to accumulate at MAMs in neurons of AD mouse models [85]. C99 augmented ER-mitochondrial tethering, promoting excessive mitochondrial accumulation of ceramide, a sphingomyelin hydrolysis product that exerts pro-apoptotic effects and impairs mitochondrial respiration [85].
Cells expressing mutant PS1 or PS2, including familial AD patient lymphoblasts with PS1 mutations [86] and patient-derived fibroblasts harboring PS1 or PS2 mutations [87] displayed disturbances in Ca2+ dynamics, independently of their roles in the γ-secretase complex. Overexpression of wild-type PS2 (but not PS1) in neuroblastoma cells and rat primary cortical neurons favored Ca2+ transfer between ER and mitochondria, which was also enhanced in PS2 mutants [88]. Consistently, PS1 and PS2 interact with IP3Rs [89] and mutant PS can sensitize IP3R to IP3, increasing IP3R-mediated Ca2+ fluxes, which in turn promote APP processing [89].
PS2 can also influence ER-mitochondrial tethering, by interacting with Mfn2, a structural MAM protein. In the presence of Mfn2, mutant PS2 expression increases ER-mitochondrial contacts and modulates ER-mitochondrial Ca2+ signaling [90]. In addition, knockdown of Mfn2 increases ER-mitochondrial contacts and Ca2+ transfer and impairs γ-secretase APP processing activity [91]. Furthermore, the contact area of ER-mitochondrial contact sites and ER-mitochondrial lipid transfer increases significantly in fibroblasts from patients with either the familial or sporadic forms of AD [92]. The emergence of altered ER-mitochondrial contacts in sporadic AD cases is important, since these are the majority of AD cases. Finally, different cell types including neurons, astrocytes and microglia contribute to AD pathogenesis, but how MAMs are dysregulated among these cell types is not well understood.
Other MAM-related disorders
Dysregulated MAM-localized Ca2+ signaling has recently been implicated in the pathogenesis of several other disorders, such as diabetes mellitus (DM), Wolfram syndrome (WS) and polycystic kidney disease (PKD), for which novel molecular mechanisms at the MAMs emerged.
Diabetes mellitus
DM is a disorder leading to the disruption of glucose homeostasis and distinguished as type 1 DM or type 2 DM (T2D) [93]. Type 1 DM is characterized by the destruction of pancreatic β cells by the immune system, impairing insulin secretion [93]. T2D is associated with lower insulin sensitivity and mostly also a decrease in insulin production, impairing cellular glucose uptake [93].
Several proteins important for both insulin signaling and cell survival are located at the MAMs and affect ER-mitochondrial Ca2+ transfer. Glycogen synthase kinase 3 β (GSK3β), a protein that inactivates glycogen synthase, is such an example. In mouse heart, GSK3β partially localizes at the MAMs, where it interacts with and stimulates IP3Rs [94]. In murine pancreatic β cells, GSK3β enhances a tissue-specific basal ER Ca2+ leak towards the mitochondria that is mediated by PS1 phosphorylated by GSK3β [95]. Another kinase, pyruvate dehydrogenase kinase 4 (PDK4) is upregulated during obesity, which led to increased MAM formation and insulin resistance [96]. At the MAMs, PDK4 binds the IP3R1-GRP75-VDAC1 complex, which enhances ER-mitochondrial Ca2+ transfer [96]. Additionally, acute glucose treatment of rat-derived pancreatic cell models was observed to stimulate ER-mitochondrial interactions and ER-mitochondrial Ca2+ transfer, depleting ER Ca2+ stores and inducing ER stress [97].
Presumably, MAMs can couple energy sensing to mitochondrial physiology and play an important role in glucose homeostasis. Hence, alterations at the level of the MAMs could play a role in the pathophysiology of DM [98]. Indeed, the number of IP3R2-VDAC1 interactions was lower in T2D patient pancreatic tissue compared to healthy controls [99]. In T2D β cells, VDAC1 levels were decreased, while IP3R2 levels were increased, indicating that specific changes in MAM-protein levels are characteristic for T2D [98, 99].
Earlier work in isolated hepatocytes revealed that IP3R1 knockdown reduces glucose production [100]. This likely involves glucagon, which recently was shown to stimulate gluconeogenesis in liver tissue via an IP3R1-dependent mechanism [101]. Elevated glucagon plasma levels in WT mice could counteract diet-induced hepatic steatosis and resistance to insulin. However, these effects are absent in mice lacking IP3R1 [101].
Wolfram syndrome
Wolfram syndrome (WS) is a rare genetic disease caused by mutations in WFS1, resulting in the more prevalent type I WS, or in CISD2, resulting in the rarer type II WS. The currently untreatable disease starts at a young age with diabetes mellitus due to pancreatic β cell dysfunction and destruction but then aggravates with severe neurological complications, resulting in early death [102].
Wolframin (Wfs1) was recently proposed to be a MAM protein [103]. Wfs1 function has been linked to ER stress and Ca2+ homeostasis [104]. Loss of Wfs1 was associated with suppressed IP3R-mediated Ca2+ release in primary neuronal models and patient fibroblasts [105, 106]. Lack of Wfs1 also evoked mild ER stress and impaired mitochondrial metabolism and dynamics as well as a decrease in ER-mitochondrial contact sites, evidenced by a severe reduction in IP3R/GRP75 and IP3R/VDAC1 interactions [105].
Recently, Wfs1 was found to reside in a complex with neuronal calcium-sensor 1 (NCS-1) [105]. NCS-1 sensitizes IP3R, thereby promoting Ca2+ oscillations in living cells [107]. NCS-1 itself directly binds IP3Rs near its N-terminal suppressor domain [108], a region critical for IP3R-channel gating. The elevated NCS-1 levels could salvage the adverse effects associated with Wfs1 deficiency, thereby augmenting mitochondrial Ca2+ uptake and improving mitochondrial respiratory chain functionality [105]. NCS-1 may couple Wfs1 to a macrocomplex with the MAM-residing complex IP3R–GRP75–VDAC1 [105]. Also, INS 832/13 cells lacking Wfs1 displayed elevated cytosolic Ca2+ levels, decreased IP3R-mediated Ca2+ release, accompanied by a reduced ER-mitochondrial Ca2+ transfer [109]. Furthermore, ibudilast, a phosphodiesterase 4 inhibitor potentially also targeting NCS-1, and calpain inhibitor XI could rescue basal cytosolic Ca2+ levels in cells lacking Wfs1, restoring insulin-producing capacity of β cells and thus opening new therapeutic possibilities for WS patients.
CISD2, the other causative gene for WS, encodes a single-pass transmembrane ER protein containing a two iron-two sulphur cluster domain, named CDGSH iron sulfur domain 2 (Cisd2). Cisd2 is specifically enriched at the MAMs [110]. Similar to Wfs1, Cisd2 impacts ER Ca2+ dynamics by interacting with IP3R1 [111] and SERCA2b [112]. Additionally, patient-derived fibroblasts carrying a mutation in CISD2 displayed increased ER-mitochondrial contacts and ER-mitochondrial Ca2+ fluxes, possibly underlying hyperfusion of the mitochondrial network [113]. Moreover, Cisd2 deficiency augmented cytosolic Ca2+ levels and modulated mitochondrial Ca2+ uptake by interacting with GTPase of immune-associated protein 5 (Gimap5), a MAM-resident protein [110]. Therefore, at the MAMs, Cisd2 may reside in a macrocomplex with IP3R, Gimap5 and perhaps other proteins regulating Ca2+ signaling [103].
Polycystic kidney disease
PKD is a collection of several monogenic diseases, primarily characterized by cyst development in kidneys and potentially other organs such as liver [114]. The autosomal-dominant form of PKD is associated with two genes, including Polycystin 2 (PC2). PC2 knockdown resulted in increased Mfn2 expression and ER-mitochondrial Ca2+ transfer, while MAM IP3R3 levels were decreased. Knockdown of Mfn2 could restore normal ER-mitochondrial Ca2+ transfer. Also, kidney sections of autosomal-dominant PKD patients displayed higher Mfn2 levels and a higher mitochondrial density, as evidenced by higher TOM20 levels per mitochondrial area. Hence, PC2 might be a key regulator of the expression of MAM-resident proteins [115].
Concluding remarks
Several disease-linked proteins recently emerged to reside and function at the MAMs as controllers of ER-mitochondrial Ca2+ transfer. Numerous studies implicate them in the pathogenesis of various diseases such as cancer, neurodegenerative diseases and other MAM-related disorders. Yet, several aspects of ER-mitochondrial Ca2+ fluxes in health and disease, including ageing, the importance in sporadic forms of neurodegenerative diseases, and integration of ER-mitochondrial Ca2+ signals with other MAM-related processes ought to be further studied [see outstanding questions box]. These recent advances also provide new targets to develop novel therapeutic strategies. Hence, future efforts should focus on translating these concepts towards disease prevention and therapy, including gene therapies as well as novel or repurposed pharmacological therapeutics. Moreover, focusing on MAM-residing proteins in the early stages of the disease conditions could help preventing disease onset and delaying disease progression. Targeting of the MAM-localized Ca2+-signaling complexes with high selectivity in a spatially restricted manner, both at the subcellular and tissue level, will be important as a potential therapeutic strategy for these disorders.
Outstanding questions.
Which proteins form the core of the IP3R tethering complex and which are cell-type or condition-dependent? Which mitochondrial targets underlie the ER-mitochondrial tethering function of IP3Rs? Which mechanisms contribute to the recruitment of IP3Rs to the MAMs?
How is the width of the cleft between ER and mitochondria regulated in physiological and in pathological conditions? Is there redundancy between various tethering proteins?
How do the different functional modules of the MAMs, e.g. lipid trafficking and Ca2+ signaling, impact each other and/or act in a concerted manner in health and disease?
Are structural or functional changes in MAMs a cause of disease or are they rather the consequence of dysregulation or represent compensatory changes? Many diseases involve distinct cell types, hence can restoring the functionality of MAMs in one cell type be sufficient to effectively counteract disease progression and outcomes? For example, is it sufficient to normalize MAM function in neurons for AD and PD?
How do ER-mitochondrial contact sites change during sporadic forms of neurodegenerative diseases?
How can we detect perturbations in MAM-resident proteins and processes early on in disease to serve as a biomarker?
Are the changes in MAMs in the context of pathology reversible, and if so, how can therapies based on pharmacological tools or gene transfer be designed to enable this reversion? How can MAM-resident proteins be targeted to restore ER-mitochondrial Ca2+ transfer in diseases?
Highlights.
Mitochondria-associated membranes (MAMs) establish signaling microdomains for the exchange of Ca2+ and lipids between ER and mitochondria
Ca2+ transfer between ER and mitochondria is critical for cellular physiology and functions, including mitochondrial metabolism and cell death
The presence, stability, levels and activity of IP3Rs, intracellular Ca2+-release channels, at the MAMs are tightly regulated by a plethora of mechanisms
Many proteins that are dysregulated or mutated in pathologies ranging from cancer to neurodegenerative disease reside at the MAMs, where they impact ER-mitochondrial Ca2+ transfer and affect cell function
Deranged ER-mitochondrial Ca2+ signaling drives pathogenesis and impacts disease outcomes.
Acknowledgements
Research in the authors’ laboratories was supported by research grants of the Research Foundation - Flanders (FWO) (G.0A34.16N, G.0901.18N and G.0818.21N to GB, grant G0E7520N to GB and IB, grant G.0C91.14N to GB and JBP, and grant G0A6919N to JBP), the Research Council - KU Leuven (OT14/101and AKUL/19/34 to GB, and C14/19/101 to GB and JBP), Central European Leuven Strategic Alliance (CELSA/18/040 to GB and AK), Stichting Alzheimer Onderzoek (SAO IP3 RECEPTOR to GB), Eye Hope Foundation/Koning Boudewijnstichting (2020-J1160630–214966 to GB), Russian Science Foundation (Grant 20–45-01004 to IB) and National Institutes of Health (Grants R01AG055577 and R01NS056224 to IB). MK obtained a doctoral fellowship from the FWO. AK is also supported by the Estonian Research Council (PRG400) and the European Regional Development Fund [2014–2020.4.01.15–0012]. IB is a holder of the Carl J. and Hortense M. Thomsen Chair in Alzheimer’s Disease Research. We apologize to all authors, whose work could not be cited due to space restricitions.
Glossary
- Autophagy
a general term that refers to the lysosomal process important for the removal of misfolded/aggregated proteins, damaged organelles and intracellular pathogens. Specific terms are used for specific organelles, e.g. mitophagy for removal of damaged mitochondria. Autophagy is a pro-survival process key for cellular homeostasis, though when deregulated can also lead to cell death.
- B-cell lymphoma-2 (Bcl-2)
founding member of the Bcl-2-protein family, which regulates cell fate by acting as an anti-apoptotic protein.
- Ca2+ signaling
the process of coupling dynamic, spatiotemporal changes in intracellular Ca2+ concentrations with the deciphering of these changes by Ca2+-dependent sensors and effectors present in the cytosol and organellar compartments.
- CDGSH iron sulfur containing domain protein 2 (Cisd2)
protein encoded by the CISD2 gene. Mutations in this gene can give rise to type II Wolfram syndrome. Cisd2 is enriched at the MAMs.
- Endoplasmic reticulum (ER)
the organelle that is not only responsible for protein synthesis & folding and lipid synthesis but also serves as the main intracellular Ca2+ store.
- Glucose-regulated protein of 75 kDa (GRP75)
a chaperone that predominantly resides in the mitochondrial matrix but also at the MAMs, where it participates in the IP3R-GRP75-VDAC1 complex.
- Inositol 1,4,5-trisphosphate (IP3)
a diffusible ligand of IP3Rs produced by phospholipase C from phosphatidylinositol 4,5-bisphosphate in response to extracellular stimuli e.g. via activation of G-protein-coupled receptors or tyrosine kinase receptors.
- Inositol 1,4,5-trisphosphate receptors (IP3Rs)
tetrameric intracellular Ca2+-release channels that mediate Ca2+ release from the endoplasmic reticulum in response to IP3. IP3Rs are also found at contact sites between ER and mitochondria, where they reside in interorganellar protein complexes involving VDAC channels.
- Mitochondria-associated ER membranes (MAMs)
a fraction of the ER membranes that can be biochemically isolated and whose protein and lipid composition represents the contact sites between ER and mitochondria.
- Mitochondrial Ca2+ Uniporter (MCU)
a pore-forming protein responsible for mitochondrial Ca2+ uptake across the inner mitochondrial membrane and whose activity is tightly controlled by several regulators.
- Mitochondrion
the organelle responsible for the Krebs cycle, oxidative phosphorylation (OXPHOS) and ultimately the production of large amounts of ATP. Due to its close proximity to the ER and the large driving force by the negative mitochondrial membrane potential, Ca2+ signals arising from the ER are also transmitted towards the mitochondria, where they boost the activity of several enzymes participating in the Krebs cycle.
- Phosphatase and tensin homologue (PTEN)
phosphatase that dephosphorylates various targets, among which PKB/Akt
- Polycystin 2 (PC2)
a gene for which mutations/deletions result in polycystic kidney disease.
- Presenilin-1 and -2 (PS1 and -2)
proteins that are part of the γ-secretase complex involved in cleavage of the amyloid precursor protein. As single proteins, presenilins are also involved in different cell functions, such as autophagy and Ca2+ homeostasis. Mutations in presenilin-1 and -2 are linked to cases of familial Alzheimer’s disease
- Protein kinase B (PKB)/Akt
pleiotropic kinase that integrates various intracellular signaling pathways.
- Sarco-/endoplasmic reticulum Ca2+ ATPase (SERCA)
ER protein that pumps Ca2+ from the cytosol back into the ER to ensure low cytosolic Ca2+ concentrations and filling of the ER.
- Voltage-dependent anion channels (VDAC)
a conductance channel located at the mitochondrial outer membrane; this channel not only permeates anions, but also permeates cations such as Ca2+, thereby transmitting IP3R-mediated Ca2+ signals towards the mitochondria.
- Wolframin (Wfs1)
protein encoded by the WFS1 gene. Mutations in this gene result in type I Wolfram syndrome. Wfs1 is linked to ER stress & Ca2+ homeostasis and is enriched at the MAMs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- 1.Prinz WA et al. (2020) The functional universe of membrane contact sites. Nature Reviews Molecular Cell Biology 21 (1), 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dolgin E (2019) How secret conversations inside cells are transforming biology. Nature 567 (7747), 162–164. [DOI] [PubMed] [Google Scholar]
- 3.Bartok A et al. (2019) IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nature Communications 10 (1), 3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Brito OM and Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456 (7222), 605–610. [DOI] [PubMed] [Google Scholar]
- 5.Filadi R et al. (2015) Mitofusin 2 ablation increases endoplasmic reticulum-mitochondria coupling. Proceedings of the National Academy of Sciences of the United States of America 112 (17), E2174–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Csordás G et al. (2018) Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends in Cell Biology 28 (7), 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Csordás G et al. (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Molecular cell 39 (1), 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giacomello M et al. (2010) Ca2+ Hot Spots on the Mitochondrial Surface Are Generated by Ca2+ Mobilization from Stores, but Not by Activation of Store-Operated Ca2+ Channels. Molecular Cell 38 (2), 280–290. [DOI] [PubMed] [Google Scholar]
- 9.Rizzuto R et al. (2012) Mitochondria as sensors and regulators of calcium signalling. Nature Reviews Molecular Cell Biology 13 (9), 566–578. [DOI] [PubMed] [Google Scholar]
- 10.Roest G et al. (2017) IP3 Receptor Properties and Function at Membrane Contact Sites. Advances in Experimental Medicine and Biology 981, 149–178. [DOI] [PubMed] [Google Scholar]
- 11.Joseph SK et al. (2019) Redox regulation of ER and mitochondrial Ca2+ signaling in cell survival and death. Cell Calcium 79, 89–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joseph SK et al. (2018) Redox regulation of type-I inositol trisphosphate receptors in intact mammalian cells. Journal of Biological Chemistry 293 (45), 17464–17476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anelli T et al. (2011) Ero1α Regulates Ca2+ Fluxes at the Endoplasmic Reticulum–Mitochondria Interface (MAM). Antioxidants & Redox Signaling 16 (10), 1077–1087. [DOI] [PubMed] [Google Scholar]
- 14.Loncke J et al. (2020) Recent advances in understanding IP3R function with focus on ER-mitochondrial Ca2+ transfers. Current Opinion in Physiology 17, 80–88. [Google Scholar]
- 15.Szabadkai G et al. (2006) Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. The Journal of Cell Biology 175 (6), 901–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu S et al. (2017) Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 136 (23), 2248–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carreras-Sureda A et al. (2019) Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nature Cell Biology 21 (6), 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filadi R et al. (2018) TOM70 Sustains Cell Bioenergetics by Promoting IP3R3-Mediated ER to Mitochondria Ca2+ Transfer. Current Biology 28 (3), 369–382.e6. [DOI] [PubMed] [Google Scholar]
- 19.Cárdenas C et al. (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142 (2), 270–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanova H et al. (2017) Endoplasmic Reticulum–Mitochondrial Ca2+ Fluxes Underlying Cancer Cell Survival. Frontiers in Oncology 7, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giorgi C et al. (2018) The machineries, regulation and cellular functions of mitochondrial calcium. Nature Reviews Molecular Cell Biology 19 (11), 713–730. [DOI] [PubMed] [Google Scholar]
- 22.Giorgio V et al. (2017) Ca2+ binding to F-ATP synthase β subunit triggers the mitochondrial permeability transition. EMBO Reports 18 (7), 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hwang MS et al. (2014) Mitochondrial Ca2+ influx targets cardiolipin to disintegrate respiratory chain complex II for cell death induction. Cell Death and Differentiation 21 (11), 1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prudent J et al. (2015) MAPL SUMOylation of Drp1 Stabilizes an ER/Mitochondrial Platform Required for Cell Death. Molecular Cell 59 (6), 941–955. [DOI] [PubMed] [Google Scholar]
- 25.Higo T et al. (2005) Subtype-Specific and ER Lumenal Environment-Dependent Regulation of Inositol 1,4,5-Trisphosphate Receptor Type 1 by ERp44. Cell 120 (1), 85–98. [DOI] [PubMed] [Google Scholar]
- 26.Li G et al. (2009) Role of ERO1-α–mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. Journal of Cell Biology 186 (6), 783–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu W et al. (2016) FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. The EMBO Journal 35 (13), 1368–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang W et al. (2016) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. eLife 5, e21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mound A et al. (2013) Molecular interaction and functional coupling between type 3 inositol 1,4,5-trisphosphate receptor and BKCa channel stimulate breast cancer cell proliferation. European Journal of Cancer 49 (17), 3738–3751. [DOI] [PubMed] [Google Scholar]
- 30.Cárdenas C et al. (2016) Selective Vulnerability of Cancer Cells by Inhibition of Ca2+ Transfer from Endoplasmic Reticulum to Mitochondria. Cell Reports 14 (10), 2313–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh A et al. (2017) Inhibition of Inositol 1, 4, 5-Trisphosphate Receptor Induce Breast Cancer Cell Death Through Deregulated Autophagy and Cellular Bioenergetics. Journal of Cellular Biochemistry 118 (8), 2333–2346. [DOI] [PubMed] [Google Scholar]
- 32.Cardenas C et al. (2020) Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum–to–mitochondria Ca2+ transfer for survival. Science Signaling 13 (640), eaay1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ando H et al. (2018) Aberrant IP3 receptor activities revealed by comprehensive analysis of pathological mutations causing spinocerebellar ataxia 29. Proceedings of the National Academy of Sciences 115 (48), 12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iommelli F et al. (2018) Inositol Trisphosphate Receptor Type 3-mediated Enhancement of EGFR and MET Cotargeting Efficacy in Non–Small Cell Lung Cancer Detected by 18F-fluorothymidine. Clinical Cancer Research 24 (13), 3126. [DOI] [PubMed] [Google Scholar]
- 35.Shibao K et al. (2010) The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 48 (6), 315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang SS et al. (2010) Caffeine-Mediated Inhibition of Calcium Release Channel Inositol 1,4,5-Trisphosphate Receptor Subtype 3 Blocks Glioblastoma Invasion and Extends Survival. Cancer Research 70 (3), 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White C et al. (2005) The endoplasmic reticulum gateway to apoptosis by Bcl-XL modulation of the InsP3R. Nature Cell Biology 7 (10), 1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan MT et al. (2006) Akt Kinase Phosphorylation of Inositol 1,4,5-Trisphosphate Receptors. Journal of Biological Chemistry 281 (6), 3731–3737. [DOI] [PubMed] [Google Scholar]
- 39.Szado T et al. (2008) Phosphorylation of inositol 1,4,5-trisphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proceedings of the National Academy of Sciences 105 (7), 2427 LP-2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giorgi C et al. (2010) PML Regulates Apoptosis at Endoplasmic Reticulum by Modulating Calcium Release. Science 330 (6008), 1247 LP-1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beretta M et al. (2020) Nox4 regulates InsP3 receptor-dependent Ca2+ release into mitochondria to promote cell survival. The EMBO Journal 39 (19), e103530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bononi A et al. (2013) Identification of PTEN at the ER and MAMs and its regulation of Ca2+ signaling and apoptosis in a protein phosphatase-dependent manner. Cell Death and Differentiation 20 (12), 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuchay S et al. (2017) PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546 (7659), 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bononi A et al. (2017) BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546 (7659), 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayashi T and Su T-P (2007) Sigma-1 Receptor Chaperones at the ER-Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 131 (3), 596–610. [DOI] [PubMed] [Google Scholar]
- 46.Avalle L et al. (2019) STAT3 localizes to the ER, acting as a gatekeeper for ER-mitochondrion Ca2+ fluxes and apoptotic responses. Cell Death & Differentiation 26 (5), 932–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu F et al. (2017) PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nature Cell Biology 19 (11), 1358–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spruce BA et al. (2004) Small Molecule Antagonists of the σ−1 Receptor Cause Selective Release of the Death Program in Tumor and Self-Reliant Cells and Inhibit Tumor Growth in vitro and in vivo. Cancer Research 64 (14), 4875. [DOI] [PubMed] [Google Scholar]
- 49.Monaco G et al. (2015) The BH4 domain of anti-apoptotic Bcl-XL, but not that of the related Bcl-2, limits the voltage-dependent anion channel 1 (VDAC1)-mediated transfer of pro-apoptotic Ca2+ signals to mitochondria. The Journal of Biological Chemistry 290 (14), 9150–9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rong Y-P et al. (2008) Targeting Bcl-2-IP3 receptor interaction to reverse Bcl-2’s inhibition of apoptotic calcium signals. Molecular Cell 31 (2), 255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bittremieux M et al. (2019) Constitutive IP(3) signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP(3) receptor disruptor BIRD-2. Cell death and differentiation 26 (3), 531–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kerkhofs M et al. (2021) BIRD-2, a BH4-domain-targeting peptide of Bcl-2, provokes Bax/Bak-independent cell death in B-cell cancers through mitochondrial Ca2+-dependent mPTP opening. Cell Calcium 94, 102333. [DOI] [PubMed] [Google Scholar]
- 53.Xie Q et al. (2018) TAT‑fused IP3R‑derived peptide enhances cisplatin sensitivity of ovarian cancer cells by increasing ER Ca2+ release. International Journal of Molecular Medicine 41 (2), 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Krajewska M et al. (2008) Bcl-B Expression in Human Epithelial and Nonepithelial Malignancies. Clinical Cancer Research 14 (10), 3011 LP-3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonneau B et al. (2013) Non-apoptotic roles of Bcl-2 family: The calcium connection. Biochimica et Biophysica Acta 1833 (7), 1755–1765. [DOI] [PubMed] [Google Scholar]
- 56.Nougarede A et al. (2018) Breast Cancer Targeting through Inhibition of the Endoplasmic Reticulum-Based Apoptosis Regulator Nrh/BCL2L10. Cancer Research 78 (6), 1404 LP-1417. [DOI] [PubMed] [Google Scholar]
- 57.Bonneau B et al. (2016) IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. eLife 5, e19896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schulman JJ et al. (2013) The Bcl-2 Protein Family Member Bok Binds to the Coupling Domain of Inositol 1,4,5-Trisphosphate Receptors and Protects Them from Proteolytic Cleavage. Journal of Biological Chemistry 288 (35), 25340–25349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moravcikova E et al. (2017) BOK displays cell death-independent tumor suppressor activity in non-small-cell lung carcinoma. International Journal of Cancer 141 (10), 2050–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Giorgi C et al. (2015) p53 at the endoplasmic reticulum regulates apoptosis in a Ca2+-dependent manner. Proceedings of the National Academy of Sciences 112 (6), 1779 LP-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ciscato F et al. (2020) Hexokinase 2 displacement from mitochondria-associated membranes prompts Ca2+-dependent death of cancer cells. EMBO Reports 21 (7), e49117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rimessi A et al. (2020) Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Seminars in Cell & Developmental Biology 98, 167–180. [DOI] [PubMed] [Google Scholar]
- 63.Huang H et al. (2013) An interaction between Bcl-XL and the voltage-dependent anion channel (VDAC) promotes mitochondrial Ca2+ uptake. The Journal of Biological Chemistry 288 (27), 19870–19881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang H et al. (2014) Mcl-1 promotes lung cancer cell migration by directly interacting with VDAC to increase mitochondrial Ca2+ uptake and reactive oxygen species generation. Cell Death & Disease 5 (10), e1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Raturi A et al. (2016) TMX1 determines cancer cell metabolism as a thiol-based modulator of ER–mitochondria Ca2+ flux. Journal of Cell Biology 214 (4), 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang X et al. (2019) Redox signals at the ER–mitochondria interface control melanoma progression. The EMBO Journal 38 (15), e100871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grayson M (2016) Parkinson’s disease. Nature 538 (7626), S1. [DOI] [PubMed] [Google Scholar]
- 68.Guardia-Laguarta C et al. (2014) α-Synuclein is localized to mitochondria-associated ER membranes. The Journal of Neuroscience 34 (1), 249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Paillusson S et al. (2017) α-Synuclein binds to the ER–mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathologica 134 (1), 129–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gautier CA et al. (2016) The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Human Molecular Genetics 25 (14), 2972–2984. [DOI] [PubMed] [Google Scholar]
- 71.Wu S et al. (2018) Mutation of hop-1 and pink-1 attenuates vulnerability of neurotoxicity in C. elegans: the role of mitochondria-associated membrane proteins in Parkinsonism. Experimental Neurology 309, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saotome M et al. (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proceedings of the National Academy of Sciences 105 (52), 20728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Niescier RF et al. (2018) MCU Interacts with Miro1 to Modulate Mitochondrial Functions in Neurons. The Journal of Neuroscience 38 (20), 4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Safiulina D et al. (2019) Miro proteins prime mitochondria for Parkin translocation and mitophagy. The EMBO Journal 38 (2), e99384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Grossmann D et al. (2019) Mutations in RHOT1 Disrupt Endoplasmic Reticulum-Mitochondria Contact Sites Interfering with Calcium Homeostasis and Mitochondrial Dynamics in Parkinson’s Disease. Antioxidants & Redox Signaling 31 (16), 1213–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shtifman A et al. (2011) Altered Ca2+ homeostasis in the skeletal muscle of DJ-1 null mice. Neurobiology of Aging 32 (1), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bonifati V et al. (2003) Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science 299 (5604), 256–259. [DOI] [PubMed] [Google Scholar]
- 78.Ottolini D et al. (2013) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum– mitochondria tethering. Human Molecular Genetics 22 (11), 2152–2168. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y et al. (2019) DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proceedings of the National Academy of Sciences 116 (50), 25322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gómez-Suaga P et al. (2019) The VAPB-PTPIP51 endoplasmic reticulum-mitochondria tethering proteins are present in neuronal synapses and regulate synaptic activity. Acta Neuropathologica Communications 7 (1), 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lane CA et al. (2018) Alzheimer’s disease. European Journal of Neurology 25 (1), 59–70. [DOI] [PubMed] [Google Scholar]
- 82.Del Prete D et al. (2017) Localization and Processing of Amyloid-β Protein Precursor in Mitochondria-Associated Membranes. Journal of Alzheimer’s Disease 55, 1549–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Area-Gomez E et al. (2009) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. The American Journal of Pathology 175 (5), 1810–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hedskog L et al. (2013) Modulation of the endoplasmic reticulum–mitochondria interface in Alzheimer’s disease and related models. Proceedings of the National Academy of Sciences 110 (19), 7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pera M et al. (2017) Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. The EMBO Journal 36 (22), 3356–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nelson O et al. (2010) Familial Alzheimer’s Disease Mutations in Presenilins: Effects on Endoplasmic Reticulum Calcium Homeostasis and Correlation with Clinical Phenotypes. Journal of Alzheimer’s Disease 21, 781–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Greotti E et al. (2019) Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 79, 44–56. [DOI] [PubMed] [Google Scholar]
- 88.Zampese E et al. (2011) Presenilin 2 modulates endoplasmic reticulum (ER)–mitochondria interactions and Ca2+ cross-talk. Proceedings of the National Academy of Sciences 108 (7), 2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cheung K-H et al. (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58 (6), 871–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Filadi R et al. (2016) Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Reports 15 (10), 2226–2238. [DOI] [PubMed] [Google Scholar]
- 91.Leal NS et al. (2016) Mitofusin-2 knockdown increases ER-mitochondria contact and decreases amyloid β-peptide production. Journal of Cellular and Molecular Medicine 20 (9), 1686–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Area-Gomez E et al. (2012) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. The EMBO Journal 31 (21), 4106–4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eizirik DL et al. (2020) Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nature Reviews Endocrinology 16 (7), 349–362. [DOI] [PubMed] [Google Scholar]
- 94.Gomez L et al. (2016) The SR/ER-mitochondria calcium crosstalk is regulated by GSK3β during reperfusion injury. Cell Death & Differentiation 23 (2), 313–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Klec C et al. (2019) Glycogen Synthase Kinase 3 Beta Controls Presenilin-1-Mediated Endoplasmic Reticulum Ca²⁺ Leak Directed to Mitochondria in Pancreatic Islets and β-Cells. Cellular Physiology and Biochemistry 52 (1), 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thoudam T et al. (2019) PDK4 Augments ER–Mitochondria Contact to Dampen Skeletal Muscle Insulin Signaling During Obesity. Diabetes 68 (3), 571–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dingreville F et al. (2019) Differential Effect of Glucose on ER-Mitochondria Ca2+ Exchange Participates in Insulin Secretion and Glucotoxicity-Mediated Dysfunction of β-Cells. Diabetes 68 (9), 1778 LP-1794. [DOI] [PubMed] [Google Scholar]
- 98.Rieusset J (2018) The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: an update. Cell Death & Disease 9 (3), 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thivolet C et al. (2017) Reduction of endoplasmic reticulum-mitochondria interactions in beta cells from patients with type 2 diabetes. Public Libary of Science One 12 (7), e0182027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang Y et al. (2012) Inositol-1,4,5-trisphosphate receptor regulates hepatic gluconeogenesis in fasting and diabetes. Nature 485 (7396), 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Perry RJ et al. (2020) Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature 579 (7798), 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Urano F (2016) Wolfram Syndrome: Diagnosis, Management, and Treatment. Current Diabetes Reports 16 (1), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Delprat B et al. (2018) Wolfram syndrome: MAMs’ connection? Cell Death & Disease 9 (3), 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fischer TT and Ehrlich BE (2020) Wolfram syndrome: a monogenic model for diabetes mellitus and neurodegeneration. Current Opinion in Physiology 17, 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Angebault C et al. (2018) ER-mitochondria cross-talk is regulated by the Ca2+ sensor NCS1 and is impaired in Wolfram syndrome. Science Signaling 11 (553), eaaq1380. [DOI] [PubMed] [Google Scholar]
- 106.Cagalinec M et al. (2016) Role of mitochondrial dynamics in neuronal development: mechanism for Wolfram syndrome. Public Library of Science Biology 14 (7), e1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Boeckel GR and Ehrlich BE (2018) NCS-1 is a regulator of calcium signaling in health and disease. Biochimica et Biophysica Acta - Molecular Cell Research 1865 (11, Part B), 1660–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nguyen LD et al. (2019) Characterization of NCS1–InsP3R1 interaction and its functional significance. Journal of Biological Chemistry 294 (49), 18923–18933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nguyen LD et al. (2020) Calpain inhibitor and ibudilast rescue β cell functions in a cellular model of Wolfram syndrome. Proceedings of the National Academy of Sciences 117 (29), 17389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang C-H et al. (2014) Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Human Molecular Genetics 23 (18), 4770–4785. [DOI] [PubMed] [Google Scholar]
- 111.Chang NC et al. (2010) Antagonism of Beclin 1-dependent autophagy by BCL-2 at the endoplasmic reticulum requires NAF-1. The EMBO Journal 29 (3), 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shen Z-Q et al. (2017) CISD2 haploinsufficiency disrupts calcium homeostasis, causes nonalcoholic fatty liver disease, and promotes hepatocellular carcinoma. Cell Reports 21 (8), 2198–2211. [DOI] [PubMed] [Google Scholar]
- 113.Rouzier C et al. (2017) A novel CISD2 mutation associated with a classical Wolfram syndrome phenotype alters Ca2+ homeostasis and ER-mitochondria interactions. Human Molecular Genetics 26 (9), 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harris PC and Torres VE (2009) Polycystic Kidney Disease. Annual Review of Medicine 60 (1), 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kuo IY et al. (2019) Polycystin 2 regulates mitochondrial Ca2+ signaling, bioenergetics, and dynamics through mitofusin 2. Science Signaling 12 (580), eaat7397. [DOI] [PMC free article] [PubMed] [Google Scholar]