

Abstract
Introduction
The etiology of sporadic Alzheimer's disease (AD) requires non‐genetically modified animal models.
Methods
The relationship of tau phosphorylation to calcium‐cyclic adenosine monophosphate (cAMP)‐protein kinase A (PKA) dysregulation was analyzed in aging rhesus macaque dorsolateral prefrontal cortex (dlPFC) and rat primary cortical neurons using biochemistry and immuno‐electron microscopy. The influence of calcium leak from ryanodine receptors (RyRs) on neuronal firing and cognitive performance was examined in aged macaques.
Results
Aged monkeys naturally develop hyperphosphorylated tau, including AD biomarkers (AT8 (pS202/pT205) and pT217) and early tau pathology markers (pS214 and pS356) that correlated with evidence of increased calcium leak (pS2808‐RyR2). Calcium also regulated early tau phosphorylation in vitro. Age‐related reductions in the calcium‐binding protein, calbindin, and in phosphodiesterase PDE4D were seen within dlPFC pyramidal cell dendrites. Blocking RyRs with S107 improved neuronal firing and cognitive performance in aged macaques.
Discussion
Dysregulated calcium signaling confers risk for tau pathology and provides a potential therapeutic target.
Keywords: aging, association cortex, calpain, impaired cognition, macaque, pyramidal cells, PKA, ryanodine receptor, tau
1. INTRODUCTION
Alzheimer's disease (AD) is a progressive disorder that causes degeneration of the association cortex and ensuing dementia. Although rare, early onset, autosomal dominant forms of the disease are caused by genetic mutations, 1 the etiology of the more common, late‐onset, sporadic AD (sAD) remains unknown, constraining therapeutic strategies. The study of sAD is beset by critical obstacles, including the rapid loss of phosphorylation sites in human brain postmortem and the necessity of genetic mutations in mouse models to induce AD‐like pathology. The primary neuropathological hallmarks of AD consist of extracellular deposits of amyloid beta (Aβ) plaques and intraneuronal neurofibrillary tangles comprising hyperphosphorylated, fibrillated tau. 2 The AD field is increasingly interested in pathological processes beyond Aβ pathology that drive AD degeneration, due to the limited success of Aβ‐targeting treatments for AD. 3 Neuropathological analyses of thousands of human brains indicate that tau pathology precedes amyloid plaque pathology by a decade, 4 , 5 suggesting that tau may be a precipitating factor in AD.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional (eg, PubMed) sources and meeting presentations on sporadic Alzheimer's disease (sAD) etiology. The importance of calcium dysregulation has been noted for decades; these relevant citations are appropriately cited.
Interpretation: Our data show that early stage tau phosphorylation arises naturally in the aging primate association cortex in concert with calcium dysregulation, that calcium can regulate tau phosphorylation, and that intracellular calcium release is associated with reduced neuronal firing and impaired cognition in aged rhesus monkeys. This hypothesis complements insights from rodent models, and is consistent with clinical findings in the public domain.
Future directions: Future research can use aging rhesus monkeys—without autosomal dominant mutations—to reveal calcium‐dependent molecular changes that initiate early stage sAD pathology, including phosphorylation at pT217‐tau, a novel blood biomarker for sAD, and to test potential therapeutics to reduce AD pathology.
Tau pathology propagates in a distinct spatial and temporal sequence in sAD, targeting glutamatergic neurons in association cortices with extensive cortico‐cortical projections, but sparing glutamatergic neurons in primary visual cortex until end‐stage disease. 4 , 6 One important factor underlying this selective vulnerability may be related to how neurons use and regulate calcium. The most common and aggressive mutations that cause autosomal dominant AD are in presenilin 1(PSEN1), a core component of the gamma secretase complex, and these induce calcium leak from the smooth endoplasmic reticulum (SER) in addition to altering amyloid precursor protein (APP) processing. 7 , 8 , 9 Calcium dysregulation also appears to develop in the absence of autosomal dominant mutations in sAD. Aberrant ryanodine receptor (RyR) function has been correlated with severity of AD pathology in hippocampus, 10 including PKA phosphorylation of RyR2 at S2808 (pS2808‐RyR2), 11 which causes calcium leak into the cytosol. 12 , 13 Furthermore, in the human dorsolateral prefrontal association cortex (dlPFC), tau pathology preferentially afflicts a subset of layer III (LIII) pyramidal cells that express high levels of the calcium‐binding protein, calbindin, in young cortex. 14 These data suggest that neurons that utilize high levels of calcium may be particularly vulnerable to AD.
Despite strong support for a role of calcium in AD, 15 the lack of an effective animal model of sAD has limited in‐depth research of early molecular events that give rise to tau pathology in the absence of genetic mutations. Rhesus monkeys provide a unique opportunity to study age‐related molecular changes and their relevance to AD‐related pathology in ways that are not possible in human samples or current rodent models. 16 Macaques have highly developed association cortices that can be analyzed with minimal post‐mortem interval (PMI) or with perfusion fixation that is not possible with human samples, thus limiting the impact of variable post‐mortem changes such as protein dephosphorylation and processing. 17 Monkeys recapitulate characteristics of human aging in many important aspects, including reduced neuronal firing and cognitive deficits, 18 , 19 amyloid plaques, 20 , 21 dysmorphic mitochondria, 22 , 23 synapse loss, 24 and the same qualitative pattern and sequence of tau pathology as humans, including tangles in the oldest animals. 20
Previous research in rhesus monkeys has shown that pyramidal cell microcircuits in LIII dlPFC, unlike those in primary visual cortex, express the cyclic adenosine monophosphate (cAMP)‐protein kinase A (PKA) signaling machinery to magnify internal calcium release near glutamate synapses. 16 Age‐related elevation in cAMP‐PKA signaling, resulting at least in part from loss of phosphodiesterase PDE4A, was shown to increase tau phosphorylation and reduce neuronal firing by opening potassium channels. 19 , 25 However, the relationship between tau phosphorylation and calcium dysregulation in the aging primate association cortex is not known. The current study examined the emergence of calcium dysregulation and tau pathology in the aging primate dlPFC, including a cohort with variable medical histories similar to human cohorts, to help identify factors that correlate with pathology. We coupled biochemical analyses with immuno‐electron microscopic (immunoEM) analyses of LIII, which contains the microcircuits critical for working memory (WM), 26 that are especially vulnerable in sAD. 14 The results suggest that calcium dysregulation in the aging association cortex is a key factor in early stage tau pathology and impaired physiology, and that reducing calcium leak can normalize task‐related neuronal firing and cognitive performance.
2. MATERIALS AND METHODS
All experiments were conducted in accordance with the guidelines of Yale University Institutional Animal Care and Use Committee, and the PHS Guide for the Care and Use of Laboratory Animals. 27 Further information is included in the Supplement.
2.1. Biochemistry
2.1.1. Animal and tissue processing for biochemistry
Rhesus monkeys used for biochemical experiments ranged in age from 8.3 to 28.6 years (N = 9 female). Animals became available during the time frame of our study, and like in human post‐mortem analysis, the subjects had varied medical and health histories. PMI was minimized to the extent possible. For the “controlled set,” tissue was collected rapidly (within 10‐20 min) from five animals by the same individual. In contrast, the full cohort of animals included four additional monkey brains that were collected by a variety of investigators, with PMIs up to 1 hour, for example, in aged animals with serious medical conditions. Further details on the cohort and tissue handling are included in the Supplement.
2.1.2. Immunoblotting
Triton‐soluble samples were collected and run on tris‐glycine gels as described. 28 Due to the size of the ryanodine receptor (565 kDa), a longer transfer was required to adequately visualize the protein. Blots were analyzed with LI‐COR equipment. Description of the process of quantification and statistical analysis is in the Supplement.
2.1.3. Primary neuron cultures
The protocol for primary cortical neuron culture preparation was adapted from Li et al 29 (full description in Supplement). Briefly, embryos were removed at E19 and brains were extracted. Neurons were dissociated with needles of increasing gauge. Cells were grown in six‐well plates coated with poly‐D‐lysine. Cultures were grown in Neurobasal media that was partially changed regularly.
Experiments were performed at 13 days‐in‐vitro (DIV). BAPTA‐AM (Millipore 196419) was dissolved in dimethyl sulfoxide (DMSO) for a stock concentration of 10 mM. The stock was diluted in culture media and cells were incubated in 10 μM BAPTA‐AM or an equivalent volume of DMSO (0.1% DMSO) for 1 hour. Following incubation, cells were rinsed with ice‐cold phosphate‐buffered saline (PBS) and then lysed in warmed 1% sodium dodecyl sulfate (SDS) buffer (see Supplement). Three independent biological replicates were utilized in the experiment and six wells per condition were analyzed in each replicate (total 18 wells per condition).
2.2. Light and electron microscopy
The brains of nine rhesus macaques (7‐30 years) were used for anatomical studies. Animal procedures including anesthesia, perfusions, and histological processing are described in the Supplement.
2.2.1. Single pre‐embedding peroxidase immunocytochemistry
Pre‐embedding techniques were utilized for optimal preservation of intracellular membranes needed to localize labeled proteins on organelles (e.g., the SER). As described, 30 sections were incubated for 72 hours at 4°C with primary antibodies in TBS, and transferred for 2 hours at room temperature to species‐specific biotinylated Fab’ or F(ab’)2 fragments in TBS. To reveal immunoperoxidase labeling, sections were incubated with the avidin‐biotin peroxidase complex (ABC) (1:300; Vector Laboratories) and then visualized in 0.025% Ni‐intensified 3,3‐diaminobenzidine tetrahydrochloride (DAB; Sigma Aldrich) as a chromogen in 100 mM phosphate buffer (PB) with the addition of 0.005% hydrogen peroxide for 10 minutes. Sections were then exposed to osmification, dehydration, and standard resin embedding following typical immunoEM procedures. Omission of primary antibodies or substitution with non‐immune serum resulted in complete lack of immunoperoxidase labeling. For single pre‐embedding gold immunocytochemistry details see the Supplement.
Labeled sections were processed for light and electron microscopy imaging as described in the Supplement.
2.3. In vivo physiology
Two aging rhesus monkeys (16‐ and 19‐year‐old males) performed an oculomotor version of the delayed response spatial working memory task while single unit recordings were made from the principal sulcal dlPFC needed to perform the task. Once delay cells were identified, S107 or 1,1′‐diheptyl‐4,4′‐bipyridinium dibromide (DHBP) was applied by iontophoresis. Additional details can be found in the Supplement and as described. 19
2.4. Cognitive assessments
The effects of systemically administered S107 were assessed in aged (18–to 31‐year‐old) rhesus macaques (one male, eight females) trained on a manual version of the delayed response test of spatial working memory. Monkeys received S107 (0.001‐0.1 mg/kg) 120 minutes before testing was administered via oral administration (p.o.). (Additional details can be found in the Supplement.)
3. RESULTS
3.1. Aged rhesus monkeys naturally develop tau pathology
Our previous work has demonstrated that aging macaques develop fibrillated tau in a pattern and sequence qualitatively similar to humans. 20 We confirmed and extended these results in the current study, with an experimentally‐controlled cohort, and monkeys with more variable medical histories. Pyramidal cells in dlPFC from very old (30 years) rhesus monkeys were labeled by antibodies that recognize key GSK3β hyperphosphorylation sites: the AT8 antibody (pS202 and pT205) currently used to diagnose AD (Figure 1A), and pT217, which shows promise as a diagnostic in cerebrospinal fluid (CSF) and plasma 31 , 32 ; [Figure 1B]). In addition to the expected AT8‐like labeling, the pT217‐tau immunolabeling of pyramidal cells showed aggregated, filamentous structures within apical dendrites, often with a twisted morphology common in neurofibrillary tangles (Figure 1B).
FIGURE 1.
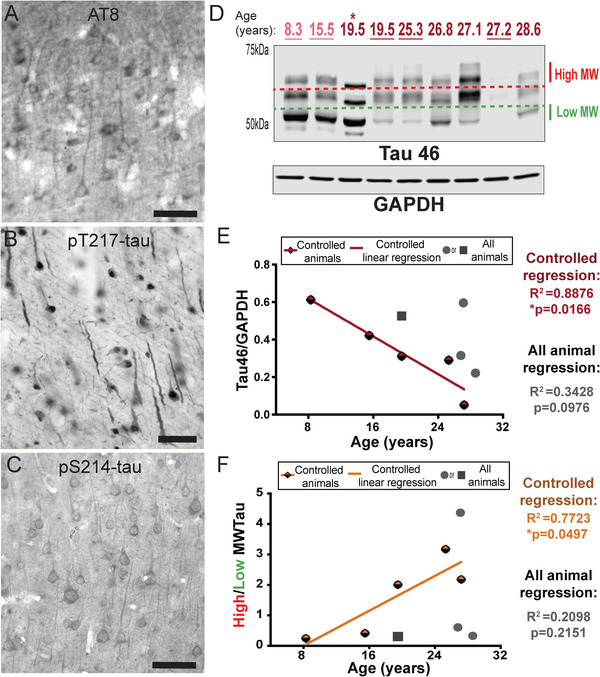
Age‐related alterations in tau phosphorylation and aggregation in monkey dlPFC. (A) Mild tau fibrillation revealed with AT8 labeling was observed in deep LIII pyramidal cells in aged monkey dlPFC (30 years), predominantly along apical dendrites. (B) Dense immunoreactivity for pT217‐tau along apical and basilar dendrites of pyramidal cells in aged monkey dlPFC (30 years) deep LIII. (C) The aged monkey dlPFC (26 years) was characterized by dense pS214‐tau immunoreactivity along apical and basal dendrites of pyramidal cells located in deep LIII, and diffuse reactivity in the neuropil. (D) Macaque dlPFC tissue (15 μg) was immunoblotted for total tau and glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH). The dotted lines represent the regions of the blot quantified for different molecular weight (MW) tau species. Low MW tau species are denoted by the green dashed line and appear at 50 kDa. High MW tau species are denoted by the red dashed line and appear > 60 kDa. Animals are labeled by their age in years: young animals are light red and aged animals are dark red. Controlled set animals are underlined. (E) Quantification of total triton‐soluble tau normalized byGAPDH are plotted by age. Controlled set animals are denoted by half‐red diamonds. First (leftmost) 19.5 animal denoted by asterisk in D is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals and labeled red (R2 = 0.8876, *P = 0.0166). Results from an all‐animal linear regression are summarized in the top right of the graph (R2 = 0.3428, P = 0.0976). (F) Quantification of the MW shift of triton‐soluble tau is represented by the ratio of high over low MW tau and plotted by age. Controlled set animals are denoted by half‐orange diamonds. First 19.5 animal denoted by asterisk in D is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals and labeled orange (R2 = 0.7723, *P = 0.0497). Results from an all‐animal linear regression are summarized in the top right of the graph (R2 = 0.2098, P = 0.2151)
Tau becomes insoluble with increasing fibrillation, and is lost from detergent‐soluble solutions, making biochemical analysis of hyperphosphorylated tau in traditional lysates more challenging. 33 , 34 However, the loss of detergent‐soluble tau can be used as an indirect index of post‐translational modifications (eg, hyperphosphorylation) that disrupt the normal solubility of the protein. For biochemical measures of tau (and phospho‐tau, see below) we used regression analyses to examine expression as a function of age for both the controlled cohort and the entire cohort. In addition, and subject to the caveat of limited numbers in the young group, we compared levels from young (<18 years) versus aged (≥18 years) animals. We found a significant (*P = 0.0166) inverse relationship between triton‐soluble tau levels and age in the dlPFC in the controlled cohort and a trending decrease with age in the full cohort (Figure 1D,E; Figure S1A). Our previous research has also shown an upward molecular weight shift of tau bands with age, consistent with increased tau modifications. 20 These higher molecular weight species have also been shown to increase the ability of tau to seed aggregates. 35 We found a significant (*P = 0.0497) positive correlation between the ratio of high‐to‐low molecular weight tau levels and age in the dlPFC in the controlled cohort and a trending increase with age in the full cohort (Figure 1D,F; Figure S1B). The natural accrual of GSK3β‐hyperphosphorylated tau and loss of detergent solubility in aged rhesus monkeys provides construct validity to humans.
Aging monkeys also express tau phosphorylation indicative of earlier stages of tau pathology, which can be captured more easily than in human given the very short PMIs possible in monkeys. PKA phosphorylation of tau at S214 is an important event, as it causes tau to detach from microtubules and primes tau for hyperphosphorylation by GSK3β. 36 Immunocytochemistry showed dense pS214‐tau along apical and basal dendrites of dlPFC deep LIII pyramidal cells in an aged monkey (26 years; Figure 1C). We confirmed a significant positive correlation with age in the controlled cohort, and overall we found a significant increase in pS214‐tau in aged animals (Figure 2A,B, S3A). EM analysis verified pS214‐tau accumulation in dendrites on microtubules and the SER, near dysmorphic “mitochondria‐on‐a‐string” (MOAS), which is characteristic of AD (Figure S2A‐B), directly over the postsynaptic membrane of asymmetric, presumed glutamatergic synapses in dendrites (Figure S2C‐D), and on the SER spine apparatus in dendritic spines near axospinous, presumed glutamatergic, asymmetric synapses (Figure S2E‐F), areas of particularly prominent calcium signaling.
FIGURE 2.
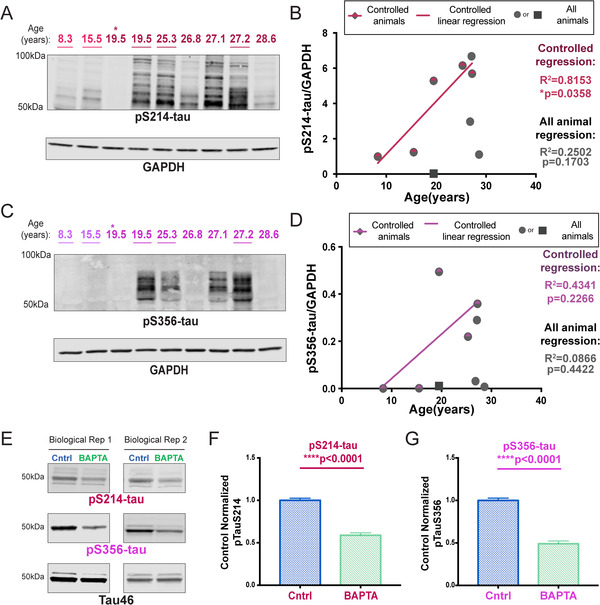
Calcium‐dependent phosphorylation of tau. (A) Macaque dlPFC tissue (20 μg) was immunoblotted for pS214‐tau, and GAPDH as a loading control. The portion of the nitrocellulose membrane was derived from the blot also used to analyze the pS2808‐RyR2 (see Figure 3E). Animals are labeled by their age in years, young animals in light red and aged animals in dark red. Controlled set animals are underlined. (B) Quantification of pS214‐tau normalized by GAPDH is plotted by age. Controlled set animals are denoted with a half‐red diamond. First 19.5 animal denoted by asterisk in blot is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals (R2 = 0.8153, *P = 0.0358). Results from an all‐animal linear regression are summarized in the bottom right of the graph (R2 = 0.2502, P = 0.1703). (C) Macaque dlPFC tissue (22 μg) was immunoblotted for pS356‐tau and GAPDH. Animals are labeled by their age in years, young animals in light magenta and aged animals in dark magenta. Controlled set animals are underlined. (D) Quantification of pS356‐tau normalized by GAPDH is plotted by age. Controlled set animals are denoted with a half‐magenta diamond. First 19.5 animal denoted by asterisk in 2C is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals (R2 = 0.4341, p = 0.2266). Results from an all‐animal linear regression are summarized in the bottom right of the graph (R2 = 0.0866, P = 0.4422). (E) Representative western blot images from two of three independent rat cortical neuron preparations collected at E19 and analyzed at 13 DIV. Neurons were incubated with BAPTA‐AM/DMSO or a DMSO “Cntrl” (0.1% DMSO in both conditions). Sodium dodecyl sulfate (SDS)‐soluble lysate was blotted for pS214‐tau (red), pS356‐tau (magenta), and total tau (black). pS214‐tau and total tau were run on the same blot (antibodies from different species). (F) Quantification of tau‐normalized pS214‐tau (18 wells per condition, three independent preparations). Values were normalized to “Cntrl” for each independent experiment. Standard error of the mean (SEM) is plotted for each group. Means were compared using a two‐tailed Mann‐Whitney test (****P < 0.0001). G) Quantification of tau‐normalized pS356‐tau. Values were normalized to “Cntrl” for each independent experiment. SEM is plotted for each group. Means between the two conditions were compared using a two‐tailed unpaired t‐test (****P < 0.0001)
Tau is also phosphorylated by calcium/calmodulin‐dependent kinase II (CaMKII) at sites associated with early stage AD tau pathology 37 ; including S356 (pS356‐tau). We were only able to detect reasonable levels of pS356‐tau in aged macaques (Figure 2C,D). Although pS356‐tau levels did not significantly correlate with age due to inherent variability, we found a significant (*P = 0.0324) increase in phosphorylation at S356 in aged monkeys in the groupwise comparison (Figure S3B).
3.2. Increased calcium signaling may directly drive tau phosphorylation
Given that CaMKII and PKA signaling can both be increased by calcium, we utilized rat primary cortical neuron cultures, collected at embryonic day 19, and tested mature neurons at 13 DIV, to assess the direct role of intracellular calcium on tau phosphorylation both at S214 and S356. Phosphorylation of both pS214‐tau and pS356‐tau was evident in neurons treated with DMSO (Figure 2E, “Cntrl”), and incubation with the cell permeable calcium chelator, BAPTA‐AM, resulted in a striking reduction in phosphorylation at both sites (Figure 2E‐G).
We examined the molecular events that may drive increases in intracellular calcium in vivo in order to assess their relationship with tau phosphorylation. PKA phosphorylation of the SER calcium channel RyR at pS2808‐RyR2 was one possible mechanism of increased intracellular calcium. Typically, RyR channels tightly regulate neuronal calcium homeostasis needed for cellular health, but pS2808‐RyR2 causes increased calcium leak into the cytosol. 12 , 13 , 38 ImmunoEM of aged LIII dlPFC revealed pS2808‐RyR2 on the SER within neurons and glia. pS2808‐RyR2 labeling in neurons was predominately within post‐synaptic elements, for example on the SER in dendrites near dysmorphic “MOAS” mitochondria (Figure 3A, Figure S4A‐B). pS2808‐RyR2 immunolabeling was also observed in spines on the SER “spine apparatus” subjacent to synapses and postsynaptic densities (Figure 3C‐D), as well as extrasynaptic locations distal from the synapse (Figure 3C). Extensive pS2808‐RyR2 labeling was also observed in astroglial leaflets ensheathing asymmetric synapses (Figure S5A‐B).
FIGURE 3.
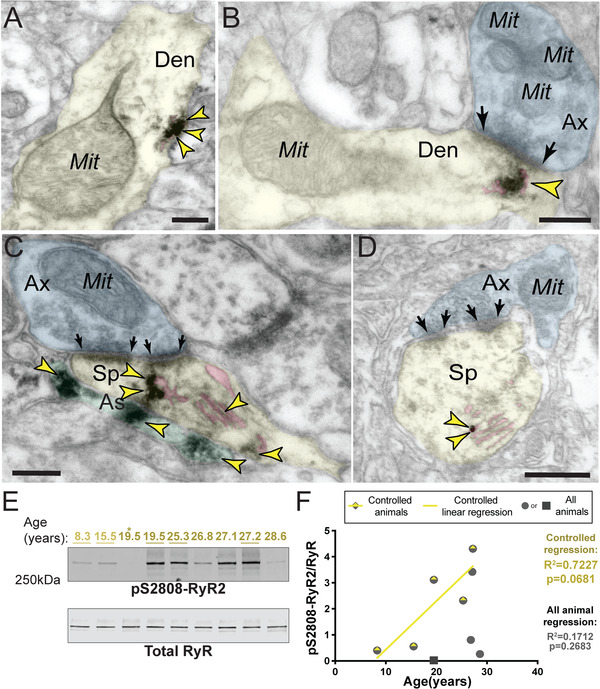
Age‐related increase in cAMP‐PKA dependent pS2808‐RyR2 in monkey dlPFC. (A) In aged macaque (26 years) dlPFC LIII, accumulation of pS2808‐RyR2 occurred in proximity to abnormal mitochondria‐on‐a‐string (MOAS) profiles in dendrites. (B) pS2808‐RyR2 immunolabeling was subjacent to synapses and the postsynaptic density in an aged macaque (26 years). In A and B, immunolabeling for pS2808‐RyR2 likely occurs in association with the SER, given preexisting literature showing that these calcium channels are primarily localized on the SER. (C‐D) Extensive pS2808‐RyR2 labeling was visualized on the SER spine apparatus (pink pseudocolored), the primary calcium‐storing organelle, within postsynaptic compartments in dendritic spines in dlPFC LIII. Both dendritic spines received axospinous glutamatergic asymmetric synapses. Precise immunolabeling for pS2808‐RyR2 in association with the spine apparatus was detected by immunoperoxidase (C) and immunogold (D) labeling. Synapses are between arrows. Color‐coded arrowheads (yellow) point to pS2808‐RyR2 immunoreactivity. Profiles are pseudocolored for clarity. Abbreviations: Ax, axon; Den, dendrite; Mit, mitochondria; Sp, dendritic spine; As, astroglia. Scale bars, 200 nm. (E) Macaque dlPFC tissue (20 μg) immunoblotted for pS2808‐RyR2 and total RyR. Animals labeled by age in years: young animals in light yellow and aged in dark yellow. Controlled animals are underlined. (F) Quantification of pS2808‐RyR2 normalized by total RyR is plotted by age. Controlled set animals are denoted with a half‐yellow diamond. First 19.5 animal denoted by asterisk in blot (Figure 3E) is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals (R2 = 0.7227, P = 0.0681). Results from an all‐animal linear regression are summarized in the bottom right of the graph (R2 = 0.1712, P = 0.2683)
Biochemical analyses of pS2808‐RyR2 levels revealed a trending (P = 0.0681) correlation between the levels of pS2808‐RyR2 and age for the controlled cohort (Figure 3E,F). Similarly, a groupwise comparison showed a trending (P = 0.0502) increase in pS2808RyR2 in aged versus young animals (Figure S3C). We took advantage of the natural variations in the aging response to examine whether phosphorylation of tau was associated with pS2808‐RyR2 in aging dlPFC. It is important to note that we found that pS2808‐RyR2 expression levels strongly correlated with tau phosphorylation at both pS214‐tau (R2 = 0.84) and pS356‐tau (R2 = 0.84) (Figure 4A,B). The constellation of pS2808‐RyR2 and pS214‐tau near dysmorphic mitochondria in dendrites highlights a nexus of early stage pathology (Figure 4C,D), consistent with human data showing that dendrites are the site of origin for cortical tau pathology in sporadic AD. 5 We also found evidence of calpain‐dependent cleavage of PKC‐α without any change in calpain levels, further suggesting elevated intracellular calcium (Figure S6).
FIGURE 4.
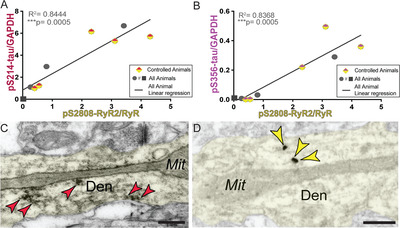
Correlation between pS2808‐RyR2 and phosphorylated tau. (A) Correlation between levels of normalized pS2808‐RyR2 (x‐axis) and pS214‐tau (y‐axis) across all animals is fit by a linear regression (R2 = 0.8444, ***P = 0.0005). Controlled animals are denoted by half‐red/half‐yellow diamonds but were not separated in analysis. First 19.5 animal denoted by asterisk in blots is denoted by a square. (B) Correlation between levels of normalized pS2808‐RyR2 (x‐axis) and pS356‐tau (y‐axis) across all animals is fit by a linear regression (R2 = 0.8368, ***P = 0.0005). Controlled animals are denoted by half‐magenta/half‐yellow diamonds but were not separated in analysis. First 19.5 animal denoted by asterisk in blots is denoted by a square. (C) Subcellular localization of pS214‐tau, revealed by immunoperoxidase labeling, along microtubules and in proximity to abnormal MOAS profiles within dendritic shafts in aged monkey (26 years) dlPFC LIII. Color‐coded arrowheads (red) point to pS214‐tau immunoreactivity. (D) Subcellular localization of pS2808‐RyR2, revealed by immunogold labeling, likely in association with SER tubules and in proximity to abnormal MOAS profiles within dendritic shafts in aged monkey (26 years) dlPFC LIII. Note the similar distribution patterns of pS214‐tau and pS2808‐RyR2 within dendritic shafts in aged monkey dlPFC, consistent with biochemistry. Color‐coded arrowheads (yellow) point to pS2808‐RyR2 immunoreactivity. Profiles are pseudocolored for clarity. Abbreviations: Den, dendrite; Mit, mitochondria. Scale bars, 200 nm
3.3. Loss of proteins that regulate calcium in aged macaque dlPFC
Cytosolic calcium can be regulated by calcium‐binding proteins such as calbindin. Calbindin‐containing pyramidal cells in LIII dlPFC become particularly vulnerable to AD tau pathology. 14 Similar to humans, we found extensive calbindin labeling within pyramidal cells in young monkey dlPFC LIII (Figure 5A). Calbindin labeling intensity was reduced in pyramidal cells from aged monkeys (Figure 5A,B), but remained in interneurons (Figure S7), similar to AD brains. 14
FIGURE 5.
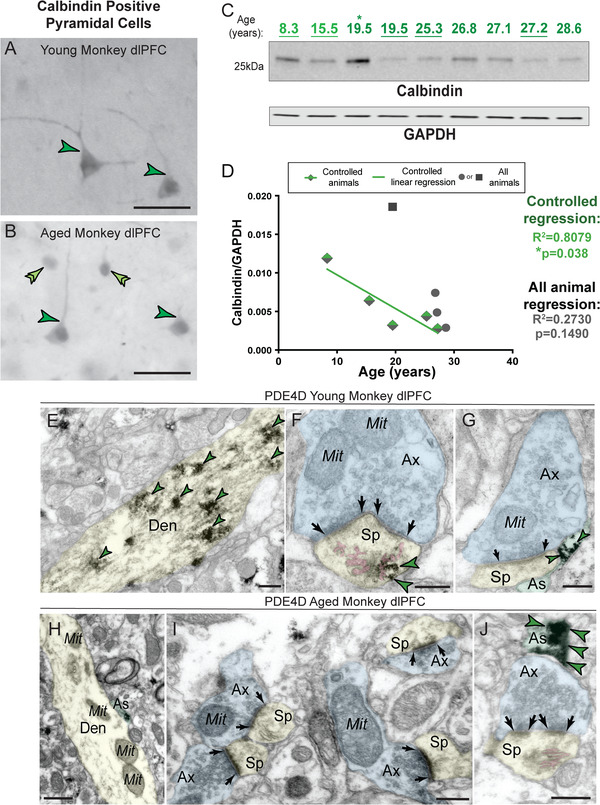
Loss of calcium‐cAMP‐PKA regulatory mechanisms in macaque dlPFC. (A) High‐magnification calbindin 28‐kDa immunoreactivity in young monkey (14 years) dlPFC deep LIII pyramidal cells (dark green single arrowheads). The labeling pattern for calbindin 28‐kDa in pyramidal cells is characterized by cytoplasmic staining of the cell body, and immunoreactivity along apical and basal dendrites, similar to healthy young humans. (B) High‐magnification calbindin 28‐kDa immunoreactivity in aged monkey (30 years) dlPFC deep LIII pyramidal cells (dark green single arrowheads). There was a decrease in calbindin 28‐kDa staining intensity and diffuse labeling pattern with advanced age in rhesus monkeys, consistent with AD patients with high neurofibrillary tangle pathology. In contrast, nearby, superficial calbindin‐containing putative GABAergic interneurons (light green double arrowheads) in dlPFC remain heavily immunoreactive to calbindin and resistant to degeneration with advanced age (additionally see Supplementary Figure S7). Scale bars, 50 μm. (C) Macaque dlPFC tissue (10 μg) immunoblotted for calbindin and GAPDH. Animals are labeled by their age in years, young animals in light green and aged animals in dark green. Controlled animals are underlined. (D) Quantification of calbindin normalized by GAPDH is plotted by age. Controlled set animals are denoted by half‐green diamonds. First 19.5 animal denoted by asterisk in C is denoted by a square. All other animals are denoted by gray circles. Linear regression is shown for the controlled set animals and labeled green (R2 = 0.8079, *P = 0.0380). Results from an all‐animal linear regression are summarized in the top right (R2 = 0.2730, P = 0.1490). (E) In young monkey (10 years) dlPFC LIII, PDE4D was predominantly concentrated in dendritic shafts in postsynaptic locations and was associated with microtubules. (F) Dense PDE4D immunolabeling was observed in postsynaptic compartments within dendritic spines, often localized in association with the SER spine apparatus (pink pseudocolored) in young monkey (11 years) dlPFC LIII. G) Immunolabeling for PDE4D was present along the plasma membrane and within the intracellular space of astroglial processes, in glial leaflets ensheathing the axospinous glutamatergic‐like asymmetric synapses. A more detailed mapping of PDE4D localization in young monkey dlPFC can be found in Datta et al, 2020. 53 (H) In aged monkey (26 years) dlPFC LIII, PDE4D immunolabeling was virtually absent from dendritic shafts. (I) PDE4D expression was absent from postsynaptic compartments within dendritic spines in aged monkey dlPFC LIII. Micrograph revealing four dendritic spines that receive axospinous glutamatergic asymmetric synapses, all immunonegative for PDE4D. (J) Subcellular localization of PDE4D was preserved within astroglial leaflets in aged macaque (26 years) dlPFC LIII. Synapses are between arrows. Color‐coded arrowheads (green) point to PDE4D immunoreactivity. Profiles are pseudocolored for clarity. Abbreviations: Ax, axon; Den, dendrite; Mit, mitochondria; Sp, dendritic spine; As, astroglial. Scale bars, 200 nm
Biochemical analyses also documented reduced calbindin in triton‐soluble lysates with advancing age (Figure 5C,D S3D). Regression analyses (Figure 5D) showed a significant (*P = 0.0380) inverse relationship between levels of calbindin in dlPFC and age within the controlled cohort. In the entire cohort there was a trending relationship between age and loss of calbindin (Figure 5D). Of special note, the 19.5‐year‐old aged monkey that had the highest level of calbindin also had reduced signs of calcium and tau modifications (denoted by asterisk and square). This animal does not have a notably different life history, but the absence of calcium‐dysregulation markers and tau phosphorylation was very striking.
We also investigated age‐related changes in the cAMP‐regulator, PDE4D. In young monkey dlPFC LIII, PDE4D was predominantly concentrated in dendritic shafts on microtubules (Figure 5E), in dendritic spines, in association with the SER spine apparatus (Figure 5F), and in astrocytes (Figure 5G). In aged monkey dlPFC LIII, PDE4D was no longer expressed in dendritic shafts (Figure 5H) or spines (Figure 5I), the same sub‐compartments where pS214‐tau accumulates in aged monkeys. However, PDE4D remained in astroglial profiles (Figure 5J), which may explain only trend‐level decreases in PDE4D protein expression using biochemical analyses of whole dlPFC with advanced age (Figure S8).
3.4. Pharmacological intervention can reduce the negative impact of dysregulated calcium signaling on neuronal physiology and working memory performance in aged monkeys
The final set of experiments tested whether blocking calcium leak from RyR would help restore neuronal firing and cognitive performance in aged monkeys. “Delay cells” in dlPFC fire throughout the delay period to maintain information in WM. Delay cell firing is reduced in aged monkey dlPFC due to dysregulated calcium‐cAMP‐PKA signaling opening K+ channels. 19 We tested whether blocking calcium leak from pS2808‐RyR2 using S107 13 , 39 might improve dlPFC neuronal physiology and enhance cognitive performance in aged monkeys. Acute iontophoresis of S107 significantly enhanced the task‐related firing of delay cells in aged monkeys (Figure 6A,B). Iontophoresis of DHBP, which blocks calcium release from the SER via RyR, also significantly enhanced the delay‐related firing of delay cells (Figure S9).
FIGURE 6.
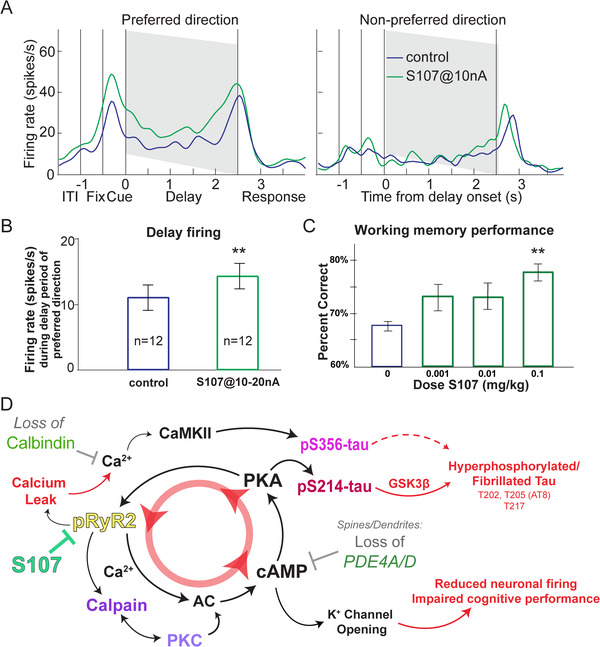
Impact of dysregulated calcium‐cAMP‐PKA signaling on macaque physiology and behavior. (A‐B) Iontophoretic application of pharmacological agent S107 enhanced delay‐related firing of dlPFC delay cell in aged monkeys performing a WM task. Mechanistically, S107 stabilizes pS2808‐RyR2 with downstream effector protein calstabin2 to suppress excess calcium leak. A single neuron example of the effect of S107 on delay‐related firing is shown. Data are shown with the spike density function of this neuron for two different conditions: control condition, blue; S107@10 nA, green. Iontophoresis of S107 increased the delay‐related firing selectively for the neuron's preferred direction. Two‐way ANOVA, Fdirectionxdrug(1,34) = 6.549, P = 0.015; Sidak's multiple comparisons: preferred direction, control vs S107, P = 0.0009 and non‐preferred direction, control vs S107, P = 0.847. (B) The mean ± SEM firing rate of 12 delay cells. S107 significantly enhanced the delay‐related firing for the preferred direction at the population level. Control vs S107, t(11) = 3.794, **P = 0.003, two‐tailed paired t test. (C) Systemic administration of S107 improves WM performance in rhesus macaques. Mean ± SEM of the behavioral response of 9 aged monkeys (18‐31 years) across the dose range of S107 tested (VEH vs 0.1 mg/kg, [**P = 0.016, F(2, 16.01) = 5.39, Greenhouse‐Geiser corrected; **P = 0.008, F(2.67, 21.36) = 5.39, Huynh‐Feldt corrected]. Pair‐wise comparisons confirmed a linear effect of dose (F(1,8) = 21.495, P = 0.002) with significant enhancement at the highest dose (**VEH vs 0.1 mg/kg, P = 0.001), and trend‐level improvement at the lower dose (P = 0.064). (D) Summary figure outlining a feedforward cycle of calcium‐cAMP‐PKA signaling that drives calcium leak through pS2808‐RyR2, subsequently leading to modifications of ion channels that disrupt normal neuronal physiology. Markers identified and characterized in other figures are denoted by a colored label. Pharmacological intervention to prevent this calcium leak (S107) is highlighted in green on the far left of the figure
A second study examined whether acute, systemic administration of S107 (p.o.; 120 minutes before testing) to aged monkeys would improve behavioral performance of a spatial WM task. We tested a wide range of doses (0.001 mg/kg – 0.1 mg/kg) and found a significant, dose‐related improvement in WM performance (Figure 6C). There was no evidence of side effects, suggesting that blocking calcium leak from RyRs might have therapeutic potential in protecting the primate dlPFC.
4. DISCUSSION
The current study demonstrated that rhesus macaques naturally develop hyperphosphorylated tau at sites relevant to AD diagnosis in humans (S202/T205, T217) and thus can be uniquely helpful for elucidating the molecular changes in aging association cortex that initiate tau pathology. We took advantage of the innate variation in age‐related changes to identify the factors associated with early stage tau pathology. We found that age‐related calcium‐cAMP‐PKA dysregulation in dlPFC was highly correlated with early‐stage tau phosphorylation. The tight correlation between calcium leak from the SER (pS2808‐RyR2) and early stage tau phosphorylation was reinforced by their similar locations within pyramidal cell dendrites, and the in vitro demonstration that tau phosphorylation at S356 and S214 was calcium dependent. The lack of tau pathology in one aging monkey with high levels of the calcium‐binding protein, calbindin, suggests that its age‐related loss from LIII dlPFC pyramidal cells may render them particularly vulnerable to the toxic effects of cytosolic calcium on tau pathology. The concurrence of multiple mechanism(s) of calcium‐cAMP‐PKA dysregulation within pyramidal cells of the dlPFC in aging macaques may help to explain why these cells are preferentially lost in sAD. 14 Finally, treatment of aged monkeys with S107 enhanced neuronal firing and cognition in aged monkeys, potentially through its pS2808‐RyR2 calcium‐leakage blocking effects, and may provide a therapeutic avenue for reducing age‐related risk of AD. Based on the current results, together with our previous data 20 , 25 and work from others, 12 , 14 , 40 , 41 we offer a model in Figure 6D in which calcium dysregulation drives aberrant tau phosphorylation and weakens neuronal firing and cognitive function.
4.1. Important limitations and avenues for further research
As noted above, aged rhesus macaques provide an excellent model for studying sAD; however, these benefits come with limitations. Given the invaluable nature of these animals to numerous studies it is very difficult to acquire high‐quality (relatively healthy, very short PMI) tissue from young and aged animals, and thus the sample sizes in non‐human primate research are necessarily limited. The smaller numbers are somewhat compensated by the increased homogeneity from control over housing conditions, diet, and much shorter PMIs than is possible with human research. The current study used a total of 29 monkeys (9 for biochemistry, 9 for microscopy, 2 for physiology, and 9 for cognitive behavior), which is relatively large for nonhuman primate studies. However, given the limitation of nine monkeys for biochemistry, with few young animals, we purposefully presented all data points, and focused on the correlative relationships with age and between multiple markers. In particular we emphasized the strong correlation between pS2808‐RyR2 and tau phosphorylation that was evident across all animals. However, we also highlighted a single 19.5‐year‐old animal who had very high levels of calbindin but low levels of calcium dysregulation and tau phosphorylation. Such individual differences may provide clues regarding factors that confer risk versus resilience in humans, where there are also large individual differences in the development of sAD. Further investigation into the relationship between calcium‐cAMP‐PKA dysregulation and AD‐related tau phosphorylation in new and expanded primate cohorts is an important avenue for future research.
4.2. Phosphorylated RyR2 drives increased calcium signaling
Calcium has long been hypothesized to play a role in AD. 15 Abnormal calcium storage and release from the SER is evident in both sporadic and inherited AD, 7 , 8 , 42 including elevated levels of pS2808‐RyR2 that increase calcium leak. 11 , 12 , 13 , 38 Calcium dysregulation has been seen early in the aging process in the entorhinal cortex in rodents, consistent with the special vulnerability of this area to AD tau pathology. 43 The current study found that pS2808‐RyR2 levels in aged primate dlPFC were highly correlated with the emergence of early stage tau pathology, and that pS2808‐RyR2 is prominently expressed on spines and dendrites near dysmorphic mitochondria, the same intracellular locations where phosphorylated tau first accumulates, and PDE4A 25 and PDE4D are lost with advancing age. Calcium leak through pS2808‐RyR2 was also shown to contribute, at least in part, to impaired dlPFC neuronal firing and WM performance in aging macaques, consistent with evidence that calcium dysregulation alters neuronal firing in aged rodent entorhinal cortex. 43 These data indicate that calcium leak from the SER could be a key factor in increasing age‐related risk for AD pathology.
4.3. Elevated calcium signaling may drive early tau phosphorylation
The current results demonstrate an effect of intracellular calcium on tau phosphorylation in vitro and suggest several direct and indirect calcium‐mediated pathways that could influence tau phosphorylation. Calcium can directly drive phosphorylation of tau at S356 by CaMKII, a site increased in AD brain, 44 , 45 and found to be highly correlated with levels of pS2808‐RyR2 in the current study. High levels of cytosolic calcium can also activate calpain, which cleaves and irreversibly activates two tau kinases, PKCα 46 and GSK3β, 47 an event that heralds accelerated tau phosphorylation in AD. 41 Cytosolic calcium can also increase cAMP‐PKA signaling, 48 thereby increasing phosphorylation of tau at S214, which primes tau for hyperphosphorylation by GSK3β at critical sites 40 : AT8 (S202/T205) sites used to diagnose AD, and T217, a potential in vivo diagnostic marker found in human CSF and blood, 31 , 32 , 49 and in aged monkey dlPFC. Further investigations into the impact of dysregulated kinases and phosphatases on tau phosphorylation, resulting from dysregulated calcium with advancing age, are an important avenue for future research. High cytosolic calcium might drive tau phosphorylation through multiple, synergistic actions, and may be especially toxic under conditions when the calcium binding protein, calbindin, is lost with age (Figure 6D).
4.4. Relevance to novel therapeutics for reducing risk of sporadic AD
Regulation of calcium signaling is a promising therapeutic avenue for reducing the risk of sAD. The finding that S107 enhanced neuronal firing and cognitive performance in aged monkeys, with no apparent side effects, indicates a benign treatment that could be initiated early in the aging process to protect cortical circuits from increased RyR2 calcium leak and/or loss of the regulatory factors calbindin, PDE4A 25 and PDE4D. It is noteworthy that PDE4D inhibitors are currently under development as potential treatments for AD 50 ; the current data, as well as recent findings showing that age‐related decreases in PDE4D in rodent PFC correlate with elevated tau phosphorylation, caution that this strategy may actually worsen rather than alleviate tau pathology in vulnerable brain regions. 51 Alternative strategies may aim to restore regulation of calcium‐cAMP signaling, for example, with GCPII inhibitors, 52 or re‐stabilizing pS2808‐RyR2 to reduce calcium leak. Chronic S107 treatment in an AD mouse model has been shown to improve behavioral performance on tests of hippocampal function. 11 Future studies are needed to determine whether chronic administration of S107 in aged rhesus macaques can reduce tau hyperphosphorylation in addition to its beneficial effects on cognitive performance.
CONFLICTS OF INTEREST
Amy F.T. Arnsten, Pasko Rakic, and Yale University receive royalties from Shire/Takeda from the US sales of Intuniv. They do not receive royalties from international sales or generic Intuniv.
AUTHOR CONTRIBUTIONS
Dibyadeep Datta and Shannon N. Leslie designed and performed the experiments in histology and biochemistry, respectively. Dibyadeep Datta collected and analyzed immunoEM and immunohistochemistry experiments. Shannon N. Leslie collected and analyzed biochemical analyses and in vitro experiments. Min Wang and Shengtao Yang conducted electrophysiological experiments. Dibyadeep Datta and Amy F.T. Arnsten analyzed behavioral experiments. Yury M. Morozov, Alvaro Duque, Shengtao Yang, Caroline Zeiss, and SueAnn Mentone contributed to the experimental design and provided technical expertise. Caroline Zeiss and Tamas L. Horvath provided invaluable tissue resources. Pasko Rakic and Christopher H. van Dyck contributed to the experimental design and revised the manuscript. Amy F.T. Arnsten and Angus C. Nairn designed the experiments, supervised the study, and critically revised the manuscript. All authors read and approved the final manuscript.
Supporting information
Supplementary information
Supplementary information
ACKNOWLEDGMENTS
We thank Dr. Andrew Marks and Armgo Pharma for the gift of S107, and Lisa Ciavarella, Tracy Sadlon, Sam Johnson, and Michelle Wilson for their technical expertise and behavioral work with the animals. Some tissue was provided by MacBrainResource, (https://medicine.yale.edu/neuroscience/macbrain/) NIMH grant R01‐MH113257 to Dr. A. Duque. This work was supported by National Institutes of Health (NIH) grants Pioneer Award DP1AG047744‐01 and R01AG061190‐02 (AFTA), Alzheimer's Association Research Fellowship AARF‐17‐533294 (Dibyadeep Datta), American Federation for Aging Research/Diamond Postdoctoral Fellowship (Dibyadeep Datta), the NIH (AG047270, AG062306, AG066508, Training Grant T32 NS41228), the State of Connecticut Department of Mental Health and Addiction Services (Angus C. Nairn), Gruber Fellowship (Shannon N. Leslie), F31AG063425 (Shannon N. Leslie), DA023999 (Pasko Rakic), AG052986 (Tamas L. Horvath) and support from the Alzheimer's Disease Research Unit (Christopher H. van Dyck).
Datta D, Leslie SN, Wang M, et al. Age‐related calcium dysregulation linked with tau pathology and impaired cognition in non‐human primates. Alzheimer's Dement. 2021;17:920–932. 10.1002/alz.12325
Dibyadeep Datta and Shannon N. Leslie contributed equally to this study.
DATA AVAILABILITY STATEMENT
All raw and quantified data will be made available by the authors upon reasonable request.
REFERENCES
- 1. Hardy J. Amyloid, the presenilins and Alzheimer's disease. Trends Neurosci. 1997;20:154‐159. [DOI] [PubMed] [Google Scholar]
- 2. Serrano‐Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Congdon EE, Sigurdsson EM. Tau‐targeting therapies for Alzheimer disease. Nat Rev Neurol. 2018;14:399‐415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol. 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 5. Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer's disease. Brain. 2015;138:2814‐2833. [DOI] [PubMed] [Google Scholar]
- 6. Lewis DA, Campbell MJ, Terry RD, Morrison JH. Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer's disease: a quantitative study of visual and auditory cortices. J Neurosci. 1987;7:1799‐1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nelson O, Supnet C, Liu H, Bezprozvanny I. Familial Alzheimer's disease mutations in presenilins: effects on endoplasmic reticulum calcium homeostasis and correlation with clinical phenotypes. J Alzheimers Dis. 2010;21:781‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mattson MP. ER calcium and Alzheimer's disease: in a state of flux. Sci Signal. 2010;3:pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Demuro A, Parker I, Stutzmann GE. Calcium signaling and amyloid toxicity in Alzheimer disease. J Biol Chem. 2010;285:12463‐12468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kelliher M, Fastbom J, Cowburn RF, et al. Alterations in the ryanodine receptor calcium release channel correlate with Alzheimer's disease neurofibrillary and beta‐amyloid pathologies. Neuroscience. 1999;92:499‐513. [DOI] [PubMed] [Google Scholar]
- 11. Lacampagne A, Liu X, Reiken S, et al. Post‐translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer's disease‐like pathologies and cognitive deficits. Acta Neuropathol. 2017;134:749‐767. [DOI] [PubMed] [Google Scholar]
- 12. Marx SO, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365‐376. [DOI] [PubMed] [Google Scholar]
- 13. Bellinger AM, Reiken S, Dura M, et al. Remodeling of ryanodine receptor complex causes “leaky” channels: a molecular mechanism for decreased exercise capacity. Proc Natl Acad Sci U S A. 2008;105:2198‐2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hof PR, Morrison JH. Neocortical neuronal subpopulations labeled by a monoclonal antibody to calbindin exhibit differential vulnerability in Alzheimer's disease. Exp Neurol. 1991;111:293‐301. [DOI] [PubMed] [Google Scholar]
- 15. Alzheimer's Association CHW. Calcium hypothesis of Alzheimer's disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178‐182.e17. [DOI] [PubMed] [Google Scholar]
- 16. Arnsten AFT, Datta D, Leslie S, Yang ST, Wang M, Nairn AC. Alzheimer's‐like pathology in aging rhesus macaques: unique opportunity to study the etiology and treatment of Alzheimer's disease. Proc Natl Acad Sci U S A. 2019;116(52):26230‐26238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang Y, Zhang Y, Hu W, et al. Rapid alteration of protein phosphorylation during postmortem: implication in the study of protein phosphorylation. Sci Rep. 2015;5:15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rapp PR, Amaral DG. Evidence for task‐dependent memory dysfunction in the aged monkey. J Neurosci. 1989;9:3568‐3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang M, Gamo NJ, Yang Y, et al. Neuronal basis of age‐related working memory decline. Nature. 2011;476:210‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paspalas CD, Carlyle BC, Leslie S, et al. The aged rhesus macaque manifests Braak stage III/IV Alzheimer's‐like pathology. Alzheimers Dement. 2018;14:680‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uno H, Alsum PB, Dong S, et al. Cerebral amyloid angiopathy and plaques, and visceral amyloidosis in aged macaques. Neurobiol Aging. 1996;17:275‐281. [DOI] [PubMed] [Google Scholar]
- 22. Morozov YM, Datta D, Paspalas CD, Arnsten AFT. Ultrastructural evidence for impaired mitochondrial fission in the aged rhesus monkey dorsolateral prefrontal cortex. Neurobiol Aging. 2017;51:9‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang L, Trushin S, Christensen TA, et al. Altered brain energetics induces mitochondrial fission arrest in Alzheimer's. Disease Sci Rep. 2016;6:18725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dumitriu D, Hao J, Hara Y, et al. Selective changes in thin spine density and morphology in monkey prefrontal cortex correlate with aging‐related cognitive impairment. J Neurosci. 2010;30(22):7507‐7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carlyle BC, Nairn AC, Wang M, et al. cAMP‐PKA phosphorylation of tau confers risk for degeneration in aging association cortex. Proc Natl Acad Sci U S A. 2014;111:5036‐5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Goldman‐Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477‐485. [DOI] [PubMed] [Google Scholar]
- 27. National Research Council CftUotGftCaUoL, Animals. Guide for the Care and Use of Laboratory Animals . In: 8th, editor. Washington (DC): National Academies Press (US) National Academy of Sciences; 2011. [Google Scholar]
- 28. Datta D, Leslie SN, Morozov YM, et al. Classical complement cascade initiating C1q protein within neurons in the aged rhesus macaque dorsolateral prefrontal cortex. J Neuroinflammation. 2020;17:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li D, Musante V, Zhou W, Picciotto MR, Nairn AC. Striatin‐1 is a B subunit of protein phosphatase PP2A that regulates dendritic arborization and spine development in striatal neurons. J Biol Chem. 2018;293:11179‐11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013;23:1643‐1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Janelidze S, Stomrud E, Smith R, et al. Cerebrospinal fluid p‐tau217 performs better than p‐tau181 as a biomarker of Alzheimer's disease. Nat Commun. 2020;11:1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barthelemy NR, Bateman RJ, Hirtz C, et al. Cerebrospinal fluid phospho‐tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer's disease and PET amyloid‐positive patient identification. Alzheimers Res Ther. 2020;12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011;70:410‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braak E, Braak H, Mandelkow EM. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994;87:554‐567. [DOI] [PubMed] [Google Scholar]
- 35. Dujardin S, Commins C, Lathuiliere A, et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer's disease. Nat Med. 2020;26:1256‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu SJ, Zhang JY, Li HL, et al. Tau becomes a more favorable substrate for GSK‐3 when it is prephosphorylated by PKA in rat brain. J Biol Chem. 2004;279:50078‐50088. [DOI] [PubMed] [Google Scholar]
- 37. Yoshimura Y, Ichinose T, Yamauchi T. Phosphorylation of tau protein to sites found in Alzheimer's disease brain is catalyzed by Ca2+/calmodulin‐dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci Lett. 2003;353:185‐188. [DOI] [PubMed] [Google Scholar]
- 38. Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Ryanodine receptor/calcium release channel PKA phosphorylation: a critical mediator of heart failure progression. Proc Natl Acad Sci U S A. 2006;103:511‐518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mei Y, Xu L, Kramer HF, Tomberlin GH, Townsend C, Meissner G. Stabilization of the skeletal muscle ryanodine receptor ion channel‐FKBP12 complex by the 1,4‐benzothiazepine derivative S107. PLoS One. 2013;8:e54208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Liu F, Liang Z, Shi J, et al. PKA modulates GSK‐3β‐ and cdk5‐catalyzed phosphorylation of tau in site‐ and kinase‐specific manners. FEBS Lett. 2006;580:6269‐6274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kurbatskaya K, Phillips EC, Croft CL, et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer's disease brain. Acta Neuropathol Commun. 2016;4:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862‐872. [DOI] [PubMed] [Google Scholar]
- 43. Gant JC, Kadish I, Chen KC, et al. Aging‐related calcium dysregulation in rat entorhinal neurons homologous with the human entorhinal neurons in which Alzheimer's disease neurofibrillary tangles first appear. J Alzheimers Dis. 2018;66:1371‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bennecib M, Gong CX, Grundke‐Iqbal I, Iqbal K. Inhibition of PP‐2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett. 2001;490:15‐22. [DOI] [PubMed] [Google Scholar]
- 45. Litersky JM, Johnson GV, Jakes R, Goedert M, Lee M, Seubert P. Tau protein is phosphorylated by cyclic AMP‐dependent protein kinase and calcium/calmodulin‐dependent protein kinase II within its microtubule‐binding domains at Ser‐262 and Ser‐356. Biochem J. 1996;316(Pt 2):655‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ekinci FJ, Shea TB. Free PKC catalytic subunits (PKM) phosphorylate tau via a pathway distinct from that utilized by intact PKC. Brain Res. 1999;850:207‐216. [DOI] [PubMed] [Google Scholar]
- 47. Goni‐Oliver P, Lucas JJ, Avila J, Hernandez F. N‐terminal cleavage of GSK‐3 by calpain: a new form of GSK‐3 regulation. J Biol Chem. 2007;282:22406‐22413. [DOI] [PubMed] [Google Scholar]
- 48. Halls ML, Cooper DM. Regulation by Ca2+‐signaling pathways of adenylyl cyclases. Cold Spring Harb Perspect Biol. 2011;3(1):a004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Barthélemy NR, Mallipeddi N, Moiseyev P, Sato C, Bateman RJ. Tau phosphorylation rates measured by mass spectrometry differ in the intracellular brain vs. extracellular cerebrospinal fluid compartments and are differentially affected by Alzheimer's disease. Front Aging Neurosci. 2019;11:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tibbo AJ, Tejeda GS, Baillie GS. Understanding PDE4's function in Alzheimer's disease; a target for novel therapeutic approaches. Biochem Soc Trans. 2019;47:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leslie SN, Datta D, Christensen KR, van Dyck CH, Arnsten AFT, Nairn AC. Phosphodiesterase PDE4D is decreased in frontal cortex of aged rats and positively correlated with working memory performance and inversely correlated with PKA phosphorylation of tau. Front Aging Neurosci. 2020;12:576723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jin LE, Wang M, Galvin VC, et al. mGluR2 versus mGluR3 metabotropic glutamate receptors in primate dorsolateral prefrontal cortex: postsynaptic mglur3 strengthen working memory networks. Cereb Cortex. 2018;28:974‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Datta D, Enwright JF, Arion D, et al. Mapping phosphodiesterase 4D (PDE4D) in macaque dorsolateral prefrontal cortex: postsynaptic compartmentalization in layer iii pyramidal cell circuits. Front Neuroanat. 2020;14:578483. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information
Data Availability Statement
All raw and quantified data will be made available by the authors upon reasonable request.