

Abstract
Background:
A growing body of evidence links maternal exposure to particulate matter < 2.5 μM in diameter (PM2.5) and deviations in fetal growth. Several studies suggest that the placenta plays a critical role in conveying the effects of maternal PM2.5 exposure to the developing fetus. These include observed associations between air pollutants and candidate placental features, such as mitochondrial DNA content, DNA methylation and telomere length. However, gaps remain in delineating the pathways linking the placenta to air pollution-related health effects, including a comprehensive profiling of placental processes impacted by maternal PM2.5 exposure. In this study, we examined alterations in a placental transcriptome-wide network in relation to maternal PM2.5 exposure prior to and during pregnancy and infant birthweight.
Methods:
We evaluated PM2.5 exposure and placental RNA-sequencing data among study participants enrolled in the Rhode Island Child Health Study (RICHS). Daily residential PM2.5 levels were estimated using a hybrid model incorporating land-use regression and satellite remote sensing data. Distributed lag models were implemented to assess the impact on infant birthweight due to PM2.5 weekly averages ranging from 12 weeks prior to gestation until birth. Correlations were assessed between PM2.5 levels averaged across the identified window of susceptibility and a placental transcriptome-wide gene coexpression network previously generated using the WGCNA R package.
Results:
We identified a sensitive window spanning 12 weeks prior to and 13 weeks into gestation during which maternal PM2.5 exposure is significantly associated with reduced infant birthweight. Two placental coexpression modules enriched for genes involved in amino acid transport and cellular respiration were correlated with infant birthweight as well as maternal PM2.5 exposure levels averaged across the identified growth restriction window.
Conclusion:
Our findings suggest that maternal PM2.5 exposure may alter placental programming of fetal growth, with potential implications for downstream health effects, including susceptibility to cardiometabolic health outcomes and viral infections.
Keywords: birthweight, air pollution, placenta, RNAseq
Background
Studies to date have established that maternal exposure to air pollution can lead to deviations in infant birthweight, an indicator of gestational quality with implications for postnatal health. Spanning high and low income countries, these studies particularly point to reductions in fetal growth due to exposure to fine particulate matter < 2.5 μM in diameter (PM2.5)(1–8). Reported findings include temporal associations between maternal PM2.5 exposure and infant birthweight, suggesting critical windows of susceptibility during gestation(9–16). Additional nuances in the overall relationship between maternal PM2.5 exposure and birth outcomes include reports of sex-specific effects(17).
The mechanism through which maternal PM2.5 exposure derails appropriate fetal development is not well characterized. The placenta, a transient gestational organ that sits at the maternal-fetal interface, is a likely candidate for conveying the effects of maternal PM2.5 exposure to the developing fetus. Studies have shown the responsiveness of placental molecular features to PM2.5 exposure, including alterations in global methylation(18), mitochondrial methylation(19), gene-specific expression(20) and gene-specific methylation(21). A few studies have further examined whether PM2.5-induced changes in birthweight may be mediated through alterations in these placental molecular features, including global methylation(22), candidate gene expression levels(22), and mitochondrial DNA content(23). However, no reports to date include a whole transcriptomic survey that comprehensively captures placental processes in the pathway between maternal PM2.5 exposure and aberrant fetal growth. We recently described a placental gene coexpression network(24) and reported associations with deviations in birthweight and trace metal exposure(25). In the current study, we leverage this placental gene network to evaluate coordinated perturbations in placental processes that may impart PM2.5-related deviations in fetal growth.
Methods
Study Population.
Women were enrolled at Women and Infants Hospital in Rhode Island between 2009 and 2013 (n=840) as part of the Rhode Island Child Health Study (RICHS). Sex and gestational age matched birthweight percentiles were generated based on the 2013 Fenton growth curve(26). Small for gestational age (SGA, ≤10th birthweight percentile) and large for gestational age (LGA, ≥90th birthweight percentile) infants were oversampled in this population and matched by sex, gestational age and maternal age to appropriate for gestational age infants (AGA, >10th and <90th birthweight percentile). Enrollment was restricted to non-pathologic, singleton, term (≥37 weeks) pregnancies without congenital/chromosomal abnormalities. Obstetric and anthropometric data were abstracted from the medical record and additional lifestyle and family history data were recorded through an interviewer-administered questionnaire.
Air Pollution Exposure.
Daily PM2.5 estimates were assigned to maternal residential addresses using hybrid spatiotemporal models as previously described(27–29). Briefly, participant addresses at the time of delivery were geocoded using ArcMap 10.1 (ESRI; Redlands, CA). Daily satellite aerosol optical depth (AOD) measurements were assigned to grid cells at a 1×1km resolution. Predicted PM2.5 values for each grid cell were based on daily calibrated models (R2=0.88), regressing ground-level PM2.5 data from monitoring sites on the assigned AOD-based PM2.5 values and additional temporal and land use predictors using mixed-effects models. The initial set of daily calibrated PM2.5 models were fit on data where both ground monitoring and AOD measurements were available. These models were then updated and applied to fill in predictions for cells with missing monitoring data as well as days with missing AOD values. Finally, the residuals of the finalized models were regressed against local land use factors at a 200 × 200m scale to map predicted grid-level exposures to residential address-specific estimates. Among participants of the RICHS cohort, PM2.5 data was available for infants born prior to 2013 and with a known residential address at the time of delivery. For each participant, we evaluated PM2.5 exposure during a window spanning 12 weeks pre-conception until birth. We focus on this exposure window as it captures the timespan most relevant to placental development. In the period immediately prior to conception, air pollution may derail maternal processes necessary to support optimal placentation(30). Following placentation, air pollution may continue to impede placental function throughout gestation as the placenta adapts to accommodate the needs of the developing fetus.
Placenta Specimen Collection.
Placental biopsies were excised exclusively on the fetal side from four quadrants within 2 cm of the cord insertion site, rinsed, and stored in RNALater. Within 72 hours, the biopsies were pooled, snap-frozen in liquid nitrogen, homogenized and stored at −80°C until further analysis.
Nucleic Acid Extraction and RNA-sequencing.
Total RNA was isolated from placental samples using an RNeasy Mini Kit. RNA yield was quantified using a NanoDrop ND-1000, and RNA integrity was assessed using an Agilent Bioanalyzer. Following rRNA depletion using a RiboZero kit, RNA libraries were prepared for sequencing on a HiSeq 2500 platform (Illumina, San Diego, CA). Sequencing runs generated single-end 50bp reads. Raw FASTQ files that passed quality control assessment using the FASTQC software were mapped to the human reference genome (hg19) using the Spliced Transcripts Alignment to a Reference (STAR) aligner. The data was filtered to remove lowly expressed genes, adjusted for GC content, corrected for library size using the trimmed mean of M values (TMM) method, and transformed to log counts per million (CPM) values. The final data-set included 12,135 genes. Placental RNASeq data was generated in a representative subset of RICHS participants (n=200) (24).
Statistical Analysis.
Daily estimated levels of PM2.5 exposure for each participant were aggregated into weekly averages, We implemented distributed lag models (DLMs) to evaluate the time-varying association between PM2.5 exposure during a given week and size for gestational age at birth (birthweight percentile, SGA vs. AGA, LGA vs. AGA). This method incorporates data from all time points simultaneously and assumes that the association between the outcome and exposure at a given time point, controlling for exposure at all other time points, varies smoothly as a function of time. The shape of the exposure-lag-response relationship is constrained by a set of basis functions (e.g., splines), with the functions defining the exposure-response relationship and the lag-response relationship combined in a bi-dimensional space of cross-basis functions. Estimation is then performed using a standard regression model, including the matrix of cross-basis functions in the model formula(31). In the current study, the exposure-response relationship was assumed to be linear, and the lag-response relationship was fit using basis spline (B-spline) functions centered at 0, with degrees of freedom selected based on model parsimony and Akaike information criterion (AIC). The continuous birthweight percentile model was fit with 2 degrees of freedom, the SGA model was fit with 2 degrees of freedom, and the LGA model was fit with 4 degrees of freedom; additional smoothing did not significantly improve the model. Sensitive windows to PM2.5 exposure were identified in regions where point estimates and 95% confidence bands do not include 0 (continuous models) or 1 (categorical models). Models were also evaluated stratified by infant sex. DLMs were performed using the dlnm R package version 2.3.9.
Weighted gene coexpression network analysis (WGCNA) was performed using placental transcriptome-wide gene expression data as previously described(24). Briefly, a similarity matrix was generated based on absolute values of pairwise Pearson correlations and transformed into an adjacency matrix using a weighted soft threshold (β =6). Genes were grouped into modules based on hierarchical clustering of their topological overlap. Each module was summarized by the first principal component of each module, defined as the module eigengene. To overlay PM2.5 exposure with the placental gene coexpression network, PM2.5 exposure was averaged across the identified growth restriction exposure window (GREW, 12 weeks prior to conception through 13 weeks gestation). Spearman correlations were calculated between average PM2.5 and the module eigengenes.
In addition to accounting for infant sex and gestational age in the Fenton growth curve-based outcome assignment, all regression models were additionally adjusted for maternal age, maternal education, and season of birth. All analyses were conducted using R version 3.6.2. R code for the presented analysis is available at https://github.com/mdeyssen/RICHS_WGCNA_AirPop.
Results
The demographic characteristics of the study population and comparisons to the full RICHS cohort are shown in Table 1. For the current study, we performed two sub-cohort analyses, one leveraging all participants with available PM2.5 exposure data (n=471) and one overlapping the PM2.5 data with available placental gene network information (n=149). Overall, both sub-cohorts are representative of the full cohort, reflecting a population of primarily of Caucasian descent (> 70%) and average PM2.5 levels across gestation of approximately 8 μg/m3. Notable differences include a slightly older maternal age, a shift in the season of birth distribution and higher birthweight percentiles among the analyzed sub-cohorts compared to the full RICHS cohort.
Table 1.
Demographic characteristics of the study population compared to the full RICHS cohort (n=799)
| p-value | ||||
|---|---|---|---|---|
| Mean (SD) | Mean (SD) | Mean (SD) | ||
| Birthweight percentile | 53.48 (34.48) | 58.18 (34.07) | 56.93 (34.14) | 0.05 |
| Gestational age (weeks) | 39.00 (0.95) | 38.99 (0.92) | 38.95 (0.96) | 0.87 |
| Maternal age (years) | 29.72 (5.47) | 30.03 (5.65) | 31.23 (4.78) | 0.01 |
| Maternal BMI (kg/m2) | 26.60 (7.00) | 26.94 (7.10) | 26.25 (6.28) | 0.53 |
| PM2.5 (ug/m3) | 7.97 (0.79) | 7.97 (0.79) | 7.99 (0.74) | 0.94 |
| N (%) | N (%) | N (%) | ||
| Birthweight Group | 0.12 | |||
| SGA | 157 (19.6) | 77 (16.3) | 22 (14.8) | |
| AGA | 456 (57.1) | 260 (55.2) | 82 (55.0) | |
| LGA | 186 (23.3) | 134 (28.5) | 45 (30.2) | |
| Sex (Male) | 398 (49.8) | 244 (51.8) | 78 (52.3) | 0.73 |
| Delivery Mode (Vaginal) | 391 (48.9) | 221 (46.9) | 82 (55.0) | 0.23 |
| Maternal Race/Ethnicity | 0.50 | |||
| White | 585 (73.2) | 361 (76.6) | 120 (80.5) | |
| Black | 60 (7.5) | 28 (5.9) | 7 (4.7) | |
| Other | 151 (18.9) | 81 (17.2) | 22 (14.8) | |
| NA | 3 (0.4) | 1 (0.2) | 0 (0.0) | |
| Maternal Education | 0.19 | |||
| Less than HS grad | 400 (50.1) | 249 (52.9) | 91 (61.1) | |
| HS grad | 144 (18.0) | 76 (16.1) | 17 (11.4) | |
| Some College | 60 (7.5) | 26 (5.5) | 5 (3.4) | |
| College grad and above | 188 (23.5) | 115 (24.4) | 34 (22.8) | |
| NA | 7 (0.9) | 5 (1.1) | 2 (1.3) | |
| Maternal Smoke status | 0.38 | |||
| No | 746 (93.4) | 441 (93.6) | 142 (95.3) | |
| Yes | 42 (5.3) | 23 (4.9) | 3 (2.0) | |
| NA | 11 (1.4) | 7 (1.5) | 4 (2.7) | |
| Season of Birth | 0.03 | |||
| Spring | 171 (21.4) | 136 (28.9) | 42 (28.2) | |
| Summer | 216 (27.0) | 111 (23.6) | 29 (19.5) | |
| Fall | 261 (32.7) | 137 (29.1) | 55 (36.9) | |
| Winter | 151 (18.9) | 87 (18.5) | 23 (15.4) |
We observe a reduction in infant birthweight percentiles due to a 1ug increase in PM2.5 sustained across all lags. However, this cumulative effect did not meet statistical significance (−4.62, [95% Confidence Interval (CI): −9.35, 0.09]). Evaluating the time-varying association between maternal PM2.5 exposure and infant birthweight percentiles, we observe a significant inverse association during an exposure window spanning 12 weeks prior to conception until 13 weeks gestation (Figure 1a). For the categorical birthweight outcomes (AGA, LGA and SGA), we observe a significant increased risk of SGA status due to a 1 ug increase in PM2.5 sustained across all lags (cumulative Risk Ratio (RR): 1.60, [95% CI: 1.03, 2.47]. In addition, an increased risk of SGA compared to AGA status is specifically observed during an exposure window spanning 2 weeks prior to conception until 14 weeks gestation (Figure 2a). There is no significant effect on LGA status due to a 1 ug increase in PM2.5 sustained across all lags (cumulative RR: 0.93, [95% CI: 0.67, 1.29]. However, a time-varying effect on LGA status due to PM2.5 is observed during two sensitive exposure windows. A decreased risk of LGA compared to AGA status is observed during an exposure window spanning 3 weeks prior to conception until 8 weeks gestation. Additionally, an increased risk of LGA compared to AGA status is observed during an exposure window spanning 29 until 32 weeks gestation (Figure 3a).
Figure 1. Association between maternal PM2.5 exposure and infant birthweight percentiles.
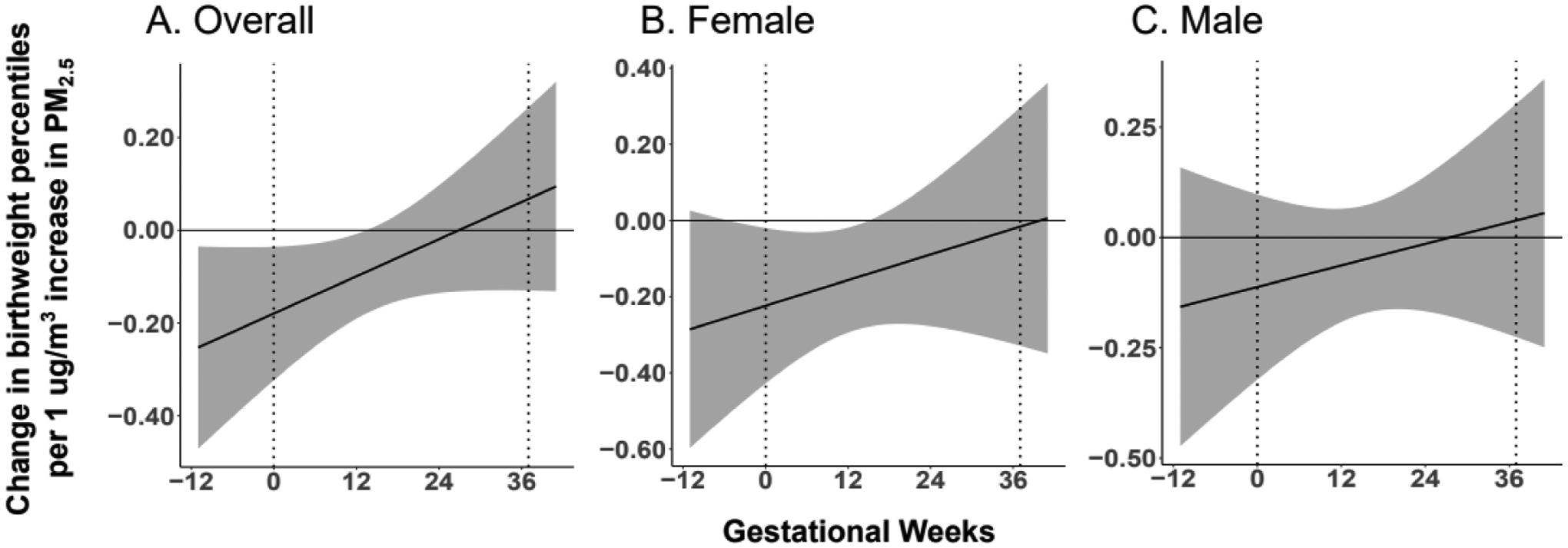
A. Maternal PM2.5 exposure from 12 weeks prior to conception until 13 weeks gestation is inversely associated with infant birthweight percentiles. B. Among female infants, maternal PM2.5 exposure from 6 weeks prior to conception until 13 weeks gestation is inversely associated with birthweight percentiles. C. Among male infants, no association is observed between maternal PM2.5 exposure and birthweight percentiles. Stratifying our analyses by sex, an overall cumulative effect on infant birthweight percentiles due to a 1 ug increase in PM2.5 sustained across all lags is observed among female infants (−7.40, [95% CI: −14.78, −0.03]) but not among male infants (−2.72, [95% CI: −9.21, 3.77]).
Figure 2. Association between maternal PM2.5 exposure and SGA status at birth.
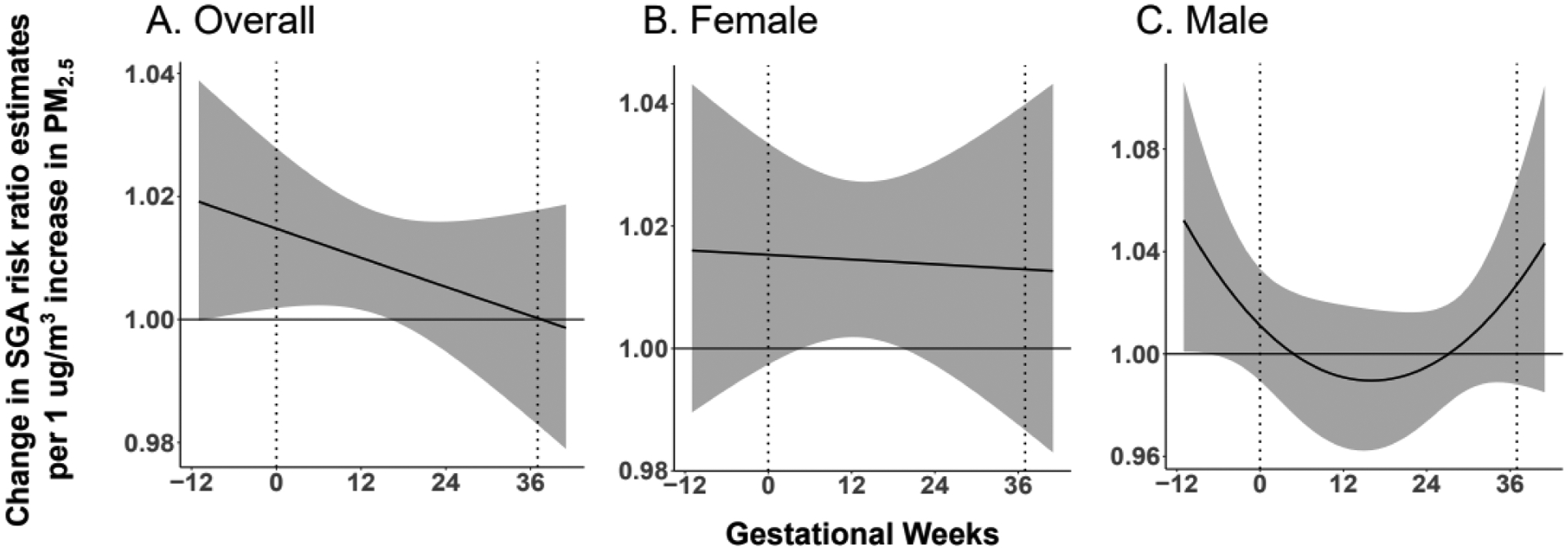
A. Maternal PM2.5 exposure from 2 weeks prior to conception until 14 weeks gestation is associated with increased risk of SGA status at birth. B. Among female infants, maternal PM2.5 exposure from 7 weeks to 16 weeks gestation is associated with increased risk of SGA status at birth. C. Among male infants, maternal PM2.5 exposure between 12 weeks to 7 weeks prior to conception is associated with increased risk of SGA status at birth.
Figure 3. Association between maternal PM2.5 exposure and LGA status at birth.
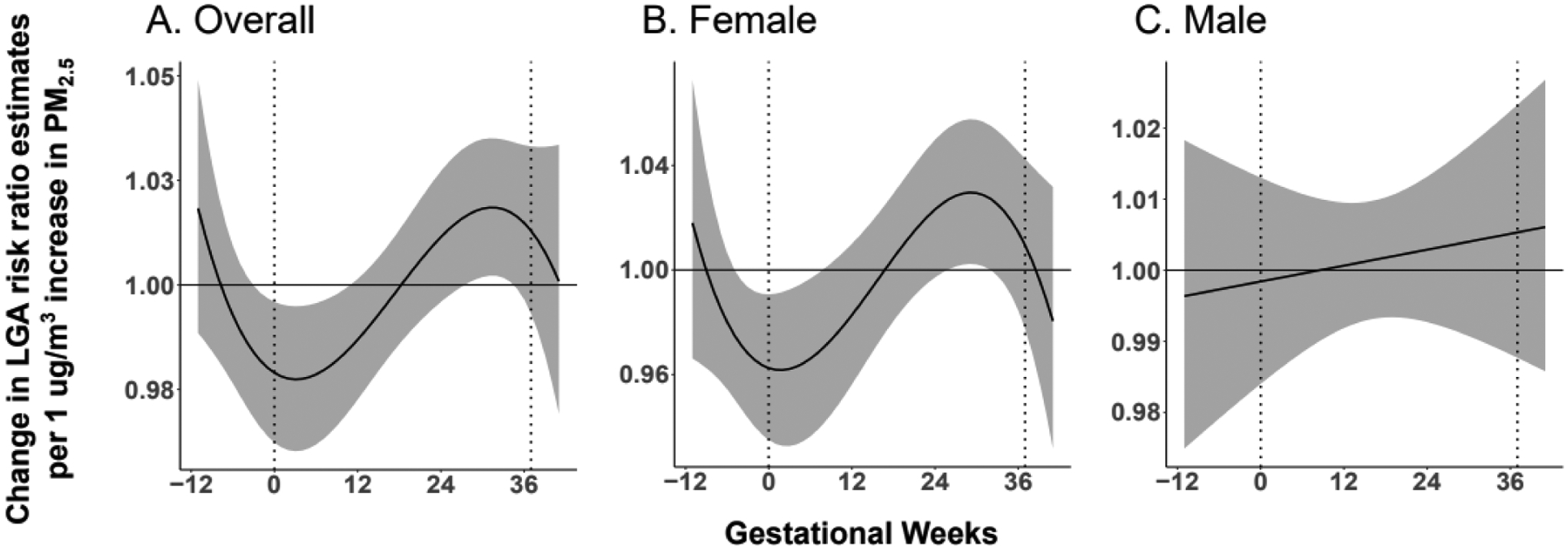
A. Maternal PM2.5 exposure from 3 weeks prior to conception until 8 weeks gestation is associated with a decreased risk of LGA status at birth. In addition, PM2.5 exposure from 29 weeks gestation until 32 weeks gestation is associated with an increased risk of LGA status at birth. B. Among female infants, PM2.5 exposure from 5 weeks prior to conception until 6 weeks gestation is associated with a decreased risk of LGA status at birth. In addition, PM2.5 exposure from 26 weeks until 30 weeks gestation is associated with an increased risk of LGA status at birth. Among male infants, no association between PM2.5 exposure and LGA status at birth is observed.
In our sex-stratified analysis, we observe a significant cumulative effect on birthweight percentiles due to a 1 ug increase in PM2.5 sustained across all lags among female infants (−7.37, [95% Confidence Interval (CI): −14.68, −0.06]). In addition, a significant inverse association is specifically observed between maternal PM2.5 exposure in the 6 weeks prior to conception until 13 weeks gestation and birthweight percentiles (Figure 1b). No significant cumulative effect across all lags (−2.71, [95% Confidence Interval (CI): −9.18, 3.77]) or sensitive window of exposure is observed among male infants (Figure 1c). Modeling birthweight as a categorical outcome, we observe a significant cumulative effect on SGA status due to a 1 ug increase in PM2.5 sustained across all lags among female infants (cumulative RR: 2.13, [95% CI: 1.09, 4.16]) but not male infants (cumulative RR: 1.64, [95% CI: 0.83, 3.24)]. We observe a time-varying effect on SGA risk due to maternal PM2.5 exposure among both male and female infants, in offset windows of exposure. Among female infants, a sensitive exposure window is detected between 7 and 16 weeks gestation (Figure 2b). Among male infants, a sensitive exposure window is detected between 12 and 7 weeks prior to conception (Figure 2c).
No significant cumulative effect on LGA status due to a 1 ug increase in PM2.5 sustained across all lags is observed among female infants (cumulative RR: 0.79, [95% CI: 0.46, 1.34]) or male infants (cumulative RR: 1.06, [95% CI: 0.68, 1.65]). However, two sensitive windows of PM2.5 exposure are observed in relation to LGA status is observed among female infants. A significant inverse association is observed in the 5 weeks prior to conception until 6 weeks gestation, and a positive association is observed between 26 weeks and 30 weeks gestation (Figure 3b). No significant sensitive window of exposure was observed in relation to LGA status among male infants (Figure 3c).
To overlay our PM2.5-related findings with our previously described placental gene coexpression network(24), we averaged maternal PM2.5 exposure from 12 weeks prior to conception and 13 weeks gestation, corresponding to the sensitive window of exposure identified in the birthweight percentile analysis, as this window also includes the growth restriction window identified in the SGA and LGA analyses. We refer to this summary measure as the PM2.5 growth-restriction exposure window (GREW) average. Differences in this PM2.5 GREW average across the birth weight groups are shown in Figure S1.
Consistent with the full cohort of participants with PM2.5 measurements (n=470), in the sub-cohort of participants with both PM2.5 and placental gene expression data (n=149), birthweight decreases as average PM2.5 levels increase, although this change did not reach statistical significance (p=0.10).
In a previous study, we identified 17 placenta gene modules in the placenta(24). Spearman correlations between these coexpression modules and the PM2.5 GREW average as well as key demographic variables are shown in Figure 4. The Gene Ontology (GO) enrichment analysis terms associated with each module are shown along the y-axis. The PM2.5 GREW average is positively correlated with modules enriched in amino acid transport (red), cellular respiration (turquoise) and cell adhesion (tan) processes and inversely correlated with modules enriched in the vasculature development (blue) and organ development (yellow) processes. As shown in the figure, the amino acid transport (red) and cellular respiration (turquoise) modules are additionally inversely correlated with birthweight percentiles. An increase in both amino acid transport and cellular respiration module activity as well as PM2.5 GREW average is linked to a decrease in birthweight percentiles. Hence, the positive correlation between the PM2.5 GREW average and these two modules is consistent with their respective inverse relationships with fetal growth.
Figure 4. Spearman correlations between placental coexpression modules and RICHS demographic characteristics.
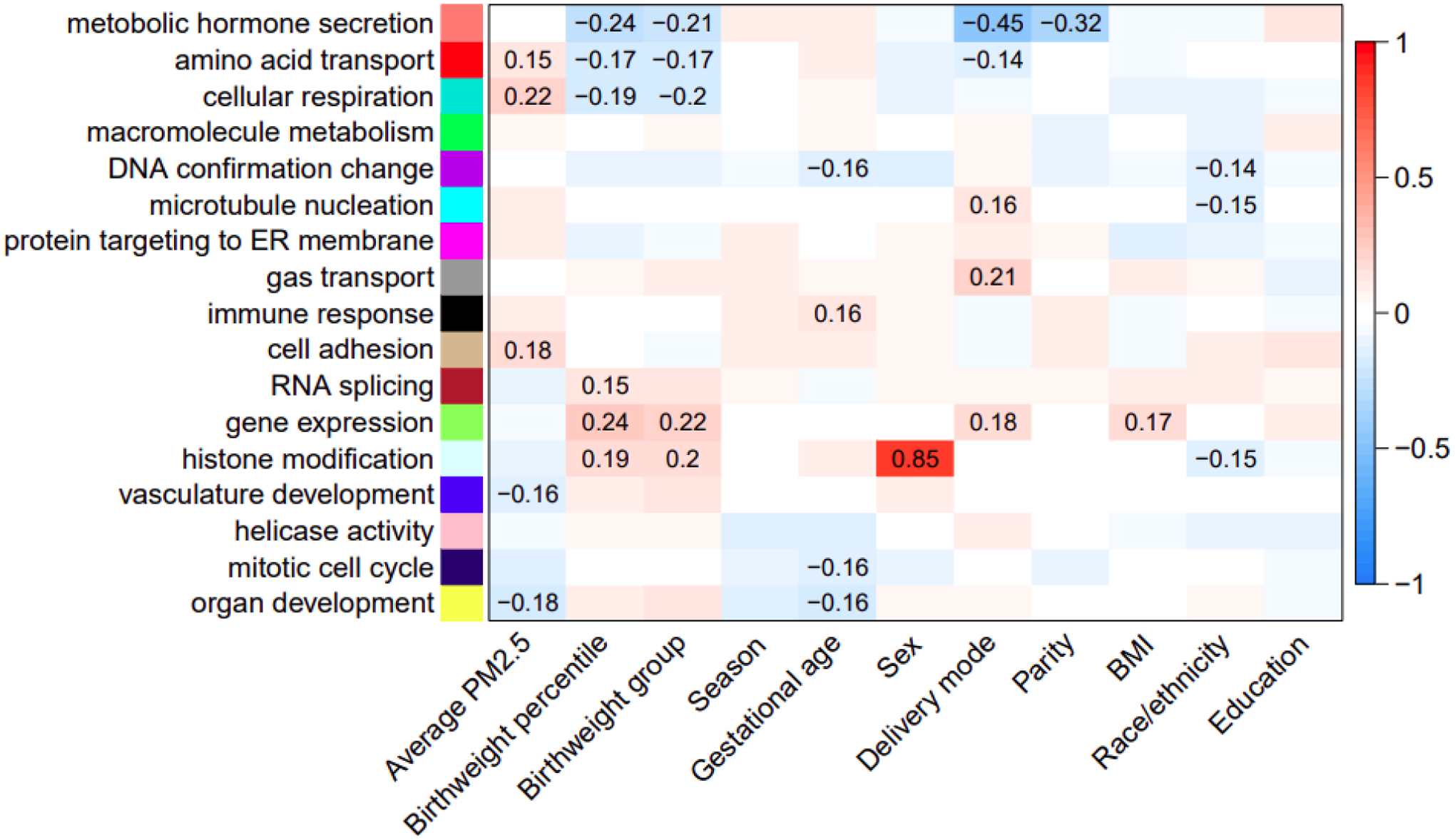
The y-axis indicates the Gene Ontology terms enriched in each module. The color gradient reflects the direction (red = positive, blue = negative) and strength of the correlation between the eigengene values of the modules (y-axis) and RICHS study participant characteristics (x-axis). Significant correlation coefficent values (p<0.05) are indicated on the plot.
Figures 5 and 6 further demonstrate the opposing direction in the correlations between genes in the cellular respiration and amino acid transport modules and the PM2.5 GREW average and birthweight percentiles. Thirty-two genes in the cellular respiration module are correlated (r > |0.20|) with both the PM2.5 GREW average and birthweight percentile, as indicated on the y-axis of each panel in Figure 5. Thirty genes (ACBD3, C14orf153, C1orf212, CCDC53, CLTB, DCTN6, DNLZ, EIF5A, FUNDC2, GNG4, HSD17B10, LLPH, NCRNA00116, NDUFA1, NDUFB10, PDE6D, PGM2, PTMA, RAB2A, SNRPG, SRP54, SSU72, SYAP1, TMUB1, TXNDC12, UBQLN1, UBXN1, UTP18, ZBTB11, ZC3H15) are positively correlated with the PM2.5 GREW average and inversely correlated with birthweight percentiles, and 2 genes (CDK18, LRRC4) are inversely correlated with the PM2.5 GREW average and positively correlated with birthweight percentiles. Additionally, as these are among the genes most highly correlated with the module eigengene value (x-axis of each plot), these genes are considered module hub genes reflective of overall module activity. In the amino acid transport module (Figure 6), only one gene, SPTY2D1, is correlated (r > |0.20|) with both the PM2.5 GREW average and birthweight percentiles. This gene is positively correlated with the PM2.5 GREW average and inversely correlated with birthweight percentiles. Given its correlation with the module eigengene value (x-axis), SPTY2D1 expression is also a module hub gene representative of overall module activity.
Figure 5. Cellular respiration module gene correlations (n=3073) with maternal PM2.5 exposure and infant birthweight percentiles.
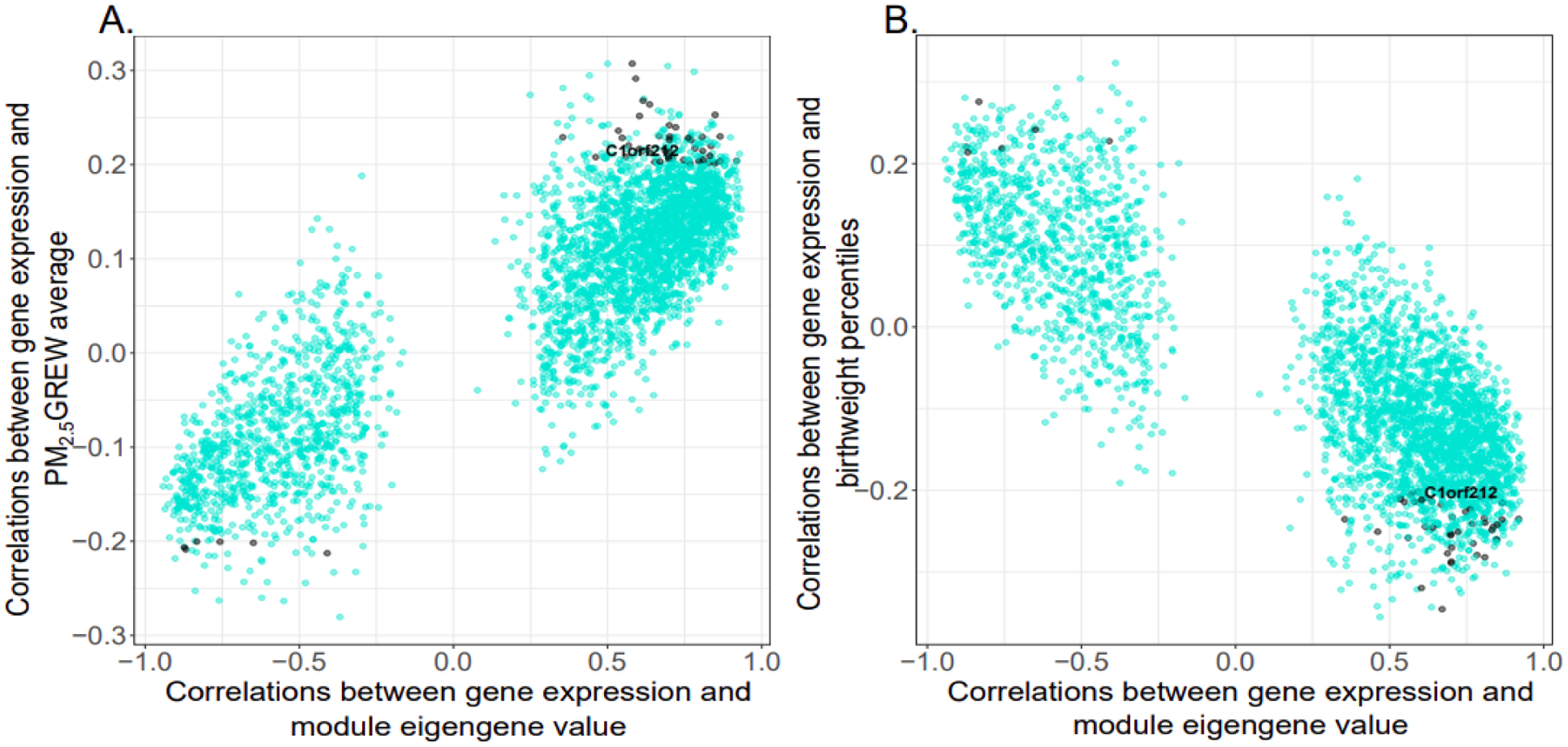
The x-axis indicates the correlation between gene expression and the module eigengene value. The y-axis indicates the correlation between genes and (A) maternal PM2.5 exposure or (B) infant birthweight percentiles. Genes with correlation coefficients > |0.20| with both maternal PM2.5 exposure and infant birthweight percentiles are indicated on the plot with black points, and all other genes are indicated as turquoise points. One locus, C1orf212, is labeled in the plot to illustrate the inverse gene correlations observed with maternal PM2.5 exposure and infant birthweight percentile.
Figure 6. Amino acid transport module gene correlations (n=486) with maternal PM2.5 exposure and infant birthweight percentiles.
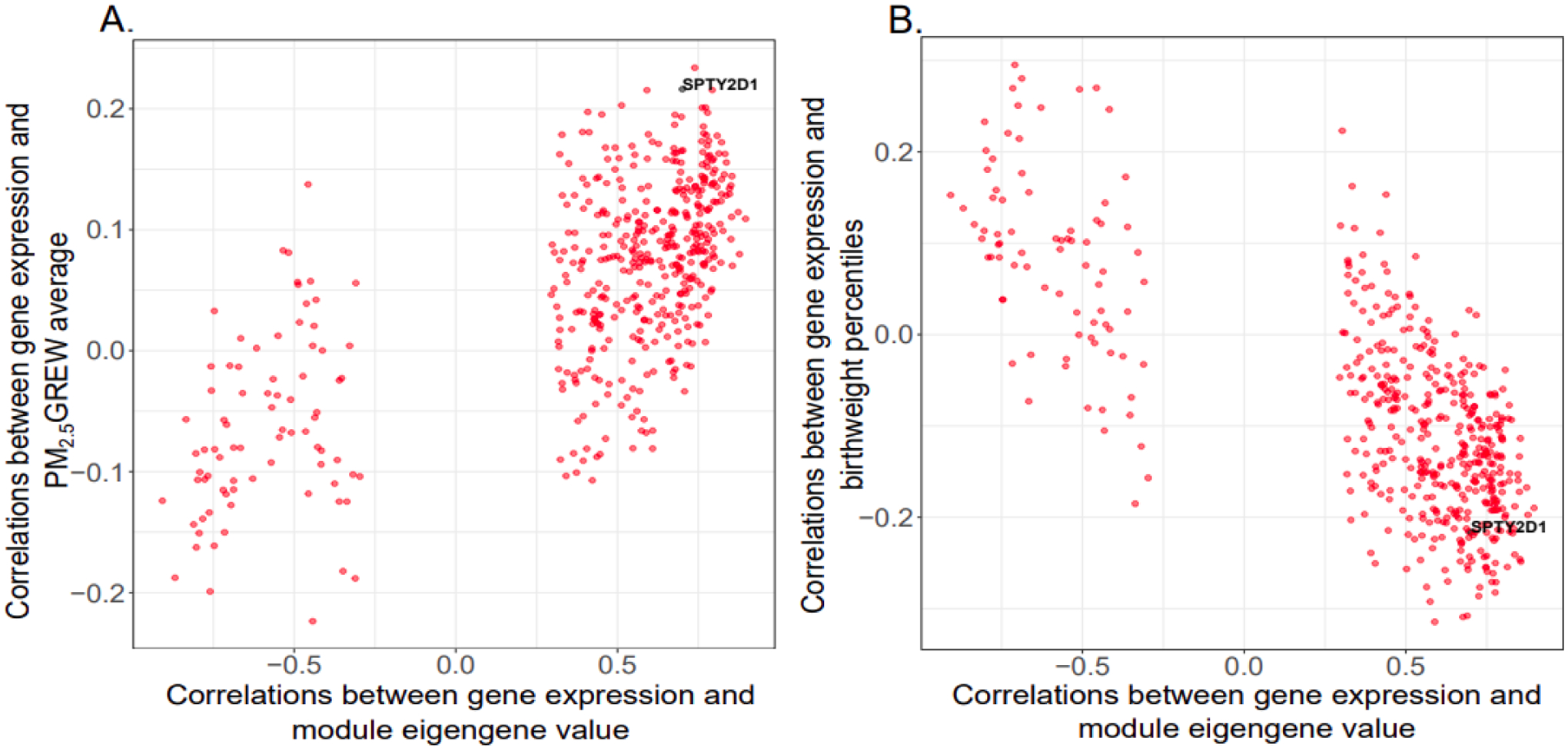
The x-axis indicates the correlation between gene expression and the module eigengene value. The y-axis indicates the correlation between genes and (A) maternal PM2.5 exposure or (B) infant birthweight percentiles. Genes with correlation coefficients > |0.20| are indicated on the plot with black points, and all other genes are indicated as red points. Only one gene, SPTY2D1, passed this threshold.
Discussion
We observed associations between maternal PM2.5 exposure extending from the preconception to the early gestational period and reductions in infant birthweight. The identified window of sensitivity was largely consistent across different parameterizations of birthweight. The window identified in association with reduced birthweight percentiles (12 weeks prior to conception – 13 weeks gestation) is consistent with the PM2.5 window in association with increased risk of SGA compared to AGA status (2 weeks prior to conception – 14 weeks gestation) and the PM2.5 window in association with decreased risk of LGA compared to AGA status (3 weeks prior to conception – 8 weeks gestation). In the LGA analysis, a PM2.5 exposure window late in gestation (29 weeks – 32 weeks gestation) is additionally associated with increased risk of LGA compared to AGA status. Stratifying by sex, the findings among female infants coincide with the overall findings while the findings among male infants are largely null.
Our findings of a PM2.5 exposure window early in gestation in association with reduced birthweight is consistent with a number of studies reported in the literature (9,10,12–14,16,32–35). We also identified an exposure window late in gestation that is associated with LGA status. Prior studies that specifically evaluated fetal overgrowth have reported similar associations (36,37). The apparent differential effect of PM2.5 exposure on birthweight in early gestation compared to late gestation could reflect a shift in how the placenta functions as it adapts to accommodate the increasing metabolic demands of the fetus. PM2.5 may impact the role of the placenta in appropriate implantation and establishment of maternal blood flow early in gestation. Later in gestation, the placenta plays a more pronounce role in growth hormone and fatty acid regulation, and PM2.5 exposure may disrupt these placental processes in support of the rapid fetal growth and changes in fetal tissue composition that occur during this period.
However, there are also studies that did not identify specific temporal windows (2,37–44) while others identified different temporal windows (45–61). It is important to note that our findings are based on weekly averages of PM2.5 while these prior reports mainly evaluated trimester-specific effects. Trimesters represent three month averages that can coincide with seasonal trends in PM2.5 levels, introducing a potential bias due to induced correlations among the averages. The implementation of a distributed lag model on weekly PM2.5 averages as applied in the current study is less susceptible to this type of bias(62). Given that not all developmental processes are neatly encapsulated within trimesters, using a more fine-scale approach of weekly averages also has the additional benefit of greater sensitivity to identify more refined windows of susceptibility that can capture perturbed biological processes that may otherwise be missed using trimester-level averages. While we did not interrogate the biological underpinnings of the preconception and early pregnancy period of susceptibility identified in the current study, this window may reflect an impact due to PM2.5 exposure on maternal physiology that compromises the ability to adequately establish and provide placental support to the developing fetus. Indeed, studies have shown that exposure to criteria air pollutants in the preconception and early pregnancy period are associated with maternal comorbid conditions, such as gestational diabetes. (63). Priming of such derailments in maternal physiology may occur as early as the pre-conception period, impeding maternal support of placental development required for adequate fetal growth.
Few studies to date have reported on sex-specific effects of maternal PM2.5 exposure on fetal growth(17,64). Contrary to the findings reported in the current study, these studies report a stronger deficit in fetal growth among male infants due to PM2.5 exposure. However, the studies differ in how PM2.5 exposure was parameterized and evaluated, limiting the comparability of the reported findings. For example, one study evaluated personal PM2.5 exposure levels averaged across a 48hr period during the second trimester, and another study evaluated ambient PM2.5 averaged across gestation and stratified by maternal obesity.”
The underpinnings of a differential impact on female infants due to preconception and early gestational PM2.5 exposure warrant further exploration.
Overlaying our previously delineated placental gene coexpression network with PM2.5 exposure averaged across the identified growth restriction window, we identified 5 coexpressed gene modules that are correlated with maternal PM2.5 exposure. Out of these five modules, modules enriched for amino acid transport and cellular respiration processes, based on Gene Ontology (GO) enrichment analysis, are additionally correlated with birthweight percentiles.
Consistent with the Gene Ontology term assigned to the cellular respiration module, ATP-dependent processes predominate among the module hub genes that are correlated with maternal PM2.5 exposure and infant birthweight percentile. These include genes that directly interact with mitochondrial processes, including ACBD3(65), C14orf153(66), DCTN6(67), DNLZ(68), FUNDC2(69), HSD17B10(70), NDUFA1(71,72), NDUFB10(72). The upregulation of mitochondrial processes fuels ATP-dependent cellular processes, with genes involved in cell motility (PDE6D)(73), muscle contractility (SYAP1, TMUB1)(74,75), intracellular vesicle transport (ACBD3, DCTN6, CCDC53) (65,76,77), and autophagy (TMUB1, UBQLN1)(78,79). Autophagy, in turn, is an important component in host response to viral infections (ACBD3, TMUB1, C1orf212, DCTN6, UBXN1) (65,78,80–82). Changes in CpG methylation levels at one of these viral infection susceptibility loci, C1orf212, was previously additionally linked with PM2.5 exposure(83). For the amino acid transport module, only one hub gene, SPTY2D1, was correlated with both maternal PM2.5 exposure and infant birthweight percentile. Prior studies most commonly link single nucleotide polymorphisms (SNPs) in this gene with overall(84,85) and sex-specific(86) differences in lipid profiles.
Recently reported human and animal studies also implicate PM2.5 exposure with alterations in the placenta, including histopathology changes(87,88), increased oxidative stress(89), increased placental aging(90), altered expression of candidate genes(20,91), mitochondrial DNA content and elevated global methylation(92). Among these studies, those that additionally evaluated placental impacts across temporal windows of exposure also identified early gestation as a critical period of sensitivity(20,92). The findings of the current study build upon these early links through a more comprehensive placental transcriptomic survey that pinpoints specific biologic processes as candidates for being dysregulated through PM2.5 exposure during the early gestational period. Taken together, these findings suggest that early maternal exposure to PM2.5 impacts fetal growth, particularly among female infants, and may program immune activation in response to viral infections and cardiometabolic outcomes later in life.
There are several limitations in this study that warrant caution in the interpretations of the findings. Complete data on maternal PM2.5 exposure, placental gene expression, and infant birthweight was only available on a subset of the cohort (n=149), limiting the power of our study. For example, formal testing of the placental network genes as mediators in the pathway between maternal PM2.5 exposure and infant birthweight was not feasible since the main effect between maternal PM2.5 exposure and infant birthweight did not reach statistical significance within the subcohort of participants with both PM2.5 and placental gene expression data. In addition, gene expression changes were evaluated in placenta collected at birth while the presented findings suggest early gestation as a sensitive window to maternal PM2.5 exposure. While it is possible that early gestational exposure impacts appropriate placentation with persistent effects detectable at term (e.g., insufficient vascularization), we cannot discern whether our identified marks reflect such a direct effect or rather a secondary adaptive placental response. A live birth bias may also impact our findings if we expect air pollution exposure to contribute to early fetal loss(93). Since early fetal loss is often unreported, this critical susceptible subset among the truly exposed is not accounted for in our study, and conditioning our analyses on data stemming from recorded birth information can introduce a selection bias. Finally, our air pollution exposure assessment specifically focused on particulate matter components while data on other criteria air pollutants (e.g., carbon monoxide, nitrogen dioxide and ozone) and indoor air pollutants were not available to be evaluated. Future studies that account for the mixed sources and composition of air pollution may better capture effects reflective of true exposure profiles.
Conclusions
This study adds to the existing body of literature pinpointing the preconception and early gestation periods as critical windows of susceptibility to maternal PM2.5 exposure induced effects on fetal growth restriction. Furthermore, this is among the first epidemiologic studies linking a critical window of susceptibility due to maternal PM2.5 exposure to potential placental programming impacts on fetal development. Our findings suggest that maternal PM2.5 exposure may alter placental programming of fetal growth, with potential implications for downstream health effects, including susceptibility to viral infections and cardiometabolic health outcomes. This adds to the growing policy impetus to reduce global air pollutant levels to protect the unborn child. Until this goal is realized, interim strategies should focus on emphasizing exposure mitigation strategies, particularly among women of childbearing age.
Supplementary Material
Highlights.
PM2.5 exposure in the periconceptional and early prenatal period is associated with reductions in fetal growth.
Female infants are particularly vulnerable to PM2.5 induced deficits in fetal growth.
Disruptions in placental processes involved in protein transport and other ATP-driven processes may play an important role in conveying the impact of PM2.5 on the developing fetus.
Funding
This research was funded by NIH-NIEHS P30ES009089, NIH-NIEHS R00ES029571, NIH-NIEHS R00ES027496, NIH-NIEHS R01ES022223, NIH-NIEHS R01ES029212, NIH-NIEHS R24ES028507, NIH-NIEHS P30ES019776, NIH-NIEHS R21ES023073.
List of Abbreviations
- AIC
Akaike information criterion
- AOD
Aerosol optical depth
- B-spline
Basis spline
- DLM
Distributed Lag Model
- GO
Gene Ontology
- GREW
Growth restriction exposure window
- PM2.5
Particulate matter < 2.5 μM in diameter
- SGA
Small for gestational age
- LGA
Large for gestational age
- AGA
Appropriate for gestational age
- STAR
Spliced Transcripts Alignment to a Reference
- RICHS
Rhode Island Child Health Study
- WGCNA
Weighted Gene Coexpression Network
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics Approval and consent to participate
All participants provided informed consent, and the protocols were approved by the Institutional Review Boards at Women and Infants Hospital and Emory University.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare that they have no competing interests.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Rosa MJ, Pajak A, Just AC, Sheffield PE, Kloog I, Schwartz J, et al. Prenatal exposure to PM2.5 and birth weight: A pooled analysis from three North American longitudinal pregnancy cohort studies. Environ Int. 2017;107:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, et al. Maternal exposure to particulate air pollution and term birth weight: A multi-country evaluation of effect and heterogeneity. Environ Health Perspect. 2013;121(3):367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao Q, Chen H, Strickland MJ, Kan H, Chang HH, Klein M, et al. Associations between birth outcomes and maternal PM2.5 exposure in Shanghai: A comparison of three exposure assessment approaches. Environ Int. 2018. August 1;117:226–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balakrishnan K, Ghosh S, Thangavel G, Sambandam S, Mukhopadhyay K, Puttaswamy N, et al. Exposures to fine particulate matter (PM2.5) and birthweight in a rural-urban, mother-child cohort in Tamil Nadu, India. Environ Res. 2018. February 1;161:524–31. [DOI] [PubMed] [Google Scholar]
- 5.Fleischer NL, Merialdi M, van Donkelaar A, Vadillo-Ortega F, Martin RV., Betran AP, et al. Outdoor air pollution, preterm birth, and low birth weight: Analysis of the world health organization global survey on maternal and perinatal health. Environ Health Perspect. 2014;122(4):425–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stieb DM, Chen L, Hystad P, Beckerman BS, Jerrett M, Tjepkema M, et al. A national study of the association between traffic-related air pollution and adverse pregnancy outcomes in Canada, 1999–2008. Environ Res. 2016. July 1;148:513–26. [DOI] [PubMed] [Google Scholar]
- 7.Pedersen M, Giorgis-Allemand L, Bernard C, Aguilera I, Andersen AMN, Ballester F, et al. Ambient air pollution and low birthweight: A European cohort study (ESCAPE). Lancet Respir Med. 2013. November;1(9):695–704. [DOI] [PubMed] [Google Scholar]
- 8.Trasande L, Wong K, Roy A, Savitz DA, Thurston G. Exploring prenatal outdoor air pollution, birth outcomes and neonatal health care utilization in a nationally representative sample. J Expo Sci Environ Epidemiol. 2013. May;23(3):315–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geer LA, Weedon J, Bell ML. Ambient air pollution and term birth weight in Texas from 1998 to 2004. J Air Waste Manag Assoc. 2012;62(11):1285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yorifuji T, Kashima S, Doi H. Outdoor air pollution and term low birth weight in Japan. Environ Int. 2015. January 1;74:106–11. [DOI] [PubMed] [Google Scholar]
- 11.Kumar N Uncertainty in the relationship between criteria pollutants and low birth weight in Chicago. Atmos Environ. 2012. March;49:171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Díaz J, Arroyo V, Ortiz C, Carmona R, Linares C. Effect of environmental factors on low weight in non-premature births: A time series analysis. PLoS One. 2016. October 1;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tapia VL, Vasquez BV., Vu B, Liu Y, Steenland K, Gonzales GF. Association between maternal exposure to particulate matter (PM2.5) and adverse pregnancy outcomes in Lima, Peru. J Expo Sci Environ Epidemiol. 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Y, Xu J, Chen D, Sun P, Ma X. The association between air pollution and preterm birth and low birth weight in Guangdong, China. BMC Public Health. 2019. January 3;19(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Z, Yang Y, Qian Z, Ruan Z, Chang J, Vaughn MG, et al. Ambient PM2.5 and birth outcomes: Estimating the association and attributable risk using a birth cohort study in nine Chinese cities. Environ Int. 2019. May 1;126:329–35. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Yuan X, Fu J, Zhang L, Hong L, Hu L, et al. Association of ambient air pollutants and birth weight in Ningbo, 2015–2017. Environ Pollut. 2019. June 1;249:629–37. [DOI] [PubMed] [Google Scholar]
- 17.Jedrychowski W, Perera F, Mrozek-Budzyn D, Mroz E, Flak E, Spengler JD, et al. Gender differences in fetal growth of newborns exposed prenatally to airborne fine particulate matter. Environ Res. 2009. May;109(4):447–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen BG, Godderis L, Pieters N, Poels K, Kiciński M, Cuypers A, et al. Placental DNA hypomethylation in association with particulate air pollution in early life. Part Fibre Toxicol [Internet]. 2013. January [cited 2015 Aug 25];10:22. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3686623&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janssen BG, Byun H-M, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIR ON AGE birth cohort study. Epigenetics [Internet]. 2015. June 3 [cited 2020 Mar 22];10(6):536–44. Available from: http://www.tandfonline.com/doi/full/10.1080/15592294.2015.1048412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saenen ND, Plusquin M, Bijnens E, Janssen BG, Gyselaers W, Cox B, et al. In utero fine particle air pollution and placental expression of genes in the brain-derived neurotrophic factor signaling pathway: An ENVIRONAGE birth cohort study. Environ Health Perspect. 2015. August 4;123(8):834–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saenen ND, Vrijens K, Janssen BG, Roels HA, Neven KY, Vanden Berghe W, et al. Lower placental leptin promoter methylation in association with fine particulate matter air pollution during pregnancy and placental nitrosative stress at birth in the ENVIRONAGE cohort. Environ Health Perspect. 2017. February 1;125(2):262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maghbooli Z, Hossein-nezhad A, Adabi E, Asadollah-pour E, Sadeghi M, Mohammad-nabi S, et al. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS One. 2018. July 1;13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clemente DBP, Casas M, Vilahur N, Begiristain H, Bustamante M, Carsin A-E, et al. Prenatal Ambient Air Pollution, Placental Mitochondrial DNA Content, and Birth Weight in the INMA (Spain) and ENVIRONAGE (Belgium) Birth Cohorts. Environ Health Perspect [Internet]. 2016. [cited 2020 Mar 22];124(5):659–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26317635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deyssenroth MA, Peng S, Hao K, Lambertini L, Marsit CJ, Chen J. Whole-transcriptome analysis delineates the human placenta gene network and its associations with fetal growth. BMC Genomics [Internet]. 2017. July 10 [cited 2017 Jul 14];18(1):520. Available from: http://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-017-3878-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deyssenroth MA, Gennings C, Liu SH, Peng S, Hao K, Lambertini L, et al. Intrauterine multi-metal exposure is associated with reduced fetal growth through modulation of the placental gene network. Environ Int [Internet]. 2018. [cited 2020 Mar 23];120:373–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30125854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fenton TR, Kim JH. A systematic review and meta-analysis to revise the Fenton growth chart for preterm infants. BMC Pediatr [Internet]. 2013. April 20 [cited 2016 Dec 5];13(1):59. Available from: http://bmcpediatr.biomedcentral.com/articles/10.1186/1471-2431-13-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kloog I, Chudnovsky AA, Just AC, Nordio F, Koutrakis P, Coull BA, et al. A new hybrid spatio-temporal model for estimating daily multi-year PM2.5 concentrations across northeastern USA using high resolution aerosol optical depth data. Atmos Environ. 2014;95:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kingsley SL, Deyssenroth MA, Kelsey KT, Awad YA, Kloog I, Schwartz JD, et al. Maternal residential air pollution and placental imprinted gene expression. Environ Int. 2017;108:204–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kingsley SL, Eliot MN, Glazer K, Awad YA, Schwartz JD, Savitz DA, et al. Maternal ambient air pollution, preterm birth and markers of fetal growth in Rhode Island: Results of a hospital-based linkage study. J Epidemiol Community Health. 2017. December 1;71(12):1131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Segal TR, Giudice LC. Before the beginning: environmental exposures and reproductive and obstetrical outcomes [Internet]. Vol. 112, Fertility and Sterility. Elsevier Inc.; 2019. [cited 2021 Mar 6]. p. 613–21. Available from: https://pubmed.ncbi.nlm.nih.gov/31561863/ [DOI] [PubMed] [Google Scholar]
- 31.Gasparrini A Modeling exposure-lag-response associations with distributed lag non-linear models. Stat Med. 2014. February 28;33(5):881–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blum JL, Chen LC, Zelikoff JT. Exposure to ambient particulate matter during specific gestational periods produces adverse obstetric consequences in mice. Environ Health Perspect [Internet]. 2017. July 1 [cited 2020 Aug 18];125(7). Available from: https://pubmed.ncbi.nlm.nih.gov/28893721/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malmqvist E, Liew Z, Källén K, Rignell-Hydbom A, Rittner R, Rylander L, et al. Fetal growth and air pollution - A study on ultrasound and birth measures. Environ Res [Internet]. 2017. January 1 [cited 2020 Aug 18];152:73–80. Available from: https://pubmed.ncbi.nlm.nih.gov/27741452/ [DOI] [PubMed] [Google Scholar]
- 34.Harris G, Thompson WD, Fitzgerald E, Wartenberg D. The association of PM2.5 with full term low birth weight at different spatial scales. Environ Res [Internet]. 2014. October 1 [cited 2020 Aug 18];134:427–34. Available from: https://pubmed.ncbi.nlm.nih.gov/25261950/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rich DQ, Demissie K, Lu SE, Kamat L, Wartenberg D, Rhoads GG. Ambient air pollutant concentrations during pregnancy and the risk of fetal growth restriction. J Epidemiol Community Health [Internet]. 2009. June [cited 2020 Aug 18];63(6):488–96. Available from: https://pubmed.ncbi.nlm.nih.gov/19359274/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shang L, Huang L, Yang L, Leng L, Qi C, Xie G, et al. Impact of air pollution exposure during various periods of pregnancy on term birth weight: a large-sample, retrospective population-based cohort study. Environ Sci Pollut Res [Internet]. 2021. January 1 [cited 2021 Apr 5];28(3):3296–306. Available from: 10.1007/s11356-020-10705-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen S, Wang S, Li T, Zhu H, Liang S, Xu K, et al. Effect of PM2.5 on macrosomia in China: A nationwide prospective cohort study. Pediatr Obes [Internet]. 2020. February 1 [cited 2020 Aug 18];15(2). Available from: https://pubmed.ncbi.nlm.nih.gov/31689002/ [DOI] [PubMed] [Google Scholar]
- 38.Brauer M, Lencar C, Tamburic L, Koehoorn M, Demers P, Karr C. A cohort study of traffic-related air pollution impacts on birth outcomes. Environ Health Perspect [Internet]. 2008. May [cited 2020 Aug 18];116(5):680–6. Available from: https://pubmed.ncbi.nlm.nih.gov/18470315/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morello-Frosch R, Jesdale BM, Sadd JL, Pastor M. Ambient air pollution exposure and full-term birth weight in California. Environ Heal A Glob Access Sci Source [Internet]. 2010. [cited 2020 Aug 18];9(1). Available from: https://pubmed.ncbi.nlm.nih.gov/20667084/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savitz DA, Bobb JF, Carr JL, Clougherty JE, Dominici F, Elston B, et al. Ambient fine particulate matter, nitrogen dioxide, and term birth weight in new york, new york. Am J Epidemiol [Internet]. 2014. February 15 [cited 2020 Aug 18];179(4):457–66. Available from: https://pubmed.ncbi.nlm.nih.gov/24218031/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basu R, Harris M, Sie L, Malig B, Broadwin R, Green R. Effects of fine particulate matter and its constituents on low birth weight among full-term infants in California. Environ Res [Internet]. 2014. [cited 2020 Aug 18];128:42–51. Available from: https://pubmed.ncbi.nlm.nih.gov/24359709/ [DOI] [PubMed] [Google Scholar]
- 42.Stieb DM, Chen L, Beckerman BS, Jerrett M, Crouse DL, Omariba DWR, et al. Associations of pregnancy outcomes and PM2.5 in a national Canadian study. Environ Health Perspect [Internet]. 2016. February 1 [cited 2020 Aug 18];124(2):243–9. Available from: https://pubmed.ncbi.nlm.nih.gov/26090691/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ebisu K, Berman JD, Bell ML. Exposure to coarse particulate matter during gestation and birth weight in the U.S. Environ Int [Internet]. 2016. September 1 [cited 2020 Aug 18];94:519–24. Available from: https://pubmed.ncbi.nlm.nih.gov/27324566/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hao J, Zhang F, Chen D, Liu Y, Liao L, Shen C, et al. Association between ambient air pollution exposure and infants small for gestational age in Huangshi, China: a cross-sectional study. Environ Sci Pollut Res [Internet]. 2019. November 1 [cited 2020 Aug 18];26(31):32029–39. Available from: https://pubmed.ncbi.nlm.nih.gov/31493084/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bell ML, Ebisu K, Belanger K. Ambient air pollution and low birth weight in Connecticut and Massachusetts. Environ Health Perspect [Internet]. 2007. July [cited 2020 Aug 18];115(7):1118–25. Available from: https://pubmed.ncbi.nlm.nih.gov/17637932/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gray SC, Edwards SE, Schultz BD, Miranda ML. Assessing the impact of race, social factors and air pollution on birth outcomes: A population-based study. Environ Heal A Glob Access Sci Source. 2014. January 29;13(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kloog I, Melly SJ, Ridgway WL, Coull BA, Schwartz J. Using new satellite based exposure methods to study the association between pregnancy pm2.5 exposure, premature birth and birth weight in Massachusetts. Environ Heal A Glob Access Sci Source [Internet]. 2012. [cited 2020 Aug 18];11(1). Available from: https://pubmed.ncbi.nlm.nih.gov/22709681/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun X, Luo X, Zhao C, Zhang B, Tao J, Yang Z, et al. The associations between birth weight and exposure to fine particulate matter (PM2.5) and its chemical constituents during pregnancy: A meta-analysis. Environ Pollut [Internet]. 2016. April 1 [cited 2020 Aug 19];211:38–47. Available from: https://pubmed.ncbi.nlm.nih.gov/26736054/ [DOI] [PubMed] [Google Scholar]
- 49.Janssen BG, Saenen ND, Roels HA, Madhloum N, Gyselaers W, Lefebvre W, et al. Fetal thyroid function, birth weight, and in Utero exposure to fine particle air pollution: A birth cohort study. Environ Health Perspect [Internet]. 2017. April 1 [cited 2020 Aug 18];125(4):699–705. Available from: https://pubmed.ncbi.nlm.nih.gov/27623605/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ye L, Ji Y, Lv W, Zhu Y, Lu C, Xu B, et al. Associations between maternal exposure to air pollution and birth outcomes: a retrospective cohort study in Taizhou, China. Environ Sci Pollut Res [Internet]. 2018. August 1 [cited 2020 Aug 19];25(22):21927–36. Available from: https://pubmed.ncbi.nlm.nih.gov/29797193/ [DOI] [PubMed] [Google Scholar]
- 51.Wang Q, Benmarhnia T, Li C, Knibbs LD, Bao J, Ren M, et al. Seasonal analyses of the association between prenatal ambient air pollution exposure and birth weight for gestational age in Guangzhou, China. Sci Total Environ [Internet]. 2019. February 1 [cited 2020 Aug 19];649:526–34. Available from: https://pubmed.ncbi.nlm.nih.gov/30179811/ [DOI] [PubMed] [Google Scholar]
- 52.Li R, Hopke PK, Dozier A, Thurston SW, Thevenet-Morrison K, Croft D, et al. Term birth weight and ambient air pollutant concentrations during pregnancy, among women living in Monroe County, New York. J Expo Sci Environ Epidemiol [Internet]. 2019. June 1 [cited 2020 Aug 19];29(4):500–9. Available from: https://pubmed.ncbi.nlm.nih.gov/30940881/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yuan L, Zhang Y, Wang W, Chen R, Liu Y, Liu C, et al. Critical windows for maternal fine particulate matter exposure and adverse birth outcomes: The Shanghai birth cohort study. Chemosphere [Internet]. 2020. February 1 [cited 2020 Aug 19];240. Available from: https://pubmed.ncbi.nlm.nih.gov/31550593/ [DOI] [PubMed] [Google Scholar]
- 54.Darrow LA, Klein M, Strickland MJ, Mulholland JA, Tolbert PE. Ambient air pollution and birth weight in full-term infants in Atlanta, 1994–2004. Environ Health Perspect [Internet]. 2011. May [cited 2020 Aug 18];119(5):731–7. Available from: https://pubmed.ncbi.nlm.nih.gov/21156397/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ha S, Hu H, Roussos-Ross D, Haidong K, Roth J, Xu X. The effects of air pollution on adverse birth outcomes. Environ Res [Internet]. 2014. [cited 2020 Aug 18];134:198–204. Available from: https://pubmed.ncbi.nlm.nih.gov/25173052/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleisch AF, Rifas-Shiman SL, Koutrakis P, Schwartz JD, Kloog I, Melly S, et al. Prenatal exposure to traffic pollution: Associations with reduced fetal growth and rapid infant weight gain. Epidemiology [Internet]. 2015. January 1 [cited 2020 Aug 18];26(1):43–50. Available from: https://pubmed.ncbi.nlm.nih.gov/25437317/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rich DQ, Liu K, Zhang J, Thurston SW, Stevens TP, Pan Y, et al. Differences in birth weight associated with the 2008 Beijing olympics air pollution reduction: Results from a natural experiment. Environ Health Perspect [Internet]. 2015. September 1 [cited 2020 Aug 18];123(9):880–7. Available from: https://pubmed.ncbi.nlm.nih.gov/25919693/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schembari A, De Hoogh K, Pedersen M, Dadvand P, Martinez D, Hoek G, et al. Ambient air pollution and newborn size and adiposity at birth: Differences by maternal ethnicity (the born in bradford study cohort). Environ Health Perspect [Internet]. 2015. November 1 [cited 2020 Aug 18];123(11):1208–15. Available from: https://pubmed.ncbi.nlm.nih.gov/25978617/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu H, Jiang B, Geng X, Zhu P, Liu Z, Cui L, et al. Exposure to fine particulate matter during pregnancy and risk of term low birth weight in Jinan, China, 2014–2016. Int J Hyg Environ Health [Internet]. 2018. March 1 [cited 2020 Aug 18];221(2):183–90. Available from: https://pubmed.ncbi.nlm.nih.gov/29097084/ [DOI] [PubMed] [Google Scholar]
- 60.Chen G, Guo Y, Abramson MJ, Williams G, Li S. Exposure to low concentrations of air pollutants and adverse birth outcomes in Brisbane, Australia, 2003–2013. Sci Total Environ [Internet]. 2018. May 1 [cited 2020 Aug 18];622–623:721–6. Available from: https://pubmed.ncbi.nlm.nih.gov/29223898/ [DOI] [PubMed] [Google Scholar]
- 61.Rhee J, Patricia Fabian M, de Cuba SE, Coleman S, Sandel M, Lane KJ, et al. Effects of maternal homelessness, supplemental nutrition programs, and prenatal PM2.5 on birthweight. Int J Environ Res Public Health [Internet]. 2019. November 1 [cited 2020 Aug 18];16(21). Available from: https://pubmed.ncbi.nlm.nih.gov/31661898/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson A, Chiu YHM, Hsu HHL, Wright RO, Wright RJ, Coull BA. Potential for Bias When Estimating Critical Windows for Air Pollution in Children’s Health. Am J Epidemiol [Internet]. 2017. December 1 [cited 2020 Aug 19];186(11):1281–9. Available from: https://pubmed.ncbi.nlm.nih.gov/29206986/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Robledo CA, Mendola P, Yeung E, Männistö T, Sundaram R, Liu D, et al. Preconception and early pregnancy air pollution exposures and risk of gestational diabetes mellitus. Environ Res. 2015; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lakshmanan A, Chiu YHM, Coull BA, Just AC, Maxwell SL, Schwartz J, et al. Associations between prenatal traffic-related air pollution exposure and birth weight: Modification by sex and maternal pre-pregnancy body mass index. Environ Res. 2015. February 1;137:268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yue X, Qian Y, Gim B, Lee I. Acyl-CoA-binding domain-containing 3 (ACBD3; PAP7; GCP60): A multi-functional membrane domain organizer [Internet]. Vol. 20, International Journal of Molecular Sciences. MDPI AG; 2019. [cited 2020 Sep 30]. Available from: https://pubmed.ncbi.nlm.nih.gov/31022988/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Melchionda L, Haack TB, Hardy S, Abbink TEM, Fernandez-Vizarra E, Lamantea E, et al. Mutations in APOPT1, encoding a mitochondrial protein, cause cavitating leukoencephalopathy with cytochrome c oxidase deficiency. Am J Hum Genet [Internet]. 2014. [cited 2020 Sep 30];95(3):315–25. Available from: https://pubmed.ncbi.nlm.nih.gov/25175347/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coppe A, Agostini C, Marino IAM, Zane L, Bargelloni L, Bortoluzzi S, et al. Genome evolution in the cold: Antarctic icefish muscle transcriptome reveals selective duplications increasing mitochondrial function. Genome Biol Evol [Internet]. 2013. [cited 2020 Sep 30];5(1):45–60. Available from: https://pubmed.ncbi.nlm.nih.gov/23196969/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vu MT, Zhai P, Lee J, Guerra C, Liu S, Gustin MC, et al. The DNLZ/HEP zinc-binding subdomain is critical for regulation of the mitochondrial chaperone HSPA9. Protein Sci [Internet]. 2012. February [cited 2020 Sep 30];21(2):258–67. Available from: https://pubmed.ncbi.nlm.nih.gov/22162012/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kraja AT, Liu C, Fetterman JL, Graff M, Have CT, Gu C, et al. Associations of Mitochondrial and Nuclear Mitochondrial Variants and Genes with Seven Metabolic Traits. Am J Hum Genet [Internet]. 2019. January 3 [cited 2020 Sep 30];104(1):112–38. Available from: https://pubmed.ncbi.nlm.nih.gov/30595373/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kotrys AV., Szczesny RJ. Mitochondrial Gene Expression and Beyond-Novel Aspects of Cellular Physiology [Internet]. Vol. 9, Cells. NLM (Medline); 2019. [cited 2020 Sep 30]. Available from: https://pubmed.ncbi.nlm.nih.gov/31861673/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Noli L, Khorsandi SE, Pyle A, Giritharan G, Fogarty N, Capalbo A, et al. Effects of thyroid hormone on mitochondria and metabolism of human preimplantation embryos. Stem Cells [Internet]. 2020. March 1 [cited 2020 Sep 30];38(3):369–81. Available from: https://pubmed.ncbi.nlm.nih.gov/31778245/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maldonado M, Padavannil A, Zhou L, Guo F, Letts JA. Atomic structure of a mitochondrial complex i intermediate from vascular plants. Elife [Internet]. 2020. August 1 [cited 2020 Sep 30];9:1–36. Available from: https://pubmed.ncbi.nlm.nih.gov/32840211/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Long H, Huang K. Transport of Ciliary Membrane Proteins [Internet]. Vol. 7, Frontiers in Cell and Developmental Biology. Frontiers Media S.A; 2020. [cited 2020 Sep 30]. Available from: https://pubmed.ncbi.nlm.nih.gov/31998723/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Davegårdh C, Hall Wedin E, Broholm C, Henriksen TI, Pedersen M, Pedersen BK, et al. Sex influences DNA methylation and gene expression in human skeletal muscle myoblasts and myotubes. Stem Cell Res Ther [Internet]. 2019. January 15 [cited 2020 Oct 1];10(1). Available from: https://pubmed.ncbi.nlm.nih.gov/30646953/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weiner CP, Mason CW, Dong Y, Buhimschi IA, Swaan PW, Buhimschi CS. Human effector/initiator gene sets that regulate myometrial contractility during term and preterm labor. Am J Obstet Gynecol [Internet]. 2010. May;202(5):474.e1–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20452493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olenick MA, Holzbaur ELF. Dynein activators and adaptors at a glance. J Cell Sci [Internet]. 2019;132(6). Available from: http://www.ncbi.nlm.nih.gov/pubmed/30877148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Derivery E, Gautreau A. Evolutionary conservation of the WASH complex, an actin polymerization machine involved in endosomal fission. Commun Integr Biol [Internet]. 2010. May;3(3):227–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20714399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol [Internet]. 2018;16(6):341–54. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29556036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tsuchiya H, Endo A, Saeki Y. Multi-Step Ubiquitin Decoding Mechanism for Proteasomal Degradation. Pharmaceuticals (Basel) [Internet]. 2020. June 23;13(6). Available from: http://www.ncbi.nlm.nih.gov/pubmed/32585960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kroeker AL, Ezzati P, Halayko AJ, Coombs KM. Response of primary human airway epithelial cells to influenza infection: a quantitative proteomic study. J Proteome Res [Internet]. 2012. August 3;11(8):4132–46. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22694362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Carnes SK, Zhou J, Aiken C. HIV-1 Engages a Dynein-Dynactin-BICD2 Complex for Infection and Transport to the Nucleus. J Virol [Internet]. 2018;92(20). Available from: http://www.ncbi.nlm.nih.gov/pubmed/30068656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu Y, O’Boyle K, Auer J, Raju S, You F, Wang P, et al. Multiple UBXN family members inhibit retrovirus and lentivirus production and canonical NFκB signaling by stabilizing IκBα. PLoS Pathog [Internet]. 2017;13(2):e1006187. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28152074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Panni T, Mehta AJ, Schwartz JD, Baccarelli AA, Just AC, Wolf K, et al. Genome-Wide Analysis of DNA Methylation and Fine Particulate Matter Air Pollution in Three Study Populations: KORA F3, KORA F4, and the Normative Aging Study. Environ Health Perspect [Internet]. 2016;124(7):983–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26731791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo T, Yin RX, Bin Y, Nie RJ, Chen X, Pan SL. Association of the SPT2 chromatin protein domain containing 1 gene rs17579600 polymorphism and serum lipid traits. Int J Clin Exp Pathol [Internet]. 2015. [cited 2020 Oct 1];8(10):12995–3010. Available from: www.ijcep.com/ [PMC free article] [PubMed] [Google Scholar]
- 85.Asselbergs FW, Guo Y, Van Iperen EPA, Sivapalaratnam S, Tragante V, Lanktree MB, et al. Large-scale gene-centric meta-analysis across 32 studies identifies multiple lipid loci. Am J Hum Genet [Internet]. 2012. November 2 [cited 2020 Oct 1];91(5):823–38. Available from: /pmc/articles/PMC3487124/?report=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guo T, Yin RX, Chen X, Bin Y, Nie RJ, Li H. Sex-specific association of the SPTY2D1 rs7934205 polymorphism and serum lipid levels. Int J Clin Exp Pathol [Internet]. 2015. [cited 2020 Oct 1];8(1):665–81. Available from: https://pubmed.ncbi.nlm.nih.gov/25755761/ [PMC free article] [PubMed] [Google Scholar]
- 87.Wylie BJ, Matechi E, Kishashu Y, Fawzi W, Premji Z, Coull BA, et al. Placental pathology associated with household air pollution in a cohort of pregnant women from Dar es Salaam, Tanzania. Environ Health Perspect [Internet]. 2017. January 1 [cited 2020 Aug 21];125(1):134–40. Available from: https://pubmed.ncbi.nlm.nih.gov/27286442/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu Y, Wang L, Wang F, Li C. Effect of fine particulate matter (PM2.5) on rat placenta pathology and perinatal outcomes. Med Sci Monit [Internet]. 2016. September 15 [cited 2020 Aug 21];22:3274–80. Available from: https://pubmed.ncbi.nlm.nih.gov/27629830/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Saenen ND, Vrijens K, Janssen BG, Madhloum N, Peusens M, Gyselaers W, et al. Placental Nitrosative Stress and Exposure to Ambient Air Pollution During Gestation: A Population Study. Am J Epidemiol [Internet]. 2016. [cited 2020 Mar 22];184(6):442–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27601048 [DOI] [PubMed] [Google Scholar]
- 90.Martens DS, Cox B, Janssen BG, Clemente DBP, Gasparrini A, Vanpoucke C, et al. Prenatal air pollution and newborns’ predisposition to accelerated biological aging. JAMA Pediatr [Internet]. 2017. December 1 [cited 2020 Aug 21];171(12):1160–7. Available from: https://pubmed.ncbi.nlm.nih.gov/29049509/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRONAGE birth cohort study. Epigenetics [Internet]. 2015. January 1 [cited 2020 Aug 21];10(6):536–44. Available from: https://pubmed.ncbi.nlm.nih.gov/25996590/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Maghbooli Z, Hossein-nezhad A, Adabi E, Asadollah-pour E, Sadeghi M, Mohammad-nabi S, et al. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS One [Internet]. 2018. July 1 [cited 2020 Aug 21];13(7). Available from: https://pubmed.ncbi.nlm.nih.gov/29979694/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liew Z, Olsen J, Cui X, Ritz B, Arah OA. Bias from conditioning on live birth in pregnancy cohorts: An illustration based on neurodevelopment in children after prenatal exposure to organic pollutants. Int J Epidemiol [Internet]. 2015. February 1 [cited 2021 Mar 9];44(1):345–54. Available from: https://pubmed-ncbi-nlm-nih-gov.ezproxy.cul.columbia.edu/25604449/ [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.