

Abstract
Previous studies have suggested that the loss of the translation initiation factor eIF4G1 homolog NAT1 induces excessive self-renewability of naïve pluripotent stem cells (PSCs). Yet the role of NAT1 in the self-renewal and differentiation of primed PSCs, is still unclear. Here we generated conditional knockout of NAT1 in primed PSCs and used the cells for the functional analyses of NAT1. Our results showed that NAT1 is required for the self-renewal and neural differentiation of primed PSCs. In contrast, NAT1 deficiency in naïve pluripotency attenuated the differentiation to all cell types. We also found that NAT1 is involved in efficient protein expression of an RNA uridyltransferase TUT7. TUT7 is involved in the neural differentiation of primed PSCs via the regulation of human endogenous retrovirus accumulation. These data demonstrated the essential roles of NAT1 and TUT7 in the precise transition of stem cell fate.
INTRODUCTION
A key question in developmental and stem cell biology is how pluripotent stem cells (PSCs) can give rise to vastly differently types and cells and tissues through differential gene expressions. The central dogma has DNA transcribed into messenger RNA (mRNA) and mature mRNA translated into protein, but each step involves a myriad of regulatory processes that modulate the protein output in space and time and ultimately give rise to higher-order biological properties. Recent studies have improved our appreciation of the extent and prevalence to which protein translation regulation contributes to the complexity of gene regulatory circuits (Taylor et al., 2019). Specifically, translation is regulated at the stage of initiation, elongation, termination, and ribosome recycling. Like transcription, the main point of regulation in translation is generally thought to be the initiation step.
The generally accepted model of translation initiation is that a translational initiation complex binds to the cap structure at the 5′ end of mRNA and scans toward the 3′ direction until it finds a methionine initiation codon (Ingolia et al., 2009; Kozak, 1989). In the translation initiation complex, a scaffold protein, designated eukaryotic translation initiation factor (eIF) 4G1, that recruits other initiation proteins, including eIF4E, eIF4A, eIF3s, and a poly A-binding protein (PABP) (Jackson et al., 2010).
Novel APOBEC-1 target 1 (NAT1, also known as death-associated protein 5 (DAP5), p97 and eIF4G2) is an evolutionally conserved (Takahashi et al., 2005), ubiquitously and abundantly expressed protein across all tissues (Thul et al., 2017; Uhlen et al., 2015; Uhlen et al., 2017). Curiously, it shares homology with the C-terminal two thirds of eIF4G1, but lacks the domains that interact with eIF4E and PABP (Imataka et al., 1997; Levy-Strumpf et al., 1997; Shaughnessy et al., 1997; Yamanaka et al., 1997). Based on the structure of NAT1 protein, it was speculated that NAT1 regulates non-canonical translation initiation via cap-independent mechanisms. Indeed, previous studies showed that NAT1 promotes the translation of specific genes with an internal ribosome entry site (IRES) in their 5’UTRs in a cap-independent fashion (Henis-Korenblit et al., 2002; Henis-Korenblit et al., 2000; Liberman et al., 2015; Marash et al., 2008; Weingarten-Gabbay et al., 2014). In addition, a recent study suggested that NAT1 promotes the translation of circular RNAs that lack m7G-cap and poly A tail (Yang et al., 2017). Furthermore, a recent study suggested that NAT1 also orchestrates non-canonical eIF4E-indepedent cap-dependent translation initiation via an alternative cap-binding protein, eIF3D (de la Parra et al., 2018), which further expanded the repertoire of mRNA targets for NAT1-mediated translation regulation. These data suggest that NAT1 plays important roles in translation regulation.
The global protein synthesis rate and translation of specific proteins in the signaling network are heavily regulated during development and differentiation (Blanco et al., 2016). Given NAT1’s proposed roles in translation regulation, the demonstrated importance of NAT1 in organism development is expected. For example, the loss of NAT1 homolog in drosophila causes severe embryonic lethality and abnormal germband extension (Yoshikane et al., 2007). Mouse genetic analysis revealed that NAT1 is essential for early embryogenesis (Yamanaka et al., 2000). Specifically, histological approaches revealed that NAT1-null mouse embryos lacked the organization of the three primary germ layers before the gastrulation stage.
During mammalian embryonic development, naïve pluripotent cells undergo multiple cycles of cleavage division and upon uterine implantation commence a primed pluripotency status before further differentiation into all three germ layers (Nichols and Smith, 2009). A recent study showed that targeted deletion of NAT1 in mouse embryonic stem cells (ESCs) blocked their differentiation, as precise translation of mitogen-activated protein kinase kinase kinase 3 (MAP3K3) was disrupted (Sugiyama et al., 2017). Importantly, these mouse ESCs have morphology, epigenetic status, and cytokine culture requirements characteristic of naïve PSCs, distinguishing them from human PSCs, which are of primed pluripotency and include human ESCs and induced PSCs (iPSCs). While the mechanism of NAT1’s function in naive PSC differentiation potential has been elucidated, its roles in primed PSC differentiation are still unknown.
In the current study, we generated conditional knockout or knockdown of NAT1 gene in human iPSCs and mouse epiblast-derived stem cells (EpiSCs) and found that NAT1 is required for the self-renewal and neural differentiation of primed PSCs. Using a combination of genetic, cellular, biochemical, and system approaches, we uncovered that NAT1 positively regulates the translation of the TUT7, which normally degrades human endogenous retrovirus type-H (HERV-H) RNAs and creates an environment permissive for the neural differentiation of primed PSCs. The NAT1-TUT7-HERV-H pathway demonstrates one important mechanism of translation initiation and RNA modification in organism development, and represents an unexplored gene regulation network of translational, post-transcriptional, and non-coding RNA control.
RESULTS
NAT1 depletion induces differentiation of human iPSCs to endoderm and mesoderm lineages, but not to neural lineage.
To understand the role of NAT1 in human PSCs, we targeted the translation start site in exon 2 by using CRISPR/Cas9 DNA digestion and a knocked-in neomycin-resistance gene (Neo) (Figures S1A–S1C). Using the heterozygous mutant human iPSC line (NAT1Neo/+), we performed a second round targeting the other NAT1 allele by knocking-in a puromycin-resistance gene (Puro) (Figure S1A). Polymerase chain reaction (PCR) screening to detect homologous recombinants indicated that all clones had Puro but not Neo even though the targeted alleles no longer had the guide RNA (gRNA) recognition site for CRISPR excision (Figures S1B and S1C). Even with neomycin and puromycin double selection in the second-round targeting, we failed to obtain homozygous targeted clones (data not shown). These results show a strong bias against the null-mutation of NAT1 and suggest that NAT1 is required for the growth and/or survival of human iPSCs. We then took a conditional knockout (cKO) approach. We introduced a doxycycline (Dox)-inducible NAT1 transgene (Tg) into NAT1Neo/+ human iPSCs and performed second round targeting in these cells (NAT1Neo/+ + Tg) in the presence of Dox (Figure S1D). Southern blotting showed that both NAT1 alleles were successfully deleted with the support of exogenous NAT1 expression (Figure S1E). We designated these cell lines NAT1 cKO human iPSCs. Western blotting confirmed the precise regulation of NAT1 transgenes by Dox (Figure S1F). Time course analysis and quantitative reverse transcription PCR (qRT-PCR) showed that the expression of NAT1 in cKO human iPSCs dropped to less than 0.5% on day 3 after Dox removal, and Dox addition quickly recovered NAT1 expression to the endogenous level within one day (Figure S1G). In sum, these data demonstrated that the NAT1 cKO human iPSC line is suitable for analyzing the loss of NAT1 function.
Next, we tested if NAT1 expression is required for human iPSCs by removing Dox in feeder-free culture condition with media containing basic fibroblast growth factor (FGF) and Activin A, which is suitable for the maintenance of primed PSCs (hereafter F/A condition). On day 0, NAT1 cKO human iPSCs expressed a pluripotency marker, OCT3/4, as well as NAT1. After 6 days of Dox removal, NAT1 cKO human iPSCs formed more compact colonies than those with Dox (Figure 1A). After passaging and culturing for another 6 days (day 12 after Dox removal), we observed massive cell death, leaving a small number of surviving cells that formed flat and spiky colonies indicative of differentiation (Figure 1A). Alkaline phosphatase (AP) activity also suggested the loss of pluripotency by NAT1 depletion on day 12 (Figure 1B). This effect was accompanied by a decreased expression of pluripotency genes such as OCT3/4 (Figures 1A and 1C). Also, we detected a significant upregulation of SOX17 (endoderm marker) and PDGFRA (mesoderm marker), but not NES (neuroectoderm marker) on days 6 and 12 after Dox removal (Figure 1C). We also confirmed that NAT1-deficient human iPSCs could not self-renew even on mitomycin C (MMC)-inactivated mouse embryonic fibroblast (MEF) feeder layer, which was used in a previous study (Figure 1D) (Yoffe et al., 2016). Global gene expression analyses revealed more than 5,000 genes were differentially expressed between the presence and absence of Dox (Figure 1E). Global gene expression analyses also showed that many mesoderm and endoderm marker genes, but not pluripotency or neuroectoderm marker genes, were upregulated after 6 days of NAT1 depletion (Figure 1F).
Figure 1 |. NAT1 is required for the self-renewal of primed human iPSCs.
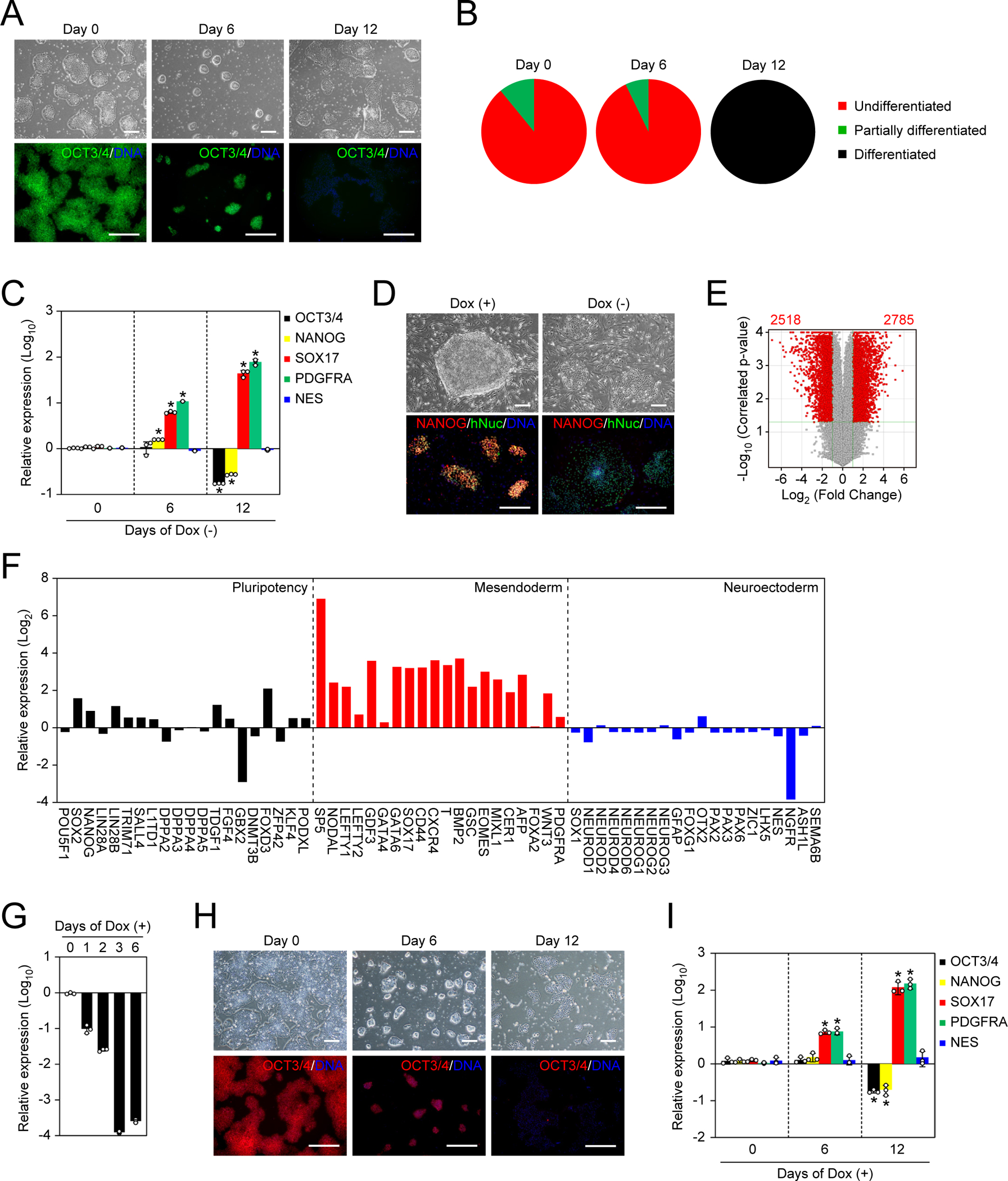
A. Representative images of NAT1 KO human iPSCs in F/A condition. Bars indicate 100 μm. See also Figure S1.
B. The loss of pluripotency by NAT1 KO was quantified by alkaline phosphatase (AP) staining.
C. Relative expression of pluripotency and differentiation markers in the time course of NAT1 KO iPSCs in F/A condition analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. day 0 by unpaired t-test. n=3.
D. Representative images of NAT1 cKO iPSCs with (+) or without (−) Dox on MEF feeders. Bars indicate 100 μm.
E. Volcano plot showing global gene expression of NAT1 cKO iPSCs maintained in F/A condition with (+) or without (−) Dox for 6 days. Red dots indicate differentially expressed genes with statistical significance (FC>2, FDR<0.05). Pearson correlation coefficient was 0.9787. n=3.
F. Relative expression of pluripotency, mesendodermal and neuroectodermal genes in NAT1 cKO iPSCs maintained in F/A condition with no Dox for 6 days compared with the same iPSCs with Dox analyzed by microarray. n=3.
G. Relative expression of NAT1 on days 0–6 of Dox addition in F/A condition by qRT-PCR. Values are normalized by GAPDH and compared with 1B4 human iPSCs. n=3.
H. Representative images of human iPSCs in the time course of NAT1 KD. Bars indicate 100 μm.
I. Relative expression of pluripotency and differentiation markers in the cells shown in Fig. 1G by qRT-PCR. Values are normalized by GAPDH and compared with 1B4 human iPSCs. *P<0.05 vs. day 0 by unpaired t-test. n=3.
To confirm whether such drastic phenotypes were caused by the loss of NAT1 expression, we utilized the CRISPR interference (CRISPRi) system, which allows efficient and specific gene silencing (Gilbert et al., 2014). We introduced a gRNA for NAT1 into a human iPSC line carrying a Dox-inducible KRAB-dCas9 expression cassette in the Adeno-Associated Virus Integration Site 1 (AAVS1) locus (Mandegar et al., 2016). The addition of Dox induced CRISPRi-mediated knockdown (KD) of NAT1 with 99.999% efficiency on day 3 (Figure 1G) and induced spontaneous differentiation judged by morphology and a decrease in OCT3/4 expression on day 12 after Dox addition (Figure 1H). We confirmed the decrease in OCT3/4 and NANOG, as well as the increase of SOX17 and PDGFRA (Figure 1I). In contrast, the expression of NES did not change. Taken together, these data demonstrated that the loss of NAT1 disrupts the self-renewal of human iPSCs with global gene expression changes including the upregulation of mesendodermal genes.
NAT1 is required for differentiation of human iPSCs to neural progenitor cells
Next, we tested the effects of NAT1 deletion on the differentiation potential of human iPSCs using the embryoid body (EB) formation. Regardless of the NAT1 expression level, human iPSCs could differentiate into alpha fetoprotein (AFP) positive (+) endoderm and smooth muscle actin (SMA) (+) mesoderm (Figure 2A). However, NAT1-deficient human iPSCs did not differentiate into βIII-TUBULIN (+) neural cells (Figure 2A). NAT1 deletion did not affect the suppression of pluripotency markers and the induction of mesendodermal markers (Figures 2A and 2B). In sharp contrast, we detected much less induction of neural markers such as MAP2 and NEUROD1 in NAT1-deficient EBs (Figure 2B).
Figure 2 |. NAT1 is required for the transition from primed pluripotency to neural progenitor fate.
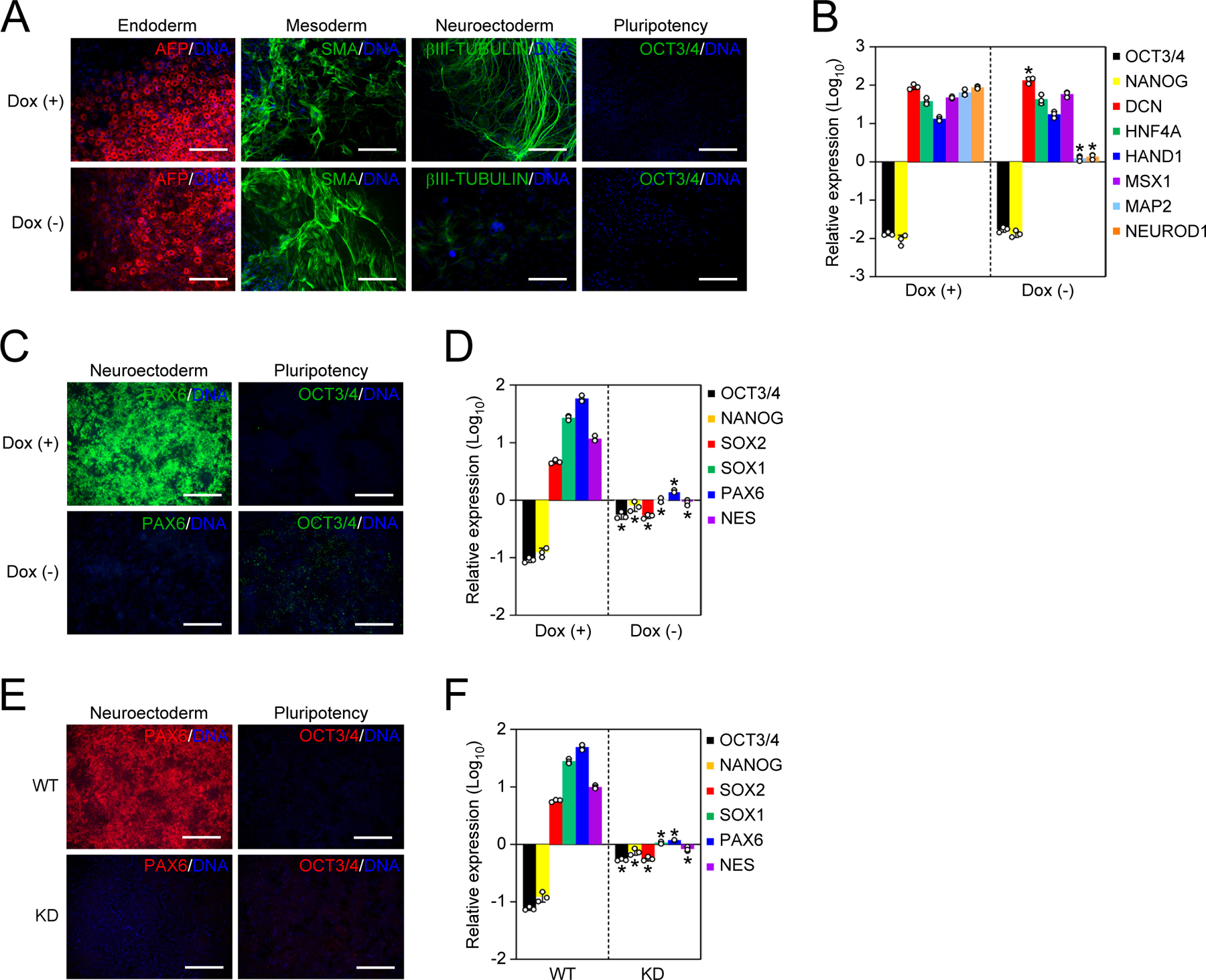
A. Immunocytochemistry of differentiated NAT1 WT and KO iPSCs by EB formation. Bars indicate 100 μm.
B. Relative expression of pluripotency and differentiation markers in the cells shown in Fig. 2A by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
C. Immunocytochemistry of differentiated NAT1 cKO iPSCs in the presence or absence of Dox by dSMADi. Bars indicate 100 μm.
D. Relative expression of pluripotency and neural genes in the cells shown in Fig. 2C analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
E. Immunocytochemistry of differentiated NAT1 WT and KD iPSCs by dSMADi. Bars indicate 100 μm.
F. Relative expression of pluripotency and neural genes in the cells shown in Fig. 2E analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 1B4 human iPSCs. *P<0.05 vs. WT by unpaired t-test. n=3.
To examine the neural differentiation potential of NAT1-depleted human iPSCs more specifically, we utilized directed neural differentiation by the dual SMAD inhibition (dSMADi) method (Chambers et al., 2009; Doi et al., 2014). With this robust method, NAT1 cKO iPSCs efficiently differentiated into PAX6 (+) neural progenitor cells (NPCs) in the presence of Dox (Figure 2C). However, without Dox, few PAX6 (+) cells emerged, although OCT3/4 expression disappeared (Figure 2C). Other NPC markers such as SOX1 and NES were also suppressed in the absence of NAT1 (Figure 2D). We also confirmed that CRISPRi-mediated NAT1 KD drastically decreased the differentiation of iPSCs into PAX6 (+) NPCs by the dSMADi (Figures 2E and 2F). These data showed that NAT1 is required for the differentiation of human iPSCs into NPCs
Increased endogenous retroviruses expression at the post-transcriptional level is responsible for defective neural differentiation of NAT1-depleted human iPSCs
To obtain clues as to how NAT1 is involved in the self-renewal and neural differentiation of human iPSCs, we looked into the results of the microarray analyses more carefully. We noticed that NAT1 depletion resulted in an increased expression of multiple genes that were overlapped with human endogenous retrovirus type H (HERV-H) (Loewer et al., 2010; Lu et al., 2014; Ohnuki et al., 2014; Santoni et al., 2012; Wang et al., 2014; Zhao et al., 2007) (Figure 3A). We confirmed increased expression of HERV-Hs on day 6 and 12 after Dox removal in NAT1 cKO human iPSCs (Figure 3B). Additionally, we detected a slight but significant increase of HERV-K, but not of HERV-W or LINE1 (Figure 3B). Since NAT1 depletion induced differentiation and cell death of human iPSCs, HERV-H level on d12 became lower than that on day 6 (Figures 1A–1C and 3B). Chromatin immunoprecipitation (ChIP) assay to examine the occupancy of RNA polymerase II (PolII) at the LTR promoter regions revealed that no significant increase in PolII occupancy, which would have explained the increased HERV-H RNAs in NAT1-depleted human iPSCs (Figure 3C). Quantification of nascently transcribed and total RNAs revealed that the increased expression of HERV-H in NAT1-depleted iPSCs was not attributable to an increase of de novo synthesis (Figure 3D). These data suggest that HERV-H RNAs are increased by NAT1 depletion at the post-transcriptional level.
Figure 3 |. Loss of NAT1 enhances HERV-Hs to inhibit neural differentiation.
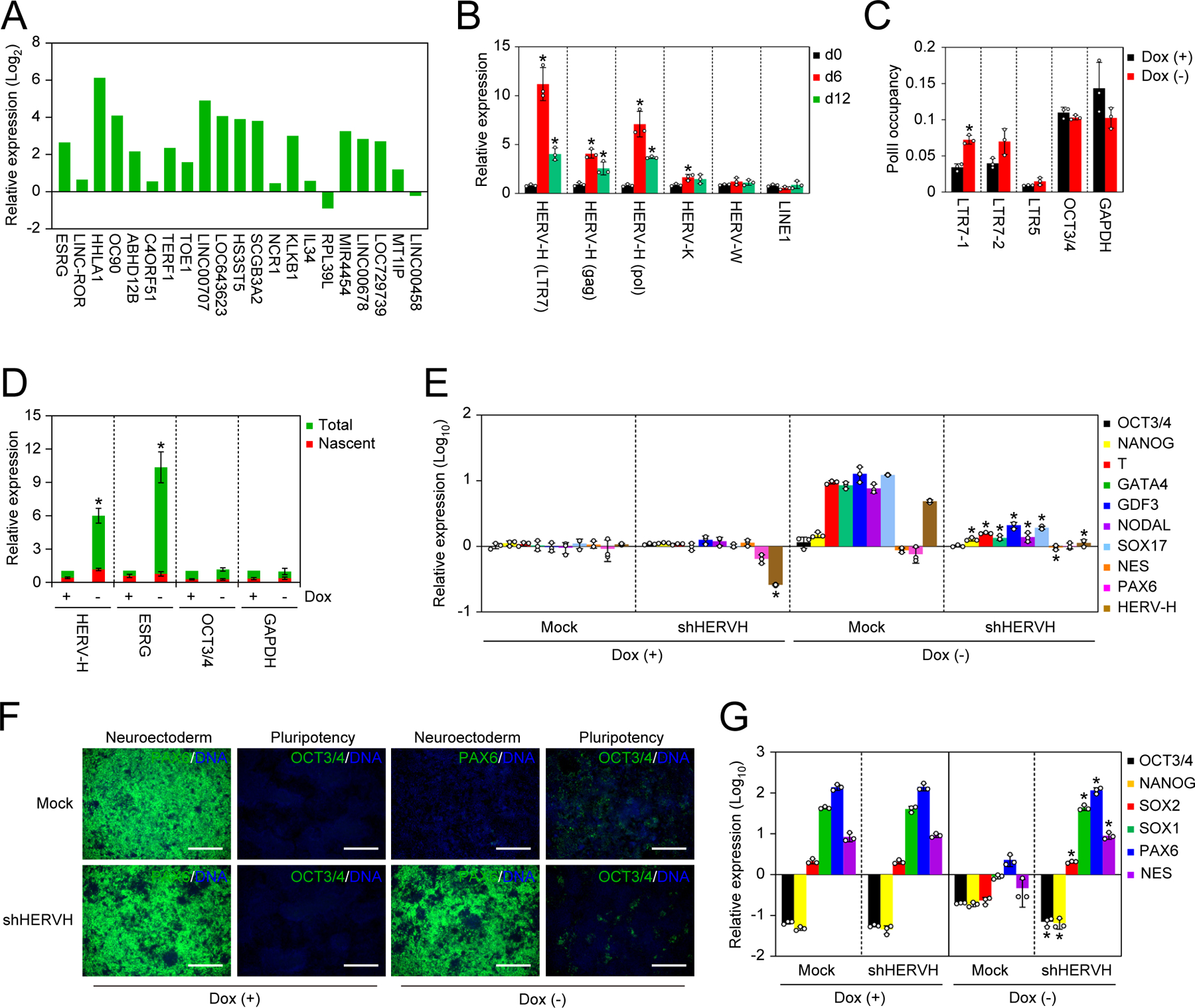
A. Relative expression of HERV-H-related transcripts in NAT1 cKO human iPSCs maintained in F/A condition with no Dox compared to those in same cell line with Dox analyzed by microarray. n=3.
B. Relative expression of transposable elements in NAT1 cKO human iPSCs maintained in F/A condition on days 0, 6 and 12 of Dox removal analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. day 0 by unpaired t-test. n=3.
C. Fold enrichment of PolII occupancy on the promoter regions of HERVs, OCT3/4 and GAPDH in NAT1 cKO human iPSCs maintained in F/A condition with or without Dox for 6 days analyzed by qPCR. n=3. Values are normalized by input control. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
D. Relative amounts of total and nascent transcribed pan HERV-Hs, ESRG, OCT3/4 and GAPDH RNAs in NAT1 cKO human iPSCs maintained in F/A condition with or without Dox for 6 days analyzed by qPCR. Values are normalized by spike RNA control and compared with the total of Dox (+). *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
E. Relative expression of pluripotency and differentiation markers in NAT1 cKO human iPSCs transfected with Mock or HERV-H shRNA (shHERVH) maintained in F/A condition with (+) or without (−) Dox for 6 days. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. Mock by unpaired t-test. n=3. See also Figure S2.
F. Immunocytochemistry of differentiated NAT1 cKO human iPSCs transfected with Mock or HERV-H shRNA (shHERVH) in the presence (+) or absence (−) of Dox by dSMADi. Bars indicate 100 μm. See also Figure S2.
G. Relative expression of pluripotency and neural genes in the cells shown in Fig. 3F analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. Mock by unpaired t-test. n=3. See also Figure S2.
We previously reported that aberrant HERV-H expression is responsible for the neural differentiation defective phenotype in human iPSCs (Koyanagi-Aoi et al., 2013; Ohnuki et al., 2014). Therefore, we hypothesized that the increased HERV-H expression contributed to the inability of NAT1-deficient human iPSCs to differentiate into neural lineage. To test this hypothesis, we suppressed the expression of HERV-H in NAT1-depleted human iPSCs by introducing a short hairpin RNA (shRNA) against a conserved LTR7 sequence (shHERVH). The HERV-H expression was decreased to ~30% of Mock-transfected control (Figure 3E). These HERV-H KD iPSCs remained undifferentiated on day 6 after the removal of Dox (NAT1) in F/A condition (Figures 3E and S2A) although they failed to self-renew by day 12 (Figure S2B). These data suggest that the upregulation of HERV-H is not responsible for the loss of self-renewal in NAT1 deficiency. As we predicted, HERV-H suppression enabled NAT1-deficient human iPSCs to differentiate into neural lineage by EB formation (Figures S2C and S2D) and dSMADi (Figures 3F and 3G). These data showed that the induction of HERV-Hs was specifically involved in the neural differentiation defective phenotype of NAT1-depleted human iPSCs.
NAT1 is required for the self-renewal and neural differentiation potential of mouse EpiSCs
To address if the role of NAT1 in primed pluripotency is conserved in different species, we generated a NAT1 cKO mouse EpiSC line, which possesses similar characteristics to human iPSCs, such as the dependency on FGF and Activin A (F/A) (Figures S3A–S3D). As with human iPSCs, mouse EpiSCs spontaneously differentiated upon the Dox depletion, as judged by OCT3/4 immunostaining (Figure 4A) and morphology (Figure 4B). Gene expression analysis revealed that some core pluripotency factors such as OCT3/4 and NANOG were downregulated in NAT1-depleted mouse EpiSCs in F/A condition, suggesting differentiation (Figure 4C). As is the case of human iPSCs, NAT1-depleted mouse EpiSCs could form EBs and differentiate into endoderm and mesoderm. However, we detected much less induction of neuroectoderm markers (Figures 4D and 4E). These data demonstrated that NAT1 is required for the self-renewal and neural differentiation of both human iPSCs and mouse EpiSCs.
Figure 4 |. NAT1 is required for the self-renewal and neural differentiation potential of mouse EpiSCs.
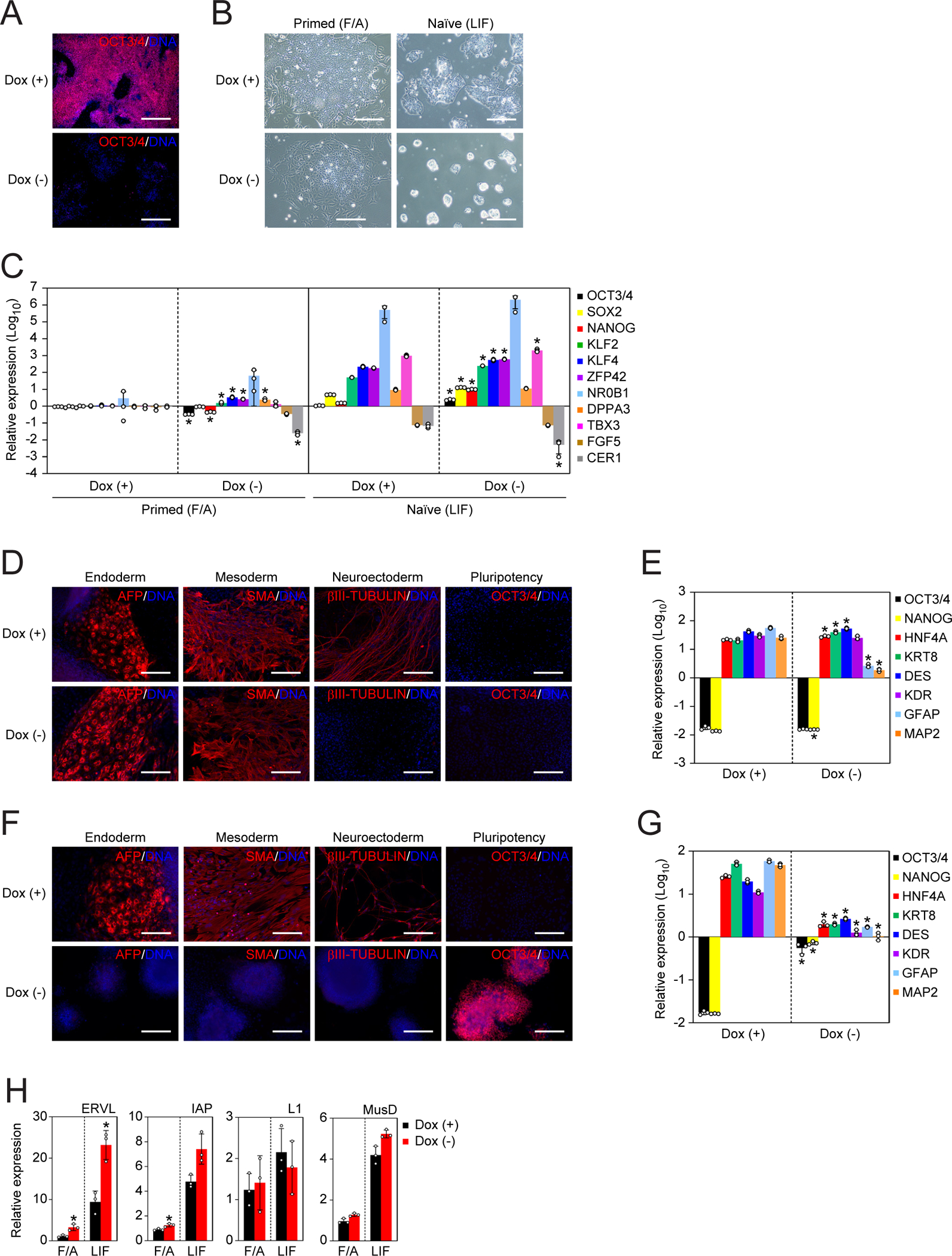
A. Immunocytochemistry of NAT1 cKO mouse EpiSCs maintained with (+) or without (−) Dox for 6 days. Bars indicate 100 μm. See also Figure S3.
B. Representative phase contrast images of NAT1 cKO primed EpiSCs maintained in F/A or naïve-converted cells in LIF-containing media with (+) or without (−) Dox. Bars indicate 100 μm.
C. Relative expression of naïve and primed pluripotency markers in the cells shown in Fig. 4B analyzed by qRT-PCR. Values are normalized by Actb and compared with X-GFP mouse EpiSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
D. Immunocytochemistry of differentiated cells derived from primed NAT1 cKO mouse EpiSCs in the presence (+) or absence (−) of Dox by EB formation. Bars indicate 100 μm.
E. Relative expression of pluripotency and differentiation markers in the cells shown in Fig. 4D analyzed by qRT-PCR. Values are normalized by Actb and compared with X-GFP mouse EpiSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
F. Immunocytochemistry of differentiated cells derived from naïve NAT1 cKO mouse EpiSCs in the presence (+) or absence (−) of Dox by EB formation. Bars indicate 100 μm.
G. Relative expression of pluripotency and differentiation markers in the cells shown in Fig. 4F analyzed by qRT-PCR. Values are normalized by Actb and compared with RF8 mouse ESCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
H. Relative expression of transposable elements in NAT1 cKO mouse primed EpiSCs (F/A) or naïve-converted cells (LIF) on day 6 of Dox removal compared to those in EpiSCs analyzed by qRT-PCR. Values are normalized by Actb and compared with X-GFP mouse EpiSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
Our previous data regarding NAT1 KO phenotypes came from mouse ESCs, which represent a more primitive state of pluripotency, so-called naïve pluripotency, than primed pluripotency, which is the phenotype of human iPSCs and mouse EpiSCs (Nichols and Smith, 2009; Sugiyama et al., 2017; Yamanaka et al., 2000). The loss of NAT1 in mouse ESCs significantly attenuated the differentiation potential for all three lineages (Yamanaka et al., 2000). To confirm the role of NAT1 in mouse naïve pluripotency, in the present study, we converted NAT1 cKO mouse EpiSCs to the naïve state by adding cytokines and chemical compounds (Kime et al., 2016). Naïve-converted mouse EpiSCs could expand in culture condition containing leukemia inhibitory factor (LIF), but not F/A, regardless of NAT1 expression, although NAT1-depeted cells formed more compact colonies than the same cell line with Dox (Figure 4B). The loss of NAT1 significantly increased core and naïve pluripotency factors (Figure 4C). In contrast to the results of primed cells, the loss of NAT1 prevented the differentiation of naïve cells to all thee lineages in EB (Figures 4F and 4G). These data confirmed our previous finding that NAT1 is required for the differentiation of naïve mouse PSCs.
We then examined the effect of NAT1 depletion on the expression of repetitive elements in mouse naïve and primed pluripotency. We found that the expression of ERVL in both conditions and IAP in F/A condition were increased in the absence of NAT1, but the expression of other transposable elements such as L1 and MusD did not alter significantly (Figure 4H). These data suggest that NAT1 regulated the expression of endogenous retroviruses not only in human, but also in mouse.
NAT1 targets TUT7 to regulate neural differentiation and HERV-H expression
Because the neural differentiation defective phenotype by the loss of NAT1 is commonly observed in primed mouse and human PSCs, we hypothesized that a NAT1 target responsible for the phenotype is conserved between the two species. Based on this hypothesis, we performed RNA immunoprecipitation and deep sequencing (RIP-seq) to identify NAT1 target RNAs using mouse EpiSCs and human iPSCs. We obtained 775 and 483 transcripts as mRNAs that interacted with NAT1 protein in mouse and human primed PSCs, respectively (Tables S1 and S2). Fifty transcripts were shared between mouse and human (Tables S1 and S2). Among them, we focused on TUT7 (also known as ZCCHC6), because of its role in RNA uridylation mediated degradation (Ustianenko et al., 2016). TUT7 mRNA is significantly enriched in NAT1-pulldown RNAs compared to negative control (Figure 5A). The loss of NAT1 in human iPSCs reduced TUT7 protein but not RNA expression (Figures 5B and 5C). Reactivation of NAT1 by adding Dox upregulated TUT7 protein expression regardless of the RNA expression level. On the other hand, NAT1 did not affect the expression of TUT4, a family gene of TUT7, which suggests the effect of NAT1 is target-dependent. CRISPRi-induced NAT1 KD was also accompanied by decrease in TUT7 protein (Figure S4). We also observed decreased TUT7 protein levels, but not mRNA levels, in NAT1-depleted mouse EpiSCs (Figures 5D and 5E). Treatment with a proteasome inhibitor, MG-132, did not alter the expression of TUT7 protein in NAT1 KO iPSCs, suggesting that NAT1 regulates TUT7 protein expression in a proteasome-mediated degradation-independent manner (Figure 5F).
Figure 5 |. TUT7 is a target of NAT1.
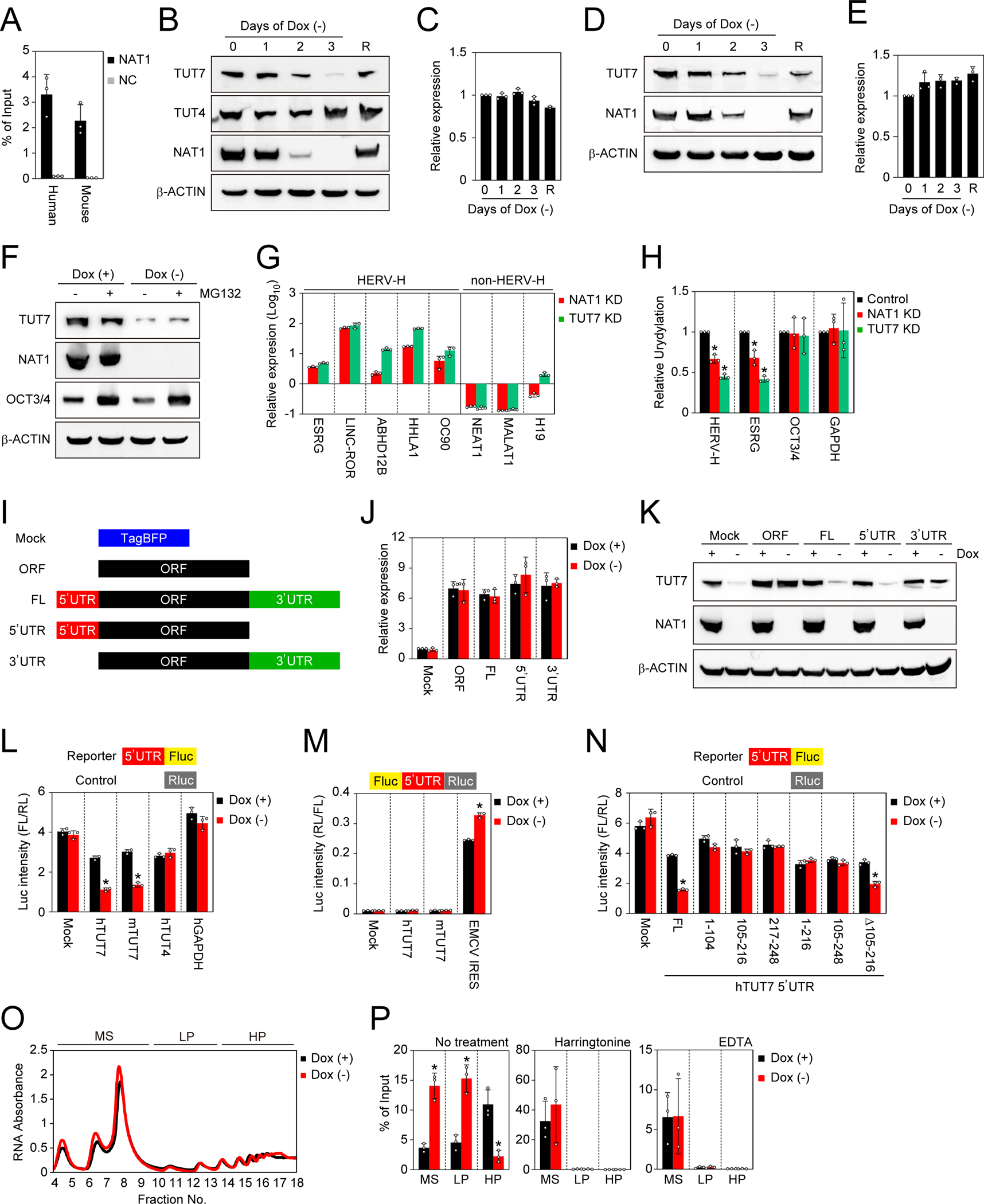
A. Pull down of TUT7 mRNA with NAT1 protein. TUT7 mRNAs in immunoprecipitants of 3xFLAG-tagged NAT1 (NAT1-FLAG) or non-tagged NAT1 (negative control, NC) human iPSCs and mouse EpiSCs purified using a FLAG antibody were quantified were by qRT-PCR. Values are normalized by input control. n=3. See also Tables S1 and S2.
B. Western blots of NAT1 cKO iPSCs in F/A condition on days 0–3 after Dox removal. On day 3 after Dox removal, we added Dox and collected the cell lysate after 1 day as rescued cells (R). See also Figure S4.
C. Relative expression of TUT7 in the cells shown in Fig. 5B analyzed by qRT-PCR. Values are normalized by GAPDH and compared with the sample on day 0. n=3.
D. Western blots of NAT1 cKO mouse EpiSCs on days 0–3 after Dox removal. On day 3 after Dox removal, we added Dox and collected the cell lysate after 1 day as rescued cells (R).
E. Relative expression of Tut7 in the cells shown in Fig. 5D analyzed by qRT-PCR. Values are normalized by Actb and compared with the sample on day 0. n=3.
F. Western blots of NAT1 cKO human iPSCs maintained in F/A condition with (+) or without (−) Dox and treated with (+) or without (−) MG132.
G. Relative expression of HERV-H and non-HERV-H non-coding RNAs in NAT1 KD or TUT7 KD human iPSCs in F/A condition analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 1B4 human iPSCs. n=3. See also Figure S5.
H. Relative uridylation levels of pan HERV-Hs, ESRG, OCT3/4 and GAPDH RNAs in NAT1 KD or TUT7 KD human iPSCs in F/A condition analyzed by qRT-PCR Values are normalized by spike RNA and compared with the control (1B4). *P<0.05 vs. control by unpaired t-test. n=3. See also Figure S5.
I. Scheme of the TUT7 RNA dissection. See also Figure S6.
J. Relative expression of TUT7 in NAT1 cKO human iPSCs transfected with the plasmids shown in Fig. 5H maintained in F/A condition with (+) or without (−) Dox. Values are normalized by GAPDH and compared with Mock. n=3. See also Figure S6.
K. Western blots of the cells shown in Fig. 5J. See also Figure S6.
L. The effects of the UTRs on the activity of firefly luciferase (Fluc) in NAT1 cKO iPSCs in F/A condition with (+) or without (−) Dox. Values of Fluc are normalized with co-transfected Renilla luciferase (Rluc) activity. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
M. The effects of the UTRs on the activity of Rluc in NAT1 cKO human iPSCs in F/A condition with (+) or without (−) Dox. Values of Rluc are normalized with Fluc activity. *P<0.05 vs. Dox (+) by unpaired t-test. n=3. n. The truncation of TUT7 5’UTR. The effects of the series of truncated human TUT7 5’UTRs on the activity of Fluc in NAT1 cKO iPSCs in F/A condition with (+) or without (−) Dox were analyzed. Values of Fluc are normalized with co-transfected Rluc activity. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
O. Representative absorbance profiling of the total fractionated RNA of NAT1 cKO human iPSCs maintained with (+) or without (−) Dox. MS, monosomes (fraction 4–9), LP, light polysomes (fraction 10–13); HP, heavy polysomes (fraction 14–18).
P. The distribution of TUT7 mRNAs in fractionated MS, LP and HP of NAT1 cKO human iPSCs on days 0 (+) and 3 (−) of Dox removal treated with or without harringtonine or EDTA analyzed by qRT-PCR. Values are normalized by spike RNA and total input RNA, and compared with the sample on day 0. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
To test if the decreased TUT7 protein expression contributed to the neural differentiation defect phenotypes in NAT1-depleted cells, we generated a Dox-inducible TUT7 cKD human iPSC line by CRISPRi with more than 99.9% efficiency (Figure S5A). TUT7 KD human iPSCs could expand normally while maintaining pluripotency marker expression (Figures S5A and S5C). In contrast, TUT7-depletion lowered the neural differentiation potential of human iPSCs (Figures S5D–S5G). On the other hand, we did not observe significant effects of TUT7-depletion on endoderm or mesoderm differentiation (Figures S5D and S5E). These data showed that TUT7 is a target of NAT1 and is involved in the neural differentiation of human iPSCs.
Next, we analyzed the effect of TUT7 on the expression of HERV-H RNAs in human iPSCs. We found that TUT7 KD significantly increased the expression of HERV-H-related genes (Figure 5G). In NAT1 KD and TUT7 KD iPSCs, the uridylation levels of HERV-Hs significantly decreased, while those of non-HERV-H genes did not change (Figure 5H). These data demonstrated that NAT1 regulates the amount of HERV-H RNAs through TUT7-mediated uridylation.
Role of NAT1 in efficient translation of TUT7
To understand how NAT1 regulates the expression of TUT7 proteins without affecting TUT7 RNA levels, we constructed a series of TUT7 expression vectors containing the open reading frame (ORF) alone, 5’UTR plus ORF, ORF plus 3’UTR, and full-length (FL) cDNA (5’UTR-ORF-3’UTR), and introduced them into NAT1 cKO human iPSCs (Figure 5I). All four vectors produced similar levels of TUT7 mRNAs (Figure 5J). However, vectors containing 5’UTR failed to produce TUT7 protein in NAT1-depeted human iPSCs (Figure 5K). In contrast, vectors without 5’UTR lead to increase in TUT7 protein. Next, we ask if these TUT7 expression vectors can rescue neural differentiation in NAT1-depleted iPSCs. As expected from the protein expression, the vector without 5’UTR rescued neural differentiation, suggesting that 5’UTR of TUT7 mRNA is responsible for the NAT1-dependent regulation (Figures S6A and S6B).
To examine whether TUT7 5’UTR is regulated by NAT1 in a context other than TUT7, we performed luciferase (luc) reporter assays in NAT1 cKO human iPSCs (Figure 5L). Both mouse and human TUT7 5’UTRs, but not control GAPDH 5’UTR, resulted in NAT1-dependent regulation (Figure 5L). However, neither mouse nor human TUT7 5’ UTR has IRES activity (Figure 5M). When we deleted a central portion of human 5’UTR, which does not exist in mouse 5’UTR, we still observed NAT1-dependent regulation (Figure 5N). However, further deletion resulted in lack of NAT1-dependetn regulation. Taken together, these data suggest that the 5’UTR of TUT7 regulates TUT7 protein expression via a NAT1-dependent, IRES-independent mechanism.
To further examine the roles of NAT1 in the translation of TUT7 mRNA, we performed polysome fractionation in NAT1 cKO human iPSCs and quantified TUT7 mRNA in each fraction. In the absence of NAT1, TUT7 mRNAs were enriched in monosome (MS) and light polysome (LP) fractions rather than in heavy polysome (HP) fractions, suggesting impaired translation (Figures 5O and 5P). Treatment with a translation elongation blocker harringtonine, or adding ethylenediaminetetraacetic acid (EDTA) as a chelate agent of magnesium ion which is required for the stability of ribosomes canceled significant differences of TUT7 mRNA profiling in NAT1 deficiency (Figure 5P). These data further support that NAT1 is crucial for the efficient translation of TUT7 mRNA.
NAT1 is dispensable for the self-renewal of NPCs, but is essential for reprogramming from NPCs to iPSCs
Next, we asked if NAT1 is required for the maintenance of NPCs. To this end, we differentiated NAT1 cKO human iPSCs into NPCs in the presence of Dox. The depletion of NAT1 by removing Dox altered neither the morphology nor the marker gene expression of NPCs (Figures 6A and 6B). Loss of NAT1 in NPCs induced changes in the expression of only a few genes compared with iPSCs in F/A condition, which exhibited significant phenotypes (Figures 1E and 6C). As observed in iPSCs, NAT1 depletion resulted in a decrease of TUT7 protein, but not TUT7 mRNA in NPCs (Figures 6D and 6E). However, HERV-Hs are silenced in NPCs and thus were not enhanced by NAT1 depletion (Figure 6B). Taken together, these data show that NAT1 is dispensable for the self-renewal of NPCs.
Figure 6 |. NAT1 is dispensable for the self-renewal of NPCs.
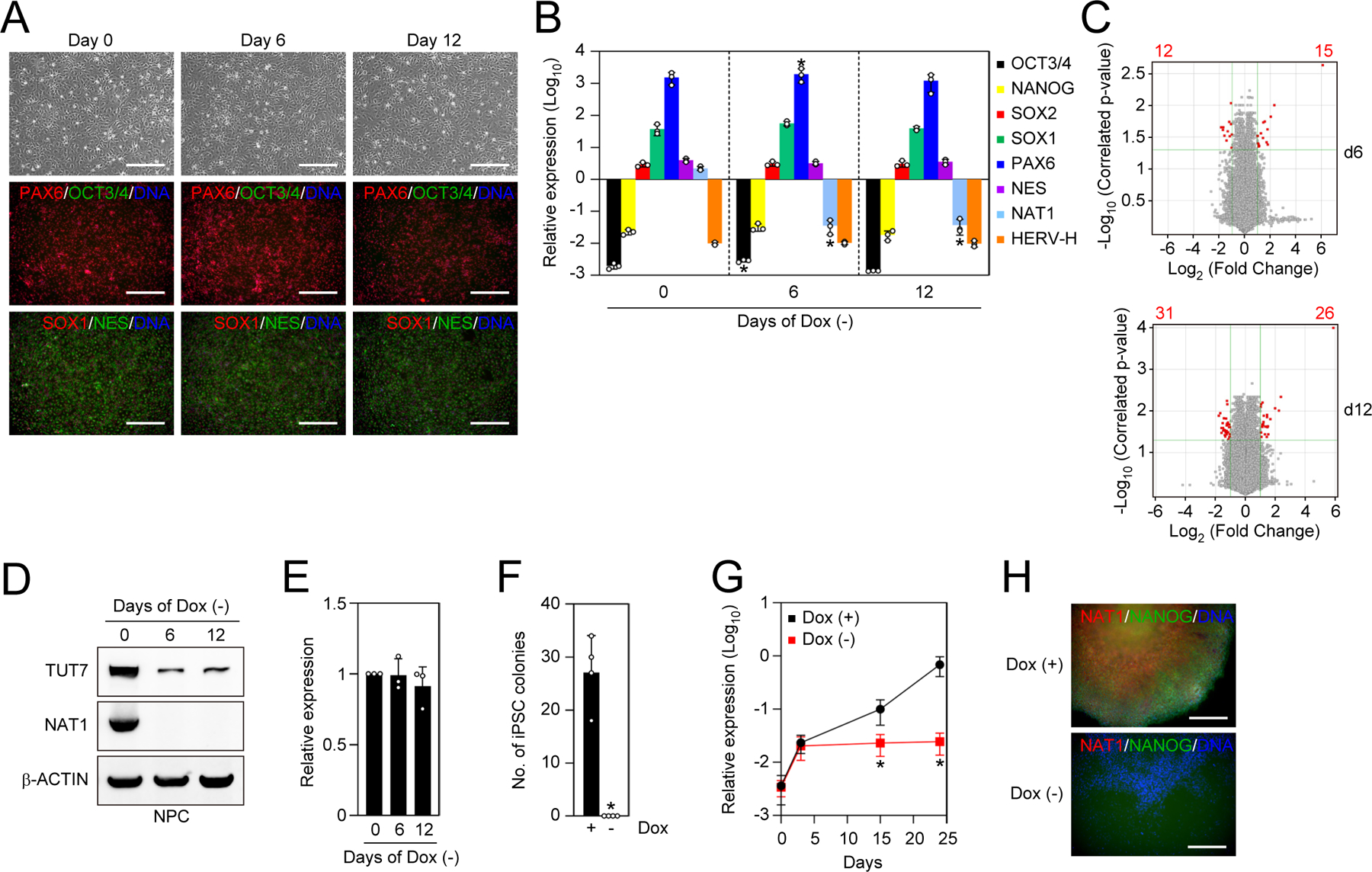
A. The generation of NAT1 cKO human NPCs under the presence of NAT1 transgene expression. Shown are representative images of NAT1 cKO human iPSC-derived NPCs cultured with (day 0) or without (days 6 and 12) Dox. Bars indicate 100 μm.
B. Relative expression of pluripotency and neural genes in NAT1 cKO human iPSC-derived NPCs cultured with (day 0) or without (days 6 and 12) Dox analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. day 0 by unpaired t-test. n=3.
C. Volcano plots showing global gene expression of NAT1 cKO human iPSC-derived NPCs cultured with or without Dox for 6 (d6) and 12 (d12) days. Red dots indicate differentially expressed genes with statistically significance (FC>2, FDR<0.05). Pearson correlation coefficients of the d6 and d12 samples were 0.9966 and 0.9969, respectively. n=3.
D. Western blots of NAT1 cKO human NPCs on days 0, 6 and 12 after Dox removal.
E. Relative expression of TUT7 in the cells shown in Fig. 6D analyzed by qRT-PCR. Values are normalized by GAPDH and compared with the sample on day 0. *P<0.05 vs. day 0 by unpaired t-test. n=3.
F. Number of AP (+) iPSC colonies from 5×104 OSKM-transduced NAT1 cKO human NPCs in the presence (+) or absence (−) of Dox on day 24. *P<0.05 vs. Dox (+) by unpaired t-test. n=4.
G. Relative expression of NANOG in the cultures of OSKM-transduced NAT1 cKO NPCs with (+) or without (−) Dox on days 0, 3, 15 and 24 analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 iPSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
H. Immunocytochemistry of the cultures of OSKM-transduced NAT1 cKO NPCs with (+) or without (−) Dox on day 24. Bars indicate 100 μm.
We then asked if NAT1 plays important roles in the reprogramming of NPCs to iPSCs. We introduced the reprogramming factors OCT3/4, SOX2, KLF4 and c-MYC (OSKM) into NAT1 cKO NPCs and then evaluated the number of iPSC colonies after 24 days in the presence or absence of Dox. The loss of NAT1 clearly inhibited iPSC generation (Figure 6F), and the increase of pluripotency genes such as NANOG was diminished (Figures 6G and 6H). These data suggest that NAT1 is required for the transition between human iPSCs and NPCs.
NAT1-depleted human iPSCs do not differentiate into neural cells in 2iLIF condition, but rather acquire naïve-like status
The dispensability of NAT1 in NPCs and our previous studies using mouse ESCs (Sugiyama et al., 2017; Yamanaka et al., 2000) suggest that the role of NAT1 is cell fate-dependent. Therefore, we hypothesized that the different NAT1 phenotypes in the two types of mouse PSCs (ESCs and EpiSCs) are caused by the different states of pluripotency. We treated NAT1 cKO human iPSCs with or without Dox with LIF and chemical inhibitors of MAPK/ERK Kinase (MEK) and Glycogen synthase kinase 3 (GSK3) (hereafter 2iL). The 2iL condition has been reported to be sufficient to maintain naïve pluripotency in mouse ESCs, but to induce neural differentiation in human PSCs (Theunissen et al., 2014; Ying et al., 2008). As expected, in the presence of Dox, NAT1 cKO human iPSCs on day 6 of 2iL treatment showed differentiation, as judged by morphology (Figure 7A) and marked increases in neural markers (Figure 7B). In sharp contrast, in the absence of Dox, cells formed compact colonies and maintained a high expression of pluripotency marker genes (Figures 7A and 7B). NAT1-depleted human iPSCs could expand, keeping tightly-packed colony formation, on MMC-treated MEFs in 2iL-containing media for more than 10 passages (Figures 7C and 7D). An inhibitor of Rho-associated coiled-coil containing protein kinase (ROCK) was dispensable for the survival of NAT1-depleted iPSCs in 2iL condition (Watanabe et al., 2007) (Figure 7E). These cells expressed naïve pluripotency markers (Figure 7F). As observed in primed state, naive-like human iPSCs showed an enhanced expression of HERV-Hs in the absence of NAT1 (Figure 7G). These data demonstrated that NAT1-depleted human iPSCs acquired a state similar to naïve-like pluripotency upon 2iL treatment.
Figure 7 |. Loss of NAT1 restricts the differentiation of naïve-like human iPSCs.
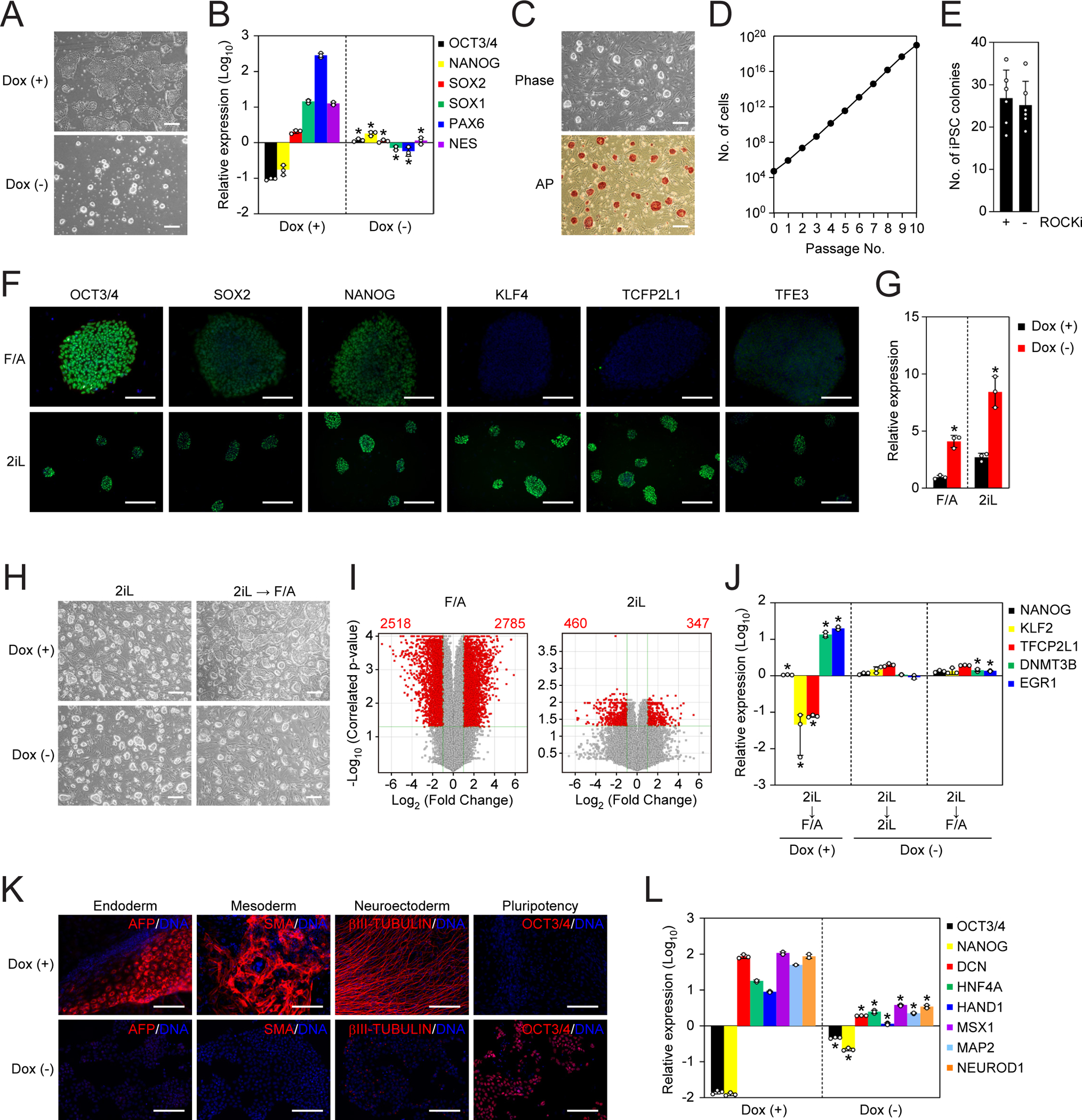
A. Representative images of NAT1 cKO human iPSCs maintained with (+) or without (−) Dox in 2iL condition for 6 days. Bars indicate 100 μm.
B. Relative expression of pluripotency and neural genes in the cells shown in Fig. 7A analyzed by qRT-PCR. Values are normalized by GAPDH and compared with 585A1 iPSCs. *P<0.05 vs. Dox (+) by unpaired ttest. n=3.
C. Representative images of NAT1 KO iPSCs converted to the naïve-like state. Phase contrast image (upper) and AP stained image (lower) are shown. Bars indicate 100 μm.
D. Growth curve of NAT1 KO iPSCs converted to the naïve-like state.
E. Colony formation efficiency of naïve-like NAT1 KO human iPSCs plated with (+) or without (−) ROCK inhibitor (ROCKi). n=6.
F. Immunocytochemistry of pluripotency markers in NAT1 cKO human iPSCs in F/A or 2iL conditions. Bars indicate 50 μm.
G. Relative expression of HERV-Hs in NAT1 cKO human iPSCs in F/A or 2iL condition with (+) or without (−) Dox. Values are normalized by GAPDH and compared with 585A1 human iPSCs. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
H. Representative phase contrast images of NAT1 cKO human iPSCs with (+) or without (−) Dox in 2iL condition or after transferring from 2iL to F/A condition. Bars indicate 100 μm.
I. Volcano plots showing global gene expression of NAT1 cKO human iPSCs with (+) or without (−) Dox in F/A (left) or 2iL (right) condition. Red dots indicate differentially expressed genes with statistical significance (FC>2, FDR<0.05). Pearson correlation coefficients of F/A and 2iL samples were 0.9787 and 0.9827, respectively. n=3. The data of F/A samples are identical to those shown in Fig. 1E.
J. Relative expression of naïve and primed pluripotency markers in the cells shown in Fig. 7H analyzed by qRT-PCR. Values are normalized by GAPDH and compared with Dox (+) in 2iL. *P<0.05 vs. 2iL by unpaired t-test. n=3.
K. Immunocytochemistry of differentiated cells derived from naïve-like NAT1 cKO human iPSCs in the presence (+) or absence (−) of Dox by EB formation. Bars indicate 100 μm.
L. Relative expression of pluripotency and differentiation markers in the cells shown in Fig. 7K compared with naïve-like human iPSCs analyzed by qRT-PCR. Values are normalized by GAPDH. *P<0.05 vs. Dox (+) by unpaired t-test. n=3.
NAT1 is required for transition of naïve-like pluripotency to primed state
After the conversion to the naïve-like state, NAT1 reactivation by Dox addition did not induce any obvious morphological changes in 2iL condition (Figure 7H). Microarray expression analyses revealed that 798 genes showed more than 2-fold difference between Dox plus and Dox minus (FDR<0.05) (Figure 7I). This effect of NAT1 on the global gene expression was much smaller than that in primed state (Figures 1E and 7I).
Notably, when the culture condition was changed from 2iL to F/A, NAT1-expressing cells changed into a flatter morphology after 6 days. In contrast, NAT1-depleted human iPSCs maintained the rounded morphology even in F/A condition (Figure 7H). Longer culture in F/A induced massive cell death in NAT1-depleted cells. We detected the downregulation of naïve pluripotency markers, such as KLF2 and TFCP2L1, and of primed pluripotency genes, such as DNMT3B and EGR1, in NAT1-expressing human iPSCs in F/A condition (Figure 7J). However, NAT1 KO human iPSCs showed similar expression patterns of these markers in 2iL and F/A conditions (Figure 7J). These data suggest that NAT1 is required for the transition of naïve-like pluripotency to primed pluripotency in human.
Next, we asked if NAT1 affects the differentiation potentials of naïve-like human iPSCs by EB formation. Naïve-like NAT1 cKO iPSCs with Dox could differentiate into all three germ layers and suppress OCT3/4 expression (Figure 7K). On the contrary, naïve-like NAT1-depleted human iPSCs poorly differentiated into any of the three lineages and failed to turn off OCT3/4 expression (Figure 7K). We also confirmed that EBs derived from naïve-like NAT1-depleted human iPSCs maintained a high expression of OCT3/4 and NANOG and low expression of differentiation markers (Figure 7L). Taken together, these data demonstrate that NAT1 is required for the transition of naïve-like to primed human pluripotency and the subsequent differentiation into somatic lineages.
DISCUSSION
Critical role of translation initiation and RNA uridylation in primed pluripotent stem cells.
The initial purpose of the current study was to determine the roles of NAT1 in human PSCs. Two independent approaches to deplete NAT1 showed that NAT1 is critical for the self-renewal and neural differentiation of human primed iPSCs. We also found that NAT1 is important for the efficient translation of TUT7 mRNA. In the presence of NAT1, TUT7 protein is efficiently produced and uridylates HERV-H RNAs, leading to their degradation. In the absence of NAT1, however, TUT7 protein is not efficiently translated, resulting in the accumulation of HERV-H RNAs. This accumulation is responsible, at least in part, for the impaired neural differentiation of NAT1-depleted human iPSCs. These data revealed important roles of translation initiation and RNA uridylation in the regulation of HERV-H expression and neural differentiation in primed PSCs.
The reverse correlation between HERV-H expression levels and neural differentiation is consistent with our previous finding that aberrant accumulation of HERV-H in human PSCs is responsible for the defect of neural differentiation (Koyanagi-Aoi et al., 2013; Ohnuki et al., 2014). HERV-Hs are expressed not only in human PSCs but also in mesendodermal cells (Xie et al., 2013). Taken together, these findings may suggest that HERV-Hs potentially contribute to the establishment of the mesendodermal lineage. Indeed, we observed an upregulation of mesendodermal genes in NAT1-deficient PSCs, and suppression of HERV-H attenuated the aberrant mesendodermal feature. The precise roles of HERV-H in cell fate determination are subject to further investigation.
Our data showed that the 5’UTR of TUT7 mRNA is required for NAT1-dependent translation regulation. Multiple mechanisms have been proposed as to how NAT1, which does not bind to the cap-binding protein eIF4E, engages in translation initiation. The most widely accepted view is the IRES-mediated mechanism (Henis-Korenblit et al., 2002; Henis-Korenblit et al., 2000; Liberman et al., 2015; Marash et al., 2008; Weingarten-Gabbay et al., 2014). However, we failed to detect IRES activity in human or mouse TUT7 5’UTR. More recently, NAT1 was reported to recognize m6A-modification of mRNA to initiate translation in a cap-independent manner (Yang et al., 2017). Alternatively, NAT1 may recognize and bind to the 5’ cap structure of mRNA through proteins other than eIF4E, such as FMR and eIF3D (Bukhari et al., 2016; de la Parra et al., 2018; Meyer et al., 2015). Further studies are required to determine how NAT1 interacts with the TUT7 5’UTR to initiate translation.
Regarding neural differentiation, the effect of TUT7 on rescuing (by forced expression) and mimicking (by KD) the NAT1 KO phenotype was partial. Also, the strength of NAT1’s effect on TUT7 translation varied in NAT1 cKO and cKD human iPSC lines, suggesting other mechanisms and/or pathways contribute to the regulation (Figure S4). Other NAT1’s targets may be involved in cell fate transition of primed PSCs to NPCs. In this study, we identified mRNAs encoding multiple proteins, such as transcription factors and RNA-binding proteins, as candidates of NAT1 targets both in mouse and human. We need to analyze these candidates to further understand precise roles of NAT1 in cell fate determination.
It remains to be determined how TUT7 specifically targets HERV-Hs, rather than other HERVs and transposable elements. One possible explanation is that TUT7 recognizes specific sequences or secondary structures that exist in HERV-H RNAs. On the other hand, in the NAT1 cKD iPSC line, we detected a significant increase of HERV-Hs despite a residual expression of TUT7 proteins (Figures 5G and S4). This suggests that other NAT1 targets are also involved in the regulation of HERV-Hs. These important issued should be analyzed in future studies.
State-specific roles of NAT1 in PSCs.
Another important finding of this study is that NAT1 plays different roles depending on the state of the PSCs. In contrast to primed state, where NAT1-depletion resulted in impaired self-renewal and neural differentiation, human and mouse naïve (-like) PSCs showed enhanced self-renewal and impaired differentiation to three germ layers. The current study clearly shows that NAT1-deficient naïve (-like) PSCs failed to transit to primed state. The poor self-renewal and survival of primed PSCs in the absence of NAT1 can explain the defect of differentiation. The failure to reprogram NAT1-deficient human NPCs to iPSCs in F/A condition also supports the requirement of NAT1 for entering the primed pluripotent state.
In both primed and naïve (-like) states, we observed a decrease in TUT7 protein, suggesting that TUT7 is a target of NAT1 regardless of the type of pluripotency. On the contrary, we found that neither TUT7 KD nor HERV-H KD could rescue the self-renewal of NAT1-deficient primed PSCs. Thus, NAT1 targets other than TUT7 should play important roles in the self-renewal of primed PSCs.
Recently, we identified MAP3K3 as a target of NAT1 in mouse ESCs (Sugiyama et al., 2017). In NAT1 KO mouse ESCs, MAP3K3 protein expression decreased, resulting in the suppression of MAPK activity. This effect explained, at least in part, the muted response of NAT1 KO mouse ESCs to MAPK inhibitor to maintain naïve pluripotency. In human, MAPK inhibitor is required for the induction and maintenance of the naïve-like state regardless of NAT1 expression, suggesting that targets other than MAP3K3 play important roles. Thus, the identification of NAT1 targets other than TUT7 and MAP3K3 is an important aim of future studies. Our results differ from a previous study that showed NAT1 KD by shRNA in human ESCs causes normal proliferation and a failure to differentiate into all lineages (Yoffe et al., 2016). The discrepancy could be explained by different levels of residual NAT1 expression. In our case, NAT1 RNA expression was <1% in cKO and <0.01% in CRISPRi-mediated KD compared to those in controls, suggesting a near complete loss of function. In contrast, shRNA-mediated KD in general cannot achieve this degree of suppression. Indeed, some western blots in the previous study detected NAT1 proteins even in KD samples (Yoffe et al., 2016). This leaky expression of NAT1 proteins could suffice to maintain self-renewal. In addition, the difference in distinguishable pluripotent states could contribute to the discrepancy. NAT1 KD human ESCs from the previous study expressed a relatively high level of REX1 (also known as ZFP42), which is a critical marker of naïve pluripotency. This fact may suggest that their cells were either heterogeneous or had partial naïve pluripotent features. If so, the studies are in fact consistent in that naïve-like NAT1 KO human iPSCs could not differentiate to any lineages. Moreover, combining the studies may suggest a dose-dependent manner of NAT1’s role in PSCs.
In this study, we identified mRNA targets of NAT1 by performing pulldown experiments in native condition. Previously, Yoffe et al. also showed the NAT1’s targets including HMGN3 which they focused on by performing immunoprecipitation with UV crosslinking (Yoffe et al., 2016). The data between these two studies was less overlapping; 3 overlapped transcripts such as MFF, MYCBP and WWP1 between Yoffe et al. (122 targets) and ours (559 targets). Possibly, the strategies of immunoprecipitation (e.g. crosslinking and antibodies) and cell culture condition affect the results.
In the current study, to elucidate the role of NAT1 in human primed pluripotency carefully, we used two different systems to suppress NAT1, namely cKO and cKD. Both approached resulted in common phenotypes, such as cell death and the defect in neural differentiation in primed pluripotent state. However, the severity of the phenotypes between these two cell lines were different. Particularly, the NAT1 cKD by CRISPRi induced cell death quicker than did cKO human iPSC line. In addition, NAT1 protein level in cKO cell line was roughly two-fold higher than that in cKD line (Figure S4). This difference can be seen in their parental iPSC lines (Figure S4). The responsiveness of TUT7 protein expression by NAT1 depletion was also different. NAT1 cKO human iPSCs showed almost complete loss of TUT7 protein by NAT1 depletion. On the other hand, TUT7 protein was still detectable after CRISPRi-induced NAT1 KD. Reasons underlying these differences remain to be determined. One hypothesis is that multiple NAT1 targets are involved in TUT7 regulation and other phenotypes with variable contributions in each cell line. In consistent with this hypothesis, Yoffe et al did not identify TUT7 as a NAT1 target by RNA-IP (Yoffe et al., 2016). Further studies are required to understand cell line-specific roles of NAT1 in cell fate determination.
Conclusion
Our study revealed crucial roles of NAT1 in PSCs. Importantly, NAT1 plays distinctive roles in the naïve and primed states of pluripotency. This dual role of NAT1 is conserved between human and mouse. We identified the RNA uridyltransferase TUT7 as a NAT1 target, a conclusion attributed to the impaired neural differentiation of NAT1-depleted primed PSCs. Considering its ubiquitous and high expression, we believe that NAT1 should have many more targets than those that have been reported. The identification of other NAT1 targets together with the role of NAT1 in other types of cells are important future tasks.
RESOURCE AVAILABILITY
Lead Contact
Kazutoshi Takahashi (kazutoshi.takahashi@gladstone.ucsf.edu)
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Gene expression microarray results are accessible in the Gene Expression Omnibus database of the National Center for Biotechnology Information website (accession number: GSE129429). Uncropped images are available in Mendeley (http://dx.doi.org/10.17632/r4wjcwyzkj.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human iPSCs were routinely maintained on recombinant laminin 511 E8 fragment (LN511, Nippi)-coated plate in StemFiT media (Ajinomoto) supplemented with 100 ng/ml recombinant basic fibroblast growth factor (bFGF, Peprotech) (hereafter F/A media) (Miyazaki et al., 2012; Nakagawa et al., 2014). For feeder culture, we prepared primary mouse embryonic fibroblasts (MEFs) from the pool of E13.5 mouse embryos (ICR strain) and maintained in Dulbecco’s Modified Eagle Medium (DMEM, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific). To inactivate proliferation, MEFs were treated with 12 μg/ml mitomycin C (MMC, Sigma-Aldrich) for 2.5 h. We plated MMC-treated MEFs at a density of 1.5 × 106 cells per a 100 mm dish coated with 0.1% gelatin (EMD Millipore). On MEF feeder, human iPSCs were maintained in DMEM/F12 (Thermo Fisher Scientific) containing 20% Knockout serum replacement (KSR, Thermo Fisher Scientific), 1% GlutaMax (Thermo Fisher Scientific), 1% MEM non-essential amino acids (NEAA, Thermo Fisher Scientific), 1 × 10−4 M 2-mercaptoethanol (2-ME, Thermo Fisher Scientific) and 8 ng/ml bFGF. Mouse EpiSCs were cultured on fibronectin (Sigma-Aldrich)-coated plate in NDiff227 media (Clontech) supplemented with 12 ng/ml bFGF and 20 ng/ml recombinant Activin A (Peprotech) (Kime et al., 2016). Mouse ESCs were maintained on gelatin-coated plates in Knockout DMEM (Thermo Fisher Scientific) containing 15% KSR, 1% GlutaMax, 1% NEAA, 1 × 10−4 M 2-ME and 1000 units/ml mouse Leukemia inhibitory factor (LIF, EMD Millipore). Established neural progenitor cells (NPCs) were maintained on Matrigel-coated plate in STEMdiff Neural Progenitor Medium (Stem Cell Technologies). All cell lines were screened for the absence of mycoplasma and confirmed retaining a normal karyotype by G-banding.
METHOD DETAILS
RNA isolation and reverse-transcription polymerase chain reaction
The cells were lysed with QIAzol lysis reagent (QIAGEN), and total RNA was purified using a miReasy Mini kit (QIAGEN). Purified RNA (0.1–1 μg) was used for single strand complementary DNA (cDNA) synthesis using a SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Thermo Fisher Scientific). Quantitative RT-PCR was performed using TaqMan Universal Master Mix II, no UNG (Thermo Fisher Scientific) or Power SYBR Green PCR Master Mix (Thermo Fisher Scientific) on either a StepOne instrument (Applied Biosystems) or an ABI7900HT Real Time PCR System (Applied Biosystems). The levels of mRNA were normalized to human GAPDH or mouse Actb expression, and then relative expression was calculated as the fold-change from the control. Primer sequences are provided in Table S3.
Transposon-mediated gene transfer
In current study, we used two kinds of transposon system such as piggybac (PB) and sleeping beauty (SB) with the plasmids encoding improved transposases (Mates et al., 2009; Yusa et al., 2011). Seven micrograms of transposon vector and 3 μg of transposase-encoding vector were co-transfected into 1 × 106 cells using a Nucleofector II (Amaxa). After two days of electroporation, the cells were selected with appropriate drugs until non-transfected cells were killed completely.
Generation of conditional knockout cell lines
For making targeting vectors for human NAT1, we introduced 5’ and 3’ homologous arms which were amplified from human genomic DNA by PCR into pBS-Neo-SV40pA and pBS-Puro-bGHpA using the In-Fusion technology (Clontech), respectively. The sequences of homologous regions are provided in Table S3. For CRIPR/Cas9-mediated gene editing, we designed guide RNA (gRNA) against exon 2 of human NAT1 gene. Primer extension was performed using a gRNA oligo and universal primer (gRNA-universal-rev). The resulting fragment was introduced into the BamHI/EcoRI site of pHL-H1-ccdB-EF1a-RiH (Li et al., 2015) using the In-Fusion technology. We co-transfected a Neo targeting vector, Cas9-expression vector and gRNA-expression vector (Li et al., 2015) into 585A1 human iPSCs (Okita et al., 2011) by electroporation using a NEPA21 electroporator (NEPAGENE). After electroporation, the cells were plated onto MMC-treated SNL feeder in Primate ESC medium supplemented with 4 ng/ml bFGF and 10 μM Y-27632 (Sigma-Aldrich). After 3 days, the cells were selected with 250 μg/ml Geneticin (Thermo Fisher Scientific). Drug-resistant colonies were isolated and screened by PCR with the primer set; SV40pA-S159/hNAT1-SCR-AS1 for a Neo targeted allele. Then, we introduced a PB vector encoding doxycycline (Dox-inducible NAT1 into NAT1+/Neo iPSC clone by electroporation. On the following day, the cells were selected with 10 μg/ml Blasticidin S (Thermo Fisher Scientific) until non-transfected cells completely died, resulting NAT1+/Neo +Tg. Then, we used this cell line for second round targeting with a Puro targeting vector in the presence of Dox. Precise targeting was confirmed by PCR with the primer set; bGHpA-S2/hNAT1-SCR-AS1 for a puro targeted allele, and southern blotting.
For mouse NAT1 targeting, we generated targeting vectors by introducing 5’ and 3’ homologous arms which were amplified from mouse genomic DNA by PCR using same strategy into pBS-Puro-bGHpA and pBS-Bsd-SV40pA, respectively (see Table S3). We first introduced a PB vector encoding Dox-inducible NAT1–3xFLAG into X-GFP EpiSC line (Bao et al., 2009). Two days later, we started the selection with 40 μg/ml Zeocin (Thermo Fisher Scientific) for 5 days. Next, we co-transfected targeting vectors (1.5 μg each) and 7 μg of a plasmid encoding Cas9 and mouse NAT1 gRNA (Slaymaker et al., 2016) into EpiSC line carrying Dox-inducible NAT1 transgenes using a Nucleofector II (Amaxa). After three days of transfection, we added 1 μg/ml Purpmycin and 10 μg/ml Blasticidin S into the media and cultured cells when the colony size became suitable for isolation. We verified homologous recombination by PCR with the primer sets; mNAT1-SCR-S1/BSD-AS91 for a BSD targeted allele and mNAT1-SCR-S1/Puro-AS135 for a Puro targeted allele. We also confirmed the loss of WT alleles by PCR with the primer set; mNAT1-WT-S1/mNAT1-WT-AS1.
Southern blot
Genomic DNA was purified using a DNeasy blood and tissue kit (QIAGEN), according to manufacturer’s protocol. Ten micrograms of purified DNA was digested using HindIII-HF (New England Biolabs) at 37°C overnight, separated on 0.8% agarose gel, and transferred to a positive charged nylon membrane (Amersham). The probe for the confirmation of homologous recombination was generated by PCR using the primer set; hNAT1-southern-probe-S/hNAT1-southern-probe-AS (see Table S3). The membrane was incubated with digoxigenin (DIG)-labeled DNA probe in DIG Easy Hyb buffer (Roche Life Science) at 42°C overnight with constant rotation. After washing, alkaline phosphatase–conjugated anti-DIG antibody (1:10000, Roche Life Science) was added to the membrane. Signals were produced with CDP-star reagent (Roche Life Science) and detected using a LAS3000 imaging system (FUJI FILM).
Western blot
Western blotting was performed as described previously (Rand et al., 2018). In brief, cells were washed once with Dulbecco’s phosphate buffered saline without calcium chloride (CaCl2) and magnesium chloride (MgCl2) (PBS, Thermo Fisher Scientific) and then lysed with RIPA buffer (Sigma-Aldrich) containing Protease/Phosphatase Inhibitor Cocktail (Cell Signaling Technology). After a 15 min incubation on ice, insoluble cell debris was removed from the cell lysates by centrifugation at 20,000 xg for 15 min at 4°C. Denatured protein samples were prepared by adding NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) and NuPAGE Sample Reducing Agent (Thermo Fisher Scientific), and heated the mixture at 70°C for 10 min. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed using the Novex NuPAGE SDS-PAGE Gel System (Thermo Fisher Scientific). Separated proteins were transferred to a nitrocellulose membrane using the iBlot transfer system (Thermo Fisher Scientific) with P0 program for 6–7 min. The membrane was blocked with 0.5x Odyssey Blocking Buffer (Li-COR Biosciences) for 30 min at room temperature with constant agitation, and then incubated with primary antibodies diluted in the blocking buffer or Signal enhancer HIKARI (Nacalai tesque) at 4°C overnight. The membrane was washed for three times with Tris buffered saline (20 mM Tris-HCl, 138 mM Sodium Chloride (NaCl), pH7.6) containing 0.1% Tween-20 (Teknova) each for 10 min. Then the membrane was incubated with secondary antibody diluted in the blocking buffer for 45 min at room temperature with protecting from the light. Images were obtained using an Odyssey Imaging system (LI-COR Biosciences). Some of analyses were performed by using the Wes system (Protein Simple) according to the manufacturer’s instruction. Antibodies and dilution rate were as follows; mouse monoclonal anti-NAT1 (1:500, 610742, BD Biosciences), rabbit polyclonal anti-ZCCHC6 (1:500, HPA020620, Sigma-Aldrich), rabbit polyclonal anti-ZCCHC6 (1:200, 25196–1-AP, Proteintech), rabbit polyclonal anti-ZCCHC11 (1:500, 18980–1-AP, Proteintech), mouse monoclonal anti-OCT3/4 (1:600, sc-5279, Santa Cruz Biotechnology), mouse monoclonal anti-β-ACTIN (1:5000, A5441, Sigma-Aldrich), rabbit polyclonal anti-β-ACTIN (1:1000, ab8227, Abcam), horseradish peroxidase (HRP)–linked mouse IgG (1:3000, #7076, Cell signaling technology), HRP–linked rabbit IgG (1:2000, #5127, Cell signaling technology), IRDye680LT anti-mouse IgG (1:15000, 926–68022, LI-COR Biosciences), IRDye680LT anti-rabbit IgG (1:15000, 925–68023, LI-COR Biosciences), IRDye800CW anti-mouse IgG (1:10000, 925–32212, LI-COR Biosciences) and IRDye800CW anti-rabbit IgG (1:10000, 925–32213, LI-COR Biosciences).
Immunocytochemistry
Immunocytochemistry was performed as described previously (Rand et al., 2018). In brief, the cells were fixed with the fixation buffer (BioLegend) for 15 min at room temperature. Then the fixed cells were treated with PBS containing 0.2% Triton X-100, 2% normal donkey serum (CHEMICON) and 1% bovine serum albumin (BSA, Thermo Fisher Scientific) for 45 min at room temperature. The cells were incubated with primary antibodies diluted in PBS containing 1% BSA at 4°C overnight. After washing with PBS (three times each for 10 min), the cells were incubated with secondary antibodies for 45 min at room temperature with protecting from the light. Nuclei were visualized with 1 μg/ml Hoechst 33342 (Thermo Fisher Scientific). Fluorescent signals were detected using a BZ-X710 imaging system (KEYENCE). Antibodies and dilution rate were as follows; mouse monoclonal anti-OCT3/4 (1:200, sc-5279, Santa Cruz Biotechnology), rabbit polyclonal anti-SOX2 (1:100, ab97959, Abcam), rabbit polyclonal anti-NANOG (1:100, ab21624, Abcam), mouse monoclonal anti-human Nuclei (1:1000, MAB4383, EMD Millipore), rabbit polyclonal anti-AFP (1:200, A0008, DAKO), mouse monoclonal anti-AFP (1:200, MAB1368, R&D Systems), mouse monoclonal anti-SMA (1:100, M0851, DAKO), mouse monoclonal anti-βIII-TUBULIN (1:1000, MAB1637, EMD Millipore), rabbit polyclonal anti-PAX6 (1:1000, 901301, BioLegend), rabbit polyclonal anti-KLF4 (1:100, sc-20691, Santa Cruz Biotechnology), goat polyclonal anti-TFCP2L1 (1:100, AF5276, R&D Systems), rabbit polyclonal anti-TFE3 (1:100, HPA023881, Sigma-Aldrich), Alexa 488-conjugated donkey anti-mouse IgG (1:500, A-21202, Thermo Fisher Scientific), Alexa 555-conjugated donkey anti-rabbit IgG (1:500, A-31572, Thermo Fisher Scientific), Alexa 647-conjugated donkey anti-mouse IgG (1:500, A-31571, Thermo Fisher Scientific), Alexa 488-conjugated anti-rabbit IgG (1:500, A-21206, Thermo Fisher Scientific), Alexa 555-conjugated donkey anti-rabbit IgG (1:500, A-31572, Thermo Fisher Scientific), Alexa 647-conjugated donkey anti-rabbit IgG (1:500, A-31573, Thermo Fisher Scientific) and Alexa 488-conjugated donkey anti-goat IgG (1:500, A-11055, Thermo Fisher Scientific).
Embryoid body (EB) differentiation
PSCs were cultured with or without Dox for three days prior to EB formation. The cells were harvested using CTK solution (ReproCELL) and transferred cell clumps onto an ultra-low binding plate (Corning) in EB media (DMEM/F12 containing 20% KSR, 1% GlutaMax, 1% NEAA and 1 × 10−4 M 2-ME). For the first 2 days, we added 10 μM Y-27362 to improve cell survival. The media was changed every other day. After 8 days of floating culture, we transferred EBs onto a plate coated with 0.1% gelatin and maintained in EB media for another 8 days.
Directed Neural differentiation by dual SMAD inhibition
Differentiation of human iPSCs into NPCs was performed as described previously (Doi et al., 2014). In brief, human iPSCs were plated at a density of 8 × 105 cells per well of LN511-coated 6-well plate in F/A media and cultured with or without Dox for 3 days. Then the media was replaced with Glasgow Minimum Essential Medium (GMEM, Thermo Fisher Scientific) containing 8% KSR, 1 mM Sodium pyruvate (Sigma-Aldrich), 1% NEAA, 1 × 10−4 M 2-ME, 100 nM LDN193189 (Stemgent) and 500 nM A83–01(Stemgent). The media was changed every other day. For establishment of NAT1 cKO NPC line, we used STEMdiff SMADi Neural Induction Kit (Stem Cell Technologies) according to manufacturer’s protocol.
Conversion of human iPSCs to naïve-like pluripotent state
The conversion of human primed iPSCs to naïve-like state was performed as previously described with slight modifications (Kime et al., 2016). NAT1 cKO iPSCs were maintained in F/A media with or without Dox for 6 days, then they were harvested using TrypLE select (Thermo Fisher Scientific) and plate single cells at 1 × 105 cells per a well of LN511-coated 6-well plate in 2iL media (StemFiT (F/A media with no bFGF) supplemented with 10 μM Y-27632, 1 μM PD0325901 (Stemgent), 3 μM CHIR99021 (Stemgent), 10 ng/ml human LIF (EMD Millipore)). After six days of conversion, the cells were transferred onto MMC-inactivated MEF feeder in 2iL media supplemented with 1 μM 1-oleoyl-2-methyl-sn-glycero-3-phosphothionate (OMPT, Avanti Polar Lipids). The cells were maintained in low oxygen condition (5%). After passage 2, we did not use Y-27632.
Conversion of mouse EpiSCs to naïve pluripotent state
Naïve conversion of mouse EpiSCs was performed as described previously (Kime et al., 2016). In brief, mouse EpiSCs were harvested using Accutase (EMD Millipore) to single cells and plated at a density of 1 × 104 cells per well of Fibronectin (Sigma-Aldrich)-coated 6-well plate in the conversion medium (1:1 mix of DMEM/F12 and Neurobasal medium (Thermo Fisher Scientific) supplemented with 0.5% N2 (Thermo Fisher Scientific), 1% B27 (Thermo Fisher Scientific), 1% GlutaMax, 0.005% BSA fraction V (Thermo Fisher Scientific), 1000 units/ml mouse LIF, 10 ng/ml Bone morphogenetic protein 4 (BMP4, R&D Systems), 64 μg/mL L-ascorbic acid 2-phosphate (Sigma-Aldrich) and 1 μM OMPT. To select converted cells, cells were cultured in conversion medium for 6–9 days, passaged, and cultured on plates coated with LN511 in 1:1 mix of the conversion medium and N2B27 medium supplemented with 1000 units/ml mouse LIF, 1 μM PD0325901 and 3 μM CHIR99021. The next day, the medium was changed to N2B27 medium supplemented with 1000 units/ml mouse LIF, 1 μM PD0325901 and 3 μM CHIR99021. After a few passages, we replaced the media with Knockout DMEM containing 15% KSR, 1% GlutaMax, 1% NEAA, 1 × 10−4 M 2-ME and 1000 units/ml mouse LIF.
CRISPR interference (CRISPRi)-mediated knockdown
For performing CRISPRi knockdown experiments, we used human iPSC line which carried Dox-inducible Krüppel associated box (KRAB) domain-fused dCas9 (KRAB-dCas9) gene expression cassette in the Adeno-Associated Virus Integration Site 1 (AAVS1) locus (CRISPRi Gen 1B clone 4, 1B4) (Mandegar et al., 2016). We chose gRNA sequences for gene of interest in the list of previous publication (Horlbeck et al., 2016). We introduced PB-U6-CNCB encoding each gRNA sequence (see Table S3) for gene of interest along with pCW-hyPBase into 1 × 106 of 1B4 iPSCs by using Nucleofection. After the selection with 10 μg/ml Blasticidin S, polyclonal populations were cultured with or without 1 μg/ml Dox for 6 days and used for the screening to identify the most effective gRNA by qRT-PCR. Then we plated transfected cells carrying the most efficient gRNA at clonal density onto LN511-coated 100 mm dish in F/A media supplemented with 10 μM Y-27632. We subcloned iPSC colonies based on fluorescence intensity and uniformity. Subsequent analyses were performed on clonal populations to obtain clean knockdowns.
iPSC Reprogramming
The reprogramming experiments were performed as described (Rand et al., 2018). In brief, to generate pantropic retroviral particles, we transfected 3μg of pMD2.G (a gift from Dr. Didier Trono) along with 6 μg of murine leukemia virus (MuLV)-based pMXs (Morita et al., 2000) encoding reprogramming factors such as OCT3/4, SOX2, KLF4 and c-MYC to 3.6 × 106 PLAT-GP cells with FuGENE 6 transfection reagent (Promega). After 24 h, the medium was replaced with 10 ml of fresh medium, and the cells were incubated for another 24 h. The following day, the virus-containing supernatants were filtered through a 0.45-μm pore size cellulose acetate filter (Corning) to remove cell debris, and concentrated using Retro-X Concentrator (Clontech). Next day, virus-containing pellets were dissolved in STEMdiff Neural Progenitor Medium containing 8 μg/ml Polybrene (EMD Millipore). Then, we mixed appropriate combinations of viruses and used them for transduction to NAT1 cKO NPCs. We designated this point as day 0. We harvested the cells on day 3 post-transduction, and re-plated them at 1 × 105 cells per a well of LN511-coated 6-well plate in STEMdiff Neural Progenitor Medium. The following day (day 4), the medium was replaced with F/A media, and the medium was changed every other day. The iPSC colonies were counted on day 24 post-transduction. We distinguished bona fide iPSC colonies from non-iPSC colonies by their morphological differences and/or alkaline phosphatase activity (Rand et al., 2018; Takahashi et al., 2007).
Luciferase reporter assay
First, we cultured NAT1 cKO human iPSCs with or without Dox for two days. One day before transfection, we plated them at a density of 1 × 105 cells per a well of LN511-coated 24-well plate in same culture condition. One microgram of pGL4.13-based reporter plasmid and 25 ng of pGL4.74 were co-transfected using FuGENE HD transfection reagent as described elsewhere. Two days after transfection, the cells were lysed by adding 0.1 ml of 1x Passive lysis buffer (Promega). Ten microliters of lysates were used for the measurement of luciferase activity. We added 25 μl of Luciferase Assay Substrate (Promega) reconstituted in Luciferase Assay Buffer II (Promega) to measure the firefly luciferase (Fluc) activity. Then, we added 25 μl of 1x Stop & Glo Substrate (Promega) diluted in Stop & Glo Buffer (Promega) to quench the Fluc activity and raise the renilla luciferase (Rluc) activity. The measurement was performed using a SpectraMax i3 (Molecular Devices) and SoftMax Pro software 6.4 (Molecular Devices).
Chromatin Immunoprecipitation (ChIP)
The ChIP assay was performed using ChIP-IT Enzymatic kit (Active Motif) according to manufacturer’s protocol. In brief, we added 0.27 ml of 37% formaldehyde solution to a 10 ml culture medium and incubate on a shaking platform for 10 min at room temperature. Immediately after fixation, cross-linking was quenched by adding 1 ml of 1.25 M glycine solution and incubating for 5 min at room temperature with constant agitation. Then the cells were washed with PBS and pelletized by spinning down. The pellet was resuspended in 1x lysis buffer and incubated for 30 min on ice. Nuclei were released by douncing on ice with 10 strokes in a dounce homogenizer. Pelletized nuclei were resuspendend in the digestion buffer and incubated at 37°C for 5 min. Then we added enzymatic sharing cocktail to pre-warmed nuclei and incubated the mixture at 37°C for 10 min. The digestion was stopped by adding ice-cold 0.5 M EDTA and chilled the reaction for 10 min on ice. Cleared supernatant after centrifugation was collected and 10% volume of the samples were removed as the input control DNAs. The rest (90%) of samples were split and used for immunoprecipitation with anti-RNA polymerase II (Abcam) or normal mouse IgG (EMD Millipore) antibodies. Three micrograms of each antibody was incubated with 50 μl of Protein G magnetic beads for 1 h at 4°C with constant rotation prior to use. After 4 h incubation, the samples were washed once with ChIP Buffer 1 and twice with ChIP Buffer 2. After washing, the beads were resuspended in 50 μl of Elution buffer and incubated for 15 min at room temperature. Then we added the Reverse cross-linking buffer to immunoprecipitated samples. Immunoprecipitated samples and input DNAs were treated with protease K and purified by using QIAquick PCR purification kit (QIAGEN). Quantification was performed by PCR using Power SYBR Green PCR Master Mix on an ABI7900HT Real Time PCR System. Primer sequences were provided in Table S3.
RNA pulldown assay
To utilize the FLAG-tag/antibody system for RNA immunoprecipitation assay, we first prepared human iPSCs expressing FLAG-tagged NAT1 by introducing SB-CAG-NAT1–3xFLAG-IP into NAT1 cKD human iPSCs. In the presence of Dox, the cells express FLAG-tagged NAT1 but not endogenous one. Because we introduced FLAG-tagged NAT1 as a transgene to generate mouse NAT1 cKO EpiSCs, we used this cell line for the assay. For both mouse and human experiments, parental cell lines such as 1B4 human iPSCs and X-GFP mouse EpiSCs which did not express FLAG-tagged NAT1 were used as negative controls. RNA immunoprecipitation was performed as previously described (Worringer et al., 2014). The cells were harvested by treating with Accutase and pelleted by centrifugation at 250 xg for 5 min at 4°C, flash frozen by soaking into liquid nitrogen. Cells were resuspended in five times volume of lysis buffer (20mM HEPES (Sigma-Aldrich), pH 7.9, 150 mM NaCl (Teknova), 0.5 mM ethylenediaminetetraacetic acid (EDTA, Thermo Fisher Scientific), 0.05% Nonidet-P 40 (Sigma-Aldrich) and Complete EDTA-free Protease Inhibitor Cocktail (Sigma-Aldrich)) and blended for 3 min on ice with the Tissue Tearer (setting 1, Biospec Products). Insoluble material was removed by centrifugation at 20,000 xg for 20 min. Collected the supernatant and adjusted to 5 mg/ml final concentration. Seventy-five microliters of Dynabeads Protein G (Thermo Fisher Scientific) and 30 μg of rat monoclonal anti-DYKDDDDK antibody (BioLegend) were mixed and rotated for 1 hr at room temperature, then rinsed four times with lysis buffer. The extract (7.5 mg) was added and the samples were rotated at 4°C overnight. Beads were washed twice with lysis buffer and then three times with wash buffer (20 mM HEPES, pH 7.9, 150 mM NaCl, 0.5 mM EDTA and Complete EDTA-free Protease Inhibitor Cocktail) each for 5 min with rotation at 4°C. Finally, the beads were resuspended in 20 μl of wash buffer supplemented with 100 μg/ml 3xFLAG peptide (Sigma-Aldrich) and 0.05% Rapigest (Waters) for the elution. Co-precipitated RNA was isolated by using QIAzol lysis reagent and a miRNeasy Mini kit with DNase-I on-column treatment.
Eighty nanograms of purified RNA was used for first-strand cDNA synthesis and following second strand cDNA synthesis with Ovation RNA-seq System V2 kit (NuGEN) according to manufacturer’s recommendation. Double stranded DNA was amplified by single primer isothermal amplification (SPIA), where polymerase initiated replication at the 3’ end of the SPIA primer, and random hexamers were used to amplify the second-strand cDNA linearly. Libraries were then prepared by using Ovation Ultralow System V2 kit (NuGEN) and analyzed their quality by Agilent 2100 Bioanalyzer (Agilent Technologies) and KAPA Library Quantification Kit illumina Platforms (KAPA Biosystems). The libraries were pooled and sequenced on a NextSeq sequencer (illumina) using NextSeq 500/550 High Output V2 kit (150 cycles) (illumina).
Raw reads were trimmed for low quality reads using default settings of fastq-mcf from ea-utils 1.12–537 (Aronesty, 2013). Trimmed reads were aligned to hg19 genome and transcriptome using STAR 2.5.2a (Dobin et al., 2013) for spliced reads, and Bowtie 2.2.4 (Langmead and Salzberg, 2012) for unspliced reads. Reads were assigned to genes using featureCounts (Liao et al., 2014) that is part of Subread suite (http://subread.sourceforge.net). The edgeR program (Robinson et al., 2010) was used to normalize raw read counts, calculate FPKM, and analyze differential expression using negative bionmial p-value following Benjamaini and Hochberg multiple testing correction. Differentially expressed genes were identified by: Log2 fold change (FC) > 0.585 for upregulated, < −0.585 for downregulated; adjusted false discovery rate (FDR) < 0.05, and counts > 3.
Nascent RNA quantification
Quantification of nascent transcribed RNA was performed using Click-iT Nascent RNA Capture Kit (Thermo Fisher Scientific) according to manufacturer’s protocol. In brief, we cultured Control iPSCs, NAT1 cKD iPSCs and TUT7 cKD iPSCs in the presence of Dox for three days to induce knockdown. Then, we added 0.2 mM of 5-ethynyl Uridine (EU) into the cultures and incubate them at 37°C for 1h. Immediately after the incubation, the cells were lysed with QIAzol lysis reagent and total RNAs were purified using a miRNeasy Mini kit. One microgram of purified RNA was used for clicking EU-labeled RNAs to biotin-azide. After the reaction, RNA was purified by ethanol precipitation. Purified RNAs were heated at 70°C for 5 min to disrupt secondary structure and biotinylated RNAs were collected by incubating with Dynabeads-Streptavidin. After washing out of unlabeled RNAs, the beads, along with 1 ng of drosophila RNA (Clontech) as a spike-in, were used for RT reaction.
Uridylated RNA detection
Quantification of uridylated RNA was performed as described previously (Pirouz et al., 2016). One microgram of purified total RNA was used for cDNA synthesis by using SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific) with random hexamers and oligo dA12 primer for total expression and uridylated RNA, respectively.
Polysome fractionation
Method for polysome fractionation was adapted from the previously published protocol (McGlincy and Ingolia, 2017). Briefly, one semiconfluent 100 mm dish of adherent cells were treated with 100 μg/ml Cycloheximide (Sigma-Aldrich) for 5 min at 37°C. For the preparation of harringtonine treated control, the cells were treated with 2 μg/ml harringtonine (Abcam) at 37°C for 3 min, then we added 100 μg/ml Cycloheximide to the media. Cells were placed on ice and gently washed twice with 5 ml ice-cold PBS. Then, the cells were scraped and dissociated in 0.4 ml freshly prepared ice-cold lysis buffer (20 mM Tris-HCl, pH7.5, 150 mM NaCl, 5 mM MgCl2 (Teknova), 1 mM dithiothreitol (DTT, Teknova), Complete EDTA-free Protease Inhibitor Cocktail, 100 μg/ml Cycloheximide, 1% TritonX-100, 25 units/ml Turbo DNase (Thermo Fisher Scientific) and 100 units/ml SUPERaseIn (Thermo Fisher Scientific) and collected into a 1.5 ml chilled DNA LoBind Tube (Eppendorf). The lysate was incubated on ice for 10 min, triturated through a 25-gauge needle (Terumo) ten times before centrifugation at 20,000 xg for 10 min at 4°C. The supernatant was collected to a new 1.5 ml tube. For the preparation of EDTA-treated control, we added 1/15 volume of 0.5 M EDTA (final concentration is 30 mM) to the cleared lysate. Samples were flash-frozen in liquid nitrogen and stored at −80°C.
A 10–45% continuous sucrose gradient contained 10% and 45% sucrose solutions (Sigma-Aldrich) respectively in 100 μg/ml Cycloheximide and 1 mM DTT in polysome buffer (25 mM Tris-HCl, pH7.5, 150 mM NaCl and 15 mM MgCl2) in a 14 × 89 mm polyclear tube (Seton) and was formed using Biocomp Gradient Master program (Biocomp). An equal amount of cell lysate of each sample (300 μl) was loaded onto the 10% to 45% continuous sucrose gradient. Polysomes were separated in sucrose gradient by centrifugation in a Beckmann ultracentrifuge using a SW-41 rotor (Beckman Coulter) at 36,000 rpm for 2.5 h at 4°C. Profile of relative RNA abundance of ribosomal subunits, monosomes, and polysomes was visualized at 254-nm wavelength, and equal-volume fractions were collected simultaneously with the Biocomp Piston Gradient Fractionator (Biocomp). For RNA analysis, an equal sample volume of TRIzol LS reagent (Thermo Fisher Scientific) was immediately added to the fractions. RNA was purified using miRNeasy mini kit according to manufacturer instruction. Purified RNAs along with 1 ng of drosophila spike RNA were used for RT reaction and following qRT-PCR.
Gene expression analysis by microarray
Microarray was performed as described previously (Rand et al., 2018). The total RNA was purified as described above and evaluated using a 2100 Bioanalyzer (Agilent Technologies). Two hundred nanograms of total RNA were labeled with Cyanine 3-CTP and used for hybridization with SurePrint G3 Human GE 8×60K ver. 3 (G4851C, Agilent Technologies) for human samples and SurePrint G3 Mouse GE 8×60K ver. 2 (G4852B, Agilent Technologies) for mouse samples with the one-color protocol. The arrays were scanned with a Microarray Scanner System (G2565BA, Agilent Technologies), and extracted signals were analyzed using the GeneSpring version 14.9.1 software program (Agilent Technologies). Gene expression values were normalized by the 75th percentile shifts. Gene ontology (GO) analyses were performed by using DAVID Bioinformatics Resources 6.7 (Huang da et al., 2009). The analyses were performed using top 300 differentially expressed genes between Dox (+) and (−) conditions with statistically significance (FC>2, FDR<0.05).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as mean ± standard deviation. Sample number (n) indicates the number of replicates in each experiment. The number of experimental repeats are indicated in figure legends. To determine statistical significance, we used unpaired t-test for the comparisons between two groups using the Excel 2016 (Microsoft) unless otherwise noted. Statistical significance in main figures was set at p < 0.05 indicated by asterisk. Error bars represent mean ± s.d.
Supplementary Material
Table S3 | Sequence information (Related to STAR Method)
The sequences of oligo DNAs used for PCR and cloning and homologous arms used for gene targeting are shown.
Table S1 | RNAs co-precipitated with FLAG-tagged NAT1 in human iPSCs (Related to Figure 5)
Shown are the gene ontology, gene name, fold change (FC) and p-value of RNA which were co-precipitated with human NAT1-FLAG protein. FC and p-value were calculated with the comparison with the results of negative control. n=3.
Table S2 | RNAs co-precipitated with FLAG-tagged NAT1 in mouse EpiSCs (Related to Figure 5)
Shown are the gene ontology, gene name, fold change (FC) and p-value of RNA which were co-precipitated with mouse NAT1-FLAG protein. FC and p-value were calculated with the comparison with the results of negative control. n=3.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-OCT3/4 | Santa Cruz Biotechnology | sc-5279, RRID:AB_628051 |
| Rabbit polyclonal anti-NANOG | Abcam | Ab21624, RRID:AB_446437 |
| Mouse monoclonal anti human Nuclei | EMD Millipore | MAB4383, RRID:AB_827439 |
| Rabbit polyclonal anti-alpha fetoprotein | DAKO | A0008, RRID:AB_2650473 |
| Mouse monoclonal anti-alpha fetoprotein | R&D Systems | MAB1368, RRID:AB_357658 |
| Mouse monoclonal anti-smooth muscle actin | DAKO | M0851, RRID:AB_2223500 |
| Mouse monoclonal anti-TUBULIN, Beta III | EMD Millipore | MAB1637, RRID:AB_2210524 |
| Rabbit polyclonal anti-PAX6 | BioLegend | 901301, RRID:AB_2565003 |
| Mouse monoclonal anti-NESTIN | Stem Cell Technologies | 60091, RRID:AB_2650581 |
| Rabbit polyclonal anti-SOX1 | Stem Cell Technologies | 60095 |
| Mouse monoclonal anti-β-ACTIN | Sigma-Aldrich | A5441, RRID:AB_476744 |
| Rabbit polyclonal anti-β-ACTIN | Abcam | ab8227, RRID:AB_2305186 |
| Rabbit polyclonal anti-SOX2 | Abcam | ab97959, RRID:AB_2341193 |
| Rabbit polyclonal anti-KLF4 | Santa Cruz Biotechnology | sc-20691, RRID:AB_669567 |
| Goat polyclonal anti-TFCP2L1 | R&D Systems | AF5726, RRID:AB_2202564 |
| Rabbit polyclonal anti-TFE3 | Sigma-Aldrich | HPA023881, RRID:AB_1857931 |
| Rabbit polyclonal anti-ZCCHC6 | Sigma-Aldrich | HPA020620, RRID:AB_1858984 |
| Rabbit polyclonal anti-ZCCHC6 | Proteintech | 25196–1-AP |
| Mouse monoclonal anti-NAT1 | BD Biosciences | 610742, RRID:AB_398065 |
| Rabbit polyclonal anti-ZCCHC11 | Proteintech | 18980–1-AP, RRID:AB_10598327 |
| Mouse monoclonal anti RNA polymerase II | Abcam | ab817, RRID:AB_306327 |
| Normal mouse IgG | EMD Millipore | 12–371, RRID:AB_145840 |
| Rat monoclonal anti-DYKDDDDK (L5) | BioLegend | 637301, RRID:AB_1134266 |
| IRDye680LT anti-mouse IgG | LI-COR Biosciences | 926–68022, RRID:AB_10715072 |
| IRDye680LT anti-rabbit IgG | LI-COR Biosciences | 925–68021 RRID:AB_10795015 |
| IRDye800CW anti-mouse IgG | LI-COR Biosciences | 925–32212, RRID:AB_2716622 |
| IRDye800CW anti-rabbit IgG | LI-COR Biosciences | 925–32213, RRID:AB_2715510 |
| Horseradish peroxidase-linked anti-mouse IgG | Cell Signaling Technology | 7076, RRID:AB_330924 |
| Horseradish peroxidase-linked anti-rabbit IgG | Cell Signaling Technology | 5127, RRID:AB_10892860 |
| Alexa 488-conjugated donkey anti-mouse IgG | Thermo Fisher Scientific | A-21202, RRID:AB_141607 |
| Alexa 555-conjugated donkey anti-mouse IgG | Thermo Fisher Scientific | A-31570, RRID:AB_2536180 |
| Alexa 647-conjugated donkey anti-mouse IgG | Thermo Fisher Scientific | A-31571, RRID:AB_162542 |
| Alexa 488-conjugated donkey anti-rabbit IgG | Thermo Fisher Scientific | A-21206, RRID:AB_2535792 |
| Alexa 555-conjugated donkey anti-rabbit IgG | Thermo Fisher Scientific | A-31572, RRID:AB_162543 |
| Alexa 647-conjugated donkey anti-rabbit IgG | Thermo Fisher Scientific | A-31573, RRID:AB_2536183 |
| Alexa 488-conjugaated donkey anti-goat IgG | Thermo Fisher Scientific | A-11055, RRID:AB_2534102 |
| Dynabeads Protein G | Thermo Fisher Scientific | 10003D |
| Anti-Digoxigenin-AP | Roche Life Science | 11093274910, RRID:AB_514497 |
| Chemicals, Peptides, Media and Recombinant Proteins | ||
| Recombinant human basic fibroblast growth factor | Peprotech | 100–18B |
| Recombinant human/murine/rat Activin A | Peprotech | 120–14E |
| Human Leukemia Inhibitory Factor | EMD Millipore | LIF1010 |
| ESGRO Recombinant Mouse LIF Protein | EMD Millipore | ESG1106 |
| Recombinant human bone morphogenetic protein 4 | R&D Systems | 314-BP-010 |
| 1-oleoyl-2-methyl-sn-glycero-3-phosphothionate | Avanti Polar Lipids | 857235 |
| Laminin-511 E8 (iMatrix-511) | Nippi | N-892012 |
| Matrigel | Corning | 356234 |
| Fibronectin bovine plasma | Sigma-Aldrich | F1141 |
| EmbryoMax 0.1% Gelatin solution | EMD Millipore | ES-006-B |
| Y-27632 | Sigma-Aldrich | Y0503 |
| Mitomycin C | Sigma-Aldrich | M4287 |
| Blasticidin S HCl | Thermo Fisher Scientific | A1113903 |
| Geneticin | Thermo Fisher Scientific | 10131035 |
| Puromycin Dihydrochloride | Thermo Fisher Scientific | A1113803 |
| Zeocin | Thermo Fisher Scientific | R25001 |
| Hygromycin B Gold | Invivogen | ant-hg-1 |
| Doxycycline hyclate | Sigma-Aldrich | D9891 |
| MG132 | Calbiochem | 474791 |
| PD0325901 | Stemgent | 04–0006 |
| CHIR99021 | Stemgent | 04–0004 |
| A83–01 | Stemgent | 04–0014 |
| LDN193189 | Stemgent | 04–0074 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | H3570 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technology | 5872 |
| Complete EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | 4693132001 |
| Drosophila melanogaster, Adult Poly A+ RNA | Clontech | 636222 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| Turbo DNase | Thermo Fisher Scientific | AM2238 |
| SUPERaseIn | Thermo Fisher Scientific | AM2694 |
| RNase-Free DNase set | QIAGEN | 79254 |
| UltraPure 0.5 M EDTA, pH8.0 | Thermo Fisher Scientific | 15575020 |
| BSA Fraction V (7.5% solution) | Thermo Fisher Scientific | 15260037 |
| Primate ESC Culture medium | ReproCELL | RCHEMD001-A |
| StemFiT Basic 02 | Ajinomoto | BASIC02 |
| NDiff 227 | Clontech | Y40002 |
| STEMdiff SMADi Neural Induction Kit | Stem Cell Technologies | 08581 |
| STEMdiff Neural Progenitor Medium | Stem Cell Technologies | 05833 |
| TrypLE select | Thermo Fisher Scientific | 12563011 |
| Accutase cell detachment solution | EMD Millipore | SCR005 |
| Dissociation solution for human ESC/iPSC (CTK) | ReproCELL | RCHETP002 |
| Dulbecco’s Modified Eagle Medium (DMEM) | Thermo Fisher Scientific | 10567014 |
| DMEM/F-12 | Thermo Fisher Scientific | 10565018 |
| Glasgow’s MEM (GMEM) | Thermo Fisher Scientific | 11710035 |
| Neurobasal Medium | Thermo Fisher Scientific | 21103049 |
| N2 supplement | Thermo Fisher Scientific | 17502048 |
| B27 supplement | Thermo Fisher Scientific | 17504044 |
| Fetal Bovine Serum | Thermo Fisher Scientific | 16000044 |
| Knockout Serum Replacement | Thermo Fisher Scientific | 10828028 |
| GlutaMax Supplement | Thermo Fisher Scientific | 35050061 |
| MEM Non-Essential Amino Acids Solution | Thermo Fisher Scientific | 11140050 |
| Sodium pyruvate solution | Sigma-Aldrich | S8636 |
| 2-mercaptoethanol | Thermo Fisher Scientific | 21985023 |
| Critical Commercial Assays | ||
| Human Stem Cell Nucleofector Kit 1 | Lonza | VAPH-5012 |
| Mouse Embryonic Stem Cell Nucleofector Kit | Lonza | VAPH-1001 |
| FuGENE HD transfection reagent | Promega | E2311 |
| FuGENE 6 transfection reagent | Promega | E2691 |
| Retro-X Concentrator | Clontech | 631455 |
| Polybrene (Hexadimethrine Bromide) | EMD Millipore | TR-1003-G |
| Alkaline Phosphatase Detection Kit | EMD Millipore | SCR004 |
| RIPA buffer | Sigma-Aldrich | R0278 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technology | #5872 |
| iBlot Transfer Stack, nitrocellulose | Thermo Fisher Scientific | IB301001 |
| NuPAGE MES SDS Running Buffer (20X) | Thermo Fisher Scientific | NP0002 |
| NuPAGE 4–12% Bis-Tris Protein Gels | Thermo Fisher Scientific | NP0327BOX |
| NuPAGE Sample Reducing Agent | Thermo Fisher Scientific | NP0004 |
| NuPAGE Antioxidant | Thermo Fisher Scientific | NP0005 |
| NuPAG LDS Sample Buffer | Thermo Fisher Scientific | NP0007 |
| Odyssey Blocking Buffer (TBS) | LI-COR Biosciences | 927–50000 |
| Immobilon Western Chemiluminescent HRP Substrate | EMD Millipore | WBKLS0500 |
| Fixation buffer for immunocytochemistry | BioLegend | 420801 |
| 10% TritonX-100 solution | Teknova | T1105 |
| Normal donkey serum | Sigma-Aldrich | D9663- |
| Sucrose | Sigma-Aldrich | S0389 |
| Harringtonine | Abcam | ab141941 |
| Rapigest SF Surfactant | Waters | 186001861 |
| QIAzol lysis reagent | QIAGEN | 79306 |
| Trizol LS reagent | Thermo Fisher Scientific | 10296028 |
| miRNeasy Mini kit | QIAGEN | 217004 |
| Direct-zol RNA kit | ZYMO Research | R2060 |
| SuperScript III First-Strand Synthesis SuperMix for qRT-PCR | Thermo Fisher Scientific | 11752050 |
| SuperScript III First-Strand Synthesis System for RT-PCR | Thermo Fisher Scientific | 18080051 |
| QIAquick PCR purification kit | QIAGEN | 28104 |
| DNeasy Blood & Tissue Kit | QIAGEN | 69504 |
| DIG Easy Hyb buffer | Roche Life Science | 11603558001 |
| CDP-Star | Roche Life Science | 11685627001 |
| KOD Xtreme Hot Start DNA Polymerase | EMD Millipore | 71975 |
| In-Fusion HD Cloning Kit | Clontech Laboratories | 639648 |
| TaqMan Universal Master Mix II, no UNG | Thermo Fisher Scientific | 4440040 |
| Power SYBR Green Master Mix | Thermo Fisher Scientific | 4367659 |
| Dual-Luciferase Reporter Assay System | Promega | E1910 |
| ChIP-IT Express Enzymatic | Active Motif | 53009 |
| Click-iT Nascent RNA Capture Kit | Thermo Fisher Scientific | C10365 |
| Q-bit BR Assay | Thermo Fisher Scientific | Q10210 |
| SurePrint G3 Human GE 8×60K v3 | Agilent Technologies | G4851C |
| SurePrint G3 Mouse GE 8×60K v2 | Agilent Technologies | G4852B |
| Ovation RNA-seq System V2 kit | NuGEN | 7102 |
| Ovation Ultralow System V2 kit | NuGEN | 0344 |
| KAPA Library Quantification Kit illumina Platforms | KAPA Biosystems | KR0405 |
| NextSeq 500/550 High Output v2 kit | illumina | FC-404–2002 |
| NAT1 (Hs00154952_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| ZCCHC6 (Hs00612265_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| POU5F1 (Hs04260367_gH) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| SOX2 (Hs01053049_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| NANOG (Hs02387400_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| SOX1 (Hs01057642_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| PAX6 (Hs00240871_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| NES (Hs04187831_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| SOX17 (Hs00751752_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| PDGFRA (Hs00998018_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| DCN (Hs00754870_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| HNF4A (Hs00230853_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| HAND1 (Hs02330376_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| MSX1 (Hs00427183_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| MAP2 (Hs00258900_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| NEUROD1 (Hs01922995_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| GDF3 (Hs00220998_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| NODAL (Hs00415443_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| T (Hs00610080_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| GATA4 (Hs00171403_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| KLF2 (Hs00360439_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| TFCP2L1 (Hs00232708_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| DNMT3B (Hs00171876_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| EGR1 (Hs00152928_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| ESRG (Hs03666618_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| LINC-ROR (Hs04332550_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| ABHD12B (Hs00997975_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| OC90 (Hs00903174_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| HHLA1 (Hs00194800_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| NEAT1 (Hs03453535_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| H19 (Hs00399294_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| MALAT1 (Hs00273907_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| GAPDH (Hs02786624_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Zcchc6 (Mm00463475_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Pou5f1 (Mm03053917_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Nanog (Mm02019550_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Sox2 (Mm03053810_s1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Klf2 (Mm00500486_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Klf4 (Mm00516104_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Tbx3 (Mm01195726_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Zfp42 (Mm03053975_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Dppa3 (Mm01184198_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Nr0b1 (Mm00431729_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Fgf5 (Mm00438919_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Cer1 (Mm00515474_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Hnf4a (Mm01247712_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Krt8 (Mm04209403_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Des (Mm00802455_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Flk1 (Mm012222421_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Gfap (Mm01253033_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Map2 (Mm00485231_m1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Actb (Mm02619580_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| gammaTub23C (Dm01841764_g1) TaqMan Assay | Thermo Fisher Scientific | 4331182 |
| Deposited Data | ||
| Raw and analyzed data (Gene expression microarray) | This paper | GSE129429 |
| Raw images | Mendeley | http://dx.doi.org/10.17632/r4wjcwyzkj.1 |
| Experimental Models: Cell Lines | ||
| 585A1 human iPSC line | (Okita et al., 2011) | RRID:CVCL_DQ06 |
| NAT1 conditional knockout human iPSC line | This study | N/A |
| X-GFP mouse EpiSC line | (Bao et al., 2009) | N/A |
| NAT1 conditional knockout mouse EpiSC line | This study | N/A |
| CRISPRi Gen B human iPSC line | (Mandegar et al., 2016) | RRID:CVCL_VM35 |
| NAT1 conditional knockdown human iPSC line | This study | N/A |
| TUT7 conditional knockdown human iPSC line | This study | N/A |
| RF8 mouse ESC line | (Meiner et al., 1996) | RRID:CVCL_T793 |
| SNL mouse embryonic fibroblast cell line | (McMahon and Bradley, 1990) | RRID:CVCL_K227 |
| Primary mouse embryonic fibroblasts | This study | N/A |
| H9 ESC-derived neural progenitor cells | Thermo Fisher Scientific | N7800200, RRID:CVCL_IU37 |
| PLAT-GP | CELL BIOLABS, INC. | RV-103 RRID:CVCL_B490 |
| Oligonucleotides | ||
| Sequences of primers, gRNA and the homologous regions for gene targeting used in this study | This study | see Table S3 |
| Recombinant DNA | ||
| PB-TRE-NAT1-CRB | This study | N/A |
| PB-TRE-NAT1–3xFLAG-CRZ | This study | N/A |
| pHL-H1-ccdB-EF1a-RiH | (Li et al., 2015) | Addgene #60601 |
| pHL-H1-EF1a-RiH: human NAT1 gRNA | This study | N/A |
| pHL-EF1a-SphcCas9-iC-A | (Li et al., 2015) | Addgene #60599 |
| eSpCas9 (1.1) | (Slaymaker et al., 2016) | Addgene #71814 |
| eSpCas9 (1.1): mouse NAT1 gRNA | This study | N/A |
| PB-U6-CNCB | This study | N/A |
| PB-U6-CNCB: human NAT1 gRNA | This study | N/A |
| PB-U6-CNCB: human TUT7 gRNA | This study | N/A |
| pCW-hyPBase | This study | N/A |
| SB-CAG-NAT1–3xFLAG-CRZ | This study | N/A |
| SB-U6-CNKH | This study | N/A |
| SB-U6-CNKH: HERV-H shRNA | This study | N/A |
| pCW-SB100X | This study | N/A |
| pMXs-OCT3/4 | Addgene | #17217 |
| pMXs-SOX2 | Addgene | #17218 |
| pMXs-KLF4 | Addgene | #17219 |
| pMXs-c-MYC | Addgene | #17220 |
| pMXs | CELL BIOLABS, INC. | RTV-010 |
| pMD2.G | Addgene | #12259, RRID:Addgene_12259 |
| pGL4.13 | Promega | E6681 |
| pGL4.13: human TUT7 5’ UTR | This study | N/A |
| pGL4.13: mouse TUT7 5’ UTR | This study | N/A |
| pGL4.13: human GAPDH 5’ UTR | This study | N/A |
| pGL4.74 | Promega | E6921 |
| pGL4.13+RL | This study | N/A |
| pGL4.13+RL: human TUT7 5’ UTR | This study | N/A |
| pGL4.13+RL: mouse TUT7 5’ UTR | This study | N/A |
| pGL4.13+RL: EMCV IRES | This study | N/A |
| pBS-Puro-bGHpA | This study | N/A |
| pBS-Neo-SV40pA | This study | N/A |
| pBS-BSD-SV40pA | This study | N/A |
| Human NAT1 targeting vector (Puro) | This study | N/A |
| Human NAT1 targeting vector (Neo) | This study | N/A |
| Mouse NAT1 targeting vector (Puro) | This study | N/A |
| Mouse NAT1 targeting vector (BSD) | This study | N/A |
| Software and Algorithms | ||
| GeneSpring version 14.9.1 | https://www.agilent.com/ | Agilent Technologies |
| DAVID Bioinformatics Resources 6.7 | https://david.ncifcrf.gov/home.jsp | (Huang da et al., 2009) |
| fastq-mcf (in ea-utils) | https://expressionanalysis.github.io/ea-utils/ | (Aronesty, 2013) |
| STAR Aligner | https://github.com/alexdobin/STAR | (Dobin et al., 2013) |
| Bowtie | http://bowtie-bio.sourceforge.net/index.shtml | (Langmead and Salzberg, 2012) |
| featureCounts | http://subread.sourceforge.net/ | (Liao et al., 2014) |
| edgeR | https://bioconductor.org/packages/release/bioc/html/edgeR.html | (Robinson et al., 2010) |
| Excel 2016 | https://www.office.com/ | Microsoft |
| GraphPad Prism 8.0.2 | https://www.graphpad.com/scientificsoftware/prism/ | GraphPad |
ACKNOWLEDGMENTS
We would like to thank A. Hotta, MA. Mandegar, K. Okita, K. Tomoda and D. Trono for providing materials, J. Cate, J. Chen, TA. Rand, D. De Silva, K. Tanabe and J. Weissman for discussions, and J. Johnson, M. Khurram, M. Lancero, M. Nakamura, D. Santos, D. Swaney, E. Tomoda and the Gladstone Core Facilities of Stem Cell and Bioinformatics for technical assistance. We are also grateful to K. Essex, R. Fujiwara, K. Higashi, H. Imagawa, K. Kamegawa, R. Kato, E. Minamitani, Y. Miyake, M. Saito, S. Takeshima and Y. Uematsu for administrative support, and P. Karagiannis for editorial assistance. This work was supported by Grants-in-Aid for Scientific Research from the Japanese Society for the Promotion of Science (JSPS) and from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT); a grant from the Funding Program for World-Leading Innovative Research and Development in Science and Technology (First Program) of the JSPS; a grant from Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine, MEXT; a grant from Japan Foundation for Applied Enzymology; and the iPS Cell Research Fund. The study was also supported by funding from Mr. Hiroshi Mikitani, Mr. Marc Benioff, the L.K. Whittier Foundation, the Roddenberry Foundation, the Gladstone Institutes, and NHLBI/NIH (U01-HL100406, U01-HL098179, R01-HL130533, R01-HL135358). The Gladstone Institutes received support from a National Center for Research Resources Grant RR18928–01.
Footnotes
DECLARATION OF INTERESTS
K.T. is on the scientific advisory board of I Peace, Inc. without salary. B.R.C. is a scientific advisor to Tenaya Therapeutics. S.Y. is a scientific advisor (without salary) of iPS Academia Japan.
REFERENCES
- Aronesty E (2013). Comparison of Sequencing Utility Programs. The Open Bioinformatics Journal 7, 1–8. [Google Scholar]
- Bao S, Tang F, Li X, Hayashi K, Gillich A, Lao K, and Surani MA (2009). Epigenetic reversion of post-implantation epiblast to pluripotent embryonic stem cells. Nature 461, 1292–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortes-Garrido R, Gkatza N, et al. (2016). Stem cell function and stress response are controlled by protein synthesis. Nature 534, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari SIA, Truesdell SS, Lee S, Kollu S, Classon A, Boukhali M, Jain E, Mortensen RD, Yanagiya A, Sadreyev RI, et al. (2016). A Specialized Mechanism of Translation Mediated by FXR1a-Associated MicroRNP in Cellular Quiescence. Molecular cell 61, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers SM, Fasano1 CA, Papapetrou EP, Tomishima M, Sadelain M, and Studer L (2009). Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature biotechnology 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Parra C, Ernlund A, Alard A, Ruggles K, Ueberheide B, and Schneider RJ (2018). A widespread alternate form of cap-dependent mRNA translation initiation. Nature communications 9, 3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi D, Samata B, Katsukawa M, Kikuchi T, Morizane A, Ono Y, Sekiguchi K, Nakagawa M, Parmar M, and Takahashi J (2014). Isolation of human induced pluripotent stem cell-derived dopaminergic progenitors by cell sorting for successful transplantation. Stem cell reports 2, 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. (2014). Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Shani G, Sines T, Marash L, Shohat G, and Kimchi A (2002). The caspase-cleaved DAP5 protein supports internal ribosome entry site- mediated translation of death proteins. Proceedings of the National Academy of Sciences of the United States of America 99, 5400–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henis-Korenblit S, Strumpf NL, Goldstaub D, and Kimchi A (2000). A novel form of DAP5 protein accumulates in apoptotic cells as a result of caspase cleavage and internal ribosome entry site-mediated translation. Mol Cell Biol 20, 496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Imataka H, Olsen HS, and Sonenberg N (1997). A new translational regulator with homology to eukaryotic translation initiation factor 4G. The EMBO journal 16, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (New York, NY) 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kime C, Sakaki-Yumoto M, Goodrich L, Hayashi Y, Sami S, Derynck R, Asahi M, Panning B, Yamanaka S, and Tomoda K (2016). Autotaxin-mediated lipid signaling intersects with LIF and BMP signaling to promote the naive pluripotency transcription factor program. Proceedings of the National Academy of Sciences of the United States of America 113, 12478–12483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi-Aoi M, Ohnuki M, Takahashi K, Okita K, Noma H, Sawamura Y, Teramoto I, Narita M, Sato Y, Ichisaka T, et al. (2013). Differentiation-defective phenotypes revealed by large-scale analyses of human pluripotent stem cells. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed]
- Kozak M (1989). The scanning model for translation: an update. J Cell Biol 108, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Strumpf N, Deiss LP, Berissi H, and Kimchi A (1997). DAP-5, a novel homolog of eukaryotic translation initiation factor 4G isolated as a putative modulator of gamma interferon-induced programmed cell death. Mol Cell Biol 17, 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T, Tanaka M, Amano N, Watanabe A, Sakurai H, et al. (2015). Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem cell reports 4, 143–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics (Oxford, England) 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liberman N, Gandin V, Svitkin YV, David M, Virgili G, Jaramillo M, Holcik M, Nagar B, Kimchi A, and Sonenberg N (2015). DAP5 associates with eIF2beta and eIF4AI to promote Internal Ribosome Entry Site driven translation. Nucleic acids research 43, 3764–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewer S, Cabili MN, Guttman M, Loh YH, Thomas K, Park IH, Garber M, Curran M, Onder T, Agarwal S, et al. (2010). Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nature genetics 42, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Sachs F, Ramsay L, Jacques PE, Goke J, Bourque G, and Ng HH (2014). The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identity. Nat Struct Mol Biol 21, 423–425. [DOI] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell stem cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marash L, Liberman N, Henis-Korenblit S, Sivan G, Reem E, Elroy-Stein O, and Kimchi A (2008). DAP5 promotes cap-independent translation of Bcl-2 and CDK1 to facilitate cell survival during mitosis. Molecular cell 30, 447–459. [DOI] [PubMed] [Google Scholar]
- Mates L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, et al. (2009). Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nature genetics 41, 753–761. [DOI] [PubMed] [Google Scholar]
- McGlincy NJ, and Ingolia NT (2017). Transcriptome-wide measurement of translation by ribosome profiling. Methods (San Diego, Calif) 126, 112–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon AP, and Bradley A (1990). The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell 62, 1073–1085. [DOI] [PubMed] [Google Scholar]
- Meiner VL, Cases S, Myers HM, Sande ER, Bellosta S, Schambelan M, Pitas RE, McGuire J, Herz J, and Farese RV Jr. (1996). Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: evidence suggesting multiple cholesterol esterification enzymes in mammals. Proceedings of the National Academy of Sciences of the United States of America 93, 14041–14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, and Jaffrey SR (2015). 5’ UTR m(6)A Promotes Cap-Independent Translation. Cell 163, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki T, Futaki S, Suemori H, Taniguchi Y, Yamada M, Kawasaki M, Hayashi M, Kumagai H, Nakatsuji N, Sekiguchi K, et al. (2012). Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nature communications 3, 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita S, Kojima T, and Kitamura T (2000). Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 7, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa M, et al. (2014). A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep 4, 3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols J, and Smith A (2009). Naive and primed pluripotent states. Cell stem cell 4, 487–492. [DOI] [PubMed] [Google Scholar]
- Ohnuki M, Tanabe K, Sutou K, Teramoto I, Sawamura Y, Narita M, Nakamura M, Tokunaga Y, Watanabe A, Yamanaka S, et al. (2014). Dynamic regulation of human endogenous retroviruses mediates factor-induced reprogramming and differentiation potential. Proceedings of the National Academy of Sciences of the United States of America 111, 12426–12431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, Hong H, Nakagawa M, Tanabe K, Tezuka K, et al. (2011). A more efficient method to generate integration-free human iPS cells. Nature methods 8, 409–412. [DOI] [PubMed] [Google Scholar]
- Pirouz M, Du P, Munafo M, and Gregory RI (2016). Dis3l2-Mediated Decay Is a Quality Control Pathway for Noncoding RNAs. Cell reports 16, 1861–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand TA, Sutou K, Tanabe K, Jeong D, Nomura M, Kitaoka F, Tomoda E, Narita M, Nakamura M, Nakamura M, et al. (2018). MYC Releases Early Reprogrammed Human Cells from Proliferation Pause via Retinoblastoma Protein Inhibition. Cell reports 23, 361–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics (Oxford, England) 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoni FA, Guerra J, and Luban J (2012). HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 9, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaughnessy JD Jr., Jenkins NA, and Copeland NG (1997). cDNA cloning, expression analysis, and chromosomal localization of a gene with high homology to wheat eIF-(iso)4F and mammalian eIF-4G. Genomics 39, 192–197. [DOI] [PubMed] [Google Scholar]
- Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, and Zhang F (2016). Rationally engineered Cas9 nucleases with improved specificity. Science (New York, NY) 351, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama H, Takahashi K, Yamamoto T, Iwasaki M, Narita M, Nakamura M, Rand TA, Nakagawa M, Watanabe A, and Yamanaka S (2017). Nat1 promotes translation of specific proteins that induce differentiation of mouse embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America 114, 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Maruyama M, Tokuzawa Y, Murakami M, Oda Y, Yoshikane N, Makabe KW, Ichisaka T, and Yamanaka S (2005). Evolutionarily conserved non-AUG translation initiation in NAT1/p97/DAP5 (EIF4G2). Genomics 85, 360–371. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Taylor DM, Aronow BJ, Tan K, Bernt K, Salomonis N, Greene CS, Frolova A, Henrickson SE, Wells A, Pei L, et al. (2019). The Pediatric Cell Atlas: Defining the Growth Phase of Human Development at Single-Cell Resolution. Developmental cell 49, 10–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theunissen TW, Powell BE, Wang H, Mitalipova M, Faddah DA, Reddy J, Fan ZP, Maetzel D, Ganz K, Shi L, et al. (2014). Systematic identification of culture conditions for induction and maintenance of naive human pluripotency. Cell stem cell 15, 471–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, Alm T, Asplund A, Bjork L, Breckels LM, et al. (2017). A subcellular map of the human proteome. Science (New York, NY) 356. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science (New York, NY) 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science (New York, NY) 357. [DOI] [PubMed] [Google Scholar]
- Ustianenko D, Pasulka J, Feketova Z, Bednarik L, Zigackova D, Fortova A, Zavolan M, and Vanacova S (2016). TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. The EMBO journal 35, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xie G, Singh M, Ghanbarian AT, Rasko T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, et al. (2014). Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature. [DOI] [PubMed]
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Muguruma K, and Sasai Y (2007). A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature biotechnology 25, 681–686. [DOI] [PubMed] [Google Scholar]
- Weingarten-Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S, Oren M, and Kimchi A (2014). The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene 33, 611–618. [DOI] [PubMed] [Google Scholar]
- Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, and Yamanaka S (2014). The let-7/LIN-41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell stem cell 14, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Poksay KS, Arnold KS, and Innerarity TL (1997). A novel translational repressor mRNA is edited extensively in livers containing tumors caused by the transgene expression of the apoB mRNA- editing enzyme. Genes & development 11, 321–333. [DOI] [PubMed] [Google Scholar]
- Yamanaka S, Zhang XY, Maeda M, Miura K, Wang S, Farese RV Jr., Iwao H, and Innerarity TL (2000). Essential role of NAT1/p97/DAP5 in embryonic differentiation and the retinoic acid pathway. The EMBO journal 19, 5533–5541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Yang Y, Chen LL, Wang Y, et al. (2017). Extensive translation of circular RNAs driven by N(6)-methyladenosine. Cell research 27, 626–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, and Smith A (2008). The ground state of embryonic stem cell self-renewal. Nature 453, 519–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoffe Y, David M, Kalaora R, Povodovski L, Friedlander G, Feldmesser E, Ainbinder E, Saada A, Bialik S, and Kimchi A (2016). Cap-independent translation by DAP5 controls cell fate decisions in human embryonic stem cells. Genes & development 30, 1991–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikane N, Nakamura N, Ueda R, Ueno N, Yamanaka S, and Nakamura M (2007). Drosophila NAT1, a homolog of the vertebrate translational regulator NAT1/DAP5/p97, is required for embryonic germband extension and metamorphosis. Development, growth & differentiation 49, 623–634. [DOI] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, and Craig NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proceedings of the National Academy of Sciences of the United States of America. [DOI] [PMC free article] [PubMed]
- Zhao M, Ren C, Yang H, Feng X, Jiang X, Zhu B, Zhou W, Wang L, Zeng Y, and Yao K (2007). Transcriptional profiling of human embryonic stem cells and embryoid bodies identifies HESRG, a novel stem cell gene. Biochemical and biophysical research communications 362, 916–922. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S3 | Sequence information (Related to STAR Method)
The sequences of oligo DNAs used for PCR and cloning and homologous arms used for gene targeting are shown.
Table S1 | RNAs co-precipitated with FLAG-tagged NAT1 in human iPSCs (Related to Figure 5)
Shown are the gene ontology, gene name, fold change (FC) and p-value of RNA which were co-precipitated with human NAT1-FLAG protein. FC and p-value were calculated with the comparison with the results of negative control. n=3.
Table S2 | RNAs co-precipitated with FLAG-tagged NAT1 in mouse EpiSCs (Related to Figure 5)
Shown are the gene ontology, gene name, fold change (FC) and p-value of RNA which were co-precipitated with mouse NAT1-FLAG protein. FC and p-value were calculated with the comparison with the results of negative control. n=3.
Data Availability Statement
Gene expression microarray results are accessible in the Gene Expression Omnibus database of the National Center for Biotechnology Information website (accession number: GSE129429). Uncropped images are available in Mendeley (http://dx.doi.org/10.17632/r4wjcwyzkj.1).