

Abstract
Chimeric antigen receptor (CAR) T cells are highly successful in the treatment of hematologic malignancies. We recently generated affinity-optimized CD38CAR T cells, which effectively eliminate multiple myeloma (MM) cells with little or no toxicities against nonmalignant hematopoietic cells. The lack of universal donors and long manufacturing times however limit the broad application of CAR T cell therapies. Natural killer (NK) cells generated from third party individuals may represent a viable source of “off the shelf” CAR-based products, as they are not associated with graft-versus-host disease unlike allogeneic T cells. We therefore explored the preclinical anti-MM efficacy and potential toxicity of the CD38CAR NK concept by expressing affinity-optimized CD38CARs in KHYG-1 cells, an immortal NK cell line with excellent expansion properties. KHYG-1 cells retrovirally transduced with the affinity-optimized CD38CARs expanded vigorously and mediated effective CD38-dependent cytotoxicity towards CD38high MM cell lines as well as primary MM cells ex vivo. Importantly, the intermediate affinity CD38CAR transduced KHYG-1 cells spared CD38neg or CD38int nonmalignant hematopoietic cells, indicating an optimal tumor nontumor discrimination. Irradiated, short living CD38CAR KHYG-1 cells also showed significant anti-MM effects in a xenograft model with a humanized bone marrow-like niche. Finally, CD38CAR KHYG-1 cells effectively eliminated primary MM cells derived from patients who are refractory to CD38 antibody daratumumab. Taken together, the results of this proof-of-principle study demonstrate the potential value of engineering affinity-optimized CD38CARs in NK cells to establish effective anti-MM effects, with an excellent safety profile, even in patients who failed to response to most advanced registered myeloma therapies, such as daratumumab.
Introduction
Chimeric antigen receptor (CAR) T cells are highly successful in the treatment of hematologic malignancies. After the initial clinical success of CD19CAR T cells in the treatment of CD19 positive B cell malignancies, several promising CAR T cell therapies targeting multiple myeloma (MM)-associated antigens such as B-cell maturation antigen (BCMA), SLAM family member 7 (SLAMF7) and G-protein coupled receptor family C group 5 member (DGPRC5D) are rapidly being developed.1–5 Focusing on the CD38 molecule, which is highly and uniformly expressed on MM cells, we have recently generated affinity-optimized CD38CAR T cells that can effectively eliminate CD38high MM cells without affecting hematopoietic cells that show intermediate or low expression of CD38.6 The clinical success of CAR-based therapies and the rapidly increasing candidate antigens for MM and other hematological malignancies increase the demand to develop more convenient, broadly applicable strategies, that would allow off-the-shelf usage for many patients without being dependent on the personalized generation of CAR based therapeutic cell products for each individual patient from their autologous T cells.7,8 Since CARs recognize their target antigens without major histocompatibility complex (MHC) restriction, off-the-shelf CAR therapy is a viable option. Nonetheless, the risk of graft-versus-host-disease (GvHD) mediated by the endogenous T cell receptor (TCR) and the lack of universal donors currently limit the off-the shelf development of CAR T cells. To tackle this important drawback, extensive efforts are being made to knock down the endogenous TCR.9,10 Another very promising option is to generate universal CAR carriers from primary Natural killer (NK) cells and even cord blood (CB)-derived (NK) cells due to the reduced risk of GvHD associated with NK cells.11–15 Next to the reduced risk of alloreactivity, NK cells can offer other benefits such as ease of expandability, reduced risk of inducing cytokine storm and antigen independent killing.16 Since NK cells exhibit a distinct and different cytokine profile than T cells and have a much shorter lifetime, it is thought that CAR NK cells should lead to less severe cytokine release syndrome and other chronic toxicities than CAR T cells.17 It is however not well established whether NK cells are suitable as CAR carriers for every type of target antigen; in particular for CD38, which is also expressed at intermediate levels on various tissues including nonmalignant hematopoietic cells.18 Even though CD38 is also expressed by a variety of nonhematopoietic cells, we have shown in a previous paper on CD38CAR T cells that CD38 levels in those tissues are much lower than those expressed in the hematopoietic system, even on nonmalignant cells.19
To address the potential efficacy and preclinical safety of CD38CAR NK cells, we now inserted our affinity-optimized CD38CARs into KHYG-1 cells. This is an immortal NK cell line, isolated from a patient with an aggressive NK cell leukemia.20 Previously, this cell line has shown to possess a high cytotoxic capacity mediated by Granzyme M.21 In addition, KHYG-1 cells express sufficient levels of E-selectin ligands, thus making the cells likely to home towards the bone marrow (BM).22,23 We evaluated KHYG-1 cells transduced with affinity-optimized CD38CARs for in vitro/ex vivo anti-MM efficacy using MM cell lines and primary MM cells derived from MM patients as target cells. We also evaluated their CD38-dependent toxicity against nonmalignant hematopoietic cells. After monitoring the effect of long-term expansion as well as irradiation to their CD38CAR-dependent effector functions we tested nonirradiated or irradiated CD38CAR KHYG-1 cells for in vivo anti MM efficacy in a unique model in which MM tumors grow in a human BM tumor microenvironment (TME). Lastly, we explored the ex vivo efficacy of CD38CAR KHYG-1 for the capacity of eliminating primary MM cells derived from patients who are refractory to a clinically applied CD38 antibody, daratumumab.
Materials and methods
Primary cells from MM patients
Mononuclear cells from BM samples containing 2%–40% MM plasma cells were isolated though Ficoll-Paque (GE Healthcare Life Sciences) density centrifugation and cryopreserved in liquid nitrogen until use. When needed, cells were thawed in RPMI-1640 medium supplemented with 50% fetal calf serum (FCS) (heat inactivated, Invitrogen/Thermo Fisher) and incubated (37°C, 5% CO2) for 4 hours prior to use in cytotoxicity assays to rest. Primary material was obtained from patients who were newly diagnosed or refractory to daratumumab. An overview of some important patient characteristics are summarized in Table 1. All patient material and clinical data used in this project has been collected according to the code of conduct for medical research developed by The Council of the Federation of Medical Scientific Societies (FEDERA, https://www.federa.org/codes-conduct).
Table 1.
Patient Characteristics.
| No. of Patients | Age | Gender | Disease Stage | Risk (Genetic Anomaly) | Refractory to | No. of Prior Treatment Lines | Time Between Last Daratumumab Treatment and Sample | ||
|---|---|---|---|---|---|---|---|---|---|
| Lenalidomide | Bortezomib | Daratumumab | |||||||
| 1 | 63 | Male | Newly diagnosed plasma cell leukemia | High risk (+1q; –17p) | NA | NA | NA | NA | NA |
| 2 | 60 | Female | NDMM | Standard risk (+1q) | NA | NA | NA | NA | NA |
| 3 | 73 | Female | NDMM | Standard risk | NA | NA | NA | NA | NA |
| 4 | 69 | Female | NDMM | Standard risk (+1q) | NA | NA | NA | NA | NA |
| 5 | 62 | Female | NDMM | Standard risk (–1p) | NA | NA | NA | NA | NA |
| 6 | 69 | Male | NDMM | Standard risk | NA | NA | NA | NA | NA |
| 7 | 76 | Female | NDMM | Standard risk (+1q) | NA | NA | NA | NA | NA |
| 8 | 70 | Male | NDMM | Standard risk | NA | NA | NA | NA | NA |
| 9 | 63 | Male | NDMM | Standard risk | NA | NA | NA | NA | NA |
| 10 | 54 | Female | NDMM | Standard risk (+1q) | NA | NA | NA | NA | NA |
| 11 | 75 | Female | RRMM (dara naive) | Standard risk | Yes | Yes | No | 4 | NA |
| 12 | 62 | Female | RRMM (dara naive) | Standard risk (+1q) | Yes | Yes | No | 4 | NA |
| 13 | 64 | Male | RRMM (dara naive) | Standard risk (–1p) | No | No | No | 4 | NA |
| 14 | 56 | Male | NDMM | Standard risk | No | No | No | NA | NA |
| 15 | 70 | Male | NDMM | Standard risk (t[11;14]) | No | No | No | NA | NA |
| 16 | 44 | Male | NDMM | Standard risk (–1p) | No | No | No | NA | NA |
| 17 | 65 | Male | RRMM (dara refractory) | ND | Yes | Yes | Yes | >5 | <1 mo |
| 18 | 59 | Female | RRMM (dara refractory) | ND | Yes | Yes | Yes | >5 | <1 mo |
| 19 | 73 | Male | RRMM (dara refractory) | Standard risk | Yes | Yes | Yes | >5 | <1 mo |
| 20 | 60 | Male | RRMM (dara refractory) | ND | Yes | Yes | Yes | 5 | <1 mo |
| 21 | 75 | Male | RRMM (dara refractory) | Standard risk | Yes | No | Yes | 5 | 3.6 mo |
| 22 | 66 | Female | RRMM (dara refractory) | ND | Yes | Yes | Yes | >5 | <1 mo |
Cell lines
KHYG-1 cells were cultured at a concentration between 0.5–1 × 106 cells/mL in complete effector cell medium (RPMI-1640, 10% FCS, 1% PenStrep (Penicillin, 10,000 U/mL; Streptomycin, 10,000 mg/mL) and supplemented by interleukin (IL)-2 (300 international unit [IU] final culture concentration; R&D), unless stated otherwise. In some experiments, KHYG-1 cells were either incubated with IL-15 (PeproTech) at various concentrations or irradiated with 10Gy prior to culture or experiments, as indicated. Target cell lines (CD38+ cell lines UM9 and THP-1; and CD38– cell line U266) were cultured in complete target cell medium (RPMI-1640, 10% FCS [Hyclone], 1% PenStrep). Effector and target cells were split twice weekly with the appropriate fresh medium.
Retroviral transduction
The generation of affinity-optimized second generation CD38CAR constructs containing a CD28 costimulatory domain and a truncated red flurorescent protein (dsRED) marker gene, all linked with 2A sequences has been described earlier.6 The MOCK-CAR consisted of an empty vector containing a truncated low-affinity nerve growth factor receptor (LNGFR) sequence. Retroviral transfection and transduction was done as described previously.24 In short, Phoenix-Ampho packaging cells were transfected with the CAR constructs, gag-pol (pHIT60) and envelope (pCOLT-GALV) vectors (Roche). Two and 3 days after transfection cell-free viral supernatant was collected, concentrated with Retro-X (Takara) according to manufacturer’s instructions (10–100X concentrated). KHYG-1 cells were transduced with the fresh concentrated viral supernatant by spinoculation in the presence of Polybrene (4 µg/mL).24 A second transduction was performed 24 hours later. Four hours after the second transduction, viral supernatant was washed away and the KHYG-1 cells were cultured as described above. Three days post transduction, CAR expression was analyzed by flow cytometry. The overall transduction efficiency/CAR expression was over 85% as deduced from the expression of marker genes LNGFR (MOCK) or dsRED (CD38CAR) (Figure S1, http://links.lww.com/HS/A161). As previously demonstrated, a soluble CD38-aided measurement of the CD38CAR shows high correlation with dsRED expression.6
Flow cytometry
Cells were washed with phosphate-buffered saline (PBS) and stained with various fluorescein-conjugated antibodies for 20 minutes in the dark at 4°C. Post staining, cells were fixated in 1% paraformaldehyde and immediately analyzed on an LSR Fortessa flow cytometer (BD). Several antibodies were used in different panels for identification of phenotype of CAR-KHYG-1 cells as well as target cells: NGFR, CD2, CD3, CD8, CD16, CD14, CD19, CD38, CD56, CD138. Viable cells were determined with a live/dead cell marker (LIVE/DEAD Fixable Near-IR; Life Technologies L10119).
Flow cytometry-based cytotoxicity assays
Flow cytometry-based cytotoxicity assays were performed, as described in detail previously.6 In short, CAR-KHYG-1 or target cells (malignant cell lines or primary bone marrow mononuclear cells [BMNC] from MM patients) were stained with VioletTrace (Thermo Fisher) for 20 minutes in the dark at 20°C, to discriminate effector and target cells. After washing away unbound dye, the cells were co-incubated at indicated effector to target (E:T) ratios in 96 well round-bottom plates for 18–24 hours (37°C, 5% CO2). Post incubation, cells were stained with appropriate markers to further distinguish the MM cells and other cells when necessary. Flow-count fluorophores (Beckman 7547053) were added, and the cells were analyzed with flow cytometry as described in the previous section. Viable single cells were enumerated by excluding dead cells, doublet events and appropriate VioletTrace staining. The percentage of cell lysis was calculated as followed: % lysis cells = (1–[absolute number of viable target cells in treated wells/absolute number of viable target cells in untreated wells]) × 100%. The lysis was only calculated when the target cell population contained at least 500 viable cells in the untreated control. An example of the gating strategy employed can be found in Figure S3 (http://links.lww.com/HS/A161).
In vivo efficacy of CD38CAR KHYG-1 cells
All animal experiments were approved by the local ethical committee for animal experimentation and were in compliance with the Dutch Animal Experimentation Act. The in vivo efficacy of CD38CAR KHYG-1 cells was tested in a humanized bone-marrow-like environment created in recombination activating gene 2 protein (RAG2)–/–γc–/– mice as described earlier.24,25 Briefly, locally bred RAG2–/–γc–/– mice, were subcutaneously inoculated with triphasic calcium phosphate particles (scaffolds) that were coated in vitro with human BM mesenchymal stromal cells (MSCs). After 6–8 weeks post implantation, mice received an IP injection with Busulfan (Busilvex, 18 mg/kg). Twenty-four hours later, Luc + UM9 MM cells (0.25–0.5 million viable cells per scaffold) were injected in the scaffolds under anesthesia. When the tumors became visible under bioluminescence imaging (BLI) (±1 wk) unmodified or irradiated CAR-KHYG-1 cells were injected in the mice IV at the indicated doses and time points together with IP injections of IL-15 (0.5 μg per injection per mouse). Tumor growth was monitored weekly by BLI analysis and by manual tumor-size measurements. Post mortem BM, spleen, blood and scaffolds were harvested; single-cell suspensions were counted, stained, and measured by flow cytometry as described elsewhere.24
Statistics
Statistical analyses were performed using GraphPad Prism software 8 with 2-tailed paired student’s t tests compared to MOCK-KHYG-1 cell values. A P < 0.05 was considered significant. Correlation analysis was done with the Pearson R correlation coefficient analysis while 1-site total was used for a nonlinear robust regression analysis.
Results
In vitro anti-tumor efficacy of CD38CAR KHYG-1 cells
We previously showed that CD38CARs with high or optimized (lower) affinities can effectively eliminate MM and acute myeloid leukemia (AML) cells with high expression of CD38.6 To evaluate the anti-MM efficacy of these CARs when expressed on NK cells, we first transduced KHYG-1 cells with affinity-optimized CD38 CARs, one having an intermediate (CD38A4) and the other a low (CD38B1) affinity to CD38, as is shown in detail in Drent et al.6 Both CARs expressed the CD28 co-stimulatory domain. All CAR transduced KHYG-1 cells expressed the CAR >85% and at high expression levels (median fluorescent intensities [MFIs]) at the cell surface (Figure S1, http://links.lww.com/HS/A161) and expanded to high numbers (Figure S2, http://links.lww.com/HS/A161). When tested against malignant cell lines, both CD38A4CAR and CD38B1CAR transduced KHYG-1 cells effectively killed the CD38+ malignant cell lines UM9 (MM) in a CD38 dependent manner as MOCK lysis levels were much lower. The lysis of THP-1 (AML) cell line by CD38B1CAR KHYG-1 cells was inferior to what was observed with CD38A4CAR KHYG-1 cells (Figure 1A). The control CD38 negative MM cell line, U266, was lysed equally by MOCK and CD38A4CAR transduced KHYG-1 cells, which was indicative of the inherent killing capacity of the KHYG-1 cells against this cell line. Importantly U266 was not lysed above MOCK-lysis levels by CD38CAR KHYG-1 cells, again indicating the CD38-dependency of the lysis of CD38+ UM9 and THP-1 cells (Figure 1A).
Figure 1.
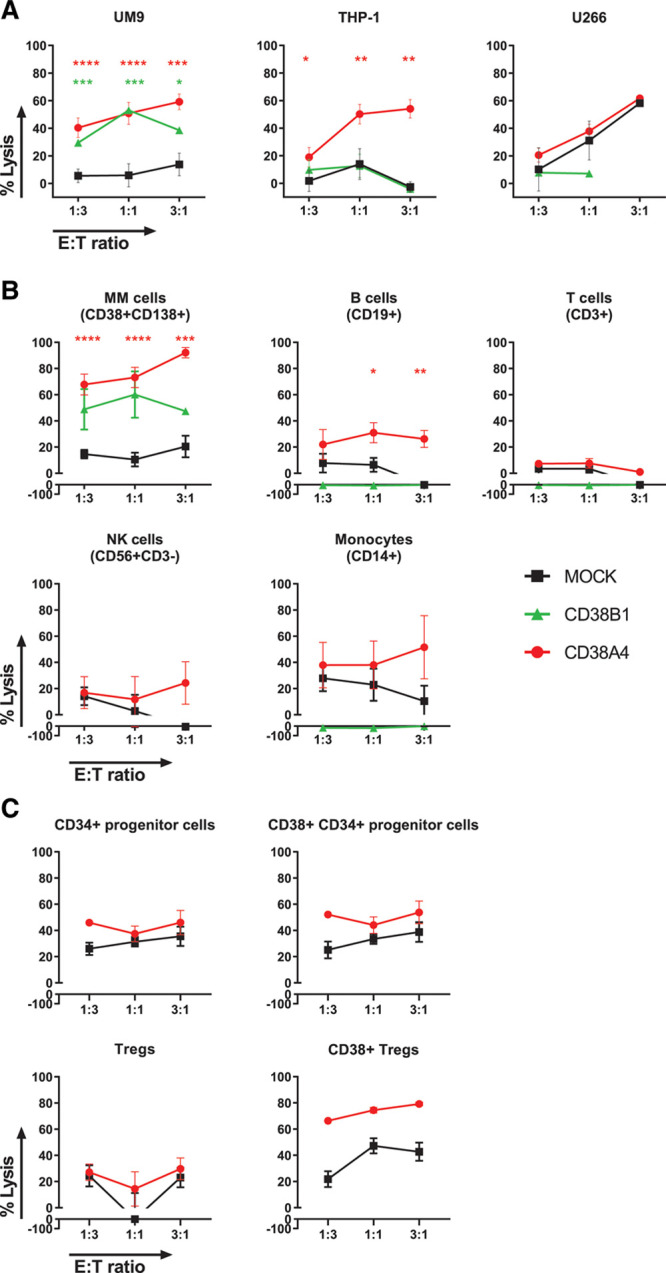
Anti-MM efficacy of KHYG-1 cells transduced with affinity-optimized CD38CARs. The cytotoxic activity of intermediate (A4) and lower affinity (B1) CD38CAR KHYG-1 cells against (A) CD38+ malignant cell lines UM9 (MM) and THP-1 (AML), and against CD38– cell line U266 in flow cytometry based 16–24 h cytotoxicity assays. The cytotoxic activity of MOCK transduced cells was also measured to assess the non-CAR dependent activity (UM9 n = 11, THP-1 n = 5, U266 n = 2). (B), The cytotoxic activity of intermediate (A4) and lower affinity (B1) CD38CAR KHYG-1 cells against malignant and nonmalignant cells in the BMMNCs of newly diagnosed MM patients (n = 16). Lysis of MM cells (CD38+CD138+) was analyzed in 13 patients. The lysis of nonmalignant cells was analyzed in less, but randomly assigned samples; T cells = CD3+ cells (n = 10), NK cells = CD56+CD3– cells (not MM cells, n = 6), B cells = CD19+CD3– cells (n = 5), monocytes = CD14+CD3– cells (n = 8). (C), The cytotoxic activity of intermediate (A4) CD38CAR KHYG-1 cells BMMNCs of newly diagnosed MM patients (n = 4). The lysis of nonmalignant cells was analyzed for T regulatory cells = CD4+CD25+CD127–, CD38+ T regulatory cells, stem cells = CD138– CD34+ and CD38+ stem cells. All data are depicted as mean + SEM of multiple experiments. Colored asterisks indicate significant differences between the relevant group and MOCK cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. AML = acute myeloid leukemia; BMMNCs = bone marrow mononuclear cells; CAR = chimeric antigen receptor; E:T = effector to target; MM = multiple myeloma; NK = natural killer; Tregs = T regulatory cells.
Anti-MM efficacy and on-target off-tumor activities of CD38CAR KHYG-1 cells in primary newly diagnosed patient-derived BMMNCs
We next tested the efficacy of CD38CAR KHYG-1 cells against primary CD138+ MM cells present at 2%–40% in the BMMNCs of MM patients. The BMMNC samples also contained CD38int or CD38neg nonmalignant hematopoietic cells, which enabled us to simultaneously evaluate the off-tumor effects of CD38CAR KHYG-1 cells. Flow cytometry-based cytotoxicity assays revealed that CD38A4CAR KHYG-1 cells mediated effective CD38-dependent lysis against primary CD38highCD138+ MM cells (n = 13), even at low E:T ratios. The MM cell lysis mediated by the lower affinity CD38B1CAR-KHYG-1 cells was significantly lower (Figure 1B). In analysis of nonmalignant cells, both CD38CAR KHYG-1 cells displayed no significant CAR-mediated lysis against T, NK cells and monocytes (Figure 1B). The CD38A4CAR KHYG-1 showed some (around 20%) CAR-dependent lysis of B cells but only at higher E:T ratios. Importantly these low lysis levels were only slightly above background lysis levels induced by MOCK cells and were significantly lower than the lysis levels of MM cells. Taken together, these results confirmed that an optimal therapeutic window can be generated using affinity-optimized CD38CARs also in NK cells as CAR carriers. In further experimentation we used primarily the CD38A4 variant since their anti-MM activity, especially against primary MM cells, was significantly better than that of CD38B1 transduced cells.
Cytotoxic activity of CD38CAR KHYG-1 cells on CD38+ fractions of regulatory T cells and hematopoietic progenitor cells
In further evaluation of their potential clinical value, we next questioned whether CD38A4CAR-KHYG-1 cells would possess, like daratumumab, any immunomodulatory effects via killing the CD38+ fractions of regulatory T cells. In addition, we tested their impact on CD38+ fraction of CD34+ progenitor cells as a potentially relevant toxicity parameter. In BM samples isolated from 4 MM patients CD38A4CAR-KHYG-1 cells, while effectively killing CD138+CD38+ MM cells, did not display significant CD38CAR-mediated lysis towards CD34+CD38+ progenitor cells, but showed elevated levels of CD38CAR-mediated lysis of CD38+ regulatory T cells (Tregs) (Figure 1C), suggesting that they may possess some immunomodulatory properties through depletion of CD38+ Tregs, which possess superior suppressor activity as compared to CD38– Tregs.18
Anti-MM activities of irradiated affinity-optimized CD38CAR KHYG-1 cells
The immortal nature of KHYG-1 cells differentiates this NK cell line from primary or cord-blood derived NK cells by enabling them to effectively expand in vitro. This property can be advantageous for “off the shelf” indications. To test this, we expanded MOCK and CD38CAR transduced cells continuously in the presence of different cytokines (Figure 2A). These assays revealed that CD38CAR KHYG-1 cells can be readily expanded in long-term cultures. Their expansion rate was even slightly better than that of MOCK transduced cells. The expansion rates were similar after addition of IL-15 at a dose of 10 mg/mL or IL-2 at doses between 100 and 900 U/mL. The combination of the cytokines did not significantly improve the expansion rates. In fact, the combination of IL-15 with the highest dose of IL-2 appeared even slightly detrimental (Figure 2A). When tested for CD38CAR-dependent cytotoxic activity, we found that even long-term cultured (>passage 140 after transduction) CD38CAR KHYG-1 cells effectively mediated CAR-dependent lysis of the MM cell line UM9 (Figure 2B).
Figure 2.
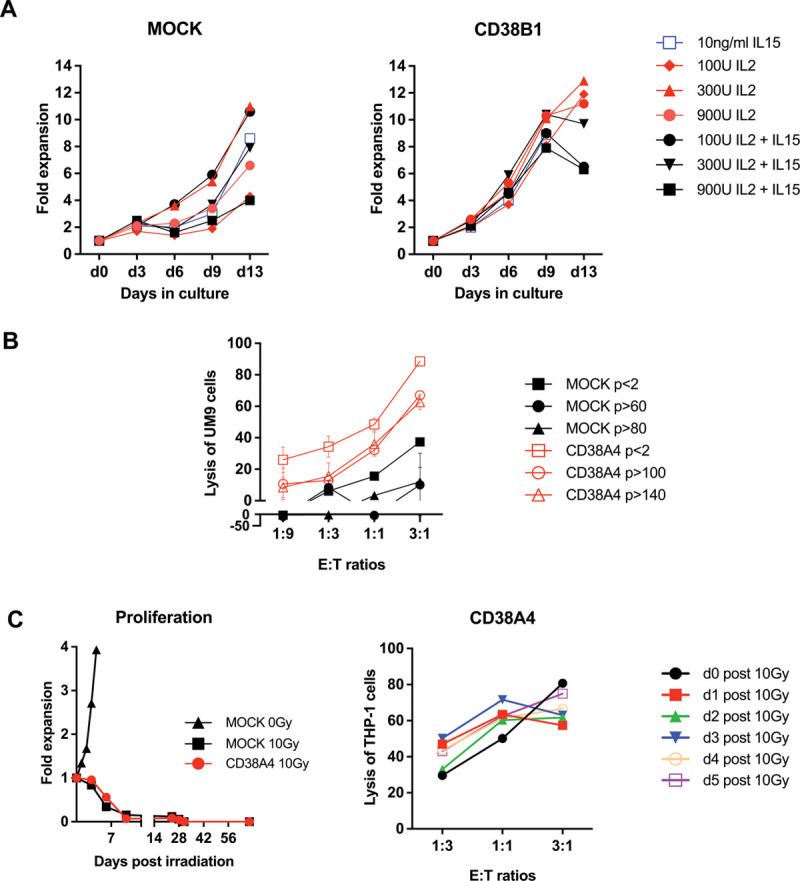
Effect of irradiation and different cytokines on effector function of CD38CAR KHYG-1 cells. (A), The expansion properties of MOCK (left) and CD38CAR KHYG-1 cells (right) in the presence of the depicted cytokines. The CD38B1 is represented as an example. (B), The cytotoxic effector function of long-term cultured MOCK and CD38A4 KHYG-1 cells at various E:T ratios. Data represent the mean ± SEM. (C), The proliferative (left) and cytotoxic activity (right) of CD38A4 transduced KYHG-1 cells after 10 Gy irradiation. Left: The cells were counted microscopically. Right: The cytotoxicity experiments were performed only with viable cells at the indicated day post irradiation. Assay was performed in duplicates. Mean and SEM are displayed. (B) and (C), Experiments were performed only with CD38A4 KHYG-1 cells as a preparation for in vivo experiments. CAR = chimeric antigen receptor; E:T = effector to target; IL = interleukin.
Although the immortal nature of KYGH-1 cells is an advantage for ex vivo expansion, before an in vivo application, they need to be rendered replication incompetent, for instance by irradiation. Short lived, nonproliferative KHYG-1 cells may also represent a better model for primary NK cells. We therefore tested the proliferative activities and functional properties of irradiated CD38CAR KHYG-1 cells (Figure 2C). Irradiation with 10 Gy killed virtually all microscopically viable MOCK or CD38CAR transduced KHYG-1 cells within a week, with cell viability of around 50% at 3 days and no detectable recovery of the cells even up to 2 months (Figure 2C). To evaluate the effect of irradiation on cytotoxic activity, we tested the lysis of THP-1 cells by irradiated but still viable CD38CAR KHYG-1 cells until 5 days after irradiation. Although the number of CD38CAR KHYG-1 cells decreased rapidly, all viable cells at the time of the cytotoxicity assays mediated effective cytotoxic activity, indicating that irradiation did not mitigate the cytotoxic activity of still viable cells (Figure 2C).
In vivo anti-MM efficacy of affinity-optimized CD38CAR KHYG-1 cells
To evaluate the in vivo efficacy of affinity-optimized CD38CAR KHYG-1 cells we used an unique xenograft mice model in which the UM9 cell derived MM tumors were grown on the back of the RAG2–/–γc–/– mice, in a humanized BM-like niche generated by SC inoculation of ceramic scaffolds coated with human MSCs.25 We first compared the activity of irradiated and nonirradiated CD38CAR KHYG-1 cells after 6 repeated injections of 10 × 106 CAR+ cells per injection (Figure 3A). In this setting, regardless of whether the cells were irradiated or not, CD38CAR KHYG-1 cells significantly delayed the tumor growth, while MOCK transduced KHYG-1 cells did not show any anti-tumor effect (Figure 3B–D). In post mortem analyses, we did not encounter any CD38CAR KHYG-1 cells in BM, blood, spleen or scaffolds regardless of irradiation (Figure S4A, http://links.lww.com/HS/A161). We also found no immediate evidence of tumor escape due to CD38 downregulation on MM tumor cell line UM9 in multiple mouse tissues (Figure S4B, http://links.lww.com/HS/A161). The results indicated the similar capacities of nonirradiated or irradiated CAR-NK cells if they are administered in a repeated fashion. Therefore, we net evaluated the effect of prolonged, repeated injections of irradiated CD38CAR KHYG-1 cells, by increasing the number of injections to 10, while the number of viable cells per dose was reduced to 4 million (Figure S5A, http://links.lww.com/HS/A161). We again observed a substantial delay in tumor growth after CD38CAR KHYG-1 cell treatment compared to the MOCK treated and untreated mice. As expected, this antitumor effect faded rapidly after stopping the injections (Figure S5B, http://links.lww.com/HS/A161; Figure S5C, http://links.lww.com/HS/A161). A nonsignificant, but slight improvement of overall survival was also detected in CD38CAR KHYG-1 treated mice (Figure S5D, http://links.lww.com/HS/A161). Again here, we did not detect surviving CAR-KHYG-1 cells in the blood, spleen, BM or scaffolds in postmortem analyses (data not shown).
Figure 3.
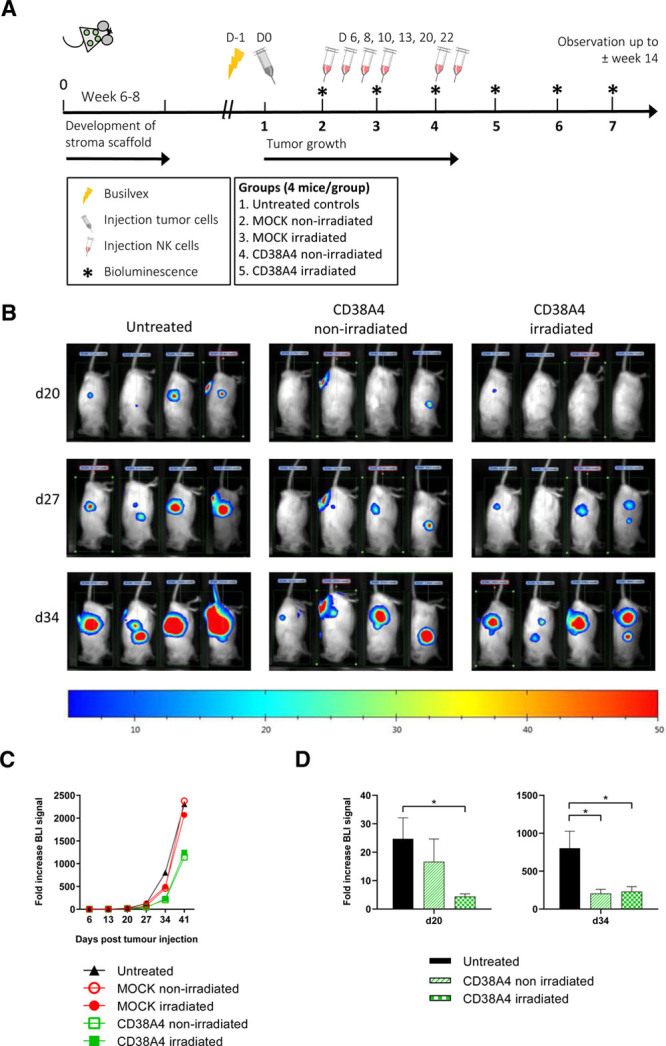
In vivo response of CD38CAR KHYG-1 cells. (A), Schematic representation of the in vivo experiment. For details refer to the relevant methods section. In short, mice were SC inoculated with human BMMSC coated scaffolds. The mice were irradiated after 6–8 wk. One day later Luc + UM9 MM cells were injected into the scaffolds. The IV CD38A4 KHYG-1 treatment started 1 week after tumor inoculation. Tumor growth was monitored weekly by BLI analysis and by manual tumor-size measurements. (B), Example BLI images from the left flanks of mice in 3 treatment groups at 3 different days. (C), Graphical representation of anti-tumor response in all groups. Mean values are displayed. (D), The graphical representation of the significant delay in tumor growth observed at day 20 and day 34 with nonirradiated or irradiated CD38A4 treated mice. (*P < 0.05, mean + SEM) as compared to nonirradiated group. MOCK treated cells showed no anti-tumor effect. BLI = bioluminescence imaging; BMMSC = bone marrow mesenchymal stromal cell; CAR = chimeric antigen receptor; MM = multiple myeloma; NK = natural killer.
Anti-MM efficacy of optimized CD38CAR KHYG-1 cells against primary MM cells derived from daratumumab refractory patients
After showing the in vitro and in vivo anti-MM potential of affinity-optimized CD38CAR KHYG-1 cells, we finally addressed whether these cells could be of benefit for patients who are refractory to another CD38-directed immunotherapy. To this end, we tested the susceptibility of CD138+ MM cells in BMMNCs of patients who became refractory to daratumumab (n = 6) (Figure 4A and Figure S6, http://links.lww.com/HS/A161). In these patients we have observed a clear, cell-dose dependent mean lysis of MM cells reaching up to 70% at E:T ratios of 1:1, which was similar to the lysis levels found in daratumumab unexposed patients. The efficiency of killing however showed a strong linear correlation with the percentage of CD38-positive cells. In addition, there was a strong nonlinear correlation between the lysis levels and CD38 expression levels (MFI) on the MM cells (Figure 4B and C) in a robust nonlinear regression analysis.
Figure 4.
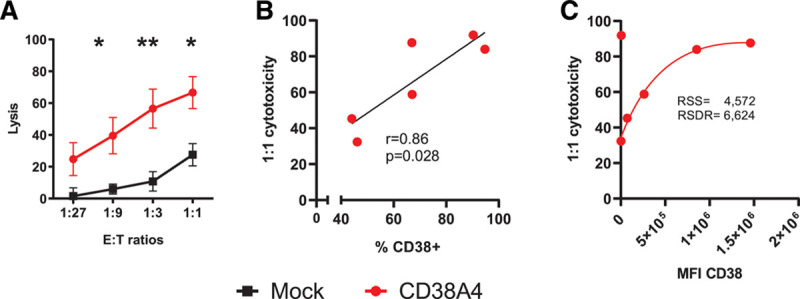
Cytotoxic activity of CD38CAR KHYG-1 cells against MM cells from daratumumab refractory patients. (A), The lysis of CD138+ MM cells from daratumumab refractory patients by CD38A4 as compared to MOCK transduced KHYG-1 cells (n = 6). Assays were performed in duplicates. Mean and SEM are displayed. *P < 0.05, **P < 0.01. Experimental details are outlined in Figure 1B and in the methods. (B), Linear correlation of the lysis (1:1 E:T ratio) with the percentage of CD38+ MM cells in the patient samples. The r and P values are indicated. (C), Nonlinear robust regression between the lysis and CD38 MFI values. The values for RSSs and the RSDRs were indicated. CAR = chimeric antigen receptor; E:T = effector to target; MFI = median fluorescent intensity; MM = multiple myeloma; RSDR = robust standard deviation of residual; RSS = robust sum of square.
Discussion
We here demonstrate the feasibility and anti-MM efficacy of engineering NK cells with affinity-optimized CD38CARs in a model system, where we used the NK cell line KHYG-1 as carrier of CD38CARs. The main reason to investigate the utility of affinity-optimized CD38CARs for MM treatment is first of all based on the fact that the expression of CD38 on MM cells is very high, homogenous and fairly stable, which makes it difficult to escape from CAR therapy, unlike CAR targeting of BCMA.26 Nonetheless, although antibody targeting of CD38 with daratumumab and isatuximab is highly efficient and safe,27–30 expression of high affinity CD38CARs in T cells induce high level of lysis of CD38int positive nonmalignant hematopoietic cells.19 Importantly, however, we have recently shown that expression of affinity-optimized CARs in T cells can generate an optimal therapeutic window with high levels of lysis against CD38high MM cells but not against CD38int nonmalignant cells6 including the CD38 fractions of CD34+ cells.6,23
In full agreement with these results, we now show that the expression of intermediate affinity CD38CARs in KHYG-1 NK cells effectively mediate anti-MM activity, while there is low or no cytotoxicity against nonmalignant cells especially against T, and NK cells, with one remarkable exception: the elimination of CD38+ regulatory T cells. These results indicate not only a favorable safety profile for CD38CAR KHYG-1 cells, but suggest that, similar to daratumumab, CD38CAR KHYG-1 cells may even have some beneficial immunomodulatory effects through the lysis of CD38+ Tregs, which are known to possess superior T cell suppressive capacities as compared to CD38– Tregs.18 In addition, we did not find significant CAR-dependent lysis of CD38+ fractions of CD34+ hematopoietic progenitor cells by CD38CAR KHYG-1 cells thus additionally supporting their favorable safety profile. On the other hand, in these experiments, we surprisingly detected some (20%–40%) innate killer activity from mock-transduced KHYG-1 cells on CD34+ cells (Figure 1C), which is not readily explained. Nevertheless, it should be noted that this observation could be a specific effect of KHYG-1 cells since primary NK cells have already been used in several clinical trials with no evidence of BM aplasia. Thus it is unlikely to expect significant toxic effects on hematopoietic progenitor cells when the CD38A4CAR will be expressed on primary NK cells for clinical application. Furthermore, when considering the potential unexpected safety issues, a major advantage of engineering CD38CARs into NK cells rather than into T cells is the fact that NK cells are short lived. It is expected that their anti-tumor effects as well as potential adverse effects will be limited with this short life span.31 Hence, even if optimized CD38CAR NK cells would cause any adverse effects, such potentially harmful effects will last much shorter as compared to CD38CAR T cells, which may develop memory and remain in the body for extended periods.32,33
Another relevant issue regarding the application of our optimized affinity CD38CAR NK cell in the clinical setting is the potential for fratricide when engineering expanded primary NK cells. This is despite the fact that in our experimental setting we did not observe significant fratricide in CD38CAR KHYG1 cells or significant lysis of resting NK cells. We have previously observed that CD38 expression is robustly upregulated during ex vivo expansion with consequent fratricide on subsequent expression of an optimized affinity CD38 CAR. This can be successfully overcome by CD38 knockout as we have shown recently.34
Currently, an important clinical category of MM patients are those patients who are refractory to daratumumab. Since daratumumab refractoriness is usually associated with CD38 downregulation in the first 3–6 months, it was relevant to test the efficacy of CD38CAR KHYG-1 cells against MM cells from daratumumab refractory patients. In a small panel (N = 6) of daratumumab refractory patients, we observed that CD38CAR KHYG-1 cells could effectively eliminate these MM cells. Interestingly, the lysis efficiency showed a nonlinear correlation with the CD38 expression levels, thus indeed suggesting that the therapeutic window of CD38CAR KHYG-1 cells can be affected by the CD38 expression levels of MM cells and it may be difficult to target MM cells with very low levels of CD38, with these intermediate affinity CD38CARs. While we showed a correlation with CD38 expression levels, a clear cut threshold level of CD38 expression required for kill could not be determined in our study, and requires the analyses of much larger cohorts. Therefore in patients where the MM cells express very low CD38 levels, it would be relevant to postpone the therapy until the CD38 levels are restored. In addition, restoring the CD38 expression by in vivo treatment with all-trans retinoic acid (ATRA), or with other agents such as Panobinostat,35 could be considered in such patients.
While in our study the KHYG-1 cells were mainly used as a model cell line, a potential advantage of using such immortal cells for therapeutic application is the possibility of an “off the shelf” application for many patients, as they can be readily expanded in large quantities. Indeed, in long-term cultures, CD38CAR transduced KHYG-1 cells vigorously expanded >2 months, even slightly better than MOCK transduced cells, and yielded very high numbers of functionally intact cell populations. In this context, another remarkable observation is the fact that even irradiated CD38CAR KHYG-1 cells could mediate effective cytotoxic activity against MM cells. This, together with their vigorous expansion capacity, suggests the possible generation of “off the shelf” products with CD38CAR KHYG-1 cells. Nonetheless, towards this end, still major improvements may be necessary, since in our in vivo experiments, we have observed that irradiated or even unmodified CD38CAR KHYG-1 cells maintain their anti-tumor effects for a brief period only, requiring multiple consecutive administrations to establish a visible anti-MM effect. The moderate in vivo anti-tumor effects may be model dependent. For instance, we cannot exclude the possibility that there is an impaired capacity of KHYG-1 cells to infiltrate the humanized tumor site in this model. It is also not well known whether KHYG-1 cells are affected by the tumor protective effects of BM-stromal cells which are present in our model. BM stromal cells are known to mitigate the CAR T and antibody-dependent cellular cytotoxicity (ADCC) effects of NK cells against MM cells.36–38 Thus, these possibilities need to be addressed in future studies. Related to this it is also noteworthy that in our in vivo experimental setting, we observe hardly any curative anti-tumor effect from CD38CAR T cells.19,24
Alternatively, the moderate in vivo anti-tumor responses could be related to the lack of in vivo expansion or persistence of nonirradiated or irradiated CD38CAR KHYG-1. In such a scenario, maximizing the cytotoxic potential of short lived KHYG-1 cells or even of primary NK cells by additional genetic engineering of cytotoxic mediators, such as TNF-related apoptosis-inducing ligand (TRAIL), may provide a solution and deserves further testing. Alternative approaches may be the combination of CAR-NK cells with immunomodulatory drugs with NK-cell activating properties. The feasibility and potential benefit of such strategies need however extensive preclinical testing.
In a future scenario, it could also be possible to engineer primary NK cells not only with CARs but with genes that allow their increased in vivo expansion and persistence. Such platforms that are in development include CB-derived or induced pluripotent stem cell-derived NK cells, especially when engineered to produce their own cytokines. Nonetheless, the long-term persistence of immune effector cells in vivo is also dependent to their susceptibility to immune regulation in the TME. Immune checkpoints expressed in the TME are known to induce exhaustion of not only T cells but also NK cells. The NK cell exhaustion is frequently related to interaction of programmed cell death protein 1 (PD-1), T cell immunoreceptor with Ig and ITIM domains (TIGIT), NKG2A and Siglec-7 with their cognate ligands, programmed death-ligand 1 (PD-L1), CD155, HLA-E and sialic acid on tumor cells respectively. Notably, KHYG-1 cells strongly express NKG2A and when co-cultured with MM cells expresses significant levels of PD-1 (unpublished data). Thus, improved CAR NK cell therapies may not only require advanced genetic engineering of NK cells but also combination therapies in which checkpoint receptors are effectively blocked.
In conclusion, the results of this proof-of-principle study indicates the benefits of engineering affinity-optimized CD38CARs into NK cells as a potential therapy for MM patients including those who are refractory to CD38-targeting antibodies.
Acknowledgments
We thank Jennemiek van Arkel, Christie Verkleij and Wassilis Bruins for their assistance in experiments and collecting the clinical data. The Rag2–/–γc–/– mice used in this study were originally obtained from the Amsterdam Medical Center (AMC, Amsterdam, The Netherlands).
Disclosures
NWCJvdD received research funding from Amgen, Novartis, and Bristol-Myers Squibb. He has membership on an entity’s Board of Directors or advisory committees in Celgene and Janssen and receives research funding from the same. SZ received research funding from Takeda, Celgene, and Janssen. She belongs to the advisory boards in Takeda, Celgene, Janssen, Sanofi, Oncopeptides, and Amgen. MO is Director and Equity Owner of ONK Therapeutics. He received research funding from Janssen, Celgene, and Bristol Myers Squibb (BMS) and is a consultant of Janssen, AbbVie. TM received research funding from Takeda, Genmab, Janssen, Novartis, and ONK Therapeutics. All the other authors have no conflicts of interest to disclose.
Sources of funding
This work is in part financially supported by ONK Therapeutics.
Supplementary Material
Footnotes
Current address for Dr Stikvoort: Karolinska Institutet, Sweden.
Supplemental digital content is available for this article.
References
- 1.Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018; 378:439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neelapu SS, Locke FL, Bartlett NL, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017; 377:2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019; 380:1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gogishvili T, Danhof S, Prommersberger S, et al. SLAMF7-CAR T cells eliminate myeloma and confer selective fratricide of SLAMF7+ normal lymphocytes. Blood. 2017; 130:2838–2847. [DOI] [PubMed] [Google Scholar]
- 5.Smith EL, Harrington K, Staehr M, et al. GPRC5D is a target for the immunotherapy of multiple myeloma with rationally designed CAR T cells. Sci Transl Med. 2019; 11:eaau7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drent E, Themeli M, Poels R, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. 2017; 25:1946–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Depil S, Duchateau P, Grupp SA, et al. ‘Off-the-shelf’ allogeneic CAR T cells: development and challenges. Nat Rev Drug Discov. 2020; 19:185–199. [DOI] [PubMed] [Google Scholar]
- 8.The quest for off-the-shelf CAR T cells. Cancer Discov. 2018; 8:787–788. [DOI] [PubMed] [Google Scholar]
- 9.Eyquem J, Mansilla-Soto J, Giavridis T, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017; 543:113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poirot L, Philip B, Schiffer-Mannioui C, et al. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015; 75:3853–3864. [DOI] [PubMed] [Google Scholar]
- 11.Oei VYS, Siernicka M, Graczyk-Jarzynka A, et al. Intrinsic functional potential of NK-cell subsets constrains retargeting driven by chimeric antigen receptors. Cancer Immunol Res. 2018; 6:467–480. [DOI] [PubMed] [Google Scholar]
- 12.Liu E, Marin D, Banerjee P, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. 2020; 382:545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Herrera L, Santos S, Vesga MA, et al. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019; 9:18729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pfefferle A, Huntington ND. You have got a fast CAR: chimeric antigen receptor NK cells in cancer therapy. Cancers (Basel). 2020; 12:E706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin MH, Kim J, Lim SA, et al. NK cell-based immunotherapies in cancer. Immune Netw. 2020; 20:e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002; 295:2097–2100. [DOI] [PubMed] [Google Scholar]
- 17.Kimpo MS, Oh B, Lee S. The role of natural killer cells as a platform for immunotherapy in pediatric cancers. Curr Oncol Rep. 2019; 21:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krejcik J, Casneuf T, Nijhof IS, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016; 128:384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drent E, Groen RW, Noort WA, et al. Pre-clinical evaluation of CD38 chimeric antigen receptor engineered T cells for the treatment of multiple myeloma. Haematologica. 2016; 101:616–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yagita M, Huang CL, Umehara H, et al. A novel natural killer cell line (KHYG-1) from a patient with aggressive natural killer cell leukemia carrying a p53 point mutation. Leukemia. 2000; 14:922–930. [DOI] [PubMed] [Google Scholar]
- 21.Suck G, Branch DR, Smyth MJ, et al. KHYG-1, a model for the study of enhanced natural killer cell cytotoxicity. Exp Hematol. 2005; 33:1160–1171. [DOI] [PubMed] [Google Scholar]
- 22.Sarkar S, Chauhan SKS, Daly J, et al. The CD38low natural killer cell line KHYG1 transiently expressing CD16F158V in combination with daratumumab targets multiple myeloma cells with minimal effector NK cell fratricide. Cancer Immunol Immunother. 2020; 69:421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suck G, Tan SM, Chu S, et al. KHYG-1 and NK-92 represent different subtypes of LFA-1-mediated NK cell adhesiveness. Front Biosci (Elite Ed). 2011; 3:166–178. [DOI] [PubMed] [Google Scholar]
- 24.Drent E, Poels R, Ruiter R, et al. Combined CD28 and 4-1BB costimulation potentiates affinity-tuned chimeric antigen receptor-engineered T Cells. Clin Cancer Res. 2019; 25:4014–4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Groen RW, Noort WA, Raymakers RA, et al. Reconstructing the human hematopoietic niche in immunodeficient mice: opportunities for studying primary multiple myeloma. Blood. 2012; 120:e9–e16. [DOI] [PubMed] [Google Scholar]
- 26.Ali SA, Shi V, Maric I, et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood. 2016; 128:1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016; 375:1319–1331. [DOI] [PubMed] [Google Scholar]
- 28.Usmani SZ, Nahi H, Plesner T, et al. Daratumumab monotherapy in patients with heavily pretreated relapsed or refractory multiple myeloma: final results from the phase 2 GEN501 and SIRIUS trials. Lancet Haematol. 2020; 7:e447–e455. [DOI] [PubMed] [Google Scholar]
- 29.Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. 2019; 394:2096–2107. [DOI] [PubMed] [Google Scholar]
- 30.Martin T, Strickland S, Glenn M, et al. Phase I trial of isatuximab monotherapy in the treatment of refractory multiple myeloma. Blood Cancer J. 2019; 9:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romee R, Rosario M, Berrien-Elliott MM, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016; 8:357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371:1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015; 7:303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurney M, Stikvoort A, Nolan E, et al. CD38 knockout natural killer cells expressing an affinity optimized CD38 chimeric antigen receptor successfully target acute myeloid leukemia with reduced effector cell fratricide. Haematologica. 2020 December 30.. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.García-Guerrero E, Gogishvili T, Danhof S, et al. Panobinostat induces CD38 upregulation and augments the antimyeloma efficacy of daratumumab. Blood. 2017; 129:3386–3388. [DOI] [PubMed] [Google Scholar]
- 36.de Haart SJ, Holthof L, Noort WA, et al. Sepantronium bromide (YM155) improves daratumumab-mediated cellular lysis of multiple myeloma cells by abrogation of bone marrow stromal cell-induced resistance. Haematologica. 2016; 101:e339–e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de Haart SJ, van de Donk NW, Minnema MC, et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin Cancer Res. 2013; 19:5591–5601. [DOI] [PubMed] [Google Scholar]
- 38.Holthof LC, Van der Horst HJ, Poels R, et al. The impact and modulation of microenvironment-induced immune resistance against CAR T cell and antibody treatments in multiple myeloma. Blood. 2019; 134(suppl_1):137. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.