

Abstract
Breast cancer (BC) has a significant heritable component but the genetic contribution remains unresolved in the majority of high-risk BC families. This study aims to investigate the monogenic causes underlying the familial aggregation of BC beyond BRCA1 and BRCA2, including the identification of new predisposing genes. A total of 11,511 non-BRCA familial BC cases and population-matched cancer-free female controls in the BEACCON study were investigated in two sequencing phases: 1303 candidate genes in up to 3892 cases and controls, followed by validation of 145 shortlisted genes in an additional 7619 subjects. The coding regions and exon–intron boundaries of all candidate genes and 14 previously proposed BC genes were sequenced using custom designed sequencing panels. Pedigree and pathology data were analysed to identify genotype-specific associations. The contribution of ATM, PALB2 and CHEK2 to BC predisposition was confirmed, but not RAD50 and NBN. An overall excess of loss-of-function (LoF) (OR 1.27, p = 9.05 × 10−9) and missense (OR 1.27, p = 3.96 × 10−73) variants was observed in the cases for the 145 candidate genes. Leading candidates harbored LoF variants with observed ORs of 2–4 and individually accounted for no more than 0.79% of the cases. New genes proposed by this study include NTHL1, WRN, PARP2, CTH and CDK9. The new candidate BC predisposition genes identified in BEACCON indicate that much of the remaining genetic causes of high-risk BC families are due to genes in which pathogenic variants are both very rare and convey only low to moderate risk.
Subject terms: Breast cancer, Cancer genetics
Introduction
The hereditary contribution to breast cancer (BC) is among the highest for solid tumours1. It is essential to identify the full repertoire of genetic risk factors to accurately inform BC risk management and interventions that can dramatically reduce risk2. However, two decades after the discovery of the BRCA1/2, only a small number of additional genes have been discovered and the genetic cause remains unresolved for the majority of hereditary BC families. Exome sequencing studies, focused on multi-case families, have revealed marked heterogeneity, which is the likely explanation for the lack of success of previous gene discovery studies where samples sizes and gene lists were generally small3,4. Consequently, we conducted the BEACCON study (hereditary BrEAst Case CONtrol study) to investigate all genes supported by either biological or empirical evidence. This resulted in a comprehensive targeted sequencing effort that examined 1303 candidate BC predisposition genes and 14 previously proposed hereditary breast and ovarian cancer (HBOC) genes in up to 5770 non-BRCA1/2 index cases and 5741 cancer-free population controls, providing a highly powered survey of the monogenic contributions to the heritable risk of BC.
Results
BEACCON study strategy
The BEACCON study was conducted in two phases (Fig. 1). Phase 1 sequenced 14 previously reported HBOC genes (CHEK2, PALB2, ATM, TP53, RAD51C, RAD51D, CDH1, BARD1, PTEN, MRE11A, BRIP1, STK11, RAD50 and NBN) and 1303 candidate genes (Supplementary Data) in up to 1990 non-BRCA1/2 familial BC index cases and 1902 female controls. The genes analysed consisted of 988 identified through previous BC germline exome studies3,5, and an additional 315 included because of a reported role in DNA damage repair6–8. Common variants are excluded (LoF MAF ≥ 0.005 and MS MAF ≥ 0.001) and the burden of remaining variants in candidate genes were compared between cases and controls to determine a short list for gene discovery purposes. A total of 145 genes from phase 1 were selected for further study based on the most statistically significant enrichment in cases (119 genes) or at a lower level of significance but with additional existing support based on current literature (26 genes). Together with the 14 HBOC genes these candidates proceeded to phase 2 and were sequenced in an additional 3780 non-BRCA1/2 cases and 3839 controls (159 genes analysed in a total of 5770 cases and 5741 controls). Subjects in both case and control cohorts were predominantly of European ancestry (95.3% cases and 98.8% controls) based on principal component analysis (PCA) (Supplementary Fig. 1).
Fig. 1. Breast cancer predisposition gene discovery and validation strategy in the BEACCON study.

Whole-exome sequencing (WES) was carried out on 150 BC affected cases from 69 high-risk BC families. Based on the data from the WES and DNA repair genes identified through literature review, 1303 candidate genes and 14 previously reported HBOC genes were screened in up to 1990 index non-BRCA1/2 familial BC cases and 1902 controls. One hundred forty-five genes selected from phase 1 and the same 14 HBOC genes are screened in an independent cohort of 3780 index non-BRCA1/2 BC cases and 3839 controls.
An average sequencing depth of 257.5 and 10× sequencing coverage of 92.4% (cases 92.0%, controls 92.9%) was achieved, with no coverage bias between the cases and controls identified at an individual gene level or between sequencing phases. PLINK identity-by-state analysis was used to identify duplicated or closely related samples.
Established moderate penetrance BC genes
Pathogenic LoF variants in CHEK2, PALB2 and ATM were observed in 1.35%, 0.90% and 0.80% of cases with ORs of 2.70, 3.47 and 2.88 (Benjamini–Hochberg adjustment, BH p = 0.0003, 0.0005 and 0.009), respectively (Fig. 2), in agreement with published evidence9,10. Rare missense (MS) variants were found in significant excess in CHEK2 (2.11%, 122 cases versus 1.24%, 71 controls; OR 1.73, 95% CI 1.27–2.35, BH p = 0.01) and ATM (5.53%, 319 cases versus 3.81%, 219 controls; OR 1.48, 95% CI 1.23–1.77, BH p = 0.0008). Applying filters for population frequency and in silico pathogenicity prediction scores to the rare MS variants, identified progressive enrichment of potentially deleterious variants in CHEK2 and ATM in the cases (Supplementary Table 1). In contrast, PALB2 showed neither an overall excess of MS variants in cases nor in any of the reported functional domains11,12.
Fig. 2. Case-control frequencies of rare LoF (MAF < 0.005) and MS (MAF < 0.001) variants in known or proposed HBOC genes (N = 14).

*Part of the data have been published previously in J. Clin. Oncol. 34(13), 1455–1459, Genet. Med. 21(4), 913–922, J. Pathol. 245(1), 53–60, J. Natl Cancer Inst. 111(12), 1332–1338, Nat. Genet. 50(10), 1346–1348 and Breast Cancer Res. Treat. 159(2), 385–392. Known pathogenic MS variants in ATM (c.7271 T > G) and TP53 (c.524 G > A, c.712 T > C, c.725 G > A, c.733 G > A, c.742 C > T and c.1009 C > T) were classified as LoF variants in this analysis.
Cancer syndrome genes
TP53, CDH1, PTEN and STK11 are rare high-risk genes that predispose to multi-cancer syndromes that include BC13–15. Pathogenic variants in TP53 were detected in nine cases (0.16%), CDH1 in four cases (0.07%) and PTEN in two cases (0.03%) but none were detected in controls for these genes and no STK11 pathogenic variants were found in cases or controls. Given the rarity of mutations, the BC risk associated with these genes, although high, could not be accurately estimated and their collective contribution to the hereditary risk of BC is small.
Proposed BC genes frequently tested on clinical multi-gene panels
There is less established evidence on the roles of RAD51C, RAD51D, BRIP1, BARD1, MRE11A, RAD50 and NBN in BC predisposition16–20; however, these genes are included on many HBOC gene panels in clinical practice. In the BEACCON study RAD51C LoF variants were highly enriched in cases; 14 cases vs 2 controls (OR 6.98, 95% CI 1.60–63.36) (Fig. 2 and previously reported in ref. 21). A non-significant excess of LoF variants were detected in the cases for RAD51D, BRIP1, BARD1 and MRE11A, and a statistically significant (unadjusted p < 0.05) excess of rare MS variants were identified in RAD51D and BRIP1. In contrast, there was no excess of variants in RAD50 and NBN which form the MRN complex with MRE11A; the LoF variant frequency in RAD50 and NBN was higher in the control group, indicating that, in the Australian population there is no evidence that these genes contribute to BC predisposition.
Identification of candidate genes from the BEACCON data
Although many of the genes tested in phase 1 showed a higher frequency of LoF and MS variants in cases (Supplementary Fig. 2), none of the 1303 candidate genes or the 14 proposed HBOC genes passed the multiple testing corrected statistical significance level. However, the adjusted frequency of rare variants detected across all the candidate genes was significantly greater in the cases (LoF: OR 1.13, p = 7.42 × 10−5; rare MS: OR 1.17, p = 8.62 × 10−55). This enrichment was confirmed for the final 145 candidate genes in phase 2 alone (LoF OR 1.09, p = 0.05; MS OR 1.26, p = 3.83 × 10−57) and combined phase 1 and 2 data (LoF OR 1.27, p = 9.05 × 10−9; MS OR 1.27, p = 3.96 × 10−73) (Supplementary Table 2).
Five candidate genes (NTHL1, CCDC60, WRN, BLM and PARP2) showed an excess of LoF variants and 37 showed an excess of MS variants in the cases (unadjusted p < 0.05; Fig. 3, Supplementary Fig. 3). Two genes (NTHL1 and WRN) were enriched for both LoF and MS variants. The 10 genes most enriched for MS variants (KMT2C, MSH3, WNK1, WRN, FANCM, RAD18, HOXD9, MC1R, RAD54B and SLEX4) remained significant upon multiple testing adjustment (Fig. 3, Supplementary Data). A high proportion of the candidate genes identified through an excess of LoF variants also exhibited an excess of MS variants, consistent with the findings for the HBOC genes. However, few of the candidate genes identified through an excess of MS variants had a corresponding excess of LoF variants (Supplementary Fig. 3).
Fig. 3. Distribution of candidate genes by ORs and p-values for LoF and MS variants.
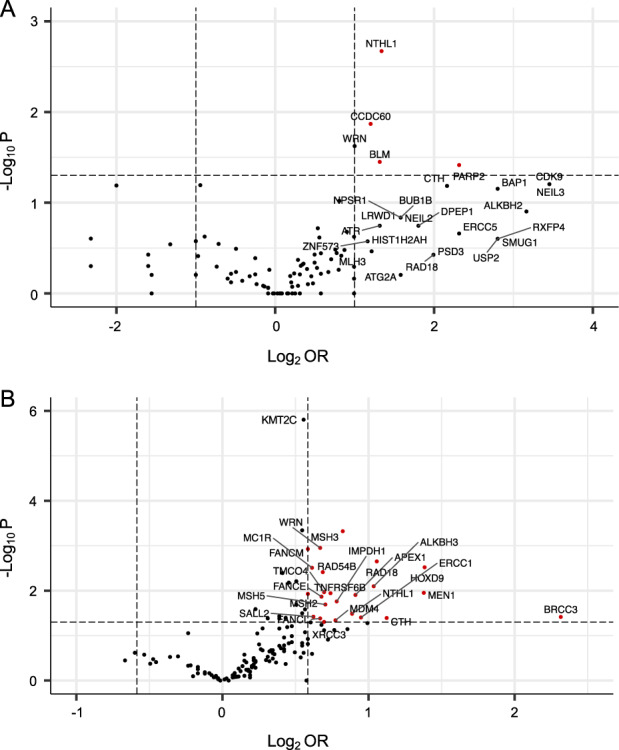
Volcano plots of the distribution of candidate genes (N = 145) by ORs and p-values in the LoF variant analysis (A) or the MS variant analysis (B). Genes are predominantly sequenced in a minimum of 4807 cases and 4782 controls with individual samples size listed in Supplementary Data. The horizontal axis is the log2(OR) and the vertical axis represents the reliability of the result (−log10(P)). The horizontal dash line signals Fisher’s exact text P-value at 0.05. Two vertical dash lines show the thresholds of ORs (OR = 2 and =0.50 for LoF variants and OR = 1.50 and =0.67 for MS variants). Each dot represents a candidate gene and the red dots represent the genes that have a minimum fold excess of variants in the cases and have passed the p-value threshold.
The top candidate genes ranked by p-values according to the excess of rare LoF variants and rare MS variants are shown in Fig. 4a and b, respectively. Details of the variant frequency of all candidate genes are summarised in Supplementary Data. The number of individuals where any LoF variant was detected was low for all of the candidate genes: only 2.75% of cases harboured one or more LoF variants in a candidate gene that had a p-value < 0.05. The estimated ORs for the leading candidates identified through LoF variants are consistent with moderate BC risk, although the confidence intervals are wide, and none of the associations are statistically significant after multiple tests correction. The frequency of MS variants detected in each gene varied widely, from less than 1% for HOXD9 and MEN1, to over 10% for KMT2C but the estimated odds ratios fell consistently within the range of low-moderate penetrance (1.33–2.61).
Fig. 4. Top candidate genes ranked by p-value according to the excess of a rare LoF and MS variants.

a (p ≤ 0.10, N = 10) and b (p ≤ 0.011, N = 14). *One LoF variant in the gene WRN (p.Arg1406Ter) located in the last exon was detected in 21 cases and 11 controls. Although this variant is reported in gnomAD at high frequency in South Asian (MAF 0.0171 for South Asians compared to 0.0015 for Europeans), the identified carriers from this study are all of European origin except for one South Asian. BLM, BAP1, WNK1 and ALKBH3 were sequenced in 5770 cases and 5741 controls, and KMT2C was sequenced in 3780 cases and 3839 controls. The remaining genes were sequenced in 4807 cases and 4782 controls.
Among the top 10 genes ranked on the basis of LoF variant enrichment, seven are involved in DNA damage repair: NTHL1 and NEIL3, which play important roles in base excision repair (BER), BLM, WRN and BAP1 in homologous recombination repair (HRR), and PARP2 and CDK9 in double strand break response. Collectively, genes involved in HRR were enriched in cases (17 genes, after exclusion of reported HBOC genes) (OR 1.48, 95% CI 1.18–1.91, p = 0.001), as were genes involved in BER (n = 17) (OR 1.32, 95% CI 1.04–1.71, p = 0.02) (Supplementary Table 3).
Identification of candidate genes from subgroup analysis
Associations of the candidate genes with specific subtypes of BC or a personal or family history of OC were assessed for 3065 cases in the ViP cohort. Cohort characteristics including age at diagnosis, hormone receptor status and family history are summarised in Supplementary Table 4. The frequency of LoF variants in the 145 candidate genes and 14 HBOC genes were examined in five cancer subgroups (ER positive, ER negative, HER2 positive, triple negative (TN) and lobular BC), as well as personal or family history of OC, in comparison to the cancer-free controls (Fig. 5). Consistent with previous reports, CHEK2 and ATM were correlated with ER positive14,15,22,23 and CDH1 with lobular BC24,25. Associations with ER negative and TN BC were identified for RAD51D, MUTYH, ERCC5, MRE11A and RAD51C21,26. A number of genes such as CHEK2, ATM and ERCC4 were correlated with HER2-positive BC. Genes associated with a personal or family history of OC included RAD51C, PALB2 and candidate genes CENPF, KIF27 and CTH.
Fig. 5. Heatmap of the associations of the candidate genes with specific subtypes of BC or a personal or family history of OC.

Only LoF variants were considered in the analysis for candidate genes and HBOC genes (N = 159) in cases from ViP cohort and controls, and genes with one or more statistically significant associations (p < 0.05, Fisher’s exact test, two-sided) are listed.
Carriers of multiple LoF variants
The study identified 188 (3.9%) cases and 111 (2.3%) of controls carrying LoF variants in multiple candidate genes (Supplementary Table 5A). The higher number of multiple LoF variant carriers in cases was not significantly different to the number expected based on the higher overall frequency of LoF variants in cases (Χ2 p-value = 0.52, Supplementary Table 5B) and the increased risk was consistent with a simple multiplicative effect: the OR increased from 1.31 to 3.16 as the number of variants carried by an individual increased from 1 to 3 (Supplementary Table 5A).
Recurrent variants in candidate genes
Seventy-six percent of the LoF variants (n = 2564/3356) and 76% of the MS variants (n = 28,385/37,342) detected were unique in the cohort, and 83% of LoF variants and 78% of MS variants have a population frequency of <0.0001 in the GnomAD database. It is possible that certain recurrent variants might be driving the signal observed for some genes; however, the effect of the majority of individual variants was impossible to assess due to their rarity. We analysed 32 LoF and 136 MS variants that were detected more frequently (>0.1% in the overall BEACCON cohort and accounted for >10% in the variants in that gene), and residual gene odds ratios were calculated with the recurrent variants excluded (Supplementary Data). This analysis showed the known CHEK2 pathogenic variant c.1100delC, with an odds ratio of 2.4, contributed 85% of all pathogenic CHEK2-carrying subjects. The residual CHEK2 odds ratio was 6.98 (p = 0.004, without adjustment for multiple testing). No individual LoF variants were observed at a similar level as the CHEK2 c.1100delC in any of the candidate genes. However a recurrent MS variants were identified in a number genes, including APEX1, FANCE and RAD54B. It should be noted that with the large number of MS variants analysed, these results would be consistent with chance findings and, with the exception of APEX1 c.50 T > C, none remained significant upon multiple testing.
Contribution to population breast cancer
To compare the relative contribution of different subgroups of coding variants to BC in the Australian population we estimated the population attributable fraction (PAF). Pathogenic variants in BRCA1 and BRCA2 were estimated (using published relative risks) to be responsible for 1.5% of BC, consistent with previous estimates27,28. Using the ORs and control frequency observed in this study, the combined contribution of pathogenic variants in PALB2, CHEK2 and ATM was estimated to be similar (PAF 1.5%) while the contribution from high-risk syndromic genes was very small (PAF 0.2%), as was the remaining HBOC panel genes (RAD51C, RAD51D, BARD1, BRIP1 and MRE11A, PAF for LoF variants 0.4%). In contrast, the excess of LoF variants in those candidate genes with at least a two-fold enrichment observed in cases versus controls (n = 26) corresponded to a PAF of 2.3%, while the collective effect of the excess of rare MS variants in phase 2 genes that showed at least a 1.5-fold enrichment summed to more than 12.3%. The same calculation applied to the published effect of polygenic risk score for BC29 in this group gives a PAF of 8.1%.
Discussion
The BEACCON study aimed to address the lack of power of previous studies to identify additional BC predisposition genes by performing extensive sequencing in 12,000 women (11,511 analysed following exclusions) and further enhancing power by using an ‘extreme phenotype’ design with enrichment of familial non-BRCA1/2 cases, compared with a control population of older women with ongoing confirmation of cancer-free status at June 2019. Three-quarters of the 1303 candidate genes screened were selected based on empirical evidence from local (69 multi-case BC families) or international whole-exome sequencing studies3, and the remainder were included to provide detailed coverage of functional pathways with established associations with BC.
While an overall enrichment in cases of LoF and MS variants was observed, this was distributed across many genes with no phase 2 candidate gene harboring LoF variants in more than 1% of the case cohort, with a median of only 0.15% (1 case in 667). The strongest candidate genes identified, including NTHL1 and WRN were characterised by an excess of both LoF and MS variants in cases, and most were involved in some aspect of DNA repair or genomic stability, particularly the HRR and BER pathways consistent with the function of the established HBOC genes30,31. An interesting exception to this is the gene CTH, which has a role in the trans-sulfuration pathway where it regulates cellular oxidative stress32. Homozygous and compound heterozygous pathogenic MS variants in CTH are observed in the recessive metabolic disorder, cystathioninuria33, indicating that these variants are associated with reduction or loss of protein function. The finding of enrichment in cases for both rare MS and LoF variants in CTH is consistent with the possibility that reduced CTH activity may predispose to BC via perturbation of cellular oxidative stress leading to increased DNA damage34.
The large majority of genes that showed an excess of MS variants did not have a corresponding excess of LoF variants in the same gene, with the strongest candidates in this group being KMT2C, WNK1, MSH3, FANCM and RAD1835,36. Of these, FANCM has been a gene of interest in multiple studies37–39; however, these reports have focused on LoF variants that were not associated with breast cancer in this study (OR 1.28, 95% CI 0.7–2.39); although more than two thirds of the LoF variants observed in FANCM were two comparatively frequent distal nonsense variants (p.Gln1701Ter and p.Arg1931Ter). An excess of MS variants in the absence of enrichment for LoF variants may indicate a false-positive result, although the finding may also reflect a genuine predisposition effect due to other mechanisms, as seen with pathogenic variants in TP53.
Genetic predisposition to specific BC subtypes is increasingly recognized and may allow identification of predisposition effects that are undetectable in the analysis of all BC. This approach was validated by the detection of the established associations of CHEK2 and ATM pathogenic variants with ER-positive tumours, and CDH1 with lobular BC. For several candidate genes, potentially pathogenic variants were enriched in a specific phenotypic sub-cohort, despite not showing evidence of an association with BC in the overall case-control cohort, including the suggested associations for ERCC5, RAD51D, MUTYH, MRE11A and TLDC1 with TN BC.
Limitations of the study may influence the interpretation of these results. An appreciation of population substructure and ethnicity is critical for studies of this nature. PCA demonstrated that BEACCON cases and controls were dominated by subjects of European origin and were directly comparable with the exception of a small difference in the Asian subgroup. In addition, while stringent quality filters were applied to select for variants of high confidence, it is possible some may still represent sequencing or alignment artefacts. A population frequency cut-off commonly used for gene discovery was implemented to prioritize a group of rare variants, which are more likely to represent moderate to high penetrance BC risk alleles (MAF ≤ 0.005 for LoF variants and MAF ≤ 0.001 for MS variants). These frequency cut-offs are arbitrary and some genuine pathogenic variants may escape this filtering and many variants below these cut-offs are likely to be non-pathogenic. Because the BEACCON study is enriched for familial cases, it is likely to overestimate the effect when compared to the general population and therefore it should be noted that the ORs detected do not equate to relative risks. Finally, the study did not investigate non-coding variants that affect gene regulation or splicing, large genomic rearrangement, or epigenetic changes that may also make important contributions to BC predisposition40.
In conclusion, the BEACCON study gives an insight into the scale required for future validation and discovery efforts that investigate rare coding variants. The low frequency of LoF and potentially pathogenic MS variants spread over a large number of different genes with apparently only moderate effect sizes meant that even with 11,511 subjects the evidence to support any candidate gene was limited. Applying the Benjamini–Hochberg adjustment for multiple comparisons, 10 candidate genes identified by an excess of MS variants, none of the candidate genes by LoF variants, and only PALB2, CHEK2 and ATM among the HBOC genes, reached conventional statistical significance. Estimation of attributable risk indicated that a substantial component of the remaining heritable contribution to BC is found in coding variation, and particularly in large numbers of rare MS variants of minor effect that are difficult both to identify and to interpret. Cohort studies at least an order of magnitude greater in size will be required if case-control data alone are to resolve the remaining monogenic causes of BC predisposition. Additional lines of evidence, such as family segregation analysis and functional studies, will still be necessary to confirm a role in BC predisposition as has been demonstrated for PALB2, ATM and RAD51C21,41,42.
Methods
Study subjects and sequencing
Case subjects are female index patients diagnosed with BC and/or ovarian cancer from 5770 HBOC families ascertained by the Variants in Practice (ViP) Study from the combined Victorian and Tasmanian Familial Cancer Centres, Australia (n = 3065), or from the Pathology North, NSW Health Pathology, Newcastle, Australia (n = 2705). All cases were assessed by a specialist Familial Cancer Clinic and determined to be eligible for clinical genetic testing for HBOC genes (≥10% chance of a pathogenic variant), but tested negative for BRCA1 and BRCA2 pathogenic variants. Pathology reports and detailed pedigrees were analysed for the cases from the ViP cohort. Controls are 5741 cancer-free female subjects who were >40 years old from the Lifepool Study (http://www.lifepool.org/). The average age of diagnosis of the cases was 49.7 years (range 19.0–94.8) and the average age of controls was 65.6 years (range 40.0–97.5). The study was approved by the Human Research Ethics Committee at the Peter MacCallum Cancer Centre (Approval # 09/29) and all participating centres. All participants provided informed consent for genetic analysis of their germline DNA.
The coding region and exon–intron boundaries (10 bp of intronic sequence at each site) of 1317 genes (phase 1) and 159 genes (phase 2) (Supplementary Data) were amplified from germline DNA using custom designed HaloPlex Targeted Enrichment Assay panels (Agilent Technologies, Santa Clara, CA) as described previously43. Full details on sequencing alignment, variant calling and variant filters are described in the supplementary online methods.
Statistical analysis
P-values were computed by Fisher’s exact test (2-sided) or Chi-squared test with Yates correction using R version 3.6.144. A p-value of <0.05 was considered as statistically significant and Benjamini–Hochberg adjustment (BH) was used for multiple test corrections45. Haldane–Anscombe correction was used to calculate the odds ratio where a variant frequency of zero. The conditional Maximum Likelihood Estimate was used for the calculation of confidence intervals. Population attributable fraction (PAF) was calculated using the frequency of variants in the Lifepool control group according to Levin’s formula46. The relative risk estimate used in PAF calculation was based on the odds ratio observed in case-control data adjusted for population background risk of breast cancer47.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank all the participants of the ViP and Lifepool studies for donating their DNA samples and clinical information. We also thank Norah Grewal, the ViP study site principal investigators Geoffrey Lindeman, Marion Harris, Tom John, Ingrid Winship and Yoland Antill, and the staff at the Peter MacCallum Cancer Centre, Royal Melbourne Hospital, Monash Health, Cabrini Health and Barwon Health Familial Cancer Centres and the Austin and Tasmanian Clinical Genetics Services, who enrolled participants and provided clinical data. We thank the following staff from Peter MacCallum Cancer Centre; Kaushalya Amarasinghe, Niko Thio and Richard Lupat from Bioinformatics core facility for helping with the bioinformatic analysis. This work was supported by the National Breast Cancer Foundation (IF-15-004, I.G.C. and P.A.J.), Cancer Australia/National Breast Cancer Foundation (PdCCRS_1107870, I.G.C. and P.A.J.), the Victorian Cancer Agency (Tumor Stream Grant, P.A.J.) and the National Health and Medical Research Council of Australia (GNT1023698, P.A.J.; GNT1041975, I.G.C.). E.K.S. is supported by NHMRC GNT1147498 and NBCF IIRS-20-025. N.L. is supported by Cancer Council Victoria.
Author contributions
N.L. contributed to study design, generating sequencing libraries, data analysis and manuscript writing; B.W.X.L. contributed to data cleaning and analysis, and manuscript writing; E.R.T. contributed to study design and data analysis; S.M.R. contributed to generating sequencing libraries and sample management; M.Z. contributed to bioinformatic analysis and plotting; S.M., L.D. and M.W.W.-B. contributed to collection of study materials or patients; D.C. and E.K.S. contributed to data interpretation and manuscript revision; R.J.S. contributed to provision of patients’ material and data interpretation; K.L.G. contributed to data interpretation and manuscript revision; P.A.J. contributed to study design, clinical interpretation, and manuscript revision; I.G.C. contributed to study design, data analysis and manuscript revision. All authors contributed to drafting, revising and final approval of the manuscript. N.L. and B.W.X.L. are co-first authors of this manuscript.
Data availability
The data generated and analysed during this study are described in the following data record: 10.6084/m9.figshare.1443945548. The sequencing data have been deposited in the European Genotype-phenotype Archive under the following accession: https://identifiers.org/ega.dataset:EGAD00001007025 (study ID: EGAS00001005043). These data include: sequencing alignment, variant calling and variant filters, principal component analysis and identity-by-state analysis. Additionally, the following data are not openly available to protect patient privacy: FCC patient database. Data requests for these data should be made to the corresponding author.
Code availability
Standard R codes were used. Code requests should be addressed to I.G.C.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Na Li, Belle W.X. Lim, Paul A. James, Ian G. Campbell.
Supplementary information
The online version contains supplementary material available at 10.1038/s41523-021-00279-9.
References
- 1.Mucci LA, et al. Familial risk and heritability of cancer among twins in Nordic Countries. JAMA. 2016;315:68–76. doi: 10.1001/jama.2015.17703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson HD, et al. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: a systematic review to update the U.S. Preventive Services Task Force recommendation. Ann. Intern. Med. 2014;160:255–266. doi: 10.7326/M13-1684. [DOI] [PubMed] [Google Scholar]
- 3.Thompson ER, et al. Exome sequencing identifies rare deleterious mutations in DNA repair genes FANCC and BLM as potential breast cancer susceptibility alleles. PLoS Genet. 2012;8:e1002894. doi: 10.1371/journal.pgen.1002894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Southey MC, et al. COMPLEXO: identifying the missing heritability of breast cancer via next generation collaboration. Breast Cancer Res. 2013;15:402. doi: 10.1186/bcr3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Complexo. et al. COMPLEXO: identifying the missing heritability of breast cancer via next generation collaboration. Breast Cancer Res. 2013;15:402–402. doi: 10.1186/bcr3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flicek P, et al. Ensembl 2013. Nucleic Acids Res. 2012;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruark E, et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature. 2013;493:406–410. doi: 10.1038/nature11725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015;41:1–8. doi: 10.1016/j.ctrv.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Lee AJ, et al. Incorporating truncating variants in PALB2, CHEK2, and ATM into the BOADICEA breast cancer risk model. Genet. Med. 2016;18:1190–1198. doi: 10.1038/gim.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buisson R, et al. Breast cancer proteins PALB2 and BRCA2 stimulate polymerase η in recombination-associated DNA synthesis at blocked replication forks. Cell Rep. 2014;6:553–564. doi: 10.1016/j.celrep.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl Acad. Sci. USA. 2009;106:7155–7160. doi: 10.1073/pnas.0811159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castera L, et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018;20:1677–1686. doi: 10.1038/s41436-018-0005-9. [DOI] [PubMed] [Google Scholar]
- 14.Buys SS, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017;123:1721–1730. doi: 10.1002/cncr.30498. [DOI] [PubMed] [Google Scholar]
- 15.Hauke J, et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018;7:1349–1358. doi: 10.1002/cam4.1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winship I, Southey MC. Gene panel testing for hereditary breast cancer. Med. J. Aust. 2016;204:188–190. doi: 10.5694/mja15.01335. [DOI] [PubMed] [Google Scholar]
- 17.Easton DF, et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015;372:2243–2257. doi: 10.1056/NEJMsr1501341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thompson ER, et al. Panel testing for familial breast cancer: calibrating the tension between research and clinical care. J. Clin. Oncol. 2016;34:1455–1459. doi: 10.1200/JCO.2015.63.7454. [DOI] [PubMed] [Google Scholar]
- 19.Suszynska M, Ratajska M, Kozlowski P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020;13:50. doi: 10.1186/s13048-020-00654-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson ER, et al. Analysis of RAD51C germline mutations in high-risk breast and ovarian cancer families and ovarian cancer patients. Hum. Mutat. 2012;33:95–99. doi: 10.1002/humu.21625. [DOI] [PubMed] [Google Scholar]
- 21.Li, N. et al. Combined tumor sequencing and case/control analyses of RAD51C in breast cancer. J. Natl Cancer Inst. 10.1093/jnci/djz045 (2019). [DOI] [PMC free article] [PubMed]
- 22.Nagel JH, et al. Gene expression profiling assigns CHEK2 1100delC breast cancers to the luminal intrinsic subtypes. Breast Cancer Res. Treat. 2012;132:439–448. doi: 10.1007/s10549-011-1588-x. [DOI] [PubMed] [Google Scholar]
- 23.Vargas AC, Reis-Filho JS, Lakhani SR. Phenotype-genotype correlation in familial breast cancer. J. Mammary Gland Biol. Neoplasia. 2011;16:27–40. doi: 10.1007/s10911-011-9204-6. [DOI] [PubMed] [Google Scholar]
- 24.Corso G, Intra M, Trentin C, Veronesi P, Galimberti V. CDH1 germline mutations and hereditary lobular breast cancer. Fam. Cancer. 2016;15:215–219. doi: 10.1007/s10689-016-9869-5. [DOI] [PubMed] [Google Scholar]
- 25.Dossus L, Benusiglio PR. Lobular breast cancer: incidence and genetic and non-genetic risk factors. Breast Cancer Res. 2015;17:37–37. doi: 10.1186/s13058-015-0546-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimelis H, et al. Triple-negative breast cancer risk genes identified by multigene hereditary cancer panel testing. J. Natl Cancer Inst. 2018;110:855–862. doi: 10.1093/jnci/djy106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong N, Ryder S, Forbes C, Ross J, Quek RG. A systematic review of the international prevalence of BRCA mutation in breast cancer. Clin. Epidemiol. 2019;11:543–561. doi: 10.2147/CLEP.S206949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fackenthal JD, Olopade OI. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer. 2007;7:937–948. doi: 10.1038/nrc2054. [DOI] [PubMed] [Google Scholar]
- 29.Mavaddat N, et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2019;104:21–34. doi: 10.1016/j.ajhg.2018.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer. 2004;4:814–819. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 32.Zhao K, Li H, Li S, Yang G. Regulation of cystathionine gamma-lyase/H(2)S system and its pathological implication. Front. Biosci. 2014;19:1355–1369. doi: 10.2741/4286. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Hegele RA. Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH) Hum. Genet. 2003;112:404–408. doi: 10.1007/s00439-003-0906-8. [DOI] [PubMed] [Google Scholar]
- 34.Chang EY, et al. Inhibition of prostaglandin reductase 2, a putative oncogene overexpressed in human pancreatic adenocarcinoma, induces oxidative stress-mediated cell death involving xCT and CTH gene expressions through 15-Keto-PGE2. PLoS ONE. 2016;11:e0147390. doi: 10.1371/journal.pone.0147390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiiski JI, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple-negative breast cancer. Proc. Natl Acad. Sci. USA. 2014;111:15172–15177. doi: 10.1073/pnas.1407909111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neidhardt G, et al. Association between loss-of-function mutations within the FANCM gene and early-onset familial breast cancer. JAMA Oncol. 2017;3:1245–1248. doi: 10.1001/jamaoncol.2016.5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Catucci I, et al. Individuals with FANCM biallelic mutations do not develop Fanconi anemia, but show risk for breast cancer, chemotherapy toxicity and may display chromosome fragility. Genet. Med. 2018;20:452–457. doi: 10.1038/gim.2017.123. [DOI] [PubMed] [Google Scholar]
- 38.Figlioli G, et al. The FANCM:p.Arg658* truncating variant is associated with risk of triple-negative breast cancer. npj Breast Cancer. 2019;5:38. doi: 10.1038/s41523-019-0127-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schubert S, et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: high frequency of FANCM pathogenic variants. Int. J. Cancer. 2019;144:2683–2694. doi: 10.1002/ijc.31992. [DOI] [PubMed] [Google Scholar]
- 40.Woodward AM, Davis TA, Silva AGS, Kirk JA, Leary JA. Large genomic rearrangements of both BRCA2 and BRCA1 are a feature of the inherited breast/ovarian cancer phenotype in selected families. J. Med. Genet. 2005;42:e31. doi: 10.1136/jmg.2004.027961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee JEA, et al. Molecular analysis of PALB2-associated breast cancers. J. Pathol. 2018;245:53–60. doi: 10.1002/path.5055. [DOI] [PubMed] [Google Scholar]
- 42.Weigelt B, et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J. Natl Cancer Inst. 2018;110:1030–1034. doi: 10.1093/jnci/djy028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson ER, et al. Prevalence of PALB2 mutations in Australian familial breast cancer cases and controls. Breast Cancer Res. 2015;17:111–111. doi: 10.1186/s13058-015-0627-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Packwood, K. et al. Breast cancer in patients with germline TP53 pathogenic variants have typical tumour characteristics: the Cohort study of TP53 carrier early onset breast cancer (COPE study). J. Pathol. Clin. Res. 10.1002/cjp2.133 (2019). [DOI] [PMC free article] [PubMed]
- 45.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995;57:289–300. [Google Scholar]
- 46.Levin ML. The occurrence of lung cancer in man. Acta Unio Int. Contra Cancrum. 1953;9:531–541. [PubMed] [Google Scholar]
- 47.Zhang J, Yu KF. What’s the relative risk? A method of correcting the odds ratio in cohort studies of common outcomes. JAMA. 1998;280:1690–1691. doi: 10.1001/jama.280.19.1690. [DOI] [PubMed] [Google Scholar]
- 48.Li, N. et al. Metadata record for the manuscript: Investigation of Monogenic Causes of Familial Breast Cancer: Data from the BEACCON Case-Control Study. figshare10.6084/m9.figshare.14439455 (2021). [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated and analysed during this study are described in the following data record: 10.6084/m9.figshare.1443945548. The sequencing data have been deposited in the European Genotype-phenotype Archive under the following accession: https://identifiers.org/ega.dataset:EGAD00001007025 (study ID: EGAS00001005043). These data include: sequencing alignment, variant calling and variant filters, principal component analysis and identity-by-state analysis. Additionally, the following data are not openly available to protect patient privacy: FCC patient database. Data requests for these data should be made to the corresponding author.
Standard R codes were used. Code requests should be addressed to I.G.C.