

Abstract
Thalamocortical neurons (TCNs) play a critical role in the maintenance of thalamocortical oscillations, dysregulation of which can result in certain types of seizures. Precise control over firing rates of TCNs is foundational to these oscillations, yet the transcriptional mechanisms that constrain these firing rates remain elusive. We hypothesized that Shox2 is a transcriptional regulator of ion channels important for TCN function and that loss of Shox2 alters firing frequency and activity, ultimately perturbing thalamocortical oscillations into an epilepsy-prone state. In this study, we used RNA sequencing and quantitative PCR of control and Shox2 knockout mice to determine Shox2-affected genes and revealed a network of ion channel genes important for neuronal firing properties. Protein regulation was confirmed by Western blotting, and electrophysiological recordings showed that Shox2 KO impacted the firing properties of a subpopulation of TCNs. Computational modeling showed that disruption of these conductances in a manner similar to Shox2’s effects modulated frequency of oscillations and could convert sleep spindles to near spike and wave activity, which are a hallmark for absence epilepsy. Finally, Shox2 KO mice were more susceptible to pilocarpine-induced seizures. Overall, these results reveal Shox2 as a transcription factor important for TCN function in adult mouse thalamus.
Keywords: epilepsy, HCN channel, oscillations, thalamus, t-type Ca2+ channel
Introduction
Rhythmic oscillations of brain activity emerge from neuronal network interactions that consist of reciprocal connections between the thalamus, the inhibitory thalamic reticular nucleus, and the cortex (Bal and McCormick 1996), commonly referred to as the thalamocortical circuit. The frequency and amplitude of these thalamocortical oscillations are dependent on brain state, for example, asleep or awake, and are important for different types of neural processing (Steriade 2000). Dysfunction of the pacing of the frequency of these oscillations that can be caused by excessive synchronization within the thalamocortical circuit underlies the distinctive spike and wave activity that occurs during absence epileptic seizures. Thus, determining molecular mechanisms that constrain the frequency of thalamocortical oscillations is important to understand the perturbations that promote epilepsy and pathological brain states.
Extensive studies have identified thalamocortical neurons (TCNs) and a complex balance of excitatory drive and inhibitory inputs to TCNs as integral components that contribute to control these oscillations (Fogerson and Huguenard 2016). Notably, TCNs are able to switch their firing states between 2 firing modes, burst or tonic firing, which are dependent on functional state. The membrane properties of TCNs and synaptic inputs contribute to these firing properties and the transition between the 2 modes (Llinas and Jahnsen 1982; Deschenes et al. 1984; Jahnsen and Llinas 1984a). Specifically, the neurons possess T-type Ca2+ currents (IT) that are important to establish a cycle of oscillatory activity (Coulter et al. 1989; Huguenard and Prince 1994). The Cav3.x family of Ca2+ channels encodes IT and is highly expressed in the nuclei of the thalamus (Kim et al. 2001a; Song et al. 2004). In addition to the current mediated by T-type channels, the H-current (IH), mediated by HCN channels, also plays a role in the generation of rhythmic activity in the thalamocortical network during sleep and epilepsy (McCormick and Bal 1997). Consequently, modulation of IT and IH is an important regulatory mechanism for modulation of oscillatory activity crucial for sleep rhythms and protection against excessive synchronization that occurs during generation of seizures. Very little is understood about the factors that contribute to expression of the ion channels that underlie these currents. At a gene transcription level, what factors may give the TCNs these unique pace-making properties, and ultimately promote resistance to seizure activity?
Shox2 is a member of the homeobox family of genes containing a 60-amino acid residue motif that represents a DNA-binding domain (De Baere et al. 1998; Clement-Jones et al. 2000; Sun et al. 2013). While homeobox genes have been characterized extensively as transcriptional regulators involved in pattern formation in both invertebrate and vertebrate species, studies indicate that they can also serve in maintenance of cellular properties (Hutlet et al. 2016; Bondos et al. 2020). We and others have shown that Shox2 plays a decisive role in the differentiation of pacemaker cells in the sinoatrial node of the heart and pulmonary vein in mice and that expression of Shox2 is a critical determinant of pacemaker properties of the mature sinoatrial node (Sun et al. 2015; Ye et al. 2015). We hypothesized that Shox2 may play a similar role in mature thalamus.
Shox2 is expressed in the embryonic and mature central nervous system and evidence suggests that it may be important to brain function. Previous studies showed that Shox2 expression in the diencephalon begins at approximately E10.5 (Rosin et al. 2015). In addition, Shox2 is directly repressed by Aristaless-related homeobox Arx, a gene implicated in intellectual disability, seizures, and autism (Fulp et al. 2008; Colasante et al. 2009), suggesting that Shox2 may also play a role in these disorders downstream of Arx. Notably, Shox2 is significantly up-regulated in an Arx mouse model of infantile spasms, and expression is decreased by treatment with estradiol, which prevents the seizures (Olivetti et al. 2014).
In this study, we found that Shox2 is expressed in TCNs in adult mice. TCNs express HCN2, HCN4, and Cav3.1 channel protein subunits that are important for firing properties of TCNs, and we hypothesized that Shox2 coordinates the expression of genes for these ion channels to affect action potential firing activity of TCNs. Using conditional KO animals, we further demonstrated that Shox2 is important for firing properties likely by affecting expression of mRNA of multiple ion channels, including Cav3.1, HCN2, and HCN4. Computational modeling of the thalamocortical circuit, including the reticular nucleus, showed that altering the currents in a manner consistent with the effects of Shox2 KO could shift a normally behaving network into an absence epilepsy state. Furthermore, behavioral studies showed that the Shox2 KO mice are more susceptible to seizure generation compared with WT mice. The present study provides novel insights into a transcription factor that contributes to TCN function and shows that transcriptional regulation by Shox2 is critical to maintain thalamic neuron function, which is fundamental to supporting physiological brain states.
Methods
Mice
All animal procedures were approved by Tulane University Institutional Animal Care and Use Committee (IACUC) according to National Institutes of Health (NIH) guidelines. Shox2 transgenic mice including Shox2Cre, Shox2LacZ, Shox2f/f, and Rosa26CreERt mice were generously donated by Dr Yiping Chen. All wild-type C57Bl/6 N mice were ordered from Charles River. Rosa26LacZ/+ (stock #003474) and Gbx2CreERt/+ breeders (stock #022135) were ordered from Jackson Lab.
In inducible KO experiments, Rosa26CreERt/+, Shox2f/f or Rosa26CreERt/CreERt, Shox2f/f female mice were crossed with Shox2−/+ male mice or, in the case of the Gbx2 animals, Gbx2CreERt/+, Shoxf/f or Gbx2CreErt/CreErt, Shoxf/f were crossed with Shox2−/+ male mice (Supplemental Fig. 1). Litters were labeled and genotyped at postnatal day 10 (P10). The KO group was the Rosa26CreERt/+, Shox2−/f mice (Gbx2CreRt/+, Shox2−/f) and the control (CR) group was the littermate Rosa26CreERt/+ (Gbx2CreErt/+), Shox2+/f. In the Shox2LacZ/+ and Shox2Cre/+ mice, the first two exons of the Shox2 allele were partially replaced by LacZ and Cre genes, respectively, in order to obtain the expression of LacZ mRNA and Cre mRNA under the control of the Shox2 promoter, while the unaffected alleles express Shox2 mRNA (Sun et al. 2013). The Rosa26CreERt/+ mouse line is a transgenic mouse line with a tamoxifen-inducible CreERt inserted in the Rosa26 loci. The Rosa26LacZ/+ mouse line is a transgenic mouse line with a floxed stop signals followed by LacZ gene inserted in the Rosa26 loci (Soriano 1999). This ‘cre reporter’ mouse strain was used to test the expression of the Cre transgene under the regulation of a specific promoter.
The Rosa26CreErt is a global KO, whereas the Gbx2CreERt mouse was used to knockdown Shox2 specifically in the medial thalamus. Our localization studies with Gbx2-promotor-driven GFP staining showed that Gbx2 promotor-driven CreERt is specifically expressed in the midline thalamus of the Gbx2CreErt adult mouse.
To induce recombination in animals bearing a CreERT2, prewarmed tamoxifen (100–160 mg/kg) was injected intraperitoneally into KO mice and CR littermates at the same time every day for five consecutive days. Tamoxifen (20 mg/mL) was dissolved in sterile corn oil (Sigma, C8267) with 10% alcohol. The littermate KO mice and CR mice of the same sex were housed together and received the same handling. Throughout experiments, the researchers were blinded to the genotype. Reverse transcription–quantitative PCR (RT-qPCR) experiments were used to confirm the efficiency of Shox2 KO in brains in every animal tested.
In order to view projections of Shox2-expressing neurons, we created the Ai27D-Shox2Cre mouse. B6.Cg-Gt (ROSA)26Sortm27.1(CAG-COP4*H134R/tdTomato)Hze/J(Ai27D) mice from Jackson labs were crossed with Shox2Cre to obtain mice with Shox2-expressing neurons labeled with tdTomato and expressing ChR2 (Fig. 2M–O).
Figure 2.
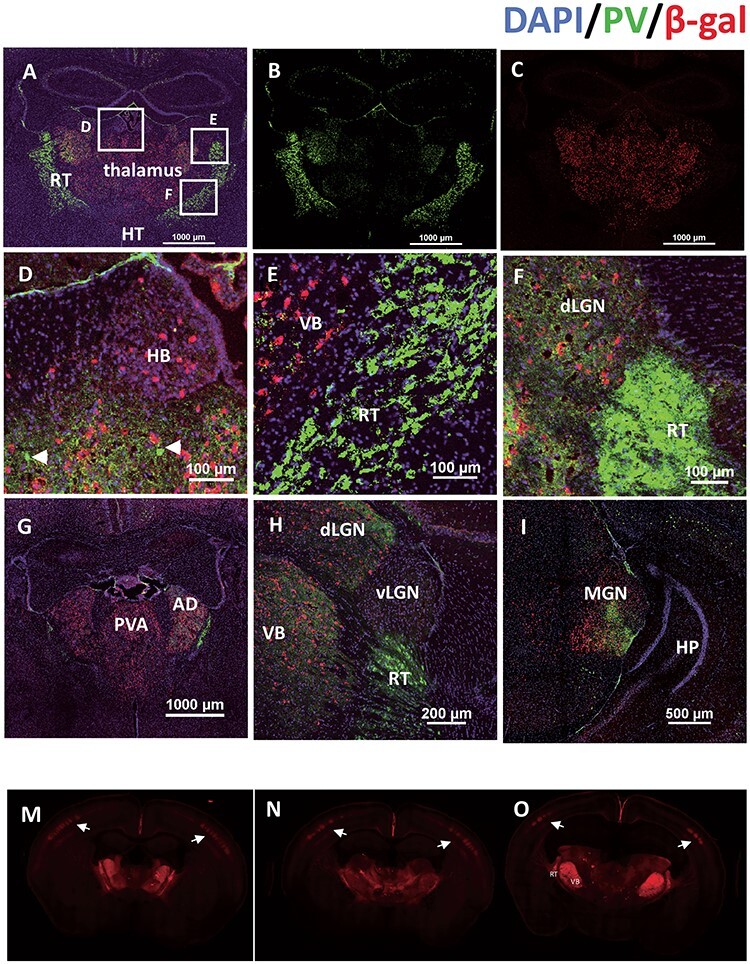
Shox2 is expressed in glutamatergic TCNs but not parvalbumin + interneurons. Coronal sections through the thalamus were costained with parvalbumin (green, A, B) and β-gal (red, A, C). Boxes in Figure A are magnified in D. (habenula), E. Ventrobasal (VB) and reticular nucleus (RT), and F. dorsal lateral geniculate nucleus (dLGN). G. Panels G-I show Shox2 expression in coronal slices of rostral to caudal thalamus (G: Paraventricular nucleus of the thalamus (PVA) relative to Bregma, approx. -1.1; H: lateral geniculate nucleus (LGN) -2.0; I: Medial geniculate nucleus (MGN) –2.9). M-O: Coronal sections from rostral (M) to caudal (O) from the Ai27D-Shox2Cre in which the presence of tdTomato indicates Shox2-expressing neurons. Shox2 is expressed in neurons throughout the thalamus and projections to the cortex, strongly targeting layer IV barrel cortex (white arrows) and layer VI.
X-gal Staining
Adult Shox2LacZ/+ or Shox2Cre/+, Rosa26LacZ/+ male mice were anesthetized by isoflurane inhalation and decapitated, and the brains were removed. Brains were sliced at 200 μm using a Vibratome Series 3000 Plus Tissue Sectioning system. Brain slices were placed into ice-cold artificial cerebrospinal fluid (aCSF) in a 24-well plate and fixed with 0.5% glutaraldehyde and 4% paraformaldehyde in phosphate-buffered saline (PBS) for 15 min. After 3X wash with ice-cold PBS, the slices were incubated with X-gal staining solution, containing X-gal (1 mg/ml), potassium ferrocyanide (4 mM), potassium ferrocyanide (4 mM), and MgCl2 (2 mM) and covered by aluminum foil at 37°C overnight. All slices were washed and post-fixed. Images were taken under a stereo microscope.
Immunohistochemistry (IHC)
Mice were deeply anesthetized by injection with ketamine (80 mg/kg) mixed with xylazine (10 mg/kg), perfused transcardially with ice-cold PBS followed by 4% paraformaldehyde in PBS and decapitated for brain collection. Mouse brains were placed in 4% paraformaldehyde in PBS at 4°C overnight for post-fixation. In order to perform cryostat sections, the brains were sequentially placed in 15 and 30% sucrose in PBS solutions at 4°C until saturation. The brain samples were embedded with optimal cutting temperature compound and stored at −20°C and cryo-sectioned in 20–50 μm coronal slices with Leica CM3050S cryostat. For IF staining, slices were washed with 50 mM Tris-Buffered Saline with 0.025% Triton X-100 (TTBS) and blocked in 2% bovine serum albumin (BSA) in TTBS for 2 h at room temperature. Primary antibodies were diluted in blocking solutions and applied on slides overnight at 4°C. Fluorescence-conjugated secondary antibodies were diluted 1:1000 in blocking solutions and applied on slides for 1 h at room temperature. 1:1000 DAPI was applied for 5 min at room temperature for nuclei staining and washed. The slices were mounted on slides with mounting media (Vector Laboratories, H-1000) and imaged under confocal microscope.
mRNA Sequencing
Thalamus mRNA was extracted from 3 KO mice and 3 CR mice and sent to BGI Americas Corporation (Cambridge, MA, USA) for RNA-seq quantification. Total RNA was isolated in tissue from the midline of the thalamus of P60 male Gbx2CreERt/+, Shox2−/f mice, and control male littermates (Gbx2CreERt/+, Shox2+/f) with the same method used for RT-qPCR RNA extraction. Around 30 million single-end 50-bp reads by BGISEQ-500 Sequencing Platform per sample were aligned to the mm10/GRCm38 mouse reference genome using Salmon v0.10.2 (Patro et al. 2017). The count data from Salmon v0.10.2 was analyzed via DESeq2 v 1.22.2 (Love et al. 2014) to identify genes differently expressed (DEGs) between KO and CR and to calculate Fragments Per Kilobase Million. Genes with adjusted P-value <0.1 were defined as DEGs and were used for further gene ontology (GO) analysis through online DAVID Bioinformatics Resources (Huang da et al. 2009a, 2009b).
Reverse Transcription–Quantitative PCR
The whole thalamus was collected from adult mouse brains (RosaCreErt-Shox2 KO) and immediately stored in RNAlater™ RNA stabilization reagent (ThermoFisher Scientific, AM7021). RNA was extracted using RNeasy Mini Kit (Qiagen, 74 104) following the standard protocol provided in the manual. The RNA concentration and quality were tested using Nanodrop Microvolume Spectrophotometers and Fluorometer as well as agarose gel investigation. Reverse transcription was conducted using iScript™ Reverse Transcription Supermix (Bio-Rad, 1 708 840). Quantitative PCR was conducted with iTaq™ Universal SYBR Green Supermix (Bio-rad, 1 725 121) in Bio-Rad CFX96 Touch™ PCR system. Data analysis was done with CFX Manager software. The expression of all the genes tested in the RT-qPCR experiments were normalized to the widely used housekeeping reference gene β-actin (Actb) and TATA-box binding protein (Tbp) (Valente et al. 2009; Li et al. 2014). All primers were designed and tested, and conditions were optimized to have an efficiency between 95% and 105%. Both the melt curves and gel investigations were used to confirm the RT-qPCR products. The primer sequences of all tested genes including reference gene Actb and Tbp are listed in Table 1.
Table 1.
Sequences of RT-qPCR primers
| Gene | Forward primer(5′- > 3′) | Reverse primer (5′- > 3′) |
|---|---|---|
| Shox2 | CCGAGTACAGGTTTGGTTTC | GGCATCCTTAAAGCACCTAC |
| actb | CTAGACTTCGAGCAGGAGAT | GATGCCACAGGATTCCATAC |
| Tbp | CCGTGAATCTTGGCTGTAAACTTG | GTTGTCCGTGGCTCTCTTATTCTC |
| Hcn1 | CTTCGTATCGTGAGGTTTAC | GTCATAGGTCATGTGGAATATC |
| Hcn2 | CTTTGAGACTGTGGCTATTG | GCATTCTCCTGGTTGTTG |
| Hcn4 | ATACTTATTGCCGCCTCTAC | TGGAGTTCTTCTTGCCTATG |
| Cacna1g | GACACCAGGAACATCACTAAC | CACAAACAGGGACATCAGAG |
| Cacna1h | TTTGGGAACTATGTGCTCTTC | TCTAGGTGGGTAGATGTCTTATC |
| Gapdh | GTCGGTGTGAACGGATTTG | TAGACTCCACGACATACTCAGCA. |
To confirm knockdown of Shox2 in the Gbx2CreERt/+ animals, adult Gbx2CreERt/+, Shox2 −/f male and female mice were anesthetized by isoflurane inhalation followed by decapitation. The brains were removed and a 1-mm thick slice through the thalamus was removed via razor blade, the location of cut is determined by Paxinos and Franklin Mouse Brain Atlas. Medial thalamus tissue is collected with 1-mm stainless steel punching tool and lateral thalamus was separated via razor blade. The collected tissues were stored in 50 μL RNA later solution and stored in −80 freezer. To homogenize collected tissues, 350 μL of RLT lysis buffer from Qiagen RNeasy Mini Kit is added to the tissue and homogenized with a pestle mortar. The homogenized tissues went through sonication with a Q55 sonicator (Qsonica) and then 350 μL cold, 70% EtoH was added to the sample. After this, the mixed solution is processed by series of spin and wash follow the instructions book from Qiagen RNeasy Mini Kit. Once RNAs are isolated from tissues, we applied qRT-PCR with Shox2 primer (Table 1) and normalized with GAPDH.
Western Blot
Thalamic tissues were collected from the adult mouse brains and immediately placed on dry ice and stored at −80°C until use. The thalamus samples were lysed with RIPA lysis buffer (150 mM sodium chloride, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris, pH 8.0) with fresh added Halt™ protease inhibitor cocktail (ThermoFisher Scientific, 78 430). Samples were centrifuged at 12000 rpm at 4°C for 20 min, and the supernatant protein samples were collected. The protein concentration of the samples was determined using the Bio-Rad DC protein assay (Bio-Rad, 500–0116). Samples were normalized with the same lysis buffer, aliquoted and stored at −80°C until use. Before loading, 5X sample buffer (ThermoFisher Scientific, 39 001) and dithiothreitol (final concentration—50 mM, DTT) were added to each protein sample. Sample mixtures were left at room temperature for 30 min. Protein (20–30 μg/well) was loaded in an SDS-PAGE gel (4% stacking gel and 8% separating gel), together with 3 μL prestained protein ladder (ThermoFisher Scientific, 26 619). The gels were run at 70 mV for 3 h, and the proteins were transferred to a preactivated PVDF membrane (Millipore, IPFL00005) at −70 mV for 3 to 4 h. SDS and methanol were added into transfer buffer at a final concentration of 0.1 and 10%, respectively. The gels were stained with Coomassie brilliant blue solution (0.1% Coomassie brilliant blue, 50% methanol, 10% glacial acetic acid) to check that no obvious proteins remained under these transfer conditions. Membranes were incubated in blocking solution with 5% nonfat dry milk and 3% BSA in TTBS at room temperature for 1 h. Primary antibody was diluted in OdysseyR Blocking Buffer in TBS and applied on the membrane at 4°C overnight. After washing the membrane with TTBS, fluorescence-conjugated secondary antibodies were diluted and applied on the membrane at room temperature for 1 h. Imaging of the stained membrane was done in an Odyssey CLx Infrared Scanner and analyzed by Image Studio Lite Ver 5.2.
Antibodies used in IHC and Western blot experiment.
| Reagent/Resource | Supplier | Details |
|---|---|---|
| Chicken anti-β-galactosidase | Abcam | Ab9361; 1:500 |
| Rabbit anti-NeuN Cy3-conjugated | Millipore Sigma | ABN78; 1:500 |
| Mouse anti-parvalbumin | Millipore Sigma | MAB1572; 1:500 |
| Rabbit-anti GFAP | PhosphoSolution | 620-GFAP; 1:300 |
| Rabbit-anti GFP | Novus Biologicals | NB 600–308; 1:300 |
| Alexa-Fluor 488 Goat anti-mouse | Life Technologies | 1:1000 |
| Alexa-Fluor 647 Goat anti-chicken | Life Technologies | 1:1000 |
| Alexa-Fluor 594 Goat anti-rabbit | Life Technologies | 1:1000 |
| Mouse anti-HCN2 | Neuromab | N71/37; 1:1000 |
| Mouse anti-HCN4 | Neuromab | N114/10; 1:1000 |
| Mouse anti-Cav3.1 | Neuromab | N178A/9; 1:1000 |
| IRDye 680RD Goat anti-mouse | Li-Cor | P/N 926–68 070; 1:10000 |
| IRDye 800CW Goat anti-rabbit IgG (H + L) | Li-Cor | P/N 926–32 211; 1:10000 |
Electrophysiology
At the same time of day (11:00 am summer and 10:00 am winter), mice were anesthetized with isoflurane and decapitated. Brains were quickly removed and immersed in oxygenated (95% O2 and 5% CO2), ice-cold N-methyl-d-glucamine (NMDG)–based slicing solution (in mM, 110 NMDG, 110 HCl, 3 KCl, 1.1 NaH2PO4, 25 NaHCO3, 25 glucose, 10 ascorbic acid, 3 pyruvic acid, 10 MgSO4, 0.5 CaCl2). The first 350 μM coronal brain section containing the most anterior paraventricular thalamus (PVA) was obtained with a Vibratome Series 3000 Plus Tissue Sectioning System. The collected brain slices were transferred and incubated in oxygenated standard aCSF (in mM, 125 NaCl, 2.5 KCl, 26 NaHCO3, 1.24 NaH2PO4, 25 dextrose, 2 MgSO4, 2 CaCl2) at 37°C for 30 min, then incubated at room temperature until use.
During the recordings, an individual slice was transferred to a recording chamber and perfused with oxygenated external solution at a speed of 1 mL/min at room temperature. Unless otherwise specified, standard aCSF was perfused as the external solution. For isolation of the specific currents, different pharmacological antagonists were applied in the external solution as stated in the results. PVA was identified as the nucleus near the border of the third ventricle enclosed by the stria medullaris. Cell-attached and whole-cell recordings were obtained using MultiClamp 700B amplifier, Digidata 1322A digitizer, and a PC running Clampex 10.3 software (Molecular Device). For cell-attached recording, glass pipettes had resistances of 2.5–3.5 MΩ filled with standard aCSF. Gigaseals were obtained in every cell by application of a small negative pressure for spontaneous action potential recording. For intracellular whole-cell patch clamp recording, glass pipettes had resistances of 3.5–6 MΩ filled with internal pipette solution (in mM): 120 Kgluconate, 20 KCl, 0.2 EGTA, 10 Hepes, 4 NaCl, 4 Mg2+ATP, 14 phosphocreatine, 0.3 Tris GTP (pH was adjusted to 7.2–7.25 by KOH, osmolarity was adjusted to 305–315 mOsm by sorbitol). Series resistance was monitored, and only cells with series resistance less than 20 MΩ and that did not change over 15% throughout the recording were further analyzed. Spontaneous action potentials were recorded in current clamp mode at membrane potential. Cells with no action potentials identified in 5 min are classified as “not active” cells.
HCN or T-type calcium currents were isolated under voltage clamp. The external solution for HCN current isolation contained 0.5 μΜ TTX, 1 mM NiCl2, 1 mM CdCl2, 2 mM BaCl2, 10 μΜ DNQX, and APV to block voltage-gated sodium channels, voltage-gated calcium channels, inwardly rectifying potassium channels, and excitatory synaptic current, respectively. NaH2PO4 was omitted to prevent precipitation with cations. A standard aCSF with 0.5 μΜ TTX (to block voltage-gated sodium channels) was used to isolate T-type calcium current. T-type calcium currents were isolated using a standard subtraction protocol (Zhang et al. 2009). Currents induced at −50 mV by step voltages ranging from −90 to 0 mV (Supplemental Fig. 2B) were subtracted from those induced by the same step voltages depolarized from a 1 s holding potential at −100 mV (Supplemental Fig. 2A) to remove the inactivation of T-type calcium current. The isolated current was completely blocked by bath application of 2 mM NiCl2 (Supplemental Fig. 2C, D), confirming the isolated current was T-type calcium current (Bhattacharjee et al. 1997; Lee et al. 1999). Since accurate measurement of T-type currents are compromised by poor voltage control, recorded currents were monitored for good voltage control. Specifically, there was no extensive delay in the onset of current; also, the onset and offset kinetics depended on voltage, not on the amplitude of current.
Computational Methods
Modeling of thalamocortical circuitry was done using a well-established computational model of the thalamocortical circuit, originally constructed by Destexhe (Destexhe et al. 1998) and implemented by Knox et al. (2018). The model is available here: https://senselab.med.yale.edu/modeldb/ShowModel?model=234233#tabs-1 and outlined extensively by Destexhe et al. (1994), Destexhe (1998), and Destexhe et al. (1998). This model has been previously described by Knox et al. (2018), but briefly, it consists of 100 TCNs, 100 cortical interneurons, 100 cortical pyramidal (PY) neurons, and 100 thalamoreticular (RE) neurons with local network connections. Cell type–specific current parameters and synaptic conductances were fit to experimental data (Destexhe et al. 1998). Here, we focus on the two currents found to be altered in Shox2 KO mice, the hyperpolarization-activated cation nonselective (HCN) current IH and the T-Type Ca2+ channel ICa,T. Previously, the network model was used to assess the effects of T-type calcium channel gene variants in RE neurons (Knox et al. 2018), and the TCN model was used to assess the effect of HCN channel modulation on TCN firing (Wang et al. 2002; Knox et al. 2018).
ICa,T is described by a Hodgkin-Huxley (1952) formalism:
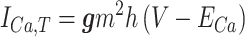 |
(1) |
where g is the conductance of the channel, m and h are the activation and inactivation gating variables of the channel, respectively, V represents the membrane potential, and ECa is the reversal potential for calcium.
The activation of T-type Ca2+ channel, m, is described by the following differential equation:
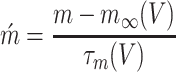 |
(2) |
where 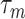 represents the inactivation time and
represents the inactivation time and 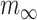 is the steady state activation:
is the steady state activation:
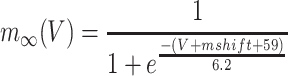 |
(3) |
Initially, we set the parameters of the model to generate nonpathological oscillations (waxing and waning sleep spindles, 8–10 Hz) in response to square pulse stimulation of 5 TCNs, as described in Knox et al. 2018. Then, we mimicked the KO deficiencies of T-type and HCN channels of 70 random TCNs and analyzed whether these changes alone perturbed the network into generating pathological oscillations (spike and wave, 3–4 Hz).
Baseline parameter settings for the nonpathological state included setting the GABAA synaptic conductance for cortical interneurons to 50% of its maximal value (Knox et al. 2018) for increased cortical excitability as in Destexhe (Destexhe et al. 1998). In order to mimic the voltage dependence of activation of the T-type Ca2+ channel in TCNs, 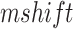 was set to −5 mV in thalamocortical cells of KO cells only. This resulted in a positive 3 mV shift in half activation, as observed in the experimental KO (see Figure 7A and B). In order to mimic the decrease in HCN current density in Shox2 KO mice, the conductance of the TCN HCN channels was reduced to 0.6 µS, which is 30% of the baseline value, 2 µS. Fast Fourier transform (FFT) analysis of the pyramidal neuron spikes times was performed by creating a histogram of spike times and analyzing it with the FFT function in python. Raster plots were generated in NEURON using the raster plot command.
was set to −5 mV in thalamocortical cells of KO cells only. This resulted in a positive 3 mV shift in half activation, as observed in the experimental KO (see Figure 7A and B). In order to mimic the decrease in HCN current density in Shox2 KO mice, the conductance of the TCN HCN channels was reduced to 0.6 µS, which is 30% of the baseline value, 2 µS. Fast Fourier transform (FFT) analysis of the pyramidal neuron spikes times was performed by creating a histogram of spike times and analyzing it with the FFT function in python. Raster plots were generated in NEURON using the raster plot command.
Figure 7.
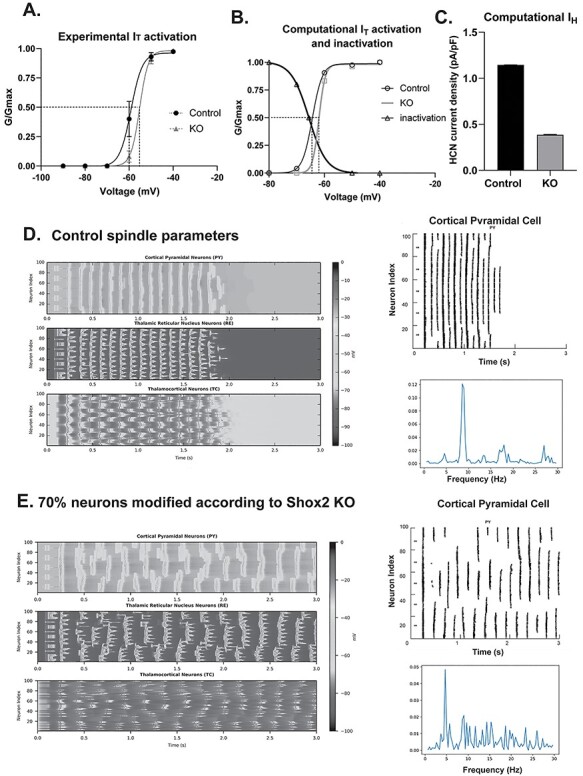
Computational model suggests that Shox2 modulation alters thalamocortical oscillation frequency. A. Plot of experimental activation and Boltzmann showing half-activation of IT for cells recorded in WT (−60 mV) and KO mice (−57 mV). B. Plot of activation of computational T-type current was generated, and Boltzmann fit showed a half activation voltage of −65 mV that shifted to −62 mV for the KO. C. Computational IH conductance was reduced by 60% as in experimental data. D. The model is set to baseline parameters that generate spindle oscillations (8 Hz). Each heat map displays activity of a cell-type population (PY, TCN, or RE) with cell index number on the y-axis and time on the x-axis. Oscillation frequency is determined by the number of PY population events per second. Right-Raster plots of computational pyramidal cell spike times. Frequency was determined by Fast Fourier transform (FFT) analysis of control (D) and KO (E) computational PY neurons in thalamocortical network reveals a peak at 8 Hz for control and ~4.5 Hz for KO paradigms.
Pilocarpine Model of Seizure Generation
Control and KO (RosaCreErt-Shox2 KO) mice were injected with methyl-scopolamine (1 mg/kg) and 30 min later received a single injection of pilocarpine (300 mg/kg). Behavioral seizures were observed and scored by a blinded observer according to the modified Racine scale (Racine et al. 1973) in 5 min blocks up to 1 h after pilocarpine injection.
Statistics
Unless otherwise noted, control and KO results were compared using a Student’s unpaired t-test.
Results
Shox2 is specifically expressed in the thalamic neurons in the brain of young adult mouse
To investigate the expression of Shox2 in postnatal mouse brain, coronal brain slices from P25 and P60 Shox2LacZ/+ mice were stained with X-gal, in which the expression of LacZ indicated Shox2 expression (Supplemental Fig. 3A and B). X-gal staining indicated that Shox2 is expressed throughout the dorsal thalamus including midline thalamus, anterior thalamus nuclei (ATN), ventrobasal nucleus (VB), dorsal lateral geniculate nucleus (dLGN), and medial lateral geniculate nucleus (MGN) but not in other regions of diencephalon including habenula, reticular nucleus of the thalamus and hypothalamus, or other regions of the nervous system like the cortex, subcortical regions of the forebrain, hippocampus, amygdala, cerebellum, and spinal cord. To determine whether the expression pattern of Shox2 changes during development, coronal brain slices from a P56 Shox2Cre/+, Rosa26LacZ/+ mouse in which LacZ is expressed in all cells that have expressed Shox2 at any time during development, were stained with X-gal (Supplemental Fig. 3C). These results showed that the expression of Shox2 is relatively restricted to the dorsal thalamus in the adult as well as during development, with sparse expression extended to habenula and superior and inferior colliculus and nuclei within the brainstem in the developing animal. We further assessed the cell type of Shox2-expressing cells.
In order to determine the cell types in which Shox2 is expressed, thalamic neurons were labeled with the neuronal nuclear protein antibody (NeuN), which is specifically expressed in mature neurons (Herculano-Houzel and Lent 2005; Gusel'nikova and Korzhevskiy 2015). Shox2 was coexpressed in most, but not all, approximately 70% of the neurons in the thalamus, NeuN-positive cells throughout the dorsal thalamus from rostral to caudal (Fig. 1A–C). Importantly, all Shox2-labeled cells were NeuN-positive, suggesting that Shox2 expression is restricted to neurons. To confirm that Shox2 was not expressed in astrocytes, the coexpression of Shox2 and Glial Fibrillary Acidic Protein (GFAP), which labels astrocytes (Eng 1985; Yang and Wang 2015), was assessed. Shox2 was not expressed in the GFAP-positive astrocytes throughout the thalamus (Fig. 1D–F). Together these results show that Shox2 is expressed in neurons and not GFAP-positive astrocytes.
Figure 1.
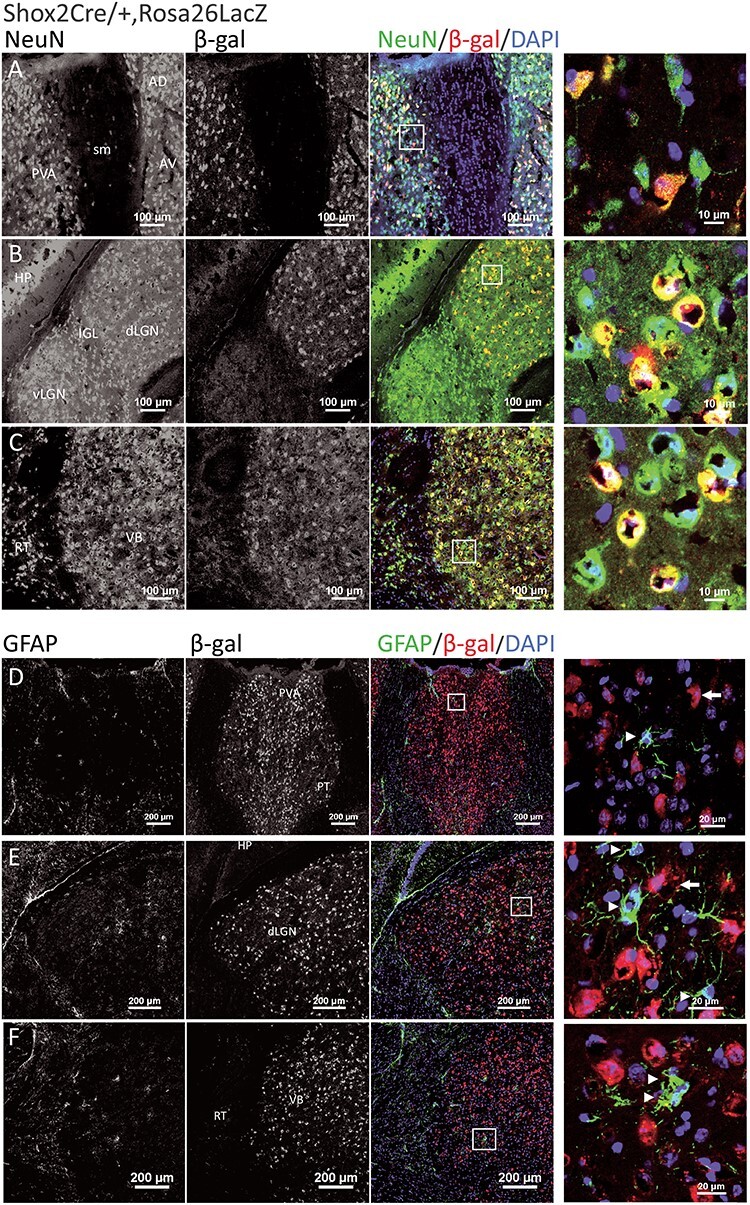
Shox2 is expressed in NeuN+ neurons in the thalamus and not in GFAP+ astrocytes. Coronal brain sections through the thalamus of Shox2Cre/+, Rosa26LacZ mice were costained with NeuN (green in last 2 columns) and β-gal, the reporter for Shox2 (red in last two columns). Three typical thalamic regions are shown, including anterior PVA (A), dorsal lateral geniculate nucleus (dLGN) (B), and ventrobasal nucleus (VB) (C). Shox2 is expressed in NeuN+ neurons (yellow, merged). Right panels are magnifications of the boxed regions, respectively, showing cells that coexpress Shox2 (red) and NeuN (green). D-F show the coexpression of astrocyte marker GFAP (green) and β-gal, the Shox2 reporter (red), in three thalamic regions: PVA (D), dLGN (E), and VB (F). Right panels in the last column are magnifications of the boxed regions. The arrowheads show the GFAP+ astrocytes, and the white arrows show Shox2-expressing cells as indicated by β-gal. No cells coexpressed GFAP and Shox2. RT: reticular thalamus; AV: anteroventral nucleus of the thalamus; AD: anterodorsal nucleus of the thalamus.
To determine if Shox2 is expressed in GABAergic neurons, immunohistochemistry (IHC) with parvalbumin (PV) on coronal brain sections from Shox2Cre/+, Rosa26LacZ/+ mice was performed (Fig. 2). Parvalbumin (PV) is highly expressed in the interneurons of the reticular nucleus of thalamus, which borders the thalamus laterally and ventrally. PV labeling delineated the reticular nucleus that defined the border of the thalamus. The PV staining results confirmed the results shown in Supplemental Figure 3 that during development, Shox2 is expressed throughout the thalamus (Fig. 2) and sparsely in the habenula (Fig. 2D) and midbrain (Fig. 2I). Importantly, Shox2 was not expressed at any point during development in cells of the cortex (Fig. 2C), reticular nucleus of the thalamus (Fig. 2E and F), hypothalamus (Fig. 2G), or hippocampus (Fig. 2I). In addition, our results showed few PV+ cells in the thalamus as previously reported (Arcelli et al. 1997), and Shox2 was not coexpressed in PV+ cells (Fig. 2G). These results suggest that Shox2 is not expressed in parvalbumin-expressing inhibitory interneurons.
Finally, the expression and projections of Shox2-expressing neurons were investigated using Shox2Cre/+, RosatdTomato-ChR2 mice. These mice allow labeling of Shox2-expressing neurons with td-Tomato and manipulation of Shox2-expressing neurons with Channelrhodopsin-2. Interestingly, we found that the Shox2-expressing neurons projected to multiple cortical areas, including retrosplenial and somatosensory cortices with a clear delineation of the barrel fields in somatosensory cortex (Fig. 2M and O). Further strong projections from Shox2-expressing neurons were observed within the thalamus, particularly the VB complex, in reticular nucleus of thalamus and internal capsule (Fig. 2M and O) projecting to somatosensory cortex. In summary, the X-gal staining and immunofluorescence results indicated that Shox2 expression was restricted to excitatory TCNs in the adult stage.
Lack of Shox2 Affects Gene Expression
To study the specific role of Shox2 in regulation of gene expression in neurons of adult thalamus, RNA sequencing was performed. For this experiment, thalamus from the inducible knockout (KO) of Shox2 in Gbx2CreERt/+, Shox2−/f mice were compared with littermate Gbx2CreERt/+, Shox2+/f control (CR) mice. The results showed 372 differentially expressed genes (DEG) between CR and KO mice, 212 of which are down-regulated and 160 are up-regulated in KO tissue (Supplemental file and Fig. 3A). Gene Ontology (GO) analysis showed Shox2 KO–affected genes in GO terms of ion channel activity, synapse function, and learning (Fig. 3B). Importantly, Shox2 KO downregulated the expression of Hcn2, Hcn4, and Cacna1g genes (Supplemental file and Fig. 3A). The protein products of these genes, HCN2, HCN4, and Cav3.1, mediate HCN current and T-type Ca2+ currents, respectively. Since these channels are significant contributors to the rhythmic firing properties of TCNs, we further pursued mRNA and protein expression of these channels.
Figure 3.
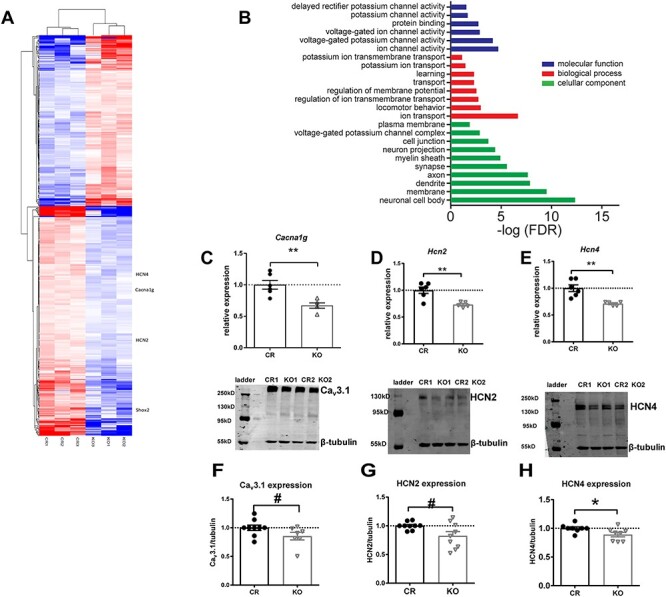
Shox2 expression affects gene expression and ion channel protein levels. RNA sequencing and analysis were performed as described in methods. A. Heatmap, made by pheatmap, saturated at 1, displays 367 DEGs (adjust P-value <0.1) in the midline thalamus between control (CR) and Gbx2CreErt, Shox2 KO mice. B. Gene ontology (GO) enrichment analysis of DEGs. All terms with an FDR (analyzed by DAVID functional annotation tool) less than 0.1 are listed. Ingenuity pathway analysis revealed that Shox2 KO-induced DEGs are highly involved in thalamus-related neurological diseases. C-E. RT-qPCR results show that Shox2 KO significantly reduced mRNA level of Cacna1g. (C), Hcn2 (D) and Hcn4 (E). F-H. Shox2 KO decreased the protein expression levels of Cav3.1 (F, ~120 kD), HCN2 (G, ~150 kD), and HCN4 (H, >200 kD) (**, P < 0.01; *, P < 0.05, #, P < 0.1). The bands around ~55–60 kD are recognized by the β-tubulin antibody.
To confirm Shox2 regulates ion channel-related genes in the whole thalamus, another transgenic mouse line, the global KO (Rosa26CreERt/+, Shox2f/− mice), in which Shox2 was reduced in the whole thalamus was used. The RNA was extracted from KO (Rosa26CreERt/+, Shox2f/− mice) and CR (Rosa26CreERt/+, Shox/f/+) mice, and RT-qPCR was performed. The Cav3 family of Ca2+ channel subunits encode IT and is highly expressed in the nuclei of the thalamus (Kim et al. 2001a; Song et al. 2004). We tested the levels of mRNA expression for Cav3.1 and Cav3.2, which are coded by Cacna1g and Cacna1h, respectively. Previous studies showed that Cav3.1 protein subunits are the primary T-type calcium channel proteins expressed in the TCNs, while Cav3.2 proteins are expressed at lower levels in the thalamus but are the prominent subunit in the reticular nucleus of the thalamus (Talley et al. 1999; Kim et al. 2001; Zhang et al. 2004; Anderson et al. 2005). Our results showed that expression of Cacna1h expression was very low in the thalamus, which confirmed the specificity of our thalamic dissection, and there was no significant difference in Cacna1h expression between CR and KO (Student’s t-test, t9 = 1.02, P = 0.34). With respect to the expression of Cacna1g, Shox2 KO significantly decreased the mRNA expression of Cacna1g (Fig. 3C; Student’s t-test, t9 = 3.85, P < 0.01) in the thalamic tissue. These results confirmed the mRNA sequencing data and suggest that Cav3.1 channels that contribute to the T-type currents are down-regulated in the thalamus.
The mRNA expression of HCN channel genes in CR and KO mice was also assessed. Previous studies reported that mouse brains express very low levels of Hcn3 (Moosmang et al. 1999), and our RNA-seq data showed no significant change in Hcn3 or expression in the KO mice; therefore, Hcn3 expression was not further investigated. The expression levels of mRNAs for Hcn1, Hcn2, and Hcn4 were investigated. Hcn2 mRNA was the most highly expressed HCN channel gene in the thalamus tissue. Hcn4 also had prominent expression, while the level of expression of Hcn1 was only about 5% of Hcn2 expression. This result is consistent with previous research indicating HCN2 and 4 channels are the most highly expressed HCN channels in the thalamus (Moosmang et al. 1999), and these results along with our sequencing results provide relative expression data of HCN mRNA expression in mouse thalamus. Hcn1 mRNA expression levels were not significantly affected by Shox2 KO in comparison to CR mice (t9 = 1.85, P = 0.10). Hcn2 and Hcn4 mRNA were significantly reduced in the Shox2 KO thalamus compared with CR mice (Fig. 3D and E, Hcn2: t9 = 3.92, P < 0.01 and Hcn4: t9 = 4.02, P < 0.01). These results also confirmed the RNA-seq results that showed that Hcn1 mRNA was not significantly affected in mouse thalamus. Together, these results show that Shox2 KO significantly reduced expression of mRNAs for HCN and Ca2+ channels. We further investigated if the proteins for these channels were also affected in the KO mice.
Western blot experiments on whole thalamus extract were performed to test the protein levels of the Cav3.1, HCN2, and HCN4 channels. The expression levels of HCN2 and HCN4 proteins were significantly decreased in the thalami of KO animals, and there was a trend toward decreased expression of Cav3.1 protein compared with CR mice (Fig. 3F–H; Cav3.1: t14 = 1.86, P = 0.08; HCN2: t16 = 2.30, P = 0.04; HCN4: t14 = 2.37, P = 0.03). The protein measurements in these Western blot staining results are consistent with sequencing and RT-qPCR results and confirmed that HCN2, HCN4, and Cav3.1 protein expression is modulated by Shox2 in the adult thalamus. While the change in expression of these channels is relatively small, it should be noted that these data are taken from the entire thalamus, including neurons and glial cells that do not express Shox2. The consistency of the sequencing, mRNA and protein expression is solid evidence that Shox2 affects expression of these ion channel genes.
Since the expression levels of the channel proteins that underlie currents important for the bursting properties of the thalamic neurons are regulated by Shox2 expression, we assessed the firing and intrinsic properties of thalamic neurons in Shox2 KO and CR mice. To best identify a single thalamic nucleus and a homogenous neuron group, the anterior PVA, the most rostral and dorsal midline nucleus, was chosen as the target region for recording. First, cell-attached voltage-clamp recordings were performed to record the spontaneous action potential currents of PVA neurons without rupturing the cell membrane. Our results indicated that a smaller percentage of cells fired spontaneous action potentials (active neurons) in KO mice compared with those in CR mice (Fig. 4A, CR: 36% vs KO: 14%; χ2 test, χ2 = 3.84, P = 0.05). We also conducted whole-cell current clamp recordings and tested spontaneous action potential firing at resting membrane potential. Slices from KO mice consistently showed a significantly lower percentage of spontaneously active neurons (CR: 47% vs KO: 18%; χ2 test, χ2 = 7.60, P < 0.01). The decreased cell excitability in PVA neurons from the KO mice was not due to significant differences in resting membrane potential between CR and KO cells (CR: −55.5 ± 1.7 (n = 35) and KO: −54.5 ± 1.7 (n = 35), P = 0.9); however, input resistance was significantly different between cells recorded from KO and CR mice (CR: 993.4 ± 52.86 (n = 30) KO: 850.3 ± 40.73 (n = 29); P = 0.04). In addition, an increased action potential threshold in KO compared with CR mice was observed (Fig. 4B; t42 = 2.0; P < 0.05). These results suggest that reduced Shox2 expression reduces the spontaneous firing of TCNs.
Figure 4.
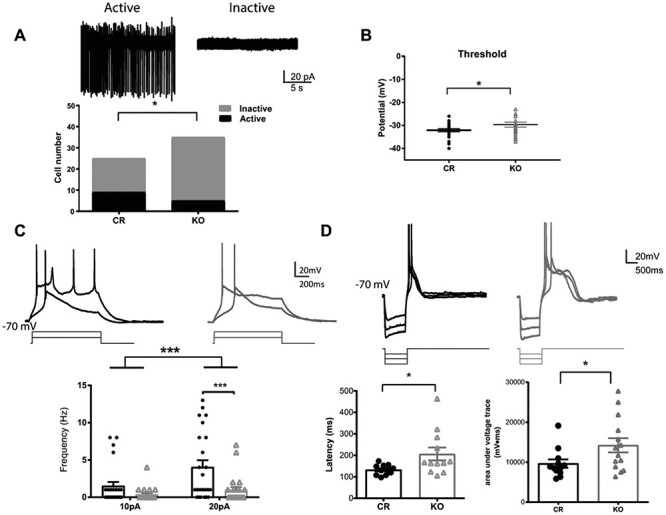
Shox2 KO decreases the ratio of cells with spontaneous action potentials in the anterior paraventricular thalamus. A. Example traces of attached-cell recordings of active cells showing spontaneous action potentials (left) and inactive cells with no action potentials (right). Bar graph representing the ratio of active and inactive cells recorded in PVA from KO and CR mice. This ratio is significantly smaller in KO than in CR mice (*, P < 0.05). B. Threshold measured from first spike of depolarization in KO and CR mice. The threshold was significantly depolarized in KO mice. C. Upper: Example trace of action potential induced by a 1 s, 10 and 20pA current injection steps, while holding membrane potential at −70 mV. Lower: Scatter plot graph showing that the number of spikes induced by 10pA and 20pA current injection at −70 mV was reduced in the KO mice (gray) relative to control mice (black). D. Upper: Example traces of rebound spikes triggered by injection of negative current steps (−50, −100 and −150pA) from a holding potential of −70 mV. Lower—left: The latency to the peak of calcium spike after current injection is significantly longer in Shox2 KO neurons than in control neurons. Lower—right: The areas under voltage traces (between spike traces and −70 mV) indicating plateau depolarizations are significantly larger in Shox2 KO neurons (arrow) than neurons from control mice (*, P < 0.05, ***; P < 0.001).
To investigate the voltage response to small depolarizations, we recorded in current clamp mode and injected 10pA and 20pA currents to evoke action potentials from resting potential and −70 mV. Action potential frequency in neurons recorded from Shox2 KO slices was significantly decreased compared with that in neurons from CR slices both at resting potentials (two-way repeat measures ANOVA: main effect of current injection, F1,44 = 19.9, P < 0.001; main effect of genotype, F1,44 = 4.90, P < 0.001; interaction, F1,44 = 7.71, P < 0.01; post-hoc Bonferroni’s test: CR vs KO at 20pA current injection, P < 0.001) and −70 mV (Fig. 4C, two-way repeated measures ANOVA: significant main effect of genotypes (CR vs KO, F(1, 60) = 4.2, P = 0.04) and current injection (10pA vs 20pA, F(1, 60) = 36.60, P < 0.001) in the number of evoked spikes in 1 s depolarization from −70 mV. These results are consistent with the spontaneous firing results that suggest that reduced Shox2 affects TCN firing properties.
In order to assess a role for the HCN and Ca2+ currents, rebound bursting was induced by injection of negative currents at −70 mV (Jahnsen and Llinas 1984a, 1984b). The latency to the peak of rebounded calcium spikes was significantly longer in neurons from Shox2 KO slices than that in neurons from CR slices (Fig. 4D; t21 = 2.29, P = 0.03). Neurons from Shox2 KO slices had a slower decay phase of the spike rebound and therefore a longer rebound duration as indicated by a larger area under the voltage trace (Fig. 4D bottom; t24 = 2.11, P = 0.05). These results suggest that Shox2 expression affects Ca2+ currents and may also affect currents important for repolarization of the depolarizations, primarily K+ currents.
Shox2 is Critical for T-Type Calcium and HCN Currents in the Thalamus
The sequencing and RT–qPCR results indicating reduced expression levels of channels and the changes in both cell excitability and intrinsic properties of PVA neurons in Shox2 KO mice suggest that Shox2 modulates intrinsic currents, especially HCN and T-type currents, which contribute to cell excitability, rebound after-hyperpolarization, and resonance. Therefore, we hypothesized that Shox2 KO impaired pacemaker-related T-type calcium current and HCN currents in PVA neurons.
First, we determined the physiological properties of T-type calcium currents in CR and Shox2 KO. T-type calcium current was isolated with a typical subtraction protocol (Zhang et al. 2009) as described in the methods (Supplemental Fig. 2). The activated current density was significantly decreased in the PVA neurons from KO mice (Fig. 5A and B, two-way repeated measures ANOVA, main effect of genotype: F1,20 = 8.70, P < 0.01; main effect of voltage: F9,180 = 63.98, P < 0.001; interaction: F9,180 = 6.38, P < 0.001). A post-hoc Bonferroni’s multiple comparisons test revealed that the current density elicited at −60 mV (−7 ± 2.7 pA) and − 50 mV (−12.3 ± 1.4 pA) in CR neurons was significantly larger than that in KO neurons (−60 mV: −0.73 ± 0.5 pA and − 50 mV: −8.1 ± 1 pA; Fig. 5B, P < 0.001). Two-way repeated measures ANOVA of normalized T-type calcium activation curve revealed a significant interaction between genotype and voltage (F9,180 = 3.27, P < 0.01) and post-hoc Bonferroni’s test showed that the normalized T-type calcium current (I/Imax) was significantly decreased at −60 mV in KO mice compared with CR mice (Fig. 5C, CR: 0.36 vs KO: 0.08, P < 0.01). This suggests that the activation of the T-type calcium current is shifted toward more depolarized membrane potentials in the neurons from the KO mice and is consistent with the delayed threshold of low-threshold spike in KO neurons in Figure 4. In addition, the time to peak of the T-type current measured at −50 mV in Shox2 KO neurons was significantly slower compared with CR neurons (Fig. 5D; t20 = 2.57, P = 0.02). The slower kinetic properties of T-type calcium current occurred during the activation (P = 0.08) but not inactivation phase (P = 0.97).
Figure 5.
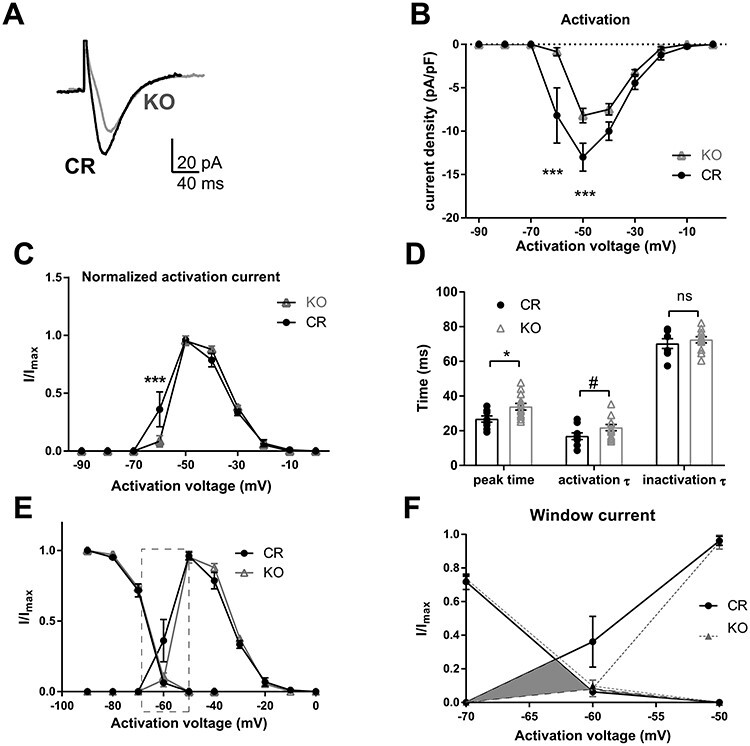
Properties of T-type calcium currents in PVA neurons of CR and Shox2 KO mice. The T-type calcium currents were isolated using voltage-clamp recordings according to methods. A. An example of T-type calcium currents recorded from PVA neurons of CR and Shox2 KO mice. T-type calcium currents in Shox2 KO mice are smaller in amplitude and slower than in CR mice. B. The current density curve of T-type calcium current activation. T-type calcium current density is smaller in PVA neurons of KO mice compared with CR mice (***, P < 0.001). C. The normalized activation curves indicate that T-type calcium (I/Imax) is larger at −60 mV in CR mice than that in KO mice (***, P < 0.001). D. Summary plot showing the time to peak (*, P < 0.05), activation and inactivation tau of the T-type current in TCNs from CR and KO mice. E. Inactivation and activation curves of T-type currents. F. Membrane potential range magnified to show T-type Ca2+ currents in −70 to −50 mV membrane potential window range.
The inactivation properties of the T-type calcium currents were determined by eliciting inactivation of the T-type calcium currents at −50 mV after a 1-second hyperpolarizing potential ranging from −90 to −40 mV (Supplemental Fig. 2). This curve confirmed that the current density was decreased in neurons from KO mice compared with neurons from CR mice (two-way ANOVA, the main effect of genotypes, F(1, 21) = 8.41, P < 0.01). However, the normalized inactivation curves of T-type calcium currents from neurons from CR mice and neurons from KO mice were not significantly different (Supplemental Fig. 2), suggesting that the inactivation kinetics of the T-type currents were not different between CR and KO mice.
Kinetics analysis revealed that the T-type current in neurons from Shox2 KO mice have a longer time to peak and slower inactivation time constant compared with neurons from CR mice. The T-type calcium current window, defined by the area under both the activation and inactivation curves, which contributes to the large amplitude and long lasting depolarization, or UP state, of the slow (<1 Hz) sleep oscillation in thalamic neurons (Williams et al. 1997; Hughes et al. 1999; Crunelli et al. 2005; Crunelli et al. 2006; Dreyfus et al. 2010), was greater in neurons from CR mice compared with neurons from KO mice (Fig. 5E). This is particularly striking in the potential range between −60 and −70 mV (Fig. 5E and F) and suggests that the Shox2-mediated reduction of the T-type current is particularly important near resting membrane potential and may contribute to slow oscillations.
Since HCN currents play a role in the firing properties of TCNs (Zobeiri et al. 2018; Zobeiri et al. 2019), and Shox2 affects expression of Hcn4 mRNA and protein, we investigated the effect of Shox2 KO on HCN current by sequential hyperpolarizations in voltage-clamp mode in the presence of BaCl2 to block inward rectifier K+ currents. The amplitude of HCN current was measured as the difference between the end current of one-second hyperpolarization and the beginning instantaneous current at −150 mV hyperpolarization (Fig. 6A). The HCN current densities in neurons from Shox2 KO mice were significantly decreased compared with neurons from CR mice (t15 = 3.1; P = 0.007; Fig. 6B). Recordings were completed in current clamp to determine the functional impact of reduced Ih, within the TCNs. Negative current injections from resting potential (Fig. 6C; 10–90 pA in 10 pA steps) induced significantly smaller voltage sags in the KO mice compared with CR mice (Fig. 6C and D; two-way ANOVA; main effect of current input F(5, 55) = 24.5; P < 0.0001 and main effect of genotype F(1, 11) = 17.26 but no interaction F(5, 55) = 0.45; P = 0.8). These results suggest that Shox2 KO reduced Ih that physiologically impacts rebound firing of TCNs. In addition, the differences in sag were revealed in the presence of BaCl2 and not in control aCSF, which suggests that an inwardly rectifying K+ current may partially compensate for the differences in Ih.
Figure 6.
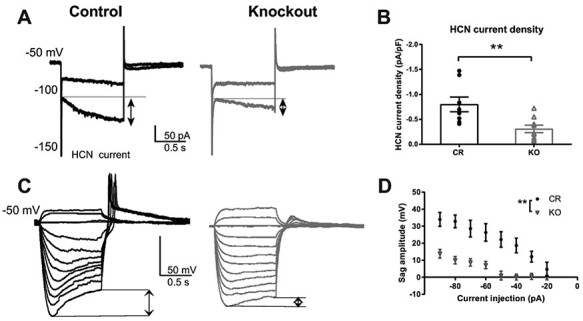
Shox2 KO decreased HCN current in anterior PVT of neurons. A. An example of HCN current elicited by hyperpolarizing cell membrane from −50 to −100 mV and −150 mV. HCN current is defined as the current difference between the current at the end of 1 s hyperpolarization and the current peak at the beginning of hyperpolarization as shown in the figure (arrows). B. Scatter plot showing that Shox2 KO (n = 9; N = 3) decreased HCN current density in TCNs (**, P < 0.01) compared with CR neurons (n = 8; N = 2). C. Example current clamp recordings demonstrating hyperpolarizing pulses and sag in CR and KO mice. D. Current voltage plot showing sag amplitude measured in response to negative current injection (90–10 pA). (**, P < 0.01; n = 7; N = 2; KO n = 6, N = 3).
Computational Modeling Predicts That Shox2 Expression Favors Normal Spindle Activity
How do these alterations in thalamocortical channel function affect the thalamocortical network? We used an established computational model of the thalamocortical network (Knox et al. 2018), to demonstrate that the Shox2-mediated alterations in currents are predicted to affect thalamocortical oscillation frequencies. We computationally mimicked the observed deficits in T-type and HCN currents in a randomly selected 70% of TCNs, as our anatomical data showed that Shox2 was expressed in approximately 70% of the TCNs.
To calibrate the model to experimental KO, we matched the shift in half activation of IT and the reduction in IH. Boltzmann equations were used to determine half activations of control (−60 mV) and KO (−57 mV) experimental T-type currents (Fig. 7A), revealing a positive shift of 3 mV in the KO. This shift was mimicked in the computational KO IT (Fig 7B, gray trace) compared with control (black trace). This half activation shift also served to reduce the current to 70%, as observed experimentally (Fig 5), because of increased overlap of the steady-state activation curve with the steady-state inactivation curve (Fig 7B). With regard to IH, based on experimental results shown in Fig 6B, we reduced IH to 30% of baseline value (Fig. 7C).
With the original, default parameters under baseline conditions, which are described in the Methods, the thalamocortical model network exhibits transient, waxing and waning, nonpathological spindle oscillations (7–8 Hz, Fig. 7D) in response to a stimulus that simulates a perturbation of the network. The heat maps in Figure 7 are generated by recording the membrane potential of the 100-neuron populations over time, with neuron index on y-axis and time on the x-axis. Dark colors indicate a membrane potential near resting while light colors indicate depolarizations and spikes. Lines of yellow and orange show a synchronous population spike. The frequency of oscillations in the PY population can be roughly quantified by counting the number of population spikes per second. When the half-activation of the TCN T-type channel is shifted by 3 mV and the current through the HCN channel is reduced to 30% in 70 random TCNs to mimic the Shox-2 KOs, PY neuron firing activity is disrupted. The population-wide oscillations slow to near 4.5 Hz (Fig. 7E), a frequency comparable to that of pathological spike and wave-frequency oscillations (~4 Hz). Moreover, the oscillatory tendencies are more sustained, indicating the network may be susceptible to absence seizure generation in the simulated knockout network (spike raster plot in the top right of Fig. 7E) compared with control (same plot in Fig. 7D). The population frequency was quantified by performing a Fast Fourier transform (FFT) on the peri-stimulus time histogram of the spikes in the PY neuron population (7D, E, right bottom). Further, we found that it was necessary to mimic both T-type and HCN deficiencies caused by Shox2 KO in TCNs to result in this near 4 Hz frequency (Supplemental Fig. 4); neither manipulation in isolation was sufficient.
Shox2 Expression Inhibits Seizure Generation
Since the computational model suggested a possible role for Shox2-mediated changes in seizure generation, we investigated the potential for seizure development in Shox2 KO mice. In order to unambiguously quantify behavioral manifestations of seizures, we used the pilocarpine model of seizure generation. While pilocarpine injection is typically used to model temporal lobe epilepsy, several studies have demonstrated a role for the thalamus in the pilocarpine-induced seizures (Bertram et al. 2001; Li et al. 2014). Further seizures can be reduced by stimulation of the anterior thalamus (Hamani et al. 2008), and these results suggest thalamic involvement in pilocarpine-induced seizures.
Analysis of seizure latency and severity showed that Shox2 KO mice spent more time in seizure (P = 0.03; Fig. 8A), and while both Shox2 KO and littermate controls increased seizure severity over time (F(5, 125) = 98.29; P < 0.0001), Shox2 KO mice developed intense seizures more rapidly compared with littermate controls (Fig. 8BF(1,25) = 7.8; P = 0.01; repeated measures ANOVA).
Figure 8.
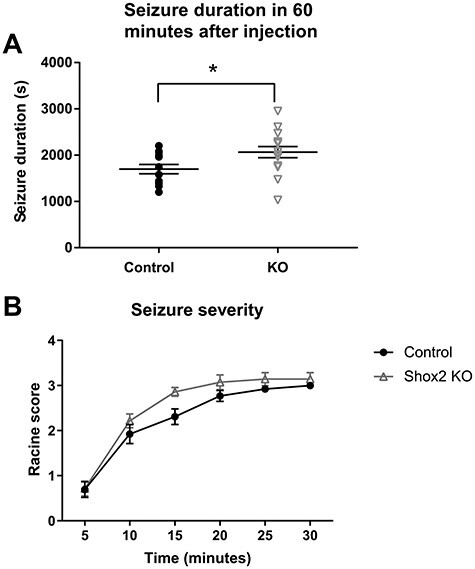
Pilocarpine induced longer duration seizures earlier compared with WT mice. Control and KO (RosaCreErt-Shox2 KO) mice were injected with methyl-scopolamine (1 mg/kg) and 30 min later received a single injection of pilocarpine (300 mg/kg). Behavioral seizures were observed and scored by a blinded observer according to the modified Racine scale in 5 min blocks up to 1 h after pilocarpine injection. A. Shox2 KO mice spent more time in seizure (P = 0.03). B. Shox2 KO mice developed seizures earlier compared with littermate controls (P = 0.01).
Discussion
This study demonstrates the importance of transcriptional activity of the homeobox protein transcription factor, Shox2, in regulation of firing properties and network function of TCNs to ultimately constrain seizure generation in adult mouse thalamus. This assertion is supported by our investigations at genetic, transcriptional, proteomic, electrophysiological, computational, and behavioral levels (Fig. 9). Genetic analysis via RNA-sequencing and Gene Ontology (GO) analysis revealed that Shox2 modulates expression of genes that encode for proteins directly associated with firing properties of TCNs, specifically voltage-gated ion channels. Further investigation using quantitative PCR and Western blotting showed that the mRNAs and proteins for several of these ion channels, namely HCN2, HCN4, and Cav3.1, are down-regulated in the thalamus of the Shox2 KO. Electrophysiological analysis revealed that Shox2 regulation of these channels and their corresponding currents contributes to the intrinsic firing properties in these neurons. Computational modeling showed that modulation of currents regulated by Shox2 is predicted to shift network oscillation frequency from spindle oscillations to a frequency associated with spike and wave patterns that characterize seizures, suggesting that Shox2 is important to maintain normal function of thalamocortical network activity. Finally, the pilocarpine model demonstrated that Shox2 KO mice exhibited a shorter latency to seizures and longer durations of seizures, suggesting that Shox2 is important for stabilization of bursting behavior within the thalamocortical circuitry.
Figure 9.
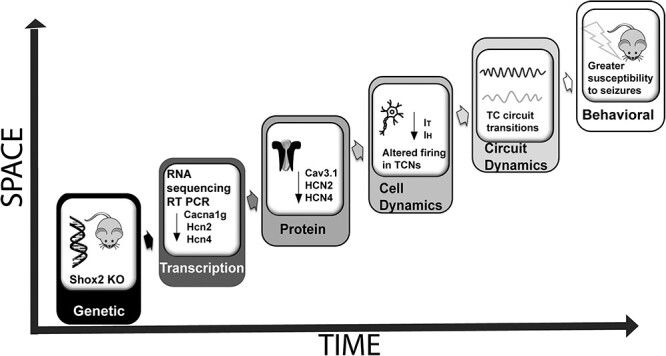
Schematic diagram of analyses accomplished in this study. We used Shox2 KO mice to determine the functional role of Shox2, demonstrating that Shox2 affects ion channel mRNA expression that is important for functional properties of the TCNs and thalamocortical circuit dynamics. This ultimately leads to a susceptibility to seizure generation.
Several types of epilepsy are characterized by 3 Hz spike-and-wave discharges on human electroencephalograms, and these patterns of activity arise from aberrant thalamocortical activity in humans (Williams 1953; Prevett et al. 1995) and animal models (Pollen et al. 1964; Avoli et al. 1983; McLachlan et al. 1984; Buzsaki et al. 1990; Inoue et al. 1993). Spindle oscillations, which are generated by thalamic circuits during sleep (Steriade et al. 1985; Steriade et al. 1987; Steriade 1993), can be gradually transformed into spike and wave discharges that ultimately result in seizures (Kostopoulos et al. 1981a; Kostopoulos et al. 1981b; McLachlan et al. 1984). In addition, other studies suggest that 5–9 Hz oscillations observed during quiet wakefulness may also progress to spike and wave activity (Pinault et al. 2001; Pinault et al. 2006). While previous experimental (Huguenard and Prince 1994; Bal et al. 1995; Destexhe et al. 1998; Castro-Alamancos 1999) and computational studies (Destexhe 1998) implicate a role for GABAergic inhibition in the transition, intrinsic properties have also been implicated (Huguenard and Prince 1992: Bal et al. 1995 #93; 1994). The computational model we used in this study was originally developed to examine circuit perturbations that promote this transition to lower frequency activity (Knox et al. 2018). Thus, it is uniquely suited for our needs. Interestingly, we found that it was necessary to mimic both T-type and HCN deficiencies in TCNs in 70% of the neurons to result in disruption of normal spindle activity and transition to spike and wave frequency activity. These results suggest that modification of intrinsic properties of specific cell function is important for transition from normal oscillation activity observed during sleep to lower hypersynchronous frequencies.
Our results show that expression of the transcription factor, Shox2, is important for maintenance of normal TCN firing activity. While regulation of other factors likely contributes to this phenotype, in this study, we investigated the IT and Ih currents that are known to contribute to thalamocortical oscillations. The channels that underlie IT, the Cav3.x channels, can mediate the low-threshold burst discharges that impact both physiological activity during slow wave sleep and pathological neuronal activity, including oscillations and hyperexcitable activity during epilepsy. Absence seizures in the thalamus are sensitive to the drug ethosuximide, which targets the Cav3.x channels (Huguenard 2002) that are highly expressed in thalamus and cortex (Talley et al. 1999). As described above, IT increases rebound bursting by deinactivation during hyperpolarizations mediated by actions of inhibitory IH activation. Several lines of evidence suggest that an increase in IT is an important mechanism that contributes to epileptiform activity. First, increased levels of IT occur before absence seizure onset (Zhang et al. 2004), suggesting that increased IT may contribute to seizure generation. Second, overexpression of Cav3.1, the predominant pore-forming subunit in the thalamo-cortical neurons, causes an absence seizure phenotype (Ernst et al. 2009), and knockout of Cav3.1 from tottering, stargazer, and lethargic mice eliminates absence seizures (Song et al. 2004). Finally, Cav3.1, but not 3.2 or 3.3, expression is down-regulated in a compensatory manner in a genetic rodent model of epilepsy, the WAG/Rij rats (Sharop et al. 2016). In addition, Cav3.1 in the thalamus was shown to be important for stabilization of sleep rhythms (Anderson et al. 2005). The above discussion suggests that dysregulation, either up- or down-regulation of the currents important for thalamic oscillations, can lead to aberrant and hyper-excitable activity and thalamic dysfunction.
The contribution to neuronal excitability of the HCN family of channels (HCN1–4) is associated with their unique properties, for example, they are both voltage- and ligand-gated. They open upon hyperpolarization and are sensitive to cyclic AMP levels. Because of the influence of HCN channels on neuronal excitability, they also play an important role in seizure generation. Several lines of evidence suggest that HCN channels are important for thalamocortical oscillations that lead to seizures. Specifically, HCN1 knockout mice are hypersensitive to pharmacologically induced seizures (Huang et al. 2009), and HCN2 knockout mice exhibit aberrant oscillations and absence seizures (Ludwig et al. 2003). Furthermore, expression of HCN channel subunits is differentially regulated in the WAG/Rij rat model of absence epilepsy (Kanyshkova et al. 2012). Our preliminary data demonstrate that HCN2, HCN4, and Cav3.1 channel expression is reduced in Shox2 KO mice, thus suggesting that Shox2 is a transcription factor that is important for expression of ion channel protein subunits that regulate oscillatory activity in the thalamus. While the physiology and intrinsic properties of thalamic relay neurons are well characterized, the genetic factors that establish the expression patterns of these channels are unknown. Shox2 represents a possible candidate that may control channel expression in the thalamic relay neurons and ultimately pattern pacemaker activity.
Several lines of evidence indicate that these studies of the role of Shox2 in pacemaker function in mice are also applicable to humans. Shox2 is a super-conserved gene with 99% amino acid identity between human SHOX2 and mouse Shox2. A recent study found that two missense mutations within the human SHOX2 gene are associated with early-onset atrial fibrillation, likely caused by a defect in pacemaker activity (Hoffmann et al. 2016; Li et al. 2018). In addition, while mice do not express the Shox gene, human SHOX and SHOX2 have 79% similar amino acid identity, and the same DNA-binding domains and putative phosphorylation sites. The functional redundancy in the regulation of heart pacemaker cells’ differentiation between human SHOX and mouse Shox2 has been demonstrated in mouse models (Liu et al. 2011; Liu et al. 2014). Therefore, investigation of Shox2 function in mouse can extend to evaluate the role of human SHOX and SHOX2 in humans. Turner syndrome (TS) is one of the most common sex chromosome abnormalities (Nielsen and Wohlert 1990; Jacobs et al. 1997) and results from the complete or partial loss of the X chromosome. Most individuals with TS have short stature, which is associated with the loss of the SHOX gene (Joseph et al. 1996; Blaschke et al. 1998; Oliveira and Alves 2011). These individuals are at increased risk for neurodevelopmental issues, including learning disabilities, visuo-spatial, social and executive function impairments (Mauger et al. 2018), and epilepsy (Puusepp et al. 2008; Jhang et al. 2014; Saad et al. 2014; Magara et al. 2015; Zhao and Lian 2015). Interestingly, the smallest chromosomal deletion associated with the neurocognitive phenotype included SHOX (Knickmeyer and Davenport 2011), “suggesting that loss of SHOX may play a role in cognitive impairments in humans.” While the mechanisms of the neurodevelopmental issues in these patients is unclear, our current study indicates that altering expression of SHOX- or SHOX2-related genes may contribute to thalamic dysfunctions and some of these neurodevelopmental impairments.
Further studies are necessary to determine the specific contribution of Shox2-expressing neurons to thalamocortical circuitry, and the role Shox2 may play beyond regulation of firing properties. In addition, future studies will investigate whether Shox2 plays a critical role during thalamus development and differentiation, the contribution of these Shox2-regulated currents to overall TCN function, and the mechanisms by which Shox2 regulates their expression.
Authorship statement
D.Y., I.F., and M.M. conceived experimental design, performed experiments, and wrote the manuscript. I.F., H.W., and C.C.C. contributed to the computational model. Y.S., X.H., I.F., C.N., E.M., and S.R. contributed data. C.S. contributed to early planning stages. Y.P.C. provided animals and reagents and L.A.S. contributed to overall design and wrote manuscript.
Funding
NIH (grant nos R21NS101482 to L.A.S., R01HL136326 to Y.P.C., NIH R01NS054281 to C.C.C.).
Notes
We thank Ruben Tikidji-Hamburyan for helpful input on model and FFT methods. Conflict of Interest: None declared.
Supplementary Material
Contributor Information
Diankun Yu, Neuroscience Program, Brain Institute, Tulane University, USA.
Isabella G Febbo, Neuroscience Program, Brain Institute, Tulane University, USA.
Matthieu J Maroteaux, Neuroscience Program, Brain Institute, Tulane University, USA.
Hanyun Wang, Neuroscience Program, Brain Institute, Tulane University, USA.
Yingnan Song, Cell and Molecular Biology, Tulane University, New Orleans, LA 70118, USA.
Xiao Han, Neuroscience Program, Brain Institute, Tulane University, USA.
Cheng Sun, Cell and Molecular Biology, Tulane University, New Orleans, LA 70118, USA.
Emily E Meyer, Neuroscience Program, Brain Institute, Tulane University, USA.
Stuart Rowe, Neuroscience Program, Brain Institute, Tulane University, USA.
Yiping Chen, Cell and Molecular Biology, Tulane University, New Orleans, LA 70118, USA.
Carmen C Canavier, Cell Biology and Anatomy, LSU Health Sciences Center, New Orleans, LA 70112, USA.
Laura A Schrader, Neuroscience Program, Brain Institute, Tulane University, USA; Cell and Molecular Biology, Tulane University, New Orleans, LA 70118, USA.
References
- Anderson MP, Mochizuki T, Xie J, Fischler W, Manger JP, Talley EM, Scammell TE, Tonegawa S. 2005. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 102:1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcelli P, Frassoni C, Regondi MC, De Biasi S, Spreafico R. 1997. GABAergic neurons in mammalian thalamus: a marker of thalamic complexity? Brain Res Bull. 42:27–37. [DOI] [PubMed] [Google Scholar]
- Avoli M, Gloor P, Kostopoulos G, Gotman J. 1983. An analysis of penicillin-induced generalized spike and wave discharges using simultaneous recordings of cortical and thalamic single neurons. J Neurophysiol. 50:819–837. [DOI] [PubMed] [Google Scholar]
- Bal T, McCormick DA. 1996. What stops synchronized thalamocortical oscillations? Neuron. 17:297–308. [DOI] [PubMed] [Google Scholar]
- Bal T, Krosigk M, McCormick DA. 1995. Synaptic and membrane mechanisms underlying synchronized oscillations in the ferret lateral geniculate nucleus in vitro. J Physiol. 483(Pt 3):641–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram EH, Mangan PS, Zhang D, Scott CA, Williamson JM. 2001. The midline thalamus: alterations and a potential role in limbic epilepsy. Epilepsia. 42:967–978. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Whitehurst RM Jr, Zhang M, Wang L, Li M. 1997. T-type calcium channels facilitate insulin secretion by enhancing general excitability in the insulin-secreting beta-cell line, INS-1. Endocrinology. 138:3735–3740. [DOI] [PubMed] [Google Scholar]
- Blaschke RJ, Monaghan AP, Schiller S, Schechinger B, Rao E, Padilla-Nash H, Ried T, Rappold GA. 1998. SHOT, a SHOX-related homeobox gene, is implicated in craniofacial, brain, heart, and limb development. Proc Natl Acad Sci U S A. 95:2406–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondos SE, Geraldo Mendes G, Jons A. 2020. Context-dependent HOX transcription factor function in health and disease. Prog Mol Biol Transl Sci. 174:225–262. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Smith A, Berger S, Fisher LJ, Gage FH. 1990. Petit mal epilepsy and parkinsonian tremor: hypothesis of a common pacemaker. Neuroscience. 36:1–14. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA. 1999. Neocortical synchronized oscillations induced by thalamic disinhibition in vivo. J Neurosci. 19:RC27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement-Jones M, Schiller S, Rao E, Blaschke RJ, Zuniga A, Zeller R, Robson SC, Binder G, Glass I, Strachan T, et al. 2000. The short stature homeobox gene SHOX is involved in skeletal abnormalities in Turner syndrome. Hum Mol Genet. 9:695–702. [DOI] [PubMed] [Google Scholar]
- Colasante G, Sessa A, Crispi S, Calogero R, Mansouri A, Collombat P, Broccoli V. 2009. Arx acts as a regional key selector gene in the ventral telencephalon mainly through its transcriptional repression activity. Dev Biol. 334:59–71. [DOI] [PubMed] [Google Scholar]
- Coulter DA, Huguenard JR, Prince DA. 1989. Calcium currents in rat thalamocortical relay neurones: kinetic properties of the transient, low-threshold current. J Physiol. 414:587–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Cope DW, Hughes SW. 2006. Thalamic T-type Ca2+ channels and NREM sleep. Cell Calcium. 40:175–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Toth TI, Cope DW, Blethyn K, Hughes SW. 2005. The 'window' T-type calcium current in brain dynamics of different behavioural states. J Physiol. 562:121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baere E, Speleman F, Van Roy N, De Paepe A, Messiaen L. 1998. Assignment of SHOX2 (alias OG12X and SHOT) to human chromosome bands 3q25-->q26.1 by in situ hybridization. Cytogenet Cell Genet. 82:228–229. [DOI] [PubMed] [Google Scholar]
- Deschenes M, Paradis M, Roy JP, Steriade M. 1984. Electrophysiology of neurons of lateral thalamic nuclei in cat: resting properties and burst discharges. J Neurophysiol. 51:1196–1219. [DOI] [PubMed] [Google Scholar]
- Destexhe A. 1998. Spike-and-wave oscillations based on the properties of GABAB receptors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 18:9099–9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Sejnowski TJ, Steriade M. 1994. A model of spindle rhythmicity in the isolated thalamic reticular nucleus. J Neurophysiol. 72:803–818. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Contreras D, Steriade M. 1998. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol. 79:999–1016. [DOI] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC, Leresche N. 2010. Selective T-type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I(T)window. The Journal of neuroscience: the official journal of the Society for Neuroscience. 30:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF. 1985. Glial fibrillary acidic protein (GFAP): the major protein of glial intermediate filaments in differentiated astrocytes. J Neuroimmunol. 8:203–214. [DOI] [PubMed] [Google Scholar]
- Ernst WL, Zhang Y, Yoo JW, Ernst SJ, Noebels JL. 2009. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. The Journal of neuroscience: the official journal of the Society for Neuroscience. 29:1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogerson PM, Huguenard JR. 2016. Tapping the brakes: cellular and synaptic mechanisms that regulate thalamic oscillations. Neuron. 92:687–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulp CT, Cho G, Marsh ED, Nasrallah IM, Labosky PA, Golden JA. 2008. Identification of Arx transcriptional targets in the developing basal forebrain. Hum Mol Genet. 17:3740–3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusel'nikova VV, Korzhevskiy DE. 2015. NeuN as a neuronal nuclear antigen and neuron differentiation marker. Acta Naturae. 7:42–47. [PMC free article] [PubMed] [Google Scholar]
- Hamani C, Hodaie M, Chiang J, Campo M, Andrade DM, Sherman D, Mirski M, Mello LE, Lozano AM. 2008. Deep brain stimulation of the anterior nucleus of the thalamus: effects of electrical stimulation on pilocarpine-induced seizures and status epilepticus. Epilepsy Res. 78:117–123. [DOI] [PubMed] [Google Scholar]
- Herculano-Houzel S, Lent R. 2005. Isotropic fractionator: a simple, rapid method for the quantification of total cell and neuron numbers in the brain. The Journal of neuroscience: the official journal of the Society for Neuroscience. 25:2518–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. 1952. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol. 117:500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Clauss S, Berger IM, Weiss B, Montalbano A, Roth R, Bucher M, Klier I, Wakili R, Seitz H, et al. 2016. Coding and non-coding variants in the SHOX2 gene in patients with early-onset atrial fibrillation. Basic Res Cardiol. 111:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4:44–57. [DOI] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM. 2009. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. The Journal of neuroscience: the official journal of the Society for Neuroscience. 29:10979–10988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes SW, Cope DW, Toth TI, Williams SR, Crunelli V. 1999. All thalamocortical neurones possess a T-type Ca2+ 'window' current that enables the expression of bistability-mediated activities. J Physiol. 517(Pt 3):805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. 2002. Block of T-type ca(2+) channels is an important action of Succinimide Antiabsence drugs. Epilepsy currents/American Epilepsy Society. 2:49–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. 1992. A novel T-type current underlies prolonged ca(2+)-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. The Journal of neuroscience: the official journal of the Society for Neuroscience. 12:3804–3817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Prince DA. 1994. Intrathalamic rhythmicity studied in vitro: nominal T-current modulation causes robust antioscillatory effects. The Journal of neuroscience: the official journal of the Society for Neuroscience. 14:5485–5502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutlet B, Theys N, Coste C, Ahn MT, Doshishti-Agolli K, Lizen B, Gofflot F. 2016. Systematic expression analysis of Hox genes at adulthood reveals novel patterns in the central nervous system. Brain Struct Funct. 221:1223–1243. [DOI] [PubMed] [Google Scholar]
- Inoue M, Duysens J, Vossen JM, Coenen AM. 1993. Thalamic multiple-unit activity underlying spike-wave discharges in anesthetized rats. Brain Res. 612:35–40. [DOI] [PubMed] [Google Scholar]
- Jacobs P, Dalton P, James R, Mosse K, Power M, Robinson D, Skuse D. 1997. Turner syndrome: a cytogenetic and molecular study. Ann Hum Genet. 61:471–483. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinas R. 1984a. Electrophysiological properties of Guinea-pig thalamic neurones: an in vitro study. J Physiol. 349:205–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinas R. 1984b. Ionic basis for the electro-responsiveness and oscillatory properties of Guinea-pig thalamic neurones in vitro. J Physiol. 349:227–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhang KM, Chang TM, Chen M, Liu CS. 2014. Generalized epilepsy in a patient with mosaic Turner syndrome: a case report. J Med Case Reports. 8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph M, Cantu ES, Pai GS, Willi SM, Papenhausen PR, Weiss L. 1996. Xp pseudoautosomal gene haploinsufficiency and linear growth deficiency in three girls with chromosome Xp22;Yq11 translocation. J Med Genet. 33:906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanyshkova T, Meuth P, Bista P, Liu Z, Ehling P, Caputi L, Doengi M, Chetkovich DM, Pape HC, Budde T. 2012. Differential regulation of HCN channel isoform expression in thalamic neurons of epileptic and non-epileptic rat strains. Neurobiol Dis. 45:450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. 2001. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type ca(2+) channels. Neuron. 31:35–45. [DOI] [PubMed] [Google Scholar]
- Knickmeyer RC, Davenport M. 2011. Turner syndrome and sexual differentiation of the brain: implications for understanding male-biased neurodevelopmental disorders. J Neurodev Disord. 3:293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox AT, Glauser T, Tenney J, Lytton WW, Holland K. 2018. Modeling pathogenesis and treatment response in childhood absence epilepsy. Epilepsia. 59:135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostopoulos G, Gloor P, Pellegrini A, Gotman J. 1981a. A study of the transition from spindles to spike and wave discharge in feline generalized penicillin epilepsy: microphysiological features. Exp Neurol. 73:55–77. [DOI] [PubMed] [Google Scholar]
- Kostopoulos G, Gloor P, Pellegrini A, Siatitsas I. 1981b. A study of the transition from spindles to spike and wave discharge in feline generalized penicillin epilepsy: EEG features. Exp Neurol. 73:43–54. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. 1999. Nickel block of three cloned T-type calcium channels: low concentrations selectively block alpha1H. Biophys J. 77:3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang ZS, Wang XH, Xu YJ, Qiao Q, Li XM, Di RM, Guo XJ, Li RG, Zhang M, et al. 2018. A SHOX2 loss-of-function mutation underlying familial atrial fibrillation. Int J Med Sci. 15:1564–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YH, Li JJ, Lu QC, Gong HQ, Liang PJ, Zhang PM. 2014. Involvement of thalamus in initiation of epileptic seizures induced by pilocarpine in mice. Neural Plast. 2014:675128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chen CH, Espinoza-Lewis RA, Jiao Z, Sheu I, Hu X, Lin M, Zhang Y, Chen Y. 2011. Functional redundancy between human SHOX and mouse Shox2 genes in the regulation of sinoatrial node formation and pacemaking function. J Biol Chem. 286:17029–17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Chen CH, Ye W, Espinoza-Lewis RA, Hu X, Zhang Y, Chen Y. 2014. Phosphorylation of Shox2 is required for its function to control sinoatrial node formation. J Am Heart Assoc. 3:e000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Jahnsen H. 1982. Electrophysiology of mammalian thalamic neurones in vitro. Nature. 297:406–408. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, et al. 2003. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 22:216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magara S, Kawashima H, Kobayashi Y, Akasaka N, Yamazaki S, Tohyama J. 2015. Rub epilepsy in an infant with Turner syndrome. Brain Dev. 37:725–728. [DOI] [PubMed] [Google Scholar]
- Mauger C, Lancelot C, Roy A, Coutant R, Cantisano N, Le Gall D. 2018. Executive functions in children and adolescents with Turner syndrome: a systematic review and meta-analysis. Neuropsychol Rev. 28:188–215. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Bal T. 1997. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 20:185–215. [DOI] [PubMed] [Google Scholar]
- McLachlan RS, Gloor P, Avoli M. 1984. Differential participation of some 'specific' and 'non-specific' thalamic nuclei in generalized spike and wave discharges of feline generalized penicillin epilepsy. Brain Res. 307:277–287. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A. 1999. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol Chem. 380:975–980. [DOI] [PubMed] [Google Scholar]
- Nielsen J, Wohlert M. 1990. Sex chromosome abnormalities found among 34,910 newborn children: results from a 13-year incidence study in Arhus. Denmark Birth Defects Orig Artic Ser. 26:209–223. [PubMed] [Google Scholar]
- Oliveira CS, Alves C. 2011. The role of the SHOX gene in the pathophysiology of Turner syndrome. Endocrinologia y nutricion: organo de la Sociedad Espanola de Endocrinologia y Nutricion. 58:433–442. [DOI] [PubMed] [Google Scholar]
- Olivetti PR, Maheshwari A, Noebels JL. 2014. Neonatal estradiol stimulation prevents epilepsy in Arx model of X-linked infantile spasms syndrome. Sci Transl Med. 6:220ra212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. 2017. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 14:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Slezia A, Acsady L. 2006. Corticothalamic 5-9 Hz oscillations are more pro-epileptogenic than sleep spindles in rats. J Physiol. 574:209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Vergnes M, Marescaux C. 2001. Medium-voltage 5-9-Hz oscillations give rise to spike-and-wave discharges in a genetic model of absence epilepsy: in vivo dual extracellular recording of thalamic relay and reticular neurons. Neuroscience. 105:181–201. [DOI] [PubMed] [Google Scholar]
- Pollen DA, Reid KH, Perot P. 1964. Micro-electrode studies of experimental 3/sec wave and spike in the cat. Electroencephalogr Clin Neurophysiol. 17:57–67. [DOI] [PubMed] [Google Scholar]
- Prevett MC, Duncan JS, Jones T, Fish DR, Brooks DJ. 1995. Demonstration of thalamic activation during typical absence seizures using H2(15) O and PET. Neurology. 45:1396–1402. [DOI] [PubMed] [Google Scholar]
- Puusepp H, Zordania R, Paal M, Bartsch O, Ounap K. 2008. Girl with partial Turner syndrome and absence epilepsy. Pediatr Neurol. 38:289–292. [DOI] [PubMed] [Google Scholar]
- Racine RJ, Burnham WM, Gartner JG, Levitan D. 1973. Rates of motor seizure development in rats subjected to electrical brain stimulation: strain and inter-stimulation interval effects. Electroencephalogr Clin Neurophysiol. 35:553–556. [DOI] [PubMed] [Google Scholar]
- Rosin JM, McAllister BB, Dyck RH, Percival CJ, Kurrasch DM, Cobb J. 2015. Mice lacking the transcription factor SHOX2 display impaired cerebellar development and deficits in motor coordination. Dev Biol. 399:54–67. [DOI] [PubMed] [Google Scholar]
- Saad K, Abdelrahman AA, Abdel-Raheem YF, Othman ER, Badry R, Othman HA, Sobhy KM. 2014. Turner syndrome: review of clinical, neuropsychiatric, and EEG status: an experience of tertiary center. Acta Neurol Belg. 114:1–9. [DOI] [PubMed] [Google Scholar]
- Sharop BR, Boldyriev OI, Batiuk MY, Shtefan NL, Shuba YM. 2016. Compensatory reduction of Cav3.1 expression in thalamocortical neurons of juvenile rats of WAG/Rij model of absence epilepsy. Epilepsy Res. 119:10–12. [DOI] [PubMed] [Google Scholar]
- Song I, Kim D, Choi S, Sun M, Kim Y, Shin HS. 2004. Role of the alpha1G T-type calcium channel in spontaneous absence seizures in mutant mice. The Journal of neuroscience: the official journal of the Society for Neuroscience. 24:5249–5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 21:70–71. [DOI] [PubMed] [Google Scholar]
- Steriade M. 1993. Cholinergic blockage of network- and intrinsically generated slow oscillations promotes waking and REM sleep activity patterns in thalamic and cortical neurons. Prog Brain Res. 98:345–355. [DOI] [PubMed] [Google Scholar]
- Steriade M. 2000. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 101:243–276. [DOI] [PubMed] [Google Scholar]
- Steriade M, Deschenes M, Domich L, Mulle C. 1985. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol. 54:1473–1497. [DOI] [PubMed] [Google Scholar]
- Steriade M, Domich L, Oakson G, Deschenes M. 1987. The deafferented reticular thalamic nucleus generates spindle rhythmicity. J Neurophysiol. 57:260–273. [DOI] [PubMed] [Google Scholar]
- Sun C, Yu D, Ye W, Liu C, Gu S, Sinsheimer NR, Song Z, Li X, Chen C, Song Y, et al. 2015. The short stature homeobox 2 (Shox2)-bone morphogenetic protein (BMP) pathway regulates dorsal mesenchymal protrusion development and its temporary function as a pacemaker during cardiogenesis. J Biol Chem. 290:2007–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Zhang T, Liu C, Gu S, Chen Y. 2013. Generation of Shox2-Cre allele for tissue specific manipulation of genes in the developing heart, palate, and limb. Genesis. 51:515–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. 1999. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. The Journal of neuroscience: the official journal of the Society for Neuroscience. 19:1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente V, Teixeira SA, Neder L, Okamoto OK, Oba-Shinjo SM, Marie SK, Scrideli CA, Paco-Larson ML, Carlotti CG Jr. 2009. Selection of suitable housekeeping genes for expression analysis in glioblastoma using quantitative RT-PCR. BMC molecular biology. 10:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Chen S, Nolan MF, Siegelbaum SA. 2002. Activity-dependent regulation of HCN pacemaker channels by cyclic AMP: signaling through dynamic allosteric coupling. Neuron. 36:451–461. [DOI] [PubMed] [Google Scholar]
- Williams D. 1953. A study of thalamic and cortical rhythms in petit mal. Brain: a journal of neurology. 76:50–69. [DOI] [PubMed] [Google Scholar]
- Williams SR, Toth TI, Turner JP, Hughes SW, Crunelli V. 1997. The 'window' component of the low threshold Ca2+ current produces input signal amplification and bistability in cat and rat thalamocortical neurones. J Physiol. 505(Pt 3):689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Wang KK. 2015. Glial fibrillary acidic protein: from intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 38:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye W, Wang J, Song Y, Yu D, Sun C, Liu C, Chen F, Zhang Y, Wang F, Harvey RP, et al. 2015. A common Shox2-Nkx2-5 antagonistic mechanism primes the pacemaker cell fate in the pulmonary vein myocardium and sinoatrial node. Development. 142:2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Bosch MA, Rick EA, Kelly MJ, Ronnekleiv OK. 2009. 17Beta-estradiol regulation of T-type calcium channels in gonadotropin-releasing hormone neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 29:10552–10562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Vilaythong AP, Yoshor D, Noebels JL. 2004. Elevated thalamic low-voltage-activated currents precede the onset of absence epilepsy in the SNAP25-deficient mouse mutant coloboma. The Journal of neuroscience: the official journal of the Society for Neuroscience. 24:5239–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Lian YJ. 2015. Epilepsy associated with Turner syndrome. Neurol India. 63:631–633. [DOI] [PubMed] [Google Scholar]
- Zobeiri M, Chaudhary R, Blaich A, Rottmann M, Herrmann S, Meuth P, Bista P, Kanyshkova T, Luttjohann A, Narayanan V, et al. 2019. The hyperpolarization-activated HCN4 channel is important for proper maintenance of oscillatory activity in the Thalamocortical system. Cereb Cortex. 29:2291–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobeiri M, Chaudhary R, Datunashvili M, Heuermann RJ, Luttjohann A, Narayanan V, Balfanz S, Meuth P, Chetkovich DM, Pape HC, et al. 2018. Modulation of thalamocortical oscillations by TRIP8b, an auxiliary subunit for HCN channels. Brain Struct Funct. 223:1537–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.