

Abstract
Background
Terrestrial hot spring settings span a broad spectrum of physicochemistries. Physicochemical parameters, such as pH and temperature, are key factors influencing differences in microbial composition across diverse geothermal areas. Nonetheless, analysis of hot spring pools from the Taupo Volcanic Zone (TVZ), New Zealand, revealed that some members of the bacterial genus, Acidithiobacillus, are prevalent across wide ranges of hot spring pHs and temperatures. To determine the genomic attributes of Acidithiobacillus that inhabit such diverse conditions, we assembled the genomes of 19 uncultivated hot spring Acidithiobacillus strains from six geothermal areas and compared these to 37 publicly available Acidithiobacillus genomes from various habitats.
Results
Analysis of 16S rRNA gene amplicons from 138 samples revealed that Acidithiobacillus comprised on average 11.4 ± 16.8% of hot spring prokaryotic communities, with three Acidithiobacillus amplicon sequence variants (ASVs) (TVZ_G1, TVZ_G2, TVZ_G3) accounting for > 90% of Acidithiobacillus in terms of relative abundance, and occurring in 126 out of 138 samples across wide ranges of temperature (17.5–92.9 °C) and pH (1.0–7.5). We recovered 19 environmental genomes belonging to each of these three ASVs, as well as a fourth related group (TVZ_G4). Based on genome average nucleotide identities, the four groups (TVZ_G1-TVZ_G4) constitute distinct species (ANI < 96.5%) of which three are novel Acidithiobacillus species (TVZ_G2-TVZ_G4) and one belongs to Acidithiobacillus caldus (TVZ_G1). All four TVZ Acidithiobacillus groups were found in hot springs with temperatures above the previously known limit for the genus (up to 40 °C higher), likely due to significantly higher proline and GC contents than other Acidithiobacillus species, which are known to increase thermostability. Results also indicate hot spring-associated Acidithiobacillus have undergone genome streamlining, likely due to thermal adaptation. Moreover, our data suggest that Acidithiobacillus prevalence across varied hot spring pHs is supported by distinct strategies, whereby TVZ_G2-TVZ_G4 regulate pH homeostasis mostly through Na+/H+ antiporters and proton-efflux ATPases, whereas TVZ_G1 mainly relies on amino acid decarboxylases.
Conclusions
This study provides insights into the distribution of Acidithiobacillus species across diverse hot spring physichochemistries and determines genomic features and adaptations that potentially enable Acidithiobacillus species to colonize a broad range of temperatures and pHs in geothermal environments.
Video Abstract
Supplementary Information
The online version contains supplementary material available at 10.1186/s40168-021-01090-1.
Keywords: Acidithiobacillus, Genome streamlining, Adaptation, Hot spring, Temperature, pH
Background
Acidithiobacillus (formerly Thiobacillus) is a gram-negative genus of rod-shaped bacteria that mostly comprises chemolithoautotrophic, obligately acidophilic (optimum pH < 4), and mesophilic or mesothermophilic species [1–3]. They are commonly distributed in acidic and sulfur-rich environments, such as acidic soil and acid mine drainage [4, 5], and recent studies show that they are widely distributed in hot springs [6, 7]. Outside of hot springs, known members of Acidithiobacillus have small differences in physiological tolerances to environmental conditions, and all grow within a pH range of 0.5–6.0, and a temperature range of 5–52 °C [3, 8–14]. Of these, A. caldus, one of the most studied species in the genus Acidithiobacillus, is the only thermoacidophile (first recovered from coal spoils) and has the highest known temperature limit (52 °C) [3, 12, 13, 15, 16]. Other members of the genus, such as A. ferrooxidans and A. thiooxidans, as well as A. caldus, are used in biohydrometallurgy to recover certain metals from sulfide ores due to their ability to oxidize sulfide and solubilize metals, and their preference for acidic environments [10, 11, 17–19]. Although members of Acidithiobacillus are best known from highly acidic settings, such as ores during bioleaching and acid mine drainage [4, 20], a search of data from almost one thousand Taupo Volcanic Zone (TVZ) hot springs, in New Zealand, showed that Acidithiobacillus are present in springs encompassing a huge range of physicochemistries, from very acidic to alkali pHs (0.6–8.94) and across an extremely wide temperature range (13.9–97.6 °C) [6, 21].
Terrestrial hot spring environments are highly heterogeneous, representing a huge spectrum of physicochemical conditions [6, 22]. Previous studies have suggested that pH and temperature are key drivers influencing microbial composition of hot springs [6, 7]. However, it has been shown that Acidithiobacillus are present in numerous hot springs that span a wide range of pHs and temperatures in the TVZ [6, 7, 23]. This suggests that this bacterial genus possesses mechanisms enabling it to inhabit diverse environmental conditions. However, it remains to be determined whether this cosmopolitanism is due to the widespread occurrence of particular Acidithiobacillus species or strains across different hot spring environments, reflecting strain or species level metabolic versatility, or whether different hot spring niches harbor phylogenetically distinct Acidithiobacillus.
To determine the phylogenetic variation and distribution of Acidithiobacillus (clades or genome-inferred species) across broad ranges of hot spring temperatures and pHs, and to elucidate the genomic features underpinning their prevalence, we collected 79 subaqueous sediment samples from various geothermal sites in the TVZ and analyzed these alongside a further 59 subaerial siliceous hot spring deposits (sinters) we previously found to be rich in the Acidithiobacillus genus [7]. Sediment-sampled hot spring pHs ranged from 2.0–7.5 and temperatures ranged from 17.5–92.9 °C, while sinter-sampled spring pHs ranged from 1.0–6.6 and temperatures ranged from 24.2–92.9 °C (Table S1). We determined the distribution of Acidithiobacillus across these samples and recovered genomes from representative samples with widespread Acidithiobacillus 16S rRNA gene amplicon sequence variants (ASVs). Using these genomes, we determined mechanisms of temperature and pH tolerance in the TVZ Acidithiobacillus groups and compared these and other genomic attributes to Acidithiobacillus from other environments. Results show that the TVZ Acidithiobacillus have distinct genomic features that potentially allow them to inhabit broad ranges of temperature and pH.
Materials and methods
Sample collection and physicochemical measurements
Fifty-nine sinter samples (with spatial replicates included) were collected from 10 hot springs across five geothermal areas (Orange Spring, Te Kopia, Parariki Stream, Rotokawa, and Tikitere) within the TVZ (Fig. 1 from [7]) in April 2018, as described in Sriaporn et al. [7]. Seventy-nine additional sediment samples (with spatial replicates included) were collected from 12 hot springs across four geothermal areas (Parariki Stream, Rotokawa, Tikitere, and Wai-O-Tapu) within the TVZ in February and November 2019. Sediment was collected from 0 to approximately a few centimeters below the water-sediment interface using sterile spatulas, and samples were placed into sterile 50 mL centrifuge tubes and transported on dry ice, before storage at − 80 °C. Hot spring pH and temperature measurements from the April 2018 sinter sampling trip were measured using portable sension™ 156 Multiparameter Meter (Hach Company, USA) and COMARK Evolution IV9001 temperature probes (COMARK, UK) [7], and pH and temperatures from the sediment sampling trips (February and November 2019) were measured using a WTW 330i handheld meter (WTW GmbH, Germany).
Fig. 1.

Plots showing the proportion of Acidithiobacillus ASVs observed in microbial communities across the TVZ hot spring sites. a Total relative abundance (%) of prokaryotic communities summed across all samples collected across six geothermal areas in the TVZ. b Boxplots showing total relative abundance of Acidithiobacillus versus other prokaryotes in each sample. Note that there are 12 samples that did not harbor Acidithiobacillus (shown at 0% in the left box plot, and 100% in the right box plot)
DNA extraction, amplicon, and metagenomic sequencing
DNA extraction was performed using the DNeasy PowerSoil® Kit (Qiagen, USA) following the manufacturer’s instructions, using 0.25–0.35 g of sinter or sediment per sample (with a no-sample negative control included). DNA quality and quantity were checked via gel electrophoresis, a NanoPhotometer (Implen, Germany), and Qubit 3.0 fluorometric quantitation (Invitrogen, USA). 16S rRNA gene amplification was performed in triplicate using 515′F/926′R primers, and 2 × 250 bp paired-end Illumina MiSeq sequencing was performed with the MiSeq Reagent Kit V2, as previously described [7], at Auckland Genomics (University of Auckland). To explore the genomic features of the TVZ Acidithiobacillus, 4 representative sinter and 18 representative sediment samples were selected for whole genome sequencing (WGS). Five to seven g of DNA was extracted per sample using the DNeasy PowerMax Soil Kit® (Qiagen, USA). Due to low DNA concentrations, multiple extractions were performed for each sample, and DNA was concentrated and cleaned by ethanol precipitation using 2× absolute ethanol, 3 M sodium acetate, and 20 μl/μg glycogen. Samples were then incubated at − 80 °C overnight before DNA pellets were washed with 70% ethanol and resuspended in 25 μl of nuclease-free water. Final DNA quality and quantity were checked using an Implen NanoPhotometer, Qubit 3.0 fluorometric quantitation with the Qubit™ dsDNA HS Assay Kit, and gel electrophoresis. WGS sequencing was performed at the Otago Genomics Facility (University of Otago, Dunedin, New Zealand). Thruplex DNA-Seq libraries (Takara Bio USA, Inc, USA) were prepared with 500–600 bp insert sizes and sequenced using the Illumina HiSeq 2500 platform with the HiSeq Rapid SBS Kit V2, yielding 2 × 250 bp reads, for the 4 sinter samples, and the HiSeq 2500 SBS Kit V4, yielding 2 × 125 bp reads, for the 18 sediment samples.
Amplicon data analysis
QIIME2 (version 2019.10) was used to process demultiplexed amplicon sequences by read-joining, quality filtering (Q score cutoff of 25) and denoising (with singletons removed), and to generate an ASV table [24–27]. Taxonomy was assigned by using the SILVA database version 132 [28–30]. R (version 3.4.4) with RStudio software (version 3.4.1), and R package ggplot2 (version 3.0.0) were used to visualize data [31–33].
Metagenomic sequence processing and genome assembly
Quality filtering and trimming of raw metagenomic sequences were undertaken using Sickle (version 1.33) with a minimum quality score threshold of 30 and a minimum retained read length of 80 bp, and FastQC version 0.11.9 was used to check quality [34, 35]. After trimming 86.5–92.2% of reads per sample were retained. Paired-end trimmed reads from each sample were then assembled separately using SPAdes version 3.11.1 with –meta -m 900 -t 16 -k 41,61,81,101,127 parameters [36]. To determine differential coverage for genome binning, reads were mapped to contigs using Bowtie version 1.2.0 with the following parameters: --phred33-quals -n 1 -l 111 --minins 100 --maxins 600 --best [37]. Contigs were binned using MetaBAT version 2.12.1 [38], MaxBin version 2.2.4 [39], and CONCOCT version 0.4.1 [40], utilizing differential coverage and tetranucleotide frequencies. The resulting bins (metagenome-assembled genomes, MAGs) were compared, and representatives were selected using DAS Tool version 1.1.1 [41]. Bin refinement was performed using VizBin with differential coverage incorporated [42]. CheckM version 1.0.12 [43] was used to estimate refined bin completeness and contamination. Estimated genome size was calculated following Castelle et al. [44]. Bins shared across the 4 sinter and 18 sediment sample assemblies were then dereplicated using dRep version 1.4.3 based on the default 99% similarity threshold to generate a set of unique representative bins [45].
Gene prediction, annotation, and genome analyses
Gene prediction was performed using Prodigal version 2.6.3 with -p meta mode [46]. Predicted coding DNA sequences (CDS) were annotated against the UniRef100 [47], UniProt [48], KEGG [49], Pfam [50], and TIGRFAM [51] databases using USEARCH version 9.0.2132 [52] with an e-value cutoff of 0.001. Prokka version 1.13.4 was used to predict tRNAs, tmRNAs, CRISPRs, and non-coding RNA [53]. Taxonomic classification was assigned based on core marker genes using GTDB-Tk version 0.2.1 [54]. The genome sequences of nine Acidithiobacillus type strains (A. caldus ATCC 51756, A. caldus SM-1, A. thiooxidans ATCC 19377, A. ferrooxidans ATCC 23270, A. ferrivorans SS3, A. albertensis DSM 14366, A. ferridurans JCM 18981, A. ferrianus MG, and A. sulfuriphilus CJ-2) and one unclassified species (Acidithiobacillus sp. UBA2486), obtained from the NCBI GenBank database, were included in these gene prediction and annotation steps for comparison with the studied TVZ Acidithiobacillus genomes.
To determine whether genomes belonged to the same species, the TVZ Acidithiobacillus bins and reference genomes were compared by calculating pairwise average nucleotide identity (ANI) values via the average nucleotide identity calculator [55] based on the approach described by Goris et al. [56] with 96.5% threshold for determining the same species [57, 58]. The Genome-to-Genome Distance Calculator (GGDC) was used to approximate in silico DNA-DNA hybridization (DDH) values to determine the likelihood that any two genomes belong to the same species using a 70% threshold [59, 60]. To examine mutation events discriminating subspecies genome clusters, GSAlign was used to detect substitutions, insertions, and deletions between genomes [61]. In addition, BLAST was used to determine the identity of 16S rRNA genes among the TVZ and other Acidithiobacillus species using the > 99% species threshold [62].
To further compare Acidithiobacillus attributes, we predicted genome minimum generation times and optimal growth temperatures using GrowthPred [63]. Genome replication rates at the time of sampling were estimated using the index of replication (iRep) [64]. Protein paralogs were determined using CD-HIT [65]. Proteins that were at least 30% identical over at least 50% of the longest sequence length were considered paralogs [66]. Genome GC contents were derived from CheckM output, and proline content was calculated from predicted protein sequences using an in-house python script [67].
To explore the genomic features of Acidithiobacillus from a wider set of environments, an additional 27 Acidithiobacillus genomes from Genome Taxonomy Database (GTDB) [68] were included for comparative analyses of genome size, optimal growth temperature, paralogs, GC contents, and proline composition (Table S2). Correlation plots between genomic features were generated using RStudio (package ggplot2), and Pearson’s correlation coefficients and t-distribution tables (df = n − 1) were used to determine the correlation coefficients and significance of correlations, respectively (via package ggpubr). A T-test was used to determine if any significant differences were present between two groups of genomes.
Extraction and reconstruction of partial and near full-length 16S rRNA gene sequences
16S rRNA gene sequences were reconstructed from each quality-filtered WGS sample using EMIRGE version 0.61.1 with a 97% clustering threshold [69]. Taxonomy was assigned using USEARCH version 11.0.667 (SINTAX algorithm) by searching sequences against the SILVA SSU non-redundant database version 132 [29, 52]. In addition, MeTaxa2 [70] was used to extract 16S rRNA genes from MAGs. In order to match independently reconstructed 16S rRNA sequences (EMIRGE), MAG-extracted 16S rRNA genes (MeTaxa2), and ASVs, relative abundance correlations were undertaken using RStudio (with package ggplot2). Pearson’s correlation coefficients and t-distribution tables (df = n − 1) were used to determine the correlation coefficient and the statistical significance, respectively (via package ggpubr) [71]. Sequences derived from the three approaches were aligned with Geneious version 11.1.2 using MUSCLE [72, 73].
Acidithiobacillus phylogenetic tree
A concatenated core gene phylogenetic tree was constructed using the TVZ Acidithiobacillus MAGs and the 10 Acidithiobacillus reference genomes obtained from the GenBank and GTDB databases [68]. Sequences were aligned using MUSCLE with Geneious [72, 73], and PhyML 3.0 was used to build and visualize a maximum-likelihood phylogenetic tree with 100 times bootstrapping [74].
Results and discussion
Hot spring characteristics
Samples were collected from sulfur-rich hot springs, which are common in the TVZ, and which spanned a broad range of water temperatures (17.5–92.9 °C) to recover mesophilic to hyperthermophilic communities, and pHs (1.0–7.5) to sample acidophilic to neutrophilic communities (Table 1, Fig. S1). Samples comprised subaerial digitate sinter and subaqueous sediments. The digitate sinters are stromatolitic siliceous deposits with protrusive features that form around hot spring margins or in shallow outflow channels just above water level (reaching approximately 1 cm above the air-water interface) [7], due to the deposition of silica from evaporative wicking of spring water [75]. In contrast, sediments (i.e., hot spring mud and unconsolidated coarser geothermal-influenced stream material) form within hot springs when underground gases react with rocks to produce clay, or are detrital (i.e., broken up pieces of sinter, surrounding rocks, and organic matter) [22], and for this study were located a few millimeters to centimeters below the water’s surface. Digitate sinter and sediment from the same pool were connected by the thermal fluids. Subaerial sinter samples were taken from five geothermal areas, which are ~ 1 to 65 km apart, with a proximal water temperature range of 24.2–92.9 °C and pHs of 1.0–6.6 (Table 1, Fig. S1). Subaqueous sediment samples were collected from four geothermal areas (~ 1 to 65 km apart) with a direct water temperature range of 17.5–92.9 °C and pH range of 2.0–7.5. It is worth noting that as the temperature and pH measurements in this study were based on hot spring fluids, they may not represent actual values for the subaerial sinter samples, but apply directly to sediment samples, which are submerged within the spring water (Table S1).
Table 1.
Site, pH and temperature ranges, and water chemistry of studied geothermal settings, including collected sample type and studied data type
| Site | pH range | Temperature range (°C) | Water chemistry | Sample type | Data type |
|---|---|---|---|---|---|
| Tikitere | 3.60–7.50 | 17.5–73.8 | Acid-sulfate-bicarbonate | Sinter, sediment | WGS, 16S |
| Parariki Stream | 1.00–2.14 | 33.4–77.0 | Acid-sulfate-chloride | Sinter, sediment | WGS, 16S |
| Rotokawa | 1.73–2.94 | 22.7–80.1 | Acid-sulfate-chloride | Sinter, sediment | WGS, 16S |
| Te Kopia | 2.14–2.22 | 43.6–92.9 | Acid-sulfate | Sinter | WGS, 16S |
| Orange Spring | 1.80–2.65 | 35.4–91.5 | Acid-sulfate | Sinter | 16S |
| Wai-O-Tapu | 2.51–5.70 | 32.4–92.9 | Acid-sulfate-chloride | Sediment | WGS, 16S |
WGS = Whole genome sequencing, 16S = 16S rRNA gene amplicon sequencing
Distribution of TVZ Acidithiobacillus across wide ranges of temperature and pH
16S rRNA gene amplicon sequencing of 138 subaerial sinter and subaqueous sediment samples produced a total of 28,204 ASV features (with 1,344,820 ASV counts). These were classified into 49 phyla, and the overall community distribution is shown in Figure S2. Of these, 1195 ASV features (95,787 ASV counts) were from genus Acidithiobacillus (order Acidithiobacillales), which comprised 7.1% in total (or on average 11.4 ± 16.8%), of prokaryotic communities across all of the hot springs sampled for this study (Fig. 1a, b). It was also the third most abundant taxon, following the archaeal orders Thermoplasmatales and Sulfolobales, and hence was the most abundant bacterial genus (Fig. 1). This corresponds well with our previous work and that of other studies, confirming the high relative abundance of Acidithiobacillus in sulfuric hot spring environments (sediment, sinter, soil, and water) in both New Zealand [6, 7, 23, 76] and elsewhere [77, 78]. One explanation for Acidithiobacillus predominance in these settings is the various genes for sulfur metabolism they characteristically possess, which enable them to oxidize sulfur, thiosulfate, sulfide, and sulfite in geothermal environments [13, 79].
Overall, we recovered more than 1000 Acidithiobacillus ASVs from 126 out of 138 (91.3% of) samples, spanning a temperature range of 17.5–92.9 °C for sediment samples and 24.2–92.9 °C for sinter samples, and a pH range of 2.0–7.5 for sediment samples and 1.0–6.6 for sinter samples (Fig. S1, Table S1, S3). Of these, just three ASVs constituted more than 90% of the total Acidithiobacillus population and were detected across 126 of the samples, suggestive of both their abundance and prevalence across a wide range of temperatures and pHs in geothermal environments (Fig. 2a–c, Fig. S3, Table S3). These are amongst the widest physicochemical ranges recorded for Acidithiobacillus (Fig. 3a, b) and are similar to findings from the TVZ 1000springs project [21], in which Acidithiobacillus was found in springs with pHs 0.6–8.94 and temperatures of 13.9–97.6 °C. However, this earlier project did not classify to species level. The only currently known thermoacidophilic Acidithiobacillus species, A. caldus (type strain ATCC 51756), has a known upper temperature limit of 52 °C (as characterized by growth experiments), which is the highest for this genus [3]. Nevertheless, more than 50% of our samples that harbored Acidithiobacillus were from sites with temperatures higher than 52 °C (Fig. S1, Table S1). These results indicate that the TVZ Acidithiobacillus may survive in conditions that exceed the formerly known upper pH limit of 6.0, which was recorded for A. albertensis (originally isolated from acidic soil) [8].
Fig. 2.

Scatter plots showing relative abundances and prevalence of the four TVZ hot spring Acidithiobacillus ASVs that correspond with recovered genomes of a A. caldus TVZ_G1; b Acidithiobacillus sp. TVZ_G2; c Acidithiobacillus sp. TVZ_G3; and d Acidithiobacillus TVZ_G4. Bubble size represents the relative abundance of each ASV per sample. The number of bubbles represents the prevalence of each Acidithiobacillus ASV
Fig. 3.

Plots showing the physicochemical ranges of TVZ Acidithiobacillus and those known for other Acidithiobacillus. Ranges are given for a temperature (with black bars indicating optimal growth temperatures calculated by GrowthPred) and b pH. Ranges shown for subaerial sinters are from the corresponding hot spring waters from which the digitate sinters form. All temperature and pH ranges shown for the TVZ species represent the springs from where they were found, while those for other Acidithiobacillus are from growth experiment characterizations
Characterization and growth experiments of TVZ Acidithiobacillus are needed to confirm the expansion of the upper physicochemical limits of Acidithiobacillus to include high temperature and circumneutral pH environments. Nevertheless, while the possibility of exogenous transport cannot be excluded, this seems unlikely given the high relative abundance of Acidithiobacillus in the studied springs, implying that these organisms are not transient. In addition, while all sampling sites were in the TVZ, each site was isolated and distant (1–65 km apart). Moreover, alpha diversity trends are consistent with expectations based on hot spring physicochemistry [7]. Shannon indices show that Tikitere had the most diverse communities overall, including the most diverse Acidithiobacillus population, likely due to the near-neutral pHs and relatively moderate temperatures (Fig. S4, Table S1). In contrast, Parariki Stream, Rotokawa, Te Kopia, and Wai-O-Tapu shared the lowest overall community diversity. Locations sampled at each of these sites were exclusively or predominately low in pH. Accordingly, results showed that, for Acidithiobacillus populations, the lowest diversity was sampled at Te Kopia. This is likely due to the very extreme temperatures and pHs (up to 93 °C and with pHs of 2.1–2.2), which are amongst the highest and lowest, respectively, in this study, and are potentially at or near Acidithiobacillus limits in these systems.
Phylogeny of TVZ Acidithiobacillus and the prevalence and predominance of distinct populations
To determine the phylogeny and genomic features that potentially explain the broad temperature and pH tolerances of the three abundant and prevalent Acidithiobacillus ASVs, we reconstructed 19 Acidithiobacillus MAGs from 22 representative samples (4 sinters and 18 sediments). All 19 MAGs were estimated to be largely or near-complete (78.2–99.4%; Table S2) with low contamination (0–2.59%; Table S2). Genomes were clustered into four groups based on an average nucleic acid similarity threshold of 99%. A partial 16S rRNA sequence was extracted from each representative MAG within each of the four groups. Additionally, EMIRGE recovered 232 near full-length 16S rRNA gene sequences from the 22 WGS samples, and three of these sequences were identical to three out of four Acidithiobacillus MAG-derived and three abundant and prevalent Acidithiobacillus ASVs based on sequence alignments (Fig. S5). The extra fourth MAG-derived 16S rRNA sequence was identical to a rarer Acidithiobacillus ASV (Table S3), which accounted for 0.4 ± 5.9% average (or 0.025% total) relative abundance. We designate these four Acidithiobacillus groups: TVZ_G1, TVZ_G2, TVZ_G3, and TVZ_G4 (Fig. 2a–d, Fig. S3, Table S3). Results also indicate a broadly positive correlation between the relative abundances of 16S rRNA sequences (amplicon and EMIRGE) and the genome coverage of the four representative MAGs (Fig. S6).
EMIRGE-derived 16S rRNA gene sequences of TVZ Acidithiobacillus shared < 99% identity with other reference Acidithiobacillus 16S rRNA gene sequences (Table S4), indicating their novelty [62]. Pairwise in silico DDH and ANI comparisons also showed that representative MAGs within the four Acidithiobacillus groups shared values below the in silico DDH (70%) and species delineation (96.5%) cut-offs [57–60] (Tables S5-6), suggesting each are a distinct species. In comparison, MAGs within each group represent conspecific populations, on the basis of DDH and ANI values of 91.1–100% and 98.8–100%, respectively—above the species threshold. Results indicate that three out of four of the groups (TVZ_G2-TVZ_G4) are novel species, while TVZ_G1 belongs to A. caldus. TVZ_G1 shares an in silico DDH value of 96% and ANI of 99.6% (based on the representative genome) with the A. caldus type strain ATCC51756 (Table S5-6). Accordingly, a concatenated core gene phylogenetic tree shows the four TVZ groups formed three distinct clades within the genus Acidithiobacillus (Fig. 4), with closely related TVZ_G3 and TVZ_G4 populations representing sub-clades of Clade III, and the six MAGS comprising TVZ_G1 representing a discrete clade within A. caldus (Clade I). In addition, while TVZ_G2 is unrelated to any formerly designated and cultivated species, its five MAGs clustered with one uncultivated bacterium from a Taiwanese hot spring, Acidithiobacillus sp. UBA2486 (Clade II, Fig. 4), which ANI and in silico DDH values indicate are the same species (Table S5-6).
Fig. 4.

A maximum-likelihood concatenated core gene phylogenetic tree with 100 times bootstrapping of TVZ and other Acidithiobacillus species. Bold script indicates the representative MAG of each species (TVZ_G2-4) or subspecies (TVZ_G1) level cluster. Thermithiobacillus tepidarius from another family and Acidiferrobacter thiooxydans from another class are used as outgroups
Of the three most abundant Acidithiobacillus populations, A. caldus TVZ_G1 was the least abundant (10.6 ± 2.7% ASV relative abundance) (Fig. 2a–c, Fig. S3, Table S3). This is contrary to previous studies, where it has been suggested that the dominant Acidithiobacillus species in geothermal settings is A. caldus [23, 78], although such classifications were based on relatively low percent identity matches (i.e., 97% partial 16S gene sequence identity). Instead, we found that the novel TVZ_G3 population was the most abundant overall, representing on average 58.8 ± 1.3% of the Acidithiobacillus population (or 41.5 ± 22.4% based on genome coverage) (Fig. 2a–d, Fig. S3, S7-8, Table S3), while its close relative, TVZ_G4, comprised only 0.4 ± 5.9% of Acidithiobacillus ASVs (or 5.3 ± 2.2% genome abundance). Of these, TVZ_G3 was also the most prevalent group, being distributed across 86.2% of samples based on the ASV (or 45.5% of the WGS subset of samples). This was followed by TVZ_G2 (52.2% ASV prevalence or 45.5% of the WGS subset) and TVZ_G1 (40.6% ASV prevalence or 36.4% of the WGS subset) (Fig. 2a–d, Fig. S3, S7-8, Table S3). In addition to its low abundance, TVZ_G4 was also the least prevalent among the four as shown by amplicon data (2.2%) and genome coverage (31.8%) (Fig. 2d, Fig. S3, S8).
Our study shows that three of the TVZ Acidithiobacillus species were present across diverse and spatially distinct hot spring environments and were therefore unconstrained by geographical isolation. However, only two genomes within TVZ_G2 were found to share 100% ANI based on their alignable fractions with the group reference MAG (Table S5). To further explore genetic diversity within the TVZ Acidithiobacillus species, we determined substitutions (single-nucleotide variants, SNVs) and insertions and deletions (indels). Results indicated a predominance of substitutions, and relatively few indels, when comparing MAG consensus sequences with the representative per group (Fig. S9). MAGs belonging to A. caldus TVZ_G1 and Acidithiobacillus TVZ_G2 exhibited the least intra-group genomic variation, with 0.005–0.15% SNVs and 0–0.002% indels (74 to 1746 substitutions and 0 to 26 indels per Mbp). The highest amount of genetic variation was detected in TVZ_G3 and TVZ_G4, with up to and 0.003% and 0.004% indels (44 and 45 events per Mbp), respectively, and up to 0.5% and 0.9% SNVs (8087 and 10,722 of substitutions per Mbp), respectively.
These intraspecies genetic diversities are small, compared to other studies where 2.6–9.7% SNVs were observed among non-hot spring A. caldus strains, and 2.5–3.5% single-nucleotide polymorphisms among A. ferrooxidans strains [80, 81]. Nevertheless, the upper values for single-nucleotide differences detected among TVZ populations are above Illumina sequencing error rates determined for metagenomics (0.04–0.12% for SNVs and 0.00028–0.00051% for indels) [82], and support for the consensus sequences is afforded by overall high genome coverages (on average 37.4 ± 45.7 × coverage) (Table S2), which indicates variations represent real biological differences. Genetic variations among MAGs recovered from unique TVZ hot springs between 1 km and 65 km apart suggests that successful colonization of these diverse habitats by cosmopolitan Acidithiobacillus species is achieved via population-level diversification, either due to environmental selection controlled by the unique chemical conditions of each hot spring or genetic drift and dispersal limitation [83, 84]. This is exemplified by habitat-differentiated ecotypes of the pelagic cosmopolitan genus Prochlorococcus [85]. Similarly, distinct clades of Sulfolobus have been found in remote hot springs [86], indicating a high degree of genetic variation driven by spatial separation.
GC contents correspond with optimal growth temperatures in Acidithiobacillus
Predicted optimal growth temperatures for the TVZ Acidithiobacillus species and other Acidithiobacillus type strains by GrowthPred are shown in Fig. 3a, along with the actual temperature ranges across which they were found. Results show that for Acidithiobacillus isolates GrowthPred predicted optimal growth temperatures near or at the reported upper temperature limits of these organisms, as expected based on the master reaction model for temperature growth curves [87, 88]. On the other hand, for TVZ populations, GrowthPred predicted optimal growth temperatures to typically fall in the middle of the temperature ranges at which they were detected, possibly due to different tolerances within populations.
In order to determine whether GC content contributes to heat tolerance, we examined the compositions of GC in the TVZ Acidithiobacillus and reference genomes. Data show that the TVZ Acidithiobacillus groups have an average GC content of 59.0 ± 2.9%, whereas the other 37 reference Acidithiobacillus have an average GC content of 56.9 ± 3.2% (Fig. 5a, d). Interestingly, A. caldus TVZ_G1 bins alone have average GC contents of 62.8 ± 0.1%. These contents are significantly higher (p = 1.2 × 10-13) than all other known Acidithiobacillus (GC content data retrieved from GTDB and also recalculated by CheckM), of which the GC contents range from 52.6 to 61.7% (Table S2). Even when compared to other strains of A. caldus, we found that the TVZ_G1 clade still had a significantly higher GC content (p = 2.27 × 10-6). Although some studies have reported that A. caldus DSM 8584 (or ATCC 51756) contains 63.9% GC [2, 3], this was based on only the 16S rRNA gene sequences, while the whole genome GC content was instead shown to be 61.7% [12].
Fig. 5.

Scatter plots of the TVZ and other Acidithiobacillus species. Plots a–c show positive correlations between a GC content (%) and optimal growth temperature (°C) (R = 0.75, p = 3.0 × 10−11); b predicted proline contents (%) and optimal growth temperature (°C) (R = 0.92, p < 2.2 × 10−16); and c percentage of paralogous proteins and coding sequences (CDS) (R = 0.63, p = 1.7 × 10−7). Plots d–f show negative correlations between d GC content (%) and estimated genome size (Mbp) (R = − 0.51, p = 7.0 × 10−5); e predicted proline content (%) and estimated genome size (Mbp) (R = − 0.81, p = 3.4 × 10−14); and f optimal growth temperature (°C) and estimated genome size (Mbp) (R = − 0.86, p < 2.2 × 10−16). g Shows a positive correlation between minimum generation time (hr) and estimated genome size (Mbp), with reference A. caldus and the Taiwanese Acidithiobacillus (UBA2486) as outliers (R = 0.36, p = 0.008). h Shows a negative correlation between number of non-coding RNA and optimal growth temperature (°C) (R = − 0.45, p = 0.00044). i Shows a positive correlation between percent of non-coding RNA and estimated genome size (Mbp) (R = 0.24, p = 0.08). Optimal growth temperatures were derived from GrowthPred calculation. The nearest (co-clustering) unclassified Acidithiobacillus species (black rimmed-circle) to TVZ_G2 is Acidithiobacillus sp. UBA2486 in all plots. Pearson’s correlation coefficients and t-distribution tables were used to determine the correlation coefficients and the significances, respectively, as described in the methods
As it has been debated whether GC content and optimal growth temperature are correlated in prokaryotes [89–92], we determined the relationship between the GC content of taxa and their preferred growth temperatures in order to verify this. Our results indicate that Acidithiobacillus GC contents and optimal growth temperatures, as predicted by GrowthPred, are positively correlated (R = 0.75, p = 3.0 × 10−11; Pearson’s correlation coefficient) (Fig. 5a). This suggests higher GC contents might contribute to heat tolerance in Acidithiobacillus from the TVZ and elsewhere. Higher thermostability is a well-known characteristic of GC-rich DNA, owing to the greater thermostability imparted by the three hydrogen bonds binding GC, in contrast to the two hydrogen bonds binding AT pairs [90, 92]. In addition, the stacking arrangement of adjacent bases is another important factor contributing to the thermal stability of DNA, as there is more favorable stacking energy for GpC/CpG pairs than for ApT/TpA pairs, such that genomes consisting of more GpC/CpG islands tend to be more thermostable than those that have fewer islands [93].
It is worth noting that GrowthPred calculates an organism’s optimal growth temperature from the frequency of a set of amino acids (IVYWREL), which is the only set (out of all amino acids) that was found to be remarkably positively correlated with optimal growth temperature [94]. The predicted optimal growth temperatures could therefore be influenced by GC-rich codons, associated with five out of seven of this set, which account for more than 40% of GC-rich codons, such as GUC and GUG (for Val, V), UGG (for Trp, W), CGC and CGG (for Arg, R), GAG (for Glu, E), and CUC and CUG (for Leu, L). Nevertheless, positive correlations between GC content and optimal growth temperatures in the TVZ Acidithiobacillus are reinforced by the high hot spring temperatures in which these species were detected (Fig. 2a–d).
High proline composition contributes to heat tolerance in TVZ Acidithiobacillus
To examine heat tolerance capability, we determined the correlation between proline content and optimal growth temperature of the TVZ Acidithiobacillus groups. Results indicated that the TVZ Acidithiobacillus genomes, along with the Taiwanese hot spring strain UBA2486, have significantly higher proportions of codons encoding proline than other Acidithiobacillus (5.5% ± 0.1 versus 5.2% ± 0.1, p = 3.8 × 10-10) (Table S2). The proline-coding contents are also positively correlated with optimal growth temperature (R = 0.92, p < 2.2 × 10−16) (Fig. 5b). Notably, proline is not one of the amino acids used by GrowthPred to calculate optimal growth temperature. Protein thermostabilization by proline substitutions is a strategy thermophiles use to cope with high-temperature environments by reducing the conformational degrees of freedom in the main polypeptide chain [95, 96]. The structure of proline is more similar to α-imino acid, rather than α-amino acid, such that its side chain is folded back to form a peptide bond with nitrogen, which results in rigid constraints on the N-C α-rotation. Alongside higher GC contents, higher proline compositions might explain the widespread occurrence of TVZ Acidithiobacillus in extremely hot environments that exceed the temperature limits of many other Acidithiobacillus.
In addition, we explored differences in heat-shock proteins (HSPs) in the TVZ genomes compared to other reference Acidithiobacillus species; however, no difference in HSP abundances (copies per genome) was observed between these groups. We believe the reason for this is that HSPs are designed initially for upshifts/downshifts in temperature. As the TVZ strains were found in high-temperature springs, this suggests adaptation to high temperatures, and no noticeable difference in HSP abundances. However, differences in expression cannot be excluded.
Evidence of genome streamlining in the TVZ Acidithiobacillus
We compared the estimated genome sizes of TVZ and other Acidithiobacillus and found the TVZ hot spring groups and the Taiwanese hot spring strain (UBA2486) had considerably smaller genome sizes on average (2.1 ± 0.3 Mbp) compared to those of the reference genomes (3.3 ± 0.4 Mbp, p = 3.2 × 10−12) (Fig. 5d–g, i). Notably, the genome sizes of the TVZ A. caldus clade were also much smaller than other A. caldus strains (2.1 ± 0.1 Mbp versus 3.1 ± 0.1 Mbp, p = 1.83 × 10-10) that are not typically associated with hot spring environments. The smaller estimated genome sizes, particularly for Acidithiobacillus spp. TVZ_G3 and TVZ_G4, are indicative of genome streamlining. During streamlining, prokaryotes minimize complexity in their cells in order to make the most use of resources required for replication and, thus, increase fitness [97]. In addition, despite the geographical distance, Acidithiobacillus sp. UBA2486, which was recovered from a hot spring environment in Taiwan (temperature = 50–85 °C and pH = 2.5) [98], shares a similar estimated genome size (along with other studied genomic attributes) with the TVZ Acidithiobacillus (Fig. 5a–i). Results are in agreement with prior research demonstrating that prokaryotes adapted to high temperatures (in this case, hot springs) tend to have smaller genome sizes, alongside lower proportions of non-coding RNA, suggestive of streamlining (Fig. 5f, h) [99]. Accordingly, we also found broadly lower proportions of non-coding RNA to estimated genome size in TVZ Acidithiobacillus spp. (Fig. 5i).
To further explore the link between adaptation to high temperatures and genome reduction in hot spring Acidithiobacillus, we plotted estimated genome size against predicted optimal growth temperature. Results illustrate a strong negative correlation between optimal growth temperature and increasing estimated genome size (R = − 0.86, p < 2.2 × 10−16; Pearson’s correlation coefficient) (Fig. 5f). Both smaller genome sizes and higher optimal growth temperatures can underpin faster genome replication rates [97, 99]. Consistent with this, we found that predicted genome replication rates were highest for the smaller hot spring Acidithiobacillus genomes, and minimum generation times were lower than for most other Acidithiobacillus (Fig. 5g, Table S2). High predicted replication rates may explain the overall high relative abundance of the TVZ Acidithiobacillus across the hot spring environments tested. In addition, the identified ranges of iRep values are consistent with those from other studies of actively growing communities [64, 100], implying that these organisms were active in the environment at the time of sampling.
Results further indicate that estimated genome size in Acidithiobacillus is negatively correlated with GC content and proline composition (R = − 0.51, p = 7.0 × 10−5 and R = − 0.81, p = 3.4 × 10−14; Pearson’s correlation coefficient) (Fig. 5d, e), even though GC content has been shown elsewhere to be positively correlated with genome size in prokaryotes [101, 102]. Although A. caldus TVZ_G1 genomes, in particular, have a relatively high GC content, this contradiction is likely due to the importance of high GC composition for thermotolerance. It is possible, therefore, that TVZ_G1 underwent genome reduction, resulting in smaller genome sizes than other A. caldus, yet preserved high GC contents as an adaptive strategy for high-temperature environments.
Finally, streamlined prokaryotes also tend to have a lower fraction of paralogs compared to CDS [57, 84]. We determined the amount of paralogous predicted protein sequences in Acidithiobacillus genomes and found that the TVZ Acidithiobacillus (along with UBA2486) have lower proportions of paralogous predicted protein sequences than other Acidithiobacillus genomes (on average 18.8% ± 4.5 versus 24.0% ± 4.2, p = 9.6 × 10−5) (Fig. 5c). Our results also show that the percent of paralogs is correlated with CDS (R = 0.63, p = 1.7 × 10−7; Pearson’s correlation coefficient) and genome size (R = 0.24, p = 0.008; Pearson’s correlation coefficient), consistent with results from other studies [66, 103].
Amino acid decarboxylases, Na+/H+ antiporters, K+ transporters, and proton-efflux ATPases contribute to habitation across wide pH ranges
All four TVZ Acidithiobacillus populations were detected across wide pH ranges exceeding the previously known limits of other Acidithiobacillus species. Here we examined genes encoding five groups of proteins involved in pH homeostasis: amino acid decarboxylases (using H+ in the decarboxylation of amino acids), K+ transporters, deiminases/deaminases (generating ammonium ions from ammonia and H+), Na+/H+ antiporters, and plasma membrane proton-efflux P-type ATPases [104–107] (Table S7). Gene prediction results show that all four TVZ populations share similar numbers of gene copies encoding proteins in K+ transporters (two to three copies each) and that this is also similar to those found in other Acidithiobacillus genomes (Fig. 6). Comparably, we found the TVZ and other Acidithiobacillus share one copy each of genes encoding proteins in the deiminases/deaminases group (agmatine deiminase, N-carbamoylputrescine amidase, and adenosine deaminase), with few exceptions (e.g., A. caldus ATCC 51756 and A. albertensis DSM 14366 lack adenosine deaminase) (Fig. 6). K+ uptake helps maintaining pH homeostasis by generating the internal positive membrane potential [104, 108], while deiminase/deaminase reactions create ammonia as a by-product which later combines with protons to produce ammonium ions [106]. Nevertheless, TVZ populations differ in the number of gene copies they respectively have that encode amino acid decarboxylases, Na+/H+ antiporters, and plasma membrane proton-efflux P-type ATPases.
Fig. 6.
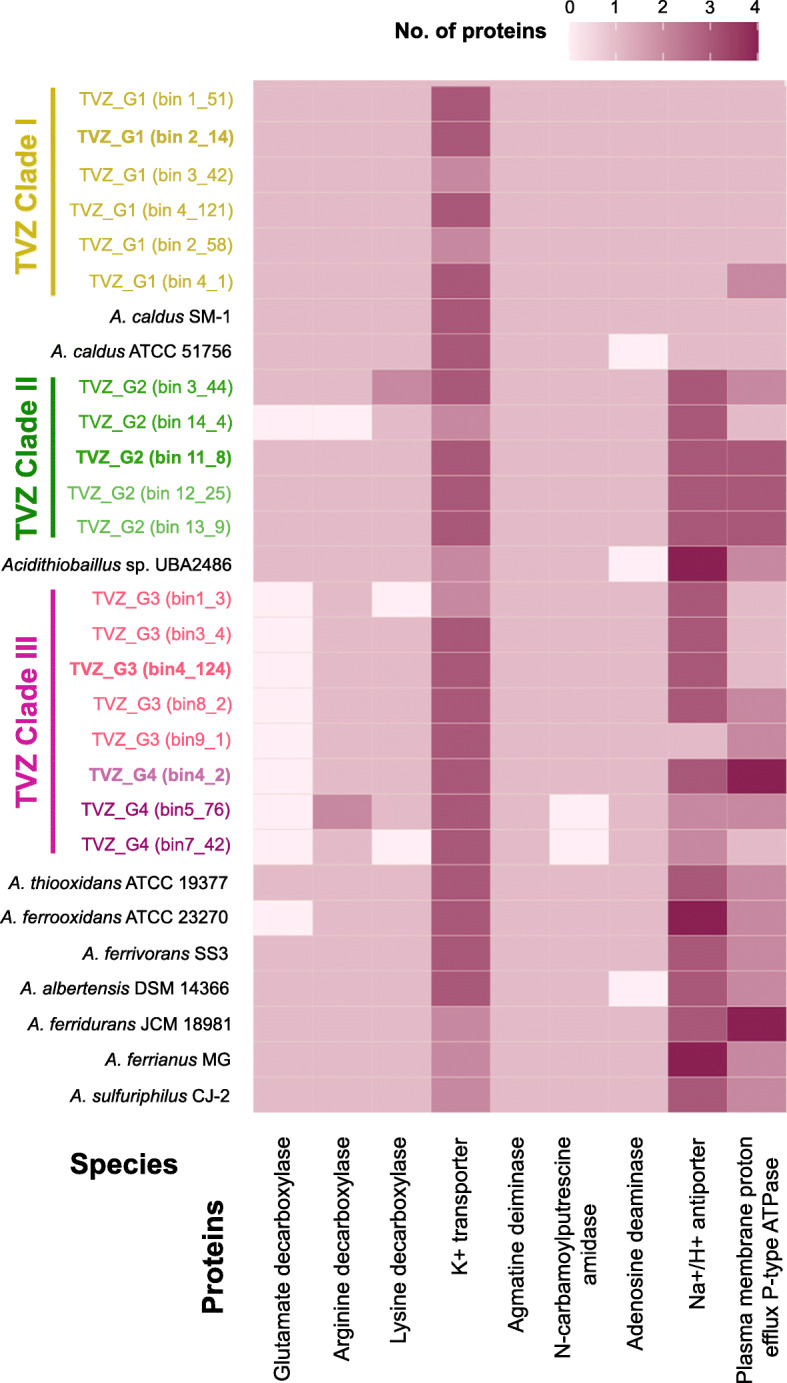
Heatmap showing the number of proteins associated with pH homeostasis in the TVZ and other Acidithiobacillus species. Bold script indicates the representative genome of the TVZ Acidithiobacillus species. Accession IDs of genes analyzed can be found in Table S7
We predict that TVZ_G1 and other A. caldus primarily rely on amino acid decarboxylases, in addition to K+ transporters, to maintain pH balances (Fig. 6). They were found to possess one copy each of genes encoding three distinct amino acid decarboxylases, and fewer copies of genes than the other TVZ groups (only one each) for Na+/H+ antiporter and plasma membrane proton-efflux ATPase. The pH homeostasis genes detected in A. caldus TVZ_G1 were similar to those of other A. caldus strains. This is likely due to their close phylogeny (Fig. 4). These genes are likely preserved by TVZ_G1, despite genome streamlining, due to their significance for inhabiting acidic environments. Although the amino acid decarboxylation indeed helps in eliminating intracellular protons, whether the decarboxylated product (e.g., GABA from glutamate decarboxylation) is exported from the cells is still being debated. Some studies claim that GABA might be kept in the cells after decarboxylation and can be further transformed and included in the TCA cycle [109, 110]. Other studies suggest that it is exported right after decarboxylation [104–107]. Either way, all 19 TVZ and 10 reference Acidithiobacillus genomes studied here were found to lack the genetic capacity to convert GABA into succinate semialdehyde and succinate (via GABA transaminase (EC 2.6.1.19) and succinate-semialdehyde dehydrogenase (EC 1.2.1.16)), and the complete glutamate decarboxylase exporting system was also not identified. Nonetheless, a previous study has shown that amino acid decarboxylation was highly expressed by A. caldus when introduced to acid stress [111].
In contrast to the A. caldus group, up to three gene copies encoding Na+/H+ antiporters and another three for plasma membrane proton-efflux ATPases are present in TVZ_G2 MAGs, alongside three more for amino acid decarboxylases. These results suggest TVZ_G2 rely on all three mechanisms to cope with excess protons to maintain their intracellular pH. MAGs in both the closely related TVZ_G3 and TVZ_G4 Clade III groups also tended to possess more than one gene copy encoding Na+/H+ antiporters (up to three). Strikingly, however, none contained genes for glutamate decarboxylase. In addition, TVZ_G3 MAGs tended to have fewer gene copies than TVZ_G2 for plasma membrane proton-efflux ATPase (one or two) (Fig. 6), while TVZ_G4 MAGs were found to possess up to four plasma membrane proton-efflux ATPase gene copies.
As for TVZ_G2-G4, Na+/H+ antiporter gene copies were also found to be high, similar to those found in A. thiooxidans, A. ferrooxidans, A. ferrivorans, and A. albertensis (Fig. 6); all except for A. ferrivorans have wide pH growth ranges (0.5–6.0) (Fig. 3b). This implies that possessing more Na+/H+ antiporters genes might be a key strategy allowing these Acidithiobacillus to live across wide pH ranges. In addition, Na+/H+ antiporters are driven by proton motive force generated by the respiratory chain, i.e., electrochemical gradients [104, 105]. They, therefore, potentially consume less energy than amino acid decarboxylation (which requires the uptake of amino acids and the exclusion of decarboxylated products) and plasma membrane proton-efflux P-type ATPases (which cost ATP) [104, 105, 112, 113]. As such, the use of antiporters for pH tolerance could also be an energy-efficient mechanism for streamlined microorganisms.
Conclusions
Four thermoacidophilic populations of Acidithiobacillus, which included three novel genome-inferred species and one new A. caldus clade, were recovered in this study. Three of these are abundant, cosmopolitan populations, widely distributed across TVZ geothermal environments and extreme ranges of temperature (17.5–92.9 °C) and pH (1.0–7.5); in both cases exceeding the previously known upper physiological limits of Acidithiobacillus. Hot spring Acidithiobacillus genomes were found to have high GC and proline-coding contents, which are interpreted to increase thermostability, potentially enabling them to live at high temperatures. Moreover, results indicate the hot spring Acidithiobacillus have undergone genome streamlining, likely as a result of thermal adaptation. In addition, we predicted that some TVZ Acidithiobacillus (TVZ_G1-G2) cope with the broad pH conditions via dependence on a higher number of amino acid decarboxylase genes. TVZ_G2-G4 also shared a roughly similar number of Na+/H+ antiporters and plasma membrane proton-efflux proteins with other reference Acidithiobacillus species that have similarly wide pH ranges, suggesting the importance of these two predicted proteins in organisms inhabiting a broad spectrum of pHs. While experimental confirmation of bioinformatic predictions is needed, results provide insights into genomic features and adaptations that we infer enable hot spring-associated Acidithiobacillus species to colonize a broad range of physicochemically diverse environments.
Supplementary Information
Acknowledgements
The authors thank B. Drake, M. Rowe, L. Steller, L. Penrose, T. Hamilton, J. Havig, M. Dobson, S. Camp, Y. Heled, M. Millan, D. P. Aparicio, and A. Hamilton for facilitating and helping with field work and geochemistry data collection. We are grateful to D. Waite, H. Sze, J. Boey, E. Gios, and C. Astudillo-García from the School of Biological Sciences at the University of Auckland for bioinformatics support. We also acknowledge Timberlands Ltd (Rotorua) for Orange Spring site access, Mercury Energy (Rotorua), Tauhara North No. 2 Trust, and the New Zealand Department of Conservation (DOC) for Parariki Stream and Lake Rotokawa site access, R. and B. McNaull for Te Kopia site access, Hell's Gate Ltd for Tikitere site access, and the Ngati Tahu-Ngati Whaoa Runanga Trust and the staff of Wai-O-Tapu Thermal Wonderland for Wai-O-Tapu site access. Finally, the authors wish to acknowledge the use of New Zealand eScience Infrastructure (NeSI) high-performance computing facilities.
Abbreviations
- ANI
Average nucleotide identity
- ASV
Amplicon sequence variant
- CDS
Coding sequence
- DDH
DNA-DNA hybridization
- GTDB
Genome Taxonomy Database
- SNV
Single-nucleotide variant
- TVZ
Taupo Volcanic Zone
- WGS
Whole genome sequencing
Authors’ contributions
CS and KMH collected samples. KAC and MVK led field trips. CS and KMH planned the study and CS conducted the laboratory work. CS and KMH analyzed and interpreted data and wrote the first draft of the manuscript. KAC and MVK revised and corrected the manuscript. All authors read and approved the final manuscript.
Funding
This study was funded by the University of Auckland Faculty Research Development Fund (grant 9856-RO-3712371), Genomics Aotearoa project 1806, and Australian Research Council discovery project DP180103204. Research was also supported by a Royal Society Te Apārangi Rutherford Discovery Fellowship awarded to KMH.
Availability of data and materials
All amplicon and WGS data from sinter samples can be accessed through NCBI under BioProject PRJNA543937. All amplicon and WGS data from sediment samples can be accessed through NCBI Bioproject PRJNA644733.
Declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no conflict of interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chanenath Sriaporn, Email: csri348@aucklanduni.ac.nz.
Kathleen A. Campbell, Email: ka.campbell@auckland.ac.nz
Martin J. Van Kranendonk, Email: m.vankranendonk@unsw.edu.au
Kim M. Handley, Email: kim.handley@auckland.ac.nz
References
- 1.Waksman SA, Joffe JS. Microörganisms concerned in the oxidation of sulfur in the soil: II. Thiobacillus thiooxidans, a new sulfur-oxidizing organism isolated from the soil. J Bacteriol. 1922;7(2):239–256. doi: 10.1128/JB.7.2.239-256.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hallberg KB, Lindström EB. Characterization of Thiobacillus caldus sp. nov., a moderately thermophilic acidophile. Microbiology. 1994;140(12):3451–3456. doi: 10.1099/13500872-140-12-3451. [DOI] [PubMed] [Google Scholar]
- 3.Kelly DP, Wood AP. Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int J Syst Evol Microbiol. 2000;50(2):511–6. 10.1099/00207713-50-2-511. [DOI] [PubMed]
- 4.Baker BJ, Banfield JF. Microbial communities in acid mine drainage. FEMS Microbiol Ecol. 2003;44(2):139–52. 10.1016/S0168-6496(03)00028-X. [DOI] [PubMed]
- 5.Jones DS, Schaperdoth I, Macalady JL. Biogeography of sulfur-oxidizing Acidithiobacillus populations in extremely acidic cave biofilms. ISME J. 2016;10(12):2879–2891. doi: 10.1038/ismej.2016.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Power JF, Carere CR, Lee CK, Wakerley GLJ, Evans DW, Button M, White D, Climo MD, Hinze AM, Morgan XC, McDonald IR, Cary SC, Stott MB. Microbial biogeography of 925 geothermal springs in New Zealand. Nat Commun. 2018;9(1):2876. doi: 10.1038/s41467-018-05020-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sriaporn C, Campbell KA, Millan M, Ruff SW, Van Kranendonk MJ, Handley KM. Stromatolitic digitate sinters form under wide-ranging physicochemical conditions with diverse hot spring microbial communities. Geobiology. 2020;18(5):619–640. doi: 10.1111/gbi.12395. [DOI] [PubMed] [Google Scholar]
- 8.Castro M, Moya-Beltrán A, Covarrubias PC, Gonzalez M, Cardenas JP, Issotta F, Nuñez H, Acuña LG, Encina G, Holmes DS, Johnson DB, Quatrini R. Draft genome sequence of the type strain of the sulfur-oxidizing acidophile, Acidithiobacillus albertensis (DSM 14366) Stand Genomic Sci. 2017;12(1):77. doi: 10.1186/s40793-017-0282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liljeqvist M, Valdes J, Holmes DS, Dopson M. Draft genome of the psychrotolerant acidophile Acidithiobacillus ferrivorans SS3. J Bacteriol. 2011;193(16):4304–4305. doi: 10.1128/JB.05373-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valdes J, Ossandon F, Quatrini R, Dopson M, Holmes DS. Draft genome sequence of the extremely acidophilic biomining bacterium Acidithiobacillus thiooxidans ATCC 19377 provides insights into the evolution of the Acidithiobacillus genus. J Bacteriol. 2011;193(24):7003–7004. doi: 10.1128/JB.06281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valdés J, Pedroso I, Quatrini R, Dodson RJ, Tettelin H, Blake R, et al. Acidithiobacillus ferrooxidans metabolism: from genome sequence to industrial applications. BMC Genomics. 2008;9(1):597. doi: 10.1186/1471-2164-9-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valdes J, Quatrini R, Hallberg K, Dopson M, Valenzuela PDT, Holmes DS. Draft genome sequence of the extremely acidophilic bacterium Acidithiobacillus caldus ATCC 51756 reveals metabolic versatility in the genus Acidithiobacillus. J Bacteriol. 2009;191(18):5877–5878. doi: 10.1128/JB.00843-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang R, Lin JQ, Liu XM, Pang X, Zhang CJ, Yang CL, Gao XY, Lin CM, Li YQ, Li Y, Lin JQ, Chen LX. Sulfur oxidation in the acidophilic autotrophic Acidithiobacillus spp. Front Microbiol. 2019;9:3290. doi: 10.3389/fmicb.2018.03290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kupka D, Rzhepishevska OI, Dopson M, Lindström EB, Karnachuk OV, Tuovinen OH. Bacterial oxidation of ferrous iron at low temperatures. Biotechnol Bioeng. 2007;97(6):1470–1478. doi: 10.1002/bit.21371. [DOI] [PubMed] [Google Scholar]
- 15.Acuña LG, Cárdenas JP, Covarrubias PC, Haristoy JJ, Flores R, Nuñez H, et al. Architecture and gene repertoire of the flexible genome of the extreme acidophile Acidithiobacillus caldus. PLoS One. 2013;8(11):e78237. 10.1371/journal.pone.0078237. [DOI] [PMC free article] [PubMed]
- 16.Zhang X, Liu X, He Q, Dong W, Zhang X, Fan F, Peng D, Huang W, Yin H. Gene turnover contributes to the evolutionary adaptation of Acidithiobacillus caldus: Insights from comparative genomics. Front Microbiol. 2016;7:1960. doi: 10.3389/fmicb.2016.01960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bosecker K. Bioleaching: metal solubilization by microorganisms. FEMS Microbiol Rev. 1997;20(3-4):591–604. doi: 10.1111/j.1574-6976.1997.tb00340.x. [DOI] [Google Scholar]
- 18.Colmer AR, Hinkle ME. The role of microorganisms in acid mine drainage: a preliminary report. Science. 1947;106(2751):253–256. doi: 10.1126/science.106.2751.253. [DOI] [PubMed] [Google Scholar]
- 19.Rohwerder T, Gehrke T, Kinzler K, Sand W. Bioleaching review part A. Appl Microbiol Biotechnol. 2003;63(3):239–248. doi: 10.1007/s00253-003-1448-7. [DOI] [PubMed] [Google Scholar]
- 20.Vera M, Schippers A, Sand W. Progress in bioleaching: fundamentals and mechanisms of bacterial metal sulfide oxidation—part A. Appl Microbiol Biotechnol. 2013;97(17):7529–7541. doi: 10.1007/s00253-013-4954-2. [DOI] [PubMed] [Google Scholar]
- 21.One Thousand Springs. The microbiology of geothermal hot springs in New Zealand. http://1000springs.org.nz. Accessed 9 December 2020.
- 22.Renaut RW, Jones, B. Hydrothermal environments, terrestrial. In: Reiner J, Thiel V (Eds.). Encyclopedia of Geobiology. Springer, Amsterdam, The Netherlands, 2011. p. 467–79. 10.1007/978-1-4020-9212-1_114.
- 23.Ward L, Taylor MW, Power JF, Scott BJ, McDonald IR, Stott MB. Microbial community dynamics in Inferno Crater Lake, a thermally fluctuating geothermal spring. ISME J. 2017;11(5):1158–1167. doi: 10.1038/ismej.2016.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;18:4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, Gregory Caporaso J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6(1):90. doi: 10.1186/s40168-018-0470-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Xu ZZ, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2(2):e00191–e00116. doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Glöckner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, Bruns G, Yarza P, Peplies J, Westram R, Ludwig W. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol. 2017;261:169–176. doi: 10.1016/j.jbiotec.2017.06.1198. [DOI] [PubMed] [Google Scholar]
- 29.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2012;41(D1):D590–D5D6. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yilmaz P, Parfrey LW, Yarza P, Gerken J, Pruesse E, Quast C, Schweer T, Peplies J, Ludwig W, Glöckner FO. The SILVA and "all-species Living Tree Project (LTP)" taxonomic frameworks. Nucleic Acids Res. 2014;42(D1):D643–D6D8. doi: 10.1093/nar/gkt1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.R Core Team . R: A language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2017. [Google Scholar]
- 32.RStudio Team . Integrated Development for R. Boston: RStudio Inc; 2015. [Google Scholar]
- 33.Wickham H. ggplot2: elegant graphics for data analysis. In: Gentlemean R, Hornik K, Parmigiani G (Eds.). Use R!. Springer, Amsterdam, The Netherlands. 2016. 10.1007/978-3-319-24277-4. [DOI]
- 34.Joshi N, Fass J. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33). 2011. Available at https://github.com/najoshi/sickle.
- 35.Andrews S. FastQC: a quality control tool for high throughput sequence data. Babraham Bioinformatics, Babraham Institute, Cambridge. 2010. Available at https://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 36.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;32(1):11–17. doi: 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165. doi: 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu YW, Tang YH, Tringe SG, Simmons BA, Singer SW. MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2(1):26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11(11):1144–1146. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 41.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3(7):836–843. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laczny CC, Sternal T, Plugaru V, Gawron P, Atashpendar A, Margossian HH, Coronado S, der Maaten L, Vlassis N, Wilmes P. VizBin - an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome. 2015;3(1):1. doi: 10.1186/s40168-014-0066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25(7):1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castelle CJ, Brown CT, Anantharaman K, Probst AJ, Huang RH, Banfield JF. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol. 2018;16(10):629–645. doi: 10.1038/s41579-018-0076-2. [DOI] [PubMed] [Google Scholar]
- 45.Olm MR, Brown CT, Brooks B, Banfield JF. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11(12):2864–2868. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11(1):119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzek BE, Huang H, McGarvey P, Mazumder R, Wu CH. UniRef: comprehensive and non-redundant UniProt reference clusters. Bioinformatics. 2007;23(10):1282–1288. doi: 10.1093/bioinformatics/btm098. [DOI] [PubMed] [Google Scholar]
- 48.Wu CH, Apweiler R, Bairoch A, Natale DA, Barker WC, Boeckmann B, et al. The Universal Protein Resource (UniProt): an expanding universe of protein information. Nucleic Acids Res. 2006;34(suppl_1):D187–DD91. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer ELL, Tate J, Punta M. Pfam: the protein families database. Nucleic Acids Res. 2013;42(D1):D222–DD30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haft DH, Selengut JD, White O. The TIGRFAMs database of protein families. Nucleic Acids Res. 2003;31(1):371–373. doi: 10.1093/nar/gkg128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 53.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 54.Chaumeil PA, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2019;36(6):1925–1927. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Average Nucleotide Identity. ANI calculator. http://enve-omics.ce.gatech.edu/ani. Kostas lab. Accessed October 2019 - December 2020.
- 56.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez-R LM, Konstantinidis KT. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 2016;4:e1900v1. doi: 10.7287/peerj.preprints.1900v1. [DOI] [Google Scholar]
- 58.Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, Pati A. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 2015;43(14):6761–6771. doi: 10.1093/nar/gkv657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meier-Kolthoff JP, Auch AF, Klenk HP, Göker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013;14(1):60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A, Rohde C, Rohde M, Fartmann B, Goodwin LA, Chertkov O, Reddy TBK, Pati A, Ivanova NN, Markowitz V, Kyrpides NC, Woyke T, Göker M, Klenk HP. Complete genome sequence of DSM 30083 T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand Genom Sci. 2014;9(1):2. doi: 10.1186/1944-3277-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin HN, Hsu WL. GSAlign: an efficient sequence alignment tool for intra-species genomes. BMC Genomics. 2020;21(1):182. doi: 10.1186/s12864-020-6569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Edgar RC. Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics. 2018;34(14):2371–2375. doi: 10.1093/bioinformatics/bty113. [DOI] [PubMed] [Google Scholar]
- 63.Vieira-Silva S, Rocha EPC. The systemic imprint of growth and its uses in ecological (meta)genomics. PLoS Genet. 2010;6(1):e1000808. doi: 10.1371/journal.pgen.1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown CT, Olm MR, Thomas BC, Banfield JF. Measurement of bacterial replication rates in microbial communities. Nat Biotechnol. 2016;34(12):1256–1263. doi: 10.1038/nbt.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 66.Swan BK, Tupper B, Sczyrba A, Lauro FM, Martinez-Garcia M, González JM, et al. Prevalent genome streamlining and latitudinal divergence of planktonic bacteria in the surface ocean. PNAS. 2013;110(28):11463–11468. doi: 10.1073/pnas.1304246110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.aa_composition.py. https://github.com/GenomicsAotearoa/environmental_metagenomics/blob/master/analysis_tools/aa_composition.py. Genomics Aotearoa. Accessed date 25 March 2020.
- 68.Parks DH, Chuvochina M, Chaumeil PA, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38(9):1–8. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 69.Miller CS, Baker BJ, Thomas BC, Singer SW, Banfield JF. EMIRGE: reconstruction of full-length ribosomal genes from microbial community short read sequencing data. Genome Biol. 2011;12(5):R44. doi: 10.1186/gb-2011-12-5-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bengtsson-Palme J, Hartmann M, Eriksson KM, Pal C, Thorell K, Larsson DGJ, Nilsson RH. metaxa2: improved identification and taxonomic classification of small and large subunit rRNA in metagenomic data. Mol Ecol Resour. 2015;15(6):1403–1414. doi: 10.1111/1755-0998.12399. [DOI] [PubMed] [Google Scholar]
- 71.Kassambara A, Kassambara MA. ggpubr: 'ggplot2' Based Publication Ready Plots. R package version 0.4.0. 2020. [Google Scholar]
- 72.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004;5(1):113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 75.Campbell KA, Guido DM, Gautret P, Foucher F, Ramboz C, Westall F. Geyserite in hot-spring siliceous sinter: Window on Earth's hottest terrestrial (paleo)environment and its extreme life. Earth-Science Rev. 2015;148:44–64. doi: 10.1016/j.earscirev.2015.05.009. [DOI] [Google Scholar]
- 76.Quatrini R, Johnson DB. Microbiomes in extremely acidic environments: functionalities and interactions that allow survival and growth of prokaryotes at low pH. Curr Opin Microbiol. 2018;43:139–147. doi: 10.1016/j.mib.2018.01.011. [DOI] [PubMed] [Google Scholar]
- 77.Hamamura N, Olson SH, Ward DM, Inskeep WP. Diversity and functional analysis of bacterial communities associated with natural hydrocarbon seeps in acidic soils at Rainbow Springs, Yellowstone National Park. Appl Environ Microbiol. 2005;71(10):5943–5950. doi: 10.1128/AEM.71.10.5943-5950.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiménez DJ, Andreote FD, Chaves D, Montaña JS, Osorio-Forero C, Junca H, et al. Structural and functional insights from the metagenome of an acidic hot spring microbial planktonic community in the Colombian Andes. PLoS One. 2012;7(12):e52069. 10.1371/journal.pone.0052069. [DOI] [PMC free article] [PubMed]
- 79.Mangold S, Valdés J, Holmes D, Dopson M. Sulfur metabolism in the extreme acidophile Acidithiobacillus caldus. Front Microbiol. 2011;2:17. doi: 10.3389/fmicb.2011.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nuñez H, Loyola D, Cárdenas JP, Holmes DS, Johnson DB, Quatrini R. Multi locus sequence typing scheme for Acidithiobacillus caldus strain evaluation and differentiation. Res Microbiol. 2014;165(9):735–742. doi: 10.1016/j.resmic.2014.07.014. [DOI] [PubMed] [Google Scholar]
- 81.Fariq A, Blazier JC, Yasmin A, Gentry TJ, Deng Y. Whole genome sequence analysis reveals high genetic variation of newly isolated Acidithiobacillus ferrooxidans IO-2C. Sci Rep. 2019;9(1):13049. doi: 10.1038/s41598-019-49213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schirmer M, D’Amore R, Ijaz UZ, Hall N, Quince C. Illumina error profiles: resolving fine-scale variation in metagenomic sequencing data. BMC Bioinform. 2016;17(1):125. doi: 10.1186/s12859-016-0976-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oakley BB, Carbonero F, van der Gast CJ, Hawkins RJ, Purdy KJ. Evolutionary divergence and biogeography of sympatric niche-differentiated bacterial populations. ISME J. 2010;4(4):488–497. doi: 10.1038/ismej.2009.146. [DOI] [PubMed] [Google Scholar]
- 84.Whittaker KA, Rynearson TA. Evidence for environmental and ecological selection in a microbe with no geographic limits to gene flow. PNAS. 2017;114(10):2651–2656. doi: 10.1073/pnas.1612346114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EM, Chisholm SW. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science. 2006;311(5768):1737–1740. doi: 10.1126/science.1118052. [DOI] [PubMed] [Google Scholar]
- 86.Whitaker RJ, Grogan DW, Taylor JW. Geographic barriers isolate endemic populations of hyperthermophilic archaea. Science. 2003;301(5635):976–978. doi: 10.1126/science.1086909. [DOI] [PubMed] [Google Scholar]
- 87.Johnson FH, Lewin I. The growth rate of E. coli in relation to temperature, quinine and coenzyme. J Cell Compa Physiol. 1946;28(1):47–75. doi: 10.1002/jcp.1030280104. [DOI] [PubMed] [Google Scholar]
- 88.Grimaud GM, Mairet F, Sciandra A, Bernard O. Modeling the temperature effect on the specific growth rate of phytoplankton: a review. Rev Environ Sci Biotechnol. 2017;16(4):625–645. doi: 10.1007/s11157-017-9443-0. [DOI] [Google Scholar]
- 89.Hurst LD, Merchant AR. High guanine–cytosine content is not an adaptation to high temperature: a comparative analysis amongst prokaryotes. P Roy Soc B-Biol Sci. 2001;268(1466):493–497. doi: 10.1098/rspb.2000.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Musto H, Naya H, Zavala A, Romero H. Alvarez-Valín F, Bernardi G. Correlations between genomic GC levels and optimal growth temperatures in prokaryotes. FEBS Lett. 2004;573(1):73–77. doi: 10.1016/j.febslet.2004.07.056. [DOI] [PubMed] [Google Scholar]
- 91.Wang HC, Susko E, Roger AJ. On the correlation between genomic G + C content and optimal growth temperature in prokaryotes: Data quality and confounding factors. Biochem Biophys Res Commun. 2006;342(3):681–684. doi: 10.1016/j.bbrc.2006.02.037. [DOI] [PubMed] [Google Scholar]
- 92.Zheng H, Wu H. Gene-centric association analysis for the correlation between the guanine-cytosine content levels and temperature range conditions of prokaryotic species. BMC Bioinform. 2010;11(S11):S7. doi: 10.1186/1471-2105-11-S11-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yakovchuk P, Protozanova E, Frank-Kamenetskii MD. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006;34(2):564–574. doi: 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zeldovich KB, Berezovsky IN, Shakhnovich EI. Protein and DNA sequence determinants of thermophilic adaptation. PLoS Comput Biol. 2007;3(1):e5. doi: 10.1371/journal.pcbi.0030005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Suzuki Y, Oishi K, Nakano H, Nagayama T. A strong correlation between the increase in number of proline residues and the rise in thermostability of five Bacillus oligo-1,6-glucosidases. Appl Microbiol Biotechnol. 1987;26(6):546–551. doi: 10.1007/BF00253030. [DOI] [Google Scholar]
- 96.Watanabe K, Suzuki Y. Protein thermostabilization by proline substitutions. J Mol Catal B-Enzym. 1998;4(4):167–180. doi: 10.1016/S1381-1177(97)00031-3. [DOI] [Google Scholar]
- 97.Giovannoni SJ, Thrash JC, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8(8):1553–65. 10.1038/ismej.2014.60. [DOI] [PMC free article] [PubMed]
- 98.Lin KH, Liao BY, Chang HW, Huang SW, Chang TY, Yang CY, Wang YB, Lin YTK, Wu YW, Tang SL, Yu HT. Metabolic characteristics of dominant microbes and key rare species from an acidic hot spring in Taiwan revealed by metagenomics. BMC Genomics. 2015;16(1):1029. doi: 10.1186/s12864-015-2230-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sabath N, Ferrada E, Barve A, Wagner A. Growth temperature and genome size in bacteria are negatively correlated, suggesting genomic streamlining during thermal adaptation. Genome Biol Evol. 2013;5(5):966–977. doi: 10.1093/gbe/evt050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tee HS, Waite D, Payne L, Middleditch M, Wood S, Handley KM. Tools for successful proliferation: diverse strategies of nutrient acquisition by a benthic cyanobacterium. ISME J. 2020;14(18):2164–2178. doi: 10.1038/s41396-020-0676-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kantor RS, Wrighton KC, Handley KM, Sharon I, Hug LA, Castelle CJ, et al. Small genomes and sparse metabolisms of sediment-associated bacteria from four candidate phyla. mBio. 2013;4(5):e00708–e00713. doi: 10.1128/mBio.00708-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mann S, Li J, Chen YPP. Insights into bacterial genome composition through variable target GC content profiling. J Comput Biol. 2010;17(1):79–96. doi: 10.1089/cmb.2009.0058. [DOI] [PubMed] [Google Scholar]
- 103.Bratlie MS, Johansen J, Sherman BT, Huang DW, Lempicki RA, Drabløs F. Gene duplications in prokaryotes can be associated with environmental adaptation. BMC Genomics. 2010;11(1):588. doi: 10.1186/1471-2164-11-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Baker-Austin C, Dopson M. Life in acid: pH homeostasis in acidophiles. Trends Microbiol. 2007;15(4):165–171. doi: 10.1016/j.tim.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 105.Krulwich TA, Sachs G, Padan E. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol. 2011;9(5):330–343. doi: 10.1038/nrmicro2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lund P, Tramonti A, De Biase D. Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol Rev. 2014;38(6):1091–1125. doi: 10.1111/1574-6976.12076. [DOI] [PubMed] [Google Scholar]
- 107.Papadimitriou K, Alegría Á, Bron PA, de Angelis M, Gobbetti M, Kleerebezem M, Lemos JA, Linares DM, Ross P, Stanton C, Turroni F, van Sinderen D, Varmanen P, Ventura M, Zúñiga M, Tsakalidou E, Kok J. Stress physiology of lactic acid bacteria. Microbiol Mol Biol Rev. 2016;80(3):837–890. doi: 10.1128/MMBR.00076-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kroll RG, Booth IR. The relationship between intracellular pH, the pH gradient and potassium transport in Escherichia coli. Biochem. 1983;216(3):709–716. doi: 10.1042/bj2160709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Karatzas KA, Brennan O, Heavin S, Morrissey J, O'Byrne CP. Intracellular accumulation of high levels of γ-aminobutyrate by Listeria monocytogenes 10403S in response to low pH: uncoupling of γ-aminobutyrate synthesis from efflux in a chemically defined medium. Appl Environ Microbiol. 2010;76(11):3529–3537. doi: 10.1128/AEM.03063-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Feehily C, Karatzas KA. Role of glutamate metabolism in bacterial responses towards acid and other stresses. J Appl Microbiol. 2013;114(1):11–24. doi: 10.1111/j.1365-2672.2012.05434.x. [DOI] [PubMed] [Google Scholar]
- 111.Mangold S, Rao Jonna V, Dopson M. Response of Acidithiobacillus caldus toward suboptimal pH conditions. Extremophiles. 2013;17(4):689–96. 10.1007/s00792-013-0553-5. [DOI] [PubMed]
- 112.Pedersen PL, Carafoli E. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. Trends Biochem Sci. 1987;12:146–50. 10.1016/0968-0004(87)90071-5.
- 113.Padan E, Zilberstein D, Schuldiner S. pH homesstasis in bacteria. Biochim Biophys Acta Biomembr. 1981;650(2-3):151–66. 10.1016/0304-4157(81)90004-6. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All amplicon and WGS data from sinter samples can be accessed through NCBI under BioProject PRJNA543937. All amplicon and WGS data from sediment samples can be accessed through NCBI Bioproject PRJNA644733.