

Cardiac hypertrophy sometimes occurs as a compensatory response to increases in left ventricle (LV) wall stress stemming from pressure overload, volume overload, or postinfarction remodeling. However, after extended periods of disease stress–induced cardiac hypertrophy, heart failure (HF) often occurs accompanied by LV hypertrophy, dilatation, and myocardial fibrosis.1 Progression from compensated hypertrophy to HF involves complex interactions among numerous cell types, including cardiomyocytes and nonmyocyte cardiac cells, acting through a wide variety of signaling pathways.2 Whereas the multifactorial nature of HF enhances difficulties in design and interpretation of mechanistic studies, it also provides multiple targets for therapeutic intervention. Thus, an comprehensive, molecular-level understanding of the natural history of HF can only be obtained through comprehensive, cell type–specific assessments of the genetic and molecular mechanisms that drive progression of pathogenic LV remodeling in failing hearts.
Single-cell RNA sequencing (scRNA-seq) is a relatively new, rapidly advancing, technology3–13 (Figure) that can comprehensively characterize gene expression and relationships in individual cells. This issue of Circulation includes a report by Ren et al14 in which the authors used scRNA-seq to monitor the transcriptomic landscape of cardiac cells during the progression from compensated hypertrophy to HF. Hypertrophy was induced in mice through transverse aortic constriction, and samples were collected at subsequent time points representing the early (0–2 weeks), intermediate (2–5 weeks), and late (5–11 weeks) stages of pathological hypertrophy; then, cardiomyocytes and noncardiomyocytes were isolated through Langendorff perfusion, and scRNA-seq analyses were conducted to investigate the dynamics of gene expression in each cell type and their corresponding contributions to disease progression. Sorting bias was minimized by using the iCell8 platform, which is conducted through robotics and precast nanowells and, unlike other platforms (eg, 10× Genomics Chromium System), is compatible with cardiomyocytes and other large cells but remains somewhat less efficient (and more expensive). Nevertheless, the number of cells sequenced (≈11 000) approached the maximum reported in other studies (20 000–30 000) and ensured that even rare cell types could be evaluated. Thus, the study provides a framework for understanding how the transcriptional activity of individual cardiac cells is altered during pathological hypertrophy, and a reservoir of clinically relevant scRNA-seq data, as well, that can be used to generate and test novel hypotheses and potential clinical interventions for the management of patients with HF.
Figure. Synopsis of the development of single-cell/nucleus RNA sequencing for cardiac applications.
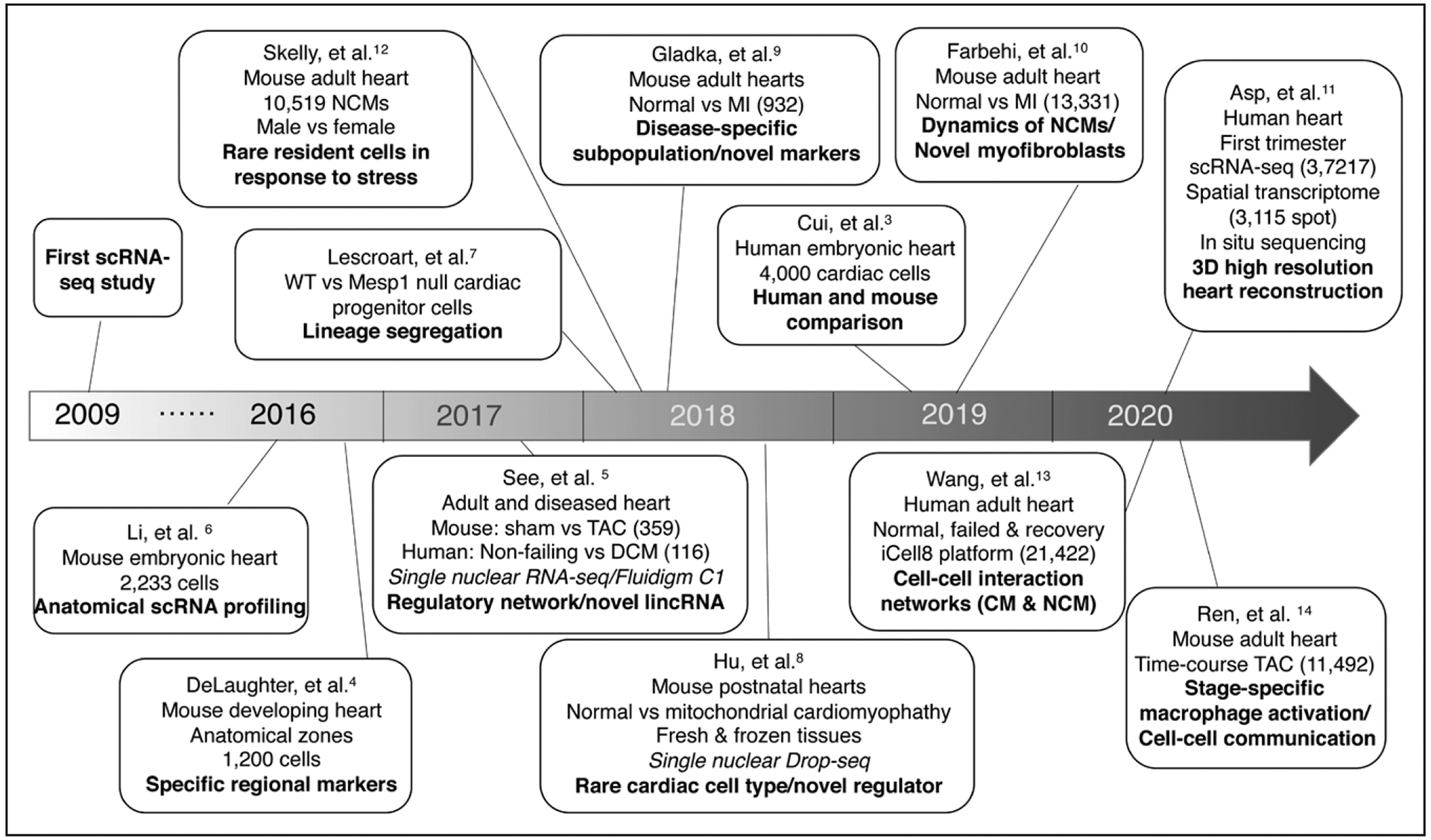
Several landmark studies that incorporated the use of single-cell or single-nucleus RNA sequencing (scRNA-seq or snRNA-seq) for investigations of heart development and disease are presented in a timeline. Seven years after the first reported transcriptome of a single cell, scRNA-seq was conducted in 1000 to 2000 cardiac cells for an investigation of murine heart development. Since then, both the number and types of cells sequenced, and the complexity of the biological processes investigated, as well, have steadily increased. Overall, the findings of these studies have demonstrated the heterogeneous response of cardiac cells to developmental signaling pathways and myocardial disease. 3D indicates 3-dimensional; CM, cardiomyocyte; DCM, dilated cardiomyopathy; Drop-seq, droplet sequencing; lincRNA, long intergenic noncoding RNA; MI, myocardial infarction; NCMs, noncardiomyocytes; TAC, transverse aortic constriction; and WT, wild-type.
RESOLVING INDIVIDUAL CELL-TYPE SUBPOPULATIONS
scRNA-seq technology separates the heart into individual cells, thereby providing a tool that can be used to magnify and sequentially reconstruct cellular connections and communication pathways. Wang et al13 previously used scRNA-seq to compare gene expression in normal human hearts and in patients with end-stage HF, but their focus was on interactions among cardiomyocytes, fibroblasts, and endothelial cells. Thus, their current report is the first large-scale evaluation of sequencing data in adult mouse hearts during the development of compensatory hypertrophy and subsequent progression to decompensation and HF.9,12 Data interpretation was facilitated by computational analysis, which enabled the authors to describe the key events and stages that occur during the development of cardiac hypertrophy; and by conducting scRNA-seq (rather than conventional bulk sequencing analysis) at multiple time points, the authors were able to investigate how heterogeneity within each cell type changed as LV dysfunction progressed. Their results indicated that interactions between cardiomyocytes and fibroblasts declined during the initial stage of pressure overload; then, activated macrophages became the dominant cellular elements during the intermediate stage by promoting inflammation and cardiac dysfunction. The late stage of severe LV dysfunction was characterized by the upregulation of molecules in fibroblasts and endothelial cells that drive fibrotic remodeling and inflammation.
The high resolution of scRNA-seq can also lead to the discovery of rare cell populations that may not precisely match the current definitions of specific cell types. For example, scRNA-seq studies are increasingly reporting the expression of noncanonical makers in a small proportion of cardiomyocytes,3,12 and Ren et al reinforce these earlier observations by identifying populations of cardiomyocytes that express fibroblast or endothelial cell markers. Furthermore, because the total number of cardiomyocytes evaluated was larger in Ren et al than in many previous studies, the number that expressed noncanonical markers was also larger and perhaps more reliable, even though they remained rare. Noncannonical marker expression has also been evaluated through immunostaining, flow cytometry, and lineage tracing, often to confirm the purity of isolated cells, but studies of the functional consequences associated with atypical marker expression are scarce. These observations also suggest that cell types cannot be conclusively defined by the expression of just one (or even several) markers and, consequently, the markers used for genetic lineage tracing must be selected with great care.
TIMING MATTERS
In patients, the progression from hypertrophy to HF can occur over several years and, consequently, management must adapt to changes in disease pathophysiology. Thus, by defining stage-specific changes in gene expression and in the activity and quantity of cell populations (and subpopulations), the finding emerging from this study may provide guidance for the timing of potential treatment strategies. The authors identified one major event of macrophage activation during the intermediate stage of cardiac hypertrophy in mice and confirmed that patterns of gene expression in samples from normal, hypertrophic, and failing human hearts were consistent with those observed in their mouse model. However, analyses in human tissues were conducted through bulk RNA-seq, so the results cannot be attributed to individual cell types. Thus, whether a similar switch in macrophage subpopulations also occurs in hypertrophic and failing human hearts has yet to be confirmed.
The authors established a key role of macrophage activation in hypertrophic cardiac dysfunction by demonstrating that measures of LV ejection fraction and fractional shortening improved significantly when mice were treated with the anti-inflammatory drugs TD139, a galectin inhibitor, and arglabin, which inhibits the NLRP3 (nucleotide-binding domain leucine-rich repeat [NLR] and pyrin domain containing receptor 3) inflammasome, during the intermediate stage (ie, when macrophage activation occurs) but not during the initial stage. Similar improvements were observed after treatment with dapagliflozin during the intermediate stage, which is consistent with a report that dapagliflozin significantly reduces the risk of worsening HF or death from cardiovascular causes among patients with HF.15 Dapagliflozin inhibits SGLT2 (sodium-glucose transport protein 2) and is indicated for managing type 2 diabetes mellitus, but its cardiovascular benefits were observed in patients irrespective of the presence of diabetes mellitus. Collectively, these observations suggest that dapagliflozin may function not only in glycemic control, but also as an inhibitor of the macrophage switch.
LIMITATIONS OF scRNA-seq
Interpretation of findings emerging from scRNA-seq analysis needs to consider biases in cell-type selection that may derive from techniques used for sample preparation, such as enzymatic digestion, or by potential cell-size limitations associated with the chosen scRNA-seq platform. Another emerging technology, single-nucleus RNA-seq, minimizes these biases5,8 but misses transcriptional information in the cytoplasm, which may be crucial for investigations of certain cell types or processes, such as cell division. Sequencing must also be conducted in multiple individuals of each species to generate comprehensive databases for data-mining applications, and as the data sets generated through scRNA-seq (and other modern sequencing techniques) continue to grow in both size and complexity, more cost-effective, efficient, and robust computational approaches must be developed to facilitate interpretation. From an individual variability perspective, integration of scRNA-seq data generated from different subjects is also needed for better data mining. Furthermore, although the insights provided by computational analyses of scRNA-seq data can be novel and profound, and additional information can be generated through other single-cell omic investigations (eg, epigenomics, proteomics, and metabolomics), the results need to be rigorously verified by using more traditional experimental techniques, and better methods for incorporating the findings from scRNA-seq analysis into the basic biological sciences and clinical studies are needed.
NEXT FRONTIER
Although scRNA-seq is a powerful method for reconstructing cellular trajectories during development and disease progression, it does not preserve the spatial information of individual cells. Thus, whether the observed heterogeneity and dynamics of cell populations can be partially attributed to anatomical location, rather than solely to disease pathology, remains unclear. A recent study evaluated cell type–specific gene expression in different regions of the heart by integrating scRNA-seq with spatial transcriptomics and in situ targeted sequencing. This novel combination of emerging technologies enabled the authors to generate 3-dimensional maps of gene expression in embryonic human hearts at 3 different time points during the first trimester after conception.11
CONCLUSIONS
scRNA-seq enables researchers to generate rich sets of gene-expression data, and the study by Ren et al uses this technology to comprehensively assess the transcriptomes of both cardiomyocytes and noncardiomyocytes as the heart progresses from compensated hypertrophy to severe LV dysfunction. Their results also identified the previously unrecognized role of macrophage subtype activation in hypertrophied and dysfunctional hearts, and illustrate the unique insights that can be gained when scRNA-seq is conducted at multiple time points to investigate the dynamics of gene expression during the progression of LV dysfunction. As single-cell analyses expand to include other types of omic data and are applied in concert with approaches that preserve the data’s spatiotemporal features, our understanding of cardiovascular biology and disease will continue to become more complete, refined, and inclusive.
Sources of Funding
This work was supported by US National Institutes of Health grants RO1 HL 99507, HL114120, HL 131017, HL149137, and UO1 HL134764.
Footnotes
Disclosures
None.
REFERENCES
- 1.Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. 2018;15:387–407. doi: 10.1038/s41569-018-0007-y [DOI] [PubMed] [Google Scholar]
- 2.Frey N, Katus HA, Olson EN, Hill JA. Hypertrophy of the heart: a new therapeutic target? Circulation. 2004;109:1580–1589. doi: 10.1161/01.CIR.0000120390.68287.BB [DOI] [PubMed] [Google Scholar]
- 3.Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, et al. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26:1934–1950. e5. doi: 10.1016/j.celrep.2019.01.079 [DOI] [PubMed] [Google Scholar]
- 4.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, et al. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.See K, Tan WLW, Lim EH, Tiang Z, Lee LT, Li PYQ, Luu TDA, Ackers-Johnson M, Foo RS. Single cardiomyocyte nuclear transcriptomes reveal a lincRNA-regulated de-differentiation and cell cycle stress-response in vivo. Nat Commun. 2017;8:225. doi: 10.1038/s41467-017-00319-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li G, Xu A, Sim S, Priest JR, Tian X, Khan T, Quertermous T, Zhou B, Tsao PS, Quake SR, et al. Transcriptomic profiling maps anatomically patterned subpopulations among single embryonic cardiac cells. Dev Cell. 2016;39:491–507. doi: 10.1016/j.devcel.2016.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lescroart F, Wang X, Lin X, Swedlund B, Gargouri S, Sànchez-Dànes A, Moignard V, Dubois C, Paulissen C, Kinston S, et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science. 2018;359:1177–1181. doi: 10.1126/science.aao4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu P, Liu J, Zhao J, Wilkins BJ, Lupino K, Wu H, Pei L. Single-nucleus transcriptomic survey of cell diversity and functional maturation in postnatal mammalian hearts. Genes Dev. 2018;32:1344–1357. doi: 10.1101/gad.316802.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, Lacraz GPA, Huibers MMH, van Oudenaarden A, van Rooij E. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138:166–180. doi: 10.1161/CIRCULATIONAHA.117.030742 [DOI] [PubMed] [Google Scholar]
- 10.Farbehi N, Patrick R, Dorison A, Xaymardan M, Janbandhu V, Wystub-Lis K, Ho JWK, Nordon RE, Harvey RP. Single-cell expression profiling reveals dynamic flux of cardiac stromal, vascular and immune cells in health and injury. Elife. 2019;8:e43882. doi: 10.7554/eLife.43882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asp M, Giacomello S, Larsson L, Wu C, Fürth D, Qian X, Wärdell E, Custodio J, Reimegård J, Salmén F, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell. 2019;179:1647–1660. e19. doi: 10.1016/j.cell.2019.11.025 [DOI] [PubMed] [Google Scholar]
- 12.Skelly DA, Squiers GT, McLellan MA, Bolisetty MT, Robson P, Rosenthal NA, Pinto AR. Single-cell transcriptional profiling reveals cellular diversity and intercommunication in the mouse heart. Cell Rep. 2018;22:600–610. doi: 10.1016/j.celrep.2017.12.072 [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, Guo G, Wang Y, Chen X, Han L, et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22:108–119. doi: 10.1038/s41556-019-0446-7 [DOI] [PubMed] [Google Scholar]
- 14.Ren Z, Yu P, Li D, Li Z, Liao Y, Wang Y, Zhou B, Wang L. Single-cell reconstruction of progression trajectory reveals intervention principles in pathological cardiac hypertrophy. Circulation. 2020;141:1704–1719. doi: 10.1161/CIRCULATIONAHA.119.043053 [DOI] [PubMed] [Google Scholar]
- 15.McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al. ; DAPA-HF Trial Committees and Investigators. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. doi: 10.1056/NEJMoa1911303 [DOI] [PubMed] [Google Scholar]