

SUMMARY
HIV monoclonal antibodies for viral reservoir eradication strategies will likely need to recognize reactivated infected cells and potently drive Fc-mediated innate effector cell activity. We systematically characterize a library of 185 HIV-envelope-specific antibodies derived from 15 spontaneous HIV controllers (HCs) that selectively exhibit robust serum Fc functionality and compared them to broadly neutralizing antibodies (bNAbs) in clinical development. Within the 10 antibodies with the broadest cell-recognition capability, seven originated from HCs and three were bNAbs. V3-loop-targeting antibodies are enriched among the top cell binders, suggesting the V3-loop may be selectively exposed and accessible on the cell surface. Fc functionality is more variable across antibodies, which is likely influenced by distinct binding topology and corresponding Fc accessibility, highlighting not only the importance of target-cell recognition but also the need to optimize for Fc-mediated elimination. Ultimately, our results demonstrate that this comprehensive selection process can identify monoclonal antibodies poised to eliminate infected cells.
In brief
Rossignol et al. characterize 185 HIV-envelope-specific antibodies derived from spontaneous HIV controllers, downselecting antibodies based on their ability to broadly recognize infected cells and potently drive Fc-mediated innate effector cell activity. This comprehensive selection process can identify monoclonal antibodies poised to eliminate infected cells for viral reservoir eradication strategies.
Graphical Abstract
INTRODUCTION
Monoclonal antibodies directed against the HIV envelope (Env) glycoprotein have been extensively characterized over the past decades. Numerous antibodies with exquisite neutralization potency and breadth against large panels of cross-clade viruses have been identified from HIV-infected individuals with chronic progressive infection but also from individuals that control HIV in the absence of anti-retroviral therapy (Freund et al., 2017; Medina-Ramírez et al., 2011; Walker et al., 2011). These antibodies target multiple epitopes on the surface of Env, and several monoclonal antibodies are in clinical development for treatment and prevention, including the CD4 binding site antibodies VRC01, 3BNC117, and VRC07–523LS; the V3-loop-directed antibodies PGT121 and 10–1074; the V1/V2 loop antibodies PGDM1400 and CAP256-VRC26.25; and the membrane-proximal region (MPER)-specific antibody 10E8 (Doria-Rose et al., 2014; Huang et al., 2012; Mouquet et al., 2012; Rudicell et al., 2014; Scheid et al., 2011; Sok et al., 2014; Walker et al., 2011; Wu et al., 2010). These antibodies, selected based on their ability to effectively bind and neutralize virions, have shown promising results in phase 1 and 2 studies by reducing plasma viremia and by delaying viral rebound during anti-retroviral therapy treatment interruptions (Bar et al., 2016; Mendoza et al., 2018; Scheid et al., 2016). However, although the anti-virion effect was clear, at least when antibodies were present at sufficiently therapeutic levels and no viral escape occurred, an effect on the size of the HIV cellular reservoir was less apparent. The elimination, or at least reduction of the reservoir, however, is the ultimate goal of HIV eradication strategies. Strategies to improve the therapeutic activity of monoclonal antibodies has included efforts to increase reservoir visibility, using immune modulators like Toll-like receptor 7 (TLR7) agonists (Borducchi et al., 2018). However, optimizing the therapeutic activity of monoclonals themselves is likely to be key for the ultimate development of an antibody-based eradication strategy.
Although the ability of a broadly neutralizing monoclonal antibody to drive neutralization is critically determined by its ability to bind tightly to highly conserved regions of the viral envelope (Gautam et al., 2016, 2018; Hessell et al., 2009; Julg et al., 2017; Rudicell et al., 2014; Xu et al., 2017), it is unclear whether this property alone is sufficient to drive cellular recognition and, more importantly, elimination. Instead, HIV reservoir cell elimination depends on both Fab- and Fc-mediated functions (Bournazos et al., 2014; Lu et al., 2016a). Upon recognition of an infected cell, antibodies must recruit innate effector cells such as natural killer cells, monocytes, and neutrophils by Fc receptors (FcγRs) expressed on these cells to drive elimination of the antibody opsonized target. Although previous studies have demonstrated broadly neutralizing antibody (bNAb) recognition and elimination of infected cells (e.g., Bruel et al., 2016, 2017; Mujib et al., 2017; Ren et al., 2018), these antibodies target neutralizing epitopes on the Env trimer that may not reflect the entirety of the epitope landscape on the infected cell surface (Moore et al., 2006). Viral assembly and budding on the infected cell surface is a dynamic process influenced by multiple viral and host components (von Bredow et al., 2015; Buttler et al., 2018; Chojnacki et al., 2017; Pezeshkian et al., 2019; Prévost et al., 2018; Richard et al., 2018), representing a potentially unique antigenic target landscape compared with that targeted by our current neutralizing antibody armamentarium. Furthermore, the ability to mediate Fc-effector functions depends on the position of the Fc in the immune complex of antibodies bound to infected cells and the resulting accessibility by FcγRs on the effector cells. Conversely, non- or poorly neutralizing antibodies, particularly those targeting the non-neutralizing GP41 immunodominant domain or CD4-induced epitopes on V1-V2, have been explored in the context of antibody-dependent cellular cytotoxicity (ADCC) (Anand et al., 2019; Davis-Gardner et al., 2017; Ferrari et al., 2011; Gohain et al., 2016; Horwitz et al., 2017; Mayr et al., 2017; Santra et al., 2015; Tyler et al., 1990; Veillette et al., 2015), demonstrating that these antibodies are able to mediate robust ADCC activity.
Interestingly, robust ADCC (Lambotte et al., 2009, 2013; Madhavi et al., 2014, 2017) and overall superior (poly) Fc functionality (Ackerman et al., 2013a, 2016) have been observed consistently in HIV elite controllers, individuals who spontaneously control viremia in the absence of anti-retroviral therapy. Although it is unclear how much these functional antibodies directly contribute to viral control, the persistent maintenance of these responses, even in the face of undetectable viremia, suggests that these highly functional humoral immune responses may be persistently stimulated and involved in surveillance of infected cells (Ackerman et al., 2016). Thus, to begin to explore whether these individuals may harbor antibodies able to selectively target and destroy infected cells, 15 HIV controllers (HCs), with robust plasma antibody Fc-effector profiles that also possessed some neutralizing antibody breadth (to capture extensively somatically hypermutated antibodies), were used as a source of potentially novel antibodies. A library of 185 monoclonal antibodies were downselected from a total of ~11,000 single B-cell receptor (BCR) sequences to broadly explore affinity-matured antibodies across a broad range of clonal families. As an alternative to current monoclonal downselection strategies, antibodies were further downselected based on their ability to recognize infected cells, across a panel of lab and clinical strains, as well as to drive robust in vitro ADCC, antibody-dependent cellular phagocytosis (ADCP), and antibody-dependent neutrophil phagocytosis (ADNP), representing a class of antibodies poised to broadly recognize and drive the destruction of infected/reactivated cells.
RESULTS
Selection of HIV monoclonal antibodies from HCs
Mounting evidence points to an enrichment of ADCC-inducing, polyfunctional antibodies, among spontaneous controllers of HIV (Ackerman et al., 2016). Although it is uncertain whether these antibodies directly contribute to virological control in vivo, these individuals may potentially represent a unique source of antibodies poised to detect and eliminate infected cells that can be exploited for therapeutic purposes. With the goal of creating a library of HC monoclonal antibodies able to recognize infected cells and drive highly effective Fc-polyfunctional anti-viral functions, we profiled the Fc-effector functional profiles of plasma across 129 spontaneous HCs. We selected HCs that exhibited high levels of Fc functions like antibody-mediated natural killer (NK) cell degranulation (CD107a, interferon gamma [IFNg], and MIP-1b expression) or antibody-mediated complement deposition (Figure 1A; ADCD gating in Figure S1). To create a diverse set of antibodies, both neutralizing and non-neutralizing, we chose HCs with different extents of neutralization breadth (e.g., high, medium, and low; Figure 1B). For the 15 selected individuals (clinical characteristics in Table S1), single-antigen-specific CD19+CD20+ immunoglobulin A (IgA−)IgM−IgG+ B cells had previously been sorted using a pool of fluorescently labeled recombinant JR-CSF gp120, JR-FL gp140, 92BR020 gp120, clade A BG505 SOSIP, and clade C IAVI C22 gp120 probes, which was aimed at broadly capturing HIV-envelope-specific binding antibodies across various targets that could potentially decorate the infected cell surface. Paired IgG heavy and light chains had been sequenced and analyzed for somatic hypermutation (SHM) by calculating mutation frequencies relative to the putative germline sequence. This dataset and the sequence analysis approach have been partially reported by Cizmeci et al. (2020) (Kepler, 2013). As high levels of SHM are suggestive of advanced affinity maturation, we hypothesized that highly mutated antibodies would demonstrate superior binding activity to the cell-surface-expressed envelope. Ultimately, 185 sequences were selected based on high rates of SHM, with a rate of 19.25% ± 6.0% in the heavy chain and 12.5% ± 4.7% in the light chain (median ± standard deviation, Figure S2) and produced in an IgG1 backbone.
Figure 1. Selection of HIV controllers for isolating monoclonal antibodies.
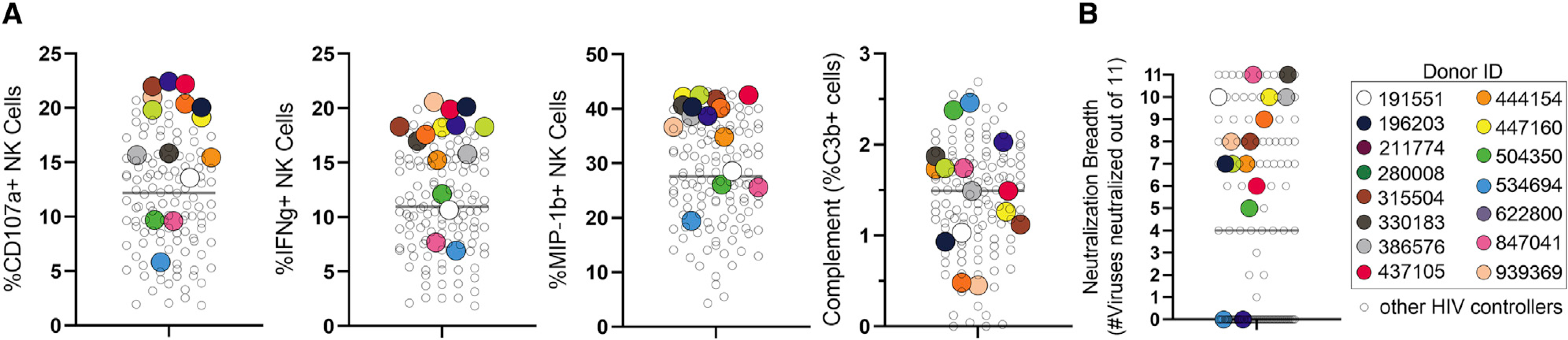
(A and B) Serum from a total of 129 HIV controllers was profiled for (A) Fc functional activity and (B) neutralization. HIV controllers chosen for B cell isolation and sorting are depicted in uniquely colored dots. The horizontal bar represents the mean.
(A) Complement and NK cell degranulation were measured.
(B) Serum neutralization breadth of a panel of 11 clade B viruses is displayed as number of viruses neutralized (at a dilution of >1:100). The viruses tested (tier 2) are as follows: QH0692.42, SC422661.8, PVO.4, TRO.11, AC10.0.29, RHPA4259.7, THRO4156.18, REJO4541.67, TRJO4551.58, WITO4160.33, and CAAN5342.A2
Recognition of infected cells by HC antibodies
We first examined the ability of the 185 HC antibodies to recognize reactivated infected cells by measuring antibody labeling of Env transcripts on the surface of the latency model cell lines ACH-2 (strain LAI) and J89-GFP (strain 89.6) in a high-throughput flow-cytometry-based assay. Although low levels of Env were detectable on both cell lines before stimulation, robust upregulation of p24+ staining and HIV-envelope expression using various Env-specific monoclonal antibodies as detectors was quantifiable upon stimulation with phorbol myristate acetate (PMA) plus ionomycin for 18 h (Figure S3). In this system, HC antibodies recognized ACH-2 cells more effectively than J89-GFP cells, with cell binding ranging 0%–85.4% and 0%–68.6%, respectively. Furthermore, antibody binding correlated across both latency cell lines and respective HIV strains LAI and 89.6 (Spearman two-tailed r = 0.438, p < 0.0001). When compared to a group of nine bNAbs that are under clinical development (Figure S4A), we observed a similar heterogeneity of ACH-2 and J89 cell-recognition magnitude (ranging 0.1%–83.5% and 5.2%–44.0%, respectively). Nevertheless, 31 and 30 HC antibodies recognized infected cells at rates equal to or exceeding the bNAb median recognition rate for both ACH-2 and J89 cells, respectively (Figure 2, top; Figure S4A), which, therefore, identified a set of monoclonals with promising cell binding potential.
Figure 2. Characterization of infected-cell binding and virus neutralization breadth of the HIV controller antibody library.

(A) Heatmap displays recognition of infected or reactivated latent (p24+) cells by antibodies. Binding scores are normalized by row (virus-infected cell). Antibodies are arranged by their overall average cellular recognition from high to low (left to right). Donors are identified by color code.
(B) Recombinant Env binding. (Top) Summary of binding to gp70 (V1-V2 construct), 13 in total. Black indicates antibodies that bound the majority (>10/13) of v1-v2 constructs. (Bottom) Antibody binding to recombinant gp120 or gp140 constructs representing multiple clades is colored by MFI. Binding scores are 3-fold background subtracted and normalized by row (gp120/140 variant). Normalization in (A) and (B) was performed by Z scoring the binding across each virus-infected cell or antigen and displaying the value on a purple-white-green heatmap.
(C) Antibody neutralization is depicted as the maximum percentage of inhibition (MPI) at 25 μg/ml. Antibodies that reached 50% inhibition or greater are indicated in dark purple, and inhibitory values between 0 (white) and 50 (purple) are shown in lighter shades.
The “average” row in each subsection summarizes the average performance, among the normalized data in (A) cell binding and (B) recombinant env binding, and the average MPI (C).
Given the limited viral diversity in these latency model cell lines, we next tested the recognition of productively infected cells by using primary CD4+ T cells or CEM-NKr-CCR5 cell lines (CEMs); the latter was used to reduce donor to donor variability (Figure S5). As the HCs were United States based and likely infected with clade B viruses, we selected several HIV clade B strains including X4 tropic NL4–3; the R5 tropic JR-CSF, and the transmitted/founder (T/F) strains WITO, CH058, CH077, and THRO (Salazar-Gonzalez et al., 2009). In all experiments, we focused on antibody recognition of infected p24+ cells with downregulated CD4 receptors (p24+CD4−) to reduce the confounder of measuring antibodies bound to CD4-receptor-virion complexes instead of the surface envelope. Seventeen HC antibodies were able to recognize over one-half of JR-CSF-infected cells, and 42 antibodies were able to recognize over one-half of NL4–3-infected cells. Overall, HC antibody recognition correlated strongly across both of these tier 2 and tier 1 viruses (Spearman two-tailed r = 0.782, p < 0.0001). Consistent with previous reports, the tested bNAbs also showed a wide variability in binding capacity (Bruel et al., 2016; Figure S4). Similarly, for the T/F-virus-infected cells, we observed 26, 25, 25, and 10 HC antibodies recognizing 25% or more of infected cells for CH058, CH077, THRO, and WITO, respectively. Top-performing HC antibodies therefore recognized clade-B-infected cells at frequencies that were comparable to, or exceeded that of, the nine tested bNAbs (Figure S4).
Recombinant Env binding breadth of the HC antibody library
To further determine binding breadth, the HC antibody library was tested against recombinant Env proteins including GP70(v1-v2) constructs and a diversity panel of gp120s and gp140s representing clades A, AE, B, BC, and C. Recognition of gp120 constructs was widespread, and 38.5% of the HC antibodies recognized all 26 cross-clade constructs at an mean fluorescent intensity (MFI) threshold of 3-fold over background, whereas 84.4% bound at least one-half of the constructs. Clade B antigens were more frequently recognized by the HC antibodies (89%), followed by clade C (82%), clade A (80%), and CRF01_AE (67%). Nine antibodies, however, did not bind any of these constructs nor did they bind infected cells, potentially suggesting that the antibodies were not HIV specific. A total of 14.5% of the HC antibody library bound GP70 constructs well, which was defined as binding greater than 10 out of 13 tested constructs, indicating that these antibodies were specific for the V1-V2 epitope (Figure 2B, V1-V2 binding; Table S2). Thus collectively, these data support reported differences in gp120 presentation and epitope availability between recombinant protein and the surface of infected cells (Bruel et al., 2017; Lewis et al., 2019).
Neutralization capacity of the HC antibody library
To further profile the functional quality of the EC antibodies, the neutralization capacity of the antibody library was tested against NL4–3 (tier 1) and JR-CSF (tier 2) as well as against a diverse panel of four tier 2 pseudoviruses (6101.1 [clade B], CH117.4 [clade BC], 001428–2.42 [Clade C], and P0402_C2_11 [clade G]) (Figure 2C). For consistency with previous cellular recognition studies, antibodies were tested at a maximum concentration of 25 μg/ml. Overall, neutralization activity and neutralization breadth were low in the HC antibody library. As expected, tier 1 (NL4–3) neutralization was most common, with 36.2% of HC antibodies (67/185 antibodies) capable of 50% neutralization, followed by JR-CSF at 20.5% (38/185 antibodies). Only a minority of HC antibodies achieved 50% neutralization of the pseudovirus panel (5.4% for 6101.1 [B], 0% for CH117.4 [BC], 4.3% for 001428–2.42 [C], and 4.3% for P0402_C2_11 [G]). Of these antibodies, the geometric mean 50% inhibitory concentration (IC50) was 1.84, 0.32, and 5.05 μg/ml for 6101.1 (B), 001428–2.42 (C), and P0402_C2_11 (G), respectively. Matched analysis of neutralization of JR-CSF and NL4–3 moderately correlated with the recognition of NL4–3- and JR-CSF-infected cells within our HC antibody library (Figures 3A and 3B; Spearman two-tailed r = 0.35, p = < 0.001 for NL4–3; and r = 0.42, p < 0.001 for JR-CSF), as previously reported (Ren et al., 2018). Interestingly, multiple antibodies with low neutralization activity demonstrated robust cell recognition, suggesting that antibodies may evolve the ability to recognize infected cells in the presence and absence of neutralization capacity (Figure 3).
Figure 3. Correlations of infected-cell recognition and neutralization activity for the HC antibodies (correlates of data shown in Figure 2).
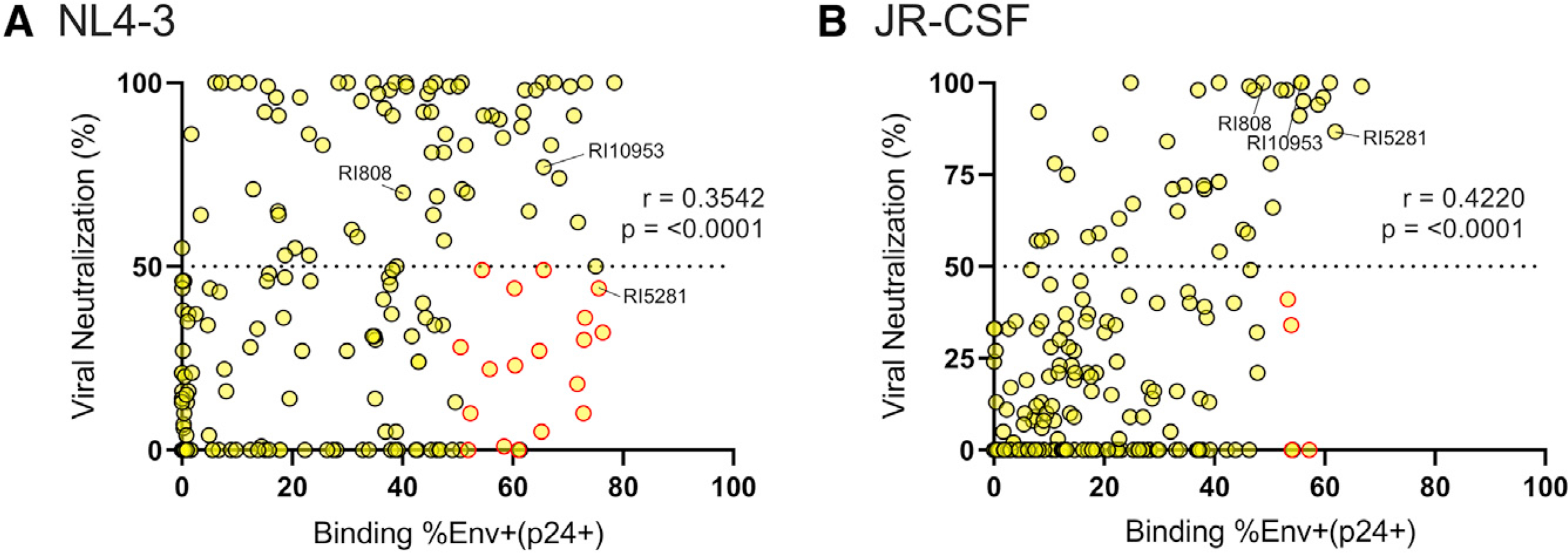
(A and B) The matched capability of the HC library to neutralize the virus as well as recognize infected primary cells was determined for (A) NL4–3 and (B) JR-CSF. Viral neutralization was measured in triplicate by a TZM-BL neutralization assay, and the average percentage of inhibition at 25 μg/ml is displayed. Antibody recognition of HIV-infected (p24+CD4−) cells was measured in duplicate across infected PBMCs from two donors, and the average is depicted. Antibodies that neutralized less than 50% of virus but bound more than 50% of infected cells are highlighted in red. Spearman two-tailed r values are displayed.
Hierarchy in recognition of cross-clade virally infected cells
The 45 HC antibodies with the highest infected-cell recognition rates and the broadest epitope binding breadth were selected for further investigation. We tested this downselected HC antibody library against primary peripheral blood mononuclear cells (PBMCs) infected with a more diverse group of HIV clades, i.e., B, C, and AE, and also included the nine bNAbs of various epitope specificities, as described before. To assay antibody binding to clinically relevant viruses, we first infected PBMCs with heterogenous primary viruses grown from the PBMCs of six antiretroviral therapy (ART)-treated HIV-infected individuals (described in Yu et al., 2019), and to explore recognition breadth within non-B-clade viruses, we also infected PBMCs with clade C and clade CRF01_AE strains. Of the top 10 antibodies with the broadest infected-cell recognition across all viruses, 7 antibodies were derived from the HCs (Figure 4). Specifically, the antibody RI5281 demonstrated the most robust binding breadth; however, the V3-specific bNAbs PGT121 and 10–1074, as well as the CD4bs 3BNC117 also exhibited broad infected-cell binding abilities. As expected, clade-dependent differences in cell recognition were observed across all tested antibodies, which is consistent with reports of clade-dependent neutralization activity; for example, CRF01_AE-infected cells were not recognized well by the tested V3-specific antibodies. Some of the broadest neutralizers, however, like the CD4bs antibodies N6 and VRC07–523LS recognized infected cells less effectively across the tested viral strains, potentially suggesting that specific targeted epitopes may be less available on the infected cell surface. Given the clear hierarchy in recognizing cross-clade virally infected cells, we selected the top three HC-derived antibodies, namely, RI5281, RI10953, and RI808, for further evaluation.
Figure 4. Relative ability of monoclonal antibodies to recognize HIV-infected cells.
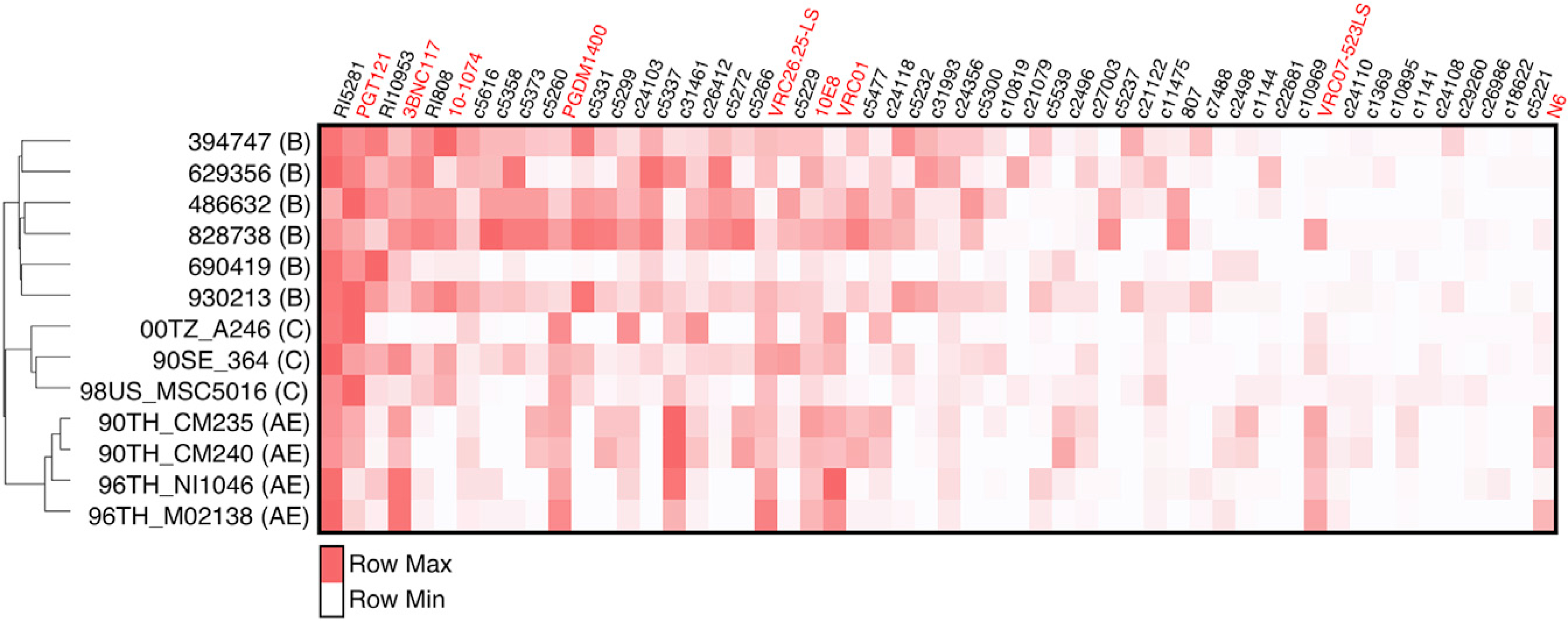
PBMCs were infected with HIV strains, and the relative ability of the top 45 HC antibodies and 9 bNAbs to recognize infected cells (p24+CD4−) is displayed. Antibodies are arranged from high to low (left to right) by average overall recognition rates of p24+CD4− among all viruses tested (including these viruses and Figures 2 and S2) cells. Antibody recognition is normalized per virus, with the brightest red highlighting the top mAb. The dendrogram displays the gp120 sequence similarity between viruses. The average recognition of duplicate values among PBMCs from two donors is depicted.
Epitope mapping of top infected-cell recognizers
To further explore the epitope region targeted by the broadest HC antibodies, negative-stain single-particle electron microscopy (nsEM) was performed on Fabs from RI5281, RI10953, and RI808 complexed with Env trimers. Fab binding was screened initially against 11 SOSIP constructs by biolayer interferometry (BLI) (Figure S6). The three Fabs showed the highest affinity for AMC018 SOSIPv4.2 (clade B, tier 2), which was chosen for further structural analysis. 2D Class averages of the Fab-AMC018 SOSIPv4.2 complexes (Figure 5A) show RI808 binding to the base of the V3 loop with high occupancy and enables a 3D reconstruction (Figure 5B). RI10953 and RI5281 exhibited lower occupancy with a maximum of one fab/trimer (Figure 5A) and heterologous binding that was unsuitable for 3D reconstruction and epitope determination. We next performed a competition ELISA to test cross-competition for gp120 binding across the same three monoclonals as well as well-defined bNAbs (Figure 5C). As expected, HC antibody RI808 binding was strongly inhibited by the V3-directed bNAbs PGT128, 447–52D, and 10–1074. Binding of HC antibodies RI5281 and RI10953 was moderately blocked by V3-directed antibody 2G12, as well as each other, but no strong inhibition was observed among other tested antibodies. Interestingly, RI10953 binding was inhibited by the CD4bs antibodies VRC01 and VRC07 as well as CD4-mimetic CD4Ig, but not 3BNC117, suggesting an epitope proximal or influenced by the CD4 binding site.
Figure 5. Epitope determination of top HC antibodies.
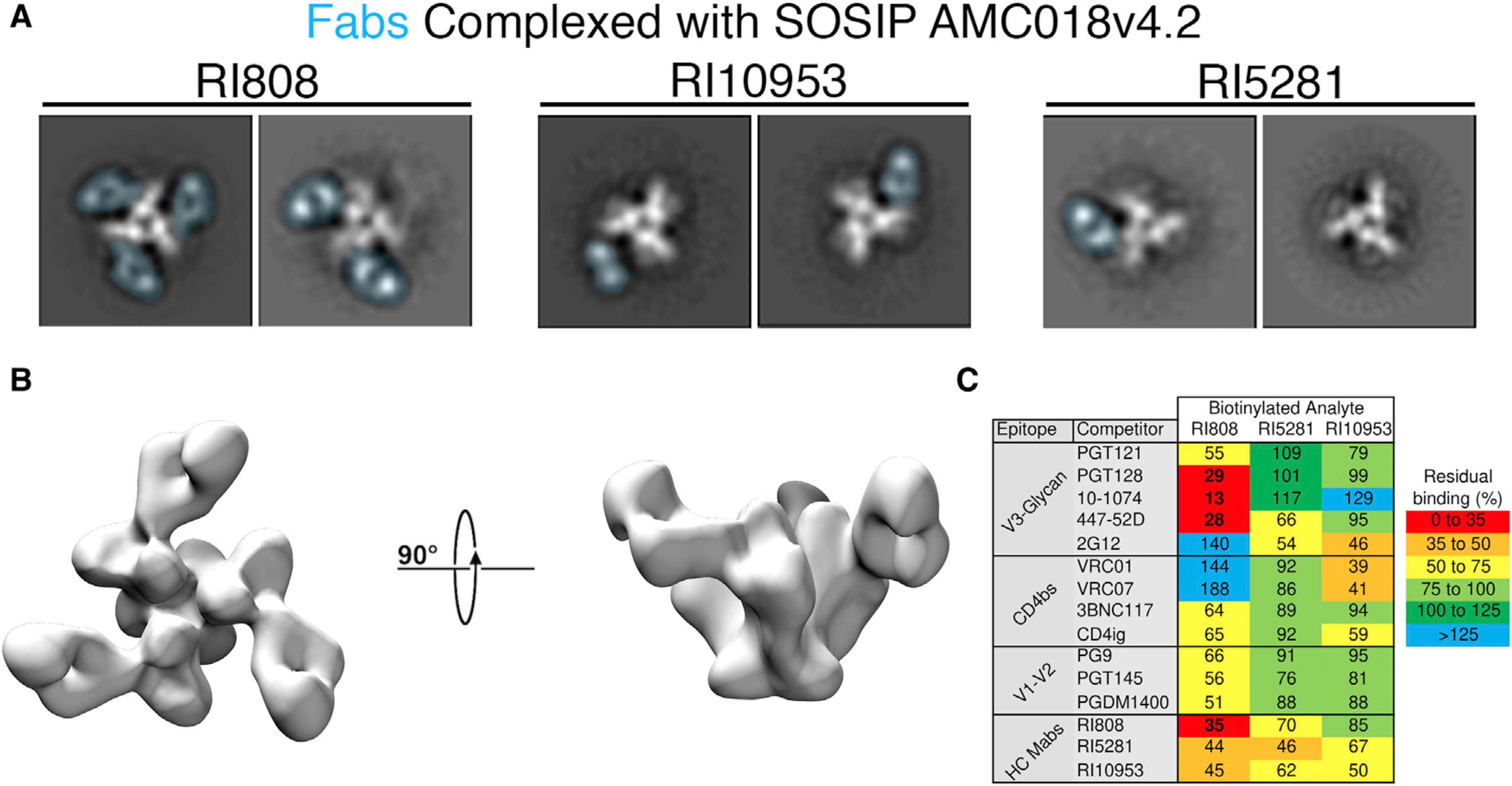
(A) Negative-stain single-particle analysis of top HC antibodies complexed with SOSIP trimers. 2D Class averages of AMC018 SOSIPv4.2 complexed with the indicated Fab. The density associated with the Fab is overlaid in blue. The structural heterogeneity and low occupancy observed in RI5281 and RI10953 were observed in all 2D class averages.
(B) A 3D negative-stain reconstruction of RI808 complexed with AMC018 SOSIPv4.2.
(C) Competition ELISAs were performed using JR-CSF gp120. The unlabeled competitor is allowed to bind (at 10 μg/ml), followed by the biotinylated antibody being analyzed (at 1 μg/ml). Values indicate %blocked/non-blocked, and numbers indicate the average of triplicate measurements from a representative experiment.
HC antibodies drive ADCC
Beyond their ability to bind, we next investigated the effectiveness of HC antibodies to drive antibody effector function. To explore the capacity of our HC monoclonal antibodies (mAbs) to eliminate infected cells, we used a high-throughput assay to measure the elimination of JR-CSF- or NL4–3-infected (p24+CD4−) CEM-NKr-CCR5 cells by isolated primary NK cells (Bruel et al., 2016). JR-CSF- or NL4–3-infected target cells were co-cultured with or without NK cells and in the presence or absence of the respective antibodies. To maintain consistency with the cellular recognition studies, antibodies were tested for ADCC at 25 μg/ml, which is consistent with maximal or near-maximal ADCC levels in other reports (von Bredow et al., 2016; Bruel et al., 2016) and in our own titration experiments (data not shown). It should be noted that these assays were performed at a relatively low effector-to-target ratio (2:1), which allowed us to test all these conditions simultaneously by using the same donors. As previously reported, tier 1 NL4–3-infected cells were more susceptible to ADCC than tier 2 JR-CSF (median infected-cell elimination of 18.6% for NL4–3 and 13.4% for JR-CSF) (von Bredow et al., 2016; Bruel et al., 2016). ADCC activity correlated with the ability of antibodies to bind infected cells, as determined in wells that did not contain effector cells (two-tailed Spearman of r = 0.75, p < 0.001 for JR-CSF-infected cells; and r = 0.79, p < 0.001 for NL4–3-infected cells) (Figure 6). ADCC scores correlated modestly across viruses as well (two-tailed Spearman r = 0.278, p = 0.035), indicating that the antibodies are able to mediate ADCC across both viruses. The top five antibodies with the highest sum total ADCC activity against both tested viruses were HC mAb RI808, followed by bNAbs VRC07–523LS and 3BNC117, and HC mAbs c5337 and c807. Overall, most (97%) of HC mAbs and bNAbs tested were able to drive ADCC activity over background levels. Of the top 10 broadest cell binders in Figure 4, the bNAbs PGT121 and 10–1074, however, only mediated ADCC activity that was in the lower half of all tested antibodies, consistent with previous data showing low ADCC despite high recognition rates (Bruel et al., 2016). Thus, our results suggest that variations in ADCC among the tested mAbs could be caused by more or less favorable binding stoichiometries and resulting Fc topology for FcγR3A-mediated NK cell activation.
Figure 6. Effector function and binding across HC antibodies and bNAbs.
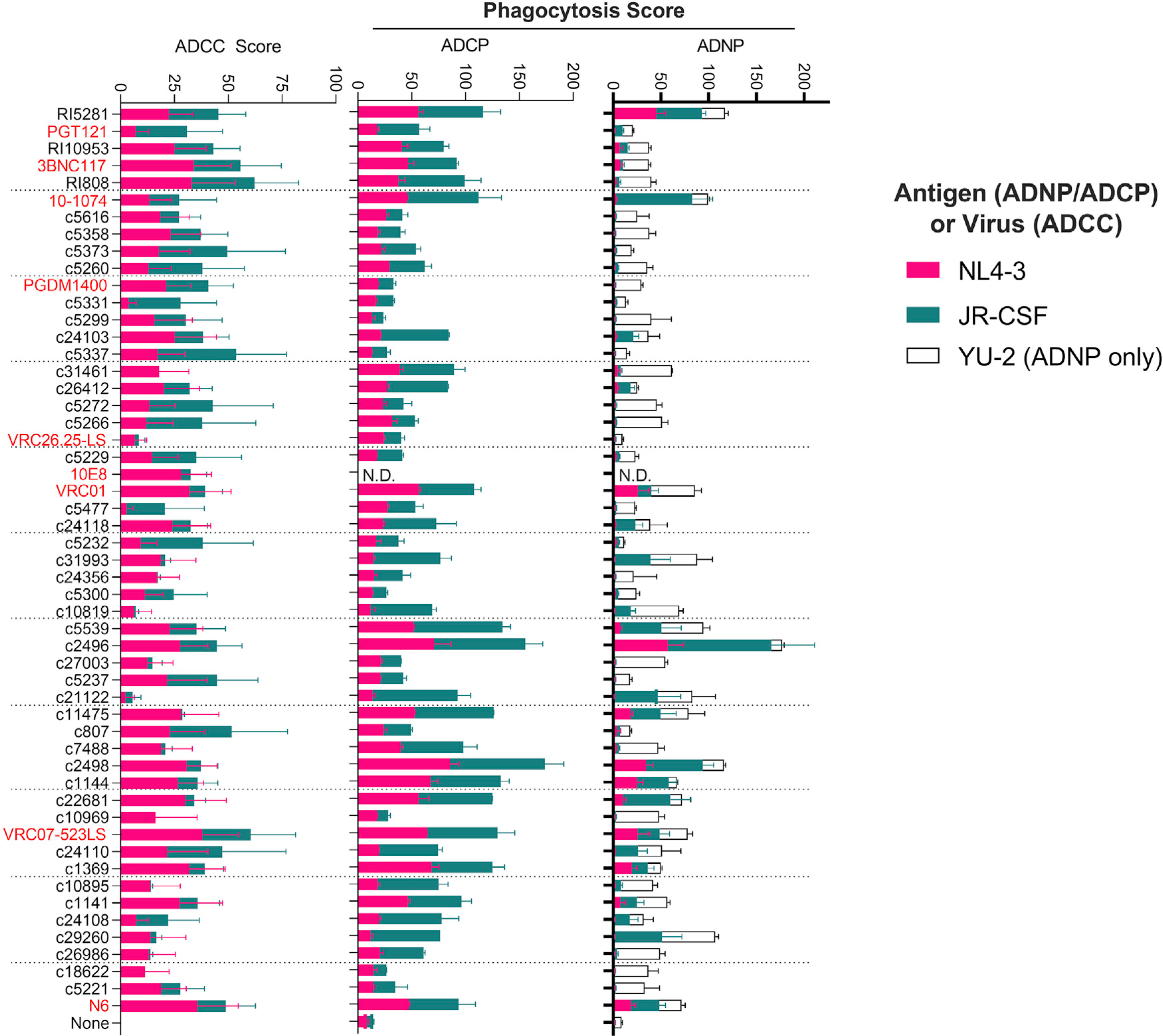
Fc function was measured for antibodies against JR-CSF- and NL4–3-infected cells (ADCC) or gp120-coated particles (ADNP and ADCP). ADCC scores are the disappearance of infected (p24+) cells relative to no-antibody controls. ADNP and ADCP scores are arbitrary units that reflect the relative ability of neutrophils (ADNP) and the pro-monocytic cell line THP-1 (ADCP) to promote phagocytosis. Antibodies are ranked from top to bottom by their ability to recognize infected cell broadly (high to low, respectively, as in Figure 4). Functional data for the MPER-binding 10E8 antibody is limited to ADCC, as beads coated with gp120 were used for the ADCP and ADNP assays. Error bars reflect standard deviation from duplicate values obtained against the indicated antigen or virus, using effector cells from two donors (ADNP and ADCC).
HC antibodies mediate antibody-dependent phagocytosis
Next, we explored the ability of the HC antibodies to induce phagocytosis of JR-CSF- or NL4–3 gp120-coated beads by primary neutrophils or by the monocyte cell line THP-1. ADNP was most efficiently mediated by two related HC antibodies sorted from the same donor (c2496 and c2498) as well as c22681 and RI5281, but also by the bNAbs 10–1074 (V3) and VRC07–523LS (CD4bs) (Figure 6). Similarly, ADCP by monocyte-like THP1 cells was most effectively driven by the same top ADNP performers c2496 and c2498, as well as other HC antibodies, i.e., c5539, c1144, c11475, and the bNAb VRC07–523LS. Antibody performance across phagocytic assays (ADNP and ADCP) was strongly correlated (Spearman two-tailed r = 0.86, p < 0.001 for NL4–3; and r = 0.80 p < 0.001 for JR-CSF), likely related to the dominant role of FcγR2 in driving phagocytosis across both cell types. Conversely, no correlation was observed across phagocytic and ADCC assays (r = 0.08–0.2), although several antibodies performed well at both phagocytosis and ADCC (Figure 6). Interestingly, when grouping the antibodies based on their original IgG isotype (e.g., IgG1, IgG2, and IgG3), antibodies that were native IgG3s tracked with higher ADCP and ADNP function than those with native IgG1s (Figure S7). Indeed, native IgG3s have been shown to mediate higher levels of ADCP than the same antibody in an IgG1 backbone (Tay et al., 2016); however, here, the HC antibodies are expressed in an identical IgG1 backbone. These data therefore suggest that the epitopes that are targeted by native IgG3 antibodies allow binding conformations that facilitate Fc-mediated phagocytic activity. Thus, binding properties related to different targets (infected cells versus gp120-coated beads) or related to differential Fc orientations required for binding to distinct Fc receptors (FcγR3 versus FcγR2) may therefore contribute to unique functional properties.
HC antibodies exhibit polyfunctional profiles
Ultimately, given the obstacles to identify and eradicate latently HIV-infected cells, the ability to recruit as many Fc functions as possible may provide a highly potent antibody with the capacity to leverage multiple arms of the innate immune system. This might be beneficial to successfully eliminate latently infected cells across different tissue compartments in which the presence and/or or access of different innate immune cells may vary. Thus, we next assessed the polyfunctional profile of each antibody by taking the sum of the average functional assay scores against JR-CSF and NL4–3. Out of the 54 HC antibodies and bNAbs tested, the top 10% polyfunctional antibodies included the HC antibodies c2496, c2498, RI5281, and c1144, but also included a known bNAb (VRC07–523LS) (Figure 6). Although the HC antibodies c2496, c2498, and c1144 showed enhanced Fc functionality relative to other tested antibodies on the selected antigens, they showed a limited ability to recognize a breadth of infected cells, binding an average of only 13.3%, 11.6%, and 9.8% of cells across the tested viruses. Although the diminished breadth potentially limits their applicability for reservoir-targeting strategies, these data suggest that the high functionality is likely the result of binding properties, i.e., to a non-conserved epitope that positions these antibodies for enhanced Fc functionality induction, relative to other epitopes.
Fc glycosylation does not explain superior polyfunctionality in HC monoclonals
The N-glycan composition of the antibody Fc domain can modulate antibody effector functions by affecting the ability of the Fc to bind various FcγRs (Li et al., 2017). We therefore analyzed Fc glycosylation by capillary electrophoresis to explore whether variations could explain differences in polyfunctionality on a selected subset of HC and bNAbs (13 and 5, respectively). Although the HC antibodies were produced on an identical IgG1 backbone in 293F cells, measurements revealed similar, although not identical, Fc glycans (Table S3). The selected bNAbs, which originate from 293F cells from multiple sources, also showed some heterogeneity; however, the overall glycosylation pattern was comparable to the HC antibodies (Table S3, HC mean compared to bNAb mean). Principally, both HC and bNAbs harbored glycans with abundant G0F at 68.91% and 72.40%, respectively, followed by G1F at 15.52 and 18.66%, respectively. Afucosylation, which has been associated with increased ADCC activity (Houde et al., 2010; Saunders, 2019; Thomann et al., 2016), was rare in both HC antibodies and bNAbs, with average levels of 1.13% and 0%, respectively. In general, no association between glycan pattern and specific Fc functions were observed, except for ADCP that tracked with the presence of bisecting glycans in the examined antibodies (two-tailed Spearman r = 0.54, p = 0.022). Thus, these data suggest that the differences in Fc functionality observed across HC antibodies and bNAbs are not related to simple differences in Fc glycosylation but rather Fc/FcγR engagement topology and antibody:antigen stoichiometric considerations.
HC antibodies are able to eliminate primary infected lymphocytes
The ability to kill primary infected cells by recruiting innate effector cells represents the ultimate therapeutic goal of a potent therapeutic antibody. Thus, we characterized the ability of the top 3 HC antibodies RI5281, RI10953, and RI808 and the top 3 bNAbs PGT121, 10–1074, and 3BNC117 to mediate elimination of NL4–3-infected primary CD4+ T cells by autologous effector PBMCs from 6 donors. NL4–3 was chosen, as it was recognized by all tested antibodies (Figure 2). Antibody-mediated clearance of infected cells was most effectively mediated by RI808, 3BNC117, and RI10953, which induced the elimination of 38.2%, 32.8%, and 21.4% of p24+CD4− cells, respectively (Figure 7A). Overall, the average antibody recognition and ADCC activity strongly correlated across the tested antibodies and dilutions (r = 0.66, p = 0.0004) (Figure 7B). Furthermore, ADCC performance tended to correlate between assays using primary target cells and the CEM reported above (in Figure 4; Figure 7C) (Spearman r = 0.82, p = 0.058). These results confirm that infected-cell recognition is critical to elimination of infected cells; however, these results also indicate that recognition does not always ensure efficient (>15%) elimination of infected primary cells, as was observed for PGT121 that strongly recognized the targets (72% of p24+CD4−) but only lead to a 14.2% reduction of infected cells. Thus, a more complex relationship between target-cell recognition, recruitment of innate immune effectors, and FcγRs interactions are likely critical to fully drive cellular cytotoxicity and suggests that cell surface binding is key but that additional Fc variables like the antibody binding topology and Fc accessibility may add essential information for the ultimate design of highly effective eradication-specific antibodies.
Figure 7. Recognition and elimination of infected primary lymphocytes by autologous effector cells.
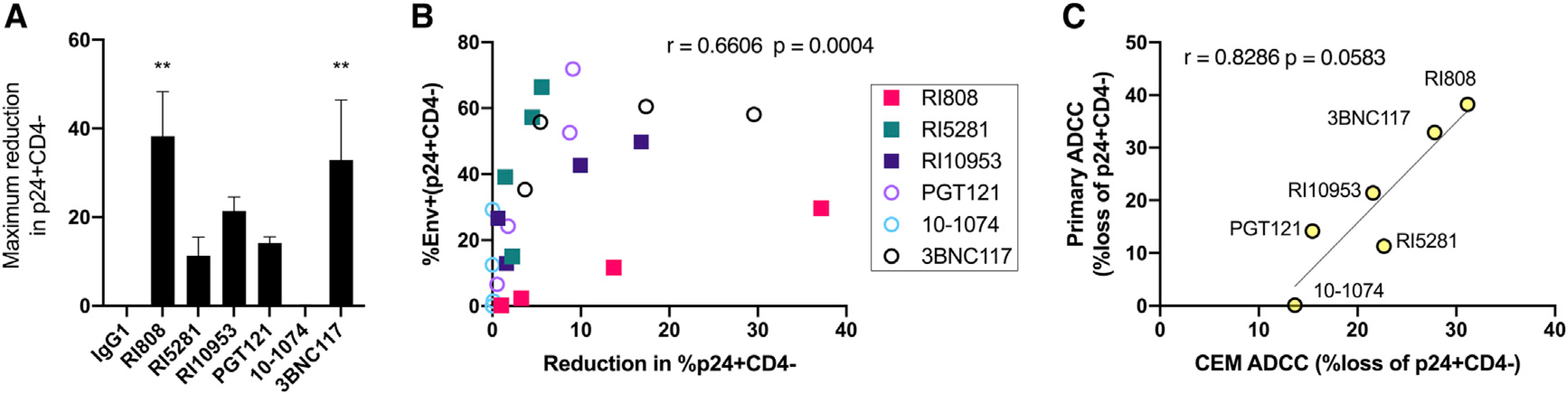
Phytohemagglutinin (PHA)-stimulated primary cells were infected with NL4–3, and antibodies were tested for their ability to bind without effectors or to drive the elimination of infected (p24+CD4−) cells by addition autologous PBMCs for 18 h. Antibodies were tested for binding and elimination over a 4-fold dilution series beginning at 25 μg/ml.
(A) Maximum elimination of infected (p24+CD4−) cells by the indicated antibody. Statistical significance is displayed relative to non-specific IgG1, and only adjusted p < 0.05 are displayed.
(B) Correlation of antibody recognition and elimination of infected cells over the dilution series. Each data point represents the mean value across three donors (ADCC) or two ADCC donors (binding) run in duplicate for the indicated antibody at a single concentration.
(C) Correlation of ADCC activity among NL4–3-infected primary target cells with an NL4–3-infected target cell line (CEM-NKr-CCR5; data in Figure 4).
Error bars in (A) indicate the standard error of the mean, and statistical significance for binding and killing was calculated independently using ANOVA with Dunnett’s multiple-comparisons test. A two-tailed Spearman correlation was used in (B) and (C). Significance is indicated as follows: *p ≤ 0.05, **p ≤ 0.01.
DISCUSSION
Antibodies poised for HIV reservoir elimination strategies will likely require the ability to detect reactivated latently infected cells across a wide breadth of viral strains and the ability to leverage innate effector killing by a Fc-FcγR interaction. Whether or not such antibodies indeed are actively contributing to viral control during natural infection is uncertain, but they are frequently enriched in spontaneous HCs (Ackerman et al., 2013a, 2013b, 2016; Johansson et al., 2011; Lambotte et al., 2009, 2013; Sadanand et al., 2018). In fact, multiple bNAbs have been isolated from HCs, which are selected based on plasma neutralization activities (Alsahafi et al., 2019; Decker et al., 2005; Ding et al., 2015; Freund et al., 2017; Guan et al., 2009, 2013; Sajadi et al., 2012, 2018; Scheid et al., 2011), but also non-neutralizing mAbs with diverse Fc effector functions have been identified (Guan et al., 2013). Here, we systematically screened a cohort of HCs, selected based on their plasma antibody functionality beyond neutralization to identify monoclonals equipped to efficiently recognize infected cells and mediate Fc functions beyond ADCC. Our results demonstrate that using this cell-recognition-focused/neutralization-agnostic approach, we were able to identify antibodies that efficiently target infected cells, with Fc domains presented in a position that supports recruitment of innate effectors and efficiently mediates anti-cellular activity.
HIV replication involves viral packaging on the plasma membrane of host cells, exposing the Env glycoprotein to the extracellular environment (Checkley et al., 2011); however, little is known about the kinetics and availability of Env on reactivated latent or actively infected cells. Our results replicate prior findings that current-generation bNAbs, which are broad and potent at virus neutralization, are highly variable in their capacity to recognize cells infected with different viral strains (Bruel et al., 2016, 2017). For example, the MPER-targeting antibody 10E8, which demonstrates exquisite neutralization breadth (Huang et al., 2012), only recognized 19% of infected cells, likely due to the limited accessibility of the target epitopes on the cell surface (Bruel et al., 2016). Furthermore, the CD4bs antibody 3BNC117 demonstrated a high capability of infected-cell recognition consistent with prior reports (Lu et al., 2016b), likely due to its ability to bind the Env trimer with higher occupancy than that of other CD4bs antibodies such as VRC01 (Nogal et al., 2017). Conversely, the CD4bs antibody N6, which has also exquisite neutralization breadth (Huang et al., 2016), did not recognize infected cells as efficiently in our assay. We can only speculate that the unique binding properties of this antibody might not be advantageous for cell-surface-exposed envelope structures. Nevertheless, of 54 monoclonal antibodies, including 45 downselected HC antibodies and the 9 bNAbs, for which infected-cell binding breadth and intensity across all 21 different HIV strains from 3 clades had been measured, multiple HC antibodies with substantial binding breadth were identified. In fact, 7 or the top 10 best cell binders were HC mAbs. Interestingly, of the top 6 binding antibodies with known epitope specificities, at least 3 (PGT121, RI808, and 10–1074) antibodies target the V3-loop, suggesting that this epitope region might be more readily accessible on the surface of infected cells. Indeed, previous studies have demonstrated that V3-glycan-targeting antibodies like PGT121, 2G12, and 10–1074 showed substantially higher levels of specific binding to cells infected with susceptible viruses compared to, i.e., CD4bs antibodies (Ren et al., 2018).
Although a correlation between the neutralization of viruses and binding to cells infected with the same viruses has been reported (Ren et al., 2018), a subset of HC antibodies bound cells at high levels but did not neutralize. This result is similar to that observed with, i.e., 2G12 or the MPER-specific bNAbs 2F5 and 4E10 that exhibited robust infected cell binding; however, they demonstrated only minimal neutralizing activity (Ren et al., 2018). Functional Env trimers, which are present on virions and the target for bNAbs, however, might be inconsistently present on the surface of productively infected cells, as the cell surface is inherently complex as well as dynamic, due to conformational changes in Env during viral assembly (Buttler et al., 2018; Murakami and Freed, 2000; Pezeshkian et al., 2019) and Env endocytosis (von Bredow et al., 2015; Egan et al., 1996). Additionally, it has been proposed that non-functional envelope proteins, such as gp41, stumps, or envelope monomers, may also be exposed as a result of gp120 shedding following interactions between CD4 receptor and Env on the surfaces of infected cells (Veillette et al., 2015). Antibodies that recognize these exposed epitopes might have strong cell binding capabilities and will likely lack neutralizing activity. In fact, structural studies revealed that the RI808 antibody Fab bound to SOSIP at high occupancy and directly competed with the V3-glycan-dependent bNAb 10–1074, suggesting potential N332-dependent binding, which is additionally supported by the lack of binding in clade-AE-infected cells (Figure 4). In contrast, RI5281 and RI10953 bound without strong competition among any tested bNAbs (Figure 5C), suggesting an unconventional epitope. Although RI5281 and RI10953 bound the SOSIP construct by BLI (Figure S6), nsEM reconstruction was hindered by binding heterogeneity and dependent low occupancy, which perhaps is a structural consequence of a flexible epitope. Indeed, RI5281 and RI10953 bound but did not excel in gp120 binding intensity analysis (Figure 2A) and yet were the broadest infected-cell-recognizing HC mAbs, suggesting differences in accessibility and/or conformation in the targeted epitope on the cell surface. Collectively, these data highlight that neutralization- or gp120-based-selection approaches would have overlooked antibodies, which effectively recognize HIV-infected cells at breadth and intensity that exceed or are competitive with clinically advanced bNAbs.
Antibody Fc functionality is driven by interactions of FcγRs on effector cells with the antibody Fc domain, and these interactions are impacted by the epitope location and the epitope density on the cell surface. It has been demonstrated that epitopes closer to the membrane mediate ADCC and complement-dependent cytotoxicity activity, whereas antibodies that target distal epitopes are efficient in inducing ADCP (Cleary et al., 2017). Many of the isolated HC monoclonals drove robust Fc functionality relative to bNAbs, consistent with the high functional profile in the donor sera. Several HC antibodies, like the related antibodies c2496 and c2498, elicited exceptional levels of effector functions (Figure 6) but lacked binding breadth, which could be consistent with the targeting of a non-conserved epitope that nonetheless allows the display of the Fc domain in a highly functional arrangement. In contrast, for some antibodies, high recognition was not sufficient to drive effector function, like in the examples of PGT121-opsonized primary cell ADCC (Figure 7A). HC antibodies with broad cellular recognition, namely, RI808 and RI5281, displayed excellent functionality at CEM-ADCC and ADCP, and in addition, RI5281 also promoted ADNP. Some of the tested bNAbs, such as 10–1074, VRC01, and VRC07–523LS, mediated robust functionality, but the latter two showed reduced cell recognition breadth, at least against the viral strains tested here. In the primary cell ADCC, RI808 and RI10953 mediated levels of infected-cell elimination that were competitive or excelled top bNAbs. As the antibodies were all IgG1 and contained the same Fc region and general glycosylation patterns, we assume that differences in their ability to mediate functions and signal through the FcγRs are likely due to differences in binding conformation of the antibody to infected cells and subsequent accessibility of the Fc region. Indeed, previous data suggested that antibody orientation on bound antigen, i.e., impacted by the epitope structure and different binding angles due to juxtaposed VH and VL contact surfaces, dramatically affects the potency of Fc-mediated effector function against HIV-1 (Acharya et al., 2014).
It should be noted that for reasons of better comparability and normalization across the tested antibodies and different viral strains used for infection, we focused on CD4-downregulated cells as antibody targets to reduce bias by the detection of CD4-virion complexes. This approach potentially excluded other relevant anti-viral cell surface targets during the infectious life cycle, and we therefore might have missed antibody responses that require other stages of the infection. In fact, multiple non-neutralizing antibodies have been described, such as A32 (Bonsignori et al., 2012; Easterhoff et al., 2020; Ferrari et al., 2011), with potent Fc functional activity. This specific antibody, however, requires CD4 expression for infected-cell recognition, and we therefore excluded the antibody from our comparative analyses. Furthermore, viruses used for cell infection might have different capacities to decrease Env expression on the cell surface by mechanisms mediated by accessory proteins Vpu and Nef, thereby preventing efficient cell recognition by antibodies (Arias et al., 2014; von Bredow et al., 2015; Prévost et al., 2018). Future research will need to focus on different stages of the replication cycle and assess how it affects the ability of diverse mAbs to robustly recognize and bind to infected cells.
Overall, our results characterize monoclonals from HCs with exceptional capacity to recognize infected cells, as well as to mediate Fc functions. These antibodies may not display the top neutralization profiles but instead efficiently recognize infected host cells and mediate innate effector functions that may be most effective for driving the elimination of primary infected cells. These antibodies may complement bNAbs in future reservoir eradication studies involving passive antibody transfer, and future work will examine the efficacy of these antibodies in vivo.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Boris Julg (bjulg@mgh.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact for non-commercial research purposes with a completed Material Transfer Agreement.
Data and code availability
3D reconstruction of AMC018 SOSIPv4.2 in complex with mAb RI808 has been deposited in the EM Data Bank (EMD-22163). The amino acid sequences of the HIV controller monoclonals, CDR3 length, somatic hypermutation levels and relative binding data displayed in Figure 2 are available in Table S2.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study participants
129 HIV-1 infected adults > 18 years old with plasma HIV-1 RNA levels < 2000 copies/ml on average, in the absence of antiretroviral therapy were included in this study and 15 individuals (12 male and 3 female, median age 51 years, range 26 to 64) were selected for downstream analysis. The clinical characteristics of the donors are summarized in Table S1. All study participants provided written informed consent prior to study participation in accordance with the Declaration of Helsinki and studies were reviewed by the institutional review board of Massachusetts General Hospital (Partners Human Research Committee (PHRC), Somerville MA 02145.
Cell lines and primary cultures
J89-GFP cells (infected with 89.6, male) were a kind gift from Dr. David Levy. CEM.NKR CCR5+ cells (CEMs, female), TZM-bl (female), ACH-2 (infected with LAV, female) were obtained from the NIH AIDS Reagent Program. THP1 (male) and HEK293T (female) cells were obtained from the American Type Culture Collection (ATCC). FreeStyle 293-F Cells (293F, female) were obtained from GIBCO. Suspension cell lines were cultured at 37°C with 5% C02 in R10 (RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine, 10 ug/ml streptomycin, and 10 IU/ml penicillin), with the exceptions of THP-1 s which were cultured in R10 with 0.05 mM 2-mercaptoethanol, and HEK293F which were cultured in Freestyle 293 expression medium (ThermoFisher). Adherent cells HEK293T and TZM-bl which were cultured in Dulbecco’s modified Eagle’s medium with 10% FBS, 10 ug/ml streptomycin and 10 IU/ml penicillin (DMEM). Buffy coats were obtained from healthy donors (Massachusetts General Hospital). HIV infection studies were performed with PBMCs isolated from buffy coats by centrifugation over histopaque-1077 (Sigma). PBMCs were cultured in R10 containing 50 units of IL-2 (R10–50) (R&D systems). Natural killer cells were enriched by negative selection from buffy coats according to the manufacturer’s instructions (StemCell Technologies), and cultured overnight in R10 containing 1 ng/ml IL-15 (R&D systems).
METHOD DETAILS
Antibody-dependent complement deposition (ADCD)
ADCD was performed as in Lofano et al. (2018). Briefly, CEM-NKr-CCR5 cells were pulsed with Gp120YU-2 (ImmuneTech), and incubated with heat-inactivated serum for 20 minutes at 37°C. Cells were washed, followed by incubation with complement source (HIV-negative donor plasma diluted 1:10 in veronal buffer with 0.1% gelatin) for 20 minutes at 37°C. The cells were then washed with 15 mM EDTA in PBS, and stained for complement with FITC anti-Human C3/C3b/iC3b- (Clone 10C7, Cedarlane). Heat-inactivated donor plasma was used as a negative control.
NK degranulation assay
The capacity of serum to promote NK degranulation was performed as in Chung et al., (2014) and Sips et al. (2016). Targets were prepared by pulsing CEM-NKr-CCR5 cells with gp120 YU-2 (ImmuneTech, 50 μg/ml) as previously described (Chung et al., 2014). After washing excess gp120, the pulsed cells were co-cultured with primary NK cells (prepared as by negative selection as above) at a ratio of 5:1 effectors: target, in the presence of diluted donor plasma (1:100 final dilution), anti-CD107a-PE-Cy5 (BD), brefeldin A (10 mg/ml, Sigma), and GolgiStop (BD) for 5 hours at 37°C. After incubation, cells were stained for surface markers with anti-CD16 APC-Cy7, anti-CD56 PE-Cy7, and anti-CD3 AF700, washed, followed by fixation and permeabilization with Fix & Perm kit (ThermoFisher). Intracellular staining was performed with anti–IFN-γ–APC (BD) and anti–MIP-1β–PE (BD). Cells were analyzed on a BD LSRII flow cytometer. NK cells were defined as CD3-negative, and CD16+ and/or CD56+.
B cell isolation and sorting
As previously described by Cizmeci et al., (2020) peripheral blood lymphocytes were isolated from buffy coats by centrifugation on histopaque (Sigma-Aldrich). CD19+ B cells were isolated by negative selection following the manufacturer’s instructions (STEMCELL Technologies). B cells were stained with fluorochrome-antibody conjugates and reagents to identify antigen-specific memory B cells using a panel consisting of propidium iodide (Life Technologies); CD3 (FITC, clone UCHT1), CD14 (FITC, clone HCD14), IgM (FITC, clone MHM-88), CD38 (PerCP-cy5.5, clone HIT2), CD20 (PE-cy7, clone 2H7), CD19 (BV421, clone HIB19), CD27 (BV510, clone O323) (All Biolegend); IgA (FITC, clone IS11–8E10) (Miltenyi Biotec). Preformed conjugates for antigen-specific B cell sorting were made as described using streptavidin conjugated to PE (Ragon provided) or AlexaFluor 647 (Life Technologies). Antigen-specific probes were made using JR-CSF gp120 (Clade B) and JRFL gp140 (Clade B) (both from ImmuneTech), as well as 92BR020 gp120 (Clade B), BG505 SOSIP (Clade A) and IAVI C22 gp120 (Clade C) (from Duke Human Vaccine Institute Protein Production Facility). Sorting was performed on a BD FACSAria. IgG+ B cells were defined as CD3/14−, CD19+, CD20+, and IgA/IgM-; antigen-specific B cells positive for probes in either PE or APC color were sorted into 96-well U-bottom plates containing cold lysis buffer. Sorted plates were frozen immediately and maintained at −80C before RT/PCR.
B cell receptor sequencing, V gene assignment, and somatic hypermutation analysis
As previously described by Cizmeci et al., (2020) natively paired variable sequences from heavy- and light- chain pairs were generated by reverse transcription, cDNA barcoding, amplification, and sequencing as described previously (DeFalco et al., 2018; Tan et al., 2014). cDNA sequences were determined by 454 Titanium sequencing. Variable genes were assigned using IgBlast (https://www.ncbi.nlm.nih.gov/projects/igblast/). Somatic hypermutation rates were calculated using SHazaM (v1.0.2) (Gupta et al., 2015). Paired heavy and light chain sequences were analyzed and ranked by somatic hypermutation (Kepler, 2013), and the top 185 unique antibodies were selected for expression.
HC antibodyexpression
The variable domains of the selected antibodies were cloned into an IgG1 backbone with an Ig kappa or lambda domain as appropriate. Monoclonal antibodies were expressed by transient transfection in HEK293F cells, followed by purification by protein A.
Viruses
Virus stocks were prepared from the clarified supernatants of transiently transfected HEK293T cells. For studies with CEM-NKr CCR5+ cells, viruses were pseudotyped by co-transfection with a plasmid encoding VSV-G. For HIV strains in which infectious molecular clones were not available, virus stocks were prepared from the clarified supernatants of PHA-stimulated infected PBMCs. HIV was titrated in TZM-BL cells using standard protocols, and infectious units were determined by X-gal staining.
HIV infection
Primary cells were activated with 3 μg/ml phytohemagglutinin (Sigma) for 3 days prior to infection. Activated cells were spinoculated with 0.01–0.1 infectious units per cell at 1,200 × g for 2 hours, and grown in R10–50 for 2–14 days. Intracellular staining was done to measure infection rate, cells infected at higher MOI (0.1 IU/cell) were used for screening 2–4DPI, while cells infected at lower MOIs were cultured for up to 14 days. CEMs were split 24 hours before infection, and spinoculated with ~0.5 infectious units per cell of pseudotyped HIV at 800 × g for 45 minutes, and cultured in R10 for 2–4 days.
Infected cell binding assay
Infected cells were washed twice in PBS, and aliquoted into 96 well plates (2×105 cells/well for PBMCs, 5×104 for cell lines). Cells were then pelleted and resuspended in PBS containing 25 μg/ml of the appropriate anti-Env antibody, and incubated for 60 minutes at RT. The plate was then washed twice with PBS, and extracellular staining was performed with Anti-CD3-AF700 (Biolegend clone SK7), Anti-CD4-BV605 (Biolegend clone OKT4), anti-CD8 FITC (BD clone HIT8a), anti-Fc APC (Biolegend clone HP6017), and Live/Dead Blue (ThermoFisher) and incubated for 20 minutes at RT. Samples were then washed twice and cells were fixed with Fix/Perm solution (BD), washed twice with Perm/Wash (BD) and resuspended in Perm/Wash containing anti-gag KC-57-RD1 (Beckman) and incubated for 20 minutes. Cells were then washed twice with Perm/Wash and resuspended in PBS for data acquisition by flow cytometry. To avoid potential differences in env expression, all HC antibodies and controls were compared for recognition on the same batch of infected cells at the same time. Infected-cell recognition rates among each virus were Z-scored using the standardize function in Microsoft Excel. Negative controls include nonspecific IgG (Biolegend), IVIG, and PBS conditions. Positive controls include polyclonal HIVIG, and combinations of bNAbs directed against multiple binding sites (such as 10E8, PGT121, and 3BNC117).
Recombinant Env binding
Binding to recombinantly expressed gp120, gp140, and V1V2 loops scaffolded on gp70 was defined by customized multiplex assay, as previously described (Brown et al., 2018). Briefly, fluorescently-coded microspheres were covalently conjugated with envelope proteins, incubated with mAb at 2.0 μg/ml, and degree of binding determined by flow cytometry. Polyclonal IgG pooled from HIV+ subjects and polyclonal IgG pooled from HIV- subjects were used as positive and negative controls, respectively.
Neutralization
Monoclonal antibody neutralization was performed at BIDMC (viral diversity panel) or the Ragon Institute (NL4–3 and JR-CSF) by a TZM-bl based luciferase assay. The viral diversity panel was assessed using Env-pseudotyped virus in a single infection cycle. Neutralization was quantitated as the relative decrease in luciferase activity over a dilution series, and neutralization curves were fit using GraphPad PRISM. To match the strains used in infection assays, NL4–3 and JR-CSF neutralization was performed using replication competent virus, and antibody potency was measured at a single concentration (25 μg/ml), in triplicate.
Antibody-dependent NK cell killing assay
ADCC was performed essentially as described by Bruel et al. (2016). CEM-NKr-CCR5 target cells were infected with pseudotyped virus at an MOI ~0.5 infectious units per cell. At two days post-infection, CEM cells were stained with CellTrace far red (Invitrogen) and 5×104 cells were added per well. Cells were washed and resuspended in diluted monoclonals for 5 minutes. After incubation, 1×105 primary NK cells (a 2:1 effector: target ratio) were added per well, cells were spun for 1 minute at 300g to promote contacts, and the cells were incubated for 4 hours at 37°C. Cells were stained for viability (blue viability dye, Life Technologies) then fixed and permeabilized for p24 staining. ADCC scores are calculated as 100 × (%p24+FarRed+ cells no antibody - %p24+FarRed+ cells antibody)/(%p24+FarRed+ cells no antibody), and any negative values are adjusted to zero. To assess optimal antibody concentration, we performed antibody titration experiments of bNAbs and a subset of HC mAbs, which showed maximal or near-maximal levels of ADCC at 25 μg/ml in our assays (data not shown).
PBMC ADCC
For killing of primary target cells, PBMCs were isolated into buffy coats and split into two aliquots. Target cells were generated by activation with 3 μg/ml of PHA for 3 days, followed by CD4 isolation via negative selection (StemCell Technologies, Inc.) and spinoculation with 0.5 IU/cell of NL4–3 (1,200 × g for 2 hours). After spinoculation, they were washed three times, and cultured for 4 days in R10–50. Before addition of effectors, infected CD4s were CellTrace far red stained (Invitrogen) for discrimination of target cells. Effector PBMCs were cultured with R10–50 with IL-15 added (1 ng/ml), and added a ratio of 5:1 E:T for 18 hours before staining. Staining was carried out using Blue Viability Dye (Invitrogen), anti-CD4-BV605 (Biolegend clone OKT4), anti-CD8 FITC (BD Biosciences, clone HIT8a), followed by fixation and permeabilization for p24 staining. ADCC score was calculated as above.
Phagocytosis assays (ADNP & ADCP)
Gp120 was purchased from ImmuneTech and biotinylated and conjugated to yellow-green Neutravidin beads (ThermoFisher). Beads were washed and resuspended in diluted monoclonal in PBS for 1 hour at 37C to allow opsonization. After incubation, beads were pelleted before resuspension with effector cells. For ADCP, 2.5×104 THP-1 cells were mixed in and incubated at 37C for 18h, followed by fixation in 4% paraformaldehyde in PBS. For ADNP, 5×104 white blood cells from peripheral blood were mixed in and incubated for 1 hour. ADNP samples were stained with CD3-AF700 (clone SK7), CD14-APC-Cy7 (clone M5E2), and Pacific blue CD66b (clone G10F5) (All Biolegend). CD3+ and CD14+ cells were excluded. Uptake of beads was determined by flow cytometry. Phagocytic scores were determined using the formula: (%FITC+) cells *(FITC MFI) / 10,000.
SOSIP and Fab protein expression
SOSIPs were expressed in HEK293F cells and purified with either PGT145 or PGT151 affinity chromatograph followed by size exclusion chromatography (SEC) using a HiLoad® 16/600 Superdex® pg200 (GE Healthcare) as described previously described (Ringe et al., 2017; de Taeye et al., 2015). Fabs were expressed in HEK293F cells and purified using affinity chromatography. Briefly, HEK293F cells (Invitrogen) were co-transfected with heavy and light chain plasmids (1:1 ratio) using PEImax (Polysciences). Transfections were performed according to the manufacturer’s protocol. Supernatants were harvested 6 days following transfection and passed through a 0.45 μm filter. Fabs were purified using CaptureSelect CH1-XL (ThermoFisher) affinity chromatography.
Biolayer interferometry
Monoclonal Fabs were loaded onto anti-human Fab-CH1 (FAB2G) biosensors (FortéBio) at a concentration of 10 μg/mL in kinetics buffer (PBS, pH 7.4, 0.01% [w/v] BSA, and 0.002% [v/v] Tween 20) until a response of 1 nanometer shift was reached. Loaded biosensors were dipped into wells containing only kinetics buffer for 1 min to acquire a baseline and then moved to wells containing 2000 nM SOSIP trimer in kinetics buffer. The trimers were allowed to associate for 180 s before the biosensor were move back to the wells containing only kinetics buffer where the baseline was acquired. Disassociation of the trimers from the Fab-loaded biosensors was recorded for 300 s. All BLI experiments were conducted at 37°C and the data were processed using the Octet System Data Analysis v9.0 (FortéBio).
Electron-microscopy
SOSIP/Fab complexes were made by mixing 15 μg AMC018 SOSIPv4.2 with 6-fold per protomer molar excess Fab and allowed to incubate for 18 to 24 hr at room temperature. Complex samples were SEC purified using a SuperoseTM 6 Increase 10/300 GL (GE Healthcare) column to remove excess Fab prior to EM grid preparation. Fractions containing the SOSIP/Fab complexes were pooled and concentrated using 10 kDa Amicon® spin concentrators (Millipore). Samples were diluted to 0.03 mg/mL in TBS (0.05 M Tris pH 7.4, 0.15 M NaCl) and adsorbed onto glow discharged carbon-coated Cu400 EM grids (Electron Microscopy Sciences) and blotted after 10 s. The grids were then stained with 3 μL of 2% (w/v) uranyl formate, immediately blotted, and stained again for 45 s followed by a final blot. Image collection and data processing was performed as described previously on a FEI Talos microscope (1.98 Å/pixel; 72,000 × magnification) with an electron dose of ~25 electrons/Å2 using Leginon (Pugach et al., 2015; Suloway et al., 2005). 2D classification, 3D sorting and 3D refinement conducted using Relion v3.0 (Zivanov et al., 2018). EM density maps were visualized using UCSF Chimera (Pettersen et al., 2004).
Competitive ELISA
Competitive ELISA was adapted from Derking et al. (2015). 384-well MaxiSorp ELISA plates were coated overnight with 2.5 ug/ml JR-52 antibody (which binds the D7 epitope of gp120; Robinson et al., 2016) in PBS. Blocking was performed with PBS containing 0.05% Tween-20 (PBST) with 5% BSA at room temperature for 2 hours. After blocking steps, wells washed with PBST and incubated with gp120 JR-CSF (ImmuneTech) diluted to 80 ng/ml in PBS for 2H at RT. After washing, unlabeled competing/blocking antibody was added at a concentration of 10 μg/ml and incubated for 1 hour. Excess antibody was washed, and biotinylated antibody was added at a concentration of 1 μg/ml for 1 hour. Plates were washed four times, and streptavidin-HRP (Pierce) was added at a dilution of 1:1000, and incubated at RT for 30 minutes. After four washes, Ultra-TMB (3,3′,5,5′-Tetramethylbenzidine, ThermoFisher) was added. Reaction was stopped by addition of 2M H2SO4 when appropriate, and absorption was measured at 450 nm on a Tecan plate reader. Competition values are calculated as the absorption in the presence of competing antibody, divided by the value in the absence of competing unlabeled antibody.
QUANTIFICATION AND STATISTICAL ANALYSIS
Figures were prepared in Microsoft Excel (v16.44) or GraphPad Prism (v9.0.0). Z-score normalization was performed in Microsoft Excel. Spearman two-tailed correlation coefficients (r) was used to correlate binding and neutralization (Figure 3), and ANOVA with Dunnett’s multiple comparisons test was used to compare ADCC function (Figure 7A). Statistical analysis was performed using GraphPad Prism. Alignment of gp120 in Figure 4 was performed in Geneious R9 (v9.1.1) using ClustalW. CDR3 loops in Table S2 were defined by Change-O (Gupta et al., 2015).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD56 PE-Cy7 | BD Biosciences | BD Biosciences Cat# 557747; RRID:AB_396853 |
| APC-Cy7 Anti-CD16 Monoclonal Antibody | BD Biosciences | BD Biosciences Cat# 557758; RRID:AB_396864 |
| Alexa Fluor® 700 anti-human CD3 antibody | BioLegend | BioLegend Cat# 344822; RRID:AB_2563420 |
| anti–IFN-γ–APC (BD) | BD Biosciences | BD Biosciences Cat# 551385; RRID:AB_398505 |
| and anti–MIP-1β–PE (BD) | BD Biosciences | BD Biosciences Cat# 550078; RRID:AB_393549 |
| Brilliant Violet 605 anti-human CD4 antibody | BioLegend | BioLegend Cat# 317438; RRID:AB_11218995 |
| FITC anti-Human CD8 Antibody | BD Biosciences | BD Biosciences Cat# 555634; RRID:AB_395996 |
| APC anti-human IgG Fc antibody | BioLegend | BioLegend Cat# 409306; RRID:AB_11149491 |
| HIV-1 core antigen-RD1 antibody | Beckman Coulter | Beckman Coulter Cat# 6604667; RRID:AB_1575989 |
| FITC anti-human CD3 antibody | BioLegend | BioLegend Cat# 300440; RRID:AB_2562046 |
| FITC anti-human CD14 antibody | BioLegend | BioLegend Cat# 325604; RRID:AB_830677 |
| FITC anti-human IgM antibody | BioLegend | BioLegend Cat# 314506; RRID:AB_493009 |
| PerCP/Cyanine5.5 anti-human CD38 antibody | BioLegend | BioLegend Cat# 303522; RRID:AB_893314 |
| PE/Cy7 anti-human CD20 antibody | BioLegend | BioLegend Cat# 302312; RRID:AB_314260 |
| Brilliant Violet 421 anti-human CD19 antibody | BioLegend | BioLegend Cat# 302234; RRID:AB_11142678 |
| Brilliant Violet 510 anti-human CD27 antibody | BioLegend | BioLegend Cat# 302836; RRID:AB_2562086 |
| IgA Antibody, FITC | Miltenyi | Miltenyi Biotec Cat# 130-114-001; RRID:AB_2726443 |
| Anti-HIV HIV Controller Monoclonals | This manuscript. See Table S2 for Sequences | N/A |
| APC/Cyanine7 anti-human CD14 antibody | BioLegend | BioLegend Cat# 301820; RRID:AB_493695 |
| Pacific Blue anti-human CD66b antibody | BioLegend | BioLegend Cat# 305112; RRID:AB_2563294 |
| Mouse Anti-Human C3 / C3b / iC3b Monoclonal Antibody, FITC Conjugated | CedarLane | CEDARLANE Cat# CL7632F; RRID:AB_10548984 |
| Anti-gp120 D7 epitope Antibody | James Robinson, jrobinso@tulane.edu | N/A |
| IgG1 Isotype Control | BioLegend | BioLegend Cat# 403502 |
| Polyclonal Anti-Human Immunodeficiency Virus Immune Globulin | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat# 3957 |
| IgG from human serum antibody | Sigma Aldrich | Sigma-Aldrich Cat# I4506; RRID:AB_1163606 |
| anti-HIV-1 Monoclonal Env 3BNC117 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat# 12474; RRID: AB_2491033 |
| Anti-HIV-1 gp120 Monoclonal PGT121 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat#12343; RRID:AB_2491041 |
| Anti-HIV-1 gp120 Monoclonal 10–1074 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat#12477; RRID:AB_2491041 |
| Anti-HIV-1 gp120 Monoclonal PGDM1400 | In house | N/A |
| Anti-HIV-1 gp120 Monoclonal VRC26.25-LS | John R Mascola, Vaccine Research Center | N/A |
| Anti-HIV-1 gp120 Monoclonal 10E8 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat#12294 |
| Monoclonal anti-HIV-1 Env VRC01 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat# 12033; RRID: AB_2491019 |
| Monoclonal anti-HIV-1 Env N6 | NIH AIDS Reagent Program; https://www.hiv.lanl.gov/ | Cat#12968 |
| Bacterial and virus strains | ||
| HIV 394747 | Yu et al., 2019 | N/A |
| HIV 629356 | Yu et al., 2019 | N/A |
| HIV 486632 | Yu et al., 2019 | N/A |
| HIV 828738 | Yu et al., 2019 | N/A |
| HIV 690419 | Yu et al., 2019 | N/A |
| HIV 930213 | Yu et al., 2019 | N/A |
| HIV 00TZ_A246 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 90SE_364 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 98US_MSC5016 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 90TH_CM235 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 90TH_CM240 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 96TH_NI1046 | NIH Aids Reagent | NIH ARP Cat#11412 |
| HIV 96TH_M02138 | NIH Aids Reagent | NIH ARP Cat#11412 |
| Biological samples | ||
| ACD-anticoagulated Blood from healthy, HIV negative donors | Massachusetts General Hospital | N/A |
| Buffy coats from healthy, HIV negative donors | Massachusetts General Hospital | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Histopaque-1077 (sigma) | Sigma | Cat#10771 |
| Fix/Perm (BD) | BD | Cat#554714 |
| FIX & PERM Cell Fixation & Cell Permeabilization Kit | ThermoFisher | Cat#GAS004 |
| Recombinant Human IL-2 Protein | R&D Systems | Cat#202-IL-050/CF |
| Recombinant Human IL-15 Protein | R&D Systems | Cat#247-ILB-025/CF |
| britelite plus Reporter Gene Assay System | Perkin Elmer | Cat#6066761 |
| Phytohemagglutinin PHA-P | Sigma | Cat#L8754 |
| LIVE/DEAD Fixable Blue Dead Cell Stain Kit, for UV excitation | Invitrogen | Cat#L34962 |
| Propidium Iodide | Invitrogen | Cat#P1304MP |
| Brefeldin A | Sigma | Cat#B5936-200UL |
| GolgiStop | BD | Cat#554724 |
| CellTrace Far Red Cell | Invitrogen | Cat#C34564 |
| Yellow-green NeutraAvidin Labeled Microspheres | Invitrogen | Cat#F8776 |
| PEI Max | Polysciences | Cat#24765 |
| Recombinant Human IL-15 Protein | R&D Systems | Cat#247-ILB |
| High Sensitivity Streptavidin-HRP | Pierce | Cat#21130 |
| gp120 JRCSF | Immune Technology Corp | Cat#IT-001-0025p |
| gp140 JR-FL | Immune Technology Corp | Cat#IT-001-0024ΔTMp |
| gp120 YU-2 | Immune Technology Corp | Cat#IT-001-0027p |
| gp120 LAI | Immune Technology Corp | Cat#IT-001-138p |
| BG505 SOSIP | Sanders et al., 2013 | N/A |
| CE-1176-A3 SOSIP | This Study | N/A |
| 246-F3 SOSIP | This Study | N/A |
| CNE55 SOSIP | This Study | N/A |
| 25710 SOSIP | This Study | N/A |
| AMC016 SOSIPv4.2 | This Study | N/A |
| AMC018 SOSIPv4.2 | This Study | N/A |
| AMC011 SOSIPv4.2 | Torrents de la Peña et al., 2019 | N/A |
| TRO-11 SOSIP | This Study | N/A |
| 20198102 SOSIP | This Study | N/A |
| TRJO SOSIPv5.3 | This Study | N/A |
| Deposited data | ||
| 3D reconstruction of AMC018 SOSIPv4.2 in complex with mAb RI808 | This manuscript | EM Data Bank accession number EMD-22163 |
| Experimental models: Cell lines | ||
| Human: HEK293T | ATCC | ATCC Cat# CRL-3216, RRID:CVCL_0063 |
| Human: Freestyle 293F | ThermoFisher | Cat#R79007; RRID: CVCL_D603 |
| Human: HeLa-derived TZM-bl | NIH Aids Reagent | NIH-ARP Cat# 8129–442; RRID:CVCL_B478 |
| Human: Jurkat-derived J89-GFP | https://doi.org/10.1128/JVI.76.17.8776-8786.2002 | N/A |
| Human: ACH-2 | NIH Aids Reagent | ACH-2 (NIH-ARP Cat# 349–443; RRID:CVCL_0138) |
| Human: CEM-NKr-CCR5 | NIH Aids Reagent | CEM.NKR-CCR5 NIH-ARP Cat# 4376–29; RRID:CVCL_X623 |
| Human: THP-1 | ATCC | ATCC Cat# TIB-202; RRID:CVCL_0006 |
| Recombinant DNA | ||
| pNL4–3 | NIH Aids Reagent | NIH ARP Cat# 114 |
| pYK-JRCSF | NIH Aids Reagent | NIH ARP Cat# 2708 |
| pCH058.c/2960 | NIH Aids Reagent | NIH ARP Cat# 11856 |
| pCH077.c/2627 | NIH Aids Reagent | NIH ARP Cat# 11752 |
| pTHRO.c/2826 | NIH Aids Reagent | NIH ARP Cat#11745 |
| pWITO.c/2474 | NIH Aids Reagent | NIH ARP Cat#11739 |
| Software and algorithms | ||
| FlowJo | BD / TreeStar Software | RRID:SCR_008520 |
| Excel | Microsoft | Microsoft Excel; RRID:SCR_016137 |
| PRISM8 | GraphPad | GraphPad Prism; RRID:SCR_002798 |
| UCSF Chimera | UCSF Chimera Team | UCSF Chimera; RRID:SCR_004097 |
| Octet System Data Analysis v9.0 | Forte Bio | N/A |
| Other | ||
| CaptureSelect CH1-XL Affinity Matrix | Thermo Scientific | Cat#2943452010 |
| Biosensor / Anti-Human Fab-CH1 2nd Generation (FAB2G) | Forte Bio | Cat#18–5125 |
Highlights.
Analysis of 185 HIV-envelope-specific antibodies from 15 spontaneous HIV controllers
Antibody downselection based on infected-cell recognition and Fc functionality
V3-loop-targeting antibodies were enriched among the top cell binders
Binding topology and Fc accessibility likely result in variable Fc functionality
ACKNOWLEDGMENTS
The study was funded by an HIV cure grant (00403 / PA-HIV-16–0061) from Gilead Sciences, Inc. to G.A. B.J. is supported by NIH grants AI138790 and AI060354. C.A.C. is supported by the NIH F31 Ruth L. Kirschstein Predoctoral Award Al131873 and by the Achievement Rewards for College Scientists Foundation. G.A. is supported by NIH grants AI060354 and AI129797. B.J. and G.A. are associate members of the I4C-HIV Martin Delaney Collaboratory (UM1AI126603). The authors gratefully acknowledge John Mascola from the NIH Vaccine Research Center for providing monoclonal antibodies. The authors thank Joshua Alex Weiner for sample and data management.
Footnotes
DECLARATION OF INTERESTS
G.A. has a financial interest (founder) in SeromYx, a company developing platform technology that describes the antibody immune response. G.A.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. The other authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109167.
REFERENCES
- Acharya P, Tolbert WD, Gohain N, Wu X, Yu L, Liu T, Huang W, Huang C-C, Kwon YD, Louder RK, et al. (2014). Structural definition of an antibody-dependent cellular cytotoxicity response implicated in reduced risk for HIV-1 infection. J. Virol 88, 12895–12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman ME, Crispin M, Yu X, Baruah K, Boesch AW, Harvey DJ, Dugast A-S, Heizen EL, Ercan A, Choi I, et al. (2013a). Natural variation in Fc glycosylation of HIV-specific antibodies impacts antiviral activity. J. Clin. Invest 123, 2183–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman ME, Dugast A-S, McAndrew EG, Tsoukas S, Licht AF, Irvine DJ, and Alter G (2013b). Enhanced phagocytic activity of HIV-specific antibodies correlates with natural production of immunoglobulins with skewed affinity for FcγR2a and FcγR2b. J. Virol 87, 5468–5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman ME, Mikhailova A, Brown EP, Dowell KG, Walker BD, Bailey-Kellogg C, Suscovich TJ, and Alter G (2016). Polyfunctional HIV-Specific Antibody Responses Are Associated with Spontaneous HIV Control. PLoS Pathog. 12, e1005315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsahafi N, Bakouche N, Kazemi M, Richard J, Ding S, Bhattacharyya S, Das D, Anand SP, Prévost J, Tolbert WD, et al. (2019). An Asymmetric Opening of HIV-1 Envelope Mediates Antibody-Dependent Cellular Cytotoxicity. Cell Host Microbe 25, 578–587.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand SP, Grover JR, Tolbert WD, Prévost J, Richard J, Ding S, Baril S, Medjahed H, Evans DT, Pazgier M, et al. (2019). Antibody-Induced Internalization of HIV-1 Env Proteins Limits Surface Expression of the Closed Conformation of Env. J. Virol 93, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, and Evans DT (2014). Tetherin antagonism by Vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 111, 6425–6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar KJ, Sneller MC, Harrison LJ, Justement JS, Overton ET, Petrone ME, Salantes DB, Seamon CA, Scheinfeld B, Kwan RW, et al. (2016). Effect of HIV Antibody VRC01 on Viral Rebound after Treatment Interruption. N. Engl. J. Med 375, 2037–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignori M, Pollara J, Moody MA, Alpert MD, Chen X, Hwang K-K, Gilbert PB, Huang Y, Gurley TC, Kozink DM, et al. (2012). Antibody-dependent cellular cytotoxicity-mediating antibodies from an HIV-1 vaccine efficacy trial target multiple epitopes and preferentially use the VH1 gene family. J. Virol 86, 11521–11532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borducchi EN, Liu J, Nkolola JP, Cadena AM, Yu W-H, Fischinger S, Broge T, Abbink P, Mercado NB, Chandrashekar A, et al. (2018). Antibody and TLR7 agonist delay viral rebound in SHIV-infected monkeys. Nature 563, 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, and Ravetch JV (2014). Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 158, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EP, Weiner JA, Lin S, Natarajan H, Normandin E, Barouch DH, Alter G, Sarzotti-Kelsoe M, and Ackerman ME (2018). Optimization and qualification of an Fc Array assay for assessments of antibodies against HIV-1/SIV. J. Immunol. Methods 455, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel T, Guivel-Benhassine F, Amraoui S, Malbec M, Richard L, Bourdic K, Donahue DA, Lorin V, Casartelli N, Noël N, et al. (2016). Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat. Commun 7, 10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruel T, Guivel-Benhassine F, Lorin V, Lortat-Jacob H, Baleux F, Bourdic K, Noël N, Lambotte O, Mouquet H, and Schwartz O (2017). Lack of ADCC Breadth of Human Nonneutralizing Anti-HIV-1 Antibodies. J. Virol 91, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttler CA, Pezeshkian N, Fernandez MV, Aaron J, Norman S, Freed EO, and van Engelenburg SB (2018). Single molecule fate of HIV-1 envelope reveals late-stage viral lattice incorporation. Nat. Commun 9, 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checkley MA, Luttge BG, and Freed EO (2011). HIV-1 envelope glycoprotein biosynthesis, trafficking, and incorporation. J. Mol. Biol 410, 582–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki J, Waithe D, Carravilla P, Huarte N, Galiani S, Enderlein J, and Eggeling C (2017). Envelope glycoprotein mobility on HIV-1 particles depends on the virus maturation state. Nat. Commun 8, 545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung AW, Ghebremichael M, Robinson H, Brown E, Choi I, Lane S, Dugast AS, Schoen MK, Rolland M, Suscovich TJ, et al. (2014). Polyfunctional Fc-effector profiles mediated by IgG subclass selection distinguish RV144 and VAX003 vaccines. Sci. Transl. Med 6, 228ra38. [DOI] [PubMed] [Google Scholar]
- Cizmeci D, Lofano G, Dugast A-S, Kim D, Cavet G, Nguyen N, Tan YC, Seaman MS, Alter G, and Julg B (2020). Distinct clonal evolution of B-cells in HIV controllers with neutralizing antibody breadth. bioRxiv. 10.1101/2020.09.02.277566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary KLS, Chan HTC, James S, Glennie MJ, and Cragg MS (2017). Antibody Distance from the Cell Membrane Regulates Antibody Effector Mechanisms. J. Immunol 198, 3999–4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis-Gardner ME, Gardner MR, Alfant B, and Farzan M (2017). eCD4-Ig promotes ADCC activity of sera from HIV-1-infected patients. PLoS Pathog. 13, e1006786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Taeye SW, Ozorowski G, Torrents de la Peña A, Guttman M, Julien JP, van den Kerkhof TLGM, Burger JA, Pritchard LK, Pugach P, Yasmeen A, et al. (2015). Immunogenicity of Stabilized HIV-1 Envelope Trimers with Reduced Exposure of Non-neutralizing Epitopes. Cell 163, 1702–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker JM, Bibollet-Ruche F, Wei X, Wang S, Levy DN, Wang W, Delaporte E, Peeters M, Derdeyn CA, Allen S, et al. (2005). Antigenic conservation and immunogenicity of the HIV coreceptor binding site. J. Exp. Med 201, 1407–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFalco J, Harbell M, Manning-Bog A, Baia G, Scholz A, Millare B, Sumi M, Zhang D, Chu F, Dowd C, et al. (2018). Non-progressing cancer patients have persistent B cell responses expressing shared antibody paratopes that target public tumor antigens. Clin. Immunol 187, 37–45. [DOI] [PubMed] [Google Scholar]
- Derking R, Ozorowski G, Sliepen K, Yasmeen A, Cupo A, Torres JL, Julien JP, Lee JH, van Montfort T, de Taeye SW, et al. (2015). Comprehensive antigenic map of a cleaved soluble HIV-1 envelope trimer. PLoS Pathog. 11, e1004767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Veillette M, Coutu M, Prévost J, Scharf L, Bjorkman PJ, Ferrari G, Robinson JE, Stürzel C, Hahn BH, et al. (2015). A Highly Conserved Residue of the HIV-1 gp120 Inner Domain Is Important for Antibody-Dependent Cellular Cytotoxicity Responses Mediated by Anti-cluster A Antibodies. J. Virol 90, 2127–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, et al. ; NISC Comparative Sequencing Program (2014). Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 509, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easterhoff D, Pollara J, Luo K, Tolbert WD, Young B, Mielke D, Jha S, O’Connell RJ, Vasan S, Kim J, et al. (2020). Boosting with AIDSVAX B/E Enhances Env Constant Region 1 and 2 Antibody-Dependent Cellular Cytotoxicity Breadth and Potency. J. Virol 94, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MA, Carruth LM, Rowell JF, Yu X, and Siliciano RF (1996). Human immunodeficiency virus type 1 envelope protein endocytosis mediated by a highly conserved intrinsic internalization signal in the cytoplasmic domain of gp41 is suppressed in the presence of the Pr55gag precursor protein. J. Virol 70, 6547–6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari G, Pollara J, Kozink D, Harms T, Drinker M, Freel S, Moody MA, Alam SM, Tomaras GD, Ochsenbauer C, et al. (2011). An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J. Virol 85, 7029–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund NT, Wang H, Scharf L, Nogueira L, Horwitz JA, Bar-On Y, Golijanin J, Sievers SA, Sok D, Cai H, et al. (2017). Coexistence of potent HIV-1 broadly neutralizing antibodies and antibody-sensitive viruses in a viremic controller. Sci. Transl. Med 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam R, Nishimura Y, Pegu A, Nason MC, Klein F, Gazumyan A, Golijanin J, Buckler-White A, Sadjadpour R, Wang K, et al. (2016). A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature 533, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam R, Nishimura Y, Gaughan N, Gazumyan A, Schoofs T, Buckler-White A, Seaman MS, Swihart BJ, Follmann DA, Nussenzweig MC, and Martin MA (2018). A single injection of crystallizable fragment domain-modified antibodies elicits durable protection from SHIV infection. Nat. Med 24, 610–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohain N, Tolbert WD, Orlandi C, Richard J, Ding S, Chen X, Bonsor DA, Sundberg EJ, Lu W, Ray K, et al. (2016). Molecular basis for epitope recognition by non-neutralizing anti-gp41 antibody F240. Sci. Rep 6, 36685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Sajadi MM, Kamin-Lewis R, Fouts TR, Dimitrov A, Zhang Z, Redfield RR, DeVico AL, Gallo RC, and Lewis GK (2009). Discordant memory B cell and circulating anti-Env antibody responses in HIV-1 infection. Proc. Natl. Acad. Sci. USA 106, 3952–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Pazgier M, Sajadi MM, Kamin-Lewis R, Al-Darmarki S, Flinko R, Lovo E, Wu X, Robinson JE, Seaman MS, et al. (2013). Diverse specificity and effector function among human antibodies to HIV-1 envelope glycoprotein epitopes exposed by CD4 binding. Proc. Natl. Acad. Sci. USA 110, E69–E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, and Kleinstein SH (2015). Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, Parren PWHI, Marx PA, and Burton DR (2009). Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat. Med 15, 951–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz JA, Bar-On Y, Lu C-L, Fera D, Lockhart AAK, Lorenzi JCC, Nogueira L, Golijanin J, Scheid JF, Seaman MS, et al. (2017). Non-neutralizing Antibodies Alter the Course of HIV-1 Infection In Vivo. Cell 170, 637–648.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D, Peng Y, Berkowitz SA, and Engen JR (2010). Post-translational modifications differentially affect IgG1 conformation and receptor binding. Mol. Cell. Proteomics 9, 1716–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Ofek G, Laub L, Louder MK, Doria-Rose NA, Longo NS, Imamichi H, Bailer RT, Chakrabarti B, Sharma SK, et al. (2012). Broad and potent neutralization of HIV-1 by a gp41-specific human antibody. Nature 491, 406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, et al. (2016). Identification of a CD4-Binding-Site Antibody to HIV that Evolved Near-Pan Neutralization Breadth. Immunity 45, 1108–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson SE, Rollman E, Chung AW, Center RJ, Hejdeman B, Stratov I, Hinkula J, Wahren B, Kärre K, Kent SJ, and Berg L (2011). NK cell function and antibodies mediating ADCC in HIV-1-infected viremic and controller patients. Viral Immunol. 24, 359–368. [DOI] [PubMed] [Google Scholar]
- Julg B, Liu PT, Wagh K, Fischer WM, Abbink P, Mercado NB, Whitney JB, Nkolola JP, McMahan K, Tartaglia LJ, et al. (2017). Protection against a mixed SHIV challenge by a broadly neutralizing antibody cocktail. Sci. Transl. Med 9, eaao4235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepler TB (2013). Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors. F1000Res. 2, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambotte O, Ferrari G, Moog C, Yates NL, Liao HX, Parks RJ, Hicks CB, Owzar K, Tomaras GD, Montefiori DC, et al. (2009). Heterogeneous neutralizing antibody and antibody-dependent cell cytotoxicity responses in HIV-1 elite controllers. AIDS 23, 897–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambotte O, Pollara J, Boufassa F, Moog C, Venet A, Haynes BF, Delfraissy JF, Saez-Cirion A, and Ferrari G (2013). High antibody-dependent cellular cytotoxicity responses are correlated with strong CD8 T cell viral suppressive activity but not with B57 status in HIV-1 elite controllers. PLoS One 8, e74855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis GK, Ackerman ME, Scarlatti G, Moog C, Robert-Guroff M, Kent SJ, Overbaugh J, Reeves RK, Ferrari G, and Thyagarajan B (2019). Knowns and unknowns of assaying antibody-dependent cell-mediated cytotoxicity against HIV-1. Front. Immunol 10, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, and Wang L-X (2017). Modulating IgG effector function by Fc glycan engineering. Proc. Natl. Acad. Sci. USA 114, 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lofano G, Gorman MJ, Yousif AS, Yu W-H, Fox JM, Dugast A-S, Ackerman ME, Suscovich TJ, Weiner J, Barouch D, et al. (2018). Antigen-specific antibody Fc glycosylation enhances humoral immunity via the recruitment of complement. Sci. Immunol 3, eaat7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C-L, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, Horwitz JA, Nogueira L, Golijanin J, Gazumyan A, et al. (2016a). Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CL, Murakowski DK, Bournazos S, Schoofs T, Sarkar D, Halper-Stromberg A, Horwitz JA, Nogueira L, Golijanin J, Gazumyan A, et al. (2016b). Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science 352, 1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi V, Wren LH, Center RJ, Gonelli C, Winnall WR, Parsons MS, Kramski M, Kent SJ, and Stratov I (2014). Breadth of HIV-1 Env-specific antibody-dependent cellular cytotoxicity: relevance to global HIV vaccine design. AIDS 28, 1859–1870. [DOI] [PubMed] [Google Scholar]
- Madhavi V, Wines BD, Amin J, Emery S, Lopez E, Kelleher A, Center RJ, Hogarth PM, Chung AW, Kent SJ, and Stratov I ENCORE1 Study Group; Sydney LTNP Study Group (2017). HIV-1 Env- and Vpu-Specific Antibody-Dependent Cellular Cytotoxicity Responses Associated with Elite Control of HIV. J. Virol 91, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr LM, Decoville T, Schmidt S, Laumond G, Klingler J, Ducloy C, Bahram S, Zolla-Pazner S, and Moog C (2017). Non-neutralizing Antibodies Targeting the V1V2 Domain of HIV Exhibit Strong Antibody-Dependent Cell-mediated Cytotoxic Activity. Sci. Rep 7, 12655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Ramírez M, Sánchez-Merino V, Sánchez-Palomino S, Merino-Mansilla A, Ferreira CB, Pérez I, González N, Alvarez A, Alcocer-González JM, García F, et al. (2011). Broadly cross-neutralizing antibodies in HIV-1 patients with undetectable viremia. J. Virol 85, 5804–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, Lehmann C, Suárez I, Oliveira TY, Lorenzi JCC, et al. (2018). Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 561, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PL, Crooks ET, Porter L, Zhu P, Cayanan CS, Grise H, Corcoran P, Zwick MB, Franti M, Morris L, et al. (2006). Nature of nonfunctional envelope proteins on the surface of human immunodeficiency virus type 1. J. Virol 80, 2515–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouquet H, Scharf L, Euler Z, Liu Y, Eden C, Scheid JF, Halper-Stromberg A, Gnanapragasam PN, Spencer DI, Seaman MS, et al. (2012). Complex-type N-glycan recognition by potent broadly neutralizing HIV antibodies. Proc. Natl. Acad. Sci. USA 109, E3268–E3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujib S, Liu J, Rahman AKMN, Schwartz JA, Bonner P, Yue FY, and Ostrowski MA (2017). Comprehensive Cross-Clade Characterization of Antibody-Mediated Recognition, Complement-Mediated Lysis, and Cell-Mediated Cytotoxicity of HIV-1 Envelope-Specific Antibodies toward Eradication of the HIV-1 Reservoir. J. Virol 91, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, and Freed EO (2000). The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. USA 97, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogal B, Bowman CA, and Ward AB (2017). Time-course, negative-stain electron microscopy-based analysis for investigating protein-protein interactions at the single-molecule level. J. Biol. Chem 292, 19400–19410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Pezeshkian N, Groves NS, and van Engelenburg SB (2019). Single-molecule imaging of HIV-1 envelope glycoprotein dynamics and Gag lattice association exposes determinants responsible for virus incorporation. Proc. Natl. Acad. Sci. USA 116, 25269–25277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prévost J, Richard J, Medjahed H, Alexander A, Jones J, Kappes JC, Ochsenbauer C, and Finzi A (2018). Incomplete Downregulation of CD4 Expression Affects HIV-1 Env Conformation and Antibody-Dependent Cellular Cytotoxicity Responses. J. Virol 92, e00484–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugach P, Ozorowski G, Cupo A, Ringe R, Yasmeen A, de Val N, Derking R, Kim HJ, Korzun J, Golabek M, et al. (2015). A native-like SOSIP. 664 trimer based on an HIV-1 subtype B env gene. J. Virol 89, 3380–3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Korom M, Truong R, Chan D, Huang SH, Kovacs CC, Benko E, Safrit JT, Lee J, Garbán H, et al. (2018). Susceptibility to Neutralization by Broadly Neutralizing Antibodies Generally Correlates with Infected Cell Binding for a Panel of Clade B HIV Reactivated from Latent Reservoirs. J. Virol 92, e00895–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J, Prévost J, Baxter AE, von Bredow B, Ding S, Medjahed H, Delgado GG, Brassard N, Stürzel CM, Kirchhoff F, et al. (2018). Uninfected Bystander Cells Impact the Measurement of HIV-Specific Antibody-Dependent Cellular Cytotoxicity Responses. mBio 9, e00358–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringe RP, Ozorowski G, Yasmeen A, Cupo A, Cruz Portillo VM, Pugach P, Golabek M, Rantalainen K, Holden LG, Cottrell CA, et al. (2017). Improving the Expression and Purification of Soluble, Recombinant Native-Like HIV-1 Envelope Glycoprotein Trimers by Targeted Sequence Changes. J. Virol 91, e00264–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JE, Hastie KM, Cross RW, Yenni RE, Elliott DH, Rouelle JA, Kannadka CB, Smira AA, Garry CE, Bradley BT, et al. (2016). Most neutralizing human monoclonal antibodies target novel epitopes requiring both Lassa virus glycoprotein subunits. Nat. Commun 7, 11544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudicell RS, Kwon YD, Ko S-Y, Pegu A, Louder MK, Georgiev IS, Wu X, Zhu J, Boyington JC, Chen X, et al. ; NISC Comparative Sequencing Program (2014). Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J. Virol 88, 12669–12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadanand S, Das J, Chung AW, Schoen MK, Lane S, Suscovich TJ, Streeck H, Smith DM, Little SJ, Lauffenburger DA, et al. (2018). Temporal variation in HIV-specific IgG subclass antibodies during acute infection differentiates spontaneous controllers from chronic progressors. AIDS 32, 443–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Lewis GK, Seaman MS, Guan Y, Redfield RR, and De-Vico AL (2012). Signature biochemical properties of broadly cross-reactive HIV-1 neutralizing antibodies in human plasma. J. Virol 86, 5014–5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajadi MM, Dashti A, Rikhtegaran Tehrani Z, Tolbert WD, Seaman MS, Ouyang X, Gohain N, Pazgier M, Kim D, Cavet G, et al. (2018). Identification of Near-Pan-neutralizing Antibodies against HIV-1 by Deconvolution of Plasma Humoral Responses. Cell 173, 1783–1795.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, et al. (2009). Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med 206, 1273–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders RW, Derking R, Cupo A, Julien J-P, Yasmeen A, de Val N, Kim HJ, Blattner C, de la Peña AT, Korzun J, et al. (2013). A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 9, e1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra S, Tomaras GD, Warrier R, Nicely NI, Liao HX, Pollara J, Liu P, Alam SM, Zhang R, Cocklin SL, et al. (2015). Human Non-neutralizing HIV-1 Envelope Monoclonal Antibodies Limit the Number of Founder Viruses during SHIV Mucosal Infection in Rhesus Macaques. PLoS Pathog. 11, e1005042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders KO (2019). Conceptual approaches to modulating antibody effector functions and circulation half-life. Front. Immunol 10, 1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TYK, Pietzsch J, Fenyo D, Abadir A, Velinzon K, et al. (2011). Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333, 1633–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu C-L, Lorenzi JCC, Feldmann A, Braunschweig M, Nogueira L, Oliveira T, et al. (2016). HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 535, 556–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sips M, Krykbaeva M, Diefenbach TJ, Ghebremichael M, Bowman BA, Dugast A-S, Boesch AW, Streeck H, Kwon DS, Ackerman ME, et al. (2016). Fc receptor-mediated phagocytosis in tissues as a potent mechanism for preventive and therapeutic HIV vaccine strategies. Mucosal Immunol. 9, 1584–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sok D, van Gils MJ, Pauthner M, Julien J-P, Saye-Francisco KL, Hsueh J, Briney B, Lee JH, Le KM, Lee PS, et al. (2014). Recombinant HIV envelope trimer selects for quaternary-dependent antibodies targeting the trimer apex. Proc. Natl. Acad. Sci. USA 111, 17624–17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, and Carragher B (2005). Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60. [DOI] [PubMed] [Google Scholar]
- Tan Y-C, Blum LK, Kongpachith S, Ju C-H, Cai X, Lindstrom TM, Sokolove J, and Robinson WH (2014). High-throughput sequencing of natively paired antibody chains provides evidence for original antigenic sin shaping the antibody response to influenza vaccination. Clin. Immunol 151, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay MZ, Liu P, Williams LD, McRaven MD, Sawant S, Gurley TC, Xu TT, Dennison SM, Liao HX, Chenine AL, et al. (2016). Antibody-Mediated Internalization of Infectious HIV-1 Virions Differs among Antibody Isotypes and Subclasses. PLoS Pathog. 12, e1005817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomann M, Reckermann K, Reusch D, Prasser J, and Tejada ML (2016). Fc-galactosylation modulates antibody-dependent cellular cytotoxicity of therapeutic antibodies. Mol. Immunol 73, 69–75. [DOI] [PubMed] [Google Scholar]
- Torrents de la Peña A, Rantalainen K, Cottrell CA, Allen JD, van Gils MJ, Torres JL, Crispin M, Sanders RW, and Ward AB (2019). Similarities and differences between native HIV-1 envelope glycoprotein trimers and stabilized soluble trimer mimetics. PLoS Pathog. 15, e1007920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler DS, Stanley SD, Zolla-Pazner S, Gorny MK, Shadduck PP, Langlois AJ, Matthews TJ, Bolognesi DP, Palker TJ, and Weinhold KJ (1990). Identification of sites within gp41 that serve as targets for antibody-dependent cellular cytotoxicity by using human monoclonal antibodies. J. Immunol 145, 3276–3282. [PubMed] [Google Scholar]
- Veillette M, Coutu M, Richard J, Batraville L-A, Dagher O, Bernard N, Tremblay C, Kaufmann DE, Roger M, and Finzi A (2015). The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J. Virol 89, 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bredow B, Arias JF, Heyer LN, Gardner MR, Farzan M, Rakasz EG, and Evans DT (2015). Envelope Glycoprotein Internalization Protects Human and Simian Immunodeficiency Virus-Infected Cells from Antibody-Dependent Cell-Mediated Cytotoxicity. J. Virol 89, 10648–10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bredow B, Arias JF, Heyer LN, Moldt B, Le K, Robinson JE, Zolla-Pazner S, Burton DR, and Evans DT (2016). Comparison of Antibody-Dependent Cell-Mediated Cytotoxicity and Virus Neutralization by HIV-1 Env-Specific Monoclonal Antibodies. J. Virol 90, 6127–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien J-P, Wang S-K, Ramos A, Chan-Hui P-Y, Moyle M, et al. ; Protocol G Principal Investigators (2011). Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yang Z-Y, Li Y, Hogerkorp C-M, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, et al. (2010). Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Pegu A, Rao E, Doria-Rose N, Beninga J, McKee K, Lord DM, Wei RR, Deng G, Louder M, et al. (2017). Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science 358, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W-H, Su D, Torabi J, Fennessey CM, Shiakolas A, Lynch R, Chun T-W, Doria-Rose N, Alter G, Seaman MS, et al. (2019). Predicting the broadly neutralizing antibody susceptibility of the HIV reservoir. JCI Insight 4, e130153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
3D reconstruction of AMC018 SOSIPv4.2 in complex with mAb RI808 has been deposited in the EM Data Bank (EMD-22163). The amino acid sequences of the HIV controller monoclonals, CDR3 length, somatic hypermutation levels and relative binding data displayed in Figure 2 are available in Table S2.