

Abstract
Cytomegalovirus (CMV) infection is a common complication of allogeneic hematopoietic cell transplantation (HCT). In this trial, we randomized adult CMV-seropositive HCT recipients without CMV viremia at screening 2:1 to receive brincidofovir or placebo until week 14 post-HCT. Randomization was stratified by center and risk of CMV infection. Patients were assessed weekly through week 15 and every third week thereafter through week 24 post-HCT. Patients who developed clinically significant CMV infection (CS-CMVi; CMV viremia requiring preemptive therapy or CMV disease) discontinued the study drug and began anti-CMV treatment. The primary endpoint was the proportion of patients with CS-CMVi through week 24 post-HCT; patients who discontinued the trial or with missing data were imputed as primary endpoint events. Between August 2013 and June 2015, 452 patients were randomized at a median of 15 days after HCT and received study drug. The proportion of patients who developed CS-CMVi or were imputed as having a primary endpoint event through week 24 was similar between brincidofovir-treated patients and placebo recipients (155 of 303 [51.2%] versus 78 of 149 [52.3%]; odds ratio, .95 [95% confidence interval, .64 to 1.41]; P = .805); fewer brincidofovir recipients developed CMV viremia through week 14 compared with placebo recipients (41.6%; P < .001). Serious adverse events were more frequent among brincidofovir recipients (57.1% versus 37.6%), driven by acute graft-versus-host disease (32.3% versus 6.0%) and diarrhea (6.9% versus 2.7%). Week 24 all-cause mortality was 15.5% among brincidofovir recipients and 10.1% among placebo recipients. Brincidofovir did not reduce CS-CMVi by week 24 post-HCT and was associated with gastrointestinal toxicity.
Keywords: Cytomegalovirus, Brincidofovir, CMX001, Antiviral, Allogeneic hematopoietic cell, transplantation, Prophylaxis
INTRODUCTION
Cytomegalovirus (CMV) infection is associated with increased morbidity and mortality in allogeneic hematopoietic cell transplantation (HCT) [1,2]. Advances in molecular detection for CMV reactivation and preemptive antiviral therapy have reduced CMV disease [3,4], yet CMV seropositivity [5–9] and early CMV reactivation remain associated with increased all-cause mortality after allogeneic HCT [9,10].
Ganciclovir and its orally bioavailable prodrug valganciclovir remain the most commonly used drugs for preemptive therapy [3,10–12]; however, ganciclovir use in CMV prophylaxis in HCT recipients has demonstrated no overall benefit over placebo due to myelosuppression and associated bacterial and fungal infections [13,14]. Therefore, prophylaxis with novel antiviral agents with acceptable safety profiles that could be started early after HCT may improve CMV-related outcomes [12,15].
Brincidofovir (CMX001) is an orally bioavailable lipid conjugate of cidofovir with demonstrated antiviral activity in vitro and in animal models against CMV [16–18] and other double-stranded DNA (dsDNA) viruses, including adenoviruses [19,20], herpesviruses [21,22], orthopoxviruses [23–28], and polyomaviruses [29–32]. Brincidofovir has a long half-life [33,34] and, unlike cidofovir, is not nephrotoxic because it is neither a substrate of organic ion transporter 1 nor concentrated in the renal proximal tubules [33–37]. These characteristics make brincidofovir an attractive candidate for antiviral prophylaxis for allogeneic HCT recipients, who are frequently affected by multiple viral infections [38,39].
In a phase 2 dose-ranging trial for CMV prevention in allogeneic HCT recipients, brincidofovir 100 mg twice weekly (BIW) significantly reduced CMV events when started after engraftment through week 13 post-HCT compared with placebo [40]. Dose-limiting toxicity occurred in the brincidofovir 200 mg BIW dose cohort. This cohort experienced severe diarrhea and other gastrointestinal adverse events (AEs), including gastrointestinal acute graft-versus-host disease (GVHD). These findings prompted the implementation of a safety monitoring and management plan (SMMP) for the brincidofovir 100 mg BIW cohort [40]. Overall tolerability and safety of brincidofovir was acceptable in cohorts dosed at ≤200 mg/week. There was no evidence of dose-dependent myelotoxicity [41] or nephrotoxicity [37,40]. Given these results, we conducted a phase 3 trial of brincidofovir for the prevention of clinically significant CMV infection in allogeneic HCT recipients (SUPPRESS Trial).
METHODS
Patients and Study Design
We enrolled CMV-seropositive patients age ≥18 years who had undergone allogeneic HCT from 44 centers in the United States, Canada, and Belgium (Appendix). Patients were eligible for participation if they were within 28 days post-transplantation, able to ingest tablets, and without detectable CMV DNA within 5 days before randomization. Exclusion criteria included body weight ≥120 kg, receipt of anti-CMV therapy post-transplantation, severe liver injury, estimated glomerular filtration rate <15 mL/min, and stage ≥2 gastrointestinal GVHD [42]. Neutrophil engraftment was not required for eligibility, given brincidofovir’s lack of myelotoxicity observed in the phase 2 trial [40]. Complete eligibility criteria are provided in Supplementary Methods, Section 1. This study was registered at ClinicalTrials.gov (NCT01769170) and at EudraCT (NCT01769170).
Eligible patients were randomized 2:1 to receive brincidofovir or placebo using an interactive Web-response system and concealed assignment, with the use of permuted blocks of 6, through week 14 (day +100) post-transplantation. Randomization was stratified by study center and risk of CMV disease progression. Patients were considered at higher risk if they received cord blood or ex vivo T cell-depleted grafts, or grafts from unrelated, mismatched, or haploidentical donors; received anti-thymocyte globulin (ATG) or alemtuzumab; or were being treated with prednisone ≥1 mg/kg/day (or equivalent). Patients who received grafts from matched-related donors without higher-risk features were considered at lower risk of CMV disease progression [3,4].
Randomized patients received one 100-mg brincidofovir tablet BIW (at alternating 3- and 4-day intervals) or one matching placebo tablet, preferably within 30 minutes after finishing a low-fat meal. Patients, study staff, and sponsors were blinded to study group assignment. Patients were permitted to use acyclovir (≤2000 mg/day) or valacyclovir (≤3000 mg/day) for herpesvirus prophylaxis or treatment following local practice.
Procedures
All patients were evaluated weekly from randomization through week 15, then every 3 weeks through week 24 post-transplantation. Plasma CMV DNA was tested at every visit in the central laboratories (lower limit of quantification, 151 copies/mL [137 IU/mL]; Supplementary Methods, Section 2). Additional plasma and urine samples were obtained for measurement of plasma brincidofovir concentrations and for future assessments of other dsDNA viruses (herpes simplex virus, varicella zoster virus, Epstein-Barr virus, human herpesvirus 6 [HHV-6], adenoviruses, and BK and JC polyomaviruses); these specimens could be used for real-time measurements if clinically indicated (Supplementary Methods, Sections 3 and 7). Patients were also evaluated at each visit for clinical manifestations of BK polyomavirus and HHV-6 (Supplementary Methods, Section 7). Detailed study assessments are presented in Supplementary Table S1.
Patients who developed clinically significant CMV infection, defined as CMV disease [43] or central laboratory CMV viremia requiring preemptive treatment [12], discontinued study drug and began anti-CMV treatment following local practice. The protocol-specified CMV DNA treatment thresholds were ≥151 copies/mL for patients at higher risk of CMV disease progression and ≥1000 copies/mL for patients who were at lower risk at the time of CMV viremia (see Supplementary Figure S2 for details).
AEs and concomitant medications were recorded through week 24 post-transplantation. AE severity was graded using the Common Terminology Criteria for Adverse Events [44]. Diarrhea, other gastrointestinal AEs, and liver enzyme elevations were managed according to the SMMP (Supplementary Methods, Section 8). The SMMP specified scenarios for study drug interruptions (up to 4 doses over 18 days) and discontinuation, and defined criteria for restarting the study drug at the randomized dose, a consolidated dose (200 mg once weekly [QW]), or a reduced dose (100 mg QW) after improvement or resolution of these AEs.
A brincidofovir population pharmacokinetic model was developed and used to explore brincidofovir exposures and their relationship with gastrointestinal AEs (Supplementary Methods, Sections 4 and 5).
Endpoints
The primary endpoint was the proportion of patients with clinically significant CMV infection through week 24 post-transplantation. Patients with missing data at week 24 for any reason (eg, death, withdrawal of consent, loss to follow-up) were imputed as having a primary endpoint event [12]. Key secondary endpoints included the incidence of clinically significant CMV infection through week 14 and time to clinically significant CMV infection. Other secondary endpoints included the incidence and time to all-cause mortality and nonrelapse mortality, incidence, and time to adjudicated BK polyomavirus disease and other dsDNA virus-related clinical disease. All endpoints were assessed at the end of the study drug treatment period (week 14) and at the end of post-treatment follow-up (week 24).
Safety endpoints included the incidence of AEs, diarrhea and other gastrointestinal events, adjudicated acute GVHD (particularly gut GVHD), hepatobiliary laboratory events, and AEs leading to dose interruption, dose reduction, or drug discontinuation.
Study Oversight
The protocol was approved by each center’s ethics committees and conducted in accordance to the International Conference on Harmonization guideline for Good Clinical Practice and the Declaration of Helsinki. An independent unblinded Data Safety Monitoring Board (DSMB) convened to review study data and provide guidance regarding study continuation. The DSMB met 8 times from November 2013 to June 2015; no changes to study design or enrollment were recommended.
Two independent blinded adjudication committees convened to review endpoint events. An Endpoint Adjudication Committee (EAC) reviewed all investigator-reported CMV disease events (and disease events for other dsDNA viruses) and all deaths to assess whether CMV disease criteria had been met [43] and whether deaths were relapse-related or attributable to CMV. A GVHD Adjudication Committee (GAC) reviewed blinded patient-level supporting information of investigator-reported acute GVHD events. The GAC assessed whether the GVHD diagnosis was likely, presumptive, or unlikely based on risk factors, clinical characteristics, biopsy results, and response to treatment. For presumptive or likely GVHD cases, the GAC assessed maximum organ stage and overall grade [42]. Endpoint adjudication decisions were done periodically, were not communicated to site investigators, and did not impact real-time patient care.
Statistical Analysis
We estimated that at least 30% of patients receiving placebo would develop clinically significant CMV infection by week 24 post-transplantation and that a 50% reduction with brincidofovir would be clinically meaningful. A 2-group continuity-corrected χ2 test with a .05 2-sided significance level would provide >85% power to detect this difference when 360 patients were allocated at a 2:1 ratio. To account for an estimated 20% dropout rate, we planned to randomize 450 patients.
The primary endpoint was compared using a 2-sided Cochran-Mantel-Haenszel test, stratified by risk of CMV disease progression at randomization, with an α level of .05, in the study’s intention-to-treat population (ie, randomized patients who received at least 1 dose of study drug). Dichotomous secondary endpoints were analyzed using the same method; missing data were imputed as not having achieved the pertinent endpoint. Time-to-event analyses were performed using the Kaplan-Meier method and log-rank tests. Secondary and exploratory analyses did not control for multiplicity of inferences. All analyses were performed using SAS version 9.1.3 (SAS Institute, Cary, NC).
RESULTS
Study Population
Between August 22, 2013, and June 5, 2015, a total of 568 patients provided consent and were assessed for eligibility (Figure 1). Of these, 110 patients were excluded, primarily due to detectable CMV DNA during screening (79 patients [71.8%]). Among the 458 randomized patients, 6 patients (4 in the brincidofovir arm and 2 in the placebo arm) were never dosed, and 5 discontinued trial participation. Death (61 patients [13.5%]) and withdrawal of consent (34 patients [7.5%]) were the most common reasons for discontinuing study participation through week 24. Deviations from eligibility criteria were documented in 17 treated patients (3.8%); 4 patients had detectable CMV DNA before randomization, 3 patients began the study drug at >28 days post-transplantation, 3 patients enrolled based on local laboratory results, 2 patients received anti-CMV medications post-HCT, 2 patients underwent CMV screening >5 days before randomization, 2 patients weighed >120 kg, and 1 patient was not screened for hepatitis C; these patients continued remained in the trial. CMV risk stratification was misclassified at randomization in 14 patients (3.1%); data from these patients were analyzed according to their verified CMV risk.
Figure 1.
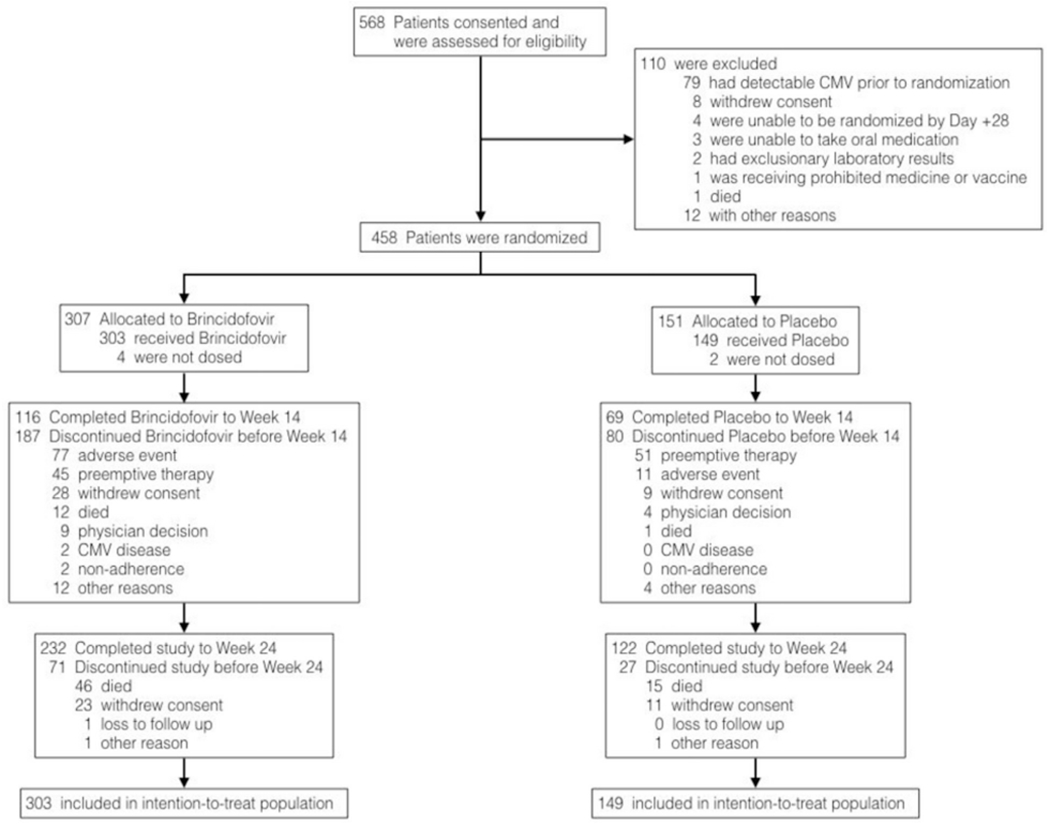
Consent, screening, enrollment, randomization, and follow-up.
The study arms were balanced in terms of baseline characteristics (Table 1) except for sex distribution; 46.2% of the brincidofovir-treated patients were female, compared with 34.2% of the placebo-treated patients. Overall, 332 patients (73.5%) were at higher risk of CMV disease progression, including 210 (46.5%) who received grafts from matched-unrelated donors, 132 (29.2%) who received ATG, and 56 (12.4%) who received T cell-depleted grafts. Myeloablative conditioning was used in 248 patients (54.9%) and tacrolimus-based GVHD prophylaxis was prescribed to 376 patients (83.2%).
Table 1.
Baseline Characteristics of Patients Who Underwent Randomization and Received the Study Drug, Intention-to-Treat Population
| Characteristic | Brincidofovir (N = 303) | Placebo (N = 149) |
|---|---|---|
| Age, yr, median (range) | 56 (18-77) | 54 (20-75) |
| Male sex, n (%) | 163 (53.8) | 98 (65.8) |
| Race, n (%) | ||
| White | 255 (84.2) | 123 (82.6) |
| African American | 24 (7.9) | 14 (9.4) |
| Asian | 17 (5.6) | 10 (6.7) |
| Other | 7 (2.3) | 2 (1.3) |
| Hispanic or Latino ethnicity, n (%) | 27 (8.9) | 13 (8.7) |
| Weight, kg, median (range) | 78.7 (42.2-122.0) | 75.3 (44.0-138.3) |
| Body mass index, kg/m2, median (range) | 26.2 (16.7-44.6) | 26.1 (17.9-43.0) |
| Indication for allogeneic HCT, n (%) | ||
| Acute myelogenous leukemia | 129 (42.6) | 64 (43.0) |
| Myelodysplastic syndrome | 52 (17.2) | 24 (16.1) |
| Non-Hodgkin lymphoma | 28 (9.2) | 18 (12.1) |
| Acute lymphocytic leukemia | 29 (9.6) | 13 (8.7) |
| Chronic myelogenous leukemia | 10 (3.3) | 6 (4.0) |
| Chronic lymphocytic leukemia | 10 (3.3) | 6 (4.0) |
| Aplastic anemia | 9 (3.0) | 7 (4.7) |
| Other diseases | 36 (11.9) | 11 (7.4) |
| Graft source, n (%) | ||
| Peripheral blood | 241 (79.5) | 113 (75.8) |
| Bone marrow | 41 (13.5) | 24 (16.1) |
| Cord blood | 19 (6.3) | 11 (7.4) |
| Other* | 2 (.7) | 7 (0.7) |
| Donor type, n(%)† | ||
| Matched unrelated | 148 (48.8) | 62 (41.8) |
| Matched related | 97 (32.0) | 52 (34.9) |
| Mismatched | 23 (7.6) | 15 (10.1) |
| Haploidentical | 14 (4.6) | 8 (5.4) |
| Donor CMV seropositive, n (%) | 154 (50.8) | 84 (56.4) |
| Myeloablative conditioning regimen, n (%) | 162 (53.5) | 86 (57.7) |
| Ex vivo T cell depletion,‡ n (%) | 36 (11.9) | 20 (13.4) |
| Alemtuzumab use, n (%) | 26 (8.6) | 12 (8.1) |
| ATG globulin use, n (%) | 85 (28.1) | 47 (31.5) |
| Acute GVHD at baseline, n (%) | 10 (3.3) | 6 (4.0) |
| Glucocorticoids at ≥1 mg/kg prednisone equivalent, n (%) | 2 (.7) | 3 (2.0) |
| Immunosuppressant use at baseline, n (%) | ||
| Tacrolimus | 251 (82.8) | 121 (81.2) |
| Mycophenolate | 87 (28.7) | 45 (30.2) |
| Cyclosporine | 43 (14.2) | 24 (16.1) |
| Sirolimus | 28 (9.2) | 10 (6.7) |
| Time to randomization after HCT, d, median (range) | 15 (3-33) | 14 (2-29) |
| Absolute neutrophil count <500 cells/μL at baseline, n (%) | 116 (38.3) | 58 (38.9) |
| CMV DNA PCR at randomization, n (%) | ||
| Not detected | 288 (95.0) | 137 (91.9) |
| Detected, <151 copies/mL§ | 15 (5.0) | 12 (8.1) |
| Risk of CMV disease progression,** n (%) | ||
| Higher risk | 223 (73.6) | 109 (73.2) |
| Lower risk | 80 (26.4) | 40 (26.8) |
Three patients from 1 site underwent allogeneic HCT using a combination of adult haploidentical peripheral blood and cord blood grafts.
Cord-blood and “other” grafts not included.
T cell depletion, CD34+ selection, and/or other.
No patient had a quantifiable CMV virus load at randomization.
Stratification criterion at randomization. Patients were considered at higher risk of CMV disease progression if they received cord blood or ex vivo T cell-depleted grafts or grafts from unrelated, mismatched, or haploidentical donors; received antithymocyte globulin or alemtuzumab; or were being treated with ≥1 mg/kg/day of prednisone (or equivalent) for acute GVHD or other conditions. Patients who received grafts from matched-related donors without any higher-risk features were considered at lower risk of CMV disease progression.
Treatment Exposure
Patients were started study drug at a median of 15 days after HCT (range, 2 to 33 days); 278 (61.5%) experienced engraftment at randomization. Study drug exposure was 54 days (range, 1-99) for brincidofovir-treated patients and 50 days (range, 1 to 100 days) for placebo-treated patients. Treatment interruptions of ≥10 days were more frequent in the brincidofovir arm (107 [35.3%] versus 13 [8.7%] for placebo). Dose modifications and reductions were implemented in 14 (4.6%) and 6 (2.0%) brincidofovir-treated patients, respectively, but not in any placebo-treated patients. Among brincidofovir-treated patients, the mean brincidofovir steady-state area under the plasma concentration-time curve (AUCss) and maximum plasma concentration (Cmax,ss) were 4283 ng·h/mL (90% confidence interval [CI], 1726 to 10,625 ng·h/mL) and 140 ng/mL (90% CI, 44.8 to 435 ng/mL), respectively.
Primary Endpoint
The proportion of patients who developed clinically significant CMV infection or were imputed as having a primary endpoint event through week 24 post-transplantation (Table 2) was not significantly different between patients treated with brincidofovir and those who received placebo (155 of 303 [51.2%] versus 78 of 149 [52.3%]); the brincidofovir treatment odds ratio for developing clinically significant CMV infection, adjusted for the risk of CMV disease progression, was .95 (95% CI, .64 to 1.41; P = .805).
Table 2.
Incidence of Clinically Significant CMV Infection through Week 14 (End of Treatment) and Week 24 (End of Study) Post-Transplantation, Intention-to-Treat Population
| Endpoints and Components | Brincidofovir (N = 303) | Placebo (N = 149) |
|---|---|---|
| Clinically significant CMV infection* through week 14, n (%) | 115 (38.0) | 69 (46.3) |
| CMV disease | 7 (2.3) | 3 (2.0) |
| Initiation of preemptive therapy for CMV infection | 67 (22.1) | 54 (36.2) |
| Death (without a previous CMV event) | 22 (7.3) | 3 (2.0) |
| Missing (other than death) | 19 (6.3) | 9 (6.0) |
| OR (95% CI) | .70 (.47-1.05) | |
| P value† | .085 | |
| Clinically significant CMV infection through week 24, n (%)‡ | 155 (51.2) | 78 (52.3) |
| CMV disease | 13 (4.3) | 5 (3.4) |
| Initiation of preemptive therapy for CMV infection | 88 (29.0) | 56 (37.6) |
| Death (without a previous CMV event) | 33 (10.9) | 6 (4.0) |
| Missing (other than death) | 21 (6.9) | 11 (7.4) |
| OR (95% CI) | .95 (.64-1.41) | |
| P value† | .805 |
For patients with more than one event, primary endpoint was assigned by the hierarchy listed here.
P values and ORs are from a Cochran-Mantel-Haenszel test stratified by likelihood of progression to CMV disease.
Trial’s primary efficacy endpoint.
Secondary Endpoints
By week 14 post-transplantation, the rate of clinically significant CMV infection was lower in the brincidofovir arm compared with the placebo arm (74 of 303 [24.4%] versus 57 of 149 [38.3%]; P = .002), but the difference was not significant when imputed events were considered (Table 2). Time to clinically significant CMV infection through week 24 is presented in Figure 2A; the probability of experiencing clinically significant CMV infections was lower in the brincidofovir group compared with the placebo group (P = .033, log-rank test). Adjudicated CMV disease by week 24 occurred in 13 patients (4.3%) in the brincidofovir arm and in 5 patients (3.4%) in the placebo arm (Table 2). Gastrointestinal CMV disease was documented in 14 (77.8%) of these patients (11 in the brincidofovir arm and 3 in the placebo arm). No deaths were attributed to CMV disease. Fewer brincidofovir-treated patients than placebo patients had quantifiable CMV viremia in central laboratory measurements through both week 14 (78 [25.7%] versus 62 [41.6%]; P < .001) and week 24 (92 [30%] versus 63 [42.3%]; P = .012; Supplementary Figure S4). No cidofovir-associated resistance mutations were observed in CMV UL54 sequences from brincidofovir-treated patients (Supplementary Methods, Section II.2.2).
Figure 2.
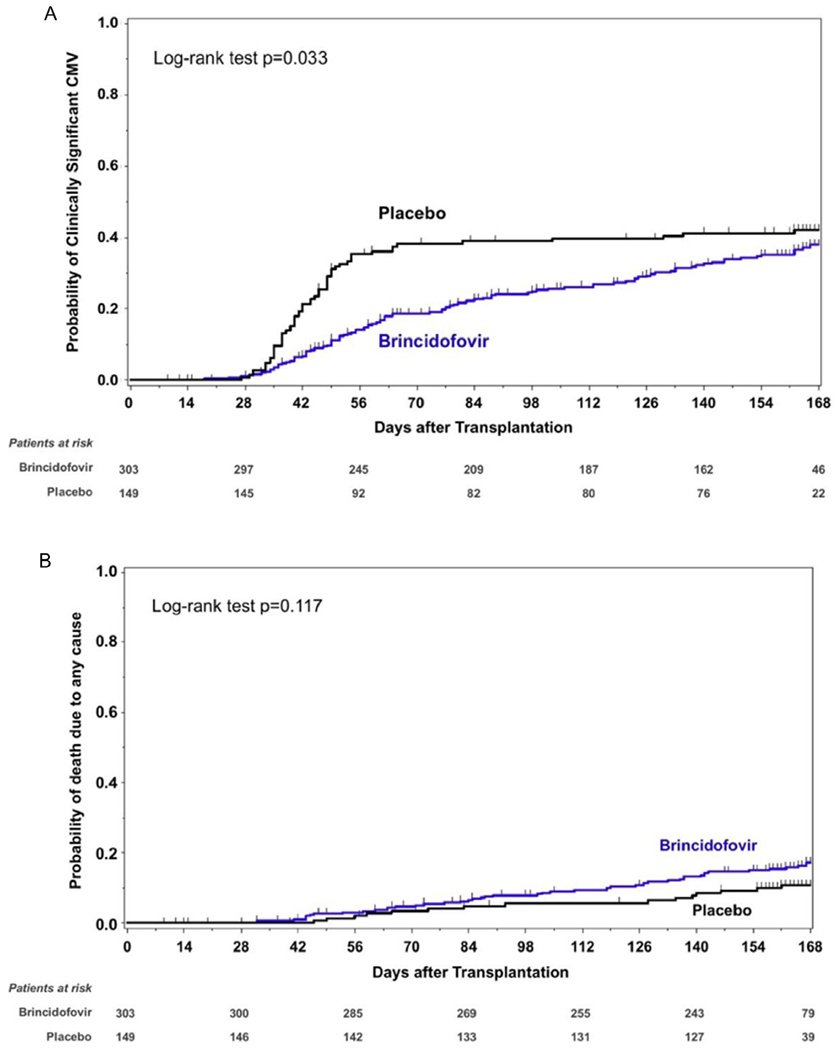
Time to clinically significant CMV infection (A) and time to all-cause mortality (B) from day of transplantation through week 24 post-transplantation, intention-to-treat population. Patients without a clinically significant CMV infection were censored at the end-of-study visit or at 24 weeks (+2-week window) after transplantation, whichever was earlier. For mortality through week 24, patients who died after week 24 post-transplantation were censored at the end-of-study visit or week 24, whichever was earlier.
Sixty-two patients died through week 24 post-transplantation, including 47 (15.5%) who received brincidofovir and 15 (10.1%) who received placebo. Time to all-cause mortality through week 24 is presented in Figure 2B; the hazard ratio of death for brincidofovir-treated patients was 1.6 (95% CI, .9 to 2.8; P = .117, log-rank test). The week 24 cumulative nonrelapse mortality was 10.2% in the brincidofovir arm and 6.7% in the placebo arm. Six patients (2.0%) in the brincidofovir arm and 4 patients (2.7%) in the placebo arm experienced graft failure through week 24.
Other dsDNA Virus Diseases
Adjudicated clinical disease caused by other dsDNA viruses different from CMV occurred in 50 (16.5%) brincidofovir-treated patients and in 21 (14.1%) placebo-treated patients through week 14 post-HCT (Supplementary Table S3); the incidence was 20.5% and 15.4%, respectively, through week 24 post-HCT. BK polyomavirus disease was the most frequent dsDNA viral disease; 40 patients (13.2%) who received brincidofovir were diagnosed with BK polyomavirus disease through week 14 post-transplantation, compared with 14 (9.4%) who received placebo (Supplementary Table S3 and Figure S3); the incidence was 14.9% and 11.4%, respectively, through week 24 post-HCT. The incidence of clinical disease caused by other dsDNA viruses was low (≤3%) and similar in the 2 study arms (Supplementary Table S3).
Efficacy Subgroup Analyses
Among patients at higher risk of CMV disease progression, the incidence of clinically significant CMV infection was lower in the brincidofovir-treated patients compared with the placebo-treated patients through week 24 post-transplantation; however, among patients at lower risk of CMV disease progression, the risk of clinically significant CMV infection was greater in the brincidofovir arm, with hazards of the 2 groups crossing around day +100 (Figure 3A and B). This difference was driven mainly by patients who underwent T cell depletion strategies (ie, use of ATG, alemtuzumab, or ex vivo T cell depletion; Figure 3C and D). Reduction in clinically significant CMV infection in brincidofovir recipients was minimal among patients who underwent myeloablative conditioning compared with those who underwent reduced-intensity conditioning (Figure 3E and F). These reductions in clinically significant CMV infection in some subgroups were not associated with improved survival (Supplementary Table S4).
Figure 3.
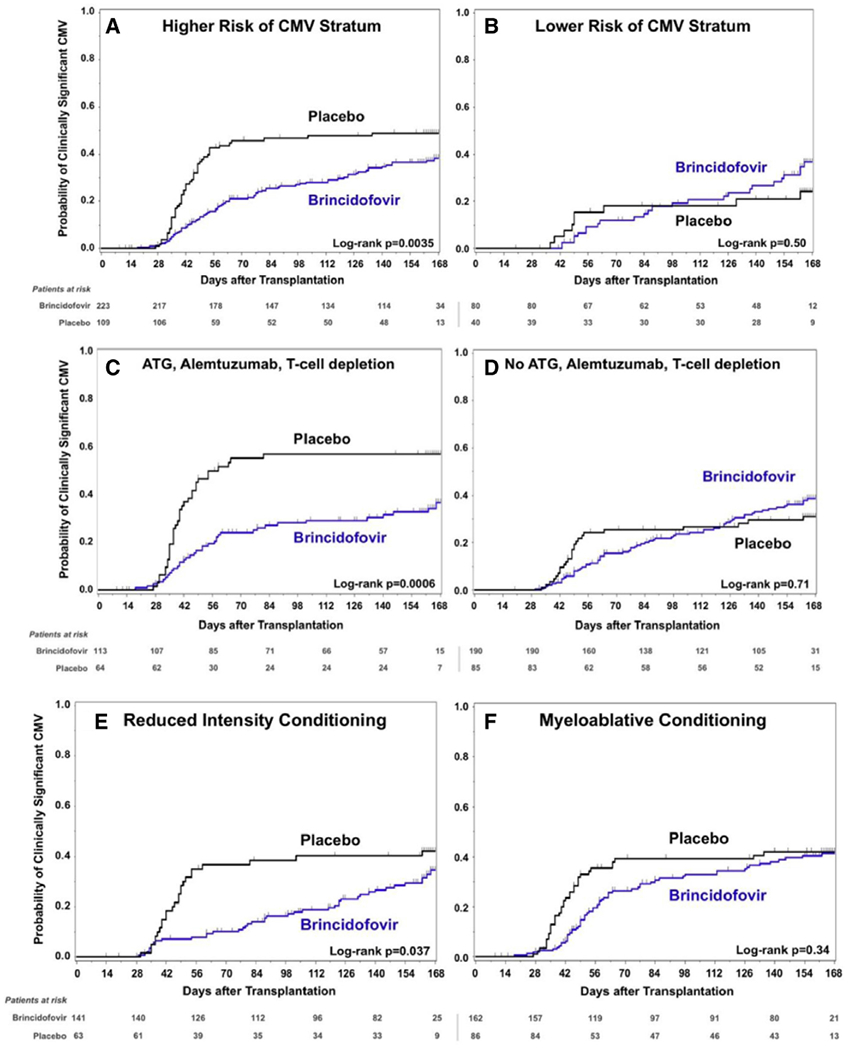
Time to clinically significant CMV infection according to several subgroups. (A and B) Results by randomization strata groups of higher and lower risk of CMV disease progression. (C and D) Post hoc results according to use of T cell depletion strategies (ATG, alemtuzumab, or ex vivo T cell depletion) (C) and all other patients (D). (E and F) Post hoc results according to conditioning regimen, either reduced-intensity (E) or myeloablative (F) conditioning.
Safety Analyses
A total of 116 (38.3%) brincidofovir-treated patients and 69 (46.3%) placebo-treated patients completed treatment through week 14 post-HCT. More patients in the placebo arm discontinued treatment to initiate preemptive therapy (51 [34.2%] versus 45 [14.9%] in the brincidofovir arm), whereas rates of treatment discontinuation due to AEs (77 [25.4%] versus 11 [7.4%]), withdrawal of consent (28 [9.2%] versus 9 [6.0%]), and on-treatment death (12 [4.0%] versus 1 [.7%]) were higher in the brincidofovir arm (Figure 1 and Supplementary Table S5). Brincidofovir-treated patients experienced more AEs grade ≥3 (Supplementary Table S7) and serious AEs (Supplementary Table S8). Gastrointestinal events were the most common AEs reported among brincidofovir-treated patients (Tables 3 and Supplementary Table S6). There was no difference in time to engraftment between the 2 study arms (Supplementary Figure S6). A total of 232 (76.6%) brincidofovir-treated patients and 122 (81.9%) placebo-treated patients completed the study.
Table 3.
Most Common AEs (Reported in ≥10% of Patients) During Study Treatment, Intention-to-Treat Population
| AE | Brincidofovir (N = 303), n(%) | Placebo (N = 149), n (%) | P Value |
|---|---|---|---|
| Any AE | 302 (99.7) | 146 (98.0) | .11 |
| Diarrhea | 184 (60.7) | 54 (36.2) | <.0001 |
| Acute GVHD | 173 (57.1) | 48 (32.2) | <.0001 |
| Abdominal pain | 104 (34.3) | 26 (17.4) | .0002 |
| Nausea | 93 (30.7) | 29 (19.5) | .013 |
| Vomiting | 74 (24.4) | 25 (16.8) | .070 |
| Decreased appetite | 67 (22.1) | 19 (12.8) | .021 |
| Peripheral edema | 52 (17.2) | 18 (12.1) | .17 |
| Hyperglycemia | 48 (15.8) | 11 (7.4) | .012 |
| Hypokalemia | 47 (15.5) | 10 (6.7) | .010 |
| Rash | 43 (14.2) | 28 (18.8) | .22 |
| Fatigue | 42 (13.9) | 28 (18.8) | .21 |
| Fever | 42 (13.9) | 27 (18.1) | .27 |
| Hypomagnesemia | 38 (12.5) | 12 (8.1) | .20 |
| Alanine aminotransferase elevation | 34 (11.2) | 9 (6.0) | .09 |
| Hypertension | 33 (10.9) | 16 (10.7) | 1.0 |
| Mucosal inflammation | 32 (10.6) | 15 (10.1) | 1.0 |
| Headache | 31 (10.2) | 21 (14.1) | .27 |
| Cough | 31 (10.2) | 20 (13.4) | .34 |
| Insomnia | 31 (10.2) | 12 (8.1) | .50 |
AEs reported here began on or after the first dose of study drug up to 7 days after the last dose. AEs were coded using MedDRA dictionary 18.0. Patients were counted only once for each AE regardless of how many events they experienced. P values were calculated using the unadjusted 2-sided Fisher’s exact test.
More brincidofovir-treated patients were diagnosed with acute GVHD (Table 3). Few patients had acute GVHD at baseline (3.3% brincidofovir, 4% placebo; Table 1). More subjects in the brincidofovir arm than in the placebo arm were adjudicated as having likely or presumptive incident acute GVHD (Supplementary Table S9) (201 [66.3%] versus 69 [46.3%]; P < .001). However, most of the excess acute GVHD among brincidofovir-treated patients was grade III GVHD (78 [25.7%] versus 8 [5.4%] in the placebo arm) with gastrointestinal involvement (174 [57.4%] versus 40 [26.8%]; P < .001) stages 1 to 4, in contrast to skin (113 [37.3%] versus 53 [35.6%]) (Supplementary Table S10). The median time from randomization to maximum gastrointestinal GVHD was 29 days in the brincidofovir arm compared to 40.5 days in the placebo arm (hazard ratio, 2.92; 95% CI, 2.05 to 4.15). A blinded review of all available gastrointestinal biopsies confirmed histopathological changes of gastrointestinal GVHD [45–47] in both study arms without the ability to distinguish brincidofovir-treated and placebo-treated patients. An excess in adjudicated liver GVHD was also noted (36 [11.9%] for brincidofovir versus 7 [4.7%] for placebo; P = .016).
The increased incidence of reported GVHD was linked to more frequent systemic glucocorticoid use in the brincidofovir arm (188 [62.0%] versus 72 [48.3%]), including prednisone (134 [44.2%] versus 44 [29.5%]) and methylprednisolone (121 [39.9%] versus 38 [25.5%]). The median cumulative glucocorticoid exposure was >8-fold higher in the brincidofovir arm through week 14 (26 versus 3 mg/kg prednisone equivalents) and remained close to 5-fold higher through week 24 (38 versus 8 mg/kg prednisone equivalents). This likely led to increased infectious disease events other than CMV in the brincidofovir arm (147 [48.5%] versus 49 [32.9%]; Supplementary Table S6) and to increased clinically significant CMV infections after week 14 (Figure 3).
More brincidofovir-treated patients had increases in alanine aminotransferase and bilirubin concentrations that persisted through week 14 (Supplementary Figure S7). More hepatobiliary AEs were reported in the brincidofovir arm (23 [7.6%] versus 5 [3.4%] for placebo; P = .11; Supplementary Table S6). Hyperbilirubinemia was more common in brincidofovir-treated patients who were receiving concomitant cyclosporine compared to those who received concomitant tacrolimus (17.1% [6 of 35] versus 2.4% [6 of 247]); the incidence of hyperbilirubinemia in patients on placebo receiving either cyclosporine or tacrolimus was .7% (1 of 138). Changes in blood cell counts were comparable across treatment arms (Supplementary Figure S7). Changes in estimated glomerular filtration rate were numerically smaller in the brincidofovir and placebo arms (−8.2 mL/min/1.73 m2 versus −13.6 mL/min/1.73 m2; P = .079) by week 14, although more acute kidney AEs were reported in the brincidofovir arm (30 [9.9%] versus 10 [6.7%]), likely secondary to diarrhea-induced prerenal azotemia.
Post Hoc Safety Analyses
The risk of having a diarrheal AE, defined as incident diarrhea grade ≥2 or gastrointestinal GVHD stage ≥1 (excluding patients with baseline diarrhea), was 73.1% (209 of 286) for the brincidofovir arm versus 34.7% (37 of 137) for the placebo arm. Events were less likely at a brincidofovir AUCss below the mean exposure (Figure 4A). There was no appreciable relationship between brincidofovir exposure and grade ≥2 alanine aminotransferase elevations.
Figure 4.
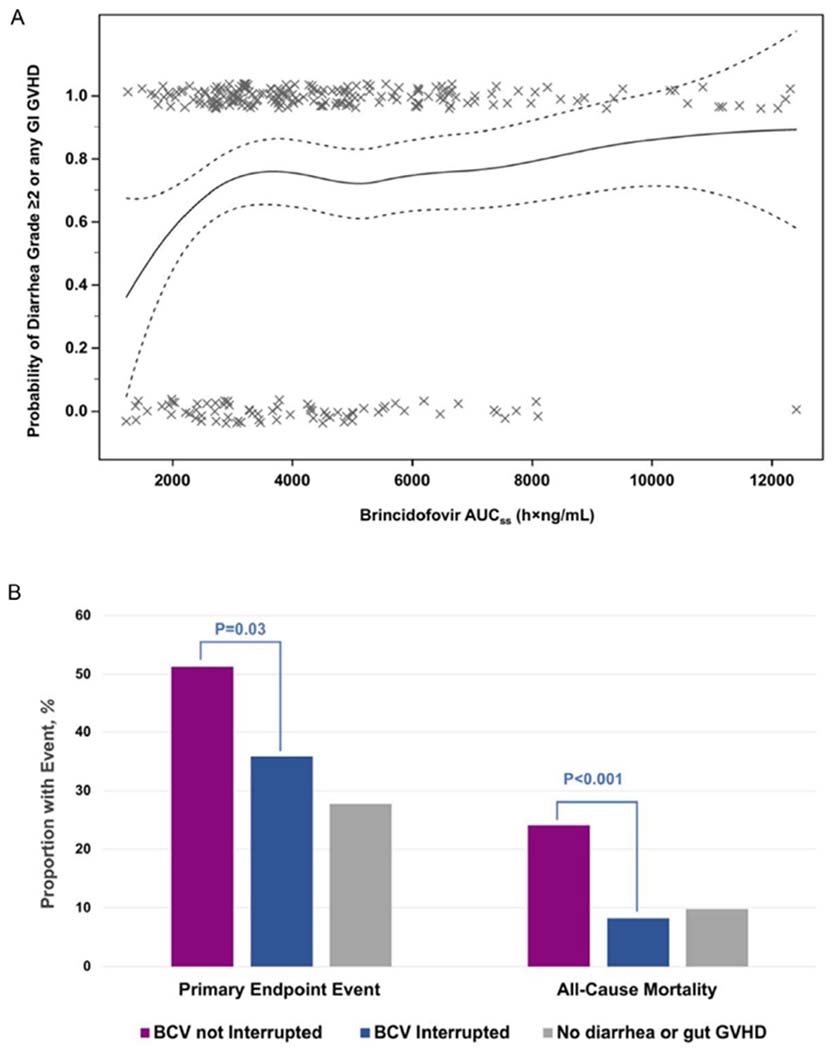
(A) Brincidofovir exposure and risk of diarrhea, Common Terminology Criteria for Adverse Events grade ≥2 or gastrointestinal GVHD stage ≥1. The solid line indicates calculated risk; dotted lines, 95% CIs of calculated risk; X, individual patients who experienced the adverse event if on 1.0 on the y-axis, who did not experience the adverse event if on 0 on the y-axis. (B) Impact of SMMP patient management on clinically significant CMV infection and all-cause mortality from week 8 through week 24 among brincidofovir-treated patients. BCV, brincidofovir; primary endpoint event, clinically significant CMV infection.
Footnote: AUC, area under the curve; BCV, brincidofovir; primary endpoint event, clinically-significant CMV infection. Panel A: solid line, calculated risk; dotted lines, 95% confidence intervals of calculated risk; X, individual patients who experienced the adverse event if on 1.0 on the y axis, who did not experience the adverse event if on 0.0 on the y axis.
We explored the impact of following the SMMP on 258 brincidofovir-treated patients who experienced any diarrhea (grade ≥1) or gastrointestinal GVHD (stage ≥1) through week 8 post-transplantation on outcomes. A total of 151 patients (58.5%) were managed according to the SMMP, 14 (5.4%) were managed more conservatively than prescribed in the SMMP, and in 93 patients (36.0%), brincidofovir was not interrupted or discontinued per the SMMP, and the patients were treated for GVHD. Both primary endpoint events and all-cause mortality were lower when patients were managed according to the SMMP or more conservatively (Figure 4B). There were no appreciable differences in brincidofovir AUCss according to SMMP management (Supplementary Figure S8). Results were similar when diarrhea grade 1 was not considered an event.
The probability of clinically significant CMV infection decreased with increasing brincidofovir exposure during treatment, but the trend reversed after treatment, with patients with higher brincidofovir exposures experiencing more events by week 24 post-transplantation (Supplementary Figure S9).
DISCUSSION
In this phase 3 trial, oral brincidofovir dosed at 100 mg BIW through week 14 post-transplantation was not superior to placebo for the prevention of clinically significant CMV infection through week 24 post-HCT. Although antiviral activity against CMV was demonstrated with fewer events of quantifiable CMV viremia and a decreased need for preemptive therapy, brincidofovir treatment led to increased gastrointestinal toxicity, including excess diagnoses and treatment of acute gastrointestinal GVHD. As a result, brincidofovir use was not associated with improved survival or overall fewer clinical diseases caused by other dsDNA viruses.
Some differences in design between the SUPPRESS phase 3 and the phase 2 trial (CMX001-201) [40] may explain the differences in efficacy and safety seen between the 2 studies. First, SUPPRESS allowed preengraftment enrollment, given the absence of dose-dependent brincidofovir myelotoxicity [40,41]; the median time to the first dose of study drug was 15 days, compared with 24 days in CMX001-201. The objective was to prevent earlier CMV and other dsDNA virus events, but this resulted in frequent initiation of brincidofovir while patients were still experiencing conditioning regimen-associated mucositis. This likely contributed to synergistic enterotoxicity [48], especially in patients who underwent myeloablative HCT, and accounts in part for the different outcomes seen according to conditioning regimen (Figure 3). Second, the primary endpoint for SUPPRESS was set at week 24 [12] and not at the end of treatment (week 14) [40] to assess for postprophylaxis CMV events [49]. This was important in determining not only postprophylaxis antiviral efficacy and potential issues of antiviral resistance, but also the longer-term consequences of the gastrointestinal toxicity observed. Third, the CMX001-201 endpoint was virologic and not linked to the initiation of preemptive therapy, such that patients who received preemptive therapy but did not have confirmed CMV viremia or disease were not considered to have met the study endpoint in CMX001-201 [40].
The potential for brincidofovir-associated diarrhea was expected. Diarrhea was a manageable event in the CMX001-201 brincidofovir 100 mg BIW cohort following the SMMP guidance. In that trial, 60% of patients completed brincidofovir treatment at this dose, compared with 54% of patients who received placebo [40], but completion rates were lower in the present study (38% brincidofovir, 46% placebo). Gastrointestinal events remained the main toxicity for brincidofovir and led to increased diagnosis and treatment of gastrointestinal GVHD. In turn, the more frequent immunosuppressive treatment for presumed grade III acute GVHD with gastrointestinal involvement likely contributed to the trends toward higher all-cause and nonrelapse mortality observed.
The use of an independent GAC was planned to address the issue of empirical treatment for gastrointestinal GVHD in patients presenting with diarrhea, but the blinded adjudication of likely or presumptive acute GVHD events was unable to distinguish patients who received brincidofovir from those who received placebo [50]. Furthermore, a blinded review of all gastrointestinal biopsy specimens was also unable to distinguish histopathological features between brincidofovir-treated and placebo-treated patients [47]. These observations suggest either that brincidofovir can induce acute GVHD events due to enteral injury or that brincidofovir enterotoxicity can cause histopathological changes that cannot be differentiated from those seen in acute GVHD, analogous to what has been described for mycophenolate-associated gastrointestinal toxicity [51,52]. A majority of the excess acute GVHD events reported in brincidofovir-treated patients were limited to the gastrointestinal tract, with otherwise a very similar incidence of reported skin GVHD, which suggests that a mechanism of enteric injury followed by an alloreactive effect could be less likely. Histopathological findings suggestive of acute GVHD have been recently reported in an autologous HCT recipient who received brincidofovir for treatment of adenovirus disease [53]. In addition, histopathological changes were attributed to mycophenolate-induced enterotoxicity in a patient who was receiving brincidofovir after kidney transplantation as part of a phase 3 prophylaxis trial [54] who experienced significant diarrhea that led to endoscopy, but was not receiving mycophenolate treatment (Chimerix, data on file).
A slight increase in adjudicated acute GVHD with liver involvement in brincidofovir-treated patients deserves mention. These events were due predominantly to an excessive number of presumptive liver stage 1 and 2 cases, based on elevated serum bilirubin concentration, in a background of diarrhea without biopsy confirmation (Supplementary Table S10). These cases may represent hyperbilirubinemia caused by brincidofovir rather than acute GVHD. The mechanisms behind these events in the present study are likely multifactorial, but an increased frequency of hyperbilirubinemia events in patients receiving concurrent brincidofovir and cyclosporine suggests a competitive interaction between brincidofovir and bilirubin for liver transport [55] mediated through OATP1B1/3 [56], because both brincidofovir and bilirubin are substrates of OATP1B1/3, and cyclosporine is a known inhibitor of both OATPs [57]
We found no evidence of an increased risk of nephrotoxicity or myelotoxicity in the brincidofovir-treated patients compared with placebo patients, confirming findings from earlier studies [37,40].
Differences in adherence to the SMMP’s recommended treatment interruptions also influenced outcomes. Treatment of brincidofovir-related diarrhea with systemic glucocorticoids in the setting of a presumptive diagnosis of acute gastrointestinal GVHD while maintaining the study drug resulted in continued or worsening diarrhea. In several instances, this was interpreted as refractory GVHD, with subsequent increases in immunosuppression with the addition of second- and third-line therapies. The blinded study, considered necessary even in diseases with objective outcome measures, including CMV viremia, certainly contributed to an increased rate of clinical diagnoses of gastrointestinal GVHD in this complex patient population. This experience also emphasizes the need to carefully evaluate HCT recipients for the etiology of AEs before increasing the doses of immunosuppressive therapy.
Although the efficacy of oral brincidofovir prophylaxis for 10 to 14 weeks in adults early after allogeneic HCT was compromised by gastrointestinal toxicity in this trial, the experience in the treatment of adenovirus disease, especially in children, has been promising [58–63]. This may be due to various factors, including starting brincidofovir treatment later post-HCT, after acute conditioning-related gastrointestinal toxicities have resolved, or possibly age-related differences in the gastrointestinal tracts of adults and children [64]. In any case, the development of an i.v. formulation of brincidofovir [65] that does not result in gastrointestinal accumulation [22] but retains the drug’s spectrum, antiviral activity, and lack of nephrotoxicity or myelotoxicity could provide a more effective and safer prevention and treatment of CMV and the multiple dsDNA viruses that frequently affect allogeneic HCT recipients.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the patients and their families and study center personnel who participated in this study. They also thank the members of the DSMB, EAC, and GAC; Hervé Momméja-Marin for his contributions to the design and conduct of the study; and Aaron Harrison and Suzanne Scott for overseeing the conduct of the study. The authors also acknowledge the invaluable contributions of Howard M. Shulman and George B. McDonald in their review of the gastrointestinal histopathology and GVHD data, respectively, and those of Donald S. David and Paul B. Watkins in their review of the hepatobiliary data.
Financial disclosure:
This study was supported by Chimerix, Inc.
Conflict of interest statement:
FMM: investigator for the SUPPRESS trial at Dana-Farber Cancer Institute and Brigham and Women’s Hospital, which were compensated for participation in the trial; member of ad hoc Chimerix advisory boards; research grant support from Ansun, Astellas, Cidara, Gilead, Merck, and Shire; consultancy fees from Fate Therapeutics, GlaxoSmithKline, Merck, Roche Molecular Systems, and Shire.
D.J.W.: investigator for the SUPPRESS trial at UCLA Medical Center, which was compensated for participation in the trial; research support from Merck, Gilead, Oxford Immunotec, and Shire.
R.F.C.: investigator for the SUPPRESS trial at University of Texas M.D. Anderson Cancer Center, which was compensated for participation in the trial; member of Chimerix advisory boards; research grant support from Merck, Novartis, Oxford Immunotec, Ansun Pharmaceuticals, Gilead, Shire, Pulmotec, Xenex, and AiCuris; consultancy fees from Merck, Oxford Immunotec, Xenex, Chimerix, Ansun Pharmaceuticals, Johnson & Johnson, and Ablynx.
K.M.M.: investigator for the SUPPRESS trial at University of Chicago, which was compensated for participation in the trial; member of Chimerix advisory boards; research support from Actelion Aicuris, Ansun, Astellas, Cidara, Finch, Gilead, Leonard Merin, Merck, Novartis, Rebiotix, Sage, Scynexis, Seres, Shire, Summit, Vertex, and Visterra; advisory boards for Astellas, Merck, GlaxoSmithKline, and Scynexis.
T.B.S.: investigator for the SUPPRESS trial at Weill Cornell Medical College–New York Presbyterian Hospital, which was compensated for participation in the trial.
G.A.P.: investigator for the SUPPRESS trial at Memorial Sloan-Kettering Cancer Center, which was compensated for participation in the trial; member of Chimerix advisory boards; research grant support and consultancy fees from Astellas, Chimerix, Merck, Oxford Immunotec, and Shire.
G.C., T.M.B., C.W., M.E.M., S.A.F., and W.G.N.: employees of and holders of stock in Chimerix.
M.J.B.: investigator for the SUPPRESS trial at Fred Hutchinson Cancer Research Center, which was compensated for participation in the trial; member of Chimerix advisory boards; research support from Astellas, Merck, and Shire; consultancy fees from Artemis Therapeutics, Astellas, Helocyte, Merck, Moderna, Oxford Immunotec, and Shire.
Footnotes
SUPPLEMENTARY DATA
Supplementary data related to this article can be found online at https://doi.org/10.1016/j.bbmt.2018.09.038.
This work was presented in part at the 2016 BMT Tandem Meetings of the Center for International Blood and Marrow Transplant Research and the American Society for Blood and Marrow Transplantation, Honolulu, Hawaii, February 18-22, 2016.
REFERENCES
- 1.Boeckh M, Nichols WG, Papanicolaou G, Rubin R, Wingard JR, Zaia J. Cytomegalovirus in hematopoietic stem cell transplant recipients: current status, known challenges, and future strategies. Biol Blood Marrow Transplant. 2003;9:543–558. [DOI] [PubMed] [Google Scholar]
- 2.Ljungman P, Hakki M, Boeckh M. Cytomegalovirus in hematopoietic stem cell transplant recipients. Infect Dis Clin North Am. 2010;24:319–337. [DOI] [PubMed] [Google Scholar]
- 3.Boeckh M, Ljungman P. How we treat cytomegalovirus in hematopoietic cell transplant recipients. Blood. 2009;113:5711–5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green ML, Leisenring W, Stachel D, et al. Efficacy of a viral load-based, risk-adapted, preemptive treatment strategy for prevention of cytomegalovirus disease after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2012;18:1687–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craddock C, Szydlo RM, Dazzi F, et al. Cytomegalovirus seropositivity adversely influences outcome after T-depleted unrelated donor transplant in patients with chronic myeloid leukaemia: the case for tailored graft-versus-host disease prophylaxis. Br J Haematol. 2001;112:228–236. [DOI] [PubMed] [Google Scholar]
- 6.Boeckh M, Nichols WG. The impact of cytomegalovirus serostatus of donor and recipient before hematopoietic stem cell transplantation in the era of antiviral prophylaxis and preemptive therapy. Blood. 2004;103:2003–2008. [DOI] [PubMed] [Google Scholar]
- 7.Marty FM, Bryar J, Browne SK, et al. Sirolimus-based graft-versus-host disease prophylaxis protects against cytomegalovirus reactivation after allogeneic hematopoietic stem cell transplantation: a cohort analysis. Blood. 2007;110:490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ljungman P, Brand R, Hoek J, et al. Donor cytomegalovirus status influences the outcome of allogeneic stem cell transplant: a study by the European Group for Blood and Marrow Transplantation. Clin Infect Dis. 2014;59:473–481. [DOI] [PubMed] [Google Scholar]
- 9.Teira P, Battiwalla M, Ramanathan M, et al. Early cytomegalovirus reactivation remains associated with increased transplant-related mortality in the current era: a CIBMTR analysis. Blood. 2016;127:2427–2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green ML, Leisenring W, Xie H, et al. Cytomegalovirus viral load and mortality after haemopoietic stem cell transplantation in the era of pre-emptive therapy: a retrospective cohort study. Lancet Haematol. 2016;3:e119–e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marty FM, Ljungman P, Papanicolaou GA, et al. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect Dis. 2011;11:284–292. [DOI] [PubMed] [Google Scholar]
- 12.Marty FM, Ljungman P, Chemaly RF, et al. Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N Engl J Med. 2017;377:2433–2444. [DOI] [PubMed] [Google Scholar]
- 13.Goodrich JM, Bowden RA, Fisher L, Keller C, Schoch G, Meyers JD. Ganciclovir prophylaxis to prevent cytomegalovirus disease after allogeneic marrow transplant. Ann Intern Med.1993;118:173–178. [DOI] [PubMed] [Google Scholar]
- 14.Winston DJ, Ho WG, Bartoni K, et al. Ganciclovir prophylaxis of cytomegalovirus infection and disease in allogeneic bone marrow transplant recipients: results of a placebo-controlled, double-blind trial. Ann Intern Med. 1993;118:179–184. [DOI] [PubMed] [Google Scholar]
- 15.Marty FM, Boeckh M. Maribavir and human cytomegalovirus-what happened in the clinical trials and why might the drug have failed? Curr Opin Virol. 2011;1:555–562. [DOI] [PubMed] [Google Scholar]
- 16.Beadle JR, Hartline C, Aldern KA, et al. Alkoxyalkyl esters of cidofovir and cyclic cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob Agents Chemother. 2002;46:2381–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kern ER, Bidanset DJ, Hartline CB, Yan Z, Zemlicka J, Quenelle DC. Oral activity of a methylenecyclopropane analog, cyclopropavir, in animal models for cytomegalovirus infections. Antimicrob Agents Chemother. 2004;48:4745–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bidanset DJ, Beadle JR, Wan WB, Hostetler KY, Kern ER. Oral activity of ether lipid ester prodrugs of cidofovir against experimental human cytomegalovirus infection. J Infect Dis. 2004;190:499–503. [DOI] [PubMed] [Google Scholar]
- 19.Hartline CB, Gustin KM, Wan WB, et al. Ether lipid-ester prodrugs of acyclic nucleoside phosphonates: activity against adenovirus replication in vitro. J Infect Dis. 2005;191:396–399. [DOI] [PubMed] [Google Scholar]
- 20.Tollefson AE, Spencer JF, Ying B, Buller RML, Wold WS, Toth K. Cidofovir and brincidofovir reduce the pathology caused by systemic infection with human type 5 adenovirus in immunosuppressed Syrian hamsters, while ribavirin is largely ineffective in this model. Antiviral Res. 2014;112:38–46. [DOI] [PubMed] [Google Scholar]
- 21.Williams-Aziz SL, Hartline CB, Harden EA, et al. Comparative activities of lipid esters of cidofovir and cyclic cidofovir against replication of herpesviruses in vitro. Antimicrob Agents Chemother. 2005;49:3724–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Quenelle DC, Lampert B, Collins DJ, Rice TL, Painter GR, Kern ER. Efficacy of CMX001 against herpes simplex virus infections in mice and correlations with drug distribution studies. J Infect Dis. 2010;202:1492–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kern ER, Hartline C, Harden E, et al. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother. 2002;46:991–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quenelle DC, Collins DJ, Herrod BP, et al. Effect of oral treatment with hexadecyloxypropyl-[(S)-9-(3-hydroxy-2- phosphonylmethoxypropyl)adenine] [(S)-HPMPA] or octadecyloxyethyl-(S)-HPMPA on cowpox or vaccinia virus infections in mice. Antimicrob Agents Chemother. 2007;51:3940–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rice AD, Adams MM, Lampert B, et al. Efficacy of CMX001 as a prophylactic and presymptomatic antiviral agent in New Zealand white rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses. 2011;3:63–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rice AD, Adams MM, Wallace G, et al. Efficacy of CMX001 as a post exposure antiviral in New Zealand White rabbits infected with rabbitpox virus, a model for orthopoxvirus infections of humans. Viruses. 2011;3:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trost LC, Rose ML, Khouri J, et al. The efficacy and pharmacokinetics of brincidofovir for the treatment of lethal rabbitpox virus infection: a model of smallpox disease. Antiviral Res. 2015;117:115–121. [DOI] [PubMed] [Google Scholar]
- 28.Grossi IM, Foster SA, Gainey MR, et al. Efficacy of delayed brincidofovir treatment against a lethal rabbitpox virus challenge in New Zealand White rabbits. Antiviral Res. 2017;143:278–286. [DOI] [PubMed] [Google Scholar]
- 29.Randhawa P, Farasati NA, Shapiro R, Hostetler KY. Ether lipid ester derivatives of cidofovir inhibit polyomavirus BK replication in vitro. Antimicrob Agents Chemother. 2006;50:1564–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Randhawa P, Zemlicka J, Sauerbrei A, et al. Anti-BK virus activity of nucleoside analogs. Antimicrob Agents Chemother. 2008;52:1519–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernhoff E, Gutteberg TJ, Sandvik K, Hirsch HH, Rinaldo CH. Cidofovir inhibits polyomavirus BK replication in human renal tubular cells downstream of viral early gene expression. Am J Transplant. 2008;8:1413–1422. [DOI] [PubMed] [Google Scholar]
- 32.Gosert R, Rinaldo CH, Wernli M, Major EO, Hirsch HH. CMX001 (1-O-hexadecyloxypropyl-cidofovir) inhibits polyomavirus JC replication in human brain progenitor-derived astrocytes. Antimicrob Agents Chemother. 2011;55:2129–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hostetler KY. Alkoxyalkyl prodrugs of acyclic nucleoside phosphonates enhance oral antiviral activity and reduce toxicity: current state of the art. Antiviral Res. 2009;82:A84–A98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Painter W, Robertson A, Trost LC, Godkin S, Lampert B, Painter G. First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-spectrum oral drug active against double-stranded DNA viruses. Antimicrob Agents Chemother. 2012;56:2726–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aldern KA, Ciesla SL, Winegarden KL, Hostetler KY. Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol Pharmacol. 2003;63:678–681. [DOI] [PubMed] [Google Scholar]
- 36.Ciesla SL, Trahan J, Wan WB, et al. Esterification of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes drug accumulation in kidney. Antiviral Res. 2003;59:163–171. [DOI] [PubMed] [Google Scholar]
- 37.Tippin TK, Morrison ME, Brundage TM, Momméja-Marin H. Brincidofovir is not a substrate for the human organic anion transporter 1: a mechanistic explanation for the lack of nephrotoxicity observed in clinical studies. Ther Drug Monit. 2016;38:777–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hill JA, Mayer BT, Xie H, et al. The cumulative burden of double-stranded DNA virus detection after allogeneic HCT is associated with increased mortality. Blood. 2017;129:2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hill JA, Mayer BT, Xie H, et al. Kinetics of double-stranded DNA viremia after allogeneic hematopoietic cell transplantation. Clin Infect Dis. 2018;66:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marty FM, Winston DJ, Rowley SD, et al. CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N Engl J Med. 2013;369:1227–1236. [DOI] [PubMed] [Google Scholar]
- 41.Papanicolaou G, Grimley M, Morrison M, Brundage T, Mommeja-Marin H. Pre-engraftment initiation of brincidofovir (CMX001) in hematopoietic cell transplant (HCT) recipients is supported by lack of myeloid toxicity. Bone Marrow Transplant. 2014;49(S1):S327. [Google Scholar]
- 42.Przepiorka D, Weisdorf D, Martin P, et al. 1994 Consensus Conference on Acute GVHD Grading. Bone Marrow Transplant. 1995;15:825–828. [PubMed] [Google Scholar]
- 43.Ljungman P, Griffiths P, Paya C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin Infect Dis. 2002;34:1094–1097. [DOI] [PubMed] [Google Scholar]
- 44.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v4.03. Available at: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40. Accessed March 17, 2018.
- 45.Sale GE, Shulman HM, McDonald GB, Thomas ED. Gastrointestinal graft-versus-host disease in man: a clinicopathologic study of the rectal biopsy. Am J Surg Pathol. 1979;3:291–299. [DOI] [PubMed] [Google Scholar]
- 46.Leisenring WM, Martin PJ, Petersdorf EW, et al. An acute graft-versus-host disease activity index to predict survival after hematopoietic cell transplantation with myeloablative conditioning regimens. Blood. 2006;108:749–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreft A, Mottok A, Mesteri I, et al. Consensus diagnostic histopathological criteria for acute gastrointestinal graft-versus-host disease improve interobserver reproducibility. Virchows Arch. 2015;467:255–263. [DOI] [PubMed] [Google Scholar]
- 48.Marty FM, Rubin RH. The prevention of infection post-transplant: the role of prophylaxis, preemptive and empiric therapy. Transpl Int. 2006;19:2–11. [DOI] [PubMed] [Google Scholar]
- 49.Razonable RR, Blumberg EA. It’s not too late: a proposal to standardize the terminology of “late-onset” cytomegalovirus infection and disease in solid organ transplant recipients. Transpl Infect Dis. 2015;17:779–784. [DOI] [PubMed] [Google Scholar]
- 50.GB McDonald. How I treat acute graft-versus-host disease of the gastrointestinal tract and the liver. Blood. 2016;127:1544–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Papadimitriou JC, Cangro CB, Lustberg A, et al. Histologic features of mycophenolate mofetil-related colitis: a graft-versus-host disease-like pattern. Int J Surg Pathol. 2003;11:295–302. [DOI] [PubMed] [Google Scholar]
- 52.Selbst MK, Ahrens WA, Robert ME, Friedman A, Proctor DD, Jain D. Spectrum of histologic changes in colonic biopsies in patients treated with mycophenolate mofetil. Mod Pathol. 2009;22:737–743. [DOI] [PubMed] [Google Scholar]
- 53.Detweiler CJ, Mueller SB, Sung AD, Saullo JL, Prasad VK, Cardona DM. Brincidofovir (CMX001) toxicity associated with epithelial apoptosis and crypt drop out in a hematopoietic cell transplant patient: challenges in distinguishing drug toxicity from GVHD. J Pediatr Hematol Oncol. 2018;40:e364–e368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.U.S. National Library of Medicine. SUSTAIN: a randomized, double-blind, multicenter, phase 3 study of the efficacy, safety, and tolerability of brincidofovir versus valganciclovir for the prevention of cytomegalovirus disease in CMV seronegative kidney allograft recipients (BCV CMV vGCV). Available at: https://clinicaltrials.gov/ct2/show/NCT02439970. Accessed April 3,2018.
- 55.Wire M, Anderson M, Arumugham T, Morrison M, Dunn J, Naderer O. Co-administration of cyclosporine (CsA) increases plasma brincidofovir (BCV) exposure in healthy volunteers. Pharmacotherapy. 2016;36:e269. [Google Scholar]
- 56.Niemi M, Pasanen MK, Neuvonen PJ. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol Rev. 2011;63:157–181. [DOI] [PubMed] [Google Scholar]
- 57.Karlgren M, Vildhede A, Norinder U, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J Med Chem. 2012;55:4740–4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Florescu DF, Pergam SA, Neely MN, et al. Safety and efficacy of CMX001 as salvage therapy for severe adenovirus infections in immunocompromised patients. Biol Blood Marrow Transplant. 2012;18:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grimley MS, Marsh RA, Bleesing JJ, et al. Cmx001 as therapy for severe adenovirus infections in immunocompromised pediatric patients: single experience in 5 patients. Biol Blood Marrow Transplant. 2012;18:S315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chittick G, Morrison M, Brundage T, Nichols WG. Short-term clinical safety profile of brincidofovir: a favorable benefit-risk proposition in the treatment of smallpox. Antiviral Res. 2017;143:269–277. [DOI] [PubMed] [Google Scholar]
- 61.Grimley MS, Chemaly RF, Englund JA, et al. Brincidofovir for asymptomatic adenovirus viremia in pediatric and adult allogeneic hematopoietic cell transplant recipients: a randomized placebo-controlled phase II trial. Biol Blood Marrow Transplant. 2017;23:512–521. [DOI] [PubMed] [Google Scholar]
- 62.Ramsay ID, Attwood C, Irish D, Griffiths PD, Kyriakou C, Lowe DM. Disseminated adenovirus infection after allogeneic stem cell transplant and the potential role of brincidofovir: case series and 10-year experience of management in an adult transplant cohort. J Clin Virol. 2017;96:73–79. [DOI] [PubMed] [Google Scholar]
- 63.Hiwarkar P, Amrolia P, Sivaprakasam P, et al. Brincidofovir is highly efficacious in controlling adenoviremia in pediatric recipients of hematopoietic cell transplant. Blood. 2017;129:2033–2037. [DOI] [PubMed] [Google Scholar]
- 64.Saffrey MJ. Aging of the mammalian gastrointestinal tract: a complex organ system. Age (Dordr). 2014;36:9603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wire MB, Morrison M, Anderson M, Arumugham T, Dunn J, Naderer O. Pharmacokinetics (PK) and safety of intravenous (IV) brincidofovir (BCV) in healthy adult subjects. Open Forum Infect Dis. 2017;4(Suppl 1). S311. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.