

Abstract
Biomaterials-based strategies have shown great promise for tissue regeneration. 3D printing technologies can deliver unprecedented control over architecture and properties of biomaterial constructs when combined with innovative material design strategies. Colloidal gels made of polymeric nanoparticles are attractive injectable and self-healing systems, but their use as bio-inks for extrusion-based printing is largely unexplored. Here, we report 3D printing of novel biomaterial constructs with shape memory behavior using photo-reactive gelatin nanoparticles as colloidal building blocks. These nanoparticles are stabilized with intraparticle covalent crosslinks, and also contain pendant methacryloyl groups as photo-reactive moieties. While non-covalent interactions between nanoparticles enable formation of colloidal gel inks that are printable at room temperature, UV-induced covalent interparticle crosslinks based on methacryloyl moieties significantly enhance mechanical properties of printed constructs. Additionally, the UV crosslinking modality enables remarkable control over swelling, degradation, and biomolecule release behavior of 3D constructs. Finally, by exploiting the mechanical properties of colloidal biomaterials after UV crosslinking, 3D constructs can be designed with shape memory properties, returning to their original programmed geometry upon re-hydration. Accordingly, these novel colloidal inks exhibit great potential to serve as bio-inks for 3D printing of biomaterials with shape-morphing features for a wide range of tissue engineering and regenerative medicine applications.
Keywords: 3D printing, Colloidal gels, Nanoparticles, Shape memory, 4D printing, Tissue engineering
1. Introduction
Damage or loss of tissues resulting from injuries or diseases are primary causes of chronic morbidity and impose a significant public health burden globally [1, 2]. During recent decades, biomaterials-based tissue engineering strategies have demonstrated great promise to serve as affordable therapeutic solutions for the regeneration of tissues such as those of the musculoskeletal system [3]. These strategies are mainly based on the use of three-dimensional (3D) constructs as scaffolds that provide a supportive environment for the formation of new tissues [4, 5]. Subsequently, the fabrication of biomaterial constructs with pre-defined internal and external architectures is a critical step for their successful application in tissue engineering [3, 4]. Traditionally, techniques such as solvent casting [6] or injection molding [7] were employed for fabrication of biomaterial constructs. During the past decade, 3D printing technologies, such as extrusion-based 3D printing [8], have become more accessible and cost-effective, revolutionizing the biomaterials field. While these techniques exhibit a remarkable ability to fabricate constructs with pre-defined architectures, such pre-fabricated constructs are typically not amenable to extensive re-shaping, which limits their clinical handling. Nonetheless, recent research progress has shown that the combination of 3D printing with innovative material design strategies can deliver unprecedented control over the properties of material constructs, e.g., producing constructs that can undergo extensive shape transformation in response to external stimuli [9, 10].
Among different biomaterial categories, hydrogels have especially received significant interest for use in 3D printing of biomedical constructs [11, 12]. These biomaterials are particularly promising for tissue engineering, regenerative medicine, and drug delivery as they can mimic various aspects of natural extracellular matrix, owing to their hydrated nature, viscoelastic response and ability to incorporate biologics such as cells and growth factors [13, 14]. Nonetheless, while several hydrogel systems are amenable to the printing process, their inferior mechanical properties and excessive swelling often limit their applications in this area [11, 15]. However, promising 3D printed scaffolds should at least exhibit sufficient mechanical stability for their clinical handling, and also controllable swelling and degradation behavior to maintain their designed architecture for a desired period under physiological conditions [3, 5, 11].
One class of hydrogels which has recently demonstrated great potential for use as biomaterials is colloidal gels. Unlike conventional (hydro)gels, colloidal gels are entirely assembled from particulate building blocks typically ranging from ~10 nm to a few microns in size [16]. While the bottom-up assembly of these materials from nano- to macro-scale can allow for hierarchical design of biomaterials [17, 18], the possibility to combine different particle types in their composition renders them as modular biomaterial platforms [19–22]. Additionally, these building blocks can be loaded or functionalized with various biomolecules or drugs [20, 23–27], expanding the biomedical utility of these systems. Several recent studies have shown that colloidal gels such as those made of gelatin nanoparticles [19, 20, 25, 26, 28, 29] are promising injectable systems for tissue regeneration.
In addition to their injectability, colloidal gels can also exhibit self-healing behavior after damage, owing to the reversibility of non-covalent interparticle bonds that can be employed for their design [20, 28, 30, 31]. This self-healing feature can enable rapid recovery of the mechanical properties of these hydrogels after destructive shearing upon their extrusion through narrow needles [31], which can possibly render these biomaterials suitable for use as bio-inks for extrusion-based 3D printing. Compared to granular hydrogels formed by jamming of larger microparticles [32–34], colloidal gels made of polymeric nanoparticles can exhibit superior mechanical performance and cohesion, as the nanoscale dimensions of their building blocks results in a higher density of interparticle bonds [26]. Previously, colloidal processing of ceramics or carbon-based materials have been employed for 3D printing of constructs, which typically involved harsh processing conditions such as high temperature [35, 36]. Nonetheless, while granular hydrogels made of polymeric microparticles have recently been demonstrated as bio-inks for extrusion-based 3D printing [15, 37–39], the potential of polymeric colloidal gels for 3D printing of biomaterial constructs is yet largely unexplored.
Here, we report 3D printing of novel biomaterial constructs with shape memory behavior using photo-reactive gelatin nanoparticles as colloidal building blocks. To this end, we synthesized gelatin nanoparticles that were stabilized with intraparticle covalent crosslinks, and modified them to contain pendant methacryloyl groups as photo-reactive functionalities. Owing to the inherent moieties present in natural gelatin and the introduced methacryloyl groups, these nanoparticles were capable of forming non-covalent and covalent interparticle bonds, respectively. While non-covalent interactions enabled the formation of a printable colloidal gel ink that could self-heal upon extrusion, ultraviolet light (UV)-induced formation of additional covalent interparticle bonds after printing could significantly enhance the mechanical properties of the printed constructs (Scheme 1). Additionally, the interparticle UV crosslinking modality in this system enabled remarkable control over swelling, degradation, and biomolecule release behavior of the 3D constructs. Finally, by exploiting the ability of these colloidal biomaterials to store a high amount of elastic energy after UV crosslinking, 3D constructs could be designed that exhibited shape memory properties, reverting their configuration from a temporary set shape to their original programmed geometry upon re-hydration. Accordingly, this study highlights the promise of colloidal gels made of methacryloyl-modified gelatin nanoparticles to serve as bio-inks for 3D printing of biomaterials with shape-morphing features with a strong potential for application in various tissue engineering and regenerative medicine strategies.
Scheme 1.
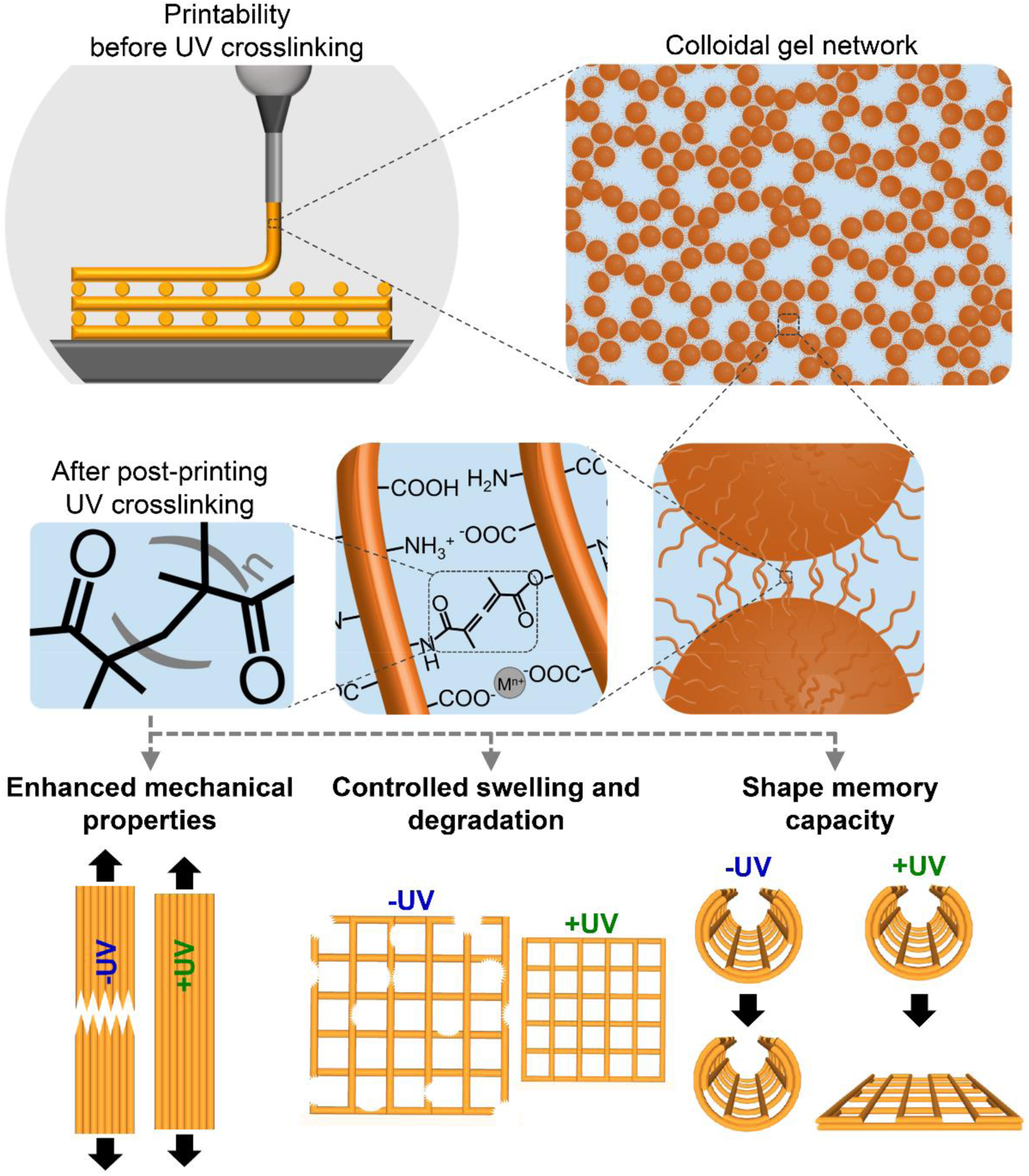
Schematic illustration of extrusion-based 3D printing of gelatin-based colloidal inks, their structure at multiple length scales, and important features of printed constructs enabled by the UV crosslinking modality in this system.
2. Materials and Methods
2.1. Materials
Gelatin powder (Type B from bovine skin; Bloom strength ~225 g), glutaraldehyde solution (25 wt% aqueous solution in water), methacrylic anhydride (containing 2,000 ppm topanol A as inhibitor; 94%), 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959; 98%), and collagenase (from clostridium histolyticum, Type IA, 0.5–5.0 FALGPA units/mg solid, ≥125 CDU/mg solid) were purchased from Sigma-Aldrich Corporation (St. Louis, MO, USA). Albumin from Bovine Serum, Alexa Fluor™ 647 conjugate (BSA-AF) was purchased from Thermo Fisher Scientific Corporation (Eugene, OR, USA). Transparent poly(ethylene terephthalate) sheets (PET; Blank Stencil Material Mylar Template Sheets; dimensions: 30 cm×30 cm; thickness: 0.15 mm) were purchased from Amazon (EG EMIGOO Store). All other chemicals were of reagent grade and were used without any purification.
2.2. Synthesis and methacryloylation of gelatin nanoparticles
Gelatin nanoparticles were synthesized based on a two-step desolvation method [20], followed by a methacryloylation step. For the first desolvation step, gelatin powder (50 g) was dissolved in Milli-Q water (1 L) at 50 °C using a magnetic stirrer. Subsequently, 1 L of acetone was added to the gelatin solution and the mixture was left for 1 h without stirring at room temperature. The supernatant was then discarded, and the precipitated high-molecular weight gelatin was re-dissolved in 0.5 L of Milli-Q at 50 °C using a magnetic stirrer. The solution was then frozen at − 80 °C, and freeze-dried. The resulting gelatin was then stored at −20 °C until further usage. Subsequently, the second desolvation step was carried out by dissolving 3.75 g of the freeze-dried gelatin in 75 mL of Milli-Q water inside a 500 mL glass beaker at 50 °C using a magnetic stirrer, after which the pH of solution was adjusted to 2.47 through dropwise addition of hydrochloric acid (1–5 M). Thereafter, acetone was loaded into four 60 mL plastic syringes (Becton-Dickinson, Franklin Lakes, NJ, USA), which were then placed into a multi-syringe infusion pump (Cole-Parmer, Vernon Hills, IL, USA). Subsequently, the gelatin solution was magnetically stirred at a stirring rate and temperature of 1200 rpm and 50 °C, respectively.
During this stirring step, 225 mL of acetone was added dropwise using the syringe pump via four Teflon tubes inserted through a Parafilm covering the beaker at a total dripping rate of 12 mL/min. During this step, the transparent gelatin solution turned into an opaque dispersion as a result of nanoparticle formation. The dispersion was then moved to another magnetic stirrer at room temperature and continued to be stirred at 600 rpm, during which 555 μL of glutaraldehyde solution (25 wt%) were added dropwise to crosslink the gelatin nanoparticles overnight. The following day, 300 mL of a guanidine hydrochloride aqueous solution (100 mM) were added to the dispersion and left to stir for 1 h to block any unreacted aldehydes. The dispersion was then filtered through a nylon cell strainer (100 μm mesh size; Fisher Scientific, Waltham, MA, USA) and washed with Milli-Q water via four centrifugation (10,000 rpm for 1 h) and redispersion cycles. A small portion (0.2 mL) of the dispersion was then air-dried at 65 °C to determine the particle concentration for the next step. To functionalize the gelatin nanoparticles with methacryloyl groups, a 1 M carbonate-bicarbonate (CB) buffer solution was prepared by dissolving 0.91 M and 0.09 M of sodium bicarbonate and sodium carbonate in Milli-Q water, respectively. The nanoparticle dispersion, the 1 M CB buffer solution and additional Milli-Q water were then mixed in an Erlenmeyer flask to reach nanoparticle and CB buffer concentrations of 10 mg/mL and 0.1 M, respectively. The mixture was then magnetically stirred at 50 °C, and its pH was adjusted to 9 by dropwise addition of a 1 M sodium hydroxide solution. Thereafter, an aliquot of methacrylic anhydride (MA) was added dropwise to the mixture at a concentration of 1.16 mL of MA per gram of gelatin nanoparticles. The flask was then covered with a Parafilm, and the reaction was left to proceed for 1 h at 50 °C using a stirring rate of 800 rpm. Thereafter, the mixture was transferred into SnakeSkin™ dialysis tubing (Thermo Fisher Scientific, Rockford, IL, USA) with a 10 kDa molecular-weight cutoff. The dialysis was then carried out against Milli-Q water at room temperature for 5 days with a daily change of the Milli-Q water. Subsequently, the dispersion was transferred into a beaker, the pH was adjusted to 7.4 using a 1 M sodium hydroxide solution, and the dispersion was flash frozen using liquid nitrogen. Finally, the frozen dispersion was freeze-dried, and then stored at −20 °C until further use. During all the steps, the samples were protected from light using aluminum foil to avoid undesired photo-induced reactions.
2.3. Characterization of gelatin nanoparticles
2.3.1. Nanoparticle tracking analysis
Nanoparticle tracking analysis (NTA) was employed to quantify the size of gelatin nanoparticles in swollen state before and after their methacryloylation using a NanoSight NS300 system (Malvern Instruments, Malvern, UK). To this end, the nanoparticle dispersions were diluted in Milli-Q water at a concentration of ~3 μg/mL. These dispersions were then injected into the measurement chamber of the instrument at room temperature using a syringe pump. Consequently, the size distribution of nanoparticles was calculated by the NanoSight NTA software based on the Brownian motion of individual nanoparticles. For these measurements, three different batches of gelatin nanoparticles were analyzed before and after methacryloylation, and more than 10,000 particle measurements were performed in each batch.
2.3.2. Scanning electron microscopy
Scanning electron microscopy (SEM) was carried out using a Quanta 400 system (Field Electron and Ion Company; Hillsboro, OR, USA). Sample preparation for imaging of nanoparticles was carried out by first mixing 0.6 mL of gelatin nanoparticle dispersion (in Milli-Q water) with 0.2 mL of acetone to minizine nanoparticle aggregation during the freeze-drying process. Thereafter, the mixture was flash frozen in liquid nitrogen and freeze-dried. Freeze-dried nanoparticles were then dispersed on adhesive carbon tapes. All the samples were sputter coated with a 5 nm gold layer prior to imaging. These images were then used for the quantification of nanoparticle size (at dry state), which was carried out by manually measuring the diameter of 100 nanoparticles per batch (three batches in total) using ImageJ software (version 1.52p).
2.3.3. Electrophoretic light scattering
Electrophoretic light scattering measurements were carried out using a Zen 3600 Zetasizer system (Malvern Instruments, Malvern, UK) to quantify the zeta potential of gelatin nanoparticles as a measure of their net surface charge. To this end, the nanoparticles were dispersed in a 5 mM HEPES buffer solution (pH=7) and the tests were performed at 21 °C with an equilibration time of 120 s. Zeta potential values were calculated from the electrophoretic mobility of nanoparticles by the Zetasizer software using the Smoluchowski model. For these measurements, three different batches of gelatin nanoparticles were analyzed before and after methacryloylation.
2.3.4. Nuclear magnetic resonance spectroscopy
Sample preparation was carried out in multiple steps involving collagenase enzyme since the gelatin nanoparticles were covalently crosslinked (using glutaraldehyde) and were therefore not readily soluble in aqueous media. To this end, 25 mg of gelatin nanoparticles, gelatin control or gelatin methacryloyl (GelMA) control were added to 0.5 mL of a phosphate-buffered saline (PBS) solution containing 2 mg/mL of collagenase. The mixtures were then subjected to 1 min of vortexing, and placed at 37 °C on a shaking table for 5 days to fully dissolve the gelatin enzymatically. The samples were then centrifuged at 13,000 rpm for 5 min to separate any possibly undissolved solids, after which 0.4 mL of each supernatant were transferred to a new Eppendorf tube. The solutions were then flash frozen with liquid nitrogen, and freeze-dried. The resulting solids were dissolved in deuterium oxide (containing 0.05 wt.% of Trimethylsilylpropanoic acid (TMSP) as a chemical shift reference) at a concentration of 25 mg/mL. Subsequently, the solutions were analyzed at 40 °C via 1H Nuclear Magnetic Resonance (1H-NMR) spectroscopy using a 600-MHz Bruker spectrometer (Bruker BioSpin, Billerica, MA, USA). The total amount (mmol) of methacryloyl groups, as well as methacrylate and methacrylamide groups, per gram of nanoparticles (or GelMA) was calculated from the 1H-NMR spectra using the following equations, as reported previously [40]:
2.4. Preparation of colloidal inks
Colloidal inks were prepared with various compositions (5–20 w/v%; see Table S1 for particle volume fractions) by adding aliquots of PBS solutions to plastic tubes containing freeze-dried methacryloyl-functionalized gelatin nanoparticles. The PBS solutions also contained different concentrations (0.1–1.0 w/v%) of a photoinitiator (Irgacure 2959) to enable further UV-induced covalent cross-linking of the inks. Moreover, for the biomolecule release experiments described in Section 2.7.3, the solutions were also supplemented with 20 μM (1.32 mg/mL) of BSA-AF as a model biomolecule. Thereafter, the tubes were subjected to vortexing and the contents were manually mixed using a spatula. Consequently, the tubes were subjected to centrifugation for 1 min at 5,000 rpm to collect the gels at the bottom of the tubes. The tubes were then covered with an aluminum foil, and refrigerated at 4 °C overnight for use in the following day.
2.5. Characterization of colloidal inks
2.5.1. Rheological evaluation of viscoelastic properties of inks
Rheological tests were carried out at room temperature using a Discovery hybrid rheometer (DHR-2, TA Instruments, Newcastle, DE, USA) equipped with a 20 mm stainless steel flat geometry and a UV Light Guide accessory (TA Instruments, Newcastle, DE, USA) using an operating gap of 300 μm (pre- and during UV crosslinking) or 260 μm (post-UV crosslinking). The UV Light Guide system was connected to an OmniCure S2000 Spot UV Curing System (Lumen Dynamics Group Inc., Mississauga, ON, Canada), which produced UV light with a primary peak at 365 nm wavelength. Frequency sweeps were performed with frequencies ranging from 0.1 to 100 Hz, at a constant strain of 1%. Storage (G′) and loss (G″) moduli values before and after UV crosslinking were determined from time sweep measurements performed for 1 min, at constant strain and frequency of 1% and 1 Hz, respectively. UV crosslinking of samples was carried out during a time sweep step (10–300 s, 1% strain, and 1 Hz frequency) at different UV intensities (2–50 mW/cm2). The initial crosslinking rate (Pa/s) was determined by calculating the slope of the G’ curve during the increasing portion of the curve until G’ reached its peak value (5–40 seconds depending on the composition). For post-UV crosslinking evaluation of the gels, the gap was adjusted from 300 μm to 260 μm to compensate for the gel network shrinkage during the crosslinking step. Three samples were analyzed for each experimental condition (n=3).
2.5.2. Determination of crosslinking depth
To determine the depth of covalent crosslinking in colloidal inks upon UV irradiation, triangle-shaped molds were first fabricated using a Form2 stereolithography-based 3D printer (Formlabs Inc., Somerville, MA, USA). A black resin was used for the fabrication of molds to enable UV crosslinking to occur only from their open side (Fig. S1). Each mold was filled with a colloidal ink and the UV crosslinking was carried out by subjecting the open side of the molds to UV irradiation (5 min at 10 mW/cm2) using the UV curing setup described in Section 2.5.1. Subsequently, each mold was disassembled, and the hydrogel piece was cut with a razor at the separation plane of two mold sections. The resulting cross-section was then imaged using an optical microscope (trinocular stereomicroscope, SM-4TZ-144A; AmScope, Irvine, CA, USA), which was followed by nanoindentation tests. The nanoindentation tests were carried out a using a UNHT3-Bio Bioindenter system (Anton Paar, Graz, Austria), equipped with an indenter with a spherical diamond tip of 200 μm radius. Indentations were carried out on the cross-section of samples at increasing distance (depth) from the UV-irradiated edge, starting at 200 μm and followed by 100 μm steps. Each indentation was performed through linear loading at a loading/unloading rate of 3000 μN/min, with a maximum load of 40 μN, and a 5.0 s pause between loading and unloading steps. The elastic modulus values were calculated by the Indentation Software (version 6.1.15; Anton Paar, Graz, Austria) using the method developed by Oliver and Pharr [41] assuming a Poisson’s ratio of 0.3 for the samples. Five hydrogel samples were analyzed for these experiments (n=5). All measurements were carried out at room temperature and in air. The samples were surrounded with hydrated tissues to minimize hydrogel drying.
2.5.3. Rheological evaluation of printability of colloidal inks
Rheological tests were carried out at 25 °C using a Discovery hybrid rheometer (DHR-2, TA Instruments, Newcastle, DE, USA) equipped with 20 mm stainless steel flat geometry using an operating gap of 500 μm. The shear-thinning behavior of the inks was determined by measuring their viscosity during flow ramps with increasing shear rates from 0.1 to 1000 1/s. The self-healing capacity of the inks upon shear-induced damage was determined using a three-step protocol at a frequency of 1 Hz: (I) time sweep at a low strain (1%), (II) gel network destruction by applying a high strain (strain sweep from 0.1% to 1000%), and (III) self-healing at a low strain (1%). The strain sweeps performed in step (II) of the self-healing experiments were then used to determine the yield stress and yield strain of each ink composition. For determination of yield stress, G′ and G″ were plotted as a function of shear stress, and the yield stress was calculated from the cross-over point of the two curves (yielding point). For determination of yield strain, G′ and G″ were plotted as a function of shear strain, and the yield strain was calculated from the cross-over point of the two curves (yielding point). Three samples were analyzed for each experimental condition (n=3).
2.5.4. Evaluation of fiber formation of colloidal inks upon extrusion
Fiber formation using different ink compositions was evaluated by recording their extrusion at room temperature through blunt needles of different sizes using a Vividia Digital Microscope (Oasis Scientific Inc., Taylors, SC, USA). To this end, colloidal inks were transferred into 10 mL plastic syringes, which were then loaded into a BioAssemblyBot® extrusion-based 3D printer (Advanced Solutions Life Sciences, Louisville, KY, USA). The extrusion pressure was then gradually increased until a continuous extrusion of an ink was achieved. The extrusion process was paused at least 10 times to capture microscopic images for evaluation of diameter and surface uniformity of fibers. These images were then thresholded and analyzed using ImageJ software (version 1.52p).
The fiber diameter was determined by measuring the diameter of the fiber in each image at 400 evenly spaced locations along the fiber length. To determine the uniformity ratio of each fiber, the total length of the surface profile on both sides of the fiber was measured in each image. This value was then divided by the total length of two straight lines drawn along the sides of the fiber, representing a perfectly uniform fiber, as reported previously [42]. For these quantifications, 10 images were analyzed per experimental group (n=10).
2.6. Fabrication of constructs
2.6.1. 3D printing
Constructs were 3D printed at room temperature using a BioAssemblyBot® extrusion-based 3D printer (Advanced Solutions Life Sciences, Louisville, KY, USA). To this end, the inks were loaded into 10 mL plastic syringes connected to a 20 G blunt needle. Construct printing was then carried out at room temperature, using a printing pressure of 25 (±10) psi and a printing speed of 8 (±2) mm/s. Construct fabrication for tensile testing was performed by printing one-layer strips (Length: 22 mm; Width: 4 mm) composed of longitudinally or transversely oriented fibers. 3D constructs were printed via deposition of two or four layers of filaments with a center-to-center inter-filament distance of 2 mm, perpendicular fiber orientation between each print layer, and an interlayer distance of 0.45 mm (75% of fiber diameter to ensure sufficient interlayer contact). UV crosslinking of constructs was carried out after printing by exposing them to 500 flashes within a UV light flash box (Otoflash Post Curing Light Pulsing Unit, EnvisionTEC Inc., Gladbeck, Germany) delivering ~60 mW/cm2 of UV light (at 230–410 nm wavelength) for a total duration of 50 s.
2.6.2. Preparation of constructs with shape memory behavior
Two- or four-layer constructs were printed onto flexible and transparent PET sheets. Two types of designs were employed and were designated as X-to-Y design (i.e., curved-to-flat and flat-to-curved), for which X indicates the temporary fixed geometry of the construct and Y indicates the intended recovered geometry.
For the curved-to-flat design, the constructs were first printed and UV crosslinked in the flat state (originally set geometry). The constructs were then curved (temporary geometry) by rolling their underlying PET sheet and placing them into cylindrical molds (Fig. S2a). Finally, the samples were flash frozen in liquid nitrogen and freeze-dried, which enabled maintenance of their temporary curved geometry.
For the flat-to-curved design, the constructs were first printed in the flat state. They were then curved by rolling their underlying PET sheet and placing them into cylindrical molds with light-permitting design (Fig. S2b), followed by UV crosslinking in this curved geometry (originally set geometry). Thereafter, the constructs were flattened (temporary geometry) by placing them between flat surfaces of a petri dish (Fig. S3), which was followed by flash freezing in liquid nitrogen and freeze-drying.
The employed cylindrical molds were 3D printed using a Fortus 450mc FDM printer (Stratasys, Eden Prairie, MN, USA). Table S2 lists the inner diameter of cylindrical molds used for re-shaping of constructs with different sizes.
2.7. Characterization of constructs
2.7.1. Tensile testing
Tensile tests were carried out using an EnduraTEC ElectroForce® 3200 uniaxial tensile testing system (Bose Corporation, Eden Prairie, MI, USA) equipped with a 1000 g (9.81 N) load cell. Sample preparation was carried out by fixing two ends of the freeze-dried constructs (fabricated as described in section 2.6.1; with dimensions of 22 mm in length and 4 mm in width) to plastic mounts using an epoxy glue (ClearWeld; JB Weld Company, Sulphur Springs, TX, USA). After fixing the mounts on the grips of the tester, the samples were re-hydrated with PBS, and their dimensions were determined by digital imaging and ImageJ analysis.
Tensile tests were performed at a deformation rate of 0.1 mm/s. The tensile modulus of the constructs was calculated from the slope of the linear region of the stress-strain curves. The tensile strength of the constructs was determined from the maximum stress measured prior to fracture of the constructs. The fracture strain of the constructs was determined from the elongation of constructs at their fracture point. In a separate set of tests, stress relaxation experiments were carried out by applying a strain of 5% using a deformation rate of 0.5 mm/s and recording the generated stress in the constructs for 120 s. Four constructs were tested per experimental condition (n=4).
2.7.2. Evaluation of in vitro swelling behavior
Two-layer constructs (10 mm height and length) were imaged using an optical microscope (trinocular stereomicroscope, SM-4TZ-144A; AmScope, Irvine, CA, USA) at multiple stages: after printing, UV crosslinking, freeze-drying, and different time points of their immersion in PBS solutions (up to 14 days) at 37 °C to study their swelling behavior. The change (%) of non-porous area and the pore size of constructs at different processing and swelling stages were quantified using ImageJ software (version 1.52p). To exclude background noise from the analysis, images (3684 pixels × 3684 pixels) were first smoothed using the Gaussian Blur filter (sigma=5.0), thresholded (auto, default, B&W, and dark background mode) and finally denoised twice using the Remove Outliers function (Radius=60 pixels; Threshold=10). The construct area at each time point was then determined using the Measure function. Pore diameter for each construct was measured using the Analyze Particles function which determined the area of each pore. The pore size was then calculated as the square root of the pore area, by assuming fully square-shaped pores. Five (n=5) and six (n=6) constructs were analyzed for non-UV-crosslinked and UV-crosslinked groups, respectively.
2.7.3. Evaluation of in vitro degradation and biomolecule release
Freeze-dried two-layer constructs (10 mm height and length; dry weight=11±3 mg) without or with inclusion of BSA-AF model biomolecules for degradation or biomolecule release experiments, respectively, were placed in Transwell® inserts with 0.4-μm pore size (Corning Life Sciences, Kennebunk, ME, USA) which were fitted into 6-well plates. Thereafter, 3 mL of freshly prepared PBS solution containing 0 or 400 ng/mL of a natural matrix metalloproteinase (MMP; collagenase) and 0.001 w/v% of sodium azide (to eliminate potential microbial growth) were added into each well. The collagenase concentration of 400 ng/mL in this work was chosen to mimic the concentration of this enzyme present under in vivo conditions, and enable comparison with previous in vitro degradation studies employing gelatin nanoparticles [18, 19, 25–27, 43, 44]. Subsequently, the plates were incubated at 37 °C on a rotating plate with a rotation rate of 90 rpm. At each time point, 2 mL of solutions were collected and stored at −20 °C inside Protein LoBind® tubes (Eppendorf AG, Hamburg, Germany) for further analysis. Thereafter, 2 mL of freshly prepared solutions were added into each well, and the plates were returned to the incubator until the next time point. For the degradation experiments, after the last time point (Day 14), the remaining constructs were freeze-dried, imaged, and then fully digested in solutions containing a high collagenase concentration (400 μg/mL) upon 2 additional days of incubation. For the release experiments, after the last time point (Day 14), collected solutions were replaced with the high collagenase solutions and the remaining constructs were fully digested. Finally, for the degradation studies, the concentration of soluble gelatin in the supernatants was measured using a bicinchoninic acid (BCA) assay (Thermo Fisher Scientific, Pierce Biotechnology, Rockford, IL USA). For the release studies, the concentration of BSA-AF molecules in the supernatants was determined by measuring the fluorescence intensity of these molecules by means of a SpectraMax M2 microplate reader (Molecular Devices, San Jose, CA, USA) using an excitation wavelength of 640 nm, emission wavelength of 670 nm, and a cutoff wavelength of 665 nm. The cumulative release of gelatin or BSA-AF molecules was determined relative to the total measurable content of each of these components. This total content was calculated from the sum of their degraded/released contents at different time points, plus the remaining contents in the constructs measured by full digestion of samples after the final time point. For degradation or release experiments, four constructs were tested per experimental condition (n=4).
2.7.4. Evaluation of constructs with shape memory behavior
After the freeze-drying step, the constructs were digitally photographed and visually inspected for their integrity. Thereafter, constructs were placed in a transparent container, to which PBS was added until the constructs were fully immersed. Accordingly, shape memory capacity of constructs was analyzed through digital imaging by evaluating the ability of re-shaped constructs to recover their original shape upon becoming in contact with the aqueous solution. Four constructs were tested per experimental condition (n=4).
2.8. Statistics
Statistical analyses were performed using GraphPad Prism 8 software. Particle size, zeta potential, tensile properties, and cell viability results were analyzed statistically using unpaired t-tests with Welch’s correction. Yield stress and yield strain of the inks were analyzed statistically using one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test. In all tests, p>0.05 was considered statistically not significant.
3. Results and Discussion
3.1. Characterization of colloidal building blocks
We first characterized the gelatin nanoparticles before (GNPs) and after their methacryloylation (MA-GNPs) to elucidate the potential effect of the methacryloylation step on the size, morphology, and surface charge of these colloidal building blocks. As shown in Fig. 1a, our results indicate that the average size of nanoparticles in swollen state remained statistically unchanged (224±49 nm for GNPs and 232±93 nm for MA-GNPs). The higher degree of polydispersity observed for MA-GNPs can be due to the formation of some aggregates during the methacryloylation step. Such aggregates were detectable in the scattering patterns of MA-GNPs during the nanoparticle tracking analysis (Fig. S4), and might arise from the screening of long-range electrostatic repulsive forces by the ionic content of the buffer used for the methacryloylation [45, 46] as well as enhanced short-range hydrophobic interactions between the nanoparticles due to the introduced hydrophobic methacryloyl moieties [47]. Upon dehydration through freeze-drying, both GNPs and MA-GNPs shrank ~2 times, while exhibiting spherical morphology (Fig. 1b). This swelling ratio and spherical morphology of gelatin nanoparticles are in agreement with a previous study, which compared the swollen and freeze-dried gelatin nanoparticles using cryogenic transmission electron microscopy and conventional SEM imaging, respectively [23].
Figure 1.
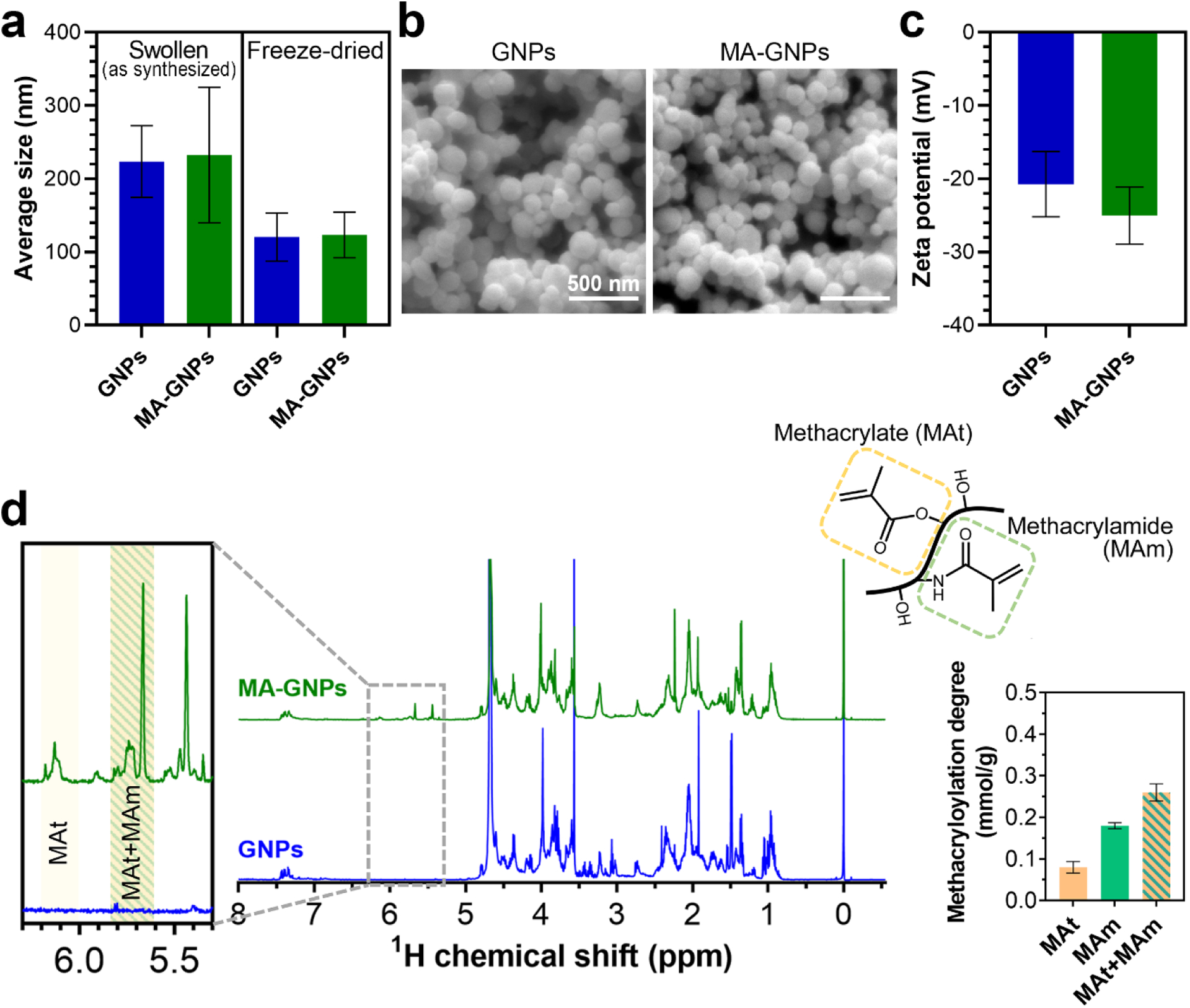
Characterization of gelatin nanoparticles before (GNPs) and after (MA-GNPs) methacryloylation. (a) Size of nanoparticles at swollen or dry states. (b) Scanning electron micrographs of freeze-dried nanoparticles. (c) Zeta potential of swollen nanoparticles at pH 7. (d) Nuclear magnetic resonance spectra and quantified methacryloyl content of nanoparticles after enzymatic digestion. All values are shown as mean ± standard deviation for n=3 samples per experimental condition.
Electrophoretic light scattering measurements indicated that both GNPs and MA-GNPs were negatively charged (−21±4 mV for GNPs and −25±4 mV for MA-GNPs) at pH 7, with no statistically significant difference (Fig. 1c). This net negative charge mainly arises from the anionic nature of the gelatin (type B) employed for nanoparticle synthesis and the consumption of positively charged amine groups by the glutaraldehyde crosslinking [28].
Next, we used 1H-NMR spectroscopy to confirm the modification of nanoparticles with methacryloyl functionalities. As the nanoparticles were chemically crosslinked with glutaraldehyde prior to methacryloylation, and were therefore not readily soluble, they were first solubilized through digestion with collagenase enzyme. To ensure successful functionalization of gelatin nanoparticles with methacryloyl groups, a 20-fold molar excess of methacrylic anhydride with respect to free amine groups present in the original gelatin was used for the methacryloylation process. As shown in Fig. 1d, NMR spectra confirmed the successful functionalization of nanoparticles with methacryloyl groups. Quantification of methacryloyl groups through integration of relevant signals indicated that MA-GNPs contained 0.26±0.02 mmol of methacryloyl per gram of nanoparticles, which consisted of 0.18±0.01 mmol/g and 0.08±0.01 mmol/g of methacrylamide and methacrylate groups, respectively. Despite the high excess of methacrylic anhydride employed for the functionalization, the methacryloylation degree of nanoparticles is comparable to that present in conventional gelatin methacryloyl (GelMA) with a lower range of methacryloyl functionalization [40, 48]. This can be explained by the lower availability of free amine groups in gelatin nanoparticles due to the glutaraldehyde crosslinking. Moreover, pH reduction was observed during the methacryloylation process from the initially set pH 9.0 to pH 6.0±0.1 after 1 h of reaction. This pH drop resulted from the byproduct of the reaction (i.e., methacrylic acid) and can hamper the methacryloylation process via protonation of free amine groups of gelatin [49]. This obstacle can potentially be overcome in future studies by NaOH titration to maintain the basic pH, and the sequential addition of methacrylic anhydride during the reaction [49, 50].
3.2. Viscoelastic and UV crosslinking behavior of colloidal inks
Next, we studied the ability of MA-GNPs to form colloidal gels as inks based on inherent non-covalent interactions that exist between gelatin nanoparticles [20, 28], and also investigated the ability of introduced methacryloyl groups to establish additional covalent interparticle crosslinks upon UV irradiation of inks in the presence of photoinitiator. To this end, we performed frequency sweep rheological measurements for inks made of varying content of MA-GNPs (5–20 w/v%; equal to 0.23–0.84 particle volume fraction, as listed in Table S1) before and after UV irradiation. As shown in Fig. 2a, 5 w/v% inks exhibited an increase in their storage moduli (G’) at frequencies higher than 10 Hz, which may be attributed to the decrease in the measurement timescale at these conditions resulting in a more elastic response by not allowing for stress relaxation in the gel network. Nonetheless, all tested compositions (Figs. 2a–d) displayed frequency-independent solid-like behavior exhibiting storage moduli (G′) higher than loss moduli (G″) over a wide range of frequencies (0.1–100 Hz). These results confirm that despite the consumption of hydroxyl and amine groups of gelatin nanoparticles during the methacryloylation step, MA-GNPs are still capable of forming robust colloid gel networks based on non-covalent interparticle interactions.
Figure 2.
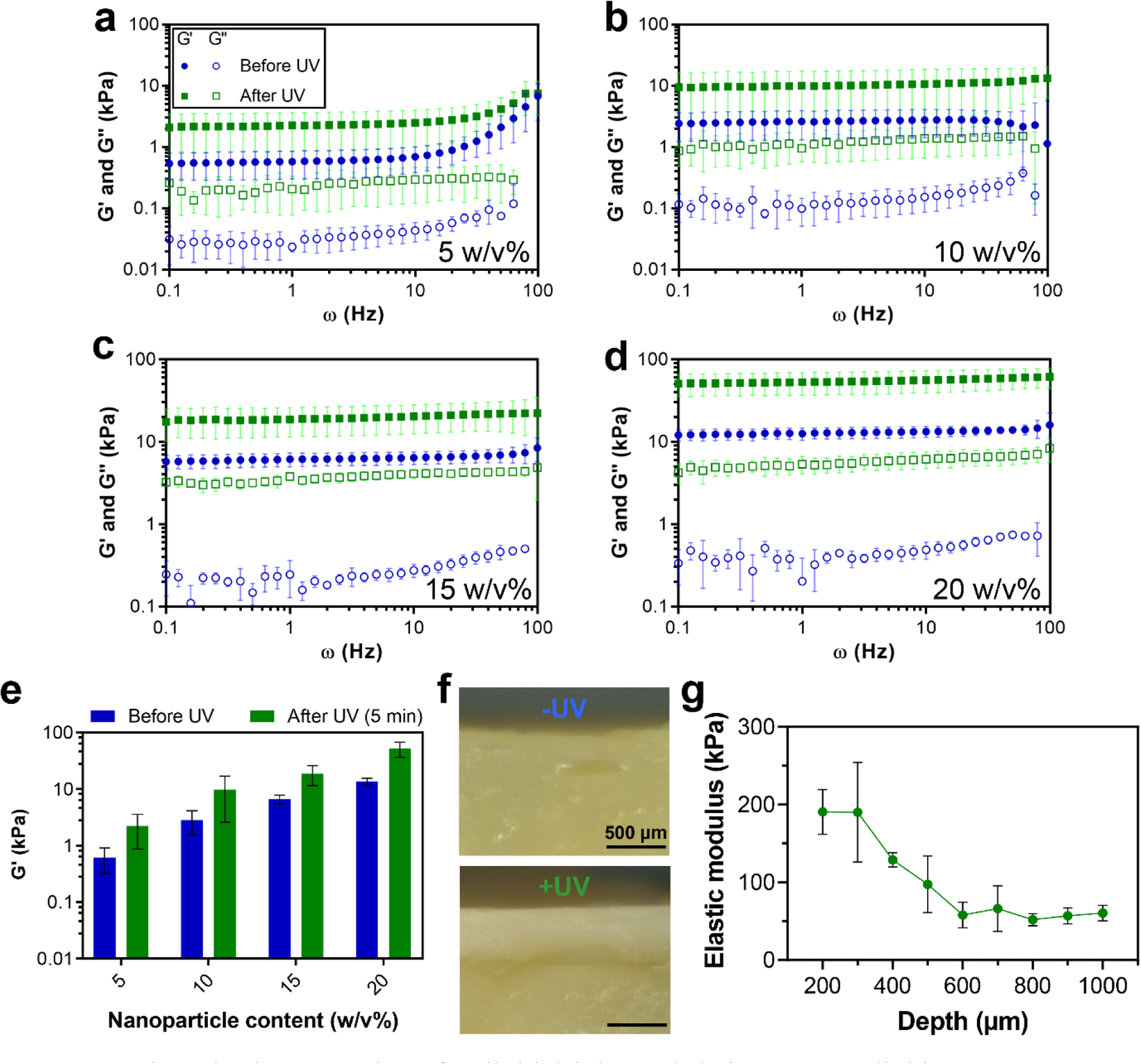
Viscoelastic properties of colloidal inks and their UV crosslinking. (a–d) Frequency dependence of storage (G′) and loss (G″) moduli of colloidal gels with different nanoparticle contents before and after UV crosslinking. (e) G’ values of different colloidal gel compositions at a frequency of 1 Hz. (f) Representative optical microscopy images of cross-section of colloidal gel samples with 20 w/v% of nanoparticle content without (−) or with (+) UV crosslinking. (g) Elastic modulus of colloidal gel samples (20 w/v%) as a function of the distance from their UV-irradiated surface (depth) measured by performing nanoindentation tests on their cross-sections. The UV crosslinking for all the samples (a–g) was performed for 5 min at a UV intensity of 10 mW/cm2. All inks contained 0.5 w/v% photoinitiator content. All values are shown as mean ± standard deviation for (a–e) n=3 or (g) n=5 samples per experimental condition.
All ink compositions displayed enhanced viscoelastic properties upon UV irradiation, revealing the ability of pendant methacryloyl groups at the nanoparticle surfaces to establish interparticle covalent bonds. As shown in Fig. 2e, the UV crosslinking for 5 min at 10 mW/cm2 resulted in ~3–4 times increase of G′ for all inks, while G″ increase was also evident from the rheological results (Fig. S5), indicating enhancement of both elastic and viscous response of the inks upon UV crosslinking.
We also investigated the impact of UV irradiation time and intensity, as well as photoinitiator content on the covalent crosslinking of the inks (Figs. S6–8), which indicated that 50 s of UV irradiation at an intensity of 10 mW/cm2 and using 0.5 w/v% of photoinitiator are indeed sufficient to achieve maximum covalent crosslinking.
To determine the applicability of this UV crosslinking process for fabrication of MA-GNP-based 3D constructs, we next studied the achievable depth of UV crosslinking in these materials. To this end, we selected the most optically non-transparent ink composition (i.e., 20 w/v%) to prepare bulk gel samples, and examined their cross-sections after exposing their surface to UV light. Covalent crosslinking upon UV irradiation of gel surfaces resulted in alteration of their apparent color from pale yellow to a whiter shade. This whiter region was apparent in optical micrographs of their cross-sections (Fig. 2f), suggesting a crosslinking depth of ~500 μm. Nanoindentation tests performed on these cross-sections at different distances from the UV-irradiated surface revealed more details about the UV crosslinking depth (Fig. 2g). These measurements showed that, after UV crosslinking, the elastic modulus remained at its maximum value (~190 kPa) for up to 300 μm in depth, followed by a gradual decrease of the modulus until reaching a minimum plateau (~ 60 kPa) at ≥600 μm. These results indicate the suitability of the UV crosslinking scheme in this material system for post-printing stabilization of extruded colloidal inks by enabling full UV crosslinking of filaments with a radius ≤300 μm.
3.3. Printability of colloidal inks
To determine extrudability of the colloidal inks for 3D printing of constructs, we then studied printability-related rheological properties of inks with compositions ranging from 10 to 20 w/v% in smaller compositional intervals of 2.5 w/v%. All compositions exhibited pseudoplastic behavior by displaying a non-Newtonian decrease of their viscosity upon an increase of shear rate (Fig. 3a; Fig. S9). These results indicate shear-thinning behavior of the inks, suggesting their suitability for extrusion through 3D printing needles.
Figure 3.
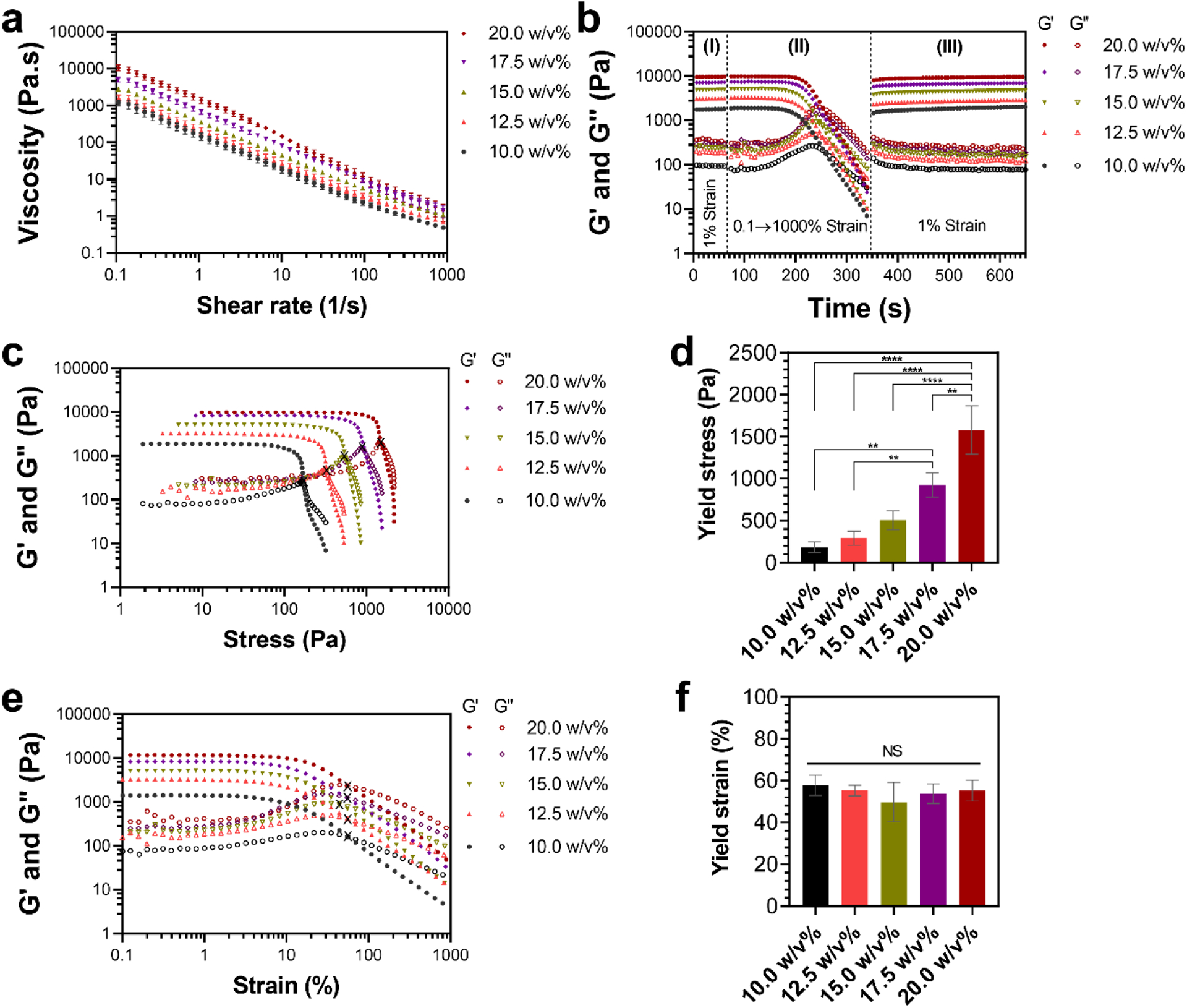
Rheological evaluation of printability of colloidal inks. (a) Shear-thinning behavior of colloidal inks during flow ramps with shear rates varying from 0.1 to 1000 1/s. (b) Self-healing behavior of colloidal inks evaluated using three characterization steps: (I) time sweep at a low strain (1%), (II) colloidal gel network destruction by applying a high strain (from 0.1% to 1000%), and (III) self-healing at a low strain (1%). (c) Viscoelastic response of colloidal inks subjected to increasing shear stress and their corresponding (d) yield stress. ** and **** indicate statistical difference with p<0.01 and p<0.0001, respectively. (e) Viscoelastic response of colloidal inks subjected to increasing shear strain and their corresponding (f) yield strain. NS indicates statistically not significant (p>0.05). X symbols in (c) and (e) indicate the cross-over point of G′ and G″ curves (yielding point) employed for determination of yield stress and yield strain of the inks, respectively. (a,d,f) All values are shown as mean ± standard deviation for n=3 samples per experimental condition.
Next, we examined the ability of colloidal inks to recover their mechanical properties after undergoing destructive shearing, which can be experienced by materials due to extensive shear forces produced during extrusion [31]. Our results indicated excellent self-healing ability of MA-GNPs inks after destructive shearing (Fig. 3b). This self-healing ability is due to the reversibility of non-covalent bonds between the nanoparticles, and is in line with the previously reported self-healing capacity of gelatin-based colloidal systems [20, 28, 29]. Quantification of the self-healing properties of the inks (Fig. S10) revealed the ability of all compositions to immediately (<15 s) recover ~70–80% of their elastic behavior (i.e., G’), followed by ~100% recovery after 5 min. These results suggest that the colloidal inks would be capable of recovering their solid-like character upon their deposition through needles during printing, enabling the maintenance of integrity of printed structures prior to further stabilization by UV crosslinking.
Previous studies have quantified yield stress and/or yield strain of hydrogels to determine their extrudability for 3D bioprinting [51, 52]. While yield stress values indicate the amount of force (pressure) required to initiate material flow, yield strain values indicate the amount of deformation necessary for this end. Although lower values of these yielding parameters could translate to their “easier” extrudability, yielding at excessively low stress or strain would compromise the shape fidelity of the bio-inks and the stability of prints as it can result in post-deposition flow of the material or brittle behavior of the constructs. Indeed, previous results suggest that suitable yield stress values for achieving successful construct printing prior to secondary post-printing crosslinking would typically be greater than 100 Pa [51]. As shown in Figs. 3c and 3d, our results showed a gradual increase of yield stress from ~200 Pa to ~1600 Pa by increasing the nanoparticle content in inks from 10.0 w/v% to 20.0 w/v%, suggesting that a higher printing pressure would be required for more concentrated ink compositions when a pneumatic extruder is employed. Nonetheless, yield strain of the inks remained unchanged among different compositions (Figs. 3e and 3f), suggesting that a similar deformation would be needed to initiate the flow in a mechanical extruder.
Stable and uniform formation of fibers (filaments) is an essential requirement for extrusion-based printing of biomaterials [53]. Therefore, we next studied the fiber-forming ability of different inks by monitoring their extrusion through a 22 gauge needle (Fig. 4a). Filter-pressing was observed for 10.0 and 12.5 w/v% compositions as initial expulsion of the liquid phase took place in the form of droplets while solid particles mainly remained in the syringe. By increasing the particle content of the inks to 15 w/v%, no filter-pressing was observed, but the fiber formation was not stable during the extrusion process. Nonetheless, uniform fibers were formed for 17.5 w/v% and 20 w/v% inks, which were then chosen for further investigations. As the next step, these two compositions were extruded through a range of larger and smaller needles to determine the diameter and uniformity of the resulting fibers. The extruded fibers from both compositions closely followed the inner diameter of the needles (7±4% diameter mismatch), producing fibers with diameters ranging from ~600 μm to ~170 μm (Fig. 4b). However, extrusion of 17.5 w/v% inks through the 30 gauge needle did not display sufficient stability, and was therefore excluded from the quantifications. Importantly, all the extruded fibers were compatible with the UV crosslinking depth of the inks discussed above, as their radii were ≤300 μm. Moreover, uniformity of the fibers was not impacted by altering the needle size (Fig. 4c), which suggests the applicability of the colloidal inks for 3D printing of constructs with a wide range of resolutions.
Figure 4.
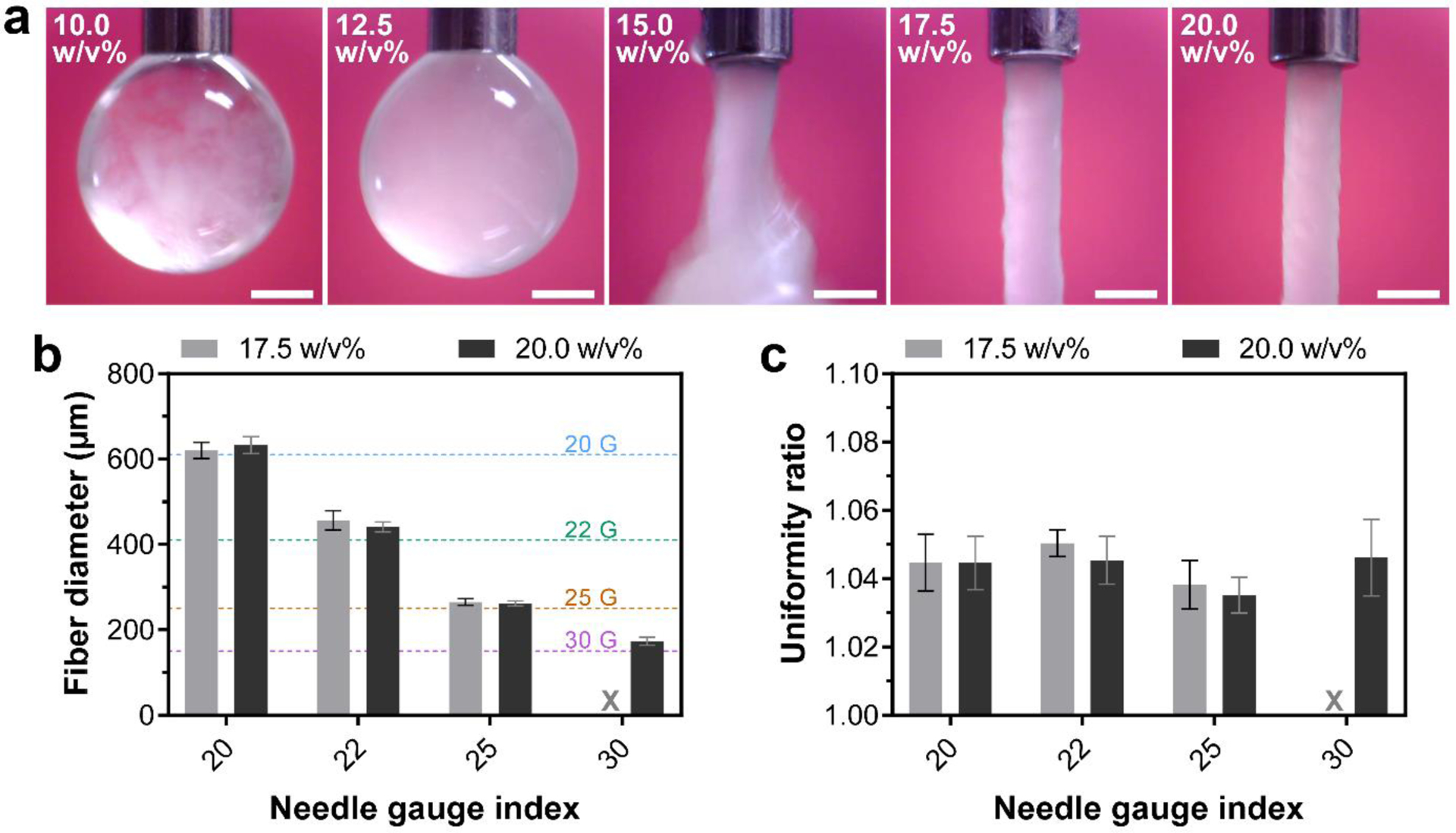
Extrusion-based fiber formation of colloidal inks. (a) Digital photographs showing ink extrusion through a 22 gauge needle for different colloidal ink compositions. Scale bars indicate 500 μm. (b) Diameter and (c) uniformity ratio of extruded fibers as a function of needle size. X indicates the condition with a low stability of fiber formation. Horizontal dashed lines in (b) indicate nominal inner diameter of different needle sizes. All values are shown as mean ± standard deviation for n=10 fibers per experimental condition.
3.4. Mechanical performance of printed constructs
Next, to determine the mechanical performance of printed constructs, we fabricated one-layer constructs by longitudinal printing of 17.5 w/v% inks and studied the effect of UV crosslinking on their mechanical properties by performing tensile tests. Non-UV-crosslinked samples were too weak to be handled after printing and transported for mechanical testing without damage. Therefore, all constructs were freeze-dried directly after printing or UV crosslinking, and were reswollen with PBS once fixed on the tensile testing grips. As shown in Fig. 5 (−UV (Long.) vs. +UV (Long.)), our results indicated that UV crosslinking significantly enhanced the mechanical properties of the constructs, increasing their tensile modulus (E) and strength (σt) to 199.6±20.0 kPa and 21.2±4.2 kPa, corresponding to 12 and 13 times increase, respectively. Strikingly, these values surpass previously reported values for silica-reinforced gelatin-based nanocomposite colloidal gels (E=40.6±6.7 kPa and σt=8.4±1.8 kPa), even though the MA-GNPs inks used for construct printing contained a lower amount of nanoparticles (particle volume fraction of 0.74 vs. 1) [20]. In addition, hydrogels assembled by packing and consequent UV-crosslinking of GelMA microbeads exhibited an E in the range of 10–30 kPa [32]. Moreover, other particle-only granular hydrogel systems used for 3D printing [33, 37, 38] produced constructs with significantly weaker mechanical properties (e.g., compressive modulus ~ 5 kPa after covalent crosslinking [37]). These results emphasize the capacity of the MA-GNP colloidal inks for 3D printing of biomaterial constructs with superior mechanical stability.
Figure 5.
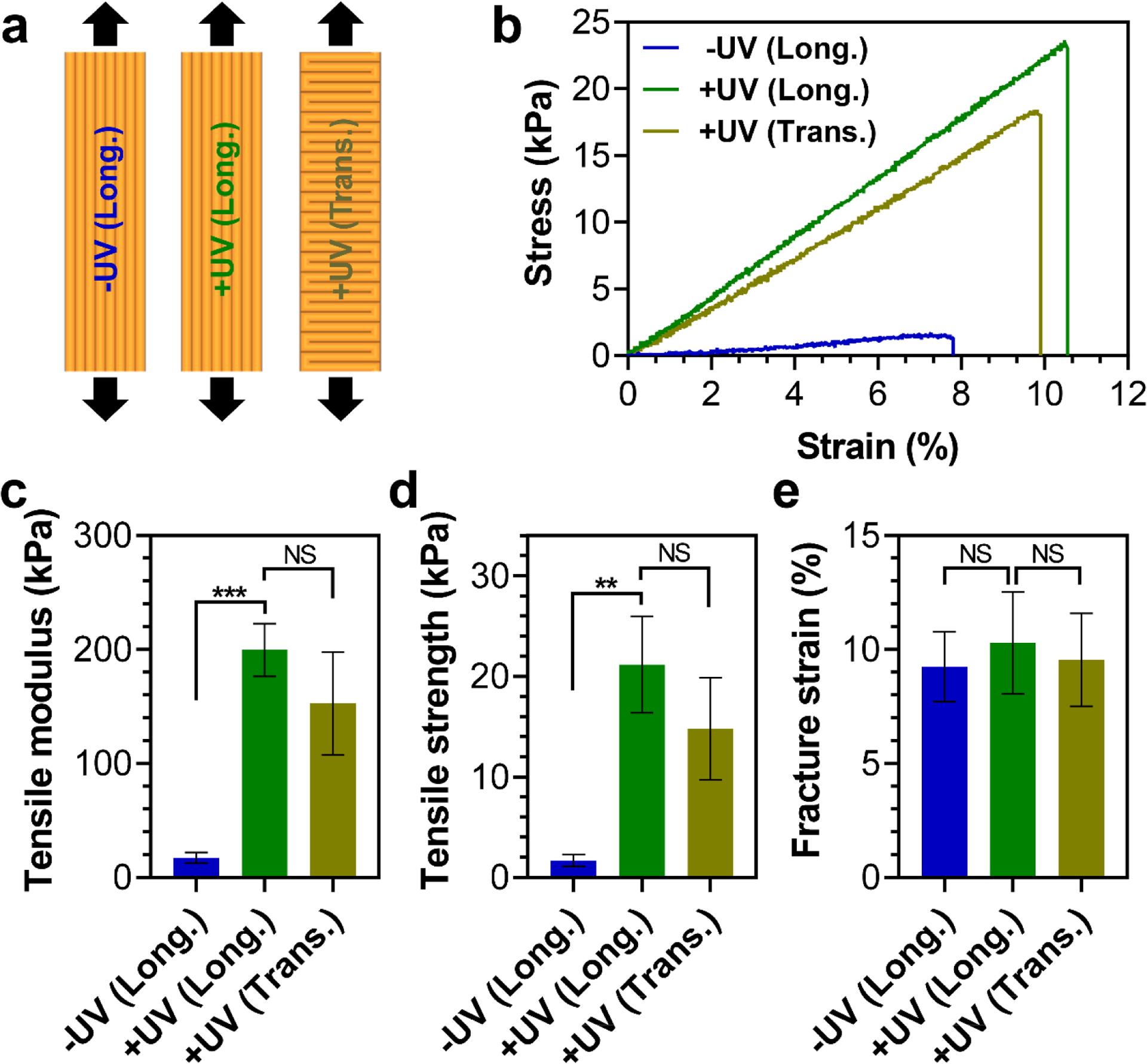
Mechanical properties of printed constructs under tension. (a) Schematic illustration of one-layer constructs printed in longitudinal (Long.) or transverse (Trans.) directions, with (+) or without (−) UV crosslinking. (b) Stress-strain curves of constructs under tensile loading, and quantified values of their (c) tensile modulus, (d) tensile strength, and (e) fracture strain. ** and *** indicate statistical difference with p<0.01 and p<0.001, respectively. NS indicates statistically not significant (p>0.05). (c,d,e) All values are shown as mean ± standard deviation for n=4 samples per experimental condition.
We also investigated the effect of printing direction on the mechanical properties of UV-crosslinked constructs by comparing the tensile performance of longitudinally and transversely printed samples (+UV (Long.) vs. +UV (Trans.)). These results revealed that printing direction had no statistically significant effect on the mechanical properties of constructs under tension (Fig. 5), indicating that inter-fiber bonds of the constructs exhibited a similar strength as the intra-fiber bonds.
3.5. In vitro swelling, degradation and release behavior of printed constructs
The compatibility of printed colloidal constructs to undergo freeze-drying and re-swelling without exhibiting significant morphological changes presents new opportunities for their clinical translatability. In particular, hydrogel-based biomaterials commonly suffer from challenges associated with their storability and sterilization, which can potentially be overcome by processing these materials into freeze-dried constructs, enabling their “off-the-shelf” applications [54, 55]. Consequently, we further investigated the morphological changes of two-layer porous constructs, with or without UV crosslinking, during different stages of post-printing processing and re-swelling at 37 °C in PBS solutions. As shown in Figs. 6a and 6b, upon UV crosslinking and freeze-drying, the constructs experienced some degree of shrinkage, which can be attributed to increased packing of nanoparticles upon covalent crosslinking and subsequent loss of water molecules upon drying. Nonetheless, these steps did not alter the average pore size of the constructs as compared to their original, as-printed design (Fig. 6c). Consequently, in view of the low shrinkage, the lack of crack formation, and the preservation of their architecture at this dry state, these freeze-dried constructs can be classified as aerogels [56, 57].
Figure 6.
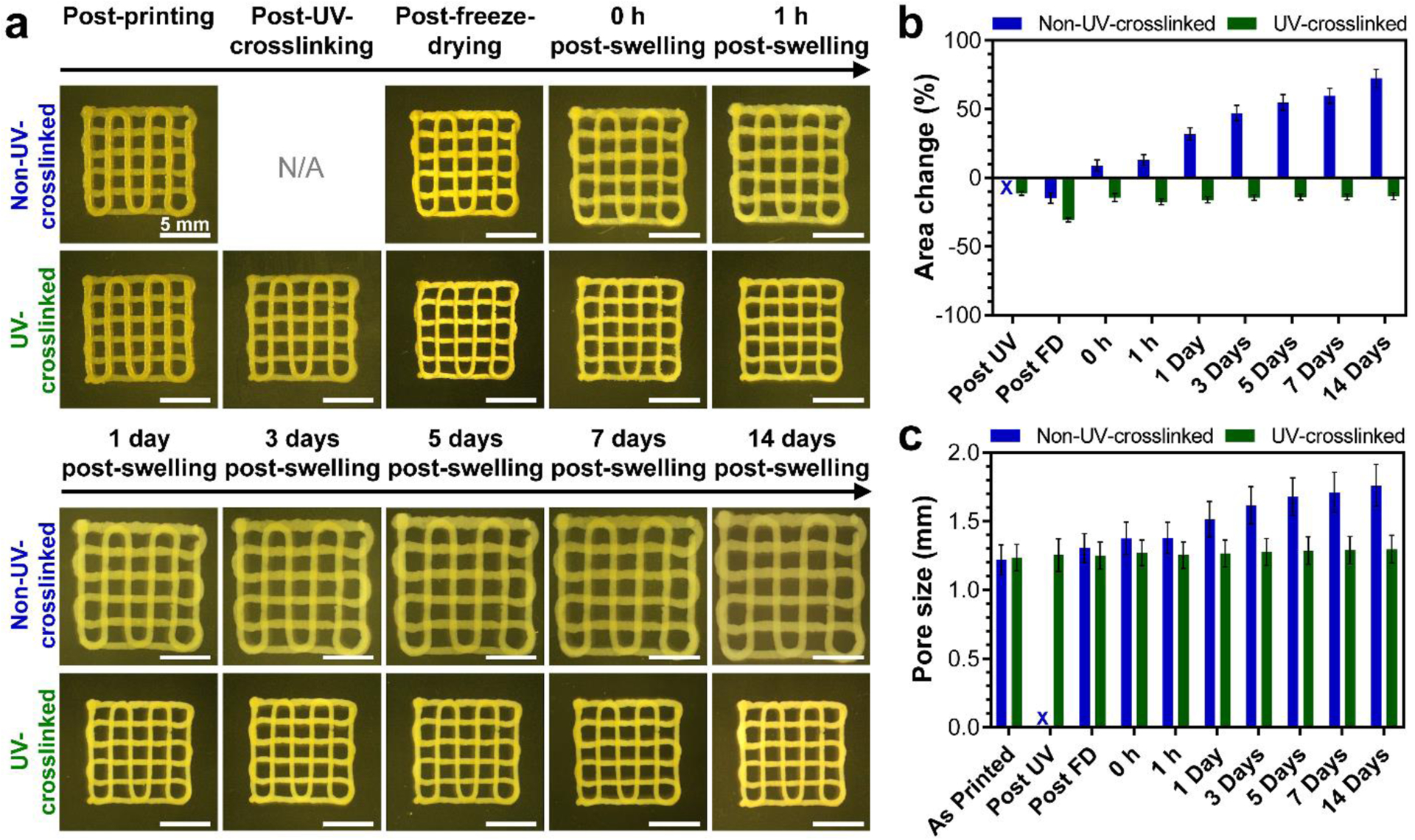
Swelling behavior of printed constructs with or without UV crosslinking. (a) Representative optical microscopy images of printed constructs at different processing and swelling stages. Quantified values of (b) change of non-porous area of constructs and (c) their pore size at different processing and swelling stages. N/A and X in (a) and (b–c), respectively, specify the lack of UV crosslinking. All values are shown as mean ± standard deviation for n=5–6 samples per experimental condition.
Upon re-swelling, non-UV-crosslinked constructs displayed significant morphological changes, exhibiting ~72% and ~44% increase of their non-porous area and pore size at day 14 of the re-swelling process, respectively. In contrast, UV-crosslinked constructs exhibited a remarkable capacity to maintain their morphology throughout the experiment; upon 14 days of incubation, the constructs matched their non-porous area at the UV crosslinking stage and no changes were observed in their pore size as compared to their as-printed morphology. This capacity signifies the potential of UV-crosslinked colloidal constructs to serve as scaffolds with pre-defined architectures under physiological conditions, and therefore to satisfy a fundamental requirement for various biomedical applications, including tissue engineering and regenerative medicine [3].
Enzymatic degradation of gelatin-based colloidal biomaterials by MMPs (i.e., collagenase) has been demonstrated previously [18, 19, 25–27]. Nonetheless, the effects of the methacryloylation process as well as UV crosslinking on the degradation behavior of the colloidal constructs were unknown. Therefore, we next studied the degradation kinetics of non-UV-crosslinked and UV-crosslinked constructs by incubating the constructs during rotation at 37 °C in solutions containing 0 or 400 ng/mL of collagenase. As shown in Fig. 7a, in the absence of collagenase, non-UV-crosslinked constructs exhibited ~65% of gelatin degradation within the first three days of incubation, which reached a plateau after this time point. In contrast, UV-crosslinked constructs exhibited near-zero gelatin degradation throughout the 14 days of the experiment. In addition, while non-UV-crosslinked constructs lost their integrity during the rotation in the incubation process, UV-crosslinked constructs could successfully preserve their architecture (Fig. S11). These results emphasize the effectiveness of the UV crosslinking modality in this system, and reveal a minimal non-enzymatic degradation for the UV-crosslinked constructs.
Figure 7.
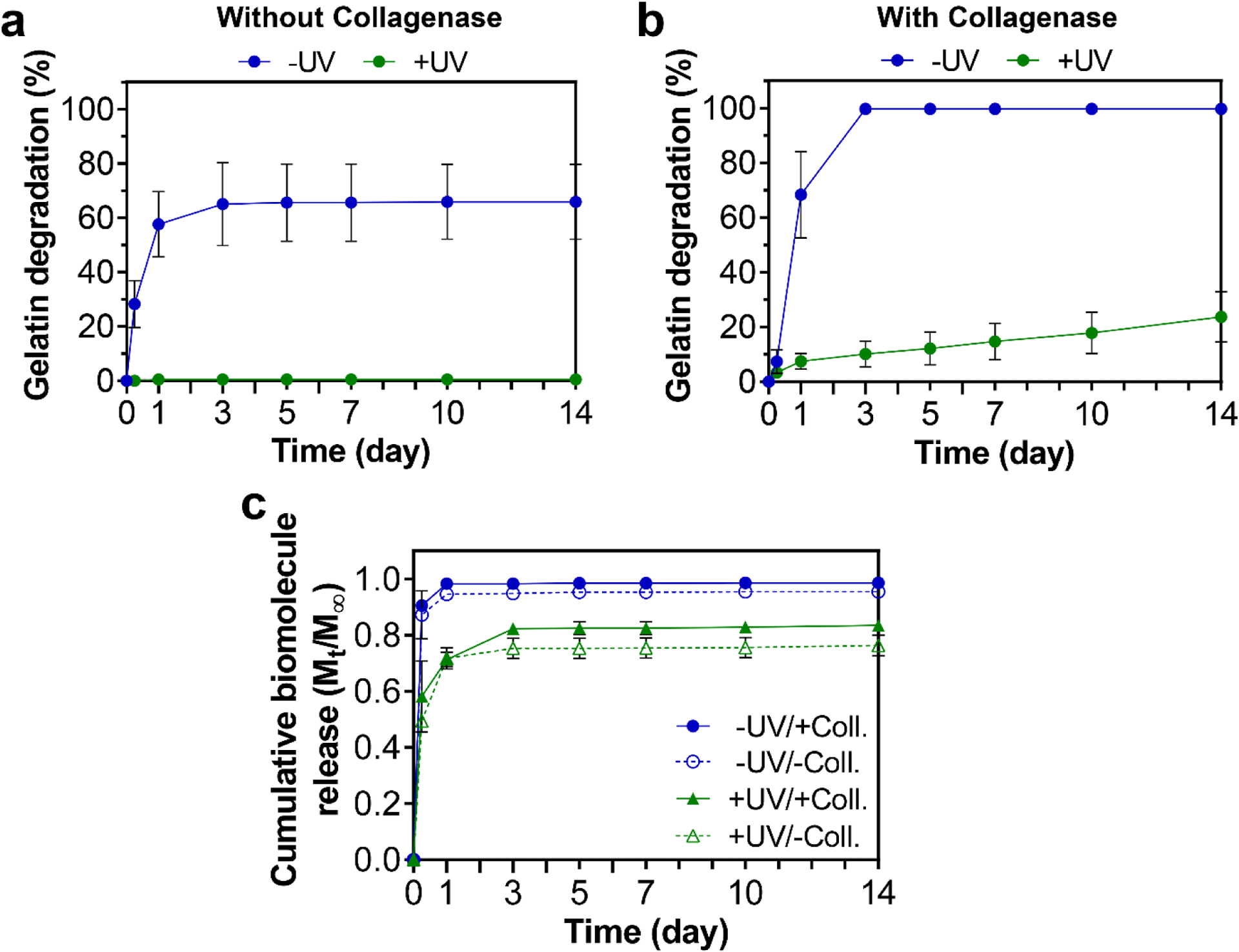
In vitro degradation and biomolecule delivery capacity of printed constructs with (+) or without (−) UV crosslinking. Degradation kinetics of constructs determined by release of soluble gelatin into solutions without (a) or with (b) collagenase enzyme. (c) Cumulative release kinetics of a fluorescently labeled model biomolecule (bovine serum albumin) from the constructs in solutions with (+Coll.) or without (−Coll.) collagenase. All values are shown as mean ± standard deviation for n=4 samples per experimental condition.
As shown in Fig. 7b, upon incubation of constructs in collagenase-containing solutions, non-UV-crosslinked constructs fully degraded within the first three days of incubation, with no traces of constructs visible at Day 14 (Fig. S11). Upon UV crosslinking, however, the constructs showed a sustained and linear degradation profile, exhibiting ~25% gelatin degradation at Day 14 while maintaining their architecture (Fig. S11). Extrapolation of this degradation profile using a linear regression fit estimates that complete enzymatic degradation of these constructs would take place upon 65 days of incubation. Consequently, these results illuminate the large tunability of degradation kinetics in this system, possibly by modulation of the UV crosslinking degree of the constructs (e.g., using different UV irradiation times), enabling them to match different application requirements. In addition, the nanoparticle building blocks used in this system can potentially be covalently functionalized with additional bioactive factors (e.g., bisphosphonate drugs [20, 23, 24]), facilitating the delivery of these functional moieties at a rate consistent with the intended degradation profile of the constructs.
On a separate note, the degradation of non-UV-crosslinked constructs, with or without collagenase, witnessed in this study (Figs. 7a and 7b) took place considerably more rapidly than the values observed previously for other gelatin-based colloidal biomaterials that were not UV-crosslinked [18, 19, 25–27]. Since gelatin nanoparticles used in previously reported systems did not undergo methacryloylation, we hypothesized that this accelerated degradation is linked to the methacryloylation process employed in this study. We therefore compared the degradation behavior of GNPs and MA-GNPs upon 3 days of incubation in solutions without or with collagenase (Fig. S12). These experiments revealed a significantly lower degradation for GNPs than for MA-GNPs, indicating the role of methacryloylation step on the enhanced degradation of nanoparticles and therefore constructs. We postulate that during the methacryloylation process, the glutaraldehyde-based covalent crosslinks of individual nanoparticles are partially compromised, likely due to the acidic environment created by methacrylic acid as the byproduct of the reaction, in which glutaraldehyde-amine bonds can exhibit a large degree of reversibility [58, 59].
Physical loading of nano- and micro-particulate gelatin carriers with biomolecules such as growth factors has been widely studied and demonstrated as a suitable pathway for controlled delivery of such bioactive factors for various biomedical applications [60, 61]. This capacity is commonly attributed to the ability of gelatin to form polyion complexes with oppositely charged biomolecules, resulting in their controlled release during enzymatic degradation of gelatin [61]. Additionally, recent research has revealed that the molecular weight of loaded biomolecules plays a major role in determining their retention in gelatin carriers [27]. To further elucidate the effect of the UV crosslinking modality in this system on the release behavior of bioactive molecules from the gelatin-based constructs, we loaded the colloidal inks with fluorescently labeled BSA as a model biomolecule, and studied the impact of UV crosslinking and enzymatic degradation on the release kinetics of these biomolecules from 3D printed constructs (Fig. 7c). These results showed that non-UV-crosslinked constructs displayed a burst and near-complete release of BSA within the first day of incubation regardless of the presence of collagenase in the degradation solutions. Interestingly, UV crosslinking could significantly decelerate the release kinetics of these model biomolecules. For these constructs, after a more gradual burst release during the first three days, the rate of release was substantially reduced, resulting in a sustained release profile for both non-enzymatic and enzymatic conditions. The retention of biomolecules observed at later time points for UV-crosslinked samples likely arises from the tight crosslinking of the colloidal gel networks in this system, which is enabled by the combinations of methacryloyl-based and glutaraldehyde-based covalent crosslinks, as well as non-covalent interactions of gelatin. The burst release profiles observed at earlier time points can be attributed to the anionic nature of albumin and the gelatin (type B) employed in this system, resulting in weak interactions between these components. Therefore, more sustained release profiles at early time points can be expected for biomolecules or gelatin matrix with appropriate physicochemical properties, such as other reported bioactive molecules with a higher binding affinity towards the selected gelatin type [18, 25–27, 60, 61]. Importantly, the inclusion of such bioactive molecules in this system requires in-depth investigations in future studies to determine the preservation of their bioactivity at different stages of 3D printing, UV crosslinking, freeze-drying and long-term storage.
3.6. Shape memory behavior of printed constructs
Shape memory biomaterials have recently emerged as promising solutions for the design of tissue engineering scaffolds that can maintain a temporary shape and revert to their original shape in response to external stimuli such as light, change of temperature, or magnetic field [62]. Clinical applications of such shape memory systems would include the design of deployable scaffolds by enabling the biomaterial to maintain a temporary small size suitable for minimally invasive delivery, followed by its expansion to fill the defect volume [63, 64]. Similarly, this ability for programmed shape transformation can be exploited for 4D printing of tissue engineering scaffolds that conform to tissue geometry in response to external stimuli [65, 66].
Hydrogels are generally flexible materials, mainly due to their high content of water molecules, which can have a plasticizing effect on their polymer chains, facilitating their molecular mobility [67]. Under frozen or dehydrated states, these materials can become rigid, retaining their shape as the mobility of their hydrophilic polymer chains is impeded by the ice crystals or the replacement of water-polymer with polymer-polymer hydrogen bonding, respectively [67, 68]. Additionally, for the UV-crosslinked constructs fabricated in this study, we witnessed a highly elastic response, with a high degree of elastic deformation upon curving and a consequent shape recovery. In view of these facts, we hypothesized that freeze-drying of re-shaped constructs can allow for a temporary fixation of their shape, and their subsequent re-hydration should enable the recovery of their original geometry. Accordingly, we investigated two types of design procedures, which were aimed to produce constructs exhibiting shape recovery from curved to flat, or from flat to curved, geometries (Fig. 8a). Since the re-shaping of the constructs after UV crosslinking can result in stress generation in their structure, and the drying of hydrated materials can result in brittle behavior, we first evaluated the ability of different construct designs (two- or four-layer prints, with three different curvature degrees) to maintain their integrity after the freeze-drying step. As shown in Table 1, all the constructs prepared with the curved-to-flat design maintained their integrity after freeze-drying. However, for the flat-to-curved design, two-layer constructs were not able to maintain their integrity, while half of the four-layer constructs could withstand the process. This high tendency of constructs with the flat-to-curved design to lose their integrity can be related to a high degree of (primarily) tensile stress generated in these constructs during the flattening process. Nonetheless, we discovered that adhesion between the samples and their underlying PET sheet (Fig. S3) played a more major role in this issue, as all four-layer constructs of this design could maintain their integrity once this PET sheet was removed after the freezing step.
Figure 8.
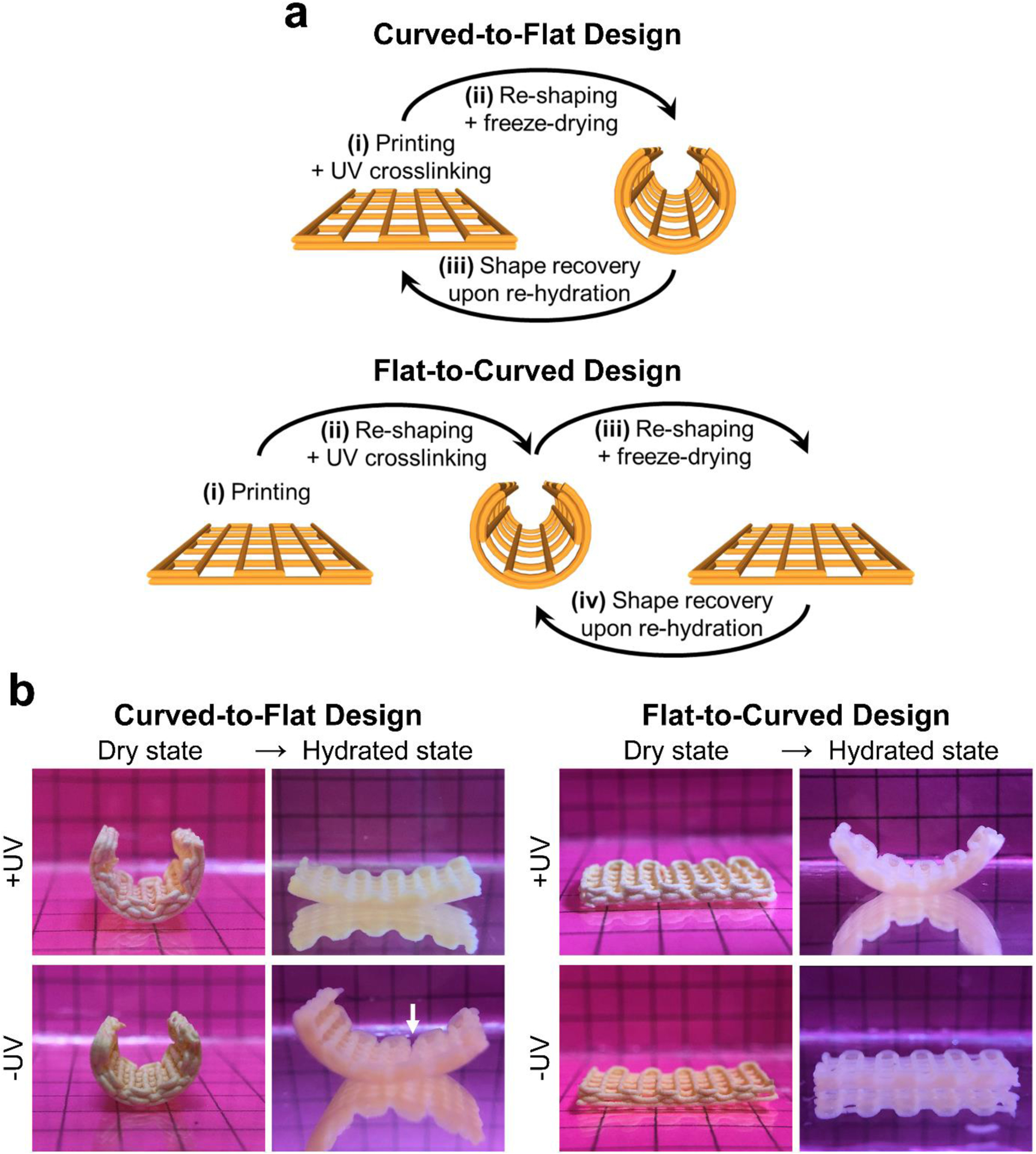
Shape memory behavior of printed constructs. (a) Schematic illustration of fabrication steps of constructs with shape memory capacity. (b) Digital photographs of different construct designs at dry state (temporary geometry) and in the hydrated state showing their shape recovery performance upon immersion in aqueous media. Constructs shown in (b) were composed of 4 layers and were 20 mm and 10 mm in length and width, respectively. Grids in the background had 5 mm × 5 mm dimensions. White arrow indicates crack formation in the construct during the re-hydration process.
Table 1.
Integrity of shape memory constructs with different designs after the freeze-drying step. All constructs were 10 mm in length and width. Each check box indicates integrity status of one construct, for n=4 samples per experimental condition.
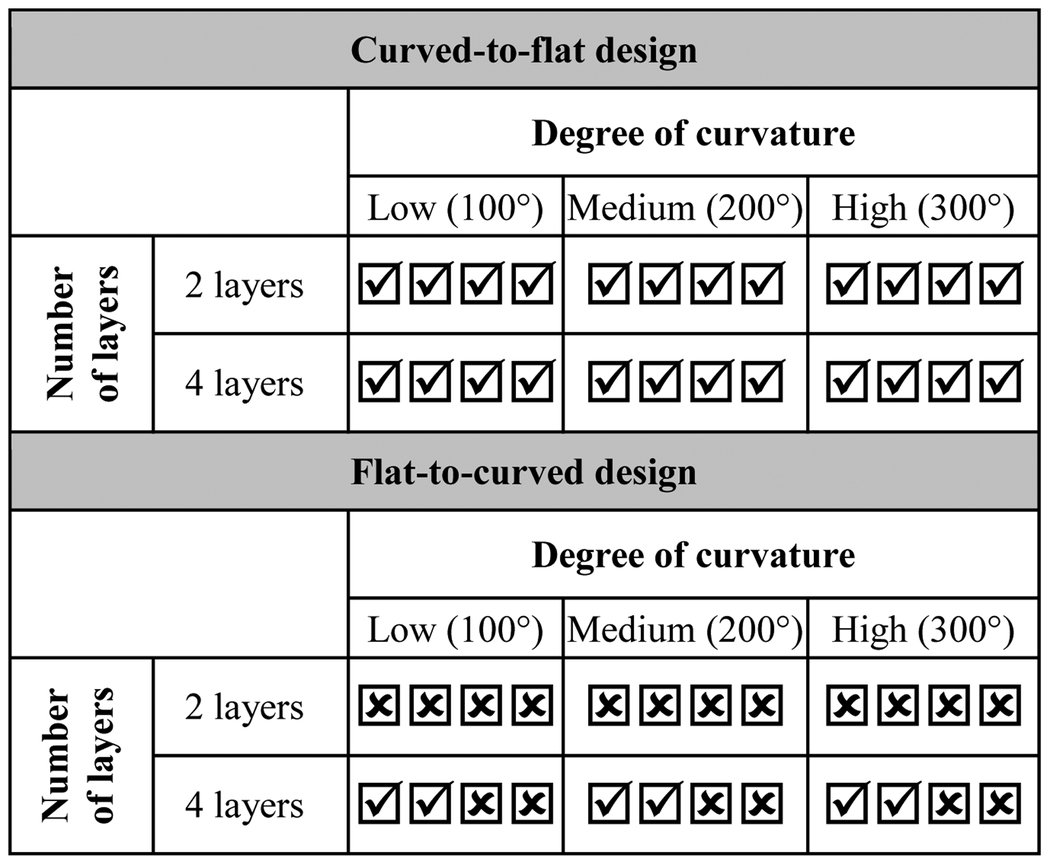
|
Construct kept its integrity;
Construct lost its integrity
We then exposed the UV-crosslinked constructs to PBS to evaluate their capacity for shape recovery, while using non-UV-crosslinked samples as a negative control. As shown in Fig. 8b, our results confirmed that UV-crosslinked constructs of both designs exhibited a high degree of shape recovery, immediately (~1 s) upon immersion in PBS. In contrast, non-UV-crosslinked samples were unable to recover their original shape, with the curved-to-flat constructs exhibiting partial or full fracture (Fig. S13) during the immersion process due to their mechanical weakness associated with the lack of covalent interparticle crosslinks in these constructs.
To elucidate the shape memory mechanism of this system, we investigated stress relaxation in printed samples with or without UV crosslinking (Fig. S14). These tests revealed that, regardless of UV crosslinking status, the colloidal constructs exhibited a remarkable ability to retain the stress generated upon deformation in their structure, showing only ~10–15% of stress relaxation after 120 s. Accordingly, we postulate that the shape memory behavior of the UV-crosslinked samples in this system arises from the combination of (i) their high elasticity, enabling the generation of a high amount of elastic energy in their structure, and (ii) their low stress relaxation behavior, allowing for the storage of this elastic energy under re-shaped and freeze-dried states. Upon re-hydration, the polymer chain mobility is revived, permitting the release of elastic energy and the recovery of constructs to their original, UV-crosslinked shape. Meanwhile, non-UV-crosslinked samples are unable to store a high amount of elastic energy due to their low elasticity and their ability to undergo plastic deformation during the re-shaping stage.
We also prepared hydrogel sheets via conventional casting using MA-GNPs or a GelMA control (Fig. S15) to determine the importance of (i) porous architecture enabled by 3D printing or (ii) the hierarchical nanoparticle-based nature of our colloidal system, respectively, for achieving rapid shape recovery (Movie S1). These experiments showed that MA-GNP-based and GelMA sheets took ~10 times (~10 s) and ~60 times (~1 min) longer than the printed constructs with curved-to-flat design to unfold upon exposure to PBS, emphasizing the key role of the 3D porous design and MA-GNP formulation in the shape memory response of the constructs.
Finally, the cytocompatibility and the suitability of gelatin-based colloidal biomaterials for use in biomedical applications are well established in various in vitro and in vivo studies [18–20, 25, 29, 69]. To additionally confirm the cytocompatibility of these biomaterial types after subjection to the methacryloylation process and UV crosslinking, we evaluated the viability and morphology of human mesenchymal stem cells encapsulated in MA-GNP inks after 1 or 24 h of culture (Fig. S16). These experiments confirmed that the methacryloylation and UV crosslinking do not compromise the cytocompatibility of this biomaterial type, highlighting the potential of MA-GNP-based constructs for use as scaffolds for tissue engineering.
4. Conclusions
We have demonstrated successful 3D printing of biomaterial constructs using photo-reactive gelatin nanoparticles as colloidal building blocks. These building blocks were prepared through methacryloyl functionalization of gelatin nanoparticles that were stabilized with intraparticle covalent crosslinks. Owing to the presence of non-covalent interactions between these nanoparticles, colloidal gels could be formed as inks that were extrudable and self-healing, and therefore printable at room temperature. The functionalization of these nanoparticles with methacryloyl groups enabled post-printing stabilization of 3D constructs via UV irradiation by establishing covalent interparticle crosslinks. The interparticle UV crosslinking modality in this system could not only significantly enhance the mechanical properties of the printed constructs, but also allowed for control over their swelling and degradation, as well as retention of loaded biomolecules within the constructs. Remarkably, these UV-induced crosslinks enabled the printed constructs to maintain their pre-designed architecture under physiological conditions involving enzymatic degradation of gelatin. Finally, owing to the elastic behavior and low stress relaxation of UV-crosslinked prints, 3D constructs could be designed that were able to uphold a temporary shape in the freeze-dried state and revert to a programmed original geometry upon re-hydration. This shape memory feature can open new avenues for the minimally invasive use of gelatin-based colloidal constructs as deployable systems, or for their use as tissue-conforming structures that can adapt to a specific tissue/defect shape once implanted in the body. Importantly, the biomaterial design strategy demonstrated in this work relies on a widely available and cost-effective natural biomaterial (i.e., gelatin) as well as relatively simple material processing methods (i.e., extrusion-based 3D printing and freeze-drying). Therefore, the introduced methodology is not limited by inaccessible and complex synthetic chemistries nor fabrication techniques. Additionally, in view of the well-established suitability of gelatin-based colloidal gels for use in tissue regeneration, 3D printed colloidal biomaterials developed in this study exhibit a great potential to serve as scaffolds with shape memory features for applications in tissue engineering.
Supplementary Material
Acknowledgments
M.D. is supported by a Rubicon postdoctoral fellowship from the Dutch Research Council (NWO; Project No. 019.182EN.004). This study was supported by the National Institutes of Health (Grant P41 EB023833). G.L.K. is supported by the Robert and Janice McNair Foundation MD/PhD Student Scholar Program. M.L.B. is supported by the National Science Foundation Graduate Research Fellowship Program. The authors would like to thank Mr. Shail Mehta for providing training for the tensile testing machine used in this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Data availability statement
The data that support the findings of this study are available from the corresponding author (A.G.M.) upon request.
References
- [1].Mock C, Cherian MN, The global burden of musculoskeletal injuries: Challenges and solutions, Clin. Orthop. Relat. Res 466 (2008) 2306. https://dx.doi.org/10.1007%2Fs11999-008-0416-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Woolf AD, Pfleger B, Burden of major musculoskeletal conditions, Bull. W. H. O 81 (2003) 646–56. [PMC free article] [PubMed] [Google Scholar]
- [3].Koons GL, Diba M, Mikos AG, Material design for bone tissue engineering, Nat. Rev. Mater 5 (2020) 584–603. 10.1038/s41578-020-0204-2. [DOI] [Google Scholar]
- [4].Du Y, Guo JL, Wang J, Mikos AG, Zhang S, Hierarchically designed bone scaffolds: From internal cues to external stimuli, Biomaterials 218 (2019) 119334. 10.1016/j.biomaterials.2019.119334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Collins MN, Ren G, Young K, Pina S, Reis RL, Oliveira JM, Scaffold fabrication technologies and structure/function properties in bone tissue engineering, Adv. Funct. Mater 2010609. 10.1002/adfm.202010609. [DOI] [Google Scholar]
- [6].Diba M, Kharaziha M, Fathi MH, Gholipourmalekabadi M, Samadikuchaksaraei A, Preparation and characterization of polycaprolactone/forsterite nanocomposite porous scaffolds designed for bone tissue regeneration, Compos. Sci. Technol 72 (2012) 716–723. 10.1016/j.compscitech.2012.01.023. [DOI] [Google Scholar]
- [7].Hiebl B, Lützow K, Lange M, Jung F, Seifert B, Klein F, Weigel T, Kratz K, Lendlein A, Cytocompatibility testing of cell culture modules fabricated from specific candidate biomaterials using injection molding, J. Biotechnol 148 (2010) 76–82. 10.1016/j.jbiotec.2010.01.006. [DOI] [PubMed] [Google Scholar]
- [8].Bittner SM, Guo JL, Melchiorri A, Mikos AG, Three-dimensional printing of multilayered tissue engineering scaffolds, Mater. Today 21 (2018) 861–874. 10.1016/j.mattod.2018.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Sydney Gladman A, Matsumoto EA, Nuzzo RG, Mahadevan L, Lewis JA, Biomimetic 4d printing, Nat. Mater 15 (2016) 413–418. 10.1038/nmat4544. [DOI] [PubMed] [Google Scholar]
- [10].Wu DJ, Bouten CVC, Dankers PYW, From molecular design to 3d printed life-like materials with unprecedented properties, Curr. Opin. Biomed. Eng 2 (2017) 43–48. 10.1016/j.cobme.2017.06.001. [DOI] [Google Scholar]
- [11].Li J, Wu C, Chu PK, Gelinsky M, 3d printing of hydrogels: Rational design strategies and emerging biomedical applications, “Mater. Sci. Eng., R” 140 (2020) 100543. 10.1016/j.mser.2020.100543. [DOI] [Google Scholar]
- [12].Bedell ML, Navara AM, Du Y, Zhang S, Mikos AG, Polymeric systems for bioprinting, Chem. Rev 120 (2020) 10744–10792. 10.1021/acs.chemrev.9b00834. [DOI] [PubMed] [Google Scholar]
- [13].Peppas NA, Hilt JZ, Khademhosseini A, Langer R, Hydrogels in biology and medicine: From molecular principles to bionanotechnology, Adv. Mater 18 (2006) 1345–1360. 10.1002/adma.200501612. [DOI] [Google Scholar]
- [14].Tibbitt MW, Anseth KS, Hydrogels as extracellular matrix mimics for 3d cell culture, Biotechnol. Bioeng 103 (2009) 655–663. 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Decante GBH, Costa J, Silva-Correia J, Collins M, Reis RL, Oliveira JM, Engineering bioinks for 3d bioprinting, Biofabrication 13 (2021) 032001. 10.1088/1758-5090/abec2c. [DOI] [PubMed] [Google Scholar]
- [16].Lu PJ, Weitz DA, Colloidal particles: Crystals, glasses, and gels, Annu. Rev. Condens. Matter Phys 4 (2013) 217–233. 10.1146/annurev-conmatphys-030212-184213. [DOI] [Google Scholar]
- [17].Rose JC, De Laporte L, Hierarchical design of tissue regenerative constructs, Adv. Healthcare Mater 7 (2018) 1701067. 10.1002/adhm.201701067. [DOI] [PubMed] [Google Scholar]
- [18].Diba M, Pape B, Klymov A, Zhang Y, Song J, Löwik DWPM, Seyednejad H, Leeuwenburgh SCG, Nanostructured raspberry-like gelatin microspheres for local delivery of multiple biomolecules, Acta Biomater. 58 (2017) 67–79. 10.1016/j.actbio.2017.05.059. [DOI] [PubMed] [Google Scholar]
- [19].Wang H, Bongio M, Farbod K, Nijhuis AWG, van den Beucken J, Boerman OC, van Hest JCM, Li Y, Jansen JA, Leeuwenburgh SCG, Development of injectable organic/inorganic colloidal composite gels made of self-assembling gelatin nanospheres and calcium phosphate nanocrystals, Acta Biomater. 10 (2014) 508–519. 10.1016/j.actbio.2013.08.036. [DOI] [PubMed] [Google Scholar]
- [20].Diba M, Wang H, Kodger TE, Parsa S, Leeuwenburgh SCG, Highly elastic and self-healing composite colloidal gels, Adv. Mater 29 (2017) 1604672. 10.1002/adma.201604672. [DOI] [PubMed] [Google Scholar]
- [21].Diba M, Camargo WA, Brindisi M, Farbod K, Klymov A, Schmidt S, Harrington MJ, Draghi L, Boccaccini AR, Jansen JA, van den Beucken JJJP, Leeuwenburgh SCG, Composite colloidal gels made of bisphosphonate-functionalized gelatin and bioactive glass particles for regeneration of osteoporotic bone defects, Adv. Funct. Mater 27 (2017) 1703438. 10.1002/adfm.201703438. [DOI] [Google Scholar]
- [22].Wang Q, Gu Z, Jamal S, Detamore MS, Berkland C, Hybrid hydroxyapatite nanoparticle colloidal gels are injectable fillers for bone tissue engineering, “Tissue Eng., Part A” 19 (2013) 2586–2593. 10.1089/ten.tea.2013.0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Farbod K, Diba M, Zinkevich T, Schmidt S, Harrington MJ, Kentgens APM, Leeuwenburgh SCG, Gelatin nanoparticles with enhanced affinity for calcium phosphate, Macromol. Biosci 16 (2016) 717–729. 10.1002/mabi.201500414. [DOI] [PubMed] [Google Scholar]
- [24].Farbod K, Curci A, Diba M, Zinkevich T, Kentgens APM, Iafisco M, Margiotta N, Leeuwenburgh SCG, Dual-functionalisation of gelatine nanoparticles with an anticancer platinum(ii)–bisphosphonate complex and mineral-binding alendronate, RSC Adv. 6 (2016) 113025–113037. 10.1039/C6RA19915A. [DOI] [Google Scholar]
- [25].Wang H, Zou Q, Boerman OC, Nijhuis AWG, Jansen JA, Li Y, Leeuwenburgh SCG, Combined delivery of bmp-2 and bfgf from nanostructured colloidal gelatin gels and its effect on bone regeneration in vivo, J. Controlled Release 166 (2013) 172–181. 10.1016/j.jconrel.2012.12.015. [DOI] [PubMed] [Google Scholar]
- [26].Wang H, Boerman OC, Sariibrahimoglu K, Li Y, Jansen JA, Leeuwenburgh SCG, Comparison of micro- vs. Nanostructured colloidal gelatin gels for sustained delivery of osteogenic proteins: Bone morphogenetic protein-2 and alkaline phosphatase, Biomaterials 33 (2012) 8695–8703. 10.1016/j.biomaterials.2012.08.024. [DOI] [PubMed] [Google Scholar]
- [27].Song J, Odekerken JCE, Löwik DWPM, López-Pérez PM, Welting TJM, Yang F, Jansen JA, Leeuwenburgh SCG, Influence of the molecular weight and charge of antibiotics on their release kinetics from gelatin nanospheres, Macromol. Biosci 15 (2015) 901–911. 10.1002/mabi.201500005. [DOI] [PubMed] [Google Scholar]
- [28].Wang H, Hansen MB, Löwik DWPM, van Hest JCM, Li Y, Jansen JA, Leeuwenburgh SCG, Oppositely charged gelatin nanospheres as building blocks for injectable and biodegradable gels, Adv. Mater 23 (2011) H119–H124. 10.1002/adma.201003908. [DOI] [PubMed] [Google Scholar]
- [29].Diba M, Polini A, Petre DG, Zhang Y, Leeuwenburgh SCG, Fiber-reinforced colloidal gels as injectable and moldable biomaterials for regenerative medicine, “Mater. Sci. Eng., C” 92 (2018) 143–150. 10.1016/j.msec.2018.06.038. [DOI] [PubMed] [Google Scholar]
- [30].Wang Q, Wang L, Detamore MS, Berkland C, Biodegradable colloidal gels as moldable tissue engineering scaffolds, Adv. Mater 20 (2008) 236–239. 10.1002/adma.200702099. [DOI] [Google Scholar]
- [31].Diba M, Spaans S, Ning K, Ippel BD, Yang F, Loomans B, Dankers PYW, Leeuwenburgh SCG, Self-healing biomaterials: From molecular concepts to clinical applications, Adv. Mater. Interfaces 5 (2018) 1800118. 10.1002/admi.201800118. [DOI] [Google Scholar]
- [32].Sheikhi A, de Rutte J, Haghniaz R, Akouissi O, Sohrabi A, Di Carlo D, Khademhosseini A, Microfluidic-enabled bottom-up hydrogels from annealable naturally-derived protein microbeads, Biomaterials 192 (2019) 560–568. 10.1016/j.biomaterials.2018.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Daly AC, Riley L, Segura T, Burdick JA, Hydrogel microparticles for biomedical applications, Nat. Rev. Mater 5 (2020) 20–43. 10.1038/s41578-019-0148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mealy JE, Chung JJ, Jeong H-H, Issadore D, Lee D, Atluri P, Burdick JA, Injectable granular hydrogels with multifunctional properties for biomedical applications, Adv. Mater 30 (2018) 1705912. 10.1002/adma.201705912. [DOI] [PubMed] [Google Scholar]
- [35].Miranda P, Saiz E, Gryn K, Tomsia AP, Sintering and robocasting of β-tricalcium phosphate scaffolds for orthopaedic applications, Acta Biomater. 2 (2006) 457–466. 10.1016/j.actbio.2006.02.004. [DOI] [PubMed] [Google Scholar]
- [36].Jiang Y, Xu Z, Huang T, Liu Y, Guo F, Xi J, Gao W, Gao C, Direct 3d printing of ultralight graphene oxide aerogel microlattices, Adv. Funct. Mater 28 (2018) 1707024. 10.1002/adfm.201707024. [DOI] [Google Scholar]
- [37].Highley CB, Song KH, Daly AC, Burdick JA, Jammed microgel inks for 3d printing applications, Adv. Sci 6 (2019) 1801076. 10.1002/advs.201801076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Xin S, Chimene D, Garza JE, Gaharwar AK, Alge DL, Clickable peg hydrogel microspheres as building blocks for 3d bioprinting, Biomater. Sci 7 (2019) 1179–1187. 10.1039/C8BM01286E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shin M, Song KH, Burrell JC, Cullen DK, Burdick JA, Injectable and conductive granular hydrogels for 3d printing and electroactive tissue support, Adv. Sci 6 (2019) 1901229. 10.1002/advs.201901229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zheng J, Zhu M, Ferracci G, Cho N-J, Lee BH, Hydrolytic stability of methacrylamide and methacrylate in gelatin methacryloyl and decoupling of gelatin methacrylamide from gelatin methacryloyl through hydrolysis, Macromol. Chem. Phys 219 (2018) 1800266. 10.1002/macp.201800266. [DOI] [Google Scholar]
- [41].Oliver WC, Pharr GM, An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments, J. Mater. Res 7 (1992) 1564–1583. 10.1557/JMR.1992.1564. [DOI] [Google Scholar]
- [42].Gao T, Gillispie GJ, Copus JS, Pr AK, Seol Y-J, Atala A, Yoo JJ, Lee SJ, Optimization of gelatin–alginate composite bioink printability using rheological parameters: A systematic approach, Biofabrication 10 (2018) 034106. 10.1088/1758-5090/aacdc7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yoshihara Y, Nakamura H, Obata K.i., Yamada H, Hayakawa T, Fujikawa K, Okada Y, Matrix metalloproteinases and tissue inhibitors of metalloproteinases in synovial fluids from patients with rheumatoid arthritis or osteoarthritis, Ann. Rheum. Dis 59 (2000) 455–461. 10.1136/ard.59.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shi M, Kretlow JD, Spicer PP, Tabata Y, Demian N, Wong ME, Kasper FK, Mikos AG, Antibiotic-releasing porous polymethylmethacrylate/gelatin/antibiotic constructs for craniofacial tissue engineering, J. Controlled Release 152 (2011) 196–205. 10.1016/j.jconrel.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Perez-Jimenez R, Godoy-Ruiz R, Ibarra-Molero B, Sanchez-Ruiz JM, The efficiency of different salts to screen charge interactions in proteins: A hofmeister effect?, Biophys. J 86 (2004) 2414–2429. 10.1016/S0006-3495(04)74298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Trefalt G, Palberg T, Borkovec M, Forces between colloidal particles in aqueous solutions containing monovalent and multivalent ions, Curr. Opin. Colloid Interface Sci 27 (2017) 9–17. 10.1016/j.cocis.2016.09.008. [DOI] [Google Scholar]
- [47].Rebers L, Granse T, Tovar GEM, Southan A, Borchers K, Physical interactions strengthen chemical gelatin methacryloyl gels, Gels 5 (2019) 4. 10.3390/gels5010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Claaßen C, Claaßen MH, Truffault V, Sewald L, Tovar GEM, Borchers K, Southan A, Quantification of substitution of gelatin methacryloyl: Best practice and current pitfalls, Biomacromolecules 19 (2018) 42–52. 10.1021/acs.biomac.7b01221. [DOI] [PubMed] [Google Scholar]
- [49].Zhu M, Wang Y, Ferracci G, Zheng J, Cho N-J, Lee BH, Gelatin methacryloyl and its hydrogels with an exceptional degree of controllability and batch-to-batch consistency, Sci. Rep 9 (2019) 6863. 10.1038/s41598-019-42186-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Hoch E, Hirth T, Tovar GEM, Borchers K, Chemical tailoring of gelatin to adjust its chemical and physical properties for functional bioprinting, Journal of Materials Chemistry B: Materials for Biology and Medicine 1 (2013) 5675–5685. 10.1039/C3TB20745E. [DOI] [PubMed] [Google Scholar]
- [51].Townsend JM, Beck EC, Gehrke SH, Berkland CJ, Detamore MS, Flow behavior prior to crosslinking: The need for precursor rheology for placement of hydrogels in medical applications and for 3d bioprinting, Progress in Polymer Science 91 (2019) 126–140. 10.1016/j.progpolymsci.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lee SC, Gillispie G, Prim P, Lee SJ, Physical and chemical factors influencing the printability of hydrogel-based extrusion bioinks, Chemical Reviews 120 (2020) 10834–10886. 10.1021/acs.chemrev.0c00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gillispie G, Prim P, Copus J, Fisher J, Mikos AG, Yoo JJ, Atala A, Lee SJ, Assessment methodologies for extrusion-based bioink printability, Biofabrication 12 (2020) 022003. 10.1088/1758-5090/ab6f0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Galante R, Pinto TJA, Colaço R, Serro AP, Sterilization of hydrogels for biomedical applications: A review, Journal of Biomedical Materials Research, Part B: Applied Biomaterials 106 (2018) 2472–2492. 10.1002/jbm.b.34048. [DOI] [PubMed] [Google Scholar]
- [55].Luong PT, Browning MB, Bixler RS, Cosgriff-Hernandez E, Drying and storage effects on poly(ethylene glycol) hydrogel mechanical properties and bioactivity, Journal of Biomedical Materials Research, Part A 102 (2014) 3066–3076. 10.1002/jbm.a.34977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ulker Z, Erkey C, An emerging platform for drug delivery: Aerogel based systems, J. Controlled Release 177 (2014) 51–63. 10.1016/j.jconrel.2013.12.033. [DOI] [PubMed] [Google Scholar]
- [57].Pierre AC, Pajonk GM, Chemistry of aerogels and their applications, Chem. Rev 102 (2002) 4243–4266. 10.1021/cr0101306. [DOI] [PubMed] [Google Scholar]
- [58].Okuda K, Urabe I, Yamada Y, Okada H, Reaction of glutaraldehyde with amino and thiol compounds, J. Ferment. Bioeng 71 (1991) 100–105. 10.1016/0922-338X(91)90231-5. [DOI] [Google Scholar]
- [59].Migneault I, Dartiguenave C, Bertrand MJ, Waldron KC, Glutaraldehyde: Behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking, BioTechniques 37 (2004) 790–802. 10.2144/04375rv01. [DOI] [PubMed] [Google Scholar]
- [60].Elzoghby AO, Gelatin-based nanoparticles as drug and gene delivery systems: Reviewing three decades of research, J. Controlled Release 172 (2013) 1075–1091. 10.1016/j.jconrel.2013.09.019. [DOI] [PubMed] [Google Scholar]
- [61].Santoro M, Tatara AM, Mikos AG, Gelatin carriers for drug and cell delivery in tissue engineering, J. Controlled Release 190 (2014) 210–218. 10.1016/j.jconrel.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Zhang K, Wang S, Zhou C, Cheng L, Gao X, Xie X, Sun J, Wang H, Weir MD, Reynolds MA, Zhang N, Bai Y, Xu HHK, Advanced smart biomaterials and constructs for hard tissue engineering and regeneration, Bone Res. 6 (2018) 31. 10.1038/s41413-018-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Montgomery M, Ahadian S, Davenport Huyer L, Lo Rito M, Civitarese RA, Vanderlaan RD, Wu J, Reis LA, Momen A, Akbari S, Pahnke A, Li R-K, Caldarone CA, Radisic M, Flexible shape-memory scaffold for minimally invasive delivery of functional tissues, Nat. Mater 16 (2017) 1038–1046. 10.1038/nmat4956. [DOI] [PubMed] [Google Scholar]
- [64].Bencherif SA, Sands RW, Bhatta D, Arany P, Verbeke CS, Edwards DA, Mooney DJ, Injectable preformed scaffolds with shape-memory properties, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 19590–19595. https://dx.doi.org/10.1073%2Fpnas.1211516109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Wang C, Yue H, Liu J, Zhao Q, He Z, Li K, Lu B, Huang W, Wei Y, Tang Y, Wang M, Advanced reconfigurable scaffolds fabricated by 4d printing for treating critical-size bone defects of irregular shapes, Biofabrication 12 (2020) 045025. 10.1088/1758-5090/abab5b. [DOI] [PubMed] [Google Scholar]
- [66].Miao S, Castro N, Nowicki M, Xia L, Cui H, Zhou X, Zhu W, Lee S.-j., Sarkar K, Vozzi G, Tabata Y, Fisher J, Zhang LG, 4d printing of polymeric materials for tissue and organ regeneration, Mater. Today 20 (2017) 577–591. 10.1016/j.mattod.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Pissis P, Kyritsis A, Hydration studies in polymer hydrogels, J. Polym. Sci. Part B: Polym. Phys 51 (2013) 159–175. 10.1002/polb.23220. [DOI] [Google Scholar]
- [68].Samouillan V, André C, Dandurand J, Lacabanne C, Effect of water on the molecular mobility of elastin, Biomacromolecules 5 (2004) 958–964. 10.1021/bm034436t. [DOI] [PubMed] [Google Scholar]
- [69].van der Stok J, Wang H, Amin Yavari S, Siebelt M, Sandker M, Waarsing JH, Verhaar JA, Jahr H, Zadpoor AA, Leeuwenburgh SC, Enhanced bone regeneration of cortical segmental bone defects using porous titanium scaffolds incorporated with colloidal gelatin gels for time- and dose-controlled delivery of dual growth factors, “Tissue Eng., Part A” 19 (2013) 2605–2614. 10.1089/ten.tea.2013.0181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.