

Summary
Low grain moisture at harvest is crucial for safe production, transport and storage, but the genetic architecture of this trait in maize (Zea mays) remains elusive. Here, we measured the dynamic changes in grain moisture content in an association‐mapping panel of 513 diverse maize inbred lines at five successive stages across five geographical environments. Genome‐wide association study (GWAS) revealed 71 quantitative trait loci (QTLs) that influence grain moisture in maize. Epistatic effects play vital roles in the variability in moisture levels, even outperforming main‐effect QTLs during the early dry‐down stages. Distinct QTL–environment interactions influence the spatio‐temporal variability of maize grain moisture, which is primarily triggered at specific times. By combining genetic population analysis, transcriptomic profiling and gene editing, we identified GRMZM5G805627 and GRMZM2G137211 as candidate genes underlying major QTLs for grain moisture in maize. Our results provide insights into the genetic architecture of dynamic changes in grain moisture, which should facilitate maize breeding.
Keywords: grain moisture, GWAS, QTL, genetic basis, moisture plasticity
Introduction
Maize (Zea mays L.) is an important staple crop worldwide. Total maize production increased 10‐fold over the past century due to efforts in hybrid breeding and germplasm improvement (Duvick, 2001). However, a critical problem remains unsolved: dry matter in maize grains spoils easily due to the excessive grain moisture at harvest. High grain moisture contents at harvest necessitate grain drying prior to the transport and storage of ears. This dry‐down process hinders mechanized harvesting, which substantially reduces farmer incomes. Accordingly, low grain moisture at harvest has become a new breeding aim worldwide, especially in temperate maize production regions (Chai et al., 2017; Wang and Li, 2017).
The final grain moisture at harvest is determined by two factors: the initial moisture content at physiological maturity and the subsequent dry‐down rate in the field environment (Chase, 1964). The grain moisture at maturity in maize is primarily controlled by genetics (Cross, 1985; De‐Jager et al., 2004; Purdy and Crane, 1967; Zhang et al., 1996), although it is also influenced by environmental factors (Magari et al., 1997). The dry‐down rate of grain is attributable mainly to temperature changes if the initial grain moisture exceeds 30%; otherwise, the dry‐down rate is mainly affected by relative humidity (Schmidt and Hallauer, 1966). In the past decade, biparental population‐based linkage analysis was commonly used to explore the genetic determinants of grain moisture in maize, and quantitative trait loci (QTLs) controlling the trait have been detected in various populations (Austin et al., 2000; Capelle et al., 2010; Li et al., 2014; Liu et al., 2020a, 2020; Mihaljevic et al., 2005; Sala et al., 2006; Wang et al., 2012; Zhang et al., 2020). Due to technical limitations, however, most experiments have involved indirect measurements of grain moisture levels at harvest based on estimated biomass, without considering the effects of the inner and outer environments on grain moisture. This indirect assessment has made it difficult to decipher the genetic underpinnings of grain moisture. Notably, no gene underlying a QTL affecting grain moisture has been cloned, likely due to the low mapping power of biparental populations and the inherent complexity of the trait. Thus, it remains quite challenging to elucidate the genetics of grain moisture levels and perform gene‐based breeding related to moisture levels in maize.
In this era of high‐throughput sequencing, genome‐wide association study (GWAS) using diverse natural populations and historical recombination has become the most popular approach to exploring quantitative agricultural traits in maize (Xiao et al., 2017). GWAS has been used to comprehensively evaluate the genetic architecture of complex traits such as oil content and flowering time (Buckler et al., 2009; Li et al., 2013) and has successfully been used to identify functional genes responsible for significant loci (Hung et al., 2012; Liu et al., 2020a, 2020; Wang et al., 2016; Yang et al., 2013).
Accurate and efficient measurement of grain moisture in maize is important for both basic research and maize breeding. Breeders often use the oven‐drying method to measure grain moisture content, but this test is time‐consuming and destructive. A mobile, flexible moisture measuring instrument has emerged as a simple, reliable tool for measuring this important trait (Austin et al., 2000; Capelle et al., 2010; Li et al., 2014; Liu et al., 2020a, 2020; Liu et al., 2020a, 2020; Mihaljevic et al., 2005; Sala et al., 2006; Wang et al., 2012; Zhang et al., 2020). Here, we used GE’s BLD5604, a digital timber‐moisture meter (Yang et al., 2010), to efficiently measure the moisture content of maize kernels in the field in real time to assess the dynamics of moisture content across kernel development (Figure 1a). Using this method, we carried out more than 750,000 measurements using 513 diverse maize inbred lines in five different environments at five developmental stages. Analysis of this dataset provided us with a comprehensive understanding of the dehydration process of maize kernels. We determined that this trait is mainly controlled by genetics and a major gene controlling grain moisture was cloned, laying the foundation for further research.
Figure 1.
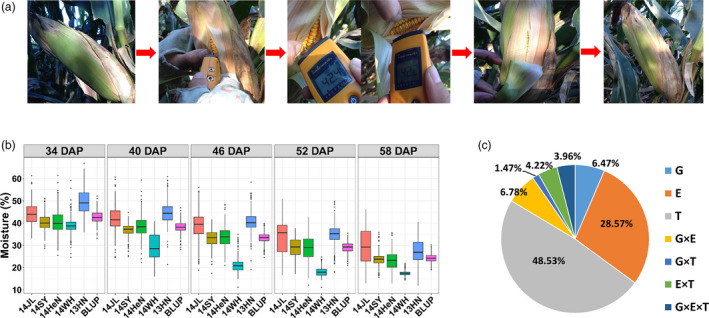
Dynamic variation in grain moisture in different stages and environments. (a) The measurement procedure using GE’s BLD5604 moisture meter. (b) Phenotypic distribution of moisture content measured for each developmental stage and environment. The different graphs show the five successive measurement stages. For each stage, the boxplots of moisture content and BLUP values were sorted by the latitude of the five geographic environments. (c) Proportion of variance in moisture content due to different components. The variance due to genotype (G), environment (E), measurement time (T) and pairwise interactions was estimated by three‐way analysis of variance (ANOVA).
Results
Maize grain moisture has strong spatio‐temporal variability
To comprehensively quantify the variability in grain moisture, we performed two measurements. First, we collected transient moisture content (MC) data at five successive stages, from 34 DAP (days after pollination) to 58 DAP. Second, we transformed the time‐series moisture data to area under the dry‐down curve (AUDDC) values; this statistical index was previously used to measure transient moisture content and dehydration rate simultaneously (Yang et al., 2010). For details about phenotypic data collection, see the Methods.
The MC data basically followed normal distribution (Figure S1), but there was variability across times series and geographical environments (Figure 1b and Table S2). In a specific environment, the average MC continuously declined from the early to late stages, but the variation in MC displayed distinct trends. For example, in the 14JL environment, the average MC kept decreasing, from 44.38% at 34 DAP to 29.27% at 58 DAP, while the variation in MC increased nearly twofold in the opposite manner. By contrast, in the 14WH environment, the average MC and variation in MC both decreased over time. These results indicate that the variability in MC in the population was simultaneously influenced by time point, growing environment and genotype–environment interaction (G × E). Three‐way analysis of variance (ANOVA) suggested that the variation in moisture was primarily affected by time point (48.53%), followed by growing environment (28.57%), indicating that moisture is highly sensitive to developmental stage and climate conditions. By contrast, genotypic effect and G × E accounted for only ~6% of overall moisture variance, followed by additional interaction effects (Figure 1c). At each time point, the broad‐sense heritability (H2) of grain moisture was high, ranging from 0.66 to 0.82 for MC data and from 0.66 to 0.82 for AUDDC data, respectively (Table S2). We estimated the best linear unbiased prediction (BLUP) value of each line across environments at each time point for MC and AUDDC data. We detected nearly consistent variation across five time points, perhaps due to reduced environmental noise (Figure 1b).
GWAS decodes significant loci underlying grain moisture in maize
To explore the genetic basis of grain moisture, we performed GWAS for MC and AUDDC traits at each time point, including all environmental and BLUP data. For 24 MC traits, we detected 58 significant influential loci (P < 2.0 × 10−6), with 1–7 loci per trait, whereas 6 MC traits had no significant loci (Figure S2 and Table S3). For 87 AUDDC traits, 130 significant loci were detected (P < 2.0 × 10−6), with 1–7 loci per trait, whereas 3 AUDDC traits had no significant loci (Figure S3 and Table S3). Each MC‐associated locus explained 1.62–17.76% of the variance, a value slightly lower than the variance (2.39–21.39%) explained by AUDDC‐associated loci (Figure S4). This result is reasonable given that AUDDC generally exhibited larger phenotypic variance than MC. Based on their physical positions, the 188 significant loci for MC and AUDDC traits were integrated into 71 unique loci (hereafter referred to as QTLs) associated with the variability in grain moisture in maize. The 71 QTLs appeared to be distributed unevenly across the whole genome (Figure 2a and Table S4). Of the 71 QTLs, 24 and 26 were uniquely detected based on MC and AUDDC traits, respectively (Figure S5), indicating that the variability in transient moisture content and dehydration rate in maize may be due to distinct genetic architectures.
Figure 2.
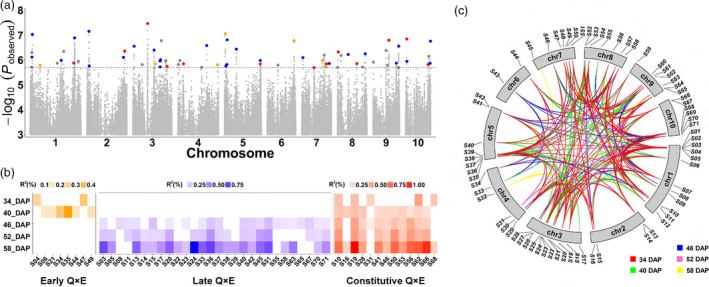
Genetic basis of the spatio‐temporal variation in moisture content. (a) Manhattan plot of GWAS for grain moisture. The 71 significant loci that influence moisture content or AUDDC value are indicated by coloured dots. The colours (orange, red and blue) represent the three classes of significant loci that interact with the environment at the early or late stage. (b) Variability of Q × E variance during the dehydration process. In the heatmap, the colour indicates how this QTL interacts with the environment, including early Q × E, later Q × E, and constitutive Q × E. The colour density indicates the variance explained by Q × E at a specific stage of dehydration. (c) Epistasis between 71 significant loci. In the circle plot, the coloured lines connecting two genomic locations indicate significant epistasis between two GWAS loci. The colour indicates the dehydration stage in which the significance of epistasis was detected.
Dynamics of the genetic effect is responsible for the variability in grain moisture
To explore the contribution of QTLs and environmental factors to the variability in grain moisture, we used a linear model encompassing all QTLs, environment and QTL–environment (Q × E) effects. The variability in grain moisture was strongly affected by environmental factors (43.88‒72.71%) at a single time point, with minor contributions by QTL (6.83‒12.41%) and Q × E (3.82‒8.39%) effects (Figure S6). Overall, the Q × E tended to progressively increase over time. When we divided the five developmental time points into the early stage (34 and 40 DAP) and late stage (46, 52 and 58 DAP), we found that 8 QTLs interacted with the environment during the early stage, whereas 28 QTLs interacted with the environment during the late stage, and 13 QTLs consistently interacted with the environment throughout the dehydration process (Figure 2b). At any developmental stage, the variance due to each Q × E was modest but obviously exceeded the variance due to the main effect of the QTL (Figure S7). These results indicate that Q × E represents a vital resource for interpreting the variability in grain moisture in addition to the main effects of QTLs. The early stage‐specific Q × E explained 0.21% to 0.49% of variance in grain moisture, and the effects of the late stage‐specific Q × E were similar (0.13% to 0.91%). Notably, the constitutive Q × E appeared to contribute 0.14% to 1.04% of variance, a value significantly higher than that of stage‐specific Q × E (P = 0.0046) (Figure 2b and Figure S8). These findings suggest that the interactions between QTLs and the environment are activated dynamically from the early to late stage of the kernel dehydration process. Moreover, the Q × E variance tended to slightly increase during the kernel dehydration process along a developmental stage gradient (Figure S9), indicating that grain moisture becomes more sensitive to the environment after entering the late stage of dehydration.
To determine how epistasis between pairwise QTLs influences grain moisture, we built a regression model that fit all QTLs and pairwise interaction terms. For simplicity, for each time point, we analysed BLUP data rather than multiple environmental data. On average, during the five time points, over half (53.13%) of the variance in moisture content could be explained by main‐effect QTLs, followed by epistasis (30.49%) (Figure S10), highlighting the crucial role of epistasis in grain moisture in maize. In contrast to Q × E, epistasis tended to explain less variance along the kernel dehydration process (Figure S10), implying that epistasis primarily regulates grain moisture during the early dehydration stage.
Across all 3,195 possible pairwise QTLs, there were 85 to 51 significant epistatic effects detected from the early to late stages of dehydration, with an average of 74 epistatic effects (Figure 2c). The majority of epistatic effects were detected at specific developmental stages, suggesting that epistasis is another mechanism that fine tunes grain moisture in addition to Q × E. At a specific developmental stage, epistasis could involve 1) two QTLs that do not interact with the environment; 2) two QTLs that interact with the environment; or 3) one QTL that interacts with the environment and one that does not. Intriguingly, the proportion of epistasis that involved Q × E substantially increased during development from the early to late stages (Figure S11 and Table S5), suggesting that more complex regulatory networks might participate in the late dry‐down stage.
Interplay of moisture plasticity with the environment and time gradients
The variation in additive effects across an environmental continuum is typically attributed to phenotypic plasticity (Lowry et al., 2019; Zan and Carlborg, 2019). To explore this notion, we focused on a GWAS locus (peak as chr9.S_93812340) that was significantly associated with moisture content in environment 14WH at 34 DAP (P = 1.59 × 10−7, MLM, n = 467) (Figure S2d). The SNP chr9.S_93812340 significantly affected moisture content between contrasting genotypes (AA/GG), in the 14 JL environment across all time points, and in 14WH during the early stage and 14HeN during the late stage, but not in the remaining environments or time points (Figure S12), indicating that this locus likely interacts with environmental and time gradients. To test this idea, we estimated the additive effect of this locus in specific environments and time points (see Materials and methods). Both spatial and temporal factors interactively influenced the additive effects of locus chr9.S_93812340 (Figure 3a). For instance, in the 14JL environment, the additive values of this locus continuously increased from 1.48 (34 DAP) to 3.17 (58 DAP). By contrast, the additive value kept decreasing from early to late development, reaching nearly zero at 58 DAP (Figure 3a).
Figure 3.
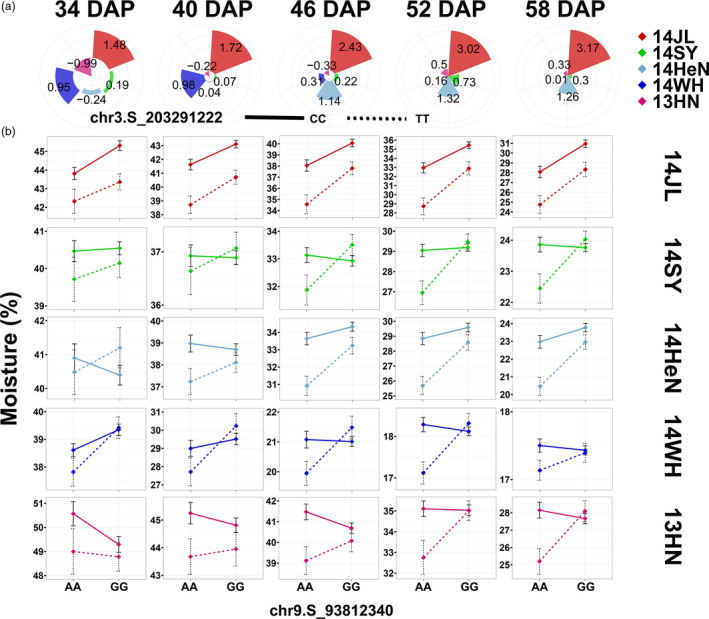
Effect spectrum of a major QTL and epistasis due to the effects of dehydration and the environment. (a) Additive effect of a major moisture‐related QTL (peak at chr9.S_93812340). The colour indicates the environment, and the size indicates the QTL effect in a specific environment. (b) Epistatic effect between two moisture‐related QTLs (peaks at chr9.S_93812340 and chr3.S_203291222). The x‐axis represents the moisture content of lines grouped by homozygous genotypes (AA/GG) at chr9.S_93812340, while the solid and dotted line represent the homozygous genotypes (CC/TT) at chr3.S_203291222. The colour of the line indicates the environment in which epistasis was detected.
To further explore the influence of interactive effects on QTL features, we selected another locus that influences moisture (peaked as chr3.S_203291222) and examined its interaction with locus chr9.S_93812340 (epistasis) in a specific environment and time point. The epistasis pattern dynamically changed in response to spatial and temporal alterations (Figure 3b). In environment 14SY, the epistatic interaction was consistently significant throughout the dehydration process. In 14JL, only one locus appeared to influence moisture additively until the late dehydration stage, but in 14WH, the loci had a significant epistatic effect during the initial dehydration process (Figure 3b). These results illustrate how epistasis dynamically regulates kernel moisture content during the dehydration process, which can be reshaped by specific environmental factors.
Genetic correlations of grain moisture with agronomic and metabolic traits
Many traits in the present maize association‐mapping population have been deciphered genetically and their underlying QTLs or genes identified (Liu et al., 2017; Wen et al., 2018; Yang et al., 2014). Of the 19 agronomic traits examined, 8 were significantly correlated with moisture content (P < 0.01) (Figure S13 and Table S6). Of the 55 primary metabolic traits, 33 were significantly correlated with moisture content (P < 0.01), including 32 positive and only 1 negative correlation (Figure S14 and Table S7). For example, early flowering was strongly correlated with a rapid moisture dry‐down rate, and larger kernel size was correlated with a higher moisture content at harvest. In total, we detected 41 moisture‐related traits (P < 0.01) associated with 35 previously detected GWAS loci associated with agronomic traits (Liu et al., 2017; Wen et al., 2018; Yang et al., 2014). Notably, 9 of the 35 loci affected moisture content during at least four stages (ANOVA, P < 0.01) (Figure 4a); these loci might also regulate kernel size, ear size or the contents of amino acids and other metabolites. For example, the GG genotype of chr1.S_170961872 simultaneously regulates both traits, as it increases kernel width by 0.76 mm and moisture content by 1.54‒2.4% relative to the AA genotype (Figure S15). We also assessed the effects of the 71 loci controlling moisture content on agronomic and metabolic traits (P < 0.01). Of these, 12 moisture‐related QTLs had absolutely no effect on any agronomic trait, while 8 QTLs did not affect any metabolic trait (Figure 4b). Notably, 56.34% of moisture‐related QTLs affected more than 15% of agronomic traits, but only 11.27% of moisture‐related QTLs affected more than 15% of metabolic traits (Figure 4b).
Figure 4.
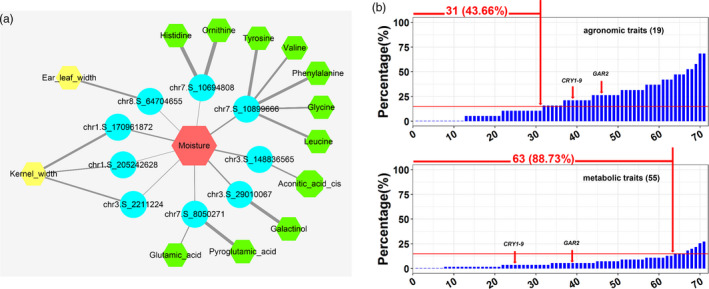
Genetic correlations of grain moisture with agronomic and metabolic traits. (a) A network of moisture content, agronomic and metabolic traits mediated by shared important loci. The circles and hexagons represent significant loci and traits, respectively. Red indicates moisture content, while yellow and green indicate agronomic and primary metabolites, respectively. The thickness of the line represents the degree of significance (–logP value). (b) The proportion of other traits affected by the detected moisture‐related QTLs. The upper panel shows the influence of each moisture‐related QTL on agronomic traits (n = 19), while the lower panel shows the metabolic traits (n = 55) (P < 0.01). The red horizontal line indicates a proportion of 15%, which was used as a threshold to determine whether a moisture‐related QTL obviously affected an agronomic or metabolic trait.
Identification of key genes controlling grain moisture in maize
A GWAS locus (peak at chr9.S_93812340) on chromosome 9 was significantly associated with moisture content (P = 1.59 × 10−7, MLM, n = 467) (Figure 5a). GRMZM5G805627, encoding a maize cryptochrome circadian regulator 1 homolog (hereafter called CRY1‐9), is the only gene within this 100 kb region (50 kb upstream and downstream of the lead SNP) according to the B73 reference genome (Figure 5b and c). B73 transcriptomic profiling data revealed that CRY1‐9 is specifically and highly expressed in maize endosperm from 6 DAP to 10 DAP (Figure S16) (Chen et al., 2014). Furthermore, GWAS detected a cis eQTL (peak at chr9.S_93812706) that regulated CYR1‐9 expression in 15 DAP kernels (P = 7.14 × 10−35, MLM, n = 341) (Figure 5D). By conditioning chr9.S_93812706 as a covariate, the cis eQTL for CYR1‐9 expression was still detected, where chr9.S_93812340 appeared to be the second most significant loci (P = 1.92 × 10−9, MLM, n = 341) (Figure 5e). These results suggested that the locus chr9.S_93812340 simultaneously affected CRY1‐9 expression (P = 4.42 × 10‐22) (Figure 5f) and moisture content (P = 9.99 × 10−4) (Figure 5g). But we’re not able to detect direct correlation between CYR1‐9 expression and moisture content (r = 0.029, P = 0.29; Figure S17). Other genetic elements, like siRNA, lncRNA within the 100kb region, may also serve as genetic factors that coordinate grain moisture content. In brief, moisture content is controlled by a major QTL on chromosome 9, which may be associated with the regulation of CYR1‐9 gene expression. Nevertheless, further experiments are needed to validate and understand the function of this gene.
Figure 5.
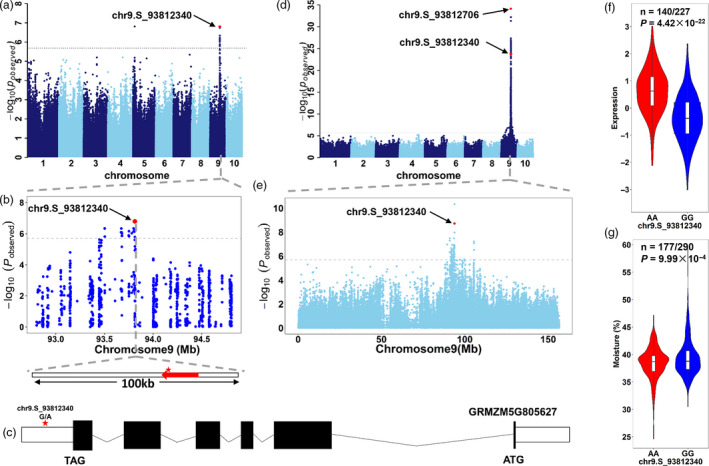
Candidate gene of a major QTL for moisture content. (a) Manhattan plot of GWAS for moisture content (34 DAP) at location 14WH. The lead SNP is indicated by a red dot. (b) Regional plot for the QTL on chromosome 9. The upper panel shows the p values of SNPs surrounding a 1‐Mb region upstream and downstream of the lead SNP. The lower panel indicates a 100 Kb region covering 50 Kb upstream and downstream of the lead SNP, where only one annotated gene (GRMZM5G805627, also named CRY1‐9) was found based on the maize reference genome. (c) Gene model of CRY1‐9. Solid boxes indicate exons, open boxes indicate untranslated regions (UTRs), and lines connecting the exons indicate introns. The red stars mark the positions of chr9.S_93812340. (d) Manhattan plot of GWAS for CRY1‐9 expression. The two key SNPs are indicated by red dots. (e) Regional Manhattan plot of conditional GWAS for CRY1‐9 expression. The GWAS for gene expression was repeated by fitting the original lead SNP (chr9.S_93812706) as a covariate. (f) The influence of chr9.S_93812340 on CRY1‐9 expression. (g) The influence of chr9.S_93812340 on moisture content. The significance of difference between two genotypes was evaluated using Student’s t‐test.
We detected another GWAS locus (peak at chr7.S_132808190) on chromosome 7 that influences AUDDC (AUDDC_5_2, AUDDC_5_3, AUDDC_5_4, AUDDC_4_3 and AUDDC_4_2) (Figure 6a and Table S4). Three genes were located within a 100 kb region of the lead SNP based on the reference genome. Of these, GRMZM2G137211 encompassed the nonsynonymous SNPs with the most significant effects (chr7.S_132808190 and chr7.S_132808253) (P = 6.34 × 10−7 and P = 8.3 × 10−7) (Figure 6b). Notably, this gene is highly expressed in immature leaf (V9) and innermost husk (R1) tissue in B73 (Sekhon et al., 2011). We selected GRMZM2G137211 as the candidate gene responsible for this significant AUDDC locus. This gene encodes a gar2‐related nucleolar protein and is therefore referred to as GAR2 hereafter. Resequencing the region near GAR2 revealed an 8‐bp InDel polymorphism (GAACATCA/‐) in the 5’ untranslated region (5’UTR) (Figure 6c) that significantly influences AUDDC (P = 2.37 × 10−7, ANOVA, n = 465) (Figure 6e). Given the high LD between this 8 bp InDel and the lead SNP, we speculated that the 8 bp InDel is likely the causal variant underlying GAR2 for AUDDC (Figure 6d).
Figure 6.
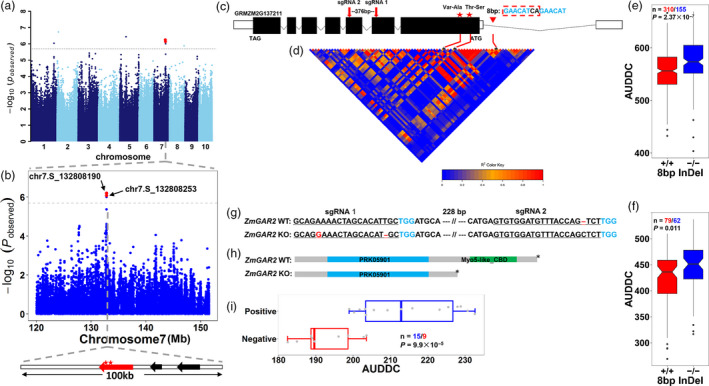
Identification of GAR2, a gene responsible for AUDDC in maize. (a) GWAS of the BLUP values for AUDDC_5_2. AUDDC_5_2 indicates the area under the dry‐down curve from 40 DAP to 58 DAP. The lead SNP underlying the QTL on chromosome 7 is indicated by a red dot. (b) Regional plot of the major QTL on chromosome 7. The top panel shows the p values of SNPs within a 15‐Mb region surrounding the lead SNP. The two most significant SNPs are indicated by red dots (P < 2.0 × 10−6). The bottom panel indicates a 100 Kb region surrounding the lead SNP that only contains three annotated genes (arrow) based on the maize reference genome. The red arrow indicates the candidate gene (GRMZM2G137211, also named GAR2) responsible for this AUDDC QTL. (c) Gene model of GAR2. Solid boxes indicate exons, while open boxes indicate untranslated regions (UTRs) and lines that connect exons represent introns. The red stars represent the two most significant SNPs that could cause amino acid substitutions (Val‐Ala and Thr‐Ser). The red triangle indicates the position of an 8 bp InDel (GAACATCA/‐) identified by gene resequencing. The red arrows indicate the positions of the two sgRNAs used for CRISPR‐Cas9 gene editing. (d) LD pattern of the GAR2 region. The heatmap indicates the pairwise r 2 values among the polymorphisms in a 0.1‐Mb region upstream and downstream of the lead SNP. The two most significant SNPs and the 8 bp InDel are indicated by red lines. (e) The effect of the 8 bp InDel on AUDDC values in the association‐mapping population. (f) The effect of the 8 bp InDel on AUDDC values in the F2:3 population. (g) Gene editing using CRISPR/Cas9. The red bases and line segment show variation and the blue bases show the PAM site. (h) Premature protein termination due to gene knockout. (i) The different AUDDC values between wild type and GAR2‐edited types via CRISPR‐Cas9. The comparison between different genotypes is based on Student’s t‐test.
To verify this notion, we constructed an F2:3 population by crossing two inbred lines, GEMS2 and ZHENG58, based on the presence or absence of the 8bp InDel. The progeny lines without the 8 bp insertion exhibited significantly higher AUDDC values than those with the 8 bp insertion (P = 0.011, ANOVA, n = 141) (Figure 6f). To further validate the function of GAR2, we performed a gene editing experiment via CRISPR/Cas9 in maize variety KN5585 (Liu et al., 2020a, 2020). Two sgRNAs were used to target the coding region of GAR2 at a 376 bp distance (Figure 6g). The positive edited lines (n = 15), carrying the premature termination of GAR2 protein and a loss‐of‐function mutation (Figure 6h), had significantly higher AUDDC values than the negative lines (n = 9) (P = 9.9 × 10−5, ANOVA, n = 24) (Figure 6i), with similar effects on moisture content (Figure S18). These results confirm the notion that GAR2 is responsible for the QTL on chromosome 7 that negatively regulates grain moisture in maize. The underlying molecular mechanism should be further explored.
Discussion
Grain moisture at harvest is influenced by grain moisture at physiological maturity and the field dry‐down rate. Grain moisture is crucial for determining the time of physiological maturity, but it is difficult to accurately estimate this parameter. Black‐layer formation (Carter and Poneleit, 1973; Daynard and Duncan, 1969) and milk‐line progression (Afuakwa and Crookston, 1984) were previously used to estimate physiological maturity in maize, but these techniques appear to be subjective and are affected by environmental conditions (Daynard, 1972). The relationship between days to silking and physiological maturity has been extensively explored to estimate the developmental status of a maize. The interval from silking to physiological maturity was estimated to be 60 days (Hanway, 1963) and 53‒61 days (Hillson and Penny, 1965). Gunn and Christensen (1965) suggested that this interval ranges from 45 days for early‐maturing hybrids to 70 days for later‐maturing hybrids. Jin et al. (2002) studied 42 maize inbred lines and 8 hybrids and determined that late and mid‐late lines reach physiological maturity at 50‒60 days after silking, and mid‐early materials reach physiological maturity at 45‒50 days after silking. These findings suggest that it may be impossible to determine the absolute time of physiological maturity because this parameter is affected by environmental factors and genetic background. Therefore, in the current study, we measured grain moisture in each line multiple times (every five days) beginning at 34 days after pollination to cover the entire period of kernel development from the early stage to the maturity or post‐maturity stage. This analysis allowed us to systematically dissect the dynamics of grain moisture during kernel development.
Moisture meter BLD5604 was used in this experiment, which enable to continuous measurement because this was a nondestructive procedure. It’s worth noting that, the accuracy of electrical conductivity based moisture meter may be biased by some factors, such as the size, composition of seed and the thickness of kernel pericarp. Therefore, it is imperative to develop simple and efficient measuring methods and instruments if we want to measure corn grain moisture more accurately and effectively.
We collected grain moisture data from five geographic locations, representing the temperate Chinese Corn Belt over a latitudinal gradient (Table S1). In general, grain moisture decreased more rapidly in the low‐latitude environment, presumably due to temperature and humidity conditions during the dehydration process (Schnidt and Hallauer, 1966). Integrating multiple GWAS data per environment and developmental stage, we identified 71 QTLs that influence grain moisture in maize. For each developmental stage, each QTL had small effects on moisture variance and jointly explained ~10% of moisture variation. These results indicate that grain moisture is controlled by many loci with minor effects, which could not have been detected using a small population. We also found that Q × E had important effects on grain moisture variation, accounting for nearly one half of the main effects of QTLs. Intriguingly, Q × E contributed more when kernel dehydration switched from early‐ to late‐stage moisture, while different sets of Q × E appeared to be specifically triggered in the early or late stage (Figure 2b and Figure S8). By contrast, pairwise QTL epistasis contributed considerably to moisture variance, that is, up to 42.61% for the early dehydration stage, which is comparable to the main effects of QTLs. Taken together, our genetic analysis based on QTL detection demonstrated that grain moisture is a typical polygenic trait that is interactively influenced by genotype and spatio‐temporal factors.
Field dry‐down occurs due to two factors: stress dehydration due to specific morphological characteristics (i.e. husk length, ear length, number of kernel rows, ear height and plant height), environmental conditions (humidity and temperature), and physiological dehydration during seed maturation due to phytohormones and dry matter in kernels; the latter is the essence of kernel dehydration. The process of kernel dehydration inhibits early embryo germination and the biosynthesis of specialized proteins that protect the embryo from the damaging effects of desiccation and enhance embryo viability during strong dehydration. The phytohormone abscisic acid (ABA) plays crucial roles in seed development and maturity and establishing embryo dormancy. White maize generally has higher grain moisture content at harvest than yellow maize (Kang and Zuber, 1989) because white maize lacks phytoene synthase (PSY), an enzyme involved in both carotenoid and ABA biosynthesis. In summary, it is important to understand the genetic basis of maize kernel dehydration; breeders use this knowledge to better select lines with lower grain moisture at harvest. However, the geographic, environmental and morphological characteristics that influence moisture dry‐down have to be considered.
In traditional breeding, rapidly dehydrating maize is empirically selected based on morphological characteristics, such as early maturity, as heading date is positively correlated with grain moisture at harvest (Figure S13) (Jin et al., 2002). It makes sense to perform this indirect selection because there are various genetic correlations between morphological traits and grain moisture. Indeed, many QTLs detected in the present study colocalize with agronomic QTLs (Figure 4a). Using the newly detected QTLs for grain moisture, it should be possible to directly select favourable alleles for the genetic improvement of maize with the rapid‐dehydration trait, especially for GAR2, which has been functionally confirmed. Surprisingly, most moisture‐related QTLs identified in this study did not influence the majority (>85%) of metabolic traits, that is GAR2 and CRY1‐9 did not affect the majority of metabolic traits but had some effects on agronomic traits (Figure 4b and Figure S19). Therefore, the QTL sets discovered in the present study could be useful for improving grain moisture without affecting the nutritional quality of kernels. Yet, more than half of these QTLs had significantly effects on agronomic traits simultaneously, likely due to QTL pleiotropy or linkage drag. The traditional approach of marker‐based selection of available allelic variants may not be suitable for utilize these QTLs that influence multiple traits. Saturation mutagenesis of gene promoters and gene editing of key QTLs or genes represent powerful approaches for manipulating multiple correlated traits for precise breeding (Eshed and Lippman, 2019; Liu et al., 2020b, 2020).
Materials and methods
Plant materials and field trials
A maize (Zea mays L.) association panel of 513 genetically diverse inbred lines (Yang et al., 2011) was used to dissect the dynamic variation in grain moisture. The 513 inbred lines were planted in a randomized block design with one‐row plots and two replications across five geographical locations in China in two years: Jilin (14JL; Gongzhuling, E 124°69′, N 43°79′), Liaoning (14SY; Shenyang, E123°43′, N 41°81′), Henan (14HeN; Xinxiang, E 113°91′, N 35°31′) and Hubei (14WH; Wuhan E 114°32′, N 30°58′) in 2014 and Hainan (13HN; Sanya, E 109°51′, N 18°25′) in 2013. Temperature and weather data were collected during data collection at all five geographical locations (Table S1). Field management followed standard procedures.
Phenotypic data collection and statistical analysis
For each line, the pollination date was recorded. For each location, the transient moisture content (MC) was measured at five successive stages – 34, 40, 46, 52 and 58 days after pollination (DAP) – using GE’s BLD5604 moisture meter. At each stage, five individuals were measured for each line, with two measurements performed per individual (Figure 1a). The mean of ten individual moisture values was used as the MC value for each line.
We also used the area under the dry‐down curve (AUDDC) (Yang et al., 2010) to efficiently quantify the dehydration rate. The formula used to calculate AUDDC is:
where, s < k and s, k ϵ {1,2,3,4,5}, and ti ϵ {34,40,46,52,58}where γ is the converted meter reading, i is the ith measured time and ti is the corresponding days after pollination. Therefore, ten AUDDC traits were obtained based on measurements at five time points: 34–40 DAP (AUDDC_2_1), 40–46 DAP (AUDDC_3_2), 46–52 DAP (AUDDC_4_3), 52–58 DAP (AUDDC_5_4), 34–46 DAP (AUDDC_3_1), 40–52 DAP (AUDDC_4_2), 46–58 DAP (AUDDC_5_3), 34–52 DAP (AUDDC_4_1), 40–58 DAP (AUDDC_5_2) and 34–58 DAP (AUDDC_5_1).
For each stage, the location term was treated as the environmental factor influencing the MC and AUDDC data. A mixed liner model was built by fitting intercept as the fixed effect and genotype and environment as random effects. The variance components of genotype and environment were estimated based on restricted maximum likelihood (REML) using the R package ‘lme4’ (R Core Team, 2013). The best linear unbiased prediction (BLUP) value for each line was obtained. The broad‐sense heritability was calculated as:
where is the genotypic variance, is the residual variance, and n is the number of environments. All data from each environment and BLUP were used for subsequent analyses, including 30 MC and 60 AUDDC traits.
Genome‐wide association analysis
Genome‐wide association study (GWAS) was performed for 90 grain moisture‐related traits using the compressed mixed linear model (Zhang et al., 2010) implemented in TASSEL V5.0 software (Bradbury et al., 2007). The mixed linear model allows the significance of a correlation between genome‐wide SNPs and target trait variation to be tested by controlling population structure and related kinship. The 1.25 million high‐quality SNPs (MAF > 5%) and the estimated population structure and kinship in the 513 diverse inbred lines were used in a previous study of agronomic traits (Gui et al., 2020; Liu et al., 2017, 2017a). To reduce the redundancy of the SNP information, we estimated the effective number of independent tests (Ne) of the 1.25 million SNPs to be 490,547 based on linkage disequilibrium using GEC software (Li et al., 2012). Thus, the p value of 2.0 × 10−6 (P = 1/Ne, Ne = 490,547) was determined to be the threshold for declaring a significant association according to the adjusted Bonferroni method (Li et al., 2012). If the significant SNPs for one trait were located close together (<50Kb), they were treated as a single locus or QTL, where the most significant SNP was considered to be the lead SNP. For each QTL detected, a 100Kb region surrounding the upstream and downstream 50 Kb regions of the lead SNP was searched for candidate genes underlying grain moisture in maize.
Publicly available RNA‐seq data for maize kernel tissue after 15 DAP were used in this study. These data were obtained from 368 lines, a subset of the 513 lines used in the present study, covering 28,769 annotated maize genes (Fu et al., 2013). Based on the expression levels of candidate genes responsible for grain moisture, we carried out expression QTL (eQTL) analysis to explore the genetic variants influencing gene expression based on the mixed linear model.
Candidate gene resequencing
The GAR2 sequence was obtained from the B73 reference sequence v2. Primers were designed using Primer‐BLAST in the NCBI website to cover the 5’ or 3’ untranslated region (UTR) of the gene (Table S8). The PCR products were sequenced to identify the candidate functional sites. The sequences were aligned using BioEdit. A total of 29 SNPs and 4 InDels were detected in the association mapping, most of which did not affect the phenotype statistically, except for an 8 bp InDel based on candidate gene association analysis (P < 0.01).
Cross‐validation using a different population
To validate the function of GAR2, we constructed an F2:3 segregation population from a cross between two inbred line (GEMS2 and Zheng 58). The parental lines were polymorphic at the 8 bp InDel located in the 5’UTR of GAR2 based on GWAS results. The segregation population of 141 lines was planted in a randomized block design with one‐row plot and one replication at Baoding in 2015 (Hebei Province, E115°47′, N 38°87′). The phenotypic data for grain moisture were collected as described above. A primer to detect the 8 bp InDel (Table S8) was used to genotype all inbred lines. Mean grain moisture was compared between the lines with or without the 8 bp insertion based on Student’s t‐test (P < 0.05).
CRISPR/Cas9‐mediated gene editing
To validate GAR2 as the causal gene responsible for the QTL on chromosome 7, we edited the gene sequence using the CRISPR‐Cas9 system. We designed two guide RNAs (gRNAs) targeting the second and third exons of GAR2 using CRISPR‐P (Liu et al., 2017b, 2017). The vector carrying the two gRNAs was introduced into immature KN5585 embryos by Agrobacterium‐mediated transformation by the WIMI Biotechnology Co., Ltd. Following self‐pollination of a T1 individual that is heterozygous at GAR2, a total of 24 T2 CRSPR‐Cas9 edited seedlings were obtained (including positive and negative control plants), which were planted in a randomized block design with one‐row plots and one replication at Sanya in 2019, Hainan province (E 109°51′, N 18°25′). DNA was extracted from each individual at the seedling stage. Primers were designed to amplify the region including the two gRNAs within the gene (Table S8). Sanger sequencing of the PCR products was used to identify the sequence variant type. Grain moisture in T2 individuals was measured at three stages after pollination and subjected to statistical analysis between edited and wild‐type plants (Student’s t‐test, P < 0.05).
Epistasis and interactions between QTLs and the environment
We subjected the newly detected QTLs to epistasis analysis based on BLUP values of grain moisture. For each developmental time, the two lead SNPs of each pair of QTLs were used to build a full regression model that included interaction term and two main‐QTL effect terms Another reduced model that merely included two main‐effect QTLs was compared with the full model to test the significance of a epistasis and the explained variance. The multiple regression model that fitted all main‐effect QTLs and all pairwise QTL terms was used to estimate the joint variance explained by overall QTLs and epistasis.
For each developmental time, we assessed the interactions between QTL and environment (Q × E) by regressing grain moisture data to the Q × E term with controlling the main‐effect QTL and environmental effect. The reduced model only fit a main‐effect QTL and environmental effect. For each QTL, the comparison between the full model and reduced model allowed us to estimate the significance and explained variance of each Q × E term. The joint variance explained by overall Q × E for grain moisture per each developmental time was estimated using a multiple regression model. All analyses of epistasis and Q × E effects were implemented with R software (R Core Team, 2013).
All genotypic and phenotypic data were deposited publicly in MAIZEGO and ZEAMAP databases. In MAIZEGO, the 1.25M SNP genotypic data was available at the section ‘Genotypic Data’ and the grain moisture data can be accessed at the section ‘Phenotypic Data’ (http://www.maizego.org/Resources.html). In ZEAMAP, the genotypic and phenotypic data can be accessed from the links (http://www.zeamap.com/ftp/02_Variants/SNPs/Maizego_1.25M_SNPs/) and (http://www.zeamap.com/ftp/03_Genetics/Phenotype/Maizego_Moisture_and_AUDDC/).
Conflict of interests
The authors declare that they have no competing interests.
Author contributions
J.Y. and Y.X. designed and supervised this study. W.L., Y.L., Y.P., Y.X. and X.L. performed the field experiments and phenotype analysis. W.L. performed the data analysis. L.W. perform the resequencing experiment. W.L., Y.Y., S.W., L.J. and J.X. perform the CRISPR‐Cas9 experiment. W.L., Y.X. and J.Y. prepared the manuscript. All authors read and approved the final manuscript.
Supporting information
Figure S1 The phenotypic distribution of moisture content for the measured and BLUP value across environments at different time points
Figure S2 Manhattan plots depicting GWAS results for MC data
Figure S3 Manhattan plots depicting GWAS results for AUDDC data
Figure S4 The distribution of effect values of all the QTLs for MC and AUDDC data
Figure S5 Colocalization of QTLs detected between AUDDC and MC data
Figure S6 Proportion of moisture variance due to different influential factors across five time points
Figure S7 The relationship between the main effect and Q × E for each QTL across different time points
Figure S8 Box plots for three types of QTL‐environment interaction effects
Figure S9 Density distribution of Q × E effects across different time points
Figure S10 Proportion of moisture variance due to QTL main effect and epistasis across different time points
Figure S11 Proportion of three types of epistasis related to interaction with environments across different time points
Figure S12 Box plot for the different phenotype between two allele of chr9.S_93812340 at different location and stages
Figure S13 The correlation between five stage moisture content and agronomic traits (only show the significantly correlated results)
Figure S14 The correlation between five stage moisture content and primary metabolite (only show the significantly correlated results)
Figure S15 Additive effect of one QTL (peaked as chr1.S_170961872) that affect moisture content and kernel width
Figure S16 The expression profile of candidate gene CYR1‐9 based on B73
Figure S17 The correlations between CYR1‐9 expression and moisture content
Figure S18 The moisture content between wild‐type and knockout plants by CRISPR/Cas9 system
Figure S19 Box plot for the different phenotype based on the 8‐bp InDel at different agronomic traits
Table S1 The information of weather condition of five locations.
Table S2 Summary of phenotype for MC and AUDDC at five locations and BLUP.
Table S3 Number of all significant loci identified in the study.
Table S4 List of candidate genes at the GWAS loci for grain moisture in maize.
Table S5 The number of different types of epistasis response to environments.
Table S6 List of the correlation between moisture content and agronomic traits.
Table S7 List of the correlation between moisture content and primary metabolites.
Table S8 List of the primers used in the study.
Acknowledgements
We thank Dr. Jiuran Zhao and his group from Beijing Academy of Agriculture & Forestry Sciences for their help with field experiments. This research was supported by the National Natural Science Foundation of China (grant nos. 31525017 and 31961133002).
Li, W. , Yu, Y. , Wang, L. , Luo, Y. , Peng, Y. , Xu, Y. , Liu, X. , Wu, S. , Jian, L. , Xu, J. , Xiao, Y. , and Yan, J. (2021) The genetic architecture of the dynamic changes in grain moisture in maize. Plant Biotechnol. J., 10.1111/pbi.13541
Contributor Information
Yingjie Xiao, Email: yxiao25@mail.hzau.edu.cn.
Jianbing Yan, Email: yjianbing@mail.hzau.edu.cn.
References
- Afuakwa, J.J. and Crookston, R.K. (1984) Using the kernel milk line to visually monitor grain maturity in maize. Crop Sci. 24, 687–691. [Google Scholar]
- Austin, D.F. , Lee, M. , Veldboom, L.R. and Hallauer, A.R. (2000) Genetic mapping in maize with hybrid progeny across testers and generations: grain yield and grain moisture. Crop Sci. 40, 30–39. [Google Scholar]
- Bradbury, P.J. , Zhang, Z. , Kroon, D.E. , Casstevens, T.M. , Ramdoss, Y. and Buckler, E.S. (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics, 23, 2633–2635. [DOI] [PubMed] [Google Scholar]
- Buckler, E.s , Holland, J.b , Bradbury, P.j , Acharya, C.b , Brown, P.j , Browne, C. , Ersoz, E. et al.(2009) The genetic architecture of maize flowering time. Science, 325, 714–718. [DOI] [PubMed] [Google Scholar]
- Capelle, V. , Remoué, C. , Moreau, L. , Reyss, A. , Mahé, A. , Massonneau, A. , Falque, M. et al.(2010) QTLs and candidate genes for desiccation and abscisic acid content in maize kernels. BMC Plant Biol. 10, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter, M.W. and Poneleit, C.G. (1973) Black layer maturity and filling period variation among inbred lines of corn (Zea mays L.). Crop Sci. 13, 436. [Google Scholar]
- Chai, Z. , Wang, K. , Guo, Y. , Xie, R. , Li, L. , Ming, B. , Hou, P. et al. (2017) Current status of maize mechanical grain harvesting and its relationship with grain moisture content. Sci. Agric. Sin. 50, 2036–2043.(in Chinese). [Google Scholar]
- Chase, S.S. (1964) Relation of yield and number of days from planting to flowering in early maturity maize hybrids of equivalent grain moisture at harvest. Crop Sci. 4, 111–112. [Google Scholar]
- Chen, J. , Zhen, B. , Zhang, M. , Xie, S. , Wang, G. , Hauck, A. and Lai, J. (2014) Dynamic trancriptome landscape of maize embryo and endosperm develoment. Plant Physiol. 166, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross, H.Z. (1985) A selection procedure for ear drying‐rates in maize. Euphytica 34, 409–418. [Google Scholar]
- Daynard, T.B. (1972) Relationships among black layer formation, grain moisture percentage, and heat unit accumulation in corn. Agron. J. 64, 716–719. [Google Scholar]
- Daynard, T.B. and Duncan, W.G. (1969) The black layer and grain maturity in corn. Crop Sci. 9, 473–476. [Google Scholar]
- De‐Jager, B. , Roux, C.Z. and Kühn, H.C. (2004) An evaluation of two collections of south african maize (Zea mays L.) germplasm: 2. the genetic basis of dry‐down rate. South African J. Plant Soil 21, 120–122. [Google Scholar]
- Duvick, D.N. (2001) Biotechnology in the 1930s: the development of hybrid maize. Nat. Rev. Genet. 2, 69–74. [DOI] [PubMed] [Google Scholar]
- Eshed, Y. and Lippman, Z.B. (2019) Revolutions in agriculture chart acoursresee for targeted breeding of old and new crops. Science 366, eaax0025. [DOI] [PubMed] [Google Scholar]
- Fu, J. , Cheng, Y. , Linghu, J. , Yang, X. , Kang, L. , Zhang, Z. , Zhang, J. et al. (2013) RNA sequencing reveals the complex regulatory network in the maize kernel. Nat. Commun. 4, 2832. [DOI] [PubMed] [Google Scholar]
- Gui, S. , Yang, L, , Li, J. , Luo, J. , Xu, X. , Yuan, J. , Chen, L. et al. (2020) ZEAMAP, a comprehensive database adapted to the maize multi‐omics era. iScience. 23, 101241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn, R.B. and Christensen, R. (1965) Maturity relationships among early to late hybrids of corn (Zea mays L.). Crop Sci. 5, 299–302. [Google Scholar]
- Hanway, J. (1963) Growth stages of corn (Zea mays, L.). Agron. J. 55, 487–92. [Google Scholar]
- Hillson, M.T. and Penny, L.H. (1965) Dry matter accumulation and moisture loss during maturation of corn grain. Agron. J. 57, 150–153. [Google Scholar]
- Hung, H‐y , Shannon, L.m , Tian, F. , Bradbury, P.j , Chen, C. , Flint‐Garcia, S.a , McMullen, M.d et al.(2012) ZmCCT and the genetic basis of day‐length adaptation underlying the postdomestication spread of maize. Proc. Natl. Acad. Sci. USA, 109, 1913–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Y. , Wang, Z. , Chang, H. , Wang, L. and Zhang, Z. (2002) Study on physiological maturityand natural drydown rate in maize. J. Northeast Agric. Univ. 9, 81–86. [Google Scholar]
- Kang, M.S. and Zuber, M.S. (1989) Combining ability for grain moisture, husk moisture, and maturity in maize with yellow and white endosperms. Cropence, 29, 689–692. [Google Scholar]
- Li, Y. , Dong, Y. , Yang, M. , Wang, Q. , Shi, Q. , Zhou, Q. , Deng, F. et al. (2014) QTL detection for grain water relations and genetic correlations with grain matter accumulation at four stages after pollination in maize. Plant Biochem. Physiol. 2, 1–9. [Google Scholar]
- Li H., Peng Z., Yang X., Wang W., Fu J., Wang J., Han Y., et al. (2013) Genome‐wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat. Genet. 45, 43–50. [DOI] [PubMed] [Google Scholar]
- Li, M.X. , Yeung, J.M. , Cherny, S.S. and Sham, P.C. (2012) Evaluating the effective numbers of independent tests and significant p‐value thresholds in commercial genotyping arrays and public imputation reference datasets. Hum. Genet. 131, 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Ding, Y. , Zhou, Y. , Jin, W. , Xie, K. and Chen, L.L. (2017b) CRISPR‐P 2.0: an improved CRISPR‐Cas9 tool for genome editing in plants. Mol. Plant, 10, 530–532. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Huang, J. , Guo, H. , Lan, L. , Wang, H. , Xu, Y. , Yang, X. et al. (2017) The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol. 175, 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H.‐J. , Jian, L. , Xu, J. , Zhang, Q. , Zhang, M. , Jin, M. , Peng, Y. et al. (2020b) High‐throughput CRISPR/Cas9 mutagenesis streamlines trait gene identification in maize. Plant Cell, 32, 1397–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Luo, X. , Niu, L. , Xiao, Y. , Chen, L. , Liu, J. , Wang, X. et al. (2017a) Distant eQTLs and non‐coding sequences play critical roles in regulating gene expression and quantitative trait variation in maize. Mol. Plant, 10, 414–426. [DOI] [PubMed] [Google Scholar]
- Liu, H.‐J. , Wang, X. , Xiao, Y. , Luo, J. , Qiao, F. , Yang, W. , Zhang, R. et al. (2020a) CUBIC: an atlas of genetic architecture promises directed maize improvement. Genome Biol. 21, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Yu, H. , Liu, Y. , Deng, S. , Liu, Q. , Liu, B. and Xu, M. (2020) Genetic dissection of grain water content and dehydration rate related to mechanical harvest in maize. BMC Plant Biol. 20, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, D.B. , Lovell, J.T. , Zhang, L. , Bonnette, J. , Fay, P.A. , Mitchell, R.B. , Lloyd‐Reilley, J. et al. (2019) QTL×environment interactions underlie adaptive divergence in switchgrass across a large latitudinal gradient. Proc. Natl. Acad. Sci. USA, 116, 12933–12941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magari, R. , Kang, M. and Zhang, Y. (1997) Genotype by environment interaction for ear moisture loss rate in corn. Crop Sci. 37, 774–779. [Google Scholar]
- Mihaljevic, R. , Schön, C.C. , Utz, H.F. and Melchinger, A.E. (2005) Correlations and QTL correspondence between line per se and testcross performance for agronomic traits in four populations of European maize. Crop Sci. 45, 114–122. [Google Scholar]
- Purdy, J. and Crane, P. (1967) Inheritance of drying rate in “mature” corn (Zea mays L.). Crop Sci. 7, 294–297. [Google Scholar]
- R Core Team . (2013) R: A Language and Environment for Statistical Computing. Vienna, Austria: R Core Team. 14, pp., 12–21. [Google Scholar]
- Sala, R.G. , Andrade, F.H. , Camadro, E.L. and Cerono, J.C. (2006) Quantitative trait loci for grain moisture at harvest and field grain drying rate in maize (Zea mays, L.). Theor. Appl. Genet. 112, 462–471. [DOI] [PubMed] [Google Scholar]
- Schmidt, J. and Hallauer, A.R. (1966) Estimating harvest date of corn in the field. Crop Sci. 6, 227–231. [Google Scholar]
- Sekhon, R.S. , Lin, H. , Childs, K.L. , Hansey, C.N. , Buell, C.R. , de Leon, N. and Kaeppler, S.M. (2011) Genome‐wide atlas of transcription during maize development. Plant J. 66, 553–563. [DOI] [PubMed] [Google Scholar]
- Wang, K. and Li, S. (2017) Analysis of influencing factors on kernel dehydration rate of maize hybrids. Sci. Agric. Sin. 50, 2027–2035.(in Chinese). [Google Scholar]
- Wang, X. , Wang, H. , Liu, S. , Serjani, A. , Li, J. , Yan, J. , Yang, X. et al. (2016) Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 48, 1233–1241. [DOI] [PubMed] [Google Scholar]
- Wang, Z. , Wang, X. , Zhang, L. , Liu, X. , Di, H. , Li, T. and Jin, X. (2012) QTL underlying field grain drying rate after physiological maturity in maize (Zea Mays L.). Euphytica, 185, 521–528. [Google Scholar]
- Wen, W. , Jin, M. , Li, K. , Liu, H. , Xiao, Y. , Zhao, M. , Alseekh, S. et al. (2018) An integrated multi‐layered analysis of the metabolic networks of different tissues uncovers key genetic components of primary metabolism in maize. Plant J. 93, 1116–1128. [DOI] [PubMed] [Google Scholar]
- Xiao, Y. , Liu, H. , Wu, L. , Warburton, M. and Yan, J. (2017) Genome‐wide association studies in maize: praise and stargaze. Mol. plant 10, 359–374. [DOI] [PubMed] [Google Scholar]
- Yang, J. , Carena, M. and Uphaus, J. (2010) Area under the dry down curve (AUDDC): a method to evaluate rate of dry down in maize. Crop Sci. 50, 2347–2354. [Google Scholar]
- Yang, X. , Gao, S. , Xu, S. , Zhang, Z. , Prasanna, B.M. , Li, L. , Li, J. et al. (2011) Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol. Breed. 28, 511–526. [Google Scholar]
- Yang, Q. , Li, Z. , Li, W. , Ku, L. , Wang, C. , Ye, J. , Li, K. et al. (2013) CACTA‐like transposable element in ZmCCT attenuated photoperiod sensitivity and accelerated the postdomestication spread of maize. Pro. Natl. Acad. Sci. USA, 110, 16969–16974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, N. , Lu, Y. , Yang, X. , Huang, J. , Zhou, Y. , Ali, F. , Wen, W. et al. (2014) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PloS Genet. 10, e1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zan, Y. and Carlborg, Ö. (2019) A polygenic genetic architecture of flowering time in the worldwide arabidopsis thaliana population. Mol. Biol. Evol. 36, 141–154. [DOI] [PubMed] [Google Scholar]
- Zhang, Z. , Ersoz, E. , Lai, C.Q. , Todhunter, R.J. , Tiwari, H.K. , Gore, M.A. , Bradbury, P.J. et al. (2010) Mixed linear model approach adapted for genome‐wide association studies. Nat. Genet. 42, 355–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Kang, M.S. and Magari, R. (1996) A diallel analysis of ear moisture loss rate in maize. Crop Sci. 36, 1140–1144. [Google Scholar]
- Zhang, J. , Zhang, F. , Tang, B. , Ding, Y. , Xia, L. , Qi, J. , Mu, X. et al. (2020) Molecular mapping of quantitative trait loci for grain moisture at harvest and field grain drying rat in maize (Zea may L.). Physiol. Plant. 169, 64–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The phenotypic distribution of moisture content for the measured and BLUP value across environments at different time points
Figure S2 Manhattan plots depicting GWAS results for MC data
Figure S3 Manhattan plots depicting GWAS results for AUDDC data
Figure S4 The distribution of effect values of all the QTLs for MC and AUDDC data
Figure S5 Colocalization of QTLs detected between AUDDC and MC data
Figure S6 Proportion of moisture variance due to different influential factors across five time points
Figure S7 The relationship between the main effect and Q × E for each QTL across different time points
Figure S8 Box plots for three types of QTL‐environment interaction effects
Figure S9 Density distribution of Q × E effects across different time points
Figure S10 Proportion of moisture variance due to QTL main effect and epistasis across different time points
Figure S11 Proportion of three types of epistasis related to interaction with environments across different time points
Figure S12 Box plot for the different phenotype between two allele of chr9.S_93812340 at different location and stages
Figure S13 The correlation between five stage moisture content and agronomic traits (only show the significantly correlated results)
Figure S14 The correlation between five stage moisture content and primary metabolite (only show the significantly correlated results)
Figure S15 Additive effect of one QTL (peaked as chr1.S_170961872) that affect moisture content and kernel width
Figure S16 The expression profile of candidate gene CYR1‐9 based on B73
Figure S17 The correlations between CYR1‐9 expression and moisture content
Figure S18 The moisture content between wild‐type and knockout plants by CRISPR/Cas9 system
Figure S19 Box plot for the different phenotype based on the 8‐bp InDel at different agronomic traits
Table S1 The information of weather condition of five locations.
Table S2 Summary of phenotype for MC and AUDDC at five locations and BLUP.
Table S3 Number of all significant loci identified in the study.
Table S4 List of candidate genes at the GWAS loci for grain moisture in maize.
Table S5 The number of different types of epistasis response to environments.
Table S6 List of the correlation between moisture content and agronomic traits.
Table S7 List of the correlation between moisture content and primary metabolites.
Table S8 List of the primers used in the study.