

Abstract
Background
Vaccines are often recognized as one of the most cost-effective public health interventions in controlling infectious diseases. Most pathogens infiltrate the body from mucosal sites, primarily from the oral and pulmonary region and reach the systemic circulation where disease manifestation starts. Traditional needle-based vaccines are usually not capable of inducing immunity at the mucosal sites where pathogen infiltrates start, but induces systemic immunity. In contrast to needle-based vaccines, mucosally administered vaccines induce immunity at both the mucosal sites and systemically. The oral route of immunization is the most convenient way to administer the vaccines. However, due to the complicated and hostile gastrointestinal structure and environment, vaccines need to overcome major hurdles while retaining their stability and immunogenicity.
Area covered
This review will briefly discuss different barriers to oral vaccine development. It gives a brief overview of different types of nano/microparticle-based oral vaccines and discusses how physicochemical characteristics of the particles influence overall immunity after oral immunization.
Expert opinion
Formulation strategies using novel lipid and polymer-based nano/microparticle platforms retain stability and antigenicity of vaccines against the harsh gastrointestinal condition. The physicochemical properties of particles can be uniquely tailored to prolong the release of antigens, and attached ligands (M-cells and APC-ligands) can precisely target uptake by immune cells. These represent viable strategies for efficient delivery of oral vaccines.
Keywords: Nanoparticles, Oral vaccines, Delivery system, Microparticles, Adjuvants, Immunology
Introduction
Vaccines are considered one of the most powerful and cost-effective approaches to combat infectious disease outbreaks (Delany et al. 2014). Vaccines work by training the body’s immune system to defend against pathogen invasion by rapidly removing pathogens or their secreted products either following neutralization or opsonization processes. An ideal vaccine should have the ability to be recognized and activated by immune cells such as B cells and T-cells, retain long-term immunological memory and provide adequate protective immunity in hosts against recall pathogens. Vaccines are prepared either from whole microorganisms (attenuated or killed) or their toxins or subunit components (proteins, nucleic acids, polysaccharides, peptides etc.) (Yadav et al. 2014). The efficacy of vaccines rely on the nature of the immunological components used in the vaccine formulations. For instance, whole pathogen-based vaccines are usually efficacious as all components of the pathogen could be immunogenic. However, the use of whole pathogen-based live/attenuated or inactivated vaccines produced by weakening or altered antigens may cause unwanted side-effects or adverse reactions and possess potential risks to reverse the original pathogenic types (Josefsberg et al. 2012). Whole pathogen-based vaccines may be especially risky in immunocompromised individuals, such as people living with HIV, tuberculosis, and cancer, the elderly, and individuals with other chronic diseases (Blaney et al. 2011). With developments in vaccine production technology, conventional vaccine production techniques using the concept of using whole pathogens as a vaccine candidate has now transitioned to using subunit components such as recombinant proteins, peptides, or polysaccharides derived from the pathogens. For instance, a particular recombinant protein, peptide or polysaccharide-based on protective epitopes, instead of the whole microorganism, can be selectively recognized by the immune system (Liljeqvist et al. 1999; Naz et al. 2007). These minimal fragments of pathogens have reduced side effects and possess an alternative solution to overcome the disadvantages of traditional vaccines. However, subunit vaccines are often less immunogenic and require coadministration of adjuvants or specific delivery systems.
Adjuvants are immunomodulatory compounds that are used with antigens in vaccine formulations to evoke robust immune responses (O’Hagan et al. 2003). Adjuvants can be classified into two major categories based on their principle functions, such as immunostimulatory adjuvants and antigen delivery systems (O’Hagan et al. 2003). Adjuvants as a vaccine delivery system can protect antigens, provide sustained release of antigens and target antigens to local lymph nodes and facilitate immune responses against delivered antigens (Zinkernagel et al. 1997). Moreover, immunomodulatory adjuvants stimulate cellular uptake of antigens from administration sites, activate antigen-presenting cells (APCs) such as B-cells and T-cells, and up-regulate cytokines and chemokines to provide a robust adaptive immune response (Fearon 1997).
The classical adjuvant used in human vaccines is based on aluminium salts, such as aluminium hydroxide, aluminium phosphate, and potassium aluminium sulphate. With the first introduction of alum in the 1920s as the first-ever human vaccine adjuvant, it still shows an excellent safety profile (Shah et al. 2017). Alum has been successfully used in malaria and tuberculosis vaccines. Only a few decades after the development of alum, researchers began to identify other novel types of adjuvants. At present, only a handful of adjuvants are approved for human use. Some of the approved adjuvants for clinical use include poly I:C, monophosphoryl lipid A, CpG oligodeoxynucleotides, incomplete Freund’s adjuvant, virus-like particles, virosomes, Imiquimod, MF59, AS03 and AS04 etc. (Apostólico et al. 2016). Most adjuvants developed at the preclinical and clinical stage were found to be unsuitable for further human use due to the high degree of toxicity and reactogenicity in humans (Reed et al. 2013). For instance, Freund’s adjuvant was highly effective as a vaccine adjuvant, however, it was highly reactive and produced serious side-effects (granuloma, abscess, ulcerative necrosis etc.) in earlier human studies which revoked its use in human (Apostólico et al. 2016).
Need for oral vaccines
Mucosal membranes and the skin provide a physical barrier against pathogenic infiltrations and serve as a major line of defence (Holmgren et al. 2005). Mucosal membranes in the aerodigestive and urogenital tracts can be invaded by pathogens as they continuously interact with foreign environments. This results in stimulation of immunoglobulin A (IgA) production that can more specifically prevent the mucosal invasion of pathogens at the site of infection. In addition to local mucosal immunity, it is also important to stimulate the production of systemic immunoglobulin G (IgG) that can neutralize pathogen replication at later stages of infections for complete protection. However, the majority of current vaccines are typically administered by the parenteral routes using subcutaneous or intramuscular injection, which are generally limited to triggering strong local mucosal immune responses (Wang et al. 2004; Lycke et al. 2010). The key step is to develop an effective vaccination strategy that could form an immunologically strong barrier at both mucosal sites and systemic sites. These barriers can be overcome by the development of effective mucosal adjuvants or delivery systems. Most clinically approved adjuvants are poor inducers of mucosal immunity and are primarily used for systemic use. Most mucosal vaccines use live/attenuated forms of pathogens as key components for induction of immunity (Miquel-Clopés et al. 2019). Unfortunately, none of the mucosal adjuvants for subunit vaccines are approved for human use. While traditional vaccine adjuvants may not be appropriate to oral vaccines, new potent mucosal adjuvants with low toxicity are critical for the development of next-generation oral vaccines (Premanand et al. 2013).
The oral route is one of the most convenient routes of administration for any therapeutic. Apart from other mucosal sites for vaccines, the oral route offers several advantages such as (1) oral vaccines are needle-free and can substantially reduce the risks associated with needle stick injuries that may transmit blood-borne pathogens by cross-transmission due to reusing contaminated or undisposed syringes and needles (Ekwueme et al. 2002), (2) oral vaccines can improve patient compliance to vaccines by avoiding ‘needle phobia’ and are painless, (3) unlike, traditional administration routes which require trained medical professionals to avoid needle-stick injuries, oral vaccines are comfortable as they can be self-administered and improve the capacity for mass immunization (Chen et al. 2011), (4) oral vaccines could avoid strict current cold-chain requirements, thus reducing the storage and transport of materials. For example, the powered delivery approach may abolish the importance of traditional cold chain transport. Besides, the needle-free delivery approach can reduce the cost of iatrogenic blood-borne pathogen transmission (Chen et al. 2011). Additionally, it is expected that oral vaccine production can be performed without requiring strict sterile conditions as for parenteral formulations, thus reducing the cost of production. The oral vaccine delivery approach can not only systemically stimulate antigen-specific antibodies such as IgG, but can also induce mucosal antigen-specific IgA antibodies in the blood (Marasini et al. 2014). Despite a large array of benefits, oral vaccines are required to overcome a series of physical and immunological barriers, such as inefficient mucosal permeability, poor immunogenicity, and antigen instability at the mucosal sites. Only a few oral vaccines are licensed for human use (Table 1). From here on, this review will discuss potential oral mucosal vaccines, barriers in oral vaccine delivery and the current on-going development of oral vaccines based on nanoparticles.
Table 1.
Current licensed oral vaccines
| Trade name | Targeting diseases | Antigens | Formulations | Dosage | Manufacturer | Reference |
|---|---|---|---|---|---|---|
| Polio Sabin™ | Polio | Live attenuated poliomyelitis virus Sabin strains Type 1 (LSc, 2ab) and Type 3 (Leon 12a, 1b) | Solution |
One dose of 0.1 ml Three doses, taken at least four weeks apart |
Multiple | Bandyopadhyay et al. (2015) |
| Vivotif® | Typhoid Fever | Live-attenuated strain, Ty21a | Enteric-coated capsule | Four doses, taken on alternate days | PaxVax, Redwood City, CA, USA | Gentschev et al. (2007) |
| Dukoral | Cholera | Inactivated whole-cell Vibrio cholerae O1 Inaba, Ogawa, classic and El Tor strains | Suspension | Two doses are taken orally (by mouth) at least one week (up to six weeks) apart | Valneva Sweden AB, Solna | Lucas et al. (2005) |
| Vaxchora | Live, oral, CVD-10-HgR | Suspension | 100 mL PO as a single dose a minimum of ten days before potential exposure to cholera | PaxVax, Redwood City, CA, USA | Chen et al. (2016) | |
| ShanChol | Formalin-killed V cholerae O1 El Tor Inaba; heat-or formalin-killed V cholerae O1 classical Ogawa; heat-killed V cholerae O1 classical Inaba; formalin-killed V cholerae O139 | Suspension | Two or three doses delivered by oral syringe | Shantha Biotecnics Ltd., India, Hyderabad | Sur et al. (2009) | |
| Euvichol-Plus®/Euvichol® | Heat inactivation of V. cholerae O1 Inaba Cairo 48 and V. cholerae O1 Ogawa Cairo 50; formalin inactivation of V. cholerae O1 Inaba Phil 6973 El Tor and V. cholerae O1 Ogawa Cairo 50; formalin inactivation of V. cholerae O139 4260B | Suspension | Two or three doses, fourteen day interval between doses | EuBiologics Co., Ltd., Seoul, Republic of Korea | Baik et al. (2015) | |
| Rotarix™ | Rotavirus | Attenuated human rotavirus (G1P strain) | Suspension | Two or three doses | GlaxoSmithKline, Wavre, Belgium | Steele et al. (2012) |
| RotaTeq™ | RV5, which consisted of five live human-bovine reassortant rotavirus strains (G1, G2, G3, G4, and P1A)a | Solution | Three doses, 4–10 weeks apart | Merck, West Point, PA, USA | Mo et al. (2017) |
aLive-attenuated pentavalent rotavirus (four human strains express G1, G2, G3, G4, and P7) from bovine rotavirus. Fifth virus expresses P1A from human and G6 from bovine rotavirus
Barriers in oral vaccine delivery
The GI tract (Gastrointestinal tract) is the primary site for the transport or uptake of antigens. The organization of the GI tract plays a vital role in the formulation of oral vaccines to enhance their efficacy. Due to the typical structure of the GI membrane, orally administered vaccines need to overcome several physiological and immunological obstacles (Fig. 1). The intestinal epithelium serves as the main physiological barrier. It is covered by mucus-secreting layers, which helps in the digestion and absorption of nutrients and provides resistance against the invasion of pathogens (Pelaseyed et al. 2014). To achieve these, the GI tract must maintain a variable pH throughout the tract that includes a highly acidic environment (stomach), have a slightly acidic or neutral environment (pH 6–7.4) in the intestine, and secrete proteolytic enzymes and bile salts for protein and fat digestion (Grant 2004). Oral vaccines, when exposed to such surroundings may degrade and lose their immunogenicity (Kompella et al. 2001). Secondly, the compact epithelial cell surface and thick mucus layer limit their absorption and the temporary residence time of antigens in the intestine limits their absorption (Mudie et al. 2010). Further, it is difficult to deliver antigens to specific induction sites orally due to the significant dilution and dispersion in mucosal secretions (Kunisawa et al. 2012). Due to the above-mentioned factors, oral vaccines require a large dose of antigens to trigger long-lasting and potent mucosal immune responses than traditional injectable routes (Ramirez et al. 2017). However, a large antigen dose is likely to pose a risk for oral tolerance, which means non-responsiveness against antigens instead of protective immunity (Pabst et al. 2012). Thirdly, the colonized commensal microbes could regulate gut immunity and have an impact on optimal mucosal immunity by modulating IgA responses in vivo (Donaldson et al. 2018). Overall, the development of oral vaccines is critical and a challenge in terms of identifying useful adjuvants and sufficiently stimulating the mucosal immune system.
Fig. 1.
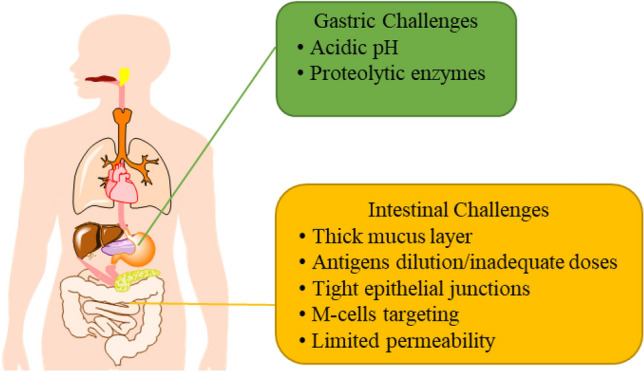
Biological barriers for efficient delivery of oral vaccines
Gastrointestinal immune system
The GI system contains the highest number of immune cells in humans compared with other mucosal systems. These cells are regulated by the gut-associated lymphoid tissue (GALT). This is one of the biggest lymphoid tissues in humans and includes both inductive and effector sites (Koziolek et al. 2015). The inductive sites in the GI tract include Peyer's patches (PPs), organized lymphoid follicles and antigen-presenting cells (APCs), while effector sites mainly involve the coordinated action of lamina propria (LP) and intestinal surface epithelium.
PPs are traditionally considered as the main inductive sites of mucosal immunity and they are the group of dome-shaped tissue that resides in the mucosal layer of the ileum and spread to the submucosa layer. PPs are surrounded by the follicle-associated epithelium (FAE), which contains both isolated and aggregated lymphoid follicles. The mucus layer of FAE is thinner than typical small intestinal villus epithelium due to the presence of goblet cells. This thinner structure is responsible for the exchange of contents between GALT and the luminal microenvironment. Microfold cells (M-cells) are an important portal of communication between foreign antigens and intestinal immunity. The M cells in FAE mainly transport luminal particulate antigens from the apical to basolateral side to awaiting APCs that trigger or inhibit the immune cascade. APCs such as dendritic cells (DCs) present in the subepithelial LP region play a critical role in immunity mediated by GALT. This region is largely populated by two types of intestinal mononuclear phagocytes; dendritic cells (DCs) and macrophages. They are located in the subepithelial dome (SED), which is below the FAE for better processing of antigen fragments so that B cells and T cells are rapidly activated.
The distribution of APCs in the GALT region is crucial for better antigen sensing and accelerating their uptake. With the administration of antigens, local lymphocytes take up the foreign antigens from the inductive sites and these antigens are processed followed by triggering their action at effective sites via cytokines or antibodies (Fig. 2). Initially, M-cell transport antigens and present them to underlying DCs, a process called ‘antigen sampling’. The fragments of antigens induce the maturation of DCs that in turn prime naïve CD4+ T cells (Brayden et al. 2005). The antigen-primed CD4+ T cells interact with B cells to stimulate the production of immunoglobulin-secreting cells (typical IgA+ B cells). These IgA+ B cells undergo isotype switching to produce antigen-specific IgA+ B cells and migrate to local mesenteric lymph nodes and finally into the systemic circulation. The IgA+ B cells further differentiate and undergo maturation into IgA+ producing plasma cells. These IgA+ producing plasma cells further bind to polymeric or dimeric Ig receptor and translocate to the luminal surface of the intestine, producing secretory IgA, responsible for local protection (Chang et al. 2014).
Fig. 2.
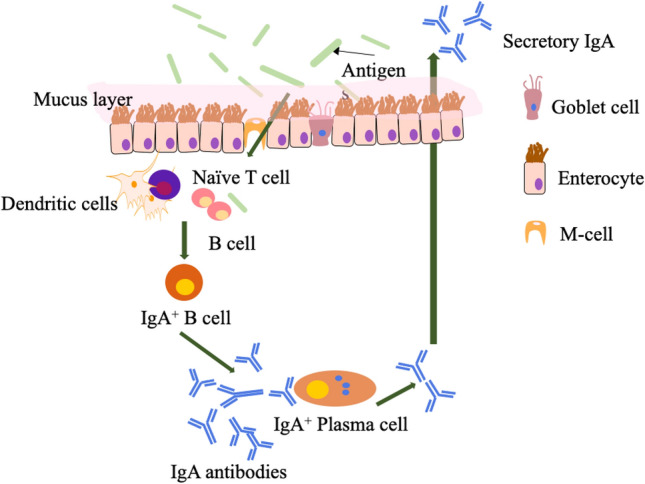
Schematic representation of mucosal immune response upon oral delivery of antigens. Antigens are translocated across gastrointestinal epithelium from Microfold cells (M-cells) and captured by dendritic cells (DC). Alternatively, antigens can be directly sampled by DC by luminal extension of dendrites. Dendritic cells process and present antigens to activate naïve T-cells. The naïve T-cells further trigger B-cell activation and induce the conversion from B cells into IgA+ plasma B cells and produce antigen-specific antibodies. The antibodies are then transported to the surface of the mucosal membrane
Nanoparticles as an oral vaccine delivery system
Oral vaccines are required to overcome two main barriers, physiological and immunological, that separates the inner milieu of the body against foreign compounds. GI epithelium forms a tight junction membrane barrier that is semipermeable and limits absorption while peptidases present in the GI tract act as the main enzyme that enzymatically degrades foreign material and blocks their invasion.
The types of antigens and delivery routes impact the efficacy of the vaccines. Since many mucosal vaccines are readily degraded in the GI tract, they require a higher dose or many repeated doses of vaccines to provide sufficient immunogenicity (Webster et al. 2003). However, administration of a large oral vaccine dose may lead to antigen tolerance in recipients (Chang et al. 2014). The likely path to overcome the tolerance issue is by delivering a low amount of antigen at a targeted site, such as underlying APCs and M-cells which produce the same level of responses as in higher doses. This can be achieved by modulation of the antigen, from soluble to particulate structures, and incorporating M-cell targeting ligands in the vaccine structure which are recognized by M-cells (Webster et al. 2003).
With the advancement in nanotechnology over the last few years, nanoparticle applications have gained increased attention with widespread use in a range of biomedical fields including sensors, medical imaging, diagnosis and delivery of drugs and vaccines (El-Sayed et al. 2020). Vaccines or antigens can be either conjugated or encapsulated into the nanoparticles or loaded on the nanoparticle surface to boost the immune response against delivered antigens (Torres-Sangiao et al. 2016). It has been shown that antigen encapsulation into nanoparticles triggers better mucosal immune responses in a mouse model than antigens-coated on the surface of the nanoparticles (Marasini et al. 2016a, b, c). Vaccine formulation materials play important roles in enhancing vaccine stability and immunogenicity. The pros and cons of each type of particles discussed in this review are highlighted in Table 2. It is important to highlight that nanomaterials used as vaccine carriers should lack immunogenicity or be non-immunogenic. Owing to their small size, nanoparticles pose a high ratio of surface area to volume, decreased clearance and increase stability. These properties provide highly efficient delivery to systemic mucosal sites such as nasal or GI tracts, and enhanced absorption and diffusion of loaded drugs (Marasini et al. 2017a, b). Overall, the significant merits of nanoparticle applications in oral vaccines are (1) Precise antigen delivery to target sites; (2) Increased stability of antigens to retain its original immunogenic properties; (3) Feasibility to deliver a high dose of antigens (4) Enhancement of antigen availability to interact with APCs (carrier-assisted transport across the intestinal membrane); (5) Immunomodulation (in cooperation with pro-inflammatory cytokines) or immunosuppression (in corporation with anti-inflammatory cytokines).
Table 2.
Pros and cons of oral nano/microparticle-based carriers
| Categories | Liposomes | Emulsion | ISCOMs | Natural polymers | Synthetic polymers |
|---|---|---|---|---|---|
| Application | DNA, peptides, proteins | Whole-cell killed, proteins, peptides | Proteins, peptides | Proteins, peptides, conjugates | Proteins, peptides, conjugates |
| Pros |
-Intrinsic adjuvant characteristics -Can accommodate both hydrophilic and lipophilic antigens -Modified liposomes have optimal stability across the intestine -Controlled release |
-Tailored immunomodulation, for example, Th1 (water-in-oil) and Th 2 (oil-in-water) -High loading efficiency of both hydrophilic (water in oil) and hydrophobic (oil-in-water) -Slow or controlled release |
-Easy modification -High antigen loading efficiency -Potent built-in adjuvant (Quil A) |
-Controlled release -Easy modification -Highly stable and adaptable |
-Controlled release -Controlled size -Engineered surface chemistry |
| Cons |
-Weak loading efficiency of hydrophilic antigens -Nonspecific interactions -Cationic liposomes could be toxic |
-Poor stability under the harsh environment in the GI system |
-Poor loading of hydrophilic antigens -Rapid clearance |
-Low antigen loading -Poor aqueous solubility |
-May have poor solubility -Insufficient antigen protection -Exposed to proteolysis in mucus |
Particle characteristics in vaccine formulations
Oral particle vaccines should aim to target M-cells in the mucosal sites, provide antigen protection in the GI tract, and enhance interaction with immune cells of interest to induce a downstream immune response (Fig. 3). Nano/microparticles usually mimic natural infections and their particulate structure can be modified to increase the possibility of antigen modulation and recognition by APCs, which is the key step to triggering the following adaptive immunity to achieve efficacy in hosts (Fahmy et al. 2008).
Fig. 3.

Strategies for nano/microparticle-based oral vaccine delivery. a Antigens can be loaded into nano/microparticles on their surface by chemical conjugation or encapsulated inside the particles. Further, the nano/microparticle surface can be modified by conjugating different targeting ligands, such as Microfold cell and dendritic cell targeting ligands. Particles can be modified on their surface by electrostatic coating with a series of cationic and anionic polyelectrolytes or pH-sensitive polymers for enhanced stability in the gastrointestinal environment. b Approaches to enhance the permeability of particles across the intestine by targeting microfold cell (M-cells)
The physicochemical properties of particles loaded with antigens influence subsequent immunological outcomes and are mainly guided by the stability, efficacy, and safety of the vaccines (Benne et al. 2016). Some physicochemical parameters such as size, surface charge, morphology, hydrophobicity, stability and bioadhesive properties of nanoparticles have a direct impact on in vivo behaviour of vaccines in the GI tract that affect the immunogenicity outcomes (Őrfi et al. 2016).
While there is a limited number of reports describing the influence of particle morphology, a recent paper showed that intravenous administration of nanoellipsoidal particles improved pharmacokinetic parameters and improved T-cell induction compared to spherical particles (Meyer et al. 2015). Mitragotri and colleagues showed that the shape of polystyrene nanoparticles have an impact on the transport and absorption across the intestinal epithelium (Banerjee et al. 2016). An in vitro study by the same group using a triple co-culture model (Caco-2 cells, HT-29 cells—similar to goblet cells to produce mucus) and Raji-B lymphocytes (similar to M cells) to mimic natural intestinal cells showed that rod-shaped particles have higher cellular uptake than sphere-shaped particles.
Similarly, Skwarczynski and colleagues prepared a lipopeptide based stable oral delivery system against hookworm, Necator Americanus using B-cell peptide epitope derived from aspartic protease Na-APR-1 (Bartlett et al. 2020). Upon oral immunization in mice, the lipopeptides that form a rod-shaped aggregate were found to be stable in the gastrointestinal conditions and induced a protective immune response against parasitic challenge studies. Both observations concluded that rod-shaped particles may have a higher impact on immunogenicity than spherical particles. However, the particle shape alone may not necessarily be the only factor in driving the immune response. Other particle characteristics, such as the size of particles may have a profound effect on the type of immune response. For instance, spherical polystyrene particles (approx. 200 nm) upon conjugation with model antigen produce a Th1-biased immune response, whereas polystyrene rod-shaped-conjugated ovalbumin particles (approx. 1500 nm) produce primarily a Th2-biased immune response (Kumar et al. 2015).
Particle size has been shown to influence interactions with immune cells in the GI tract and their circulation period in vivo (Duan et al. 2013). Based on diffusivity studies in intestinal mucus, smaller molecules such as capsid virus-like particles were shown to pass through the mucus barrier rather than larger molecules, such as proteins (Cone 2009). The mucus mesh network displayed a heterogeneous pore size, ranging from 20 to 1800 nm. However, nanoparticles should avoid the rapid clearance by mucosal fluids, which is a fluid with viscosity and elastic forces. Thus, smaller molecules can be transported easily (Lai et al. 2009). It is therefore reasonable to predict that the transport rate is limited by the pore size.
Particle surface charge is another factor that influences the interaction with immune cells. Negatively charged particles were shown to have decreased uptake by APCs, and subsequently could not stimulate the following cellular process effectively (Xu et al. 2015). Besides, the hydrophilicity/hydrophobicity properties of the surface of nano/microparticulate carrier was shown to affect the mucosal permeability of the antigens (Witten et al. 2018). For instance, hydrophobic nanoparticles were easily recognized as invaders and recognized by the immune cells located within the reticuloendothelial system (Wen et al. 2016). Another study has shown that mucoadhesive properties of chitosan and sodium alginate enhance antigen interaction with immune cells by prolonging their residence time in the mucosal sites to slow the release of antigens and avoiding GI mucosal clearance (Jiao et al. 2016). Overall, the physicochemical properties for nanoparticles are highly associated with immunogenic efficacy of the vaccines.
Types of nano/microparticles
Liposomes
Liposomes are extensively explored in mucosal vaccine delivery and some of the liposome-based vaccine formulations against hepatitis A (Epaxal) and influenza (Inflexal V) are already in the market (Marasini et al. 2017a, b). Liposomes are spherical vesicles with self-assembled phospholipid bilayers and inner aqueous cores. This unique amphiphilic structure of liposomes enables them to entrap hydrophilic antigens in the inner aqueous core and lipophilic antigens in the lipid bilayer (Bernasconi et al. 2016). Liposomes are usually formed as unilamellar or multi-lamellar structures with, sizes ranging from tens of nanometers to micrometres depending upon their method of preparation (Marasini et al. 2017a, b). Liposomes can be modified to enhance their stability in the mucosal tract, protect antigens from degradation and improve circulation times (Marasini et al. 2019). Liposomes are well suited for the delivery of antigens in extreme conditions in the GIT. Similarly, Liu and coworkers showed that liposome-encapsulated DNA vaccines were able to induce both humoral and cellular immunity following oral immunization in mice and these immune responses were protective against respiratory challenge infections (Liu et al. 2014).
Liposomes can also be customized for the efficient delivery of complex antigens. Studies have shown that liposome modification with polyethylene glycol (PEG) can trigger local mucosal and systemic immunity upon oral or sublingual administration (Minato et al. 2003; Oberoi et al. 2016). Similarly, in another study, vaccine formulations were prepared by entrapping monovalent detergent split influenza (A/Victoria/210/2009 H3N2) into unmodified liposomes, PEG-modified liposomes, and phospholipid-PEG conjugates and methyl glycol chitosan modified liposomes. It was revealed that methyl glycol chitosan-coated phospholipid-PEG bearing liposomes (bearing both mucoadhesive and neutral charge) induced the most efficient flu-specific mucosal and systemic immunity (Oberoi et al. 2016). Interestingly, another study revealed that the dose of antigens and the number of liposomes dictate the types of the humoral immune response (mucosal vs systemic) upon oral immunization (Minato et al. 2003). A small number of PEG-modified liposomes encapsulating the antigens was shown to be better at inducing mucosal immunity, whereas a larger number of liposomes encapsulating a similar dose of antigen tends to improve systemic immunity.
Liposomes have also shown the potential to be used as cold-chain free vaccines. In one study, liposomes were modified to include a targeting moiety on the surface with a polymeric spacer such as PEG (Wang et al. 2014). It was shown that the inclusion of mannose-PEG-cholesterol conjugates in liposomes can efficiently entrap both the adjuvant (lipid A) and antigens and produce a stable liposome even after lyophilization. This vaccine formulation was able to induce potent mucosal and systemic immunity when given orally to a mouse model.
Another approach to facilitate the delivery of antigens is by coating the surface of the liposomes using a layer-by-layer (LBL) approach via polyelectrolytes (Marasini et al. 2016a, b, c). Using this approach, liposomes are usually coated with oppositely charged polymers which are mediated by electrostatic attraction between opposite charges. This approach protects the antigen from possible degradation, increase GI membrane permeation and retain the formulation at the site if mucoadhesive polymers are used. In a prior study, liposomes were engineered with chitosan and alginate and their efficiency to deliver lipopeptide vaccines was evaluated in mice (Marasini et al. 2016a, b, c). It was observed that the liposomes coated with cationic trimethyl chitosan produced a significant increase in antigen-specific serum IgG titers and antigen-specific mucosal IgA titers as compared to lipopeptides alone or lipopeptide-encapsulated liposomes. Similarly, another study using LBL to coat liposomes using vitamin B12-chitosan and alginate showed enhanced uptake of LBL vaccine formulation by a macrophage cell line and was followed by significant enhancement of humoral immunity (Verma et al. 2016).
Emulsion (oil-in-water)
Emulsions are employed as a safe and effective colloid for vaccine delivery against many infectious diseases. Emulsions are formed by amphiphilic surfactants formed via dispersing two immiscible liquids (Singh et al. 2017). Either water-in-oil or oil-in-water emulsions can be used to target mucosal membranes (Singh et al. 2017). Comparatively, water-in-oil emulsions have a better encapsulation and delivery ability than oil-in-water type, since the former can efficiently incorporate hydrophobic antigens (Singh et al. 2017). Freund’s adjuvants is a classic example of a water-in-oil- emulsion adjuvant, which is usually emulsified with antigens. MF59, AF03 and AS03 are squalene-based emulsion adjuvants, which have been already developed for influenza vaccines in the past 10 years. A study has shown that emulsion of Toll-like receptor agonists (TLRa) with squalene achieve modified pharmacokinetic and immunogenicity profiles against Neisseria meningitidis (Lodaya et al. 2018). However, none of these emulsions are used as carriers for oral delivery of vaccines.
Other lipids
Non-ionic surfactant vesicle stabilized bile salts (bilosomes) have been evaluated for oral delivery of peptides and proteins. The bilosome-antigen conjugates have been reported as potential oral vaccines against several diseases including hepatitis B, influenza and tetanus (Mann et al. 2006; Shukla et al. 2010; Corthésy et al. 2018). Incorporation of diphtheria toxoid (DTx) in bilosomes enhanced anti-DTx IgG and mucosal IgA levels without tolerance after oral immunization in mice (Shukla et al. 2011). A study showed that a bilosome-based vaccine prepared by encapsulating recombinant baculovirus VP1 (Bac-VP1) produced a significant amount of serum IgG and mucosal IgA titers as compared to bilosome not associated with Bac-VP1 (Premanand et al. 2013).
Immunostimulatory complexes (ISCOMs)
ISCOMs are cage-like particles made from antigens, cholesterol, phospholipid, and Quil A saponins (Lovgren et al. 1988). Typically, they are spherical and micellar particles and are approx 40 nm in diameter, representing icosahedral symmetry (Özel 1989). ISCOMs can significantly induce both Th1 and Th2-biased immune responses (Lövgren Bengtsson et al. 2011). The combination of ISCOMs with Quil A (Quillaja) saponin gives them a particulate nature and provides immunomodulatory properties. Quillaja saponin is a plant glycoside composed of aglycone and two sugar chains (Hu et al. 2001). They can be used for hydrophobic proteins and peptides. ISCOMs are widely used in mucosal vaccine delivery. For example, the inclusion of ISCOMs with cholera toxin and S. aureus protein A (CTA1-DD) showed strong humoral immune responses upon oral administration (Mowat et al. 2001). ISCOMATRIX is a similar delivery system but without antigen. Some vaccines based on ISCOMATRIX are approved for clinical use (Siegrist 2008).
Natural polymers
Chitosan is a naturally occurring linear polysaccharide derived from the exoskeleton of crustaceans and exists as copolymers of β-(1-4)-linked glucosamine and N-acetylglucosamine (Sudarshan et al. 1992). Natural chitosan is largely insoluble at a pH > 6 which limits its biological application as a vaccine delivery vehicle. One of the methods to increase its aqueous solubility is by methylating its primary amino group (Marasini et al. 2016a, b, c). Chitosan is a mucoadhesive polymer that increases antigen residence time in the mucosa. It can also open tight epithelial junctions by translocating tight junction proteins that enable smooth translocation of antigens (Smith et al. 2004). A chitosan-based nasal vaccine delivery platform (ChiSys®) has been approved for human use as a mucosal adjuvant (Watts et al. 2014) and it has shown good efficacy in a large number of diseases, including tuberculosis, influenza and chlamydia (Wu et al. 2017).
Ting and colleagues studied the feasibility of oral delivery of DNA vaccines using an M-cell targeting approach (Ye et al. 2014). A chitosan nanoparticle conjugated to M-cell targeting peptide ligand (CPE30) and encapsulated with plasmid encoding CVB3 predominant antigen pVP1 was prepared and tested in mice after oral administration. The chitosan-peptide antigen formulation (CPE30-chitosan-pVP1) showed increased mucosal and M-cell uptake compared to other formulations without chitosan. Except for fecal SIgA and mucosal expression of T-cells, there was no significant difference in antigen-specific serum IgG levels and T-cell responses. Another study prepared GI stable nanoparticles using tripolyphosphate, mannosylated chitosan and Eudragit® and encapsulated bovine serum albumin. Upon oral immunisation in rats, the nanoparticles targeted Peyer's patches and showed antigen-specific system IgG and mucosal IgA responses (Xu et al. 2018).
Alginate
Alginate is a non-toxic, biocompatible, and biodegradable polymer with widespread use in drug and vaccine delivery. Its unique property to resist the low pH in the GI tract makes it a suitable carrier for oral delivery of protein, DNA and pH-sensitive antigens (Sosnik 2014). Alginate microparticles have been examined as potential oral powder delivery systems that could replace the current practice of cold chain storage for injection. For instance, the whole attenuated pathogen V. cholerae was encapsulated into alginate and Eudragit® L30 D-55 microparticles by spray drying. These microparticles showed improved GI stability and retained their ability to induce strong antibody titers following oral immunization in mice (Pastor et al. 2013). Additionally, another study showed the feasibility of incorporating Bacille Calmette–Guerin (BCG) into alginate microparticles. After a single oral immunisation with BCG- encapsulated microparticles, they produced significant IgG and IgA titres in the lungs (Hosseini et al. 2015). In a recent study by Li and coworkers, alginate and chitosan-coated calcium phosphate nanoparticles encapsulating albumin were prepared and tested for stability in the GI tract. The alginate-coated nanoparticles were found to be stable and their oral immunization in mice showed a significant increase in mucosal IgA and serum IgG titres (Cao et al. 2020).
Hyaluronic acid (HA) is a linear polysaccharide formed by two sugar moieties: glucuronic acid and acetyl glucosamine monomers. Hyaluronic acid is biocompatible, biodegradable, and possesses mucoadhesive properties (Nashchekina et al. 2018). HA can be tailored to prepare stable nanoparticles loaded with antigens at a size close to 100 nm (Bussio et al. 2019). HA has been shown to trigger dendritic cell maturation and enhance T-cell activation (Mummert 2005). There are limited reports on the use of HA as an oral vaccine delivery system. However, they have shown great potential as a nasal vaccine against influenza and Yersinia pestis (Singh et al. 2001; Fan et al. 2015) and only have a ‘potential use’ in oral vaccines.
Glucan particles (GP) are porous and hollow microspheres with 2 to 4-micron cell wall shells produced by 1,3-D-glucan and trace amounts of chitin of Saccharomyces cerevisiae (Baker’s yeast) with a series of alkaline, acid, and solvent extraction processes (De Smet et al. 2014). GP has been shown to have promising aspects in the delivery of oral vaccines. For instance, a study showed that albumin loaded GP particles were efficiently internalized by intestinal cells (Caco-2 and HT-29) and these particles downregulated TGF- β expression (indicator for oral tolerance suggesting feasibility for oral immunisation at large doses). Upon oral administration of these particles, mice showed increased production of intestinal mucosal IgA antibodies (De Smet et al. 2013). In addition to their ability to deliver proteins, GP is also known to facilitate the targeted delivery of DNA antigens to phagocytic cells (Soto et al. 2008).
Synthetic polymers
The application of synthetic polymers to oral vaccines has been extensively investigated in recent years. Poly(lactic-co-glycolic acid) (PLGA) is a copolymer of polylactic acid (PLA) and polyglycolic acid (PGA). PLGA is a US FDA (Food and drug administration) approved biocompatible and biodegradable polymer widely used in the delivery of drugs and antigens (Silva et al. 2016). It can be tailored to precisely control the release kinetics of antigens and adjuvants in mucosal membranes (Silva et al. 2016). For instance, the ratio of lactic and glycolic acids (increasing ratio of PGA leads to faster degradation) and molecular weight of polymers (low molecular weight gives faster degradation-burst release) could be easily adjusted for customized delivery of antigens (Silva et al. 2016).
PLGA has been explored as a suitable carrier for the co-delivery of antigens and adjuvants. For instance, a study encapsulated toll-like receptor 4 (TLR4) ligand monophosphoryl lipid A (MPLA) with OVA antigens and evaluated their immunological efficacy upon oral administration in mice. With single immunization, PLGA nanoparticles encapsulating both the adjuvants and OVA antigens generated significant systemic IgG and mucosal IgA titers compared to their controls (PLGA+OVA antigen) and a buffer group (Sarti et al. 2011). PLGA can be functionalized to conjugate M-cell targeting ligands and be co-delivered with adjuvants. Wang and colleagues showed that PLGA can be conjugated to M-cell targeting ligands, ulex europaeus agglutinin-1 (UEA-1) and co-delivered with MPLA and OVA antigens as nanoparticles. These nanoparticles were stable in the GI tract, targeted M-cells and dendritic cells and produced efficient humoral responses upon oral administration in mice (Ma et al. 2014).
It has been shown that the efficacy of PLGA-based nanoparticles as an oral vaccine carrier is further enhanced by formulating with hydroxypropyl methylcellulose phthalate HP55 (Tan et al. 2017). This special polymer has gastric resistant properties and degrades at pH 5.5 or higher. The immunogenicity of PLGA nanoparticle-based vaccines incorporating H. Pylori recombinant antigen CCF was increased when nanoparticles were formulated with HP55. For example, HP55 based PLGA nanoparticles enhanced the Th1/Th17-bias protection against Helicobacter pylori infection without gastric acid degradation (Tan et al. 2017).
Others (cell-membrane coating technology)
More recently, cell-membrane coating technology has become increasingly popular in the oral delivery of subunit vaccines. This technology allows the insertion of antigenic materials (cell-membrane containing toxins) on the surface of core–shell particles and protects the antigenic components against degradation during their journey in the GIT. For instance, one study assessed the delivery efficacy of magnesium and titanium dioxide core–shell particles coated with antigenic materials, mucoadhesive and pH-resistant polymers (Wei et al. 2019). The outer layer stabilized the particles against the harsh stomach pH and efficiently delivered antigens to the intestinal wall by improving retention time and permeability across the membrane. Upon oral immunization in mice, this technology was able to generate a high level of antigen-specific mucosal antibody titers.
Conclusion and future direction
Vaccine targeted at mucosal sites protects against pathogen invasion at both the mucosal sites and in the systemic circulation. While lots of pathogens infect the host via mucosal entry, there are only a handful of oral vaccines available for human use (Table 1). These include vaccines against polio, rotavirus, typhoid, and cholera. Most of these vaccines are formulated as live attenuated strains of microorganisms, except for a vaccine against cholera (Dukoral®), which is formulated as a mixture of cholera toxin B subunit strains and inactivated V. cholerae 01 whole cells (New 2019). A licensed vaccine based on nano/microparticles is still lacking, although few nano/microparticle-based vaccines are being trialled in humans. While current research is mostly focused on the development of gold standard mucosal adjuvants (like alum for parenteral vaccines), the only questions remaining are whether these adjuvants would be potent enough to cover various types of antigens targeting different diseases and whether these adjuvants are safe in humans. The nano/microparticle-based vaccine carriers currently utilized for oral vaccine delivery are summarized in Table 3.
Table 3.
Summary of nano/microparticle-based carrier utilized for oral delivery of vaccines
| Particles | Antigens | Immune responses | Reference |
|---|---|---|---|
| Liposomes | M1 gene of influenza A virus | Enhanced humoral and cellular immunity; protective immunity upon challenge studies | Liu et al. (2014) |
| Modified liposome (chitosan-coated phospholipid-PEG bearing liposomes) | Monovalent detergent split influenza | Enhanced mucosal and systemic immunity | Oberoi et al. (2016) |
| Modified liposomes (PEG-modified) | Ovalbumin (OVA) | Enhanced mucosal and systemic immunity | Minato et al. (2003) |
| Modified liposomes layer-by-layer polymer-coated liposomes | Peptide (J14) | Enhanced mucosal and systemic immunity | Marasini et al. (2016a, b, c) |
| Modified liposomes (Mannose-PEG-cholesterol conjugate (MPC) entrapped lipid A) | Ovalbumin (OVA) | Enhanced systemic and mucosal immunity | Wang et al. (2014) |
| Bilosomes | Tetanus toxoid | Enhanced mucosal and systemic immunity | Mann et al. (2006) |
| Bilosomes | Diphtheria toxoid (DTx) | Enhanced mucosal and systemic immunity | Shukla et al. (2011) |
| Bilosomes | Recombinant baculovirus displaying VP1 (Bac-VP1) | Enhanced mucosal and systemic immunity | Premanand et al. (2013) |
| Bilosomes | Tetanus toxoid | Enhanced mucosal and systemic immunity. Induced Th2-biased immune responses | Mann et al. (2006) |
| ISCOMs | Cholera toxin-CTA1-DD/ OVA323–339 peptide epitope | Enhanced antigen-specific serum IgG antibodies | Mowat et al. (2001) |
| Chitosan (CS) conjugated peptide | CPE30 peptide | Production of antigen-specific fecal SIgAand mucosal T cell immune responses | Ye et al. (2014) |
| Eudragit® L100-coated-mannosylated chitosan nanoparticles | Bovine serum albumin (BSA) | Enhanced mucosal and systemic immunity | Xu et al. (2018) |
| Alginate | Heat killed V. cholerae | Production of IgG and IgM responses and protective response against cholera | Pastor et al. (2013) |
| Alginate-encapsulated bacille Calmette-Guerin (BCG) | Bacille Calmette-Guerin (BCG) | Enhanced mucosal and systemic immunity | Hosseini et al. (2015) |
| Alginate and chitosan-coated calcium phosphate nanoparticles | Bovine serum albumin (BSA) | Enhanced mucosal and systemic immunity | Cao et al. (2020) |
| Glucan particles | Ovalbumin (OVA) | Enhanced mucosal immunity | De Smet et al. (2013) |
| Ulex europaeus agglutinin-1 (UEA-1) conjugated PLGA NP containing monophosphoryl lipid A | Ovalbumin (OVA) | Enhanced humoral immunity | Ma et al. (2014) |
| PLGA + monophosphoryl lipid A (MPLA) | Ovalbumin (OVA) | Enhanced mucosal and systemic immunity | Sarti et al. (2011) |
| Acid-resistant HP55/PLGA nanoparticle | H. pylori recombinant antigen CCF | Induced potentTh1/Th17-bias immune response | Tan et al. (2017) |
| Cell membrane coating technology. (Mg-TiO2-RBC-toxin-chitosan-entric coated particles) | Toxoids | Production of antigen-specific mucosal antibody titers | Wei et al. (2019) |
The primary hurdle in oral vaccine development is retaining antigen stability against the harsh conditions of the GI tract, enhancing antigen transport from the apical to basolateral side in the intestine and targeting antigens to underlying immune cells. The use of a novel nanotechnological platform provides a promising prospect for enhancing the immunogenicity of oral vaccines (Fig. 3). Formulation strategies include the use of novel lipid and polymer-based nano/microparticle platforms where antigens can be conjugated or encapsulated into particles and protected against the GI environment. The physicochemical properties can be tailored to enhance GI transport, prolong the release of antigens and attach ligands (M-cells and APC-ligands) to precisely allow site specific targeting by immune cells. Another novel way to enhance the immunogenicity is by linking TLR agonists with peptide or protein-based antigens.
Liposomes and chitosan-based delivery systems appear to be the most preferred delivery systems for oral vaccines. As these delivery systems have already progressed into the clinic for other modes of administration, there is high hope that they represent the “gold standard” for most antigens intended for oral delivery. Additionally, the development of new classs of pH-sensitive polymers and modification of the liposomes-based particles with those polymers provides an effective antigens protection and tailored release of antigens. The reports on optimum physicochemical properties of nano/microparticles for oral delivery of vaccines are conflicting. However, studies show that rod-shaped, nano/micron size (< 2 microns), cationic and hydrophobic properties are the preferred physicochemical properties for oral nano/microparticle-based vaccines. Further research and development of nanocarriers and vaccine design should ultimately lead to identifying the “gold standard” oral vaccine delivery system. This generic system is expected to provide an easy solution to roll out effective vaccination strategies in resource-limited developing countries.
Acknowledgements
NM was supported by a UQ development fellowship and internal funding from the School of Biomedical Science, The University of Queensland.
Declarations
Conflict of interest
All authors (M. Li, L.M. Kaminskas and N. Marasini) declare that they have no conflict of interest.
Research involving human and animal rights
This article does not contain any studies with human and animal subjects performed by any of the authors.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Apostólico JDS, VaS L, Coirada FC, Boscardin SB, Rosa DS. Adjuvants: classification, modus operandi, and licensing. J Immunol Res. 2016;2016:1459394–1459394. doi: 10.1155/2016/1459394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baik YO, Choi SK, Olveda RM, Espos RA, Ligsay AD, Montellano MB, Yeam JS, Yang JS, Park JY, Kim DR. A randomized, non-inferiority trial comparing two bivalent killed, whole cell, oral cholera vaccines (Euvichol vs Shanchol) in the Philippines. Vaccine. 2015;33:6360–6365. doi: 10.1016/j.vaccine.2015.08.075. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay AS, Garon J, Seib K, Orenstein WA. Polio vaccination: past, present and future. Future Microbiol. 2015;10:791–808. doi: 10.2217/fmb.15.19. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Qi J, Gogoi R, Wong J, Mitragotri S. Role of nanoparticle size, shape and surface chemistry in oral drug delivery. J Control Release. 2016;238:176–185. doi: 10.1016/j.jconrel.2016.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett S, Eichenberger RM, Nevagi RJ, Ghaffar KA, Marasini N, Dai Y, Loukas A, Toth I, Skwarczynski M. Lipopeptide-based oral vaccine against hookworm infection. J Infect Dis. 2020;221:934–942. doi: 10.1093/infdis/jiz528. [DOI] [PubMed] [Google Scholar]
- Benne N, Van Duijn J, Kuiper J, Jiskoot W, Slütter B. Orchestrating immune responses: How size, shape and rigidity affect the immunogenicity of particulate vaccines. J Control Release. 2016;234:124–134. doi: 10.1016/j.jconrel.2016.05.033. [DOI] [PubMed] [Google Scholar]
- Bernasconi V, Norling K, Bally M, Höök F, Lycke NY. Mucosal vaccine development based on liposome technology. J Immunol Res. 2016 doi: 10.1155/2016/5482087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaney JE, Wirblich C, Papaneri AB, Johnson RF, Myers CJ, Juelich TL, Holbrook MR, Freiberg AN, Bernbaum JG, Jahrling PB. Inactivated or live-attenuated bivalent vaccines that confer protection against rabies and Ebola viruses. J Virol. 2011;85:10605–10616. doi: 10.1128/JVI.00558-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayden DJ, Jepson MA, Baird AW. Keynote review: intestinal Peyer's patch M cells and oral vaccine targeting. Drug Discov Today. 2005;10:1145–1157. doi: 10.1016/S1359-6446(05)03536-1. [DOI] [PubMed] [Google Scholar]
- Bussio JI, Molina-Perea C, González-Aramundiz JV. Hyaluronic acid nanocapsules as a platform for needle-free vaccination. Pharmaceutics. 2019;11:246. doi: 10.3390/pharmaceutics11050246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao P, Han FY, Grøndahl L, Xu ZP, Li L. Enhanced oral vaccine efficacy of polysaccharide-coated calcium phosphate nanoparticles. ACS Omega. 2020;5:18185–18197. doi: 10.1021/acsomega.0c01792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang S-Y, Ko H-J, Kweon M-N. Mucosal dendritic cells shape mucosal immunity. Exp Mol Med. 2014;46:e84–e84. doi: 10.1038/emm.2014.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Fernando GJ, Crichton ML, Flaim C, Yukiko SR, Fairmaid EJ, Corbett HJ, Primiero CA, Ansaldo AB, Frazer IH. Improving the reach of vaccines to low-resource regions, with a needle-free vaccine delivery device and long-term thermostabilization. J Control Release. 2011;152:349–355. doi: 10.1016/j.jconrel.2011.02.026. [DOI] [PubMed] [Google Scholar]
- Chen WH, Cohen MB, Kirkpatrick BD, Brady RC, Galloway D, Gurwith M, Hall RH, Kessler RA, Lock M, Haney D. Single-dose live oral cholera vaccine CVD 103-HgR protects against human experimental infection with Vibrio cholerae O1 El Tor. Clin Infect Dis. 2016;62:1329–1335. doi: 10.1093/cid/ciw145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RA. Barrier properties of mucus. Adv Drug Deliv Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Corthésy B, Bioley G. Lipid-based particles: versatile delivery systems for mucosal vaccination against infection. Front Immunol. 2018;9:431. doi: 10.3389/fimmu.2018.00431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smet R, Demoor T, Verschuere S, Dullaers M, Ostroff GR, Leclercq G, Allais L, Pilette C, Dierendonck M, De Geest BG. β-Glucan microparticles are good candidates for mucosal antigen delivery in oral vaccination. J Control Release. 2013;172:671–678. doi: 10.1016/j.jconrel.2013.09.007. [DOI] [PubMed] [Google Scholar]
- De Smet R, Allais L, Cuvelier CA. Recent advances in oral vaccine development: yeast-derived β-glucan particles. Hum Vaccin Immunother. 2014;10:1309–1318. doi: 10.4161/hv.28166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delany I, Rappuoli R, De Gregorio E. Vaccines for the 21st century. EMBO Mol Med. 2014;6:708–720. doi: 10.1002/emmm.201403876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson G, Ladinsky M, Yu K, Sanders J, Yoo B, Chou W-C, Conner M, Earl A, Knight R, Bjorkman P. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science. 2018;360:795–800. doi: 10.1126/science.aaq0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9:1521–1532. doi: 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]
- Ekwueme DU, Weniger BG, Chen RT. Model-based estimates of risks of disease transmission and economic costs of seven injection devices in sub-Saharan Africa. Bull World Health Organ. 2002;80:859–870. [PMC free article] [PubMed] [Google Scholar]
- El-Sayed A, Kamel M. Advances in nanomedical applications: diagnostic, therapeutic, immunization, and vaccine production. Environ Sci Pollut Res. 2020;27:19200–19213. doi: 10.1007/s11356-019-06459-2. [DOI] [PubMed] [Google Scholar]
- Fahmy TM, Demento SL, Caplan MJ, Mellman I, Saltzman WM. Design opportunities for actively targeted nanoparticle vaccines. Nanomedicine. 2008 doi: 10.2217/17435889.3.3.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Sahdev P, Ochyl LJ, Akerberg JJ, Moon JJ. Cationic liposome–hyaluronic acid hybrid nanoparticles for intranasal vaccination with subunit antigens. J Control Release. 2015;208:121–129. doi: 10.1016/j.jconrel.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon DT. Seeking wisdom in innate immunity. Nature. 1997;388:323–324. doi: 10.1038/40967. [DOI] [PubMed] [Google Scholar]
- Gentschev I, Spreng S, Sieber H, Ures J, Mollet F, Collioud A, Pearman J, Griot-Wenk ME, Fensterle J, Rapp UR. Vivotif®–a ‘magic shield’for protection against typhoid fever and delivery of heterologous antigens. Chemotherapy. 2007;53:177–180. doi: 10.1159/000100515. [DOI] [PubMed] [Google Scholar]
- Grant J. Anatomy and physiology of the gastrointestinal tract. In: Matarese L, Steiger E, Seidner D, editors. Intestinal failure and rehabilitation: a clinical guide. Boca Raton: CRC Press; 2004. pp. 5–23. [Google Scholar]
- Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11:S45–S53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- Hosseini M, Dobakhti F, Pakzad S, Ajdary S. Immunization with single oral dose of alginate-encapsulated BCG elicits effective and long-lasting mucosal immune responses. Scand J Immunol. 2015;82:489–497. doi: 10.1111/sji.12351. [DOI] [PubMed] [Google Scholar]
- Hu K-F, Lövgren-Bengtsson K, Morein B. Immunostimulating complexes (ISCOMs) for nasal vaccination. Adv Drug Deliv Rev. 2001;51:149–159. doi: 10.1016/s0169-409x(01)00165-x. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Pang X, Liu M, Zhang B, Li L, Zhai G. Recent progresses in bioadhesive microspheres via transmucosal administration. Colloids Surf B. 2016;140:361–372. doi: 10.1016/j.colsurfb.2015.12.049. [DOI] [PubMed] [Google Scholar]
- Josefsberg JO, Buckland B. Vaccine process technology. Biotechnol Bioeng. 2012;109:1443–1460. doi: 10.1002/bit.24493. [DOI] [PubMed] [Google Scholar]
- Kompella UB, Lee VH. Delivery systems for penetration enhancement of peptide and protein drugs: design considerations. Adv Drug Deliv Rev. 2001;46:211–245. doi: 10.1016/s0169-409x(00)00137-x. [DOI] [PubMed] [Google Scholar]
- Koziolek M, Grimm M, Becker D, Iordanov V, Zou H, Shimizu J, Wanke C, Garbacz G, Weitschies W. Investigation of pH and temperature profiles in the GI tract of fasted human subjects using the Intellicap® system. J Pharm Sci. 2015;104:2855–2863. doi: 10.1002/jps.24274. [DOI] [PubMed] [Google Scholar]
- Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. Shape and size-dependent immune response to antigen-carrying nanoparticles. J Control Release. 2015;220:141–148. doi: 10.1016/j.jconrel.2015.09.069. [DOI] [PubMed] [Google Scholar]
- Kunisawa J, Kurashima Y, Kiyono H. Gut-associated lymphoid tissues for the development of oral vaccines. Adv Drug Deliv Rev. 2012;64:523–530. doi: 10.1016/j.addr.2011.07.003. [DOI] [PubMed] [Google Scholar]
- Lai SK, Wang Y-Y, Wirtz D, Hanes J. Micro-and macrorheology of mucus. Adv Drug Deliv Rev. 2009;61:86–100. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liljeqvist S, Ståhl S. Production of recombinant subunit vaccines: protein immunogens, live delivery systems and nucleic acid vaccines. J Biotechnol. 1999;73:1–33. doi: 10.1016/s0168-1656(99)00107-8. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu J, Wang B, Zeng S, Qi F, Lu C, Kimura Y, Liu B. Oral vaccination with a liposome-encapsulated influenza DNA vaccine protects mice against respiratory challenge infection. J Med Virol. 2014;86:886–894. doi: 10.1002/jmv.23768. [DOI] [PubMed] [Google Scholar]
- Lodaya RN, Brito LA, Wu TY, Miller AT, Otten GR, Singh M, O'hagan DT, Stable nanoemulsions for the delivery of small molecule immune potentiators. J Pharm Sci. 2018;107:2310–2314. doi: 10.1016/j.xphs.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Lövgren Bengtsson K, Morein B, Osterhaus AD. ISCOM technology-based Matrix MTM adjuvant: success in future vaccines relies on formulation. Expert Rev Vaccines. 2011;10:401–403. doi: 10.1586/erv.11.25. [DOI] [PubMed] [Google Scholar]
- Lovgren K, Morein B. The requirement of lipids for the formation of immunostimulating complexes (iscoms) Biotechnol Appl Biochem. 1988;10:161–172. [PubMed] [Google Scholar]
- Lucas ME, Deen JL, Von Seidlein L, Wang X-Y, Ampuero J, Puri M, Ali M, Ansaruzzaman M, Amos J, Macuamule A. Effectiveness of mass oral cholera vaccination in Beira, Mozambique. N Engl J Med. 2005;352:757–767. doi: 10.1056/NEJMoa043323. [DOI] [PubMed] [Google Scholar]
- Lycke N, Bemark M. Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol. 2010;3:556–566. doi: 10.1038/mi.2010.54. [DOI] [PubMed] [Google Scholar]
- Ma T, Wang L, Yang T, Ma G, Wang S. M-cell targeted polymeric lipid nanoparticles containing a toll-like receptor agonist to boost oral immunity. Int J Pharm. 2014;473:296–303. doi: 10.1016/j.ijpharm.2014.06.052. [DOI] [PubMed] [Google Scholar]
- Mann JF, Scales HE, Shakir E, Alexander J, Carter KC, Mullen AB, Ferro VA. Oral delivery of tetanus toxoid using vesicles containing bile salts (bilosomes) induces significant systemic and mucosal immunity. Methods. 2006;38:90–95. doi: 10.1016/j.ymeth.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Marasini N, Kaminskas LM. Subunit-based mucosal vaccine delivery systems for pulmonary delivery—are they feasible? Drug Dev Ind Pharm. 2019;45:882–894. doi: 10.1080/03639045.2019.1583758. [DOI] [PubMed] [Google Scholar]
- Marasini N, Skwarczynski M, Toth I. Oral delivery of nanoparticle-based vaccines. Expert Rev Vaccines. 2014;13:1361–1376. doi: 10.1586/14760584.2014.936852. [DOI] [PubMed] [Google Scholar]
- Marasini N, Giddam AK, Ghaffar KA, Batzloff MR, Good MF, Skwarczynski M, Toth I. Multilayer engineered nanoliposomes as a novel tool for oral delivery of lipopeptide-based vaccines against group A Streptococcus. Nanomedicine. 2016;11:1223–1236. doi: 10.2217/nnm.16.36. [DOI] [PubMed] [Google Scholar]
- Marasini N, Giddam AK, Khalil ZG, Hussein WM, Capon RJ, Batzloff MR, Good MF, Toth I, Skwarczynski M. Double adjuvanting strategy for peptide-based vaccines: trimethyl chitosan nanoparticles for lipopeptide delivery. Nanomedicine. 2016;11:3223–3235. doi: 10.2217/nnm-2016-0291. [DOI] [PubMed] [Google Scholar]
- Marasini N, Khalil ZG, Giddam AK, Ghaffar KA, Hussein WM, Capon RJ, Batzloff MR, Good MF, Skwarczynski M, Toth I. Lipid core peptide/poly(lactic-co-glycolic acid) as a highly potent intranasal vaccine delivery system against group A streptococcus. Int J Pharm. 2016;513:410–420. doi: 10.1016/j.ijpharm.2016.09.057. [DOI] [PubMed] [Google Scholar]
- Marasini N, Ghaffar KA, Skwarczynski M, Toth I. Micro and Nanotechnology In Vaccine Development. New York: William Andrew Publishing; 2017. pp. 221–239. [Google Scholar]
- Marasini N, Skwarczynski M, Toth I. Intranasal delivery of nanoparticle-based vaccines. Ther Deliv. 2017;8:151–167. doi: 10.4155/tde-2016-0068. [DOI] [PubMed] [Google Scholar]
- Meyer RA, Sunshine JC, Perica K, Kosmides AK, Aje K, Schneck JP, Green JJ. Biodegradable nanoellipsoidal artificial antigen presenting cells for antigen specific T-cell activation. Small. 2015;11:1519–1525. doi: 10.1002/smll.201402369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato S, Iwanaga K, Kakemi M, Yamashita S, Oku N. Application of polyethyleneglycol (PEG)-modified liposomes for oral vaccine: effect of lipid dose on systemic and mucosal immunity. J Control Release. 2003;89:189–197. doi: 10.1016/s0168-3659(03)00093-2. [DOI] [PubMed] [Google Scholar]
- Mo Z, Mo Y, Li M, Tao J, Yang X, Kong J, Wei D, Fu B, Liao X, Chu J. Efficacy and safety of a pentavalent live human-bovine reassortant rotavirus vaccine (RV5) in healthy Chinese infants: a randomized, double-blind, placebo-controlled trial. Vaccine. 2017;35:5897–5904. doi: 10.1016/j.vaccine.2017.08.081. [DOI] [PubMed] [Google Scholar]
- Mowat AM, Donachie AM, Jägewall S, Schön K, Löwenadler B, Dalsgaard K, Kaastrup P, Lycke N. CTA1-DD-immune stimulating complexes: a novel, rationally designed combined mucosal vaccine adjuvant effective with nanogram doses of antigen. J Immunol. 2001;167:3398–3405. doi: 10.4049/jimmunol.167.6.3398. [DOI] [PubMed] [Google Scholar]
- Mudie DM, Amidon GL, Amidon GE. Physiological parameters for oral delivery and in vitro testing. Mol Pharm. 2010;7:1388–1405. doi: 10.1021/mp100149j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummert ME. Immunologic roles of hyaluronan. Immunol Res. 2005;31:189–205. doi: 10.1385/IR:31:3:189. [DOI] [PubMed] [Google Scholar]
- Nashchekina YA, Raydan M. Noninvasive penetration of 5 nm hyaluronic acid molecules across the epidermal barrier (in vitro) and its interaction with human skin cells. Skin Res Technol. 2018;24:129–134. doi: 10.1111/srt.12400. [DOI] [PubMed] [Google Scholar]
- Naz RK, Dabir P. Peptide vaccines against cancer, infectious diseases, and conception. Front Biosci. 2007;12:1833–1844. doi: 10.2741/2191. [DOI] [PubMed] [Google Scholar]
- New RRC. Formulation technologies for oral vaccines. Clin Exp Immunol. 2019;198:153–169. doi: 10.1111/cei.13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohagan DT, Singh M. Microparticles as vaccine adjuvants and delivery systems. Expert Rev Vaccines. 2003;2:269–283. doi: 10.1586/14760584.2.2.269. [DOI] [PubMed] [Google Scholar]
- Oberoi HS, Yorgensen YM, Morasse A, Evans JT, Burkhart DJ. PEG modified liposomes containing CRX-601 adjuvant in combination with methylglycol chitosan enhance the murine sublingual immune response to influenza vaccination. J Control Release. 2016;223:64–74. doi: 10.1016/j.jconrel.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Őrfi E, Szebeni J. The immune system of the gut and potential adverse effects of oral nanocarriers on its function. Adv Drug Deliv Rev. 2016;106:402–409. doi: 10.1016/j.addr.2016.09.009. [DOI] [PubMed] [Google Scholar]
- Özel M, Hℷlund S, Gelderblom HR, Morein B. Quaternary structure of the immunostimulating complex (iscom) J Ultrastruct Mol Struct Res. 1989;102:240–248. doi: 10.1016/0889-1605(89)90018-9. [DOI] [PubMed] [Google Scholar]
- Pabst O, Mowat A. Oral tolerance to food protein. Mucosal Immunol. 2012;5:232–239. doi: 10.1038/mi.2012.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor M, Esquisabel A, Talavera A, Fernández S, Cedré B, Infante J, Callicó A, Pedraz J. An approach to a cold chain free oral cholera vaccine: in vitro and in vivo characterization of Vibrio cholerae gastro-resistant microparticles. Int J Pharm. 2013;448:247–258. doi: 10.1016/j.ijpharm.2013.02.057. [DOI] [PubMed] [Google Scholar]
- Pelaseyed T, Bergström JH, Gustafsson JK, Ermund A, Birchenough GM, Schütte A, Van Der Post S, Svensson F, Rodríguez-Piñeiro AM, Nyström EE. The mucus and mucins of the goblet cells and enterocytes provide the first defense line of the gastrointestinal tract and interact with the immune system. Immunol Rev. 2014;260:8–20. doi: 10.1111/imr.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premanand B, Prabakaran M, Kiener TK, Kwang J. Recombinant baculovirus associated with bilosomes as an oral vaccine candidate against HEV71 infection in mice. PLoS ONE. 2013;8:e55536. doi: 10.1371/journal.pone.0055536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez JEV, Sharpe LA, Peppas NA. Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev. 2017;114:116–131. doi: 10.1016/j.addr.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- Sarti F, Perera G, Hintzen F, Kotti K, Karageorgiou V, Kammona O, Kiparissides C, Bernkop-Schnürch A. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials. 2011;32:4052–4057. doi: 10.1016/j.biomaterials.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Shah RR, Hassett KJ, Brito LA. Overview of vaccine adjuvants: introduction, history, and current status. Methods Mol Biol. 2017;1494:1–13. doi: 10.1007/978-1-4939-6445-1_1. [DOI] [PubMed] [Google Scholar]
- Shukla A, Bhatia A, Amarji B, Singh B, Katare O, Vyas S. Nano-bilosomes as potential vaccine delivery system for effective combined oral immunization against tetanus and hepatitis B. J Biotechnol. 2010;150:98–99. [Google Scholar]
- Shukla A, Singh B, Katare O. Significant systemic and mucosal immune response induced on oral delivery of diphtheria toxoid using nano-bilosomes. Br J Pharmacol. 2011;164:820–827. doi: 10.1111/j.1476-5381.2011.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist C-A. Vaccine immunology. Vaccines. 2008;5:17–36. [Google Scholar]
- Silva A, Soema P, Slütter B, Ossendorp F, Jiskoot W. PLGA particulate delivery systems for subunit vaccines: linking particle properties to immunogenicity. Hum Vaccin Immunother. 2016;12:1056–1069. doi: 10.1080/21645515.2015.1117714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Briones M, O’Hagan DT. A novel bioadhesive intranasal delivery system for inactivated influenza vaccines. J Control Release. 2001;70:267–276. doi: 10.1016/s0168-3659(00)00330-8. [DOI] [PubMed] [Google Scholar]
- Singh Y, Meher JG, Raval K, Khan FA, Chaurasia M, Jain NK, Chourasia MK. Nanoemulsion: concepts, development and applications in drug delivery. J Control Release. 2017;252:28–49. doi: 10.1016/j.jconrel.2017.03.008. [DOI] [PubMed] [Google Scholar]
- Smith J, Wood E, Dornish M. Effect of chitosan on epithelial cell tight junctions. Pharm Res. 2004;21:43–49. doi: 10.1023/b:pham.0000012150.60180.e3. [DOI] [PubMed] [Google Scholar]
- Sosnik A. Alginate particles as platform for drug delivery by the oral route: state-of-the-art. ISRN Pharm. 2014 doi: 10.1155/2014/926157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto ER, Ostroff GR. Characterization of multilayered nanoparticles encapsulated in yeast cell wall particles for DNA delivery. Bioconjug Chem. 2008;19:840–848. doi: 10.1021/bc700329p. [DOI] [PubMed] [Google Scholar]
- Steele AD, Neuzil KM, Cunliffe NA, Madhi SA, Bos P, Ngwira B, Witte D, Todd S, Louw C, Kirsten M. Human rotavirus vaccine RotarixTM provides protection against diverse circulating rotavirus strains in African infants: a randomized controlled trial. BMC Infect Dis. 2012;12:1–8. doi: 10.1186/1471-2334-12-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudarshan N, Hoover D, Knorr D. Antibacterial action of chitosan. Food Biotechnol. 1992;6:257–272. [Google Scholar]
- Sur D, Lopez AL, Kanungo S, Paisley A, Manna B, Ali M, Niyogi SK, Park JK, Sarkar B, Puri MK. Efficacy and safety of a modified killed-whole-cell oral cholera vaccine in India: an interim analysis of a cluster-randomised, double-blind, placebo-controlled trial. Lancet. 2009;374:1694–1702. doi: 10.1016/S0140-6736(09)61297-6. [DOI] [PubMed] [Google Scholar]
- Tan Z, Liu W, Liu H, Li C, Zhang Y, Meng X, Tang T, Xi T, Xing Y. Oral Helicobacter pylori vaccine-encapsulated acid-resistant HP55/PLGA nanoparticles promote immune protection. Eur J Pharm Biopharm. 2017;111:33–43. doi: 10.1016/j.ejpb.2016.11.007. [DOI] [PubMed] [Google Scholar]
- Torres-Sangiao E, Holban AM, Gestal MC. Advanced nanobiomaterials: vaccines, diagnosis and treatment of infectious diseases. Molecules. 2016;21:867. doi: 10.3390/molecules21070867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AK, Sharma S, Gupta P, Singodia D, Kansal S, Sharma V, Mishra PR. Vitamin B12 grafted layer-by-layer liposomes bearing HBsAg facilitate oral immunization: effect of modulated biomechanical properties. Mol Pharm. 2016;13:2531–2542. doi: 10.1021/acs.molpharmaceut.6b00274. [DOI] [PubMed] [Google Scholar]
- Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, Zganiacz A, Hitt M, Xing Z. Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. J Immunol. 2004;173:6357–6365. doi: 10.4049/jimmunol.173.10.6357. [DOI] [PubMed] [Google Scholar]
- Wang N, Wang T, Zhang M, Chen R, Niu R, Deng Y. Mannose derivative and lipid A dually decorated cationic liposomes as an effective cold chain free oral mucosal vaccine adjuvant-delivery system. Eur J Pharm Biopharm. 2014;88:194–206. doi: 10.1016/j.ejpb.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Watts P, Smith A, Hinchcliffe M. Mucosal delivery of biopharmaceuticals. In: Neves JD, Sarmento B, editors. Biology, challenges and strategies. New York: Springer; 2014. pp. 499–516. [Google Scholar]
- Webster DE, Gahan ME, Strugnell RA, Wesselingh SL. Advances in oral vaccine delivery options. Am J Drug Deliv. 2003;1:227–240. [Google Scholar]
- Wei X, Beltrán-Gastélum M, Karshalev E, Esteban-Fernández De Ávila B, Zhou J, Ran D, Angsantikul P, Fang RH, Wang J, Zhang L. Biomimetic micromotor enables active delivery of antigens for oral vaccination. Nano Lett. 2019;19:1914–1921. doi: 10.1021/acs.nanolett.8b05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Waltman A, Han H, Collier JH. Switching the immunogenicity of peptide assemblies using surface properties. ACS Nano. 2016;10:9274–9286. doi: 10.1021/acsnano.6b03409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten J, Samad T, Ribbeck K. Selective permeability of mucus barriers. Curr Opin Biotechnol. 2018;52:124–133. doi: 10.1016/j.copbio.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Zhao H, Li M, Yue Y, Xiong S, Xu W. Intranasal vaccination with mannosylated chitosan formulated DNA vaccine enables robust IgA and cellular response induction in the lungs of mice and improves protection against pulmonary mycobacterial challenge. Front Cell Infect Microbiol. 2017;7:445. doi: 10.3389/fcimb.2017.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Ensign LM, Boylan NJ, SchöN A, Gong X, Yang J-C, Lamb NW, Cai S, Yu T, Freire E. Impact of surface polyethylene glycol (PEG) density on biodegradable nanoparticle transport in mucus ex vivo and distribution in vivo. ACS Nano. 2015;9:9217–9227. doi: 10.1021/acsnano.5b03876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zhang W, Chen Y, Xu Y, Wang B, Zong L. Eudragit® L100-coated mannosylated chitosan nanoparticles for oral protein vaccine delivery. Int J Biol Macromol. 2018;113:534–542. doi: 10.1016/j.ijbiomac.2018.02.016. [DOI] [PubMed] [Google Scholar]
- Yadav DK, Yadav N, Khurana SMP. Animal biotechnology. Amsterdam: Elsevier; 2014. pp. 491–508. [Google Scholar]
- Ye T, Yue Y, Fan X, Dong C, Xu W, Xiong S. M cell-targeting strategy facilitates mucosal immune response and enhances protection against CVB3-induced viral myocarditis elicited by chitosan-DNA vaccine. Vaccine. 2014;32:4457–4465. doi: 10.1016/j.vaccine.2014.06.050. [DOI] [PubMed] [Google Scholar]
- Zinkernagel RM, Ehl S, Aichele P, Oehen S, Kündig T, Hengartner H. Antigen localisation regulates immune responses in a dose-and time-dependent fashion: a geographical view of immune reactivity. Immunol Rev. 1997;156:199–209. doi: 10.1111/j.1600-065x.1997.tb00969.x. [DOI] [PubMed] [Google Scholar]